
Bioinspired peptide/polyamino acid assemblies as quorum sensing inhibitors for the treatment of bacterial infections
Yanan
Jiang
a,
Fanying
Meng
b,
Zhenghong
Ge
 a,
Yuxiao
Zhou
*b,
Zhen
Fan
*ab and
Jianzhong
Du
*abc
a,
Yuxiao
Zhou
*b,
Zhen
Fan
*ab and
Jianzhong
Du
*abc
aSchool of Materials Science and Engineering, Tongji University, Shanghai 201804, China
bDepartment of Gynaecology and Obstetrics, Shanghai Key Laboratory of Anesthesiology and Brain Functional Modulation, Clinical Research Center for Anesthesiology and Perioperative Medicine, Translational Research Institute of Brain and Brain-Like Intelligence, Shanghai Fourth People's Hospital, School of Medicine, Tongji University, Shanghai 200434, China
cSchool of Materials Science and Engineering, East China University of Science and Technology, Shanghai 200237, China
First published on 16th October 2024
Abstract
Insufficient development of new antibiotics and the rise in antimicrobial resistance are putting the world at risk of losing curative medicines against bacterial infection. Quorum sensing is a type of cellular signaling for cell-to-cell communication that plays critical roles in biofilm formation and antimicrobial resistance, and is expected to be a new type of effective target for drug resistant bacteria. In this review we highlight recent advances in bioinspired peptide/polyamino acid assemblies as quorum sensing inhibitors across various microbial communities. In addition, existing obstacles and future development directions of peptide/polyamino acid assemblies as quorum sensing inhibitors were proposed for broader clinical applications and translations. Overall, quorum sensing peptide/polyamino acid assemblies could be vital tools against bacterial infection and antimicrobial resistance.
1. Introduction
1.1 Bacterial infection
Bacterial infections resulting from surgery, trauma, or congenital diseases are a significant concern and a leading cause of high mortality.1 Bacteria can invade the body through wounds and induce bacteremia or sepsis resulting in multi-organ failure and death.2 Bacterial infections not only pose a serious health threat to patients but also impose a substantial medical burden. For instance, in Europe, patients with wound infections occupy a large number of emergency beds, and the annual cost of treating these infections in the UK reaches billions of pounds.3,4 The most effective method for treating wound infections remains the use of antimicrobial agents. These agents typically inhibit or kill most microorganisms; however, a small number of bacteria with inherent or acquired resistance can survive and proliferate. Unfortunately, the overuse of antimicrobials and non-compliance with prescribed treatment regimens have led to the emergence of antimicrobial resistance (AMR).5,6 Consequently, even with the preemptive use of antimicrobials, infection rates for wounds such as open fractures and soft tissue injuries remain as high as 24.15–37%.71.2 Antimicrobial resistance
Antimicrobial drugs, including antibiotics, antiviral drugs, antifungal agents, and antiparasitic medications, are essential for preventing and treating infections in humans, animals, and plants.8 However, AMR has been a significant global health challenge, leading to high morbidity and mortality rates. AMR occurs when bacteria, viruses, fungi, and parasites change over time and no longer respond to medications, making infections harder to treat and increasing the risk of disease spread, severe illness, and death.9,10 The increasing prevalence of drug-resistant pathogens has diminished the effectiveness of antibiotics and other antimicrobial agents, making infections more challenging or even impossible to treat.11 Currently, resistance has been detected in all antibiotics used in clinical practice, with only a few new drugs currently in development. The emergence and spread of resistant pathogens, especially those with new resistance mechanisms, continue to threaten our ability to treat common infections.12,13 Of particular concern is the rapid global spread of multi-resistant and pan-resistant bacteria, often referred to as “superbugs,” which cause infections that are not treatable with existing antimicrobial drugs.14 Most antibiotics work through selective toxicity, binding with high affinity for critical bacterial cell targets, inhibiting essential cellular functions, and leading to growth inhibition or cell death.15 If these primary targets are altered or protected through chemical modifications, antibiotic binding becomes inefficient, resulting in resistance.16 AMR refers to the ability of microorganisms to withstand the effects of various antimicrobial agents. Once AMR develops, drugs that once killed or inhibited the microorganisms lose their efficacy. AMR poses a severe global threat to human health, particularly in surgical procedures, chronic wound infections, and organ transplantation.17 A study from 2022 estimated that nearly 5 million deaths per year are attributed to resistant bacteria, with the burden being more significant in low- and middle-income countries.18 Understanding the mechanisms of AMR is crucial for developing new antimicrobial drugs.191.3 Quorum sensing (QS)
Bacterial biofilms are found in the majority of chronic, hard-to-heal wounds compared to normal wounds and are considered a major factor in delayed wound healing.20,21 Compared to planktonic bacteria, bacteria within biofilms exhibit a resistance to antibiotics that is increased by 10 to 1000 times.22 Abnormal microenvironments in biofilms, such as nutrient deficiencies, production of reactive oxygen species, and low pH, can lead to altered metabolic pathways, resulting in the development of drug resistance in bacteria.23 Simultaneously, QS in biofilms allows bacteria to communicate with each other by secreting signal molecules.24,25 This communication mechanism can further regulate drug resistance mechanisms such as efflux pumps and bacterial secretion systems, facilitating the exchange of information and the sharing of resistance genes among bacteria to promote AMR.26 This process enhances the overall drug resistance of bacteria within the biofilm.271.4 Quorum sensing inhibitors (QSIs)
Given the essential role of QS in biofilm formation and drug resistance, targeting QS in bacteria is anticipated to effectively inhibit and remove biofilms from drug-resistant bacteria.28–30 Over the past twenty years, extensive research has been conducted on bacterial intercellular communication, and many excellent research papers have been published.31 Several distinct signaling systems have already been identified, and it is highly promising that more QS-related monomers and polymers will be identified in the future.32 Small, diffusible signaling molecules produced by bacteria, which play a crucial role in population sensing, are known as autoinducers.33 Autoinducers allow bacteria to monitor changes in their population density and coordinate their activities by regulating gene expression.34 When the concentration of an autoinducer surpasses a specific threshold, it prompts the bacteria to either activate or repress the expression of certain genes.35 This regulation facilitates coordinated behaviors within the bacterial community, such as biofilm formation, production of virulence factors, synthesis of antibiotics, and fluorescein production.36,37 There are several major types of autoinducers: (i) acyl homoserine lactones (AHLs): used by many Gram-negative bacteria to regulate processes such as biofilm formation and virulence.38 (ii) Autoinducing peptides (AIPs): used by Gram-positive bacteria to control activities like sporulation and virulence factor production.39 (iii) Autoinducer-2 (AI-2): a universal signaling molecule used by both Gram-negative and Gram-positive bacteria for interspecies communication.40 (iv) Diffusible signal factors (DSFs): fatty acid derivatives synthesized by certain Gram-negative bacteria.41,42QSIs are compounds that disrupt bacterial communication systems by interfering with or blocking QS mechanisms. By preventing the synthesis, release, or perception of autoinducers, QSIs inhibit bacterial collective behaviors, thereby controlling their virulence and drug resistance.43–45 The current range of QSIs is diverse, including small molecule compounds, natural products, synthetic peptides, enzyme inhibitors, and antibodies or nucleic acid aptamers. Many QSIs are designed to block signaling from autoinducers by mimicking their structures to competitively bind to QS receptors. For example, AHL mimetics are QSIs that imitate the structure of AHL molecules, allowing them to competitively bind to AHL receptors and inhibit QS signaling.46 Similarly, AIP mimics can interfere with the QS systems of Gram-positive bacteria by imitating the structure of AIPs.47,48 QSIs combat AMR through various mechanisms, including blocking biofilm formation, reducing the production of virulence factors, inhibiting horizontal gene transfer, enhancing the effects of conventional antimicrobial drugs, and developing novel therapeutic strategies.32 With ongoing research and advancements in applied technologies, QSIs are expected to become crucial tools in the fight against bacterial resistance, offering new solutions for public health.24
1.5 Quorum sensing peptides (QSPs)
QSPs are short chains of amino acids that bacteria produce and secrete.49 These peptides can be either simple linear forms or more complex cyclic structures. They play a crucial role in altering gene expression by binding to specific receptors on the bacterial cell surface or within the cell, thus initiating signal transduction cascades.50In bacteria, QSPs are synthesized as precursor proteins within the cytoplasm and subsequently processed into their mature forms. For example, competence-stimulating peptides in Streptococcus pneumoniae (S. pneumoniae) are first produced as longer precursor proteins, which are then cleaved to generate active signaling molecules.51 Once released, these peptides accumulate in the environment.52,53 When their concentration reaches a threshold level, they are detected by sensor proteins or receptor kinases, triggering downstream reactions. For instance, in Staphylococcus aureus (S. aureus), the AgrD peptide binds to AgrC receptors, leading to the activation of virulence gene expression.54,55 In this review we will delve into the interactions of various QSPs within different microbial communities and how these interactions can inform the development of novel antimicrobial therapies to treat bacterial infections.
1.6 Peptide self-assembly
A peptide usually suffers from low in vivo stability and bioavailability, which limits its broader biomedical applications. One practical approach to combating this is to construct self-assembly nanoparticles with a peptide monomer as the building block to increase its in vivo stability. Peptide self-assembly is the process in which peptides spontaneously organize into nanostructures with well-defined architectures through non-covalent interactions.56,57 This phenomenon plays a crucial role in various biological processes and has significant applications in nanotechnology, materials science, and medicine.58 Peptides typically assemble via hydrogen bonds between backbone amides, resulting in secondary structures such as α-helices and β-sheets.59 Hydrophobic amino acid residues tend to aggregate, minimizing their contact with water, thereby driving the formation of stable structures like micelles and vesicles.60 Charged amino acid residues contribute to the overall structure and stability of the self-assembly through electrostatic attractions or repulsions.61 Linear peptides can form diverse structures, including fibers and sheets, while cyclic peptides often form more rigid and stable rings or tubes.62 Amphiphilic peptides, containing both hydrophilic and hydrophobic regions, can assemble into various structures, such as micelles and bilayers, in aqueous environments.63–65Peptide assemblies offer significant advantages over monomeric peptides, making them valuable in various fields.66 They exhibit enhanced stability due to non-covalent interactions, increasing resistance to degradation and extending half-life in biological environments. Peptide assemblies provide improved functional diversity, allowing the combination of different peptides to form multifunctional nanostructures for drug delivery, antimicrobial therapy, and tissue engineering.67,68 Their size and shape can be precisely controlled, creating structures with specific properties. Multivalency enhances biological activity, improving target affinity and efficacy. Additionally, they achieve specific targeting and efficient delivery, feature controllable release profiles, and are biocompatible and biodegradable, ensuring safe degradation and clearance in the body.69
In this review, we summarize innovative approaches to combat bacterial infections using peptides and polyamino acid assemblies as QSIs. As QS plays critical roles in biofim formation and AMR,24 interfering with QS with inhibitors could inhibit biofilm formation, virulence factor production, and antibiotic resistance. By targeting QS, the metabolism and communication of bacteria can be disturbed and made more susceptible to conventional treatments. This paper provides an overview of recent advances in the development of peptide/polyamino acid-based QSIs (Fig. 1). Various strategies for designing these peptides or polyamino acid assemblies are included, including the incorporation of functional groups to enhance their binding affinity toward QS receptors and the use of mimetic sequences that resemble natural QS molecules. Synthetic methods such as solid-phase peptide synthesis and self-assembly techniques are also detailed, highlighting their roles in creating effective QSIs.70 Additionally, existing studies demonstrate the significant effects of peptide/multi-amino acid assemblies in inhibiting the QS activity of pathogenic bacteria. These studies illustrate the practical application of QSIs in reducing biofilm formation and virulence, with examples in S. aureus and S. pneumoniae.71,72 Moreover, existing obstacles and future perspectives of quorum sensing peptides or polyamino acid assemblies are also proposed for broader clinical applications and translations.
 | ||
| Fig. 1 Schematic diagram of bioinspired peptide/polyamino acid assemblies as quorum sensing inhibitors for the treatment of bacterial infections. | ||
2. QS signal transduction pathways
2.1 Two-component signal transduction system (TCSTS)
Gram-positive bacteria utilize several distinct signaling pathways based on peptide signaling molecules and receptor characteristics.73 Some species employ multiple signaling pathways to achieve QS effects. TCSTS is one of the most extensively studied circuit types and is found in numerous bacterial species, both Gram-positive and Gram-negative.74 Among Gram-positive bacteria, this system was identified in S. pneumoniae (Com system).75 It has also been discovered in S. aureus (Agr system) and Enterococcus faecalis (Fsr system).76 The system comprises two primary components: the sensing kinase and the response regulator.77 The sensing kinase is a membrane-bound protein that detects specific environmental stimuli and is divided into three parts: the extracellular domain which senses external signals, the transmembrane domain, and the intracellular kinase domain.78 The response regulator features a receiver domain that accepts phosphoryl groups from the sensing kinase and an output domain that mediates the cellular response by regulating gene expression through DNA binding.73 When a specific stimulus is detected, the sensing kinase autophosphorylates on conserved histidine residues in its kinase domain.79 This phosphate group is subsequently transferred to a conserved aspartic acid residue on the response regulator, leading to its activation.80 Activated response regulators can then alter gene expression or other cellular activities by binding to DNA or interacting with other cellular components, enabling the cell to adjust to the environmental changes detected by the kinase.2.2 Rgg/Rap, RNPP, NprR, PlcR, PrgX (RRNPP)
The RRNPP pathway is a prevalent intercellular communication and regulatory system found in Gram-positive bacteria.81 It controls bacterial group behaviors and physiological functions using small peptide signaling molecules and their corresponding receptor proteins.82 The name “RRNPP” derives from several representative family members: Rgg, Rap, NprR, PrgX, and Phr.83 In contrast to TCSTS, small peptide signaling molecules in the RRNPP pathway are secreted outside the cell.84 They accumulate to a specific concentration and are then either reabsorbed by the cell or directly interact with intracellular receptor proteins.85 The receptor protein binds to the signal peptide and directly regulates gene expression.86 The RRNPP pathway and the TCSTS both play crucial roles in bacterial signal transduction and the regulation of group behaviors. Each system exhibits distinct mechanisms of signal detection and transmission, reflecting their unique roles in bacterial adaptation and survival strategies.3. Peptide-based QS system
3.1 Agr QS system
The accessory gene regulator (agr) QS system is the principal regulatory mechanism linked to staphylococcal virulence, playing a crucial role in pathogenesis.93 When the agr-system is activated, it leads to corresponding changes in gene expression and the production of several virulence factors, including nucleases, lipases, and exoenzymes. The agr-system includes two separate transcripts, RNAII and RNAIII, which are each regulated by the P2 and P3 promoters, respectively.94
RNAII is actually a transcript composed of four genes: agrD, agrB, agrC, and agrA, which encode the main functional proteins of the agr system (Fig. 2).71 The agrD transcript encodes AgrD, a brief polypeptide referred to as the precursor autosensing peptide (pro-AIP). AgrB plays a crucial role in the functionality of the QS system of S. aureus by transforming the pro-AIP into its active form, known as the mature AIP. This protein, AgrB, is embedded in the bacterial cell membrane, where it not only processes pro-AIP but also facilitates its export from the cell.95 Upon the attachment of mature AIP to the AgrC receptor on a nearby bacterial cell, AgrB first executes the cleavage at the C-terminus of AgrD. Subsequently, it facilitates the thioesterification process between the C-terminal carboxylate and the thiol group of a conserved cysteine, positioned four amino acids from the C-terminus. In the final step, the type I signal peptidase SpsB carries out the N-terminal cleavage of AgrD, enabling the release of an active AIP from the cell surface.96
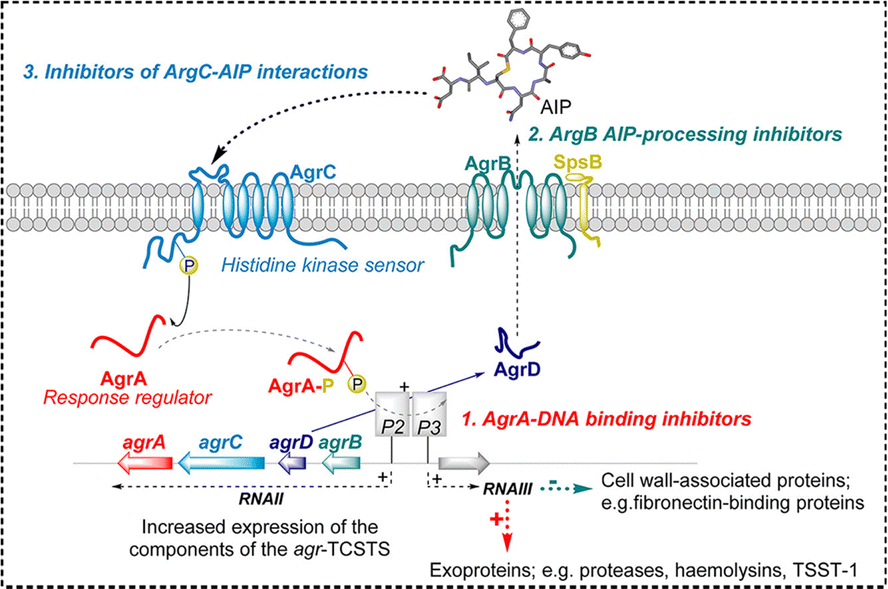 | ||
| Fig. 2 A schematic representation of the Agr quorum sensing circuit in Staphylococcus aureus.71 Reproduced from ref. 71 with permission from American Chemical Society, copyright 2020. | ||
The P3 transcript directly generates RNAIII, a crucial regulatory RNA in S. aureus crucial for expressing its virulence.97 Activated by the P3 promoter within the agr system, RNAIII orchestrates the bacterium's virulence traits through several mechanisms, notably by regulating the expression of various virulence genes. Beyond its regulatory functions, RNAIII also codes for δ-hemolysin, a cytotoxic peptide, amplifying its significance in the bacterial pathogenicity.
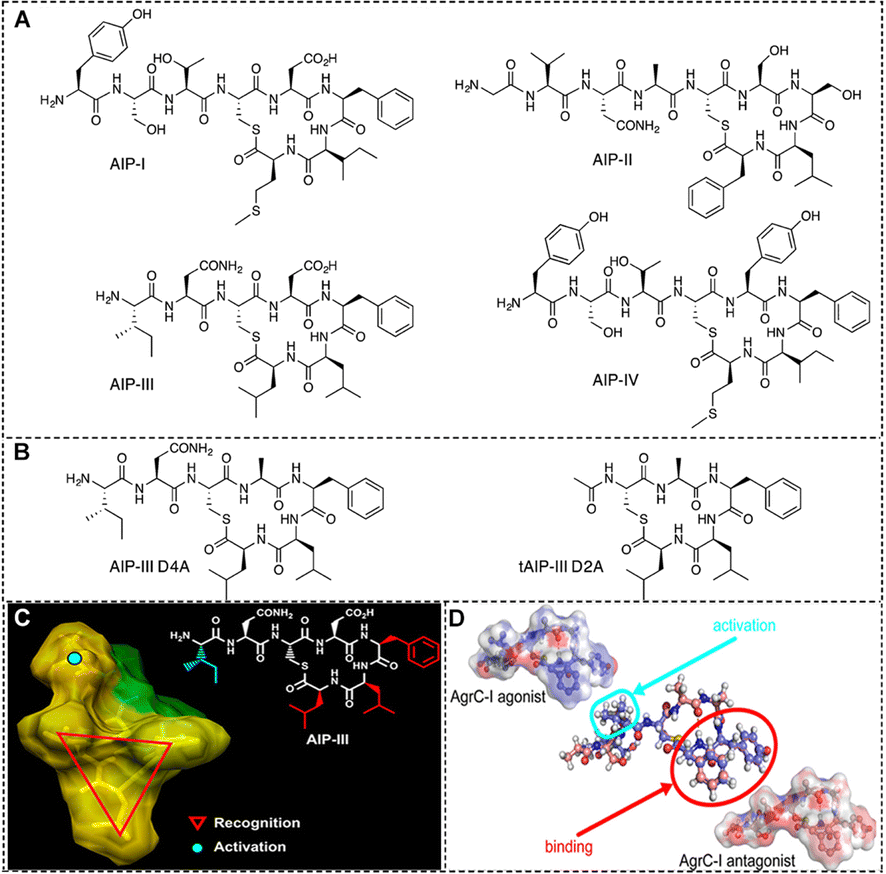 | ||
| Fig. 3 (A) Structures of the native AIPs (I–IV) and (B) two representative AIP-III analogues used by S. aureus for QS.100 Reproduced from ref. 100 with permission from American Chemical Society, copyright 2013. (C) A tri-residue hydrophobic “knob” essential for both activation and inhibition and a fourth anchor point on the exocyclic tail needed for receptor activation.100 Reproduced from ref. 100 with permission from American Chemical Society, copyright 2013. (D) Activation of AgrC-I requires the presence and specific orientation of at least two hydrophobic residues within the internal loop as well as a hydrophobic group at the C-terminus.98 Reproduced from ref. 98 with permission from American Chemical Society, copyright 2020. | ||
Inhibiting AIP:AgrC interactions offers a direct approach to disrupt S. aureus QS, effectively diminishing bacterial virulence. Over the past two decades, non-natural peptides and small molecules designed to inhibit AgrC have been the focus of extensive research.101 To date, four distinct AIP pairs have been characterized, leading to the classification of S. aureus into four different specificity groups (I–IV) (Fig. 3(A)).100 Additionally, the researchers discovered that each of the four natural AIPs could cross-inhibit the other three non-homologous AgrC receptors.
Initially, groups I and II were the most prevalent in human S. aureus infections, leading most efforts to develop modulators for disrupting AIP:AgrC interactions to focus on AIP-I and AIP-II signaling.102 By examining the synthetic peptides, it was observed that the synthetic thiolactone cyclic peptides from S. aureus groups I and II activated their respective homologous agr systems while inhibiting the non-homologous systems. This confirms the necessity of thioester macrocycles in maintaining the functional integrity of the peptides.
Recent research has revealed that the presence of group III S. aureus in infections is more common than previously thought. This finding suggests that the distribution of S. aureus groups in clinical settings may be shifting, necessitating a broader focus in the development of modulators for AIP III:AgrC interactions. Blackwell's group identifies AIP-III as an excellent peptide scaffold for designing AgrC inhibitors.100 The structure activity relationship (SAR) of the AIP-III peptide identifies two main features in its backbone: (1) a thioester macrocycle formed by the cysteine sulfhydryl group and the C-terminal end of the peptide, along with a unique hydrophobic triangular ‘knob’ comprising the three inner-ring side chains (Phe5, Leu6, and Leu7); and (2) a short chain of 2–4 amino acids at the N-terminal end of the peptide. Subsequently, they replaced thioester bonds with amide bonds to create amide-linked AIP analogues, significantly enhancing their hydrolytic stability and solubility in aqueous media (Fig. 3(B) and (C)).100
Most peptide-based QSIs are obtained using solid-phase peptide synthesis (SPPS).103,104 The basic approach of SPPS involves attaching the C-terminal amino acid of the peptide to a solid-phase resin, followed by a series of protection and deprotection, coupling, and washing steps. Each amino acid monomer is sequentially coupled from the C-terminus to the N-terminus, until the desired peptide chain is synthesized. The entire process can be automated, significantly improving the efficiency of peptide synthesis.105
Blackwell et al. further developed a solid-phase synthesis method for AIP amide analogues, facilitating experiments to evaluate their hydrolytic stability and antimicrobial properties.106 The synthesis of straight-chain peptides was accomplished through a standard Fmoc SPPS technique. This method employed 2-chlorotrityl chloride resin as the support, Fmoc-protected amino acids as the building blocks, and 1-hydroxybenzotriazole/N,N′-diisopropylcarbodiimide as the coupling agents. Instead of using cysteine, L-diaminopropionic acid was utilized on the resin for the synthesis. Once the straight-chain peptides were synthesized on the resin, AIP peptide analogues were prepared in the liquid phase. The cyclization was achieved using benzotriazol-1-yl-oxytripyrrolidinium hexafluorophosphate and N,N-diisopropylethylamine. Following cyclization, trifluoroacetic acid was employed to remove the side-chain protecting groups, resulting in the formation of the AIP peptide analogues.
Currently, machine learning and computational analysis have been developed as suitable tools for identifying target peptides and assessing the likelihood of a given sequence being an AIP.107 The Quorumpeps database is currently the only available experimental QSP database. Each peptide entry contains detailed annotation information, including the source species, sequence information, functional description, and detailed information about the associated QS system.108 QSPpred is the pioneering web server for the prediction and design of QSPs, with the majority of its peptide data sourced from the Quorumpeps database. QSPpred is an accessible tool for identifying targetable AIPs in S. aureus. It also highlights key motifs, structural domains, and amino acids within these AIPs that can be used as primary targets.109
AIP signaling presents a promising basis for developing tools to investigate Gram-positive QS and for designing potential therapeutic compounds. AIPs work by interacting with membrane-bound proteins, meaning AIP analogs can exert their effects without entering the cell. This offers significant benefits in reducing toxicity, enhancing delivery efficiency and specificity, and minimizing resistance. Given that the interaction of AIP with its receptor is biologically active, the development of modulators can focus on refining the backbone to improve this interaction, rather than starting from scratch. This method greatly expedites the development of therapeutic agents.
3.2 Com-QS system
The natural transformation process in S. pneumoniae takes place solely when the bacterium reaches a competent state, which is controlled by environmental conditions and the density of the bacterial population. Competence stimulating peptide (CSP) is a small signaling molecule composed of 17 amino acid residues.112 To date, two main groups of S. pneumoniae have been identified, each with distinct signal peptides (CSP1 and CSP2) and corresponding receptors (ComD1 and ComD2). The CSP precursor, ComC, is processed intracellularly and secreted by the ABC transporter protein (ComAB) into its mature form. When the bacterial population reaches a critical density, the concentration of CSP increases, activating the Com system through an auto-inducing mechanism. CSP binds to the ComD histidine kinase receptor on the cell membrane, activating its autophosphorylation activity. The phosphorylated ComD then transfers the phosphate group to the intracellular ComE, thereby activating it. Phosphorylated ComE functions as a transcription factor, leading to the self-induction of the QS circuit (ComABCDE) and the expression of comX, which encodes a QS-regulated phenotype (Fig. 4).113
 | ||
| Fig. 4 General streptococcal QS pathway.113 Reproduced from ref. 113 with permission from American Chemical Society, copyright 2017. | ||
Competent bacteria capture exogenous DNA through DNA-binding proteins, such as ComGC, on their membranes. The formation of uptake complexes on the bacterial surface enables DNA to pass through the cell membrane into the cytoplasm.72 Typically, the exogenous DNA entering the cell is double-stranded; one strand is degraded during membrane passage, while the other strand enters the cell. The single-stranded DNA that reaches the cytoplasm is integrated into the bacterial genome by homologous recombination mediated by the RecA protein. RecA recognizes homologous regions within the DNA sequence and facilitates the exchange between the exogenous DNA and the bacterium's own DNA. This process ensures the stable integration of exogenous DNA into the bacterial genome, thereby introducing new genetic information.
Lau and Tal-Gan's teams have discovered two primary CSP variants in pneumococci, designated as CSP1 and CSP2, each associated with distinct spherical phenotypes.114,115 These CSP variants bind to their specific receptors, ComD1 and ComD2, respectively, triggering different signaling pathways. Given that the key components of the destructive capacity regulator inhibit DNA transformation and decrease virulence, synthetic analogs that competitively inhibit CSP could serve as effective agents for managing pneumococcal infections and limiting horizontal gene transfer during infection.116 Lau et al. substituted and deleted the first three amino acid residues at the N-terminus and the last two amino acids at the C-terminus to create a series of synthetic CSP analogues. They found that CSP1-E1A was the only variant that outperformed CSP1 in inhibiting DNA transformation development in a time- and concentration-dependent fashion. This study also revealed that the first and third amino acid residues of CSP1 are crucial for inducing competence (Fig. 5(A) and (B)).115 In 2017, Tal-Gan and colleagues extensively investigated the SAR of CSP1 and CSP2 through thorough alanine and D-amino acid scans of natural signals. Their findings underscored a strong correlation between alpha helicity and the bioactivity of CSP1 analogs, suggesting that the alpha helix also serves as the bioactive conformation for CSP2. The study reaffirmed previous observations regarding the pivotal role of the N-terminal region in CSP signaling for ComD activation, and emphasized the importance of hydrophobic residues within the peptide center region for ComD recognition. Moreover, they demonstrated that the C-terminal regions of CSP1 and CSP2 are dispensable without compromising signaling activity.113
 | ||
| Fig. 5 Average heavy atom configuration of CSP1 (A) CSP1-E1A, (B) CSP1-K6A, (C) CSP2-d10, and (D) CSP2-E1Ad10.115 Reproduced from ref. 115 with permission from American Chemical Society, copyright 2018. (E) The dnCSP is a dual-action CSP that inhibits the group 1 pneumococcus competence regulon while activating the group 2 pneumococcus competence regulon.117 Reproduced from ref. 117 with permission from American Chemical Society, copyright 2022. | ||
Additionally, the researchers identified several synthetic CSP analogs with distinct activity profiles: analysis of CSP1 led to the discovery of CSP1-K6A, the first pan-motif ComD activator, which exhibited comparable potency against both ComD receptors (Fig. 5(C)). Subsequently, the CSP2 analogue CSP2-d10 was found to have ∼20-fold higher agonistic activity on ComD than natural CSP2 (Fig. 5(D)). Meanwhile, investigation of CSP2 yielded CSP2-E1Ad10, the most potent ComD2 inhibitor identified to date. While CSP2-E1Ad10 showed weaker inhibitory activity against ComD1, it still exhibited significant potential, suggesting its utility as a starting point for developing potent pan-group ComD inhibitors.115 In 2019, Tal-Gan engineered cyclic dominant-negative competence stimulating peptide (dnCSP) analogs through a rational design approach coupled with conformational optimization. Among these analogs, csp1-E1A-cyc (Dap6E10) emerged as a promising lead compound, demonstrating enhanced resistance to enzymatic degradation without inducing toxicity. Importantly, this potent dnCSP significantly reduced mortality in mice afflicted with acute pneumonia caused by both group 1 and group 2 pneumococcal strains.118 They chose to use urea bridge chemistry to facilitate side chain-to-side chain peptide cyclization. Structural analysis revealed that this method stabilized the bioactive alpha-helix conformation. The first pneumococcal double-acting lead, dnCSP CSP1-E1A-cyc (Dab6Dab10), was found to inhibit group 1 pneumococcal competence regulators while activating group 2 pneumococcal competence regulators. Additionally, the cyclic peptide boasts a much longer half-life than the linear natural CSP, while still being non-toxic (Fig. 5(E)).117
3.3 PlcR-QS system
QS signaling in Bacillus cereus is regulated by the AIPs PapR and its receptor PlcR. PlcR, a key regulatory protein from the family of transcriptional activators, plays a pivotal role in bacterial community sensing.120 When PlcR binds to specific DNA sequences, it triggers the expression of genes associated with virulence factors, extracellular enzymes, and other metabolites. The activity of PlcR is regulated by the signal peptide PapR, a precursor protein secreted by bacteria through the Sec mechanism. In an external environment, PapR is cleaved by proteases to form an active signal peptide. The mature PapR signaling peptide is reabsorbed by the bacteria through the oligopeptide permease Opp and binds to PlcR, forming an activation complex that triggers the regulatory functions of PlcR (Fig. 6).120 The genes activated by the PlcR-PapR complex include those encoding various virulence factors (e.g., hemolysin, phospholipase C) and extracellular enzymes (e.g., proteases, lipases). The expression of these genes allows the bacteria to efficiently utilize environmental resources, thereby enhancing their pathogenicity and survival within the host. PlcR is autoregulated and negatively controlled by Spo0A-P. CodY positively regulates the expression of PlcR-dependent genes through an unknown mechanism. Through the PlcR-PapR system, Bacillus cereus can coordinate the expression of its virulence factors in response to suitable environmental conditions, thereby increasing its pathogenic potential.

3.4 Rgg-QS system
To date, all sequenced strains of Streptococcus pyogenes have been found to contain four rgg paralogues, named rgg1, rgg2, rgg3, and rgg4.125 Regulatory genes of the glucosyltransferase (Rgg) family, such as Rgg2 and Rgg3, play a crucial role in controlling gene expression through a population-sensing system. This system influences various aspects of bacterial physiological activity, virulence, and biofilm formation. Rgg proteins are prevalent in Gram-positive bacteria, including Streptococcus pyogenes, Streptococcus thermophilus, and Streptococcus agalactiae. SHP (short hydrophobic peptide) is a small peptide encoded by the shp gene, which is typically located near the rgg gene.126 SHPs interact specifically with Rgg proteins to modulate their regulatory functions. Each Rgg protein usually corresponds to one or more specific SHP, facilitating precise regulatory control within the bacterial cell.
 | ||
| Fig. 7 Schematic representation of the Rgg/SHP system in S. pneumoniae D39.127 Reproduced from ref. 127 with permission from American Society for Microbiology, copyright 2017. | ||
In 2007, researchers discovered a novel modified peptide in Streptococcus pyogenes LMD-9, named Pep1357C. This peptide consisted of nine amino acids (AKGDGWKVM) but exhibited a 2 Da loss during post-translational modification, hypothesized to result from the loss of two hydrogens due to a linkage between Lys2 and Trp6 residues, forming a cyclic peptide. Additionally, in mutants with inactivated synthesis of short hydrophobic peptides, transcriptional regulators, or oligopeptide transport systems, the transcription of the gene encoding Pep1357C was significantly reduced. This led to the hypothesis that the transcription of the Pep1357C gene is controlled by a novel population-sensing system. The researchers then identified the mature form of Streptococcus pyogenes-produced Streptococcus lactis, SHP1555, with the sequence DILIIVGG. Streptococcus lactis was also found to encode two peptides (DIIIIVGG/DILIIVGG); however, Streptococcus pyogenes encoded a more varied amino acid sequence (DIIIIVGG/ETIIIIGGG). Therefore, they hypothesized that all mature SHPs should have either Asp or Glu at their N termini, as these conserved residues seem to be necessary for protease recognition.128
3.5 NPrR-QS system
Before spore formation begins, NprR primarily exists unbound to NprX, forming a flexible dimer that can bind to Spo0F. Consequently, spore-forming kinases cannot phosphorylate Spo0F when it is bound to NprR, keeping Spo0A inactive. This preservation of the PlcR regulon results in the production of extracellular virulence factors such as hemolysins, degradative enzymes, and enterotoxins, allowing the bacteria to remain virulent and ultimately causing host death. After the host's death, the bacterial density in the cadaver increases, raising the concentration of mature intracellular NprX enough to lock NprR into a tight superhelical conformation that is incompatible with Spo0F binding, thus triggering the sporulation phosphorylation chain. Concurrently, the NprR–NprX complex forms a tetramer and binds to DNA, activating the transcription of NprR-regulated genes involved in the bacteria's necrotrophic lifestyle. This indicates that NprR allows the bacteria to survive and eventually produce spores in the host carcass, enhancing their ability to spread in the environment (Fig. 8).130 In conclusion, the pathogenic and necrotrophic lifestyle of Bt is tightly regulated by two sequentially acting population-sensing systems during infection.

4. Peptide/polyamino acid assemblies against or targeting QS
4.1 Acyl homoserine lactones (AHLs)
AHLs are the most prevalent class of autoinducer in Gram-negative bacteria and the most comprehensively studied group of QS molecules.32 AHLs generally exhibit similar structural features: (i) homoserine lactone ring: this is a five-membered lactone ring derived from the amino acid homoserine. (ii) Acyl side chain: attached to the nitrogen atom of the homoserine lactone ring is a variable acyl chain, which can differ in length (typically from 4 to 14 carbons) and can include functional groups like 3-oxo or 3-hydroxy groups. The variation in the acyl chain determines the specificity and function of the AHL molecule in QS. (iii) Functional groups: the acyl chain can be saturated or unsaturated, and modifications such as the presence of 3-oxo or 3-hydroxy groups further diversify the signaling capabilities of AHLs.32,134,135In the 1980s, the QS molecule, N-3-oxohexanoyl-L-homoserine lactone (3OC6-HSL), was indentified in the luminescent marine bacterium Vibrio fischeri.136,137 QS systems that utilize this molecule exhibit luminescence at high cell densities but not at low cell densities.38,138
LasR is an important QS receptor primarily found in Pseudomonas aeruginosa and other Gram-negative bacteria. AHL molecules are synthesized by the LasI enzyme in the QS system and accumulate as bacterial density increases.139 When the concentration of AHL reaches a certain threshold, it binds to the LasR receptor, activating the expression of a series of genes that regulate bacterial behavior. Currently, inhibitors targeting LasR (such as small-molecule compounds) are being developed to disrupt signaling pathways, reducing bacterial virulence and biofilm formation.139–141
In 2009, Yang et al. conducted a structure-based virtual screening for LasR inhibitors in the SuperDrug and SuperNatural databases. The results revealed that salicylic acid exhibited significant QS inhibition activity against Pseudomonas aeruginosa, indicating its potential as an anti-virulence drug. Picolylamine is an organic base that has recently been explored for designing diagnostic probes. Both salicylic acid and this base share a common characteristic: metal coordination and chelation with metals.140 In 2023, Sharma designed a novel peptide, N-salicyl-AAn-picolamide peptides containing two scaffolds (salicylic acid and picolylamine) at the relatively terminal positions. Biological evaluation indicated that this conjugated peptide exhibited quorum sensing inhibition against Pseudomonas aeruginosa.141
Tropolone is a non-benzenoid aromatic molecule that is a component of many troponoid natural products. It possesses various biological activities, including antibacterial, anti-inflammatory, antitumor, and antiviral effects. In 2022, Sharma et al. rationally designed alklyamionotroponyl sulfone derivatives through Cu-catalyzed C(sp2)–H functionalization at the tropone ring and screened their anti-QS activity against Pseudomonas aeruginosa. Ultimately, the two sulfones significantly downregulate the lasI/R QS genes, and these molecules also inhibit swarming motility, biofilm formation, and the production of pyocyanin, thereby reducing the virulence of Pseudomonas aeruginosa in cells.142
4.2 Self-assembly of AHL
AHLs exhibit an amphiphilic structure, with polar nonionic L-homoserine head groups and nonpolar alkyl tails.143 This dual nature allows AHL molecules to spontaneously organize themselves in aqueous environments. The polar head groups interact with water molecules, while the nonpolar tails aggregate away from water, forming various nanostructures such as micelles, bilayers, or vesicles.143 This self-assembly process enhances their stability and facilitates their diffusion and transport within bacterial communities, aiding in communication and coordination among bacterial cells.144 David et al. employed biophysical characterization and atomic molecular dynamics simulations to examine the self-assembly behavior of 12 structurally related AHLs. Their research demonstrated that C12-AHL and 3-oxo-C12-AHL generally form spherical aggregates with diameters of approximately 5–6 nm, whereas C14-AHL, C16-AHL, 3-oxo-C14-AHL, and 3-oxo-C16-AHL are more likely to form bilayer vesicles. This study provides valuable insights into the self-assembly mechanisms of this important class of nonionic amphiphiles and establishes a potential basis for understanding how the chemical structure of AHLs affects bacterial processes such as biofouling and toxin production.1444.3 Assemblies of QSPs
Certain peptides involved in QS function as assemblies. Before spore formation, Bt transitions to a necrotrophic lifestyle, a process regulated by the sensor NprR. In the presence of its signaling peptide NprX, NprR adopts a tetrameric conformation, allowing it to bind specific DNA sequences and activate genes essential for bacterial survival in insect carcasses.130 Without NprX, NprR remains as a dimer, negatively controlling spore formation independently of its transcription factor activity.132 The tetrameric structure of the NprR–NprX complex suggests that NprR can bind to both DNA binding sites, whereas, in the absence of the binding peptide, NprR dissociates into a dimer and loses its ability to bind DNA.1304.4 Other QSP assemblies
Ji et al. proposed a novel therapeutic strategy to combat biofilm-associated infections by combining the QSI curcumin (Cur) with conventional antibiotics (Fig. 9).145 The initial step involved preparing Cur-DA nanoparticles (NPs) through electrostatic self-assembly of Cur-loaded poly(amidoamine) dendritic polymer (PAMAM) and 2,3-dimethylmaleic anhydride (DA)-modified biotin-polyethylene glycol-polylysine (biotin-PEG-PLys). These Cur-DA NPs were then further modified to target CD54 through biotin-affinity interaction, creating anti-CD54@Cur-DA NPs. Given that anti-CD54 can selectively bind to inflamed vascular endothelial cells in infected tissues, these anti-CD54@Cur-DA NPs can effectively target and accumulate in bacterial-infected tissues in vivo. Additionally, anti-CD54@Cur-DA NPs are sensitive to the acidic pH of the biofilm microenvironment, which induces charge reversal and size reduction. This pH sensitivity facilitates deeper biofilm penetration and enhances curcumin accumulation in infected tissues. Compared with free Cur, anti-CD54@Cur-DA NPs could effectively inhibit the expression of QS-related genes and secretion of QS signaling molecules more significantly. This study provides an innovative strategy to improve the therapeutic efficacy of QSIs through active targeting and enhanced biofilm penetration, which is very promising in the treatment of infections associated with bacterial QS.145 | ||
| Fig. 9 A schematic representation of the construction of anti-CD54@Cur-DA NPs and their application in inhibiting quorum sensing within biofilms and enhancing antibiotic therapy against biofilm-associated infections.145 Reproduced from ref. 145 with permission from American Chemical Society, copyright 2023. | ||
Ho et al. developed an innovative nanocarrier system based on a newly synthesized amphiphilic lipid, squalene hydrogen sulfate (SqHS), to enable the simultaneous delivery of the polycationic antibiotic tobramycin (Tob), a frontline therapy widely used for treating Pseudomonas aeruginosa (PA) infections, and a hydrophobic QSI. This nanocarrier system can effectively disrupt PA virulence factor expression through the QSI function without affecting cell growth, while also exhibiting excellent antibacterial activity through Tob. The synergistic effect of both components allows for the eradication of PA biofilms with approximately 16 times lower Tob concentration compared to using the drug alone. This dual-drug delivery method provides high drug bioavailability at the infection site and within the infected tissue, enhancing antibacterial activity across various stages of infection (Fig. 10).146
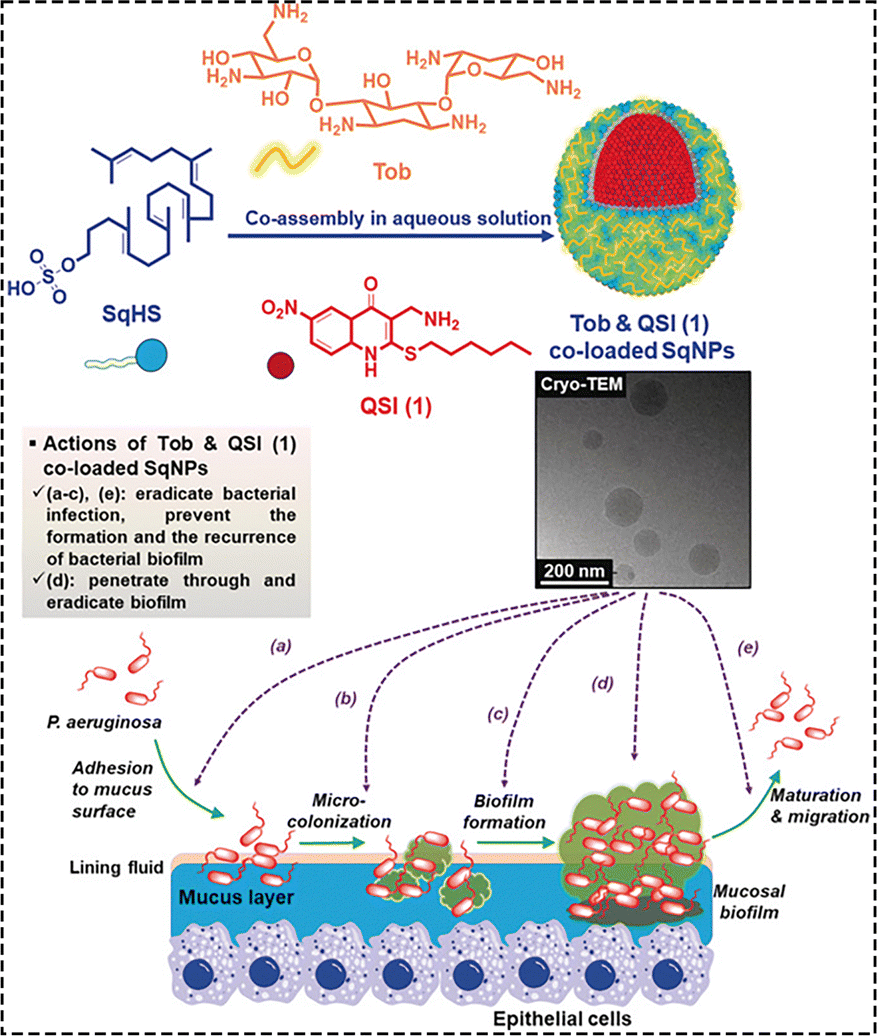 | ||
| Fig. 10 A schematic representation of the co-assembled SqNPs loaded with Tob and QSI to interfere with quorum sensing and eradicate bacterial infection.146 Reproduced from ref. 146 with permission from Wiley-VCH, copyright 2020. | ||
In addition, Li et al. investigated the impact of hydroxyl groups on the anti-quorum sensing (anti-QS) and antibiofilm activities of structurally similar cyclic dipeptides, including cyclo(L-Pro-L-Tyr), cyclo(L-Hyp-L-Tyr), and cyclo(L-Pro-L-Phe), in Pseudomonas aeruginosa PAO1. The results showed that cyclo(L-Pro-L-Phe), which lacks a hydroxyl group, exhibited strong inhibitory effects on virulence factors and cytotoxicity, but was less effective in inhibiting biofilm formation compared to its hydroxylated counterparts. In contrast, cyclo(L-Pro-L-Tyr) and cyclo(L-Hyp-L-Tyr) suppressed gene expression in the las and rhl systems, contributing to the inhibition of biofilm formation and the reduction of virulence factors. Additionally, the introduction of hydroxyl groups significantly enhanced the self-assembly capabilities of these peptides. At the highest tested concentrations, both cyclo(L-Pro-L-Tyr) and cyclo(L-Hyp-L-Tyr) were able to form assembled particles. This indicates that the presence of hydroxyl groups plays a crucial role in modulating the antimicrobial activities and self-assembly properties of cyclic dipeptides. Further studies revealed that the QS system of Pseudomonas aeruginosa involves three main pathways: LAS, RHL, and PQS. The LAS and RHL systems primarily control the production of pyocyanin, the expression of elastase and protease, and the maturation stage of biofilm formation, while the PQS system is mainly involved in the early stages of biofilm formation. Cyclo(L-Pro-L-Tyr) and cyclo(L-Hyp-L-Tyr) inhibit biofilm formation by affecting the las and rhl systems, thereby reducing the production of virulence factors, whereas cyclo(L-Pro-L-Phe) mainly targets the rhl system. To investigate the potential inhibitory mechanism of these cyclic dipeptides on the QS system of Pseudomonas aeruginosa PAO1, molecular docking was employed to explore the interactions between the three cyclic dipeptides and the QS receptor LasR. The docking data indicated that these cyclic dipeptides could bind to LasR in the same region as the autoinducer 3OC12-HSL, with binding affinities slightly higher than that of the natural ligand. These findings suggest that hydroxyl groups are crucial for the antimicrobial activity and self-assembly properties of cyclic dipeptides, providing a basis for the design and optimization of anti-QS compounds (Fig. 11).147
 | ||
| Fig. 11 A schematic representation of the effect of cyclic dipeptides on anti-QS and anti-biofilm activities of Pseudomonas aeruginosa PAO1.147 Reproduced from ref. 147 with permission from Elsevier, copyright 2023. | ||
5. Conclusions and outlook
Highlighted in this review are recent advances in bioinspired peptide/polyamino acid assemblies as QSIs across various microbial communities. Based on the characteristics of signal transduction molecules and their receptors, bacterial QS signalling pathways are classified into two groups, TCSTS represented by Agr and Com and RRNPP pathways represented by PlcR and Rgg. Regarding different QS systems, the corresponding quorum sensing peptide/polyamino acid targeting various receptors was cited and introduced here. In addition, to overcome the relatively low biostability and bioavailability of the peptide/polyamino acid, self-assembly of the peptide/polyamino acid was developed against drug resistant bacteria and reviewed.Despite the considerable advantages of peptide/polyamino acid and its assemblies as QSIs, their practical applications still encounter several hurdles. For instance, the current peptide and polyamino acid synthesis still suffer from drawbacks such as complexity, length limitation, undesired purity and yield, high cost and environmental and safety concerns. High yield and cost effective scalable production of peptide and polyamino acid have been highly desired to promote clinical translation. During the last decade, flow chemistry utilizes channels to conduct a reaction in a continuous stream rather than in a flask with precise control on reaction parameters to enhance reactivity, which has received much attention. The continuous advancements in flow chemistry technologies and methodologies show promise for further improving peptide and polyamino acid synthesis. Innovations such as integrated microfluidic devices, advanced catalysts, and in-line purification systems are expected to enhance the efficiency, scalability, and versatility of peptide and polyamino acid synthesis using flow chemistry. In addition, through integrating flow chemistry with nanomorphological characterization techniques, it could combine the synthesis and self-assembly of peptide/polyamino acids together. In summary, flow chemistry could be an efficient approach to synthesize a series of peptide/polyamino acid assemblies with the desired morphology and biofunctions for translational study.
In addition, to meet the demands of complicated physiological environments, it would be necessary to incorporate multiple capabilities within peptide/polyamino acid assemblies. Infection therapy typically performs multiple tasks such as targeted bacterial population sensing, controlling drug release, inhibiting horizontal gene transfer and reducing virulence factor production. One obstacle is how to design peptide/polyamino acid assemblies to support multifunctionality without interfering with each other. One possible approach is to construct peptide/polyamino acid assemblies with precise dimensions and nanostructures to ensure the multifunctionality. For example, implementing multiple secondary structures or compartments within peptide/polyamino acid assemblies could be applied to separate different functional components. Besides the above traditional chemical approach to realize the multiple capabilities within peptide/polyamino acid assemblies, machine learning has been widely applied recently to aid the design and synthesis of biomaterials. In addition, the highly dimensional design space for peptide/polyamino acid assemblies, combined with the need for compatibility and efficacy in complex biological environments, results in the development of multifunctional peptide/polyamino acid assemblies being burdened by time-consuming trial-and-error experimentation. The use of machine learning-driven data science could greatly speed up developments of peptide/polyamino acid assemblies as quorum sensing inhibitors. The applications of machine learning extend beyond peptide and polyamino acid design, encompassing process optimization and enhancing our understanding of structure–property relationships in these complex systems. Overall, accessing the next generation of pepide/polyamino acid assemblies as quorum sensing inhibitors will more than likely require some combination of the above approaches, which could greatly speed up the development of treatment approaches against antimicrobial resistance.
Author contributions
Yanan Jiang: conceptualization and writing – original draft. Fanying Meng: conceptualization. Zhenghong Ge: conceptualization. Yuxiao Zhou: supervision and writing – review & editing. Zhen Fan: conceptualization, supervision, and writing – review & editing. Jianzhong Du: supervision and writing – review & editing.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
This research was supported by the National Natural Science Foundation of China (52222306, 22075212, 22335005 and 21925505), the Innovation Program of Shanghai Municipal Education Commission (2023ZKZD28), and the Shanghai international scientific collaboration fund (23520710900). J. D. is the recipient of the National Science Fund for Distinguished Young Scholars (21925505).References
- C. Lindholm and R. Searle, Int. Wound J., 2016, 13(Suppl. 2), 5–15 CrossRef PubMed.
- A. W. Pountain, P. Jiang, T. Yao, E. Homaee, Y. Guan, K. J. C. McDonald, M. Podkowik, B. Shopsin, V. J. Torres, I. Golding and I. Yanai, Nature, 2024, 626, 661–669 CrossRef CAS PubMed.
- J. Posnett, F. Gottrup, H. Lundgren and G. Saal, J. Wound Care, 2009, 18, 154–161 CrossRef CAS PubMed.
- J. F. Guest, N. Ayoub, T. McIlwraith, I. Uchegbu, A. Gerrish, D. Weidlich, K. Vowden and P. Vowden, BMJ Open, 2015, 5, e009283 CrossRef PubMed.
- A. Uberoi, A. McCready-Vangi and E. A. Grice, Nat. Rev. Microbiol., 2024, 22, 507–521 CrossRef CAS PubMed.
- K. J. Y. Wu, B. I. C. Tresco, A. Ramkissoon, E. V. Aleksandrova, E. A. Syroegin, D. N. Y. See, P. Liow, G. A. Dittemore, M. Yu, G. Testolin, M. J. Mitcheltree, R. Y. Liu, M. S. Svetlov, Y. S. Polikanov and A. G. Myers, Science, 2024, 383, 721–726 CrossRef CAS PubMed.
- B. A. Lloyd, C. K. Murray, W. Bradley, F. Shaikh, D. Aggarwal, M. L. Carson and D. R. Tribble, Mil. Med., 2017, 182, 346–352 CrossRef PubMed.
- A. Parmanik, S. Das, B. Kar, A. Bose, G. R. Dwivedi and M. M. Pandey, Curr. Microbiol., 2022, 79, 388 CrossRef CAS PubMed.
- R. Goldburg, S. Roach, D. Wallinga and M. Mellon, Science, 2008, 321, 1294 CrossRef CAS PubMed.
- G. Taubes, Science, 2008, 321, 356–361 CrossRef CAS PubMed.
- B. Nogrady, Nature, 2023, 624, S30–S32 CrossRef CAS PubMed.
- D. van Duin and D. L. Paterson, Infect. Dis. Clin. North Am., 2016, 30, 377–390 CrossRef PubMed.
- W. P. J. Smith, B. R. Wucher, C. D. Nadell and K. R. Foster, Nat. Rev. Microbiol., 2023, 21, 519–534 CrossRef CAS PubMed.
- B. Plackett, Nature, 2022, 612, S33 CrossRef CAS PubMed.
- D. Hyun, Nature, 2022, 612, S32 CrossRef CAS PubMed.
- E. R. Mega, Nature, 2019 DOI:10.1038/d41586-019-02861-5.
- F. Prestinaci, P. Pezzotti and A. Pantosti, Pathog. Global Health, 2015, 109, 309–318 CrossRef PubMed.
- S. Ajulo and B. Awosile, PLoS One, 2024, 19, e0297921 CrossRef CAS PubMed.
- M. J. Shepherd, T. Fu, N. E. Harrington, A. Kottara, K. Cagney, J. D. Chalmers, S. Paterson, J. L. Fothergill and M. A. Brockhurst, Nat. Rev. Microbiol., 2024, 22, 650–665 CrossRef CAS PubMed.
- S. Darvishi, S. Tavakoli, M. Kharaziha, H. H. Girault, C. F. Kaminski and I. Mela, Angew. Chem., Int. Ed., 2020, 134, e202112218 CrossRef PubMed.
- J. W. Costerton, K. J. Cheng, G. G. Geesey, T. I. Ladd, J. C. Nickel, M. Dasgupta and T. J. Marrie, Annu. Rev. Microbiol., 1987, 41, 435–464 CrossRef CAS PubMed.
- V. Choi, J. L. Rohn, P. Stoodley, D. Carugo and E. Stride, Nat. Rev. Microbiol., 2023, 21, 555–572 CrossRef CAS PubMed.
- O. Ciofu, C. Moser, P. Ø. Jensen and N. Høiby, Nat. Rev. Microbiol., 2022, 20, 621–635 CrossRef CAS PubMed.
- M. Whiteley, S. P. Diggle and E. P. Greenberg, Nature, 2017, 551, 313–320 CrossRef CAS PubMed.
- B. M. Mony, P. MacGregor, A. Ivens, F. Rojas, A. Cowton, J. Young, D. Horn and K. Matthews, Nature, 2014, 505, 681–685 CrossRef CAS PubMed.
- A. A. Mashruwala, B. Qin and B. L. Bassler, Cell, 2022, 185, 3966–3979 CrossRef CAS PubMed.
- H. Koo, R. N. Allan, R. P. Howlin, P. Stoodley and L. Hall-Stoodley, Nat. Rev. Microbiol., 2017, 15, 740–755 CrossRef CAS PubMed.
- S. Mukherjee and B. L. Bassler, Nat. Rev. Microbiol., 2019, 17, 371–382 CrossRef CAS PubMed.
- A. Vashistha, N. Sharma, Y. Nanaji, D. Kumar, G. Singh, R. P. Barnwal and A. K. Yadav, Bioorg. Chem., 2023, 136, 106551 CrossRef CAS PubMed.
- M. J. Eickhoff and B. L. Bassler, Cell, 2018, 174, 1328 CrossRef CAS PubMed.
- K. L. Maxwell, Cell, 2019, 176, 7–8 CrossRef CAS PubMed.
- N. Amara, B. P. Krom, G. F. Kaufmann and M. M. Meijler, Chem. Rev., 2011, 111, 195–208 CrossRef CAS PubMed.
- W. C. Fuqua, S. C. Winans and E. P. Greenberg, J. Bacteriol., 1994, 176, 269–275 CrossRef CAS PubMed.
- F. Rojas, E. Silvester, J. Young, R. Milne, M. Tettey, D. R. Houston, M. D. Walkinshaw, I. Perez-Pi, M. Auer, H. Denton, T. K. Smith, J. Thompson and K. R. Matthews, Cell, 2019, 176, 306–317 CrossRef CAS PubMed.
- J. E. Silpe and B. L. Bassler, Cell, 2019, 176, 268–280 CrossRef CAS PubMed.
- H. Yazdani-Ahmadabadi, K. Yu, K. Gonzalez, H. D. Luo, D. Lange and J. N. Kizhakkedathu, ACS Appl. Mater. Interfaces, 2024, 16, 38631–38644 CrossRef CAS PubMed.
- N. Khadraoui, R. Essid, B. Damergi, N. Fares, D. Gharbi, A. M. Forero, J. Rodriguez, G. Abid, E. B. Kerekes, F. Limam, C. Jimenez and O. Tabbene, Biofilm, 2024, 8, 100205 CrossRef PubMed.
- A. Eberhard, A. L. Burlingame, C. Eberhard, G. L. Kenyon, K. H. Nealson and N. J. Oppenheimer, Biochemistry, 1981, 20, 2444–2449 CrossRef CAS PubMed.
- L. S. Håvarstein, G. Coomaraswamy and D. A. Morrison, Proc. Natl. Acad. Sci. U. S. A., 1995, 92, 11140–11144 CrossRef PubMed.
- X. Chen, S. Schauder, N. Potier, A. Van Dorsselaer, I. Pelczer, B. L. Bassler and F. M. Hughson, Nature, 2002, 415, 545–549 CrossRef CAS PubMed.
- R. Trirocco, M. Pasqua, A. Tramonti, B. Colonna, A. Paiardini and G. Prosseda, Sci. Rep., 2023, 13, 13170 CrossRef CAS PubMed.
- E. M. Bosire, C. R. Eade, C. J. Schiltz, A. J. Reid, J. Troutman, J. S. Chappie and C. Altier, Infect. Immun., 2020, 88, e00226-20 CrossRef PubMed.
- K. H. J. West, S. V. Ma, D. A. Pensinger, T. Tucholski, T. N. Tiambeng, E. L. Eisenbraun, A. Yehuda, Z. Hayouka, Y. Ge, J. D. Sauer and H. E. Blackwell, Biochemistry, 2023, 62, 2878–2892 CrossRef CAS PubMed.
- M. Murali, F. Ahmed, H. G. Gowtham, J. O. Aribisala, R. A. Abdulsalam, A. A. Shati, M. Y. Alfaifi, R. Z. Sayyed, S. Sabiu and K. N. Amruthesh, Sci. Rep., 2023, 13, 15505 CrossRef CAS PubMed.
- Z. Y. Miao, X. Y. Zhang, M. H. Yang, Y. J. Huang, J. Lin and W. M. Chen, J. Med. Chem., 2023, 66, 15823–15846 CrossRef CAS PubMed.
- C. Fuqua and E. P. Greenberg, Nat. Rev. Mol. Cell Biol., 2002, 3, 685–695 CrossRef CAS PubMed.
- P. H. Marques, A. K. Jaiswal, F. A. de Almeida, U. M. Pinto, A. B. Ferreira-Machado, S. Tiwari, S. C. Soares and A. D. Paiva, Mol. Diversity, 2023 DOI:10.1007/s11030-023-10722-7.
- M. Magri, E. M. Bouricha, M. Hakmi, R. E. Jaoudi, L. Belyamani and A. Ibrahimi, Bioinform. Biol. Insights, 2023, 17, 11779322231212755 CrossRef PubMed.
- K. R. Leistikow, D. S. May, W. S. Suh, G. Vargas Asensio, A. J. Schaenzer, C. R. Currie and K. R. Hristova, mSystems, 2024, 9, e0071224 CrossRef PubMed.
- E. L. Eisenbraun, T. D. Vulpis, B. N. Prosser, A. R. Horswill and H. E. Blackwell, J. Am. Chem. Soc., 2024, 146, 15941–15954 CrossRef CAS PubMed.
- T. A. Milly and Y. Tal-Gan, RSC Chem. Biol., 2020, 1, 60–67 RSC.
- A. H. Broderick, D. M. Stacy, Y. Tal-Gan, M. J. Kratochvil, H. E. Blackwell and D. M. Lynn, Adv. Healthcare Mater., 2014, 3, 97–105 CrossRef CAS PubMed.
- Y. Tal-Gan, M. Ivancic, G. Cornilescu, T. Yang and H. E. Blackwell, Angew. Chem., Int. Ed., 2016, 55, 8913–8917 CrossRef CAS PubMed.
- J. K. Vasquez and H. E. Blackwell, ACS Infect. Dis., 2019, 5, 484–492 CrossRef CAS PubMed.
- R. Inagaki, A. Koshiba, E. Nasuno and N. Kato, Biochem. Biophys. Res. Commun., 2024, 711, 149912 CrossRef CAS PubMed.
- Y. Li, X. He, P. Wang, B. Yuan, Y. Pan, X. Hu, L. Lu, A. Wu and J. Li, Small, 2024, 20, e2308621 CrossRef PubMed.
- H. Li, X. Qian, H. Mohanram, X. Han, H. Qi, G. Zou, F. Yuan, A. Miserez, T. Liu, Q. Yang, H. Gao and J. Yu, Nat. Nanotechnol., 2024, 19, 1141–1149 CrossRef CAS PubMed.
- H. Liu and H. Wang, Adv. Drug Delivery Rev., 2024, 209, 115327 CrossRef CAS PubMed.
- K. Sakamoto, Y. Yamamoto, H. Inaba and K. Matsuura, ACS Synth. Biol., 2024, 13, 1842–1850 CrossRef CAS PubMed.
- R. Shi, H. Li, X. Jin, X. Huang, Z. Ou, X. Zhang, G. Luo and J. Deng, Acta Biomater., 2024, 184, 473–476 CrossRef PubMed.
- W. Zhang, Y. Zeng, Q. Xiao, Y. Wu, J. Liu, H. Wang, Y. Luo, J. Zhan, N. Liao and Y. Cai, Nat. Commun., 2024, 15, 5670 CrossRef CAS PubMed.
- J. D. Hartgerink, E. Beniash and S. I. Stupp, Science, 2001, 294, 1684–1688 CrossRef CAS PubMed.
- H. Xu, K. Qi, C. Zong, J. Deng, P. Zhou, X. Hu, X. Ma, D. Wang, M. Wang, J. Zhang, S. M. King, S. E. Rogers, J. R. Lu, J. Yang and J. Wang, Small, 2024, 20, e2304424 CrossRef PubMed.
- C. Huang, Y. C. Liu, H. Oh, D. S. Guo, W. M. Nau and A. Hennig, Chemistry, 2024, 30, e202400174 CrossRef CAS PubMed.
- S. Li, Z. Wang, S. Song, Y. Tang, J. Zhou, X. Liu, X. Zhang, M. Chang, K. Wang and Y. Peng, ACS Infect. Dis., 2024, 10, 1839–1855 CrossRef CAS PubMed.
- N. J. Sinha, M. G. Langenstein, D. J. Pochan, C. J. Kloxin and J. G. Saven, Chem. Rev., 2021, 121, 13915–13935 CrossRef CAS PubMed.
- A. Levin, T. A. Hakala, L. Schnaider, G. J. L. Bernardes, E. Gazit and T. P. J. Knowles, Nat. Rev. Chem., 2020, 4, 615–634 CrossRef CAS.
- J. Wang, Y. Li and G. Nie, Nat. Rev. Mater., 2021, 6, 766–783 CrossRef CAS PubMed.
- X.-Y. Guo, L. Yi, J. Yang, H.-W. An, Z.-X. Yang and H. Wang, Chem. Commun., 2024, 60, 2009–2021 RSC.
- V. C. Kalia, Biotechnol. Adv., 2013, 31(2), 224–245 CrossRef CAS PubMed.
- A. R. Horswill and C. P. Gordon, J. Med. Chem., 2020, 63, 2705–2730 CrossRef CAS PubMed.
- S. Syed, L. Viazmina, R. Mager, S. Meri and K. Haapasalo, FEBS Lett., 2020, 594, 2570–2585 CrossRef CAS PubMed.
- V. Monnet and R. Gardan, Mol. Microbiol., 2015, 97, 181–184 CrossRef CAS PubMed.
- J. J. van Rensburg, K. R. Fortney, L. Chen, A. J. Krieger, B. P. Lima, A. J. Wolfe, B. P. Katz, Z. Y. Zhang and S. M. Spinola, Antimicrob. Agents Chemother., 2015, 59, 3789–3799 CrossRef CAS PubMed.
- Y. Li, W. Hutchins, X. Wu, C. Liang, C. Zhang, X. Yuan, D. Khokhani, X. Chen, Y. Che, Q. Wang and C. H. Yang, Mol. Plant Pathol., 2015, 16, 150–163 CrossRef CAS PubMed.
- L. C. Cook and M. J. Federle, FEMS Microbiol. Rev., 2014, 38, 473–492 CrossRef CAS PubMed.
- K. Kurabayashi, Y. Hirakawa, K. Tanimoto, H. Tomita and H. Hirakawa, J. Bacteriol., 2015, 197, 861–871 CrossRef PubMed.
- S. N. Joslin, C. Pybus, M. Labandeira-Rey, A. S. Evans, A. S. Attia, C. A. Brautigam and E. J. Hansen, Infect. Immun., 2015, 83, 146–160 CrossRef PubMed.
- G. S. A. Wright, A. Saeki, T. Hikima, Y. Nishizono, T. Hisano, M. Kamaya, K. Nukina, H. Nishitani, H. Nakamura, M. Yamamoto, S. V. Antonyuk, S. S. Hasnain, Y. Shiro and H. Sawai, Sci. Signaling, 2018, 11, eaaq0825 CrossRef PubMed.
- Q. Wang, M. Chen and W. Zhang, J. Vet. Med. Sci., 2018, 19, 260–270 Search PubMed.
- D. Perez-Pascual, V. Monnet and R. Gardan, Front. Microbiol., 2016, 7, 706 CrossRef PubMed.
- S. Parthasarathy, L. D. Jordan, N. Schwarting, M. A. Woods, Z. Abdullahi, S. Varahan, P. M. S. Passos, B. Miller and L. E. Hancock, J. Bacteriol., 2020, 202, e00063-20 CrossRef PubMed.
- M. B. Neiditch, G. C. Capodagli, G. Prehna and M. J. Federle, Annu. Rev. Genet., 2017, 51, 311–333 CrossRef CAS PubMed.
- A. K. Kotte, O. Severn, Z. Bean, K. Schwarz, N. P. Minton and K. Winzer, Microbiology, 2020, 166, 579–592 CrossRef CAS PubMed.
- J. Feng, W. Zong, P. Wang, Z. T. Zhang, Y. Gu, M. Dougherty, I. Borovok and Y. Wang, Biotechnol. Biofuels, 2020, 13, 84 CrossRef CAS PubMed.
- H. Do and M. Kumaraswami, J. Mol. Biol., 2016, 428, 2793–2804 CrossRef CAS PubMed.
- J. M. Wozniak, R. H. Mills, J. Olson, J. R. Caldera, G. D. Sepich-Poore, M. Carrillo-Terrazas, C. M. Tsai, F. Vargas, R. Knight, P. C. Dorrestein, G. Y. Liu, V. Nizet, G. Sakoulas, W. Rose and D. J. Gonzalez, Cell, 2020, 182, 1311–1327 CrossRef CAS PubMed.
- H. Nishimasu, L. Cong, W. X. Yan, F. A. Ran, B. Zetsche, Y. Li, A. Kurabayashi, R. Ishitani, F. Zhang and O. Nureki, Cell, 2015, 162, 1113–1126 CrossRef CAS PubMed.
- G. Koch, A. Yepes, K. U. Forstner, C. Wermser, S. T. Stengel, J. Modamio, K. Ohlsen, K. R. Foster and D. Lopez, Cell, 2014, 158, 1060–1071 CrossRef CAS PubMed.
- L. Deng, F. Costa, K. J. Blake, S. Choi, A. Chandrabalan, M. S. Yousuf, S. Shiers, D. Dubreuil, D. Vega-Mendoza, C. Rolland, C. Deraison, T. Voisin, M. D. Bagood, L. Wesemann, A. M. Frey, J. S. Palumbo, B. J. Wainger, R. L. Gallo, J. M. Leyva-Castillo, N. Vergnolle, T. J. Price, R. Ramachandran, A. R. Horswill and I. M. Chiu, Cell, 2023, 186, 5375–5393 CrossRef CAS PubMed.
- V. Lazar, O. Snitser, D. Barkan and R. Kishony, Nature, 2022, 610, 540–546 CrossRef CAS PubMed.
- J. A. N. Alexander, L. J. Worrall, J. Hu, M. Vuckovic, N. Satishkumar, R. Poon, S. Sobhanifar, F. I. Rosell, J. Jenkins, D. Chiang, W. A. Mosimann, H. F. Chambers, M. Paetzel, S. S. Chatterjee and N. C. J. Strynadka, Nature, 2023, 613, 375–382 CrossRef CAS PubMed.
- L. Tan, Y. Huang, W. Shang, Y. Yang, H. Peng, Z. Hu, Y. Wang, Y. Rao, Q. Hu, X. Rao, X. Hu, M. Li, K. Chen and S. Li, Front. Microbiol., 2022, 13, 700894 CrossRef PubMed.
- S. C. Jordan, P. R. Hall and S. M. Daly, Sci. Rep., 2022, 12, 1251 CrossRef CAS PubMed.
- S. O. Lee, S. Lee, J. E. Lee, K. H. Song, C. K. Kang, Y. M. Wi, R. San-Juan, L. E. Lopez-Cortes, A. Lacoma, C. Prat, H. C. Jang, E. S. Kim, H. B. Kim and S. H. Lee, Sci. Rep., 2020, 10, 20697 CrossRef CAS PubMed.
- C. Huber, I. Stamm, W. Ziebuhr, G. Marincola, M. Bischoff, B. Strommenger, G. Jaschkowitz, T. Marciniak, C. Cuny, W. Witte, J. Doellinger, C. Schaudinn, A. Thurmer, L. Epping, T. Semmler, A. Lubke-Becker, L. H. Wieler and B. Walther, Sci. Rep., 2020, 10, 14787 CrossRef CAS PubMed.
- J. M. Jiang, G. Chen, Y. Y. Chen, S. J. Wan, S. M. Chen, H. G. Ren, Z. X. Lin, H. Feng, H. Zhang and H. X. Xu, Food Funct., 2022, 13, 5050–5060 RSC.
- J. K. Vasquez, K. H. J. West, T. Yang, T. J. Polaske, G. Cornilescu, M. Tonelli and H. E. Blackwell, J. Am. Chem. Soc., 2020, 142, 750–761 CrossRef CAS PubMed.
- P. Williams, P. Hill, B. Bonev and W. C. Chan, Microbiology, 2023, 169, 001381 CrossRef CAS PubMed.
- Y. Tal-Gan, M. Ivancic, G. Cornilescu, C. C. Cornilescu and H. E. Blackwell, J. Am. Chem. Soc., 2013, 135, 18436–18444 CrossRef CAS PubMed.
- C. P. Gordon, S. D. Olson, J. L. Lister, J. S. Kavanaugh and A. R. Horswill, J. Med. Chem., 2016, 59, 8879–8888 CrossRef CAS PubMed.
- K. Vadakkan, K. Sathishkumar, S. Kuttiyachan Urumbil, S. Ponnenkunnathu Govindankutty, A. Kumar Ngangbam and B. Devi Nongmaithem, Bioorg. Chem., 2024, 148, 107465 CrossRef CAS PubMed.
- R. B. Merrifield and J. M. Stewart, Nature, 1965, 207, 522–523 CrossRef CAS PubMed.
- I. Coin, M. Beyermann and M. Bienert, Nat. Protoc., 2007, 2, 3247–3256 CrossRef CAS PubMed.
- J. M. Collins, S. K. Singh, T. A. White, D. J. Cesta, C. L. Simpson, L. J. Tubb and C. L. Houser, Nat. Commun., 2023, 14, 8168 CrossRef CAS PubMed.
- Y. Tal-Gan, M. Ivancic, G. Cornilescu, T. Yang and H. E. Blackwell, Angew. Chem., Int. Ed., 2016, 55, 8913–8917 CrossRef CAS PubMed.
- S. Kumar, R. D. A. Balaya, S. Kanekar, R. Raju, T. S. K. Prasad and R. K. Kandasamy, Comput. Struct. Biotechnol. J., 2023, 21, 1995–2008 CrossRef CAS PubMed.
- E. Wynendaele, A. Bronselaer, J. Nielandt, M. D’Hondt, S. Stalmans, N. Bracke, F. Verbeke, C. Van De Wiele, G. De Tré and B. De Spiegeleer, Nucleic Acids Res., 2013, 41, D655–D659 CrossRef CAS PubMed.
- A. Rajput, A. K. Gupta and M. Kumar, PLoS One, 2015, 10, e0120066 CrossRef PubMed.
- A. Gómez-Mejia, G. Gámez and S. Hammerschmidt, Int. J. Med. Microbiol., 2018, 308, 722–737 CrossRef PubMed.
- G. Salvadori, R. Junges, D. A. Morrison and F. C. Petersen, Front. Cell. Infect. Microbiol., 2019, 9, 1009 Search PubMed.
- E. Gil, M. Noursadeghi and J. S. Brown, Front. Cell. Infect. Microbiol., 2022, 12, 929483 CrossRef PubMed.
- Y. Yang, B. Koirala, L. A. Sanchez, N. R. Phillips, S. R. Hamry and Y. Tal-Gan, ACS Chem. Biol., 2017, 12, 1141–1151 CrossRef CAS PubMed.
- L. Zhu and G. W. Lau, PLoS Pathog., 2011, 7, e1002241 CrossRef CAS PubMed.
- Y. Yang, G. Cornilescu and Y. Tal-Gan, Biochemistry, 2018, 57, 5359–5369 CrossRef CAS PubMed.
- B. Koirala, J. Lin, G. W. Lau and Y. Tal-Gan, ChemBioChem, 2018, 19, 2380–2386 CrossRef CAS PubMed.
- M. Lella, M. W. Oh, S. H. Kuo, G. W. Lau and Y. Tal-Gan, J. Med. Chem., 2022, 65, 6826–6839 CrossRef CAS PubMed.
- Y. Yang, J. Lin, A. Harrington, G. Cornilescu, G. W. Lau and Y. Tal-Gan, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 1689–1699 CrossRef CAS PubMed.
- L. Slamti and D. Lereclus, J. Bacteriol., 2005, 187, 1182–1187 CrossRef CAS PubMed.
- L. Slamti, S. Perchat, E. Huillet and D. Lereclus, Toxins, 2014, 6, 2239–2255 CrossRef CAS PubMed.
- R. Grenha, L. Slamti, M. Nicaise, Y. Refes, D. Lereclus and S. Nessler, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 1047–1052 CrossRef CAS PubMed.
- M. Gorgan, S. Vanunu Ofri, E. R. Engler, A. Yehuda, E. Hutnick, Z. Hayouka and M. A. Bertucci, Res. Microbiol., 2023, 174, 104139 CrossRef CAS PubMed.
- M. J. Stoltzfus, R. E. Workman, N. C. Keith and J. W. Modell, Nat. Microbiol., 2024, 9, 2410–2421 CrossRef CAS PubMed.
- E. P. Armitage, G. de Crombrugghe, A. J. Keeley, E. Senghore, F. E. Camara, M. Jammeh, A. Bittaye, H. Ceesay, I. Ceesay, B. Samateh, M. Manneh, B. Kampmann, C. E. Turner, A. Kucharski, A. Botteaux, P. R. Smeesters, T. I. de Silva, M. Marks and M. S. S. Group, Lancet Microbe, 2024, 5, 679–688 CrossRef PubMed.
- A. Vieira, Y. Wan, Y. Ryan, H. K. Li, R. L. Guy, M. Papangeli, K. K. Huse, L. C. Reeves, V. W. C. Soo, R. Daniel, A. Harley, K. Broughton, C. Dhami, M. Ganner, M. A. Ganner, Z. Mumin, M. Razaei, E. Rundberg, R. Mammadov, E. A. Mills, V. Sgro, K. Y. Mok, X. Didelot, N. J. Croucher, E. Jauneikaite, T. Lamagni, C. S. Brown, J. Coelho and S. Sriskandan, Nat. Commun., 2024, 15, 3916 CrossRef CAS PubMed.
- O. Xie, C. Zachreson, G. Tonkin-Hill, D. J. Price, J. A. Lacey, J. M. Morris, M. I. McDonald, A. C. Bowen, P. M. Giffard, B. J. Currie, J. R. Carapetis, D. C. Holt, S. D. Bentley, M. R. Davies and S. Y. C. Tong, Nat. Commun., 2024, 15, 3477 CrossRef CAS PubMed.
- R. Junges, G. Salvadori, S. Shekhar, A. Åmdal Heidi, N. Periselneris Jimstan, T. Chen, S. Brown Jeremy and C. Petersen Fernanda, mSphere, 2017, 2, e00324-17 CrossRef PubMed.
- B. Fleuchot, A. Guillot, C. Mézange, C. Besset, E. Chambellon, V. Monnet and R. Gardan, PLoS One, 2013, 8, e66042 CrossRef CAS PubMed.
- T. Dubois, K. Faegri, S. Gélis-Jeanvoine, S. Perchat, C. Lemy, C. Buisson, C. Nielsen-LeRoux, M. Gohar, P. Jacques, N. Ramarao, L. Slamti, A.-B. Kolstø and D. Lereclus, PLoS Pathog., 2016, 12, e1006049 CrossRef PubMed.
- S. Perchat, A. Talagas, S. Poncet, N. Lazar, I. Li de la Sierra-Gallay, M. Gohar, D. Lereclus and S. Nessler, PLoS Pathog., 2016, 12, e1005779 CrossRef PubMed.
- L. Wu, X. Guo, X. Liu and H. Yang, Front. Microbiol., 2017, 8, 1968 CrossRef PubMed.
- S. Zouhir, S. Perchat, M. Nicaise, J. Perez, B. Guimaraes, D. Lereclus and S. Nessler, Nucleic Acids Res., 2013, 41, 7920–7933 CrossRef CAS PubMed.
- J. Rocha, V. Flores, R. Cabrera, A. Soto-Guzmán, G. Granados, E. Juaristi, G. Guarneros and M. de la Torre, Appl. Microbiol. Biotechnol., 2012, 94, 1069–1078 CrossRef CAS PubMed.
- B. Fu, Y. Xing, C. Gong and H. Zhao, Environ. Sci.: Water Res. Technol., 2022, 8, 1211–1222 RSC.
- L. Ziesche, L. Wolter, H. Wang, T. Brinkhoff, M. Pohlner, B. Engelen, I. Wagner-Döbler and S. Schulz, Mar. Drugs, 2019, 17, 20 CrossRef CAS PubMed.
- J. Engebrecht and M. Silverman, Proc. Natl. Acad. Sci. U. S. A., 1984, 81, 4154–4158 CrossRef CAS PubMed.
- J. Engebrecht, K. Nealson and M. Silverman, Cell, 1983, 32, 773–781 CrossRef CAS PubMed.
- H. Nealson Kenneth, T. Platt and J. W. Hastings, J. Bacteriol., 1970, 104, 313–322 CrossRef PubMed.
- M. N. Hurley, M. Camara and A. R. Smyth, Eur. Respir. J., 2012, 40, 1014–1023 CrossRef CAS PubMed.
- L. Yang, M. T. Rybtke, T. H. Jakobsen, M. Hentzer, T. Bjarnsholt, M. Givskov and T. Tolker-Nielsen, Antimicrob. Agents Chemother., 2009, 53, 2432–2443 CrossRef CAS PubMed.
- S. S. Panda, S. Kumari, M. Dixit and N. K. Sharma, ACS Omega, 2023, 8, 30349–30358 CrossRef CAS PubMed.
- S. Meher, S. Kumari, M. Dixit and N. Sharma, Chem. – Asian J., 2022, 17, e202200866 CrossRef CAS PubMed.
- J. D. Moore, F. M. Rossi, M. A. Welsh, K. E. Nyffeler and H. E. Blackwell, J. Am. Chem. Soc., 2015, 137, 14626–14639 CrossRef CAS PubMed.
- C. G. Gahan, S. J. Patel, M. E. Boursier, K. E. Nyffeler, J. Jennings, N. L. Abbott, H. E. Blackwell, R. C. Van Lehn and D. M. Lynn, J. Phys. Chem. B, 2020, 124, 3616–3628 CrossRef CAS PubMed.
- Y. Chen, Y. Gao, Y. Huang, Q. Jin and J. Ji, ACS Nano, 2023, 17, 10019–10032 CrossRef CAS PubMed.
- D. K. Ho, X. Murgia, C. De Rossi, R. Christmann, A. G. Hufner de Mello Martins, M. Koch, A. Andreas, J. Herrmann, R. Muller, M. Empting, R. W. Hartmann, D. Desmaele, B. Loretz, P. Couvreur and C. M. Lehr, Angew. Chem., Int. Ed., 2020, 59, 10292–10296 CrossRef CAS PubMed.
- L. Li, Z. Xu, R. Cao, J. Li, C. J. Wu, Y. Wang and H. Zhu, iScience, 2023, 26, 107048 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2024 |