
Dual photoresponsive & water-triggered nitric oxide releasing materials based on rhodium-based metal–organic polyhedra†
Francisco J.
Carmona
 a,
Thiago
Negrão Chuba
ac,
Elí
Sánchez-González
a,
Jenny
Pirillo
d,
Yuh
Hijikata
e and
Shuhei
Furukawa
*ab
a,
Thiago
Negrão Chuba
ac,
Elí
Sánchez-González
a,
Jenny
Pirillo
d,
Yuh
Hijikata
e and
Shuhei
Furukawa
*ab
aInstitute for Integrated Cell-Material Sciences (WPI-iCeMS), Kyoto University, Yoshida, Sakyo-ku, Kyoto 606-8501, Japan. E-mail: shuhei.furukawa@icems.kyoto-u.ac.jp
bDepartment of Synthetic Chemistry and Biological Chemistry, Graduate School of Engineering, Kyoto University, Katsura, Nishikyo-ku, Kyoto 615-8510, Japan
cGraduate School of Advanced Integrated Studies in Human Survivability, Kyoto University, Yoshida, Sakyo-ku, Kyoto 606-8306, Japan
dDepartment of Chemistry and Biotechnology, School of Engineering, and Department of Materials Chemistry, Graduate School of Engineering, Nagoya University, Chikusa-ku, Nagoya 464-8603, Japan
eResearch Center for Net Zero Carbon Society, Institute of Innovation for Future Society, Nagoya University, Chikusa-ku, Nagoya, 464-8601, Japan
First published on 28th November 2023
Abstract
The exogenous administration of nitric oxide (NO) is considered a potential therapeutic treatment against a great variety of diseases due to its significant role in multiple physiological functions. Due to the gaseous nature, short lifetime and dose- and tissue-dependent activity of this molecule, the development of new administration procedures is required to control the NO delivery in terms of dosage, timing, and location. In this work, we propose a new molecular material based on robust metal–organic polyhedra (MOPs) for controlled NO release. We select dirhodium paddlewheel complex-based cuboctahedral MOPs (RhMOP), in which NO can chemically coordinate to the open-metal sites at the axial sites of dirhodium paddlewheel moieties. We further prepare amorphous coordination polymer particles (CPPs) by connecting RhMOP with bis(imidazole) linkers at the external axial sites. Both molecular MOPs and polymeric CPPs show relevant NO payloads and the release of NO can be triggered by two different stimuli: light and humidity. We show that imidazole ligands coordinating to the external axial sites of the paddlewheel moieties tune the light-triggered NO release property. We further demonstrate that the size and the extrinsic pores of CPPs are important for enhanced NO release.
Introduction
Nitric oxide (NO) is a gaseous signalling molecule endogenously produced in mammals to regulate cellular functions with a vast impact on several physiological processes including vasodilation, neurotransmission, and immunity, among others.1,2 In the physiological environment, NO is a highly reactive gas that readily diffuses across cell membranes and tissues in the body. In addition, its biological activity is tissue-specific and dose-dependent, being toxic at high concentrations. Consequently, NO is endogenously produced on the target site, at the specific dose and at any time when necessary, through the enzymatic degradation of L-arginine by nitric oxide synthase (NOS).3Due to its ubiquitous physiological activity in humans, the biological roles of NO and its potential as a drug have been the focus of intense cell biology and medical research during the last few decades.4 The development of materials that enable the on-demand generation of exogenous NO with a spatiotemporally controlled gas release is a key aspect for the successful progression of both research fields. For instance, in cell biology, the localized NO release toward intracellular targets would favour the elucidation of the biological role of NO inside of specific cells of interest.5 From the viewpoint of medical research, NO has been demonstrated to have a beneficial activity against a great variety of diseases.4,6,7 Therefore, the safe and effective administration of exogenous NO to patients would favour the implementation of NO in clinical treatments.
With the aim to generate NO in a controlled manner, small molecular NO donors that spontaneously release NO after self-decomposition in physiological media, or in response to external stimuli such as heat or light, were first proposed.8 However, the control over the spatial release of NO from these molecules relies on the solubility and distribution of the NO donor in physiological media, making it difficult to localize the NO molecules in specific targets. To circumvent these shortcomings, the assembly of NO donors, or NO gas itself, into solid-state materials has been proposed. This approach allows for restricting the dispersion of NO donors in solution and for tuning the NO-release rate.4,9 In addition, solid-state materials can be easily handled and shaped by microfabrication techniques, which facilitates the manufacture of materials enabling localization of the NO release in target tissues.5
Among the different solids tested to host NO, metal–organic frameworks (MOFs) have shown the highest drug payloads, thanks to their large surface areas and elevated pore volumes. In particular, these porous materials possess accessible metal ions inside their cavities with unsaturated coordination sites, called open metal sites, which reversibly chemisorb NO,9,10 being the NO released by means of a ligand exchange reaction with water molecules.11–14
By cutting out of the smallest pore unit from MOFs, one can design a molecular counterpart called metal–organic polyhedra (MOPs).15 This cage-shaped supramolecule with an intrinsic cavity can possess open metal sites in their structures, which allow for their surface functionalization to not only trap guest molecules by chemisorption (e.g., NO) but also to deliberately tune specific properties of the cage (e.g., chemical stability or hydrophilic/hydrophobic character) or even to regulate their assembly in the solid state.16–18 Unlike insoluble MOF solids, MOPs can be dissolved in solvent while maintaining their porous structure. Such molecular nature allows their characterization, processing, and use in solution. Although these characteristics make MOPs ideal candidates to manufacture novel materials for generating NO on-demand, either as discrete molecules or as building blocks for constructing solid-state materials, the study of MOPs in this field remains unexplored.
In this work, we demonstrate the potential of MOPs to prepare novel NO-releasing materials. We take advantage of the open metal sites in a robust dirhodium paddlewheel complex-based cuboctahedral MOP of [Rh2(bdc-C12)2]12 (C12RhMOP; H2bdc-C12 = 5-dodecoxybenzene-1,3-dicarboxylic acid) to chemisorb NO. The resulting NO-loaded molecule releases NO in response to two different external stimuli: light and humidity. In addition, we functionalize the MOP by means of two different approaches with the aim of gaining control over the NO release (Fig. 1). One is to introduce a monotopic imidazole linker (1-benzylimidazole, biz) in the external surface of the cage to modify the coordination sphere of the dirhodium paddlewheel units. The other is to use a ditopic linker (1,4-bis(imidazol-1-ylmethyl)benzene, bix), to assemble the cages into amorphous extended structures known to be coordination polymer particles (CPPs) with submicrometer particle size. We determine the impact of the external functionalization of the cages on their NO-releasing profiles.
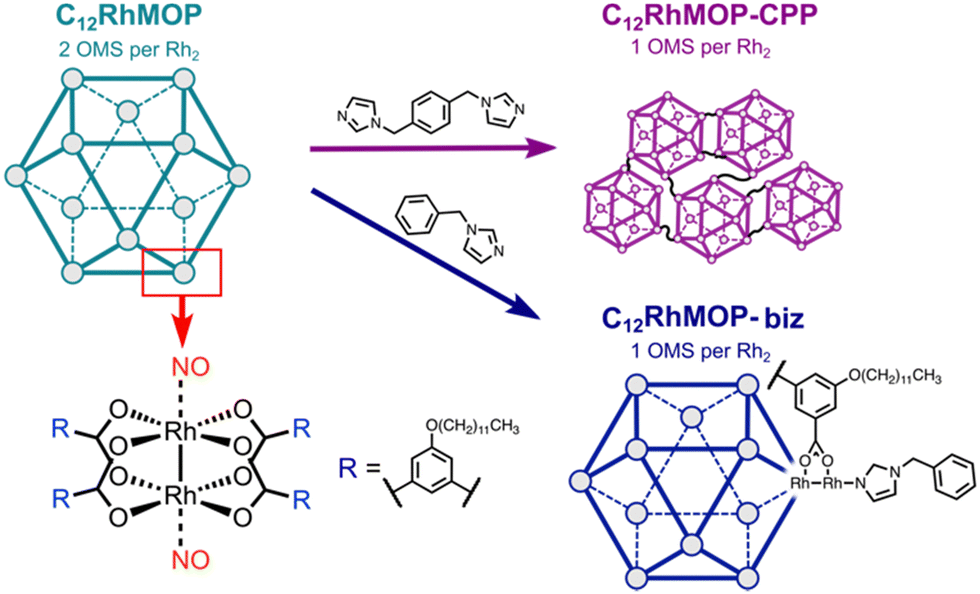 | ||
| Fig. 1 Schematic representation of the cuboctahedron structure of C12RhMOP (green) indicating the organic ligands employed to prepare the compounds C12RhMOP-biz (blue) and the C12RhMOP-CPP (violet). The chemical structure of the dirhodium paddlewheel motif, along the site where NO coordinates, as well as of the linker forming the MOP are also indicated. | ||
Results and discussion
Design, synthesis, and characterization of C12RhMOP-based materials
We selected C12RhMOP to explore the potential of MOPs to prepare novel NO-releasing materials because of the following three reasons. (i) C12RhMOP displays permanent porosity after solvent removal due to the robust [Rh2(COO)4] moiety with open metal sites, which can be coordinated by NO molecules. (ii) The open metal sites located on the external surface allow for the introduction of functional groups via coordination bonds. (iii) The hydrophobic –O(CH2)11CH3 groups on the external surface of the cage confer a high solubility in a variety of organic solvents and enable its characterization, processing and use in solution (Fig. 1).In the first step, C12RhMOP was prepared according to a previously reported protocol and its chemical structure was confirmed by 1H-NMR (Fig. S2, ESI†).19 Then, we functionalized the external surface of C12RhMOP with a monotopic ligand biz to give rise to a discrete cage (C12RhMOP-biz)19,20 allowing us to further evaluate the impact of the coordination sphere in the dirhodium paddlewheel units on the NO release properties of the cage. We chose biz as a functionalization molecule because of two main reasons: a high affinity of imidazole moiety for soft Rh(II) ions and its coordination only to the external open metal sites due to its larger molecular size than pore apertures of MOP. The latter ensures the availability of the internal open metal sites in the cage to be loaded with NO.
As shown in Fig. 2a, and as we previously reported,19 the addition of an excess of the monotopic ligand biz (13 eq. mol) to a solution of C12RhMOP in DMF led to a blue shift in the band of its visible absorption spectrum (λmax at 596 nm to 560 nm for C12RhMOP and C12RhMOP-biz, respectively), with the concomitant colour change of the solution from green to purple. This band is characteristics of dirhodium paddlewheel complexes and it is assigned to the π* → σ* electronic transition in the metal–metal bonded Rh2 core, which is highly dependent on the nature of the axial ligands.21,22 Thus, these results pointed to the successful functionalization of the external surface of the cage with biz. A quantitative analysis by NMR indicates that the resulting compound C12RhMOP-biz possesses the composition [Rh2(bdc-C12)2(biz)0.68]12, in which 68% of the external open sites of the cage are occupied by biz molecules. This estimation provides 1.32 available open metal sites per the paddlewheel moiety (Fig. S3, ESI†). This value is later confirmed by NO chemisorption (vide infra). In addition, dynamic light scattering (DLS) analysis of the solutions of C12RhMOP and C12RhMOP-biz in DMF results in a weight-number distribution of 2.56 ± 0.37 nm and 2.58 ± 0.38 nm, respectively (Fig. 2b). Both values are in agreement with the size of C12RhMOP,16,19 confirming the presence of non-aggregated MOP molecules in solution.
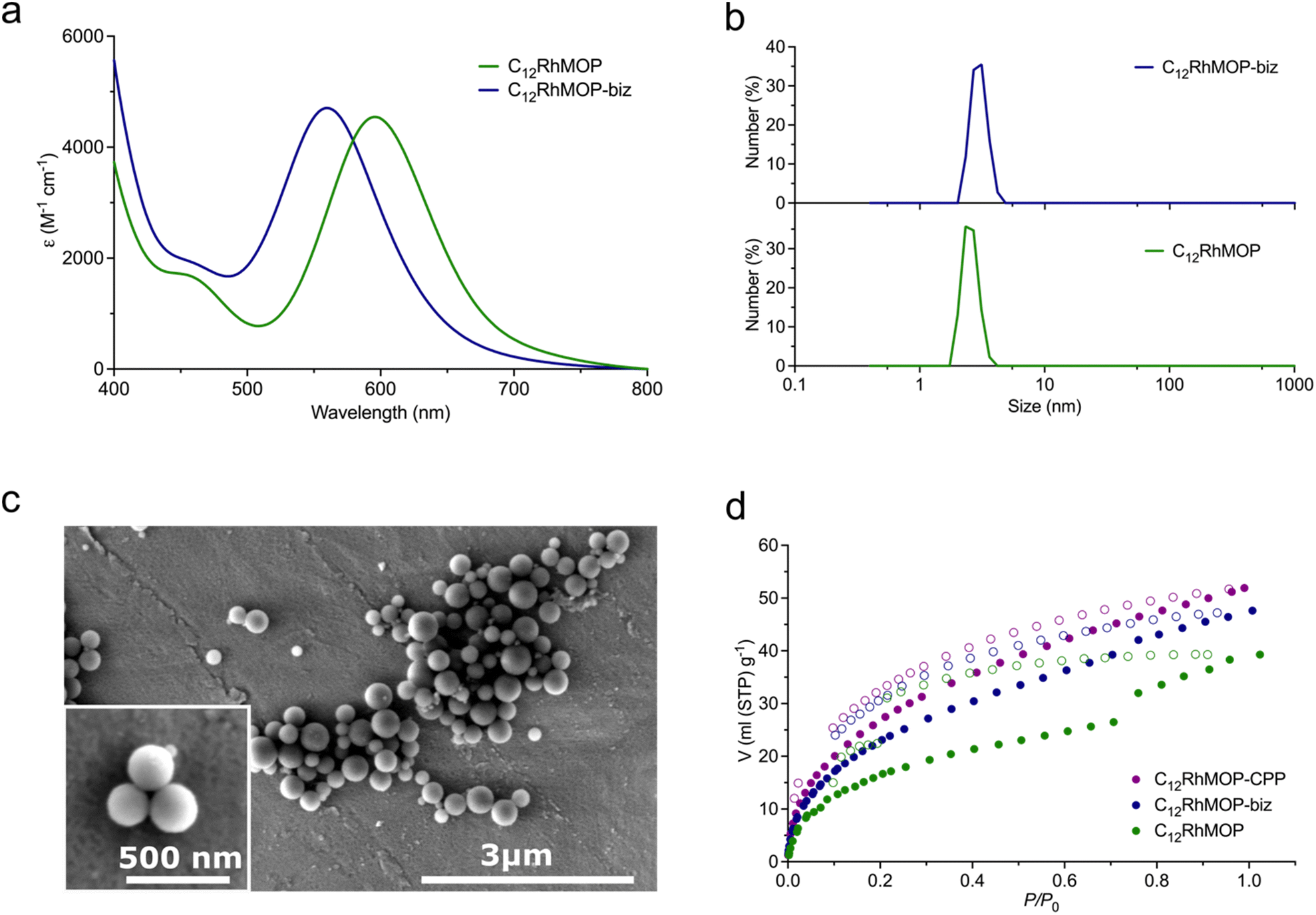 | ||
| Fig. 2 (a) Visible absorption spectra of the solution of C12RhMOP (green curve) and C12RMOP-biz (blue curve) in DMF, (b) number size distribution of C12RhMOP (green curve) and C12RMOP-biz (blue curve) in DMF, determined by DLS, (c) SEM images of C12RhMOP-CPP, (d) adsorption isotherms of CO2 for C12RhMOP (green curve), C12RMOP-biz (blue curve) and C12RhMOP-CPP (violet curve) at 195 K. | ||
To fabricate solid-state materials platform for the controlled release of NO, we linked C12RhMOP with a ditopic ligand on the external surface for assembling them into extended polymeric structures. In particular, we choose bix as a linker, which was shown to produce submicrometer CPPs,19 as analogous coordination spheres to a molecular counterpart of C12RhMOP-biz.
The addition of 6 eq. mol of bix to a C12RhMOP solution in DMF resulted in the formation of CPP (named here C12RhMOP-CPP) with an average particle size of 252 ± 63 nm (Fig. 2c). A quantitative NMR analysis confirms that all the external metal sites in the MOP monomers are saturated with the ditopic ligands (Fig. S4, ESI†), while the internal open metal sites inside the cavities remain available to be loaded with NO molecules (1 available open metal sites per the paddlewheel moiety).
Finally, we confirmed that the three materials displayed permanent porosity by CO2 adsorption at 195 K (Fig. 2d). Noteworthy, this aspect is of paramount importance because the materials must be thermally activated to make available the open metal sites in the cages before being loaded with NO.
Preparation and characterization of the NO-loaded materials
We evaluated their ability to load NO by chemisorption. The NO adsorption isotherms shown in the Fig. 3a indicate that all materials adsorb NO at 298 K, suggesting a strong affinity towards NO. Foremost, the amount of NO adsorbed by each compound is in full agreement with the number of available open metal sites (Table S1, ESI†).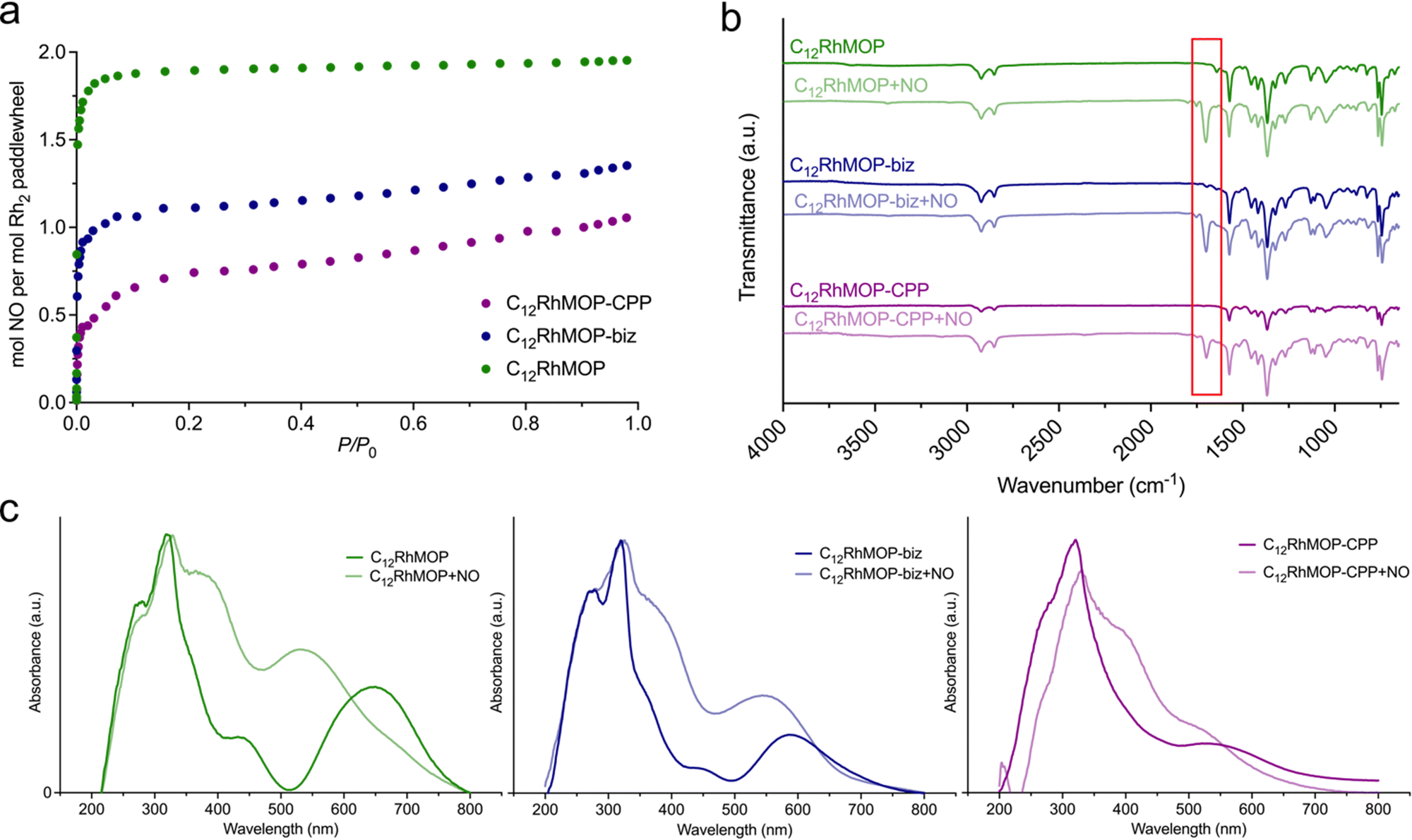 | ||
| Fig. 3 (a) Adsorption isotherm of NO for C12RhMOP (green curve), C12RMOP-biz (blue curve) and C12RhMOP-CPP (violet curve) at 298 K, (b) FTIR spectra of all materials before and after being loaded with NO. The red frame highlights the new band appearing at 1698 cm−1 in the material loaded with nitric oxide, (c) diffuse reflectance spectra for all materials before and after being loaded with nitric oxide. | ||
Additionally, we analysed the NO-adsorbed solids by infrared spectroscopy (FTIR) and diffuse-reflectance spectroscopy. The FTIR spectra of all samples show a new band appearing at 1698 cm−1 assigned to the N–O stretching vibration. This band is shifted to lower energy in comparison with the characteristics band of free gaseous NO at 1900 cm−1, ruling out the physisorption of NO molecules inside the cage cavities. The diffuse-reflectance spectroscopy revealed that the loading of NO leads to a blue shift of the lowest energy band of all three materials. Indeed, a concomitant change in the colour of the solids was observed from green for C12RhMOP, blue for C12RhMOP-biz or purple for C12RhMOP-CPP, to red for all NO-loaded compounds (Fig. 3c). As anticipated above, this absorption band is highly dependent on the chemical nature of the ligands coordinated to the axial position of dirhodium paddlewheel moieties. Therefore, altogether, these results confirm the feasibility of loading the three compounds with NO molecules by the saturation of their available open metal sites (named here, C12RhMOP + NO, C12RhMOP-biz + NO, C12RhMOP-CPP + NO, respectively). While both internal and external open sites in C12RhMOP are saturated with NO molecules (2 mmol of NO per the paddlewheel), the C12RhMOP-CPP can host one molecule of NO per the paddlewheel at the available open metal sites inside the cavities of the cage. In the case of C12RhMOP-biz, where the external open sites are partially occupied with biz molecules, both available external and internal sites are saturated with molecules of NO. The resulting NO payloads for C12RhMOP + NO, C12RhMOP-biz + NO and C12RhMOP-CPP + NO, expressed as mmol of NO adsorbed per gram of material, were estimated to be 2.1, 1.3 and 1.0 mmol g−1, respectively. Noteworthy, these values are in line with the comparable NO loadings observed for MOFs with open metal sites (Table S2, ESI†). For instance, the MIL-88 family of MOFs composed by iron(III) carboxylates (FeOX(L)3, L = fumarate, terephthalate, 2-aminoterephthalate or 2-nitroterephthalate) and the copper(II)-based MOF known as HKUST-1 (Cu3(trimesate)2) show uptakes in the range of 1–2.5 mmol g−1.11,13 The isoreticular CPO-27 series (M2(dhtp), dhtp = 2,5-dioxido-1,4-benzenedicarboxylate, M = Co2+, Ni2+ and Fe2+) have proved to possess higher payloads (6–7 mmol of NO per gram),12,14 due to the large density of open metal sites in their structures.
Study of NO-release
Apart from storing relevant quantities of NO, an ideal NO-releasing material should release it in response to an external stimulus or to a specific physiological change. Among the NO-releasing materials with open metal sites, water or moisture is the main chemical stimulus used for triggering the NO release. The mechanism of the NO release relies on the binding of the water molecules to the metal sites, which leads to the release of the NO molecules by a ligand-exchange reaction. Although this strategy has already been successfully exploited and it offers certain control on the NO release (e.g., by modulating the relative humidity of an airflow),23 it is highly desirable the development of systems with alternative or complementary approaches for controlling the gas release. Such an increase in the materials’ versatility widens their potential fields of applications. Light is a non-invasive stimulus enabling exquisite spatial control on the NO release. In addition, the feasibility of manipulating several parameters such as the power of irradiation or wavelength allows to gain control of the release kinetics. Several metal-nitrosyl complexes have proved to release NO by the excitation of their metal to ligand charge transfer band with UV-visible light.24 This fact, together with the photoactivity of dirhodium tetracarboxylates compounds,25,26 prompts us to assess if the NO-loaded C12RhMOP-based materials offer a photoresponsive NO release.In the first step, we took advantage of the feasibility of soluble MOPs and evaluated the photostability of discrete C12RhMOP + NO and C12RhMOP-biz + NO molecules in solution. We monitored the evolution of the UV-Vis absorption spectra of the solutions of both molecules after their irradiation with visible light (300–600 nm) and compared the changes with those in the dark. Toluene was chosen as a solvent due to its non-coordinating character, avoiding an unwanted solvent-induced ligand-exchange reaction.
The UV-vis spectra of both C12RhMOP + NO and C12RhMOP-biz + NO solutions in toluene remained unaltered when they were kept in the dark, confirming their stability in absence of water and under dark conditions (Fig. S5, ESI†). By contrast, the irradiation with visible light of both solutions led to a progressive red shift of the lowest-energy absorption band for both compounds (Fig. S6, ESI†). Given that this absorption band is sensitive to changes in the nature of the ligands coordinating to the axial sites of the dirhodium paddlewheel moieties, these results suggest that the NO molecules are gradually removed from the MOPs. Indeed, the UV-Vis spectra of C12RhMOP + NO after the longer irradiation time correspond to the spectra of the unloaded counterpart, supporting that the NO-chemisorption process is reversible by photoactivation in solution (Fig. S7, ESI†). The UV-vis absorption spectrum of C12RhMOP-biz + NO did not change after 30 min of irradiation while the spectrum of C12RhMOP + NO was gradually changing even after 240 min of irradiation. This evidence suggests that the imidazole ligand bonded to the axial site of the paddlewheel moieties weakens the Rh–NO bond through the Rh–Rh bond.
To understand its mechanism, we performed computational analysis using density functional theory. We used the model compounds Rh2(HCO2)4(NO)2 and Rh2(HCO2)4(NO)(1-methyl imidazole) for the dirhodium moiety of C12RhMOP + NO and C12RhMOP-bix + NO, respectively. The DFT analysis shows a binding energy of 35.7 kcal mol−1 for each NO ligand in Rh2(HCO2)4(NO)2, and a binding energy of 19.5 kcal mol−1 for the NO ligand in Rh2(HCO2)4(NO)(1-methyl imidazole). These results indicate that the substitution of one molecule of NO by an imidazole ligand weakens the remaining Rh–NO bond, supporting the experimental results (Fig. S8 and Table S3, ESI†)
To take advantage of assembling states of MOPs, we further studied the release from the solid-state materials of C12RhMOP + NO, C12RhMOP-biz + NO and C12RhMOP-CPP + NO by NO analyser that selectively detects gaseous NO with ozone-assisted chemiluminescence. To perform these experiments, the materials were first spin-coated on a glass substrate and introduced in a sealed chamber with a quartz window. Then, the samples were irradiated with visible light (300–600 nm) while a nitrogen flow went through the sample chamber carrying the NO generated from the samples to the analyser (Fig. 4a, Fig. S10a and S11a, ESI†).
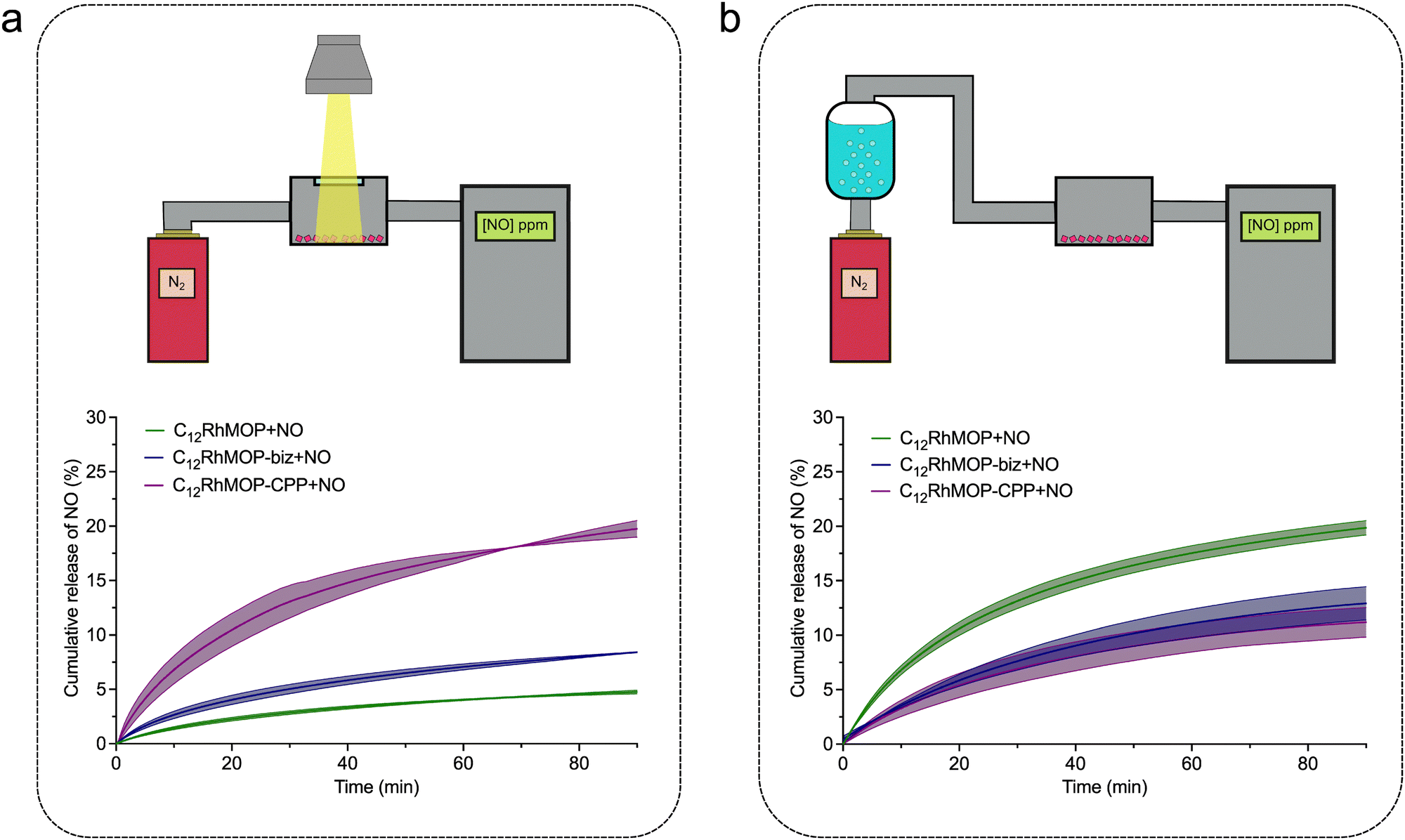 | ||
| Fig. 4 Schematic representation of the experimental setup and cumulative release of NO from C12RhMOP + NO (green curves), C12RhMOP-biz + NO (blue curves) and C12RhMOP-CPP + NO (violet curves) in solid state: (a) under photoactivation with visible light and (b) in contact with a nitrogen flow saturated with water. All the measurements were performed by triplicate (translucent area). | ||
The results shown in Fig. 4a demonstrate that all materials released NO under photoactivation in the solid state. In addition, as in the experiments performed in solution, C12RhMOP + NO shows a slower NO-release rate than C12RhMOP-bix + NO. Note that the assembled material of C12RhMOP-CPP + NO showed the fastest and most efficient photoresponsive NO-release in the solid state among the three compounds. Given that the dirhodium paddlewheel units in both C12RhMOP-bix + NO and C12RhMOP-CPP + NO have a similar coordination environment, the difference in photoresponsive NO-release kinetics observed is explained by following two main reasons. (i) The effectiveness of the photoactivation of the materials in the solid state is highly dependent on the penetration depth of light.27 Therefore, C12RhMOP-CPP + NO, which forms spherical particles with an average diameter of 250 nm shows a more effective photo-triggered NO-release than C12RhMOP-biz + NO, which is only obtained as uncontrollable micrometre-sized ill-defined particles (Fig. S9, ESI†). (ii) The external voids in C12RhMOP-CPP + NO generated by the crosslinking of the cages allow a better diffusion of the NO molecules released, compared to the case of C12RhMOP-biz + NO particles, where the cages are closely aggregated in the solid state. These results bring to light the importance of the controlled assembly of the cages into extended polymeric structures for preparing efficient light-triggered NO-releasing platforms in the solid state.
Finally, given that all porous materials with open metal sites exploited as NO-releasing materials show a water-triggered gas release,10 we evaluated the ability of the three materials to release NO by water-exchange in the solid state. The experimental setup used for these experiments similar to the one for the photoresponsive studies except the use of the nitrogen flow saturated with water before passing through the sample chamber and the measurement in the dark to avoid an unwanted light-triggered release (Fig. 4b, Fig. S10b and S11b, ESI†). All materials released NO by ligand exchange reaction with water; C12RhMOP-NO shows the most efficient NO release while both C12RhMOP-biz + NO and C12RhMOP-CPP + NO behaves similarly. This difference can be explained by the accessibility of water to NO coordination sites. In the case of C12RhMOP-biz + NO and C12RhMOP-CPP + NO, NO molecules are encapsulated inside the cavity of MOPs, to which water molecules are hardly accessible. By contrast, C12RhMOP + NO also possesses NO molecules at the external surface of MOP, with which water molecules easily replace.
Note that any of the materials studied did not desorb the total amount of NO-loaded in the solid state. This phenomenon was previously observed for other porous materials with open metal sites loaded with NO such as HKUST-1 (Table S4, ESI†) and the reason was rationalized by the strong coordination bond between copper ion and NO molecules. In addition, there are specific reasons that explain the low desorption of NO observed during the experiments in the solid state. Regarding the water-triggered NO release, the hydrophobic groups in the external surface of the cages most likely limit the diffusion of the water molecules inside the solids to reach the metal sites, hindering the ligand exchange reaction. In the case of the photoresponsivity studies, the low NO-release observed in the solid state can be attributed to the penetration depth of the light. The particle size of the three materials, even in the case of the coordination polymer particles, can be influenced by the penetration depth of the visible light, preventing the irradiation of the whole sample, and limiting the amount of NO released. The similar particle size dependency was previously observed for photoactive carbon monoxide releasing materials.28
Conclusions
We demonstrated that the robust metal–organic polyhedra C12RhMOP ([Rh2(bdc-C12)2]12) with open metal sites load NO by chemisorption and release them in response to two different stimuli: light and water. In addition, we proved that the functionalization of the external surface of the cages modulates their NO-release properties. The coordination of 1-benzylimidazole to the external open sites of the cages weakens the Rh–NO bond, leading to a more efficient photoresponsive NO release, both in solution and in the solid state. Furthermore, the assembly of the C12RhMOPvia the surface functionalization with a bis(imidazole) linker resulted in extended polymeric particles and showed a more effective light-triggered NO release than the NO-loaded molecular counterparts, thanks to the extrinsic pores generated between the linked MOPs. On the contrary, the functionalization of the cages with imidazole linkers on the surface limits the NO binding only inside the cavity, to which water molecules are more difficult to access, leading to the less efficient water-triggered NO release. Notwithstanding, the amount of NO released from the three C12RhMOP-based materials under two different triggers, light and water, is biologically relevant, paving the way for the use of these materials in cell biology or medical research.Author contributions
F. J. C. and S. F. conceived and designed the project. F. J. C., T. N. C. and E. S. G. performed all synthetic and characterization experiments. J. P. and Y. H. performed DFT calculation. F. J. C., T. N. C. and S. F. analysed the data and wrote the manuscript. All authors discussed the results and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
T. N. C. is grateful to the MEXT scholarship. This study was supported by JSPS KAKENHI grant number 21K18192 (Challenging Research (Pioneering)) for S. F. The authors thank the iCeMS Analysis Center for access to analytical instruments.Notes and references
- R. Wang, Trends Biochem. Sci., 2014, 39, 227–232 CrossRef CAS PubMed.
- J. O. Lundberg and E. Weitzberg, Cell, 2022, 185, 2853–2878 CrossRef CAS PubMed.
- P. N. Coneski and M. H. Schoenfisch, Chem. Soc. Rev., 2012, 41, 3753–3758 RSC.
- T. Yang, A. N. Zelikin and R. Chandrawati, Adv. Sci., 2018, 5, 1701043 CrossRef PubMed.
- S. Furukawa, Cell-Inspired Materials and Engineering, Springer, Cham, 2021, pp. 125–133 Search PubMed.
- Y. Wo, E. J. Brisbois, R. H. Bartlett and M. E. Meyerhoff, Biomater. Sci., 2016, 4, 1161–1183 RSC.
- A. W. Carpenter and M. H. Schoenfisch, Chem. Soc. Rev., 2012, 41, 3742–3752 RSC.
- P. G. Wang, M. Xian, X. Tang, X. Wu, Z. Wen, T. Cai and A. J. Janczuk, Chem. Rev., 2002, 102, 1091–1134 CrossRef CAS PubMed.
- Y. Zhou, T. Yang, K. Liang and R. Chandrawati, Adv. Drug Delivery Rev., 2021, 171, 199–214 CrossRef CAS PubMed.
- A. Carné-Sánchez, F. J. Carmona, C. Kim and S. Furukawa, Chem. Commun., 2020, 56, 9750–9766 RSC.
- B. Xiao, P. S. Wheatley, X. Zhao, A. J. Fletcher, S. Fox, A. G. Rossi, I. L. Megson, S. Bordiga, L. Regli, K. M. Thomas and R. E. Morris, J. Am. Chem. Soc., 2007, 129, 1203–1209 CrossRef CAS PubMed.
- A. C. McKinlay, B. Xiao, D. S. Wragg, P. S. Wheatley, I. L. Megson and R. E. Morris, J. Am. Chem. Soc., 2008, 130, 10440–10444 CrossRef CAS PubMed.
- A. C. McKinlay, J. F. Eubank, S. Wuttke, B. Xiao, P. S. Wheatley, P. Bazin, J. C. Lavalley, M. Daturi, A. Vimont, G. De Weireld, P. Horcajada, C. Serre and R. E. Morris, Chem. Mater., 2013, 25, 1592–1599 CrossRef CAS.
- E. D. Bloch, W. L. Queen, S. Chavan, P. S. Wheatley, J. M. Zadrozny, R. Morris, C. M. Brown, C. Lamberti, S. Bordiga and J. R. Long, J. Am. Chem. Soc., 2015, 137, 3466–3469 CrossRef CAS PubMed.
- V. Guillerm, D. Kim, J. F. Eubank, R. Luebke, X. Liu, K. Adil, M. S. Lah and M. Eddaoudi, Chem. Soc. Rev., 2014, 43, 6141–6172 RSC.
- S. Furukawa, N. Horike, M. Kondo, Y. Hijikata, A. Carné-Sánchez, P. Larpent, N. Louvain, S. Diring, H. Sato, R. Matsuda, R. Kawano and S. Kitagawa, Inorg. Chem., 2016, 55, 10843–10846 CrossRef CAS PubMed.
- J. Troyano, S. Horike and S. Furukawa, J. Am. Chem. Soc., 2022, 144, 19475–19484 CrossRef CAS PubMed.
- A. Khobotov-Bakishev, L. Hernández-López, C. von Baeckmann, J. Albalad, A. Carné-Sánchez and D. Maspoch, Adv. Sci., 2022, 9, 1–19 Search PubMed.
- A. Carné-Sánchez, G. A. Craig, P. Larpent, T. Hirose, M. Higuchi, S. Kitagawa, K. Matsuda, K. Urayama and S. Furukawa, Nat. Commun., 2018, 9, 1–8 CrossRef.
- A. C. Ghosh, A. Legrand, R. Rajapaksha, G. A. Craig, C. Sassoye, G. Balázs, D. Farrusseng, S. Furukawa, J. Canivet and F. M. Wisser, J. Am. Chem. Soc., 2022, 144, 3626–3636 CrossRef CAS PubMed.
- E. Warzecha, T. C. Berto, C. C. Wilkinson and J. F. Berry, J. Chem. Educ., 2019, 96, 571–576 CrossRef CAS.
- B. Boyar and D. Robinson, Coord. Chem. Rev., 1983, 50, 109–208 CrossRef.
- P. S. Wheatley, A. R. Butler, M. S. Crane, S. Fox, B. Xiao, A. G. Rossi, I. L. Megson and R. E. Morris, J. Am. Chem. Soc., 2006, 128, 502–509 CrossRef CAS PubMed.
- M. J. Rose, M. M. Olmstead and P. K. Mascharak, J. Am. Chem. Soc., 2007, 129, 5342–5343 CrossRef CAS.
- P. M. Bradley, B. E. Bursten and C. Turro, Inorg. Chem., 2001, 40, 1376–1379 CrossRef CAS PubMed.
- P. K. L. Fu, P. M. Bradley and C. Turro, Inorg. Chem., 2001, 40, 2476–2477 CrossRef CAS PubMed.
- C. Kim, S. Diring, S. Furukawa and S. Kitagawa, Dalton Trans., 2015, 44, 15324–15333 RSC.
- S. Diring, A. Carné-Sánchez, J. Zhang, S. Ikemura, C. Kim, H. Inaba, S. Kitagawa and S. Furukawa, Chem. Sci., 2017, 8, 2381–2386 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: 1H-NMR spectra, UV-Vis spectra, diffuse reflectance spectra, SEM images, tables comparing the materials prepared with already reported systems, DFT calculations. See DOI: https://doi.org/10.1039/d3tb02162a |
| This journal is © The Royal Society of Chemistry 2024 |