
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Enhancing ionic conductivity and suppressing Li dendrite formation in lithium batteries using a vinylene-linked covalent organic framework solid polymer electrolyte†
Jin
Yang‡
 a,
Chenxiao
Lin‡
be,
Yonglei
Wang
b,
Yaolin
Xu
b,
Duong Tung
Pham
b,
Xiangqi
Meng
b,
Khanh Van
Tran
b,
Sijia
Cao
bd,
Nikolay
Kardjilov
b,
André
Hilger
b,
Jan Dirk
Epping
a,
Ingo
Manke
b,
Arne
Thomas
*a and
Yan
Lu
*bcd
a,
Chenxiao
Lin‡
be,
Yonglei
Wang
b,
Yaolin
Xu
b,
Duong Tung
Pham
b,
Xiangqi
Meng
b,
Khanh Van
Tran
b,
Sijia
Cao
bd,
Nikolay
Kardjilov
b,
André
Hilger
b,
Jan Dirk
Epping
a,
Ingo
Manke
b,
Arne
Thomas
*a and
Yan
Lu
*bcd
aDepartment of Chemistry/Functional Materials, Technische Universität Berlin, Hardenbergstraße 40, 10623 Berlin, Germany. E-mail: arne.thomas@tu-berlin.de
bInstitute of Electrochemical Energy Storage, Helmholtz-Zentrum Berlin für Materialien und Energie, Hahn-Meitner-Platz 1, 14109 Berlin, Germany. E-mail: yan.lu@helmholtz-berlin.de
cInstitute for Technical and Environmental Chemistry, Friedrich-Schiller-Universität Jena, Philosophenweg 7b, 07743 Jena, Germany
dHelmholtz Institute for Polymers in Energy Applications (HIPOLE), Philosophenweg 7b, 07743 Jena, Germany
eCollege of New Energy, Ningbo University of Technology, 315336 Ningbo, China
First published on 5th December 2023
Abstract
The growing demand for energy-dense and safe batteries drives research towards all-solid-state lithium (Li) batteries. Existing poly(ethylene oxide) (PEO)-based solid polymer electrolytes (SPEs) suffer from low Li+ conductivity and Li dendrite penetration. Covalent organic frameworks (COFs) as highly crystalline, porous, and chemically diverse organic materials show great potential to address these problems. However, extensively studied imine-linked COFs show insufficient electrochemical stability against reactive Li metal, limiting their application for Li batteries. Herein, we develop a chemically stable vinylene-linked covalent organic framework (VCOF)-based SPE. By incorporating <4 wt% VCOF-1, a 25% improvement in ionic conductivity and a 46% increase in Li+ transference number at 60 °C are achieved. DFT calculations reveal that VCOF-1 facilitates Li+ transport through its cylindrical pores aided by PEO. In situ X-ray tomography confirms that VCOF-1 substantially suppresses Li dendrite growth in the VCOF-SPE-based Li metal batteries attributed to the enhanced Li-ion conduction and 12-fold improved mechanical strength. VCOF-SPEs also exhibit a high capacity of ∼145 mA h g−1 at 0.1C in LiFePO4|Li coin cells. Notably, the LiFePO4|Li pouch cell withstands abuse test conditions such as folding, cutting, and nail penetration. These results demonstrate the potential of VCOFs in future all-solid-state Li metal batteries for energy storage.
1 Introduction
Lithium (Li) metal batteries are particularly useful devices for sustainable energy storage due to their high energy density.1–3 Nevertheless, numerous challenges remain regarding their widespread use, e.g., the safety risks related to hazardous Li dendrite growth and the high flammability of conventional organic liquid electrolytes.4 The increasing demand for safe Li metal batteries has been driving the research toward solid-state electrolytes.2,3,5–9 Polyethylene oxide (PEO) based solid polymer electrolytes (SPEs) are considered promising electrolytes for all-solid-state Li metal batteries due to their electrochemical stability against Li metal and mechanical flexibility.2,3,10–12 However, Li dendrite growth and penetration through the PEO-based SPEs is a critical issue due to their limited mechanical strength, which causes battery failure and safety threats.13,14 Besides, the high crystallinity of PEO results in poor ionic conductivity (10−8 to 10−7 S cm−1) at room temperature and an inferior Li-ion transference number, far from meeting the practical application requirements.15 An efficient approach to deal with the issues of PEO-based SPEs mentioned above is to add plasticizers or polymers into PEO membranes.16 The Ooyama's group found that the ionic conductivity of PEO/LiClO4 blended with polyethylenimine (PEI) is much higher (∼10−4 S cm−1 at room temperature) than that of PEO/LiClO4 electrolyte.17 The introduction of PEI could efficiently suppress the crystallization of PEO and thus sharply increase the Li-ion mobility and ionic conductivity.18Recently, covalent organic frameworks (COFs) as crystalline and highly porous organic materials have demonstrated distinct advantages as electrolyte and electrode materials in lithium batteries.3,19,20 By choosing diverse functional organic building blocks, the chemical and physical properties and functionality of COFs can be tuned precisely, e.g., to facilitate redox reactions and the chemical interaction of different components in the batteries.19 When used in SPEs, it was reported that the ordered, micro- or mesopores of the COFs can provide pathways for Li-ion transport.21 Moreover, COFs are typically fine powders that can intimately interact with the electrodes or electrolytes of the batteries, facilitating the interfacial reactions. However, typically, COFs are synthesized from highly reversible linkages (bonds formed to connect the building blocks) such as boroxines or imines. The reversibility of these linkages allows error correction of the crystal structures, which facilitates the high crystallinity but compromises the chemical stability of the COFs.22,23 In particular, the electro-/chemical stability of the most often reported imine-linked COFs against reactive Li metal is insufficient, largely compromising their potential for applications in Li metal batteries.24,25
When searching for COFs with improved electro-/chemical stability, vinylene-linked 2D COFs (VCOFs), which are chemically stable owing to their non-polar vinylene bond, have emerged as particularly promising COF materials.26–35 The non-polar vinylene bond also yields a fully in-plane π-conjugation, which could enhance the charge transfer of VCOFs compared with that of COFs constructed by heteroatom-containing linkages.36 Zhang's group reported the preliminary Li+ conduction ability of a novel VCOF-based SPE.32 Thus, VCOFs may potentially assist the ionic transport process and enhance the ionic conductivity of VCOF-based SPEs. However, to the best of our knowledge, VCOF-based SPEs have not been explored in rechargeable batteries.32
Here, we report a VCOF-SPE that is composed of VCOF-1,26 polyethylene oxide (PEO), polyvinylidene fluoride (PVDF), and lithium bis(trifluoromethanesulfonyl)imide (LiTFSI) as a solid-state electrolyte for Li batteries.15 VCOF-1 was synthesized from commercially available raw materials in conventional autoclaves following a facile and scalable solvothermal method.26,35 The addition of VCOF-1 in the VCOF-SPE is demonstrated to enhance the Li+ conductivity and transference number, benefiting from the short hopping distances of Li+ between neighboring VCOF-1 layers. The great potential of VCOFs as functional additives to advance SPEs towards their application in all-solid-state Li metal batteries is demonstrated.
2 Results and discussion
2.1 Characterization of VCOF-1 and VCOF-SPE
VCOF-1 was synthesized from 2,4,6 trimethyl s-triazine (TMT) and terephthalaldehyde via the solvothermal method in an autoclave.26 The successful synthesis of VCOF-1 with high crystallinity and porous 2D structures (Fig. 1b) was confirmed by a combination of powder X-ray diffraction (PXRD), Fourier transform infrared spectroscopy (FTIR), 13C solid-state cross-polarization magic-angle spinning NMR spectra (13C ss-NMR), and N2 gas sorption analyses (Fig. 1 and S2†). VCOF-1 is a fluffy powder with a bright yellow color (inset of Fig. 1c). The experimental PXRD patterns of VCOF-1 showed intense and sharp diffraction peaks, which matched well with the simulated AA stacking patterns (Fig. 1b and c). FTIR results confirmed the completion of the reaction and the formation of vinylene bonds within the COF from the aldol condensation reaction (Fig. S2a†). The 13C ss-NMR peaks of the COFs were assigned. The peak at about 170 ppm corresponds to the triazine ring, while the peak at 140 ppm was assigned to one of the carbon atoms in the newly formed vinylene linkages, further confirming the chemical structures of VCOF-1 (Fig. S2b†). From the N2 (at 77 K) sorption measurements, VCOF-1 showed a Brunauer–Emmett–Teller (BET) specific surface area of 1364 m2 g−1 (Fig. S2c†), and the pore size distribution was determined to be centered at 1.50 nm (Fig. S2f†). The morphology of the VCOFs was observed by scanning electron microscopy (SEM) and transmission electron microscopy (TEM) measurements, showing a spherical morphology with a 2D layered substructure (Fig. S2d and e†).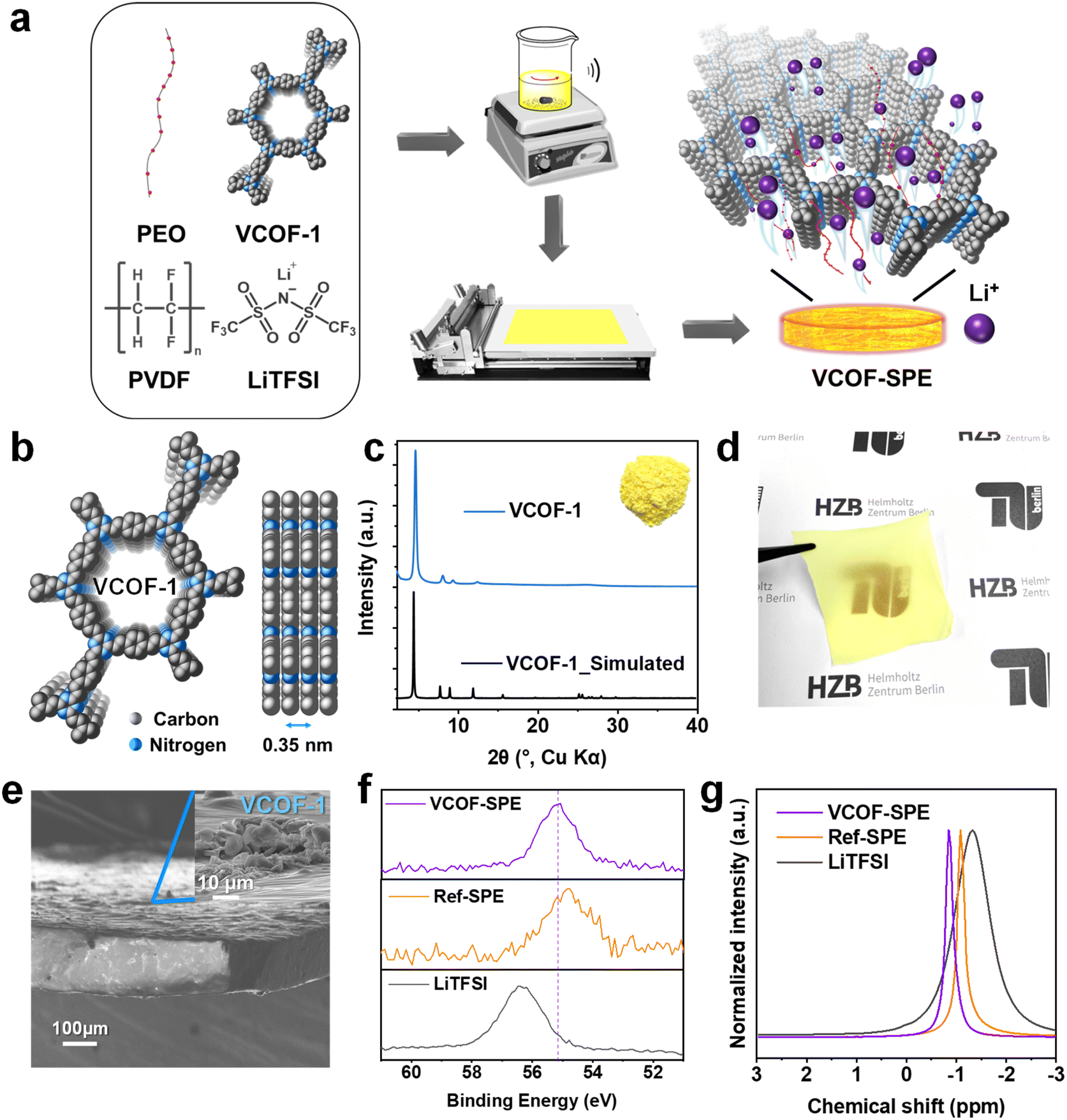 | ||
| Fig. 1 Synthesis and characterization of the SPEs. (a) The preparation procedure of the SPEs. (b) Crystal structure of VCOF-1 (c and a direction view). (c) Comparison of the experimental and simulated AA stacking PXRD patterns of VCOF-1 (inset: photograph of VCOF-1). (d) Photograph and (e) cross-sectional SEM images of the prepared VCOF-SPE. (f) High-resolution Li 1s XPS spectra, (g) static 7Li ss-NMR spectra of the VCOF-SPE and Ref-SPE compared with LiTFSI. Ref-SPE represents reference SPE with the same composition as VCOF-SPE but without adding VCOF-1. | ||
After confirming its ordered and uniform structure, the synthesized VCOF-1 was applied to PEO/lithium bis(trifluoromethanesulfonyl)imide (LiTFSI) based SPE (Fig. 1a). The content of VCOF-1 was adjusted to 3.85 wt% based on the electrochemical measurements (vide infra). Reference SPE (Ref-SPE) with the same composition but without adding VCOF-1 was also prepared as the comparative sample. PVDF was introduced in order to enhance the mechanical properties of the SPEs. The incorporation of PVDF aimed to address the issue of stickiness and difficulty in peeling off SPEs prepared solely from PEO and LiTFSI, particularly when adhered to the PTFE plate. In addition, PVDF also reduces the crystallinity of PEO and thereby enhances the mobility of Li ions.37 The prepared VCOF-SPE formed flexible and homogeneous self-standing membranes with the same color as that of VCOF-1 (Fig. 1c and d). Cross-sectional SEM images of the VCOF-SPE further showed that the VCOF-1 particles were uniformly distributed in the membrane (Fig. 1e, with the full image in S3c†), as no large gathered VCOF-1 can be observed both on the surface and inside the membrane. The PXRD pattern of VCOF-SPE revealed the (100) diffraction peak of VCOF-1, which confirmed the retention of VCOF-1's crystallinity in VCOF-SPE (Fig. S3b†). The disappearance of the diffraction peaks at 19.2° and 23.3° (2 theta degree) of PEO revealed that the crystalline contents of PEO have been largely amorphized, which releases more PEO chains to facilitate the Li+ conduction.38,39 The sharp diffraction peaks of LiTFSI also disappeared in VCOF-SPE, indicating that LiTFSI had been homogeneously dispersed within the PEO and VCOF-1 networks. The chemical environment of Li ions in the SPEs was investigated by FTIR, high-resolution X-ray photoelectron spectroscopy (XPS), and 7Li ss-NMR measurements. FTIR generally confirmed the interaction of LiTFSI, PEO, and VCOF in the SPE (Fig. S4, Table S1†).40 The –CF3 asymmetric stretching at 1197 cm−1 in LiTFSI shifted to 1182 cm−1 and became broader in VCOF-SPE, while the –CF3 symmetric stretching band at 1244 cm−1 was observed at 1227 cm−1 in Ref-SPE and 1229 cm−1 in VCOF-SPE, respectively. Meanwhile, the asymmetric –SO2– stretching band shifted from 1325 cm−1 to 1327 cm−1 in VCOF-SPE. These shifts in –SO2– and –CF3 groups suggested a strong interaction between VCOF-1, PEO, and LiTFSI. The chemical state of the Li ions was then probed by high-resolution XPS (Fig. 1f and S5†). The binding energy (BE) of Li in VCOF-SPE is located at 55.2 eV, which is different from the BE of Li in LiTFSI (56.3 eV) and Ref-SPE (54.8 eV).41,42 This indicates that Li ions merge and actively interact with VCOF-1 in VCOF-SPE. Finally, the 7Li ss-NMR in Fig. 1g unambiguously proves the distinct Li chemical environment in VCOF-SPE compared to in Ref-SPE and LiTFSI.43,44 The 7Li signals of VCOF-SPE and Ref-SPE become narrower and shift to a lower field compared to those of LiTFSI, indicating that PEO acts as a vital component to promote the movement of Li+. In addition, the higher mobility of Li+ in VCOF-SPE than in Ref-SPE can be inferred from the 7Li spin-lattice relaxation time (T1), as shorter T1 times positively correlate with better Li+ conductivity in these composites (Fig. S6†). The fitted 7Li T1 times are reduced from 0.357 s in Ref-SPE to 0.280 s in VCOF-SPE (21.5% faster), corroborating the superior Li+ conductivity of VCOF-SPE in comparison to that of Ref-SPE. Thermogravimetric analysis (TGA) showed a first weight loss for VCOF-SPE at ∼355 °C, indicating the excellent thermal stability of VCOF-SPE (Fig. S7†). The thermal behavior of VCOF-SPE was also investigated using differential scanning calorimetry (DSC). Fig. S8† shows a glass transition temperature (Tg) of around −37.9 °C. The second event, Tm1 appeared at about 47.5 °C and can be assigned to the gradual melting of the intermediate crystalline compound. The melting (Tm2) of crystalline PVDF at approximately 135 °C is observed.
2.2 Electrochemical characterization and theoretical simulations of VCOF-SPE
As the key feature of the electrochemical properties of SPEs, the ionic conductivities (σ, S cm−1) of VCOF-SPE and Ref-SPE were determined by electrochemical impedance spectroscopy (EIS). Here, stainless steel|SPEs|stainless steel (SS|SPEs|SS) symmetric coin cells were assembled to obtain impedance spectra (Fig. S9†). Fig. 2a shows the temperature dependence of ionic conductivities of VCOF-SPE and Ref-SPE in the temperature range of 10–80 °C. A sharp conductivity increase was observed from 10 to 80 °C for both the VCOF-SPE and Ref-SPE. The measured conductivities of VCOF-SPE at 10 °C and 25 °C are 1.53 × 10−7 S cm−1 and 2.11 × 10−6 S cm−1, respectively, which then increases to 2.66 × 10−4 S cm−1 at 60 °C and 7.79 × 10−4 S cm−1 at 80 °C, respectively. These values are higher than those of Ref-SPE (9.27 × 10−8 S cm−1 at 10 °C, 2.12 × 10−4 S cm−1 at 60 °C, and 6.14 × 10−4 S cm−1 at 80 °C). The ionic conductivity is increased by 25% at 60 °C by adding only 3.85 wt% of VCOF-1 into the SPE membrane. The conductivity of VCOF-SPE showed an increase with the increasing content of VCOF-1 (Fig. S10†), reaching an optimum at 3.85 wt%. Beyond this content, the conductivity started to gradually decrease. This decrease can be attributed to the combined effect of increasing Li+-free VCOF-1 and decreased Li+-containing LiTFSI, which tends to reduce conductivity. The ionic conductivities were simulated based on different SPE models. Aligning with the experimental results, a similar trend of the simulated ionic conductivity was observed for VCOF-SPEs and Ref-SPEs as the temperature changed (Fig. S11†). Also, in the atomistic simulation, VCOF-SPE exhibits a higher ionic conductivity than Ref-SPEs, which can be attributed to the stronger specific binding of TFSI anions to the VCOF-1 fragments (Fig. S12†). The nanopores in VCOFs facilitate Li-ion transport since the highly porous structure of VCOF-1 allows for the occurrence of Li-ion hopping or/and the vehicle mechanisms.45 Besides, the ordered, cylindrical micro- or mesopores of the COFs can provide pathways for Li-ion transport when VCOFs are used in SPEs.46 Above all, the introduced VCOF-1 effectively decoupled the transport of Li cations and TFSI anions, leading to decreased anion mobility and thereafter enhanced cation diffusivity in electrolytes.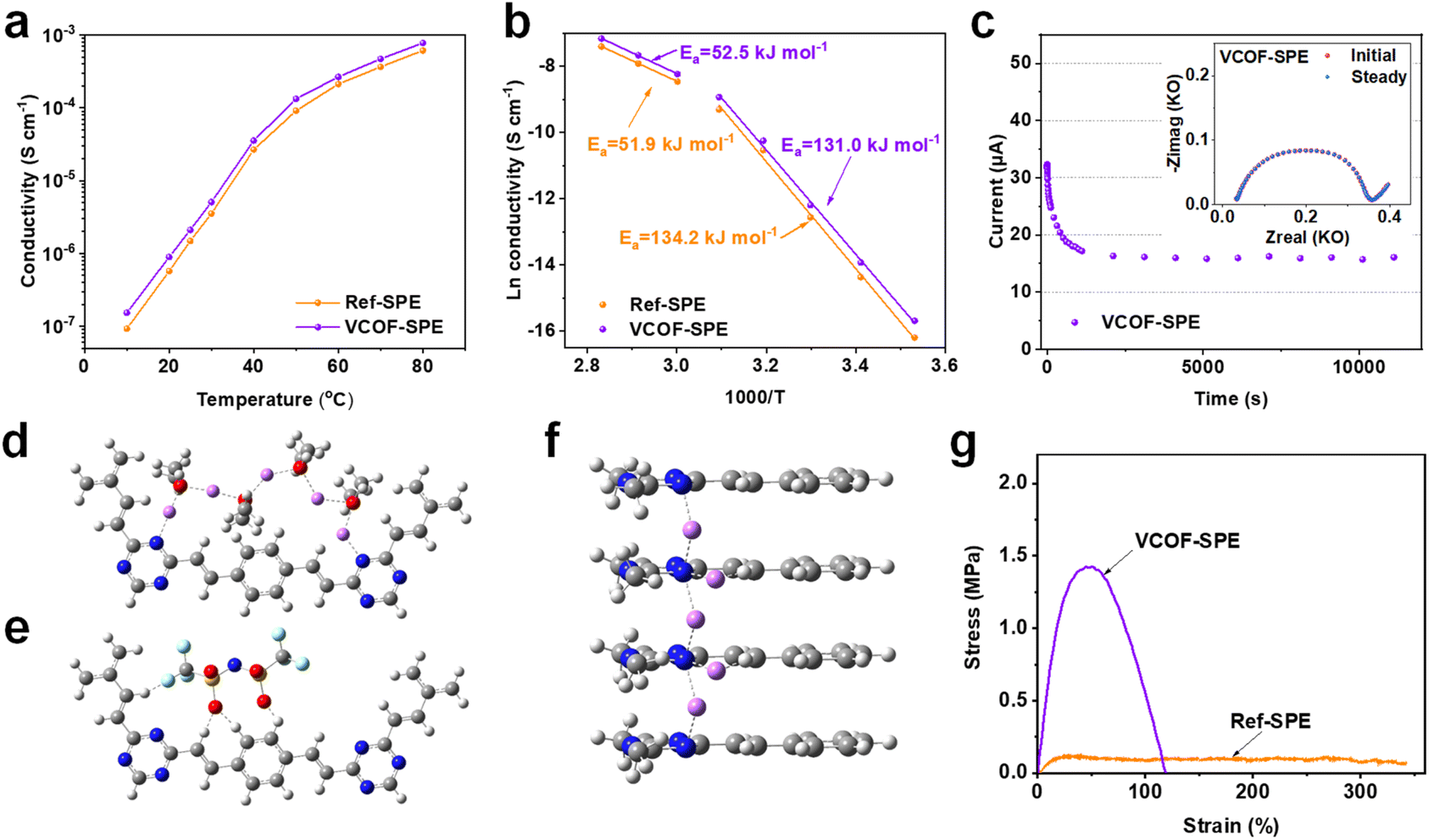 | ||
| Fig. 2 (a) Temperature-dependent conductivity of VCOF-SPE and the Ref-SPE membrane. (b) Arrhenius plots of SPEs in the temperature range of 10–80 °C. (c) Chronoamperometry of the Li|VCOF-SPE|Li cell at a potential of 10 mV (inset shows the comparison of the AC impedance of the cells before and after DC polarization at 60 °C). The preferred migration pathways of Li+ (magenta) along with planar (d) and axial (f) directions of VCOF-1 structures and preferential interactions (e) of TFSI anions with VCOF-1 layers (C: gray; N: blue; H: white; O: red; S: yellow; F: cyan; gray stick: covalent bond; dash stick: coordinate bond). (g) Stress–strain curves of VCOF-SPE and Ref-SPE. | ||
The enhanced Li+ conductivity of VCOF-SPE can also be attributed to the enhanced Li+ mobility as confirmed by XPS and ss-NMR results (Fig. 1f and g). Additionally, we found that the relationship between ln![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) σ and 1000/T followed the Arrhenius behavior (Ea = −bR, where Ea is the activation energy for ion transport, b is the slope of the regressed linear lnσ–1000/T plots, and R is the gas constant of 8.314 J mol−1 K−1). As shown in Fig. 2b, the fitted curves are divided into two linear parts. The curves align with the experimental data fitted by using the Arrhenius formula, demonstrating the typical features of ion conduction in the SPEs. The inflection points of the two curves correlated with the melting temperature of crystalline PEO from DSC results (Fig. S8†). The Ea of VCOF-SPE in the temperature range of 10–40 °C and 50–80 °C was calculated to be 131.0 kJ mol−1 and 52.5 kJ mol−1, respectively. Theoretically, the activation energy is defined as the energy barrier between adjacent coordinate sites for Li+ conduction. The change in the activation energy with the temperature can be explained by the improved motion of polymer chains and the gradual melting of the intermediate crystalline compound above 50 °C.
σ and 1000/T followed the Arrhenius behavior (Ea = −bR, where Ea is the activation energy for ion transport, b is the slope of the regressed linear lnσ–1000/T plots, and R is the gas constant of 8.314 J mol−1 K−1). As shown in Fig. 2b, the fitted curves are divided into two linear parts. The curves align with the experimental data fitted by using the Arrhenius formula, demonstrating the typical features of ion conduction in the SPEs. The inflection points of the two curves correlated with the melting temperature of crystalline PEO from DSC results (Fig. S8†). The Ea of VCOF-SPE in the temperature range of 10–40 °C and 50–80 °C was calculated to be 131.0 kJ mol−1 and 52.5 kJ mol−1, respectively. Theoretically, the activation energy is defined as the energy barrier between adjacent coordinate sites for Li+ conduction. The change in the activation energy with the temperature can be explained by the improved motion of polymer chains and the gradual melting of the intermediate crystalline compound above 50 °C.
DFT calculations were further performed to elucidate the migration pathway of Li+ inside the VCOF-1 pores by comparing the migration energy barriers at rate-determining steps in two types of anisotropic pathways that are parallel and perpendicular to axially stacked representative VCOF-1 structures (denoted as axial and planar pathways, respectively). A notable finding was that Li+ migration in the axial pathway requires a much lower migration energy barrier (∼5.7 kcal mol−1 as shown in Fig. 2f) than the in-plane one (∼26.7 kcal mol−1 as shown in Fig. 2d). This preferred through-plane Li+ migration pathway along the axial direction (Fig. 2f) is essentially attributed to the short hopping distances of Li+ between neighboring VCOF-1 layers, promoted by N atoms that are adjacently aligned in the VCOF-1 crystalline structure. In addition, the H atoms in the VCOF-1 layers and O atoms in TFSI− and PEO polymers can considerably enhance the thermodynamic stability of Li+ intermediates along with the diffusion of Li+ in the axial pathway (Fig. 2f). The hopping of Li+ along the planar pathway (Fig. 2d) is essentially mediated by the O atoms in PEO polymers with the aid of cation–anion interactions. The Li+ intermediates in the planar pathway are thermodynamically less favorable due to the long hopping distances between neighboring aromatic rings, and the mobility of ionic groups (PEO chains and TFSI anions) nearby further weakens the stability of Li+ intermediates (Fig. 2e). These theoretical results demonstrate the directional Li+ conduction along the VCOF-1 channel pores, in which the N atoms in the VCOF-1 framework play a significant role. In summary, Li+ aided by PEO can shuttle along the cylindrical pores of VCOF-1, which promotes the Li+ conductivity in VCOF-SPEs leading to a higher ionic conductivity of VCOF-SPE.
Besides the high ionic conductivity, the Li+ transference number (tLi+) is another crucial factor for SPEs.47,48Fig. 2c and S13† show the DC polarization curve and the AC impedance spectra for the Li|VCOF-SPE|Li symmetric cell setup at 60 °C, respectively. The value of tLi+ was calculated to be 0.38, which is 46% higher than that of the Ref-SPE without VCOF-1 (tLi+ = 0.26). The enhancement of the Li+ transference number with the addition of VCOF-1 could be attributed to the immobilization of TFSI anions by VCOF-1, as it possesses a much stronger binding energy with the TFSI anion (−428.6 kJ mol−1) than what is observed for PEO with the TFSI anion (−238.9 kJ mol−1) (Fig. S12†). Moreover, the mechanical properties of the SPE were evaluated by tensile tests. The stress–strain curves in Fig. 2g show that VCOF-SPE possesses a tensile strength of 1.43 MPa, which is 11.9 times that of Ref-SPE (0.12 MPa). The superior mechanical properties of VCOF-SPE should also enhance the stability of the finally assembled batteries.
In summary, experimentally, PXRD identified PEO amorphization in VCOF-SPE, which is crucial for improved Li+ conduction. FTIR, XPS, and 7Li ss-NMR revealed the distinct chemical environment of Li+ within VCOF-SPE, while 7Li ss-NMR also showcased VCOF-SPE's superior Li+ mobility compared to Ref-SPE. Additionally, DFT showed that axial Li+ transportation, i.e., along the VCOF-1 cylindrical pores, is markedly more efficient than planar Li-ion conduction. This efficiency is attributed to the shorter interlayer distance, which is enhanced by adjacent N atoms and assisted by PEO.
2.3 In situ observation of the Li dendrites
Fig. S14† shows the cross-section views of the tomography cell and corresponding average thickness at different temperatures. The average thickness increased merely by 1.6% (from 618 to 628 μm) when the cell temperature increased from ambient to 70 °C, indicating the good dimensional stability of VCOF-SPE at an elevated temperature. The growth of Li dendrites at the interface of the SPEs was directly observed by in situ X-ray tomography. The measurements were conducted by applying symmetric Li|SPE|Li tomography cells (ESI Section 3.2†) at a working temperature of 60 °C. The cell was charged and discharged at 0.1 mA cm−2 for 1 hour repetitively to allow the Li stripping plating processes. Fig. 3a and b show the time-dependent voltage profiles of the cells with VCOF-SPE and Ref-SPE over 300 hours, respectively. The VCOF-SPE cell exhibited impressively high stability throughout the cycling process (Fig. 3a). In contrast, the cycling profile of the Ref-SPE-based cell in Fig. 3b shows a steady voltage increase versus the running time. This increasing stripping/plating voltage is similar to the liquid electrolyte behavior, typically resulting from the unstable Li deposition and continuous solid electrolyte interphase (SEI) formation at the electrode/electrolyte interface.49 The large polarization of the Ref-SPE-based cell indicated its much higher internal resistance and lower ionic mobility as well. Short-circuit occurred in the Ref-SPE-based cell after 300 hours, indicating the Li dendrite penetration through the Ref-SPE due to its poor mechanical strength.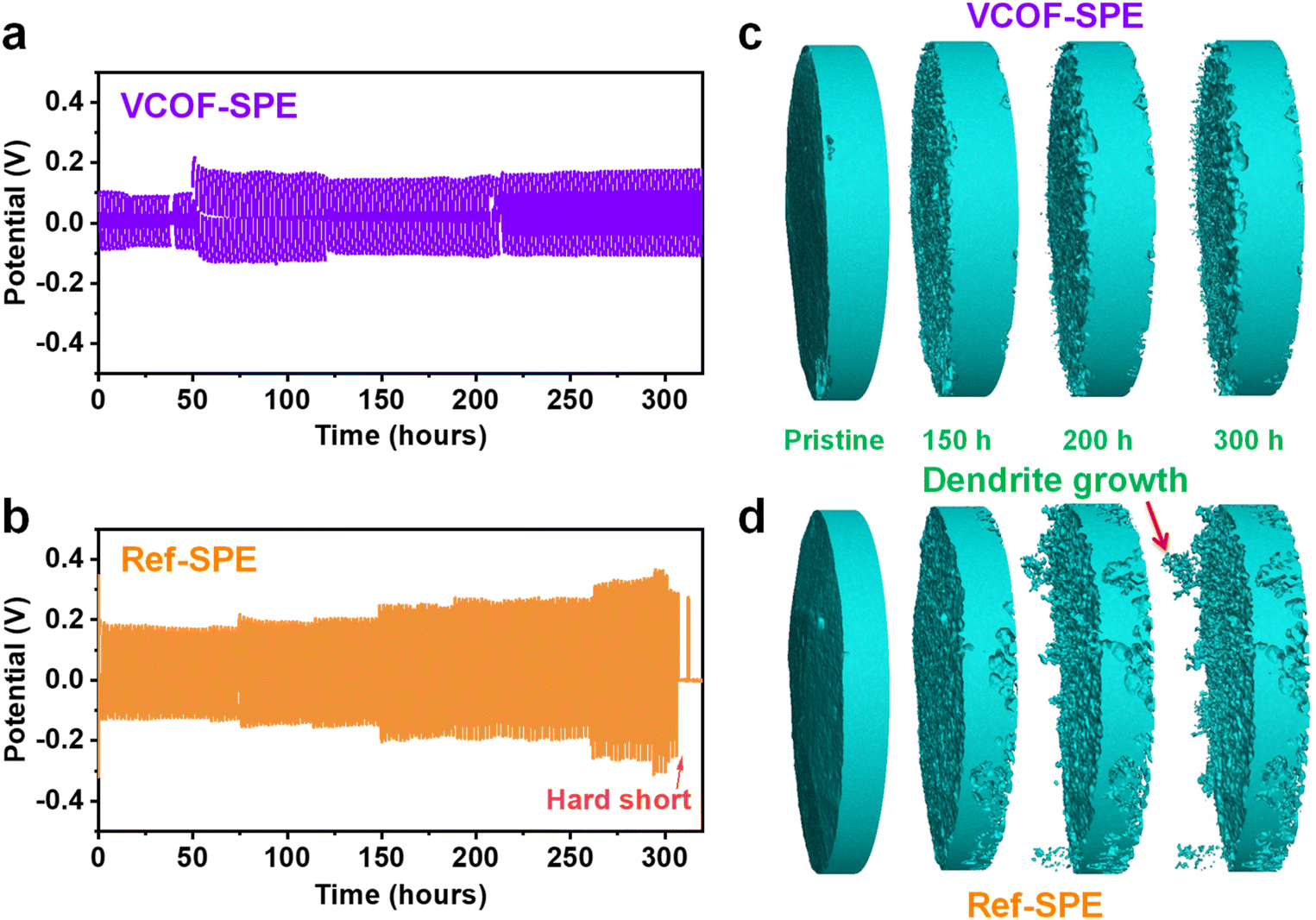 | ||
| Fig. 3 Inhibition of Li dendrites by VCOF-SPE. Stripping/plating profiles of (a) VCOF-SPE and (b) Ref-SPE samples over 300 hours at 60 °C. Tomography images of lithium electrodes inside a tomography-cell using (c) VCOF-SPE and (d) Ref-SPE after 0, 150, 200, and 300 hours of stripping/plating at a current density of 0.1 mA cm−2 and working temperature of 60 °C. | ||
In situ X-ray tomography (see details in ESI Section 3.2†) illustrated the morphological evolution of the Li foil at the original state and after 150, 200, and 300 hours of Li plating/stripping using the VCOF-SPE and Ref-SPE (Fig. 3c and d). Remarkably, the addition of VCOF-1 substantially suppressed dendrite formation throughout the 300 hour striping/plating process (Fig. 3c). In contrast, the Ref-SPE showed evident growth of Li dendrites on the electrode after 200 hours (Fig. 3d). This observation is consistent with the voltage profile of the Ref-SPE-based cell in Fig. 3b, in which a short circuit occurred after 310 hours due to the problematic Li dendrite growth and penetration through the SPE. Thus, the advantage of VCOF-SPE over the Ref-SPE toward Li dendrite suppression is revealed, which could be attributed to the synergistic effects of enhanced mechanical strength and improved Li-ion conductivity and Li-ion transference number. The enhanced mechanical strength (Fig. 2g) could restrain the growth of Li dendrites, and the significantly improved Li-ion conductivity and Li-ion transference number inhibit the formation of a Li+ gradient at the electrolyte–electrode interface. Fig. S15† demonstrates the galvanostatic cycling for SPEs at various current densities to determine the critical current density (CCD) for VCOF-SPE and Ref-SPE. As expected, the voltage of the cells increased with increasing current densities. The critical current density of VCOF-SPE is 0.55 mA cm−2, which is higher than that of Ref-SPE (0.30 mA cm−2) at 60 °C, indicating a stable interface between the lithium metal anode and VCOF-SPE membrane. Fig. S16† shows the rate performance of Li|VCOF-SPE|Li and Li|Ref-SPE|Li cells at different current densities of 0.05, 0.1, and 0.2 mA cm−2. The voltage increased as expected with increasing current densities for the VCOF-SPE and Ref-SPE membranes. The Li|VCOF-SPE|Li cell shows stable cycling with low voltage hysteresis throughout a 900 min cycle, indicating the favourable Li plating/stripping kinetics and superior interface properties. In comparison, the Li|Ref-SPE|Li cell exhibits stable cycling with high voltage hysteresis.
2.4 Half-cell performance of VCOF-SPE in a Li|VCOF-SPE|LFP coin cell
The performances of VCOF-SPE were further investigated in a coin cell made of LiFePO4 (LFP) as the cathode, VCOF-SPE as the electrolyte, and Li metal as the anode. The working temperature was maintained at 60 °C during electrochemical testing. Fig. S17† shows the CV curves of the cell at different cycles. A cathodic peak and an anodic peak were observed. The CV curves overlap after three cycles, suggesting its excellent electrochemical reversibility. Fig. S18† shows the linear sweep voltammetry (LSV) curves of VCOF-SPE and Ref-SPE. LSV was recorded by using an electrochemical workstation between 0 V and 6 V at a scanning rate of 0.1 mV s−1. As seen, VCOF-SPE is stable in the voltage range from 2.5 to 4.0 V and starts to decompose at around 4.6 V. Fig. 4a illustrates the discharge/charge profiles of the VCOF-SPE-based coin cell at 0.1C. The cell was tested to achieve a high capacity of ∼145 mA h g−1. The first cycle showed a discharging capacity of 142.6 mA h g−1, which then decreased slightly in the subsequent 100 cycles while maintaining an excellent coulombic efficiency of ∼100% (Fig. 4b). In comparison, the battery based on Ref-SPE exhibited much lower capacities of 109.2 mA h g−1 at 0.1C, which further decreased to 62.2 mA h g−1 after 68 cycles (Fig. S19†). Fig. 4c reveals the battery capacities at various current densities for the VCOF-SPE-based battery, in which 136.3, 127.4, 120.4, 115.2, and 111.2 mA h g−1 discharge capacities were recorded at 0.2, 0.4, 0.6, 0.8, and 1.0C, respectively. The Ref-SPE based battery exhibited much lower discharge capacity compared to that with VCOF-SPE at different current densities. The results prove the excellent rate ability of VCOF-SPE to adapt to various current densities. To investigate the stability of VCOF-SPE against repeated Li+ intercalation/de-intercalation, the cell was maintained at a high current density of 0.5C for 120 cycles (Fig. 4d). A positive output discharge capacity was maintained at an average of ∼120 mA h g−1 with 85% capacity retention. The discharge capacity of the LFP|VCOF-SPE|Li cell is much higher than that of LFP|Ref-SPE|Li at 0.5C due to the higher conductivity of VCOF-SPE. When cycling at 0.5C, the capacity of the Ref-SPE-based battery first increases before decreasing, which indicates a process of interfacial contact improvement. In comparison, the VCOF-SPE-based battery exhibited gradual and gentle capacity decay, indicating that the interfacial contact is stabilized within the first cycle. This can be attributed to the good compatibility of VCOF-SPE, which contributes to a stable interface between VCOF-SPE and electrodes. Besides, the LiNi0.8Co0.1Mn0.1O2(NCM811)|VCOF-SPE|Li and LiNi0.5Mn1.5O4 (LNMO)|VCOF-SPE|Li cells were further assembled to investigate the scope of use of VCOF-SPE. However, these cells cannot work normally. It is reported that pure PEO starts to oxidize at a voltage above 3.9 V versus Li/Li+.50 Besides, the electrochemical stability window of the polymer electrolyte, as determined by the LSV method, is overestimated.51 Since the charging voltage of the NCM811 and LNMO based half-cell is higher than 4.0 V, a Ni-based cathode with strong oxidizing properties could result in the decomposition of PEO-based SPEs.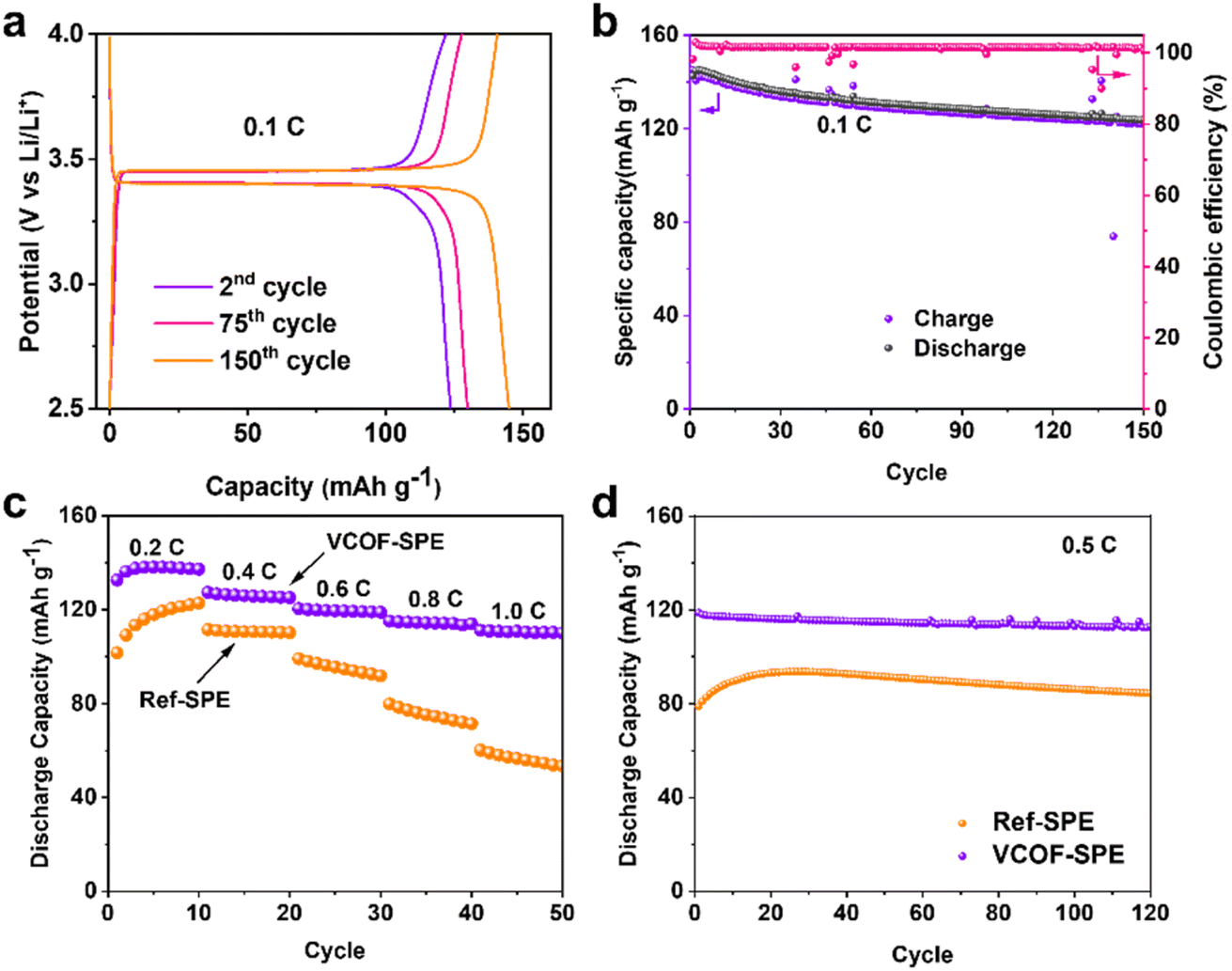 | ||
| Fig. 4 Half-cell performance of VCOF-SPE in a Li|VCOF-SPE|LFP coin cell. (a) Charge/discharge profile, (b) cycling ability at 0.1C current density, (c) rate performance at different C-rates, and (d) cycling performance at a high current density of 0.5C (1C = 170 mA g−1). | ||
2.5 Pouch-cell performance and the abuse test of VCOF-SPE
To further demonstrate the potential of VCOF-SPE for practical applications, LFP|VCOF-SPE|Li pouch cells were assembled and tested. The electrochemical performances of the pouch cells were tested at 0.1C (Fig. 5a and b). The delivered capacity reached 80 mA h g−1 at the initial cycle and stabilized at 131.5 mA h g−1 after 20 cycles. The capacity increase is associated with an electrochemical activation process of the cell, mainly attributed to the gradual improvement of interfacial contacts between the electrodes and the SPE, caused by the lack of stacking pressure in pouch cells. After activation, the cell showed excellent cycling stability, with ∼100% retention over 100 cycles (Fig. 5b). The assembled LFP|VCOF-SPE|Li pouch cells also demonstrated high flexibility and safety, which was revealed by abuse tests. As shown in Fig. 5c, the pouch cell was connected to a red light-emitting diode (LED). After slightly and completely folding the pouch cell, the LED remained lit (Fig. 5d and e). Moreover, the cell could still function and light the LED after nail penetration (Fig. 5g) and even cut by more than half (Fig. 5h), demonstrating its high safety.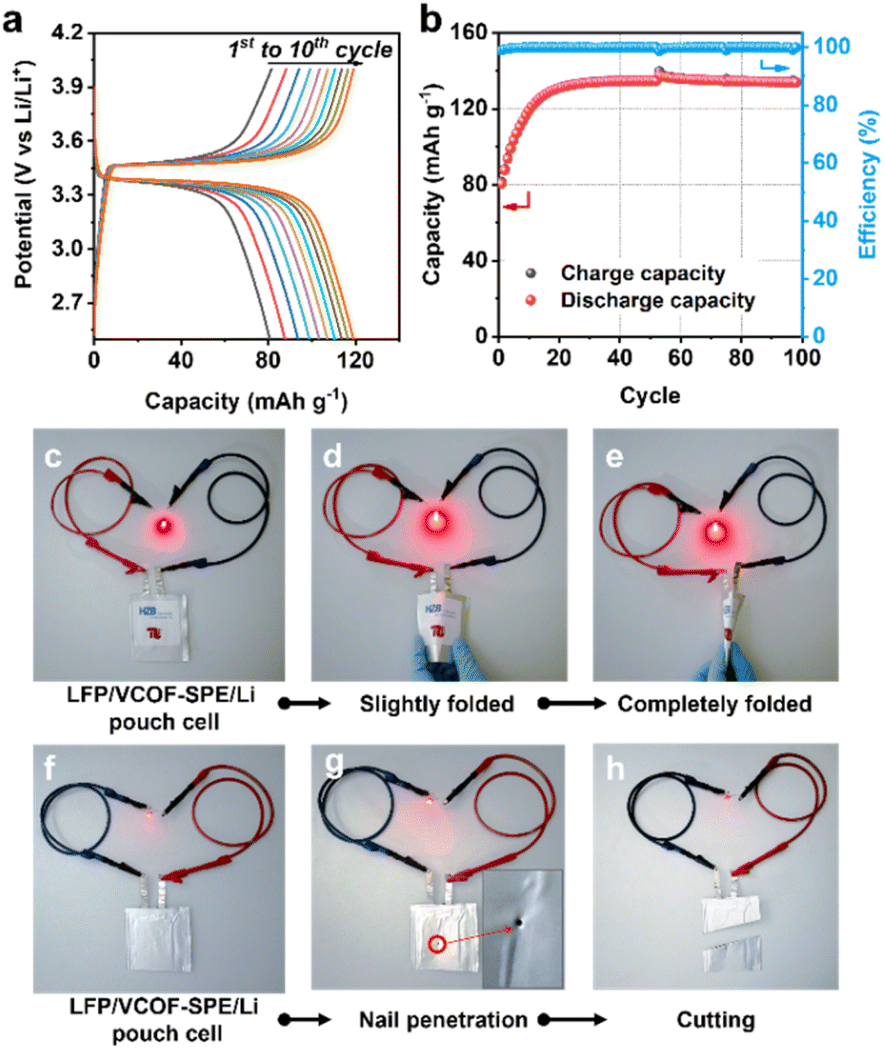 | ||
| Fig. 5 Pouch-cell performance and the abuse test of VCOF-SPE. Electrochemical performance of the LFP cathode in a Li metal pouch-cell using the VCOF-SPE: (a) charge/discharge profiles and (b) specific capacity and coulombic efficiency at 0.1C current density. (c and f) The LFP|VCOF-SPE|Li pouch cell lights a LED bulb. Abuse tests of the LFP|VCOF-SPE|Li pouch cell after being (d) slightly folded, (e) completely folded, (g) pierced, and (h) cut. | ||
3 Conclusions
In summary, we developed a chemically stable vinylene-linked COF functionalized SPE (VCOF-SPE) that is composed of VCOF-1/PEO/LiTFSI/PVDF. The addition of VCOF-1 enabled favorable migration of Li+ along the cylindrical channels in VCOF-1, facilitating Li+ conduction in SPE. The growth of Li dendrites at the Li/VCOF-SPE interface was remarkably inhibited, benefiting from the synergistic effects of improved Li-ion conductivity and transference number and greatly enhanced mechanical strength of the VCOF-SPE. Moreover, all-solid-state Li metal pouch cells with VCOF-SPE showed superior cycling performance and cycling stability compared to those with the Ref-SPE and could survive abuse tests such as folding, cutting, and nail penetration, demonstrating their operational safety. These results demonstrated the great potential of VCOFs as chemically stable and easy-to-synthesize COFs for advanced SPEs, pushing forward the development of all-solid-state Li metal batteries for future energy storage.Author contributions
Dr Jin Yang and Dr Chenxiao Lin contributed equally to this work. Jin Yang: conceptualization, methodology, validation, formal analysis, investigation, writing-original draft. Chenxiao Lin: conceptualization, methodology, formal analysis, investigation, resources, writing – original draft. Yonglei Wang: software, formal analysis, writing – original draft. Yaolin Xu: formal analysis, writing-review & editing. Duong Tung Pham: conceptualization, methodology, formal analysis, writing – original draft. Xiangqi Meng: investigation, formal analysis, writing-original draft. Khanh Van Tran: conceptualization, methodology, software, formal analysis, investigation, writing – original draft. Sijia Cao: investigation. Nikolay Kardjilov: investigation, formal analysis. André Hilger: investigation, formal analysis. Jan Dirk Epping: investigation, formal analysis. Ingo Manke: writing – review & editing. Arne Thomas: supervision, writing – review & editing. Yan Lu: supervision, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
A. Acharjya is acknowledged for his suggestions on the synthesis of VCOF-1. J. Yang acknowledges support from the IMPRS for Elementary Processes in Physical Chemistry. Support from the BMBF within the Fördermaßnahme Batterie 2020, Förderkennzeichen: 03XP0410, Dialysorb is acknowledged. Y. Lu thanks the support from the German Ministry of Education and Research (BMBF) within the research program Batterie 2020 (Förderkennzeichen: 03XP0527C, FestPoLiS)References
- S. Randau, D. A. Weber, O. Kötz, R. Koerver, P. Braun, A. Weber, E. Ivers-Tiffée, T. Adermann, J. Kulisch, W. G. Zeier, F. H. Richter and J. Janek, Nat. Energy, 2020, 5, 259–270 CrossRef CAS.
- Z. Xue, D. He and X. Xie, J. Mater. Chem. A, 2015, 3, 19218–19253 RSC.
- W.-H. Huang, X.-M. Li, X.-F. Yang, X.-X. Zhang, H.-H. Wang and H. Wang, Mater. Chem. Front., 2021, 5, 3593–3613 RSC.
- J. Wang, W. Huang, A. Pei, Y. Li, F. Shi, X. Yu and Y. Cui, Nat. Energy, 2019, 4, 664–670 CrossRef.
- X. Ke, Y. Wang, L. Dai and C. Yuan, Energy Storage Mater., 2020, 33, 309–328 CrossRef.
- X. Ke, Y. Wang, G. Ren and C. Yuan, Energy Storage Mater., 2020, 26, 313–324 CrossRef.
- F. Zheng, M. Kotobuki, S. Song, M. O. Lai and L. Lu, J. Power Sources, 2018, 389, 198–213 CrossRef.
- Q. Zhao, X. Liu, S. Stalin, K. Khan and L. A. Archer, Nat. Energy, 2019, 4, 365–373 CrossRef.
- T. Famprikis, P. Canepa, J. A. Dawson, M. S. Islam and C. Masquelier, Nat. Mater., 2019, 18, 1278–1291 CrossRef CAS PubMed.
- B. Qiu, F. Xu, J. Qiu, M. Yang, G. Zhang, C. He, P. Zhang, H. Mi and J. Ma, Energy Storage Mater., 2023, 60, 102832 CrossRef.
- B. Qiu, K. Liang, W. Huang, G. Zhang, C. He, P. Zhang and H. Mi, Adv. Energy Mater., 2023, 13, 2301193 CrossRef CAS.
- Y.-N. Liu, Z. Xiao, W.-K. Zhang, J. Zhang, H. Huang, Y.-P. Gan, X.-P. He, G. G. Kumar and Y. Xia, Rare Met., 2022, 41, 3762–3773 CrossRef CAS.
- X. Chen, B. Zhao, C. Yan and Q. Zhang, Adv. Mater., 2021, 33, 2004128 CrossRef CAS PubMed.
- M. Jia, P. Wen, Z. Wang, Y. Zhao, Y. Liu, J. Lin, M. Chen and X. Lin, Adv. Funct. Mater., 2021, 31, 2101736 CrossRef CAS.
- J. Li, K. Zhu, J. Wang, K. Yan, J. Liu, Z. Yao and Y. Xu, Mater. Technol., 2022, 37, 240–247 CrossRef CAS.
- P. Chen, Q. Zeng, Q. Li, R. Zhao, Z. Li, X. Wen, W. Wen, Y. Liu, A. Chen, Z. Li, X. Liu and L. Zhang, Chem. Eng. J., 2022, 427, 132025 CrossRef CAS.
- R. Tanaka, M. Sakurai, H. Sekiguchi, H. Mori, T. Murayama and T. Ooyama, Electrochim. Acta, 2001, 46, 1709–1715 CrossRef CAS.
- Y. Jiang, X. Yan, Z. Ma, P. Mei, W. Xiao, Q. You and Y. Zhang, Polymers, 2018, 10, 1237 CrossRef PubMed.
- D. Zhu, G. Xu, M. Barnes, Y. Li, C. Tseng, Z. Zhang, J. Zhang, Y. Zhu, S. Khalil, M. M. Rahman, R. Verduzco and P. M. Ajayan, Adv. Funct. Mater., 2021, 31, 2100505 CrossRef CAS.
- X. Zhao, P. Pachfule and A. Thomas, Chem. Soc. Rev., 2021, 50, 6871–6913 RSC.
- G. Zhang, Y. L. Hong, Y. Nishiyama, S. Bai, S. Kitagawa and S. Horike, J. Am. Chem. Soc., 2019, 141, 1227–1234 CrossRef CAS.
- R. Liu, K. T. Tan, Y. Gong, Y. Chen, Z. Li, S. Xie, T. He, Z. Lu, H. Yang and D. Jiang, Chem. Soc. Rev., 2021, 50, 120–242 RSC.
- K. T. Tan, S. Ghosh, Z. Wang, F. Wen, D. Rodríguez-San-Miguel, J. Feng, N. Huang, W. Wang, F. Zamora, X. Feng, A. Thomas and D. Jiang, Nat. Rev. Methods Primers, 2023, 3, 1 CrossRef CAS.
- Z. Zhao, W. Chen, S. Impeng, M. Li, R. Wang, Y. Liu, L. Zhang, L. Dong, J. Unruangsri, C. Peng, C. Wang, S. Namuangruk, S. Y. Lee, Y. Wang, H. Lu and J. Guo, J. Mater. Chem. A, 2020, 8, 3459–3467 RSC.
- C. Niu, W. Luo, C. Dai, C. Yu and Y. Xu, Angew. Chem., Int. Ed., 2021, 60, 24915–24923 CrossRef CAS PubMed.
- A. Acharjya, P. Pachfule, J. Roeser, F. Schmitt and A. Thomas, Angew. Chem., Int. Ed., 2019, 58, 14865–14870 CrossRef CAS PubMed.
- H. Lyu, C. S. Diercks, C. Zhu and O. M. Yaghi, J. Am. Chem. Soc., 2019, 141, 6848–6852 CrossRef CAS PubMed.
- T. Jadhav, Y. Fang, W. Patterson, C. Liu, E. Hamzehpoor and D. F. Perepichka, Angew. Chem., Int. Ed., 2019, 58, 13753–13757 CrossRef CAS.
- S. Wei, F. Zhang, W. Zhang, P. Qiang, K. Yu, X. Fu, D. Wu, S. Bi and F. Zhang, J. Am. Chem. Soc., 2019, 141, 14272–14279 CrossRef CAS.
- S. Xu, M. Richter and X. Feng, Acc. Mater. Res., 2021, 2, 252–265 CrossRef CAS.
- S. Bi, F. Meng, D. Wu and F. Zhang, J. Am. Chem. Soc., 2022, 144, 3653–3659 CrossRef.
- F. Meng, S. Bi, Z. Sun, B. Jiang, D. Wu, J. Chen and F. Zhang, Angew. Chem., Int. Ed., 2021, 60, 13614–13620 CrossRef.
- S. Bi, Z. Zhang, F. Meng, D. Wu, J. Chen and F. Zhang, Angew. Chem., Int. Ed., 2022, 61, e202111627 CrossRef.
- S. Xu, Z. Liao, A. Dianat, S. Park, M. A. Addicoat, Y. Fu, D. L. Pastoetter, F. G. Fabozzi, Y. Liu, G. Cuniberti, M. Richter, S. Hecht and X. Feng, Angew. Chem., Int. Ed., 2022, 134, e202202492 CrossRef.
- A. Acharjya, L. Longworth-Dunbar, J. Roeser, P. Pachfule and A. Thomas, J. Am. Chem. Soc., 2020, 142, 14033–14038 CrossRef.
- T. He, K. Geng and D. Jiang, Trends Chem., 2021, 3, 431–444 CrossRef.
- V. St-Onge, M. Cui, S. Rochon, J.-C. Daigle and J. P. Claverie, Commun. Mater., 2021, 2, 83 CrossRef CAS.
- S. Xue, Y. Liu, Y. Li, D. Teeters, D. W. Crunkleton and S. Wang, Electrochim. Acta, 2017, 235, 122–128 CrossRef CAS.
- W. A. Henderson and S. Passerini, Electrochem. Commun., 2003, 5, 575–578 CrossRef CAS.
- H. Chen, D. Adekoya, L. Hencz, J. Ma, S. Chen, C. Yan, H. Zhao, G. Cui and S. Zhang, Adv. Energy Mater., 2020, 10, 2000049 CrossRef CAS.
- H. Xu, P. H. Chien, J. Shi, Y. Li, N. Wu, Y. Liu, Y. Y. Hu and J. B. Goodenough, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 18815–18821 CrossRef CAS.
- K. N. Wood and G. Teeter, ACS Appl. Energy Mater., 2018, 1, 4493–4504 CrossRef CAS.
- C. Gerbaldi, J. R. Nair, M. A. Kulandainathan, R. S. Kumar, C. Ferrara, P. Mustarelli and A. M. Stephan, J. Mater. Chem. A, 2014, 2, 9948–9954 RSC.
- D. A. Vazquez-Molina, G. S. Mohammad-Pour, C. Lee, M. W. Logan, X. Duan, J. K. Harper and F. J. Uribe-Romo, J. Am. Chem. Soc., 2016, 138, 9767–9770 CrossRef CAS PubMed.
- H. Yang and N. Wu, Energy Sci. Eng., 2022, 10, 1643–1671 CrossRef CAS.
- G. Zhang, Y. L. Hong, Y. Nishiyama, S. Bai, S. Kitagawa and S. Horike, J. Am. Chem. Soc., 2019, 141, 1227–1234 CrossRef CAS PubMed.
- O. Sheng, J. Zheng, Z. Ju, C. Jin, Y. Wang, M. Chen, J. Nai, T. Liu, W. Zhang, Y. Liu and X. Tao, Adv. Mater., 2020, 32, 2000223 CrossRef CAS.
- P. G. Bruce and C. A. Vincent, J. Electroanal. Chem. Interfacial Electrochem., 1987, 225, 1–17 CrossRef CAS.
- Z. Liang, G. Zheng, C. Liu, N. Liu, W. Li, K. Yan, H. Yao, P. C. Hsu, S. Chu and Y. Cui, Nano Lett., 2015, 15, 2910–2916 CrossRef CAS.
- J. Qiu, X. Liu, R. Chen, Q. Li, Y. Wang, P. Chen, L. Gan, S. Lee, D. Nordlund, Y. Liu, X. Yu, X. Bai, H. Li and L. Chen, Adv. Funct. Mater., 2020, 30, 1909392 CrossRef CAS.
- A. Méry, S. Rousselot, D. Lepage and M. Dollé, Materials, 2021, 14, 3840 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ta04822e |
| ‡ Jin Yang and Chenxiao Lin contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |