
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTotal synthesis of [β-HIle]2-nodupetide: effect of ester to amide substitution on its antimicrobial activity†
Harra Ismi Farahac,
Unang Supratman abc,
Ace Tatang Hidayatabc and
Rani Maharani*abc
abc,
Ace Tatang Hidayatabc and
Rani Maharani*abc
aDepartment of Chemistry, Faculty of Mathematics and Natural Science, Universitas Padjadjaran, Jatinangor, West Java, Indonesia. E-mail: r.maharani@unpad.ac.id
bCentral Laboratory, Universitas Padjadjaran, Jalan Raya Bandung-Sumedang KM 21, Jatinangor 45363, West Java, Indonesia
cPharmacy Faculty, Universitas Mulawarman, Samarinda 75242, East Kalimantan, Indonesia
First published on 9th July 2024
Abstract
The increasing prevalence of deaths due to multidrug-resistant bacteria (MDRB) in infectious disease therapy has become a global health concern. This led to the development of new antimicrobial therapeutic agents that can combat resistance to pathogenic bacteria. The utilization of natural peptide compounds as potential antimicrobial agents is very promising. Nodupetide, a cyclodepsipeptide with very strong antimicrobial activity against Pseudomonas aeruginosa was isolated from the fermentation of Nodulisporium sp. Unfortunately, one of its residues (3S,4S)-3-hydroxy-4-methylhexanoic acid (HMHA) is not commercially available and the synthesis strategies applied have not been successful. Hence, we synthesized its cyclopeptide analogue [β-HIle]2-nodupetide by replacing HMHA with isoleucine homologue. A combination of solid- and solution-phase peptide synthesis was successfully carried out to synthesize [β-HIle]2-nodupetide with an overall yield of 10.4%. The substitution of HMHA with β-homoisoleucine (β-HIle) changed the ester bond into an amide bond in nodupetide's backbone. The analogue was considerably inactive against Pseudomonas aeruginosa. It can be concluded that the ester bond is crucial for the antimicrobial activity of nodupetide.
Introduction
Multidrug-resistant bacteria (MDRB), triggered by inappropriate and prolonged use of antibiotics, is currently a global health concern. In recent years, there has been an increasing number of annual mortality cases caused by multidrug-resistant Pseudomonas aeruginosa (MDRP) and methicillin- and vancomycin-resistant Staphylococcus aureus (MRSA and VRSA), which are major antibiotics used for the treatment of infectious diseases.1 Thus, to reduce the prevalence of MDRB cases, it is important to develop new antimicrobial agents with a broad mechanism of action.Nodupetide 1 (Fig. 1), which is one of the natural cyclodepsipeptides, was isolated from the fermentation of the fungi Nodulisporium sp. and exhibited strong antimicrobial activity against Pseudomonas aeruginosa. Nodupetide has two non-proteogenic amino acid (NPAA) residues, namely (3S,4S)-3-hydroxy-4-methylhexanoic acid (HMHA) and D-Leu residues.2 The presence of these NPPA residues can increase the stability and permeability of peptides.3 Bioactive natural peptides can be intrinsically used as drug candidates; however, their low bioavailability and short plasma time often hinder their therapeutic use and require structural optimization.4 A strategy to address this challenge is by approaching the chemical synthesis of peptides. However, the peptide synthesis approach is also immensely challenging. It begins with the synthesis of precursors followed by the purification of the target molecule, which often present many challenges and require stupendous efforts.5
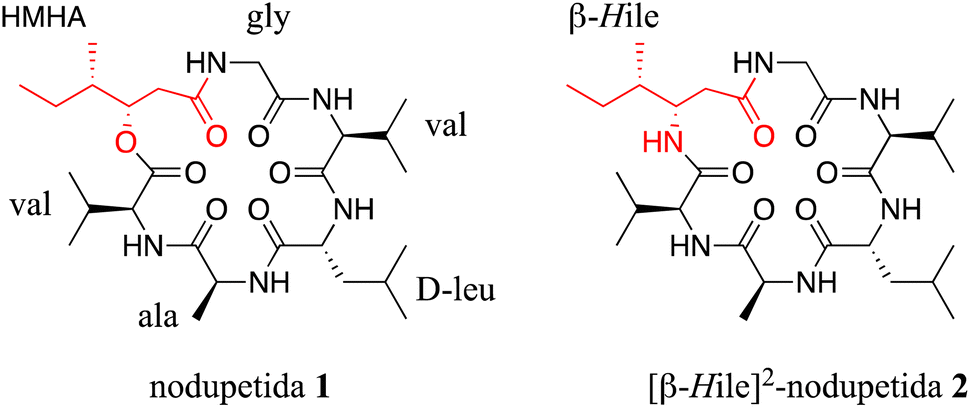 | ||
| Fig. 1 Structure of nodupetida 1 and its analogue [β-HIle]2-nodupetide 2. | ||
The challenges in the preparation of NPAA precursors are associated with stereoselectivity and low yield.4 In our study to synthesize nodupetide, we faced difficulties in synthesizing HMHA precursor. The synthesis procedure we followed involved low reproducibility, instability, and high cost, which were obstacles in the synthesis of HMHA. In addition, HMHA is not commercially available. Therefore, nodupetide has not been synthesized since HMHA is not successfully synthesized.
These obstacles lead us to replace HMHA with β-homoisoleucine (β-HIle), which is a readily available residue. β-HIle is an isoleucine homologue and also classified as NPAA, which provide the advantage of resistance to proteolysis enzymes.6 The replacement with this residue converts the ester bond into an amide bond in the nodupetide backbone.
Alteration of the peptide backbone would affect the stereochemistry, conformation and physicochemical properties of the analogue, which consequently affects its biological activity.7–9 Either it increases or decreases its antimicrobial activity. The mechanism of antimicrobial action itself is a complex process with multiple factors influencing the growth inhibition or death of microbes.10,11 As mentioned in the synthesis of daptomycin analogues, the change of ester to amide decreased their antibacterial activity.8 Chen and co-workers reported that increasing the hydrophobicity of α-helical antimicrobial peptides can increase its antimicrobial activity, but a further increase in hydrophobicity can dramatically decrease its activity.7
A combination of solid- and solution-phase peptide synthesis is an effective strategy in the development of cyclopeptide compounds.12–14 The linear peptide is synthesized in the solid phase and cyclized in the liquid phase with a very dilute concentration to prevent side reactions during the cyclization process. The synthesis of nodupetide and its analogues has not been reported previously.
Hence, in this study, we designed and synthesized [β-HIle]2-nodupetide 2, which is analogue of nodupetide 1 (Fig. 1) in which HMHA is replaced with β-HIle. In addition, the purpose of this study is to evaluate the alteration of the ester-to-amide bond in the nodupetide backbone on its antimicrobial activity.
Result and discussion
Synthesis of [β-HIle]2-nodupetide (2)
[β-HIle]2-nodupetide 2 was synthesized using a combination of solid- and solution-phase peptide synthesis (Fig. 2). Linear [β-HIle]2-nodupetide was synthesized in the solid phase, and cyclization was carried out in the solution phase. The cyclization sites chosen were glycine as C-terminus and valine as N-terminus. Glycine as C-terminus can provide an advantage to the cyclization process, as it can induce the desired conformation for cyclization. Therefore, the sequence of amino acid residues of [β-HIle]2-nodupetide is [Gly]1, [β-HIle]2, [Val]3, [Ala]4, [D-Leu]5, [Val]6.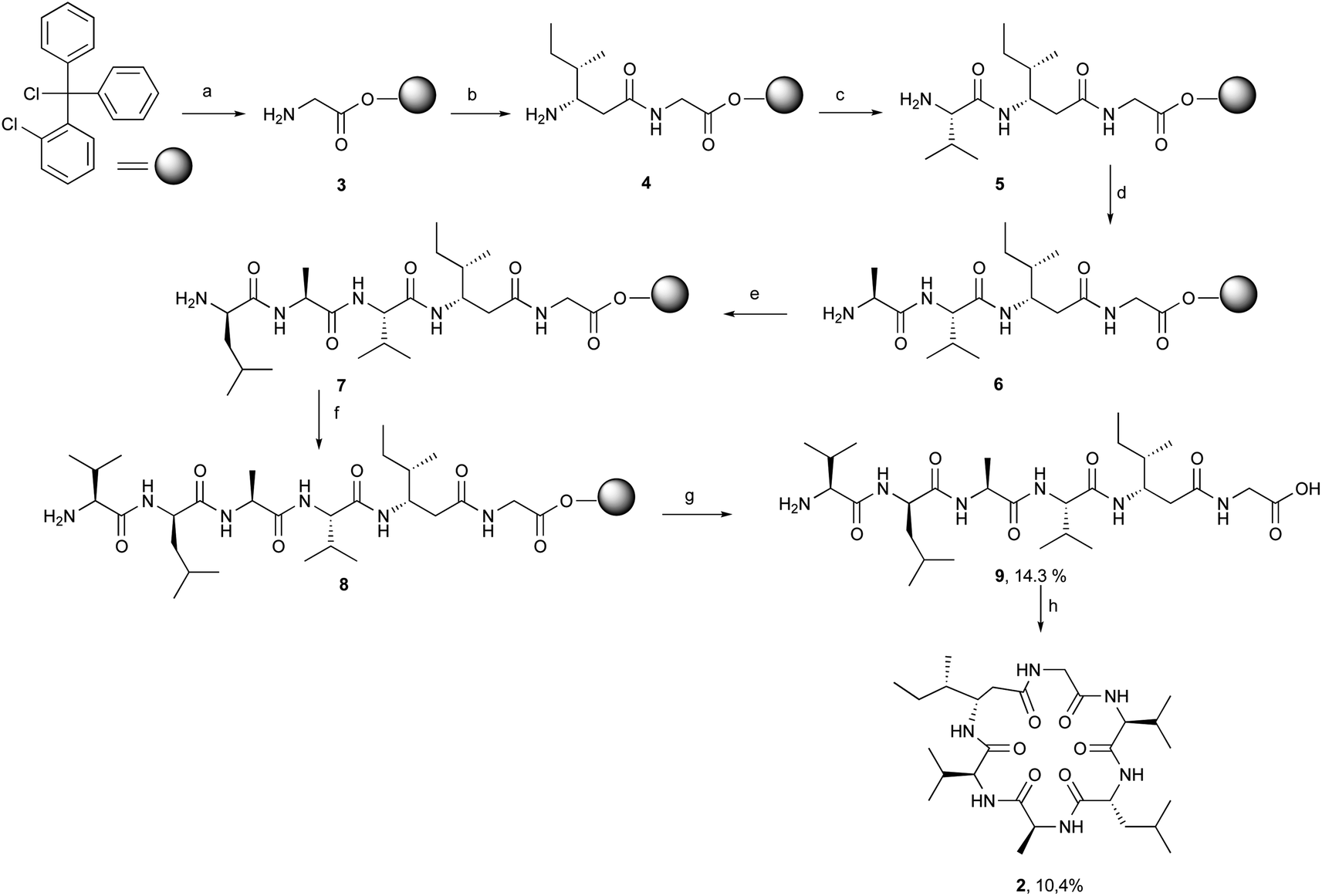 | ||
Fig. 2 General procedure of Fmoc-strategy SPPS and solution-phase macrocyclization, ((a)–(g) in solid phase) (a) (1) Fmoc-Gly-OH, DIPEA, DCM, 4 h, rt; (2) MeOH/DCM/DIPEA (15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :80:5); (3) 20% piperidine in DMF, (b) (1) Fmoc-L-β-HIle-OH, DIC, OxymaPure, DMF, 2 h, rt; (2) 20% piperidine in DMF, (c) (1) Fmoc-L-Val-OH, DIC, OxymaPure, DMF, 2 h, rt; (2) 20% piperidine in DMF, (d) (1) Fmoc-L-Ala-OH, DIC, OxymaPure, DMF, 2 h, rt; (2) 20% piperidine in DMF, (e) (1) Fmoc-D-Leu-OH, DIC, OxymaPure, DMF, 2 h, rt; (2) 20% piperidine in DMF, (f) (1) Fmoc-L-Val-OH, DIC, OxymaPure, DMF, 2 h; (2) 20% piperidine in DMF, (g) 20% TFA in DCM, 2 × 10 min, rt, (h) (solution phase) HATU, DIPEA, DCM, 7 days, rt. :80:5); (3) 20% piperidine in DMF, (b) (1) Fmoc-L-β-HIle-OH, DIC, OxymaPure, DMF, 2 h, rt; (2) 20% piperidine in DMF, (c) (1) Fmoc-L-Val-OH, DIC, OxymaPure, DMF, 2 h, rt; (2) 20% piperidine in DMF, (d) (1) Fmoc-L-Ala-OH, DIC, OxymaPure, DMF, 2 h, rt; (2) 20% piperidine in DMF, (e) (1) Fmoc-D-Leu-OH, DIC, OxymaPure, DMF, 2 h, rt; (2) 20% piperidine in DMF, (f) (1) Fmoc-L-Val-OH, DIC, OxymaPure, DMF, 2 h; (2) 20% piperidine in DMF, (g) 20% TFA in DCM, 2 × 10 min, rt, (h) (solution phase) HATU, DIPEA, DCM, 7 days, rt. | ||
Linear hexapeptide 9 was synthesized by SPPS, initiated by attaching the first amino acid residue Fmoc-Gly-OH onto 2-chlorotrityl chloride (2-CTC) resin. The use of 2-CTC resin can suppress racemization, and prevent DKP formation, and resin release can be carried out under mild acidic conditions.15 The loading resin value was obtained as 0.542 mmol g−1. In order to avoid loading resin value of over 1 mmol g−1, a reaction for small residues such as glycine can be accomplished with a shorter reaction time. The high-loading resin value might induce the peptide chain to form aggregates through hydrophobic interactions, which can inhibit subsequent coupling reactions.16 The resin was then capped to mask the remaining chlorine group on the resin to prevent early rupture of the peptide chain.
The coupling reaction for peptide chain elongation of [β-HIle]2-nodupetide linear 9 was accomplished using a combination of DIC and an additive of OxymaPure with a rapid reaction time, which only took 2 h for each coupling reaction. A combination of DIC and OxymaPure has demonstrated superior performance in amino acid activation for peptide synthesis.17,18 The use of OxymaPure as an additive is excellent, as it is known to suppress racemization and can increase the efficiency of the coupling reaction.18,19 The elongation of the linear peptide chain began with the attachment of the second residue Fmoc-L-β-HIle-OH on peptidyl-resin 3 and generated the dipeptidyl-resin 4. The successful completion of the coupling reaction was monitored using the chloranil test and showed a negative result, in which the colour of the resin did not change. Before further amino acid coupling, deprotection of the Fmoc group was firstly carried out using 20% piperidine in DMF, a successful deprotection was indicated by the positive result of the chloranil test, which is the resin turning red. The coupling of the third to sixth residues, Fmoc-L-Val-OH, Fmoc-L-Ala-OH, Fmoc-D-Leu-OH, and Fmoc-L-Val-OH respectively followed by the removal of the Fmoc group repetitively and generated the intermediates 5, 6, 7 and hexapeptidyl-resin 8, respectively. All coupling and deprotection reactions were monitored by the chloranil test.
The hexapeptidyl-resin 8 was further detached from the resin using 20% TFA in DCM, as the peptide chain has no side chain protecting group. The crude linear precursor of [β-HIle]2-nodupetide 9 gave a yield of 80.8%, based on the loading resin value. The linear hexapeptide crude was purified by flash column chromatography using C-18 silica gel with methanol:water (6:4) as the eluent. The desired linear hexapeptide 9 appeared as a major peak at a retention time of 4.43 min (Fig. S1†) and was obtained as a white powder of 5.3 mg, yield of 14.3% with >97% purity. The HR-ToF-MS spectra showed [M + H]+ with m/z 585.3973, which is consistent with the calculated mass data showing a [M + H]+ with m/z 585.3976 for C28H53N6O7 (Fig. S2†). The 1H-NMR and 13C-NMR data showed the number of protons and carbons that corresponded to the structure of linear hexapeptide 9 (Fig. S3 and S4†).
Linear hexapeptide 9 cyclization is carried out under highly dilute conditions, to avoid the formation of side reaction products. Cyclization was carried out using HATU as a coupling reagent in dichloromethane at a very dilute concentration (1 mM). The cyclization reaction took up to 7 days to afford the desired cyclic crude product 2. In addition, we also tried the cyclization using PyBOP coupling reagent in DMF (1.25 mM) with the addition of NaCl salt. The addition of NaCl salt initiated dipole interaction between sodium ions and oxygen atoms on the carbonyl group of amino acid residue resulting in a closer C-terminal with N-terminal, thereby the cyclization process will be easier.14,20 The addition of NaCl salt is also an attempt to increase the solubility of peptide 9.14,20 The cyclization reaction with PyBOP and NaCl salt also requires a reaction time of 7 days. The reaction was monitored using TLC. The cyclic crude obtained using the PyBOP/NaCl coupling reagent in DMF produced a lesser crude product compared to the cyclic crude using HATU. This was probably due to the workup process after the reaction, which needed to be extracted using brine solution and organic solvent. After the cyclization reaction was completed, the reaction mixture was then concentrated with a rotary evaporator. The crude product was further purified using semipreparative RP-HPLC with 50% acetonitrile in water with 1% TFA, a flow rate of 2 mL min−1 and yielded 10.4% [β-HIle]2-nodupetide with 93.5% of purity. Analytical RP-HPLC results showed a major peak at the retention time of 9.17 min (Fig. S5†).
[β-HIle]2-nodupetide 2 was characterized by HR-ToF-MS, 1H-NMR, and 13C-NMR. The HR-ToF-MS spectrum of [β-HIle]2-nodupetide showed a molecular ion peak [M + H]+ m/z 567.3860, which is consistent with the calculated mass [M + H]+ m/z 567.3870 for C28H51N6O6 (Fig. S6†). Furthermore, the characterization results of [β-HIle]2-nodupetide 2 by 1H-NMR and 13C-NMR (spectra in Fig. S7 and S8†) are presented in Table 1, and compared with nodupetide isolate from the literature.2 The 1H-NMR spectrum of [β-HIle]2-nodupetide 2 showed the presence of six alpha protons at the chemical shifts of 4.41, 4.22, 4.02, 3.96, 3.91 and 2.44 ppm. 13C-NMR spectrum showed the presence of six carbonyl signals at chemical shifts of 173.5, 173.2, 173.1, 172.7, 171.9 and 170.5 ppm. In addition, six alpha carbons were confirmed at chemical shifts of 60.7, 59.9, 51.8, 48.9, 42.0, and 36.9 ppm. There are chemical shift differences both in 1H- and 13C-NMR data of [β-HIle]2-nodupetide 2, compared to its parent compound 1. The differences might be due to the change of the ester-to-amide bond in the peptide backbone which transformed the environment around the proton and carbon nuclei, subsequently leading to differences in chemical shifts.21 Furthermore, the use of different deuterated solvents also causes the alterations in proton and carbon chemical shifts of the analogue.
| Assignment | [β-HIle]2-nodupetide (CD3OD, 1H-NMR 500 MHz, 13C-NMR 125 MHz) | Nodupetide (DMSO-d6, 1H-NMR 400 MHz, 13C-NMR 100 MHz) | ||
|---|---|---|---|---|
| 1H, δ in ppm (ΣH, m) | 13C (ppm) | 1H, δ in ppm (ΣH, m) | 13C (ppm) | |
| HMHA* | ||||
| CO | 169.8 | |||
| α-CH2 | 2.25 (2H, dd, J = 14.4, 2.0) | 37.7 | ||
| β-CH | 4.92 (1H, ddd, J = 8.8, 4.0, 2.0) | 75.3 | ||
| γ-CH | 1.58 (1H, m) | 38.0 | ||
| δ-CH2 | 1.06 (2H, m) | 24.4 | ||
| σ-CH3 | 0.83 (3H, t, J = 7.6) | 14.3 | ||
| σ′-CH3 | 0.84 (3H, d, J = 6.8) | 11.5 | ||
|
||||
| Gly4 | ||||
| CO | 170.5 | 169.0 | ||
| α-CH2 | 3.91 (2H, d, J = 6.0) | 42.0 | 4.07 (2H, dd, J = 6.4, 16.8) | 41.9 |
|
||||
| β-HIle2 | ||||
| CO | 171.9 | |||
| α-CH2 | 2.44 (2H, d, J = 4.5) | 36.9 | ||
| β-CH | 3.78 (1H, d, J = 12.0) | 52.6 | ||
| γ-CH | 1.68–1.72 (1H, m) | 39.1 | ||
| δ-CH2 | 1.15–1.21 (2H, m) | 29.6 | ||
| σ-CH3 | 0.99 (3H, t, J = 7.8) | 14.2 | ||
| σ′-CH3 | 1.00 (3H, d, J = 6.7) | 11.8 | ||
|
||||
| Val3 | ||||
| CO | 172.7 | 170.7 | ||
| α-CH | 3.96 (1H, d, J = 5.5) | 59.9 | 4.08 (1H, m) | 57.7 |
| β-CH | 2.26–2.33 (1H, m) | 29.8 | 2.07 (1H, m) | 29.5 |
| γ-CH3 | 0.95 (3H, d, J = 4.6) | 18.2 | 0.85 (3H, d, J = 6.8) | 18.7 |
| γ′-CH3 | 0.97 (3H, d, J = 4.6) | 22.0 | 0.88 (3H, d, J = 6.8) | 20.9 |
|
||||
| Ala4 | ||||
| CO | 173.1 | 171.8 | ||
| α-CH | 4.22 (1H, q, J = 7.0) | 48.9 | 4.19 (1H, q, J = 7.2) | 47.9 |
| β-CH3 | 1.40 (3H, d, J = 6.5) | 17.7 | 1.22 (3H, d, J = 7.2) | 17.4 |
|
||||
| D-Leu5 | ||||
| CO | 173.2 | 171.2 | ||
| α-CH | 4.41 (1H, q, J = 5.5) | 51.8 | 4.02 (1H, d, J = 6.4) | 51.8 |
| β-CH2 | 1.46–1.52 (2H, m) | 37.2 | 1.48 (2H, ddd, J = 9.6, 6.4, 4.0) | 38.6 |
| γ-CH | 1.78–1.82 (1H, m) | 25.5 | 1.63 (1H, m) | 24.1 |
| δ-CH3 | 1.00 (3H, d, J = 6.0) | 20.1 | 0.88 (3H, d, J = 6.8) | 19.0 |
| δ′-CH3 | 0.92 (3H, d, J = 6.0) | 24.5 | 0.80 (3H, d, J = 6.8) | 23.0 |
|
||||
| Val6 | ||||
| CO | 173.5 | 171.7 | ||
| α-CH | 4.02 (1H, d, J = 6.0) | 60.7 | 4.03 (1H, m) | 58.7 |
| β-CH | 2.00–2.05 (1H, m) | 29.6 | 1.86 (1H, m) | 29.5 |
| γ-CH3 | 0.96 (3H, d, J = 4.9) | 18.5 | 0.86 (3H, d, J = 6.4) | 18.9 |
| γ′-CH3 | 0.98 (3H, d, J = 4.9) | 17.9 | 0.84 (3H, d, J = 6.4) | 17.5 |
Biological activity of [β-HIle]2-nodupetide
[β-HIle]2-nodupetide and its linear precursor were tested for their antimicrobial activity against four pathogenic microbes (Table 2). Based on the growth inhibition data, it was shown that both peptides are inactive against the four pathogenic microbes tested, where both showed MIC values >1000 μg mL−1 and are considered inactive.22 The result was in line with a report from Moreira and co-workers also explaining, the replacement of threonine residue to (2S,3S-DABA) and its amide isomer, which caused a decrease in their antimicrobial activity. They explained the possibility of specific chiral interaction with the membrane.8| Peptides | MIC (μg mL−1) | |||
|---|---|---|---|---|
| Candida albican (fungi) | Pseudomonas aeruginosa (Gram-negative) | Staphylococcus aureus (Gram-positive) | Escherichia coli (Gram-negative) | |
| Nodupetide 12 | — | 2.83 | — | — |
| [β-HIle]2-nodupetide 2 | >1000 | >1000 | >1000 | >1000 |
| Linear [β-HIle]2-nodupetide 9 | >1000 | >1000 | >1000 | >1000 |
| Ciprofloxacin | 0.156 | 0.005 | 0.078 | 15.625 |
The conversion of ester to amide bond on the nodupetide backbone might also lead to a conformational change and result in a dramatic decrease in its antimicrobial activity. As previously reported by Hosono and team, the change of amide to ester bond in some cyclic hexapeptides affected the membrane permeability which was related to conformational change. The conformational alteration is probably caused by the rearrangement of the intramolecular hydrogen bonding network or even the loss of the hydrogen bonding network. These conditions probably caused the change in the conformational preferences of the cyclic peptides in lipophilic media.23 It seems likely that conformation is a critical factor in differentiating target engagement and whole-cell activity.24
Ester bond is regarded as susceptible to enzymatic degradation. Thus, the presence of an ester bond is often considered to be a drawback because it reduces the stability of the peptide. Nevertheless, a study also conducted by Hosono and team showed that the cyclodepsipeptides had similar stability in plasma as cyclopeptides. This might be due to the macrocyclization shielding the ester bond from the enzyme degradation and it is influenced by the sequences and ring size of the cyclic peptide.23
The substitution of HMHA with β-HIle, seemed to cause an increase in the hydrophobicity of nodupetide analogue 2. The mechanism of action of antimicrobial peptides is also affected by the hydrophobicity of the peptides. Increasing the hydrophobicity results in an increase in the antibacterial activity. However, increased hydrophobicity beyond the optimum led to a decrease in antibacterial activity. This is probably due to increased dimerization, which prevents access to the membrane in prokaryotic cells.7 The mechanism of action of antimicrobial peptides is a very complex process with multiple factors, which need to be evaluated to enhance antimicrobial activity.
Conclusions
The conversion of ester to amide bond in nodupetide backbone resulted in a dramatic decrease in antimicrobial activity, in which the [β-HIle]2-nodupetide was inactive. The ester bond seemed to be crucial for the antimicrobial activity of nodupetide. In the present study, it was necessary to synthesize several analogues of nodupetide to evaluate the influencing factor of their antimicrobial activity. Despite the inactivity, the application of combination synthesis strategies of the solid- and solution-phase peptide synthesis was successfully carried out to synthesize [β-HIle]2-nodupetide with an overall yield of 10.4% with 93.5% purity.Materials and methods
Materials
The peptides were characterized using 1H-NMR and 13C-NMR on an Agilent NMR 500 MHz (1H) and 125 MHz (13C) equipment using CD3OD solvent. Mass spectroscopy spectra were measured using Waters HR-ToF-MS Lockspray. The loading resin absorbance was measured on the TECAN Infinite Pro 200 UV-Vis spectrophotometer. Analysis of linear and cyclic peptides was performed by Waters 2998 Photodiode Array Detector (PDA), LiChrospher 100 C-18, COSMOSIL 5 μM (4.6 × 250 mm) column RP-HPLC at the wavelength of 210, 240, and 254 nm. The mobile phase used was a gradient eluent consisting of deionized water (H2O) (A) with 0.1% TFA and acetonitrile B with 0.1% TFA. All amino acids, coupling reagents, and resin were purchased from GL-Biochem, Shanghai, China. They included 2-chlorotrityl chloride resin, N-[(dimethylamino)-1H-1,2,3-triazolo-4,5-[b]pyridine-1-yl-methelene]-N-methylmethanaaminium hexafluorophosphate N-oxide (HATU), benzotriazole-1-yloxytripyrrolidinophosphonium hexafluorophosphate (PyBOP), Fmoc-L-Ala-OH, Fmoc-Gly-OH, Fmoc-D-Leu-OH, Fmoc-L-Val-OH, Fmoc-L-β-HIle-OH. The other reagents such as dichloromethane (DCM), N,N′-dimethylformamide (DMF), N,N′-diisopropylethylamine (DIPEA), N,N′-diisopropylcarbodiimide (DIC), OxymaPure, piperidine, methanol, n-hexane, ethyl acetate, acetic acid, and trifluoroacetic acid (TFA) were pro-analysis grade. The microbes that were used for the assays were supplied by Thermo Scientific (Staphylococcus aureus ATCC 6538, Escherichia coli ATCC 11229, Candida albicans ATCC 10231) and Pseudomonas aeruginosa was provided by central laboratory Universitas Padjadjaran.Synthesis of [β-HIle]2-nodupetide (2)
:DCM:DIPEA 15:80:5 (10 mL) twice for 15 min. Then, the Fmoc-AA1-resin was deprotected by 20% piperidine in DMF (5 mL) for 2 × 5 min to afford the free amine group. For the next coupling reaction, the free amine group was coupled to the second Fmoc-protected amino acid (Fmoc-AA2-OH) Fmoc-L-β-HIle-OH (4 eq.) using DIC (4 eq.) and OxymaPure (4 eq.) as coupling agents in DMF (4 mL) for 2 h at room temperature. The Fmoc group was removed using 20% piperidine in DMF (5 mL) for 2 × 5 min to yield the resin-AA1-AA2-NH2. The cycle of coupling reaction and removing the Fmoc groups was repeated with subsequent Fmoc-protected amino acids to yield the resin-coupled hexapeptide. Finally, the last step was to cleave the resin using TFA 20% in DCM (10 mL) for 2 × 10 min, followed by filtration. The filtrate was evaporated and washed with DCM repeatedly, then dried under the vacuum to yield the crude peptide. The crude peptide was purified by column chromatography on silica C-18 ODS (eluted with methanol:water 6:4) to afford the desired linear peptide 5.3 mg, 14.3% as a white powder with >97% purity. The purity of linear peptide was analyzed using analytical RP-HPLC (20–80% acetonitrile in water for 50 min, flow rate 1 mL min−1, λ 240 nm).The spectral data of 9: HR-ToF-MS m/z [M + H]+ 585.3973 (calcd m/z [M + H]+ 585.3976 C28H53N6O7) (Fig. S2†); 1H NMR (500 MHz, CD3OD, δ, ppm, J (Hz)) (Fig. S3†), 3.72–3.75 (2H, m, Gly1 Hα), 2.45 (2H, dd, J = 4.0; 14.5 β-HIle2 Hα), 4.33–4.36 (1H, q, J = 6.3 β-HIle2 Hβ), 1.56–1.59 (1H, m, β-HIle2 Hγ), 1.48–1.52 (2H, m, β-HIle2 Hδ), 0.87 (3H, t, J = 7.4 β-HIle2 CH3), 0.89 (3H, d, J = 6.9 β-HIle2 H3), 4.19–4.22 (1H, m, Val3 Hα), 2.05–2.12 (1H, m, Val3 Hβ), 0.94 (3H, d, J = 6.3 Val3 CH3), 1.00 (3H, d, J = 6.3 Val3 CH3), 4.26 (1H, d, J = 7.5 Ala4 Hα), 1.41 (3H, d, J = 7.5 Ala4 CH3), 4.14 (1H, d, J = 8.0 D-Leu5 Hα), 1.62–1.66 (2H, m, D-Leu5 Hβ), 1.70–1.72 (1H, m, D-Leu5 Hγ), 1.09 (3H, d, J = 6.8 D-Leu5 CH3), 1.06 (3H, d, J = 6.8, D-Leu5 CH3), and 3.61–3.65 (1H, m, Val6 Hα), 2.18–2.23 (1H, m, Val6 Hβ), 0.88 (3H, d, J = 6.3 Val6 CH3), 0.91 (3H, d, J = 6.3 Val6 CH3). 13C-NMR (125 MHz, CD3OD, δ, ppm) (Fig. S4†) 173.2 (Gly1 C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 38.9 (Gly Cα), 173.2 (β-HIle2 CO), 26.2 (β-HIle2 Cα), 49.6 (β-HIle2 Cβ), 37.1 (β-HIle2 Cγ), 25.0 (β-HIle2 Cδ), 14.2 (β-HIle2 CH3), 10.2 (β-HIle2 CH3), 171.6 (Val3 CO), 58.5 (Val3 Cα), 30.2 (Val3 Cβ), 21.7 (Val3 CH3), 20.6 (Val3 CH3), 173.0 (Ala4 CO), 50.7 (Ala4 Cα), 16.4 (Ala4 CH3), 172.0 (D-Leu5 CO), 52.7 (D-Leu5 Cα), 38.7 (D-Leu5 Cβ), 23.9 (D-Leu5 Cγ), 25.0 (D-Leu5 CH3), 24.6 (D-Leu5 CH3), 169.4 (Val6 CO), 58.5 (Val6 Cα), 30.6 (Val6 Cβ), 17.4 (Val6 CH3), 17.7 (Val6 CH3).
O), 38.9 (Gly Cα), 173.2 (β-HIle2 CO), 26.2 (β-HIle2 Cα), 49.6 (β-HIle2 Cβ), 37.1 (β-HIle2 Cγ), 25.0 (β-HIle2 Cδ), 14.2 (β-HIle2 CH3), 10.2 (β-HIle2 CH3), 171.6 (Val3 CO), 58.5 (Val3 Cα), 30.2 (Val3 Cβ), 21.7 (Val3 CH3), 20.6 (Val3 CH3), 173.0 (Ala4 CO), 50.7 (Ala4 Cα), 16.4 (Ala4 CH3), 172.0 (D-Leu5 CO), 52.7 (D-Leu5 Cα), 38.7 (D-Leu5 Cβ), 23.9 (D-Leu5 Cγ), 25.0 (D-Leu5 CH3), 24.6 (D-Leu5 CH3), 169.4 (Val6 CO), 58.5 (Val6 Cα), 30.6 (Val6 Cβ), 17.4 (Val6 CH3), 17.7 (Val6 CH3).Biological assay
Data availability
Data supporting this study are included within the article and ESI.†Author contributions
The manuscript was written through the contribution of all authors. Conceptualisation, methodology, writing original draft, and writing-review and editing, H. I. F., R. M.; supervision, A. T. H., and U. S; funding acquisition, H. I. F., R. M., and A. T. H. All authors have read and agreed to the published version of the manuscript. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful to Balai Pembiayaan Pendidikan Tinggi (BPPT) Kemendikbudristek through Beasiswa Pendidikan Indonesia (BPI), the Indonesia Endowment Fund for Education (LPDP) for rendering financial support, as well as Universitas Padjadjaran, for the Academic Leadership Grant (ALG)-No. 1405/UN6.3.1/PT.00/2024).References
- T. Ito, N. Matsunaga, M. Kurashima, Y. Demizu and T. Misawa, Enhancing Chemical Stability through Structural Modification of Antimicrobial Peptides with Non-Proteinogenic Amino Acids, Antibiotics, 2023, 12(8), 1326, DOI:10.3390/antibiotics12081326.
- H. M. Wu, L. P. Lin, Q. L. Xu, W. B. Han, S. Zhang, Z. W. Liu, Y. N. Mei, Z. J. Yao and R. X. Tan, Nodupetide, a potent insecticide and antimicrobial from Nodulisporium sp. associated with Riptortus pedestris, Tetrahedron Lett., 2017, 58, 663–665, DOI:10.1016/j.tetlet.2017.01.009.
- C. Domhan, P. Uhl, C. Kleist, S. Zimmermann, F. Umstätter, K. Leotta, W. Mier and M. Wink, Replacement of L-amino acids by D-amino acids in the antimicrobial peptide ranalexin and its consequences for antimicrobial activity and biodistribution, Molecules, 2019, 24, 2987, DOI:10.3390/molecules24162987.
- Y. Ding, J. P. Ting, J. Liu, S. Al-Azzam, P. Pandya and S. Afshar, Impact of non-proteinogenic amino acids in the discovery and development of peptide, Amino Acids, 2020, 52, 1207–1226, DOI:10.1007/s00726-020-02890-9.
- R. Maharani, R. T. C. Brownlee, A. B. Hughes and B. M. Abbott, A total synthesis of a highly N-methylated cyclodepsipeptide [2S,3S-Hmp]-aureobasidin L using solid-phase methods, Tetrahedron, 2014, 70, 2351–2358, DOI:10.1016/j.tet.2014.02.036.
- T. Narancic, S. A. Almahboub and K. E. O'Connor, Unnatural amino acids: production and biotechnological potential, World J. Microbiol. Biotechnol., 2019, 35(4), 67, DOI:10.1007/s11274-019-2642-9.
- Y. Chen, M. T. Guarnieri, A. I. Vasil, M. L. Vasil, C. T. Mant and R. S. Hodges, Role of peptide hydrophobicity in the mechanism of action of α-helical antimicrobial peptides, Antimicrob. Agents Chemother., 2007, 51, 1398–1406, DOI:10.1128/AAC.00925-06.
- R. Moreira, G. Barnawi, D. Beriashvili, M. Palmer and S. D. Taylor, The effect of replacing the ester bond with an amide bond and of overall stereochemistry on the activity of daptomycin, Bioorg. Med. Chem., 2019, 27, 240–246, DOI:10.1016/j.bmc.2018.12.004.
- Q. Y. Zhang, Z. Bin Yan, Y. M. Meng, X. Y. Hong, G. Shao, J. J. Ma, X. R. Cheng, J. Liu, J. Kang and C. Y. Fu, Mil. Med. Res., 2021, 8(48), 1–25, DOI:10.1186/s40779-021-00343-2.
- X. Li, S. Zuo, B. Wang, K. Zhang and Y. Wang, Antimicrobial Mechanisms and Clinical Application Prospects of Antimicrobial Peptides, Molecules, 2022, 27, 2675, DOI:10.3390/molecules27092675.
- C. Zhang and M. Yang, Antimicrobial Peptides: From Design to Clinical Application, Antibiotics, 2022, 11, 349, DOI:10.3390/antibiotics11030349.
- R. Maharani, M. I. Muhajir, J. M. Dirgantara, A. Hardianto, T. Mayanti, D. Harneti, N. Nurlelasari, K. Farabi, A. T. Hidayat, U. Supratman and T. Siahaan, Synthesis and anticancer evaluation of [d-Ala]-nocardiotide A, RSC Adv., 2024, 14, 4097–4104, 10.1039/d4ra00025k.
- M. Muhajir, A. Hardianto, J. Al Anshori, D. Sumiarsa, T. Mayanti, N. Nurlelasari, D. Harneti, A. Hidayat, U. Supratman and R. Maharani, Total Synthesis of Nocardiotide A by Using a Combination of Solid- and Solution-Phase Methods, ChemistrySelect, 2021, 6, 12941–12946, DOI:10.1002/slct.202103441.
- R. Maharani, H. N. A. Yayat, A. T. Hidayat, J. Al Anshori, D. Sumiarsa, K. Farabi, T. Mayanti, Nurlelasari, D. Harneti and U. Supratman, Synthesis of a Cyclooctapeptide, Cyclopurpuracin, and Evaluation of Its Antimicrobial Activity, Molecules, 2023, 28(12), 4779, DOI:10.3390/molecules28124779.
- M. Ieronymaki, M. E. Androutsou, A. Pantelia, I. Friligou, M. Crisp, K. High, K. Penkman, D. Gatos and T. Tselios, Use of the 2-Chlorotrityl Chloride Resin for Microwave-Assisted Solid Phase Peptide Synthesis, Pept. Sci., 2015, 104, 506–514, DOI:10.1002/bip.22710.
- B. Merrifield, The Role of the Support in Solid Phase Peptide Synthesis, Br. Polym. J., 1984, 16, 173–178 CrossRef CAS.
- S. R. Manne, B. G. De La Torre, A. El-Faham and F. Albericio, OxymaPure Coupling Reagents: Beyond Solid-Phase Peptide Synthesis, Synthesis, 2020, 52, 3189–3210, DOI:10.1055/s-0040-1706296.
- R. Subirós-Funosas, R. Prohens, R. Barbas, A. El-Faham and F. Albericio, Oxyma: an efficient additive for peptide synthesis to replace the benzotriazole-based HOBt and HOAt with a lower risk of explosion, Chem.–Eur. J., 2009, 15, 9394–9403, DOI:10.1002/chem.200900614.
- A. El-Faham, Z. Al Marhoon, A. Abdel-Megeed and F. Albericio, OxymaPure/DIC: an efficient reagent for the synthesis of a novel series of 4-[2-(2-acetylaminophenyl)-2-oxo-acetylamino] benzoyl amino acid ester derivatives, Molecules, 2013, 18, 14747–14759, DOI:10.3390/molecules181214747.
- M. Liu, Y. C. Tang, K. Q. Fan, X. Jiang, L. H. Lai and Y. H. Ye, Cyclization of several linear penta- and heptapeptides with different metal ions studied by CD spectroscopy, J. Pept. Res., 2005, 65, 55–64, DOI:10.1111/j.1399-3011.2004.00199.x.
- A. C. Conibear, K. J. Rosengren, C. F. W. Becker and H. Kaehlig, Random coil shifts of posttranslationally modified amino acids, J. Biomol. NMR, 2019, 73, 587–599, DOI:10.1007/s10858-019-00270-4.
- E. Strandberg, P. Wadhwani and A. S. Ulrich, Antibiotic Potential and Biophysical Characterization of Amphipathic β-Stranded [XZ]n Peptides with Alternating Cationic and Hydrophobic Residues, Front. Med. Technol., 2021, 3, 622096, DOI:10.3389/fmedt.2021.622096.
- Y. Hosono, S. Uchida, M. Shinkai, C. E. Townsend, C. N. Kelly, M. R. Naylor, H. W. Lee, K. Kanamitsu, M. Ishii, R. Ueki, T. Ueda, K. Takeuchi, M. Sugita, Y. Akiyama, S. R. Lokey, J. Morimoto and S. Sando, Amide-to-ester substitution as a stable alternative to N-methylation for increasing membrane permeability in cyclic peptides, Nat. Commun., 2023, 14, 1416, DOI:10.1038/s41467-023-36978-z.
- Y. Li, N. P. Lavey, J. A. Coker, J. E. Knobbe, D. C. Truong, H. Yu, Y. S. Lin, S. L. Nimmo and A. S. Duerfeldt, Consequences of Depsipeptide Substitution on the ClpP Activation Activity of Antibacterial Acyldepsipeptides, ACS Med. Chem. Lett., 2017, 8, 1171–1176, DOI:10.1021/acsmedchemlett.7b00320.
- R. Maharani, E. F. Yanti, M. D. I. Melati and D. Sihotang, Synthesis of Trypsin-modulating Oostatic Factor (TMOF) and its Analogues by Solid-phase Peptide Synthesis Using DIC/Oxyma as Coupling Reagent, Procedia Chem., 2015, 17, 125–131, DOI:10.1016/j.proche.2015.12.124.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra04401k |
| This journal is © The Royal Society of Chemistry 2024 |