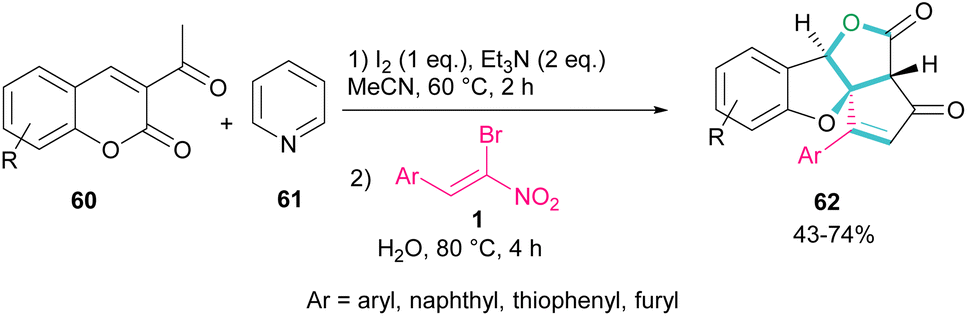DOI:
10.1039/D4RA02474E
(Review Article)
RSC Adv., 2024,
14, 14835-14846
Developments and applications of α-bromonitrostyrenes in organic syntheses
Received
1st April 2024
, Accepted 29th April 2024
First published on 8th May 2024
Abstract
The presence of the bromo and nitro groups in the structure of α-bromonitrostyrene makes them highly reactive and versatile reagents in organic syntheses. α-Bromonitrostyrenes act as an effective dielectrophile in the reaction with various nucleophiles. In these reactions, the bromo and nitro groups behave as good leaving groups for the assembly of a diverse range of heterocyclic compounds, such as dihydrofurans, dihydropyranes, furans, pyrroles, pyrazoles, isooxazolines, spiropyrrolidines, etc. In the current review, we have focused on the transformations of α-bromonitrostyrenes under organocatalysis, metal catalysis, and base-catalysis systems as well as catalyst-free conditions, since 2010.
1. Introduction
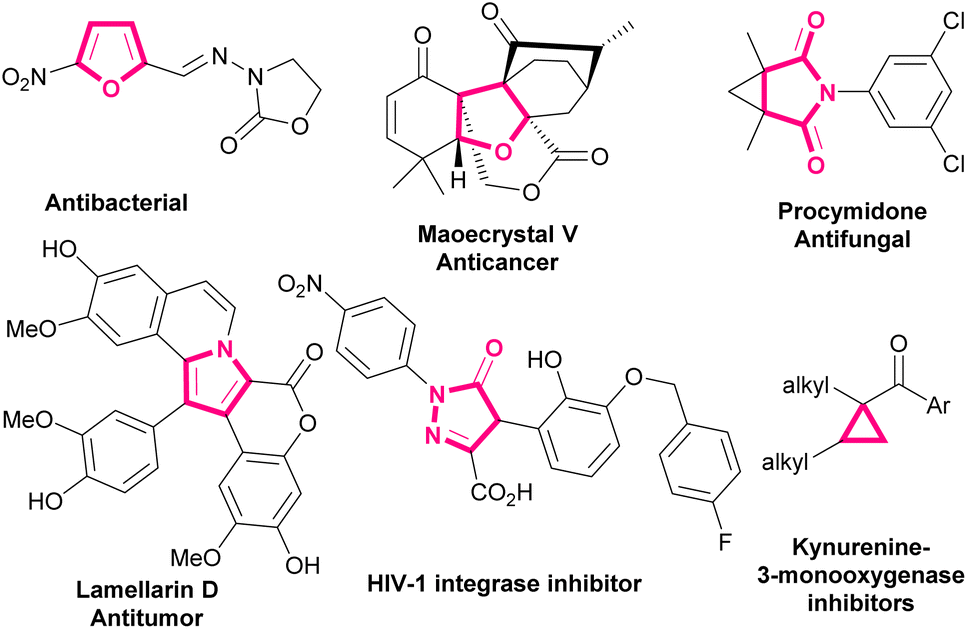
Bromonitroalkenes, especially bromonitrostyrenes are versatile building blocks for the construction of biological and pharmaceutical molecules and optically active compounds, such as dihydrofurans,1,2 pyrroles,3–6 and pyrazoles.7–9 Selected examples of biologically active molecules containing pyrrole, furan, dihydrofuran, or cyclopropane cores are shown in Scheme 1.
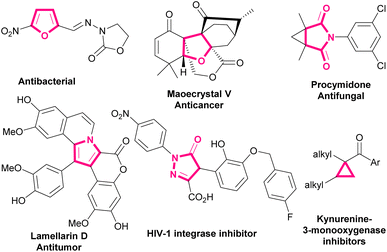 |
| | Scheme 1 Selected examples of biologically active molecules containing pyrrole, furan, dihydrofuran, or cyclopropane rings. | |
Due to the presence of both nitro and bromo groups, α-bromonitrostyrenes exhibit peculiar properties in comparison with nitrostyrenes and bromostyrenes.10–14 These substituents can behave as good leaving groups in the nucleophilic substitution reactions. Because of this feature, α-bromonitrostyrenes can easily be used in Michael addition reactions as a dipolarophile to make various functionalized dihydrofurans, pyrroles, pyrazoles, bicyclic oximes, triazoles, imidazoles, cyclopropanes, etc.15 In such reactions, α-bromonitroalkenes act as a 1,2-bielectrophile in a diverse range of cascade reactions with binucleophiles, such as 1,3-dicarbonyl compounds, enamines, aldehydes, alkenes, phenols, diazo compounds and other miscellaneous reagents.
Michael addition reactions of α-bromonitroalkenes can be performed in the presence of organocatalysts, metal catalysts, and bases, as well as catalyst-free systems. Among them, advances in enantioselective organocatalytic Michael reactions have provided various enantiomerically enriched compounds and biologically active molecules.16–19 On the other hand, domino Michael/Henry reactions are known as powerful methods for rendering complex molecular architectures through constructing the carbon–carbon bonds in a one-pot procedure without isolating intermediates.20,21 Recently, progress in asymmetric domino reactions utilizing chiral organocatalysts has been well revealed and interesting achievements has been gained.22–24
Recently, various research teams, such as Jakubec,25 Gao,26 Lee,27 Halimehjani,28 Shen,29 and Zhang30 reported the organic transformations of nitrostyrenes. In this review, we have specifically discussed the use of α-bromonitrostyrenes in various types of catalytic systems, such as organocatalysis, metal catalysis and base catalysis as well as catalyst-free reactions. We have also highlighted important features of the reactions and mechanisms to familiarize the readers with the reactivity and behavior of α-bromonitrostyrenes in organic syntheses.
2. Transformations of bromonitrostyrenes
2.1. Organocatalyst-catalyzed transformations of bromonitrostyrenes
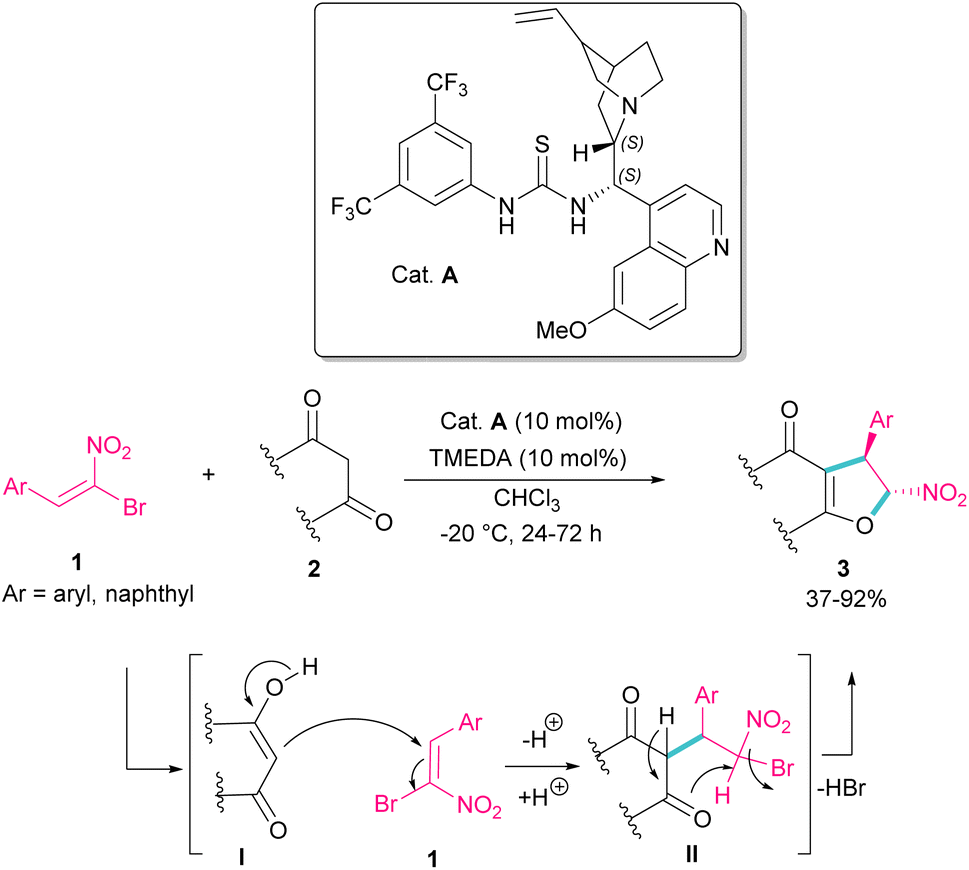
In 2010, Rueping et al. used (E)-β-bromonitrostyrenes 1 for the synthesis of dihydrofurans 3 from 1,3-dicarbonyl compounds 2 (Scheme 2).31 In this reaction, the chiral thiourea catalyst A can close two substrates together by hydrogen-bonding interaction and facilitate Michael addition of diketone 2 as a dinucleophile to bromonitrostyrene 1 as an electrophile to generate intermediate II. Then, an intramolecular nucleophilic addition delivered product 3 by the release of HBr. The same organocatalyst was used in the reaction of bromonitrostyrene, as a dielectrophile and 1,3-indandione as a dinucleophile to make spiro-nitrocyclopropane in 57% yield with 35% ee.32
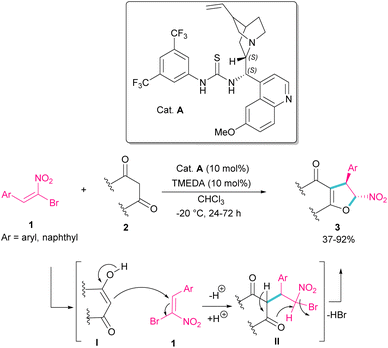 |
| | Scheme 2 Organocatalysis reaction of (E)-β-bromonitrostyrenes with 1,3-dicarbonyl compounds. | |
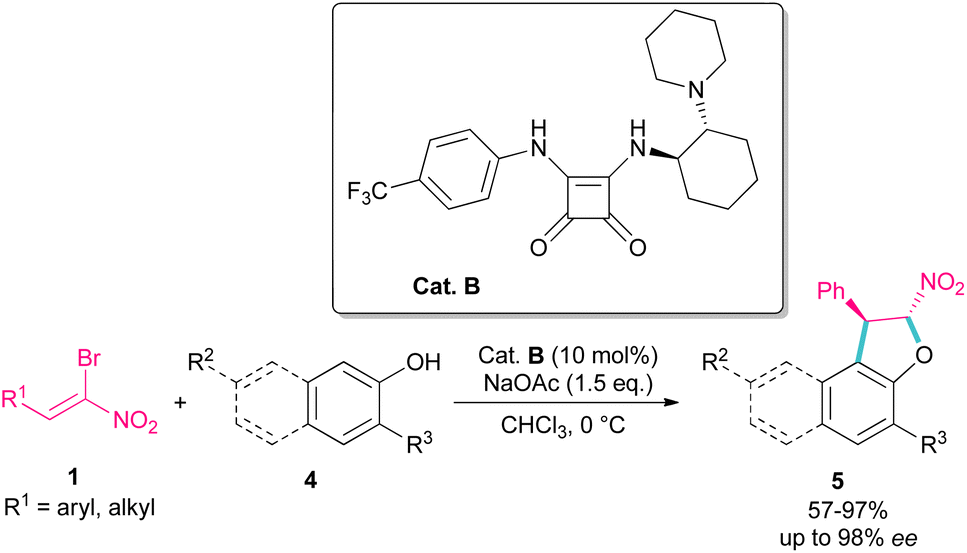
Parra and co-workers used an organocatalysis system to synthesize enantioselective dihydroarylfuran derivatives 5 from the cyclization reaction between (Z)-bromonitroalkenes 1 and naphthol 4 (Scheme 3).33 The hydrogen bonding interaction of chiral organocatalyst B with both substrates was responsible for the suitable orientation for Michael–Friedel–Crafts reaction and subsequent SN2 reaction on the carbon attached bromide. It should be noted that HBr resulting from the reaction can be neutralized with a stoichiometric amount of a base.
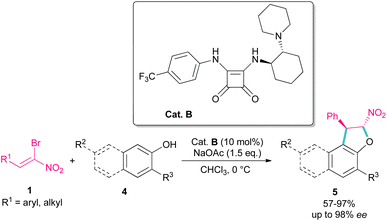 |
| | Scheme 3 Reaction of (Z)-bromonitroalkenes and naphthols catalyzed by a chiral organocatalyst. | |
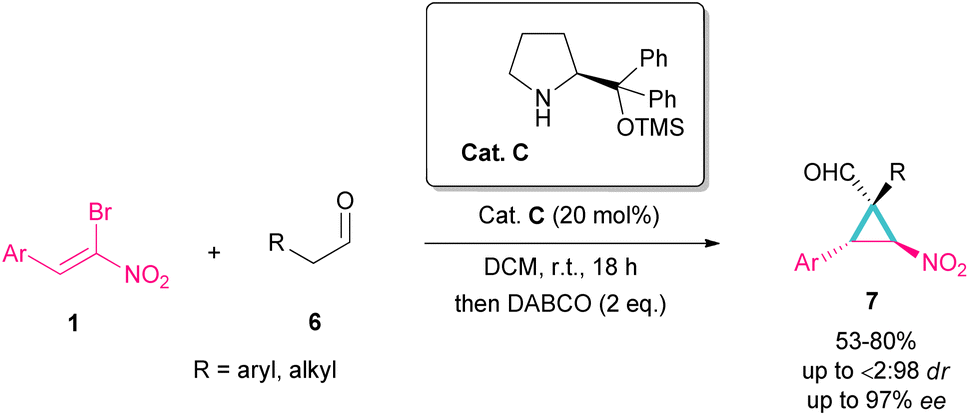
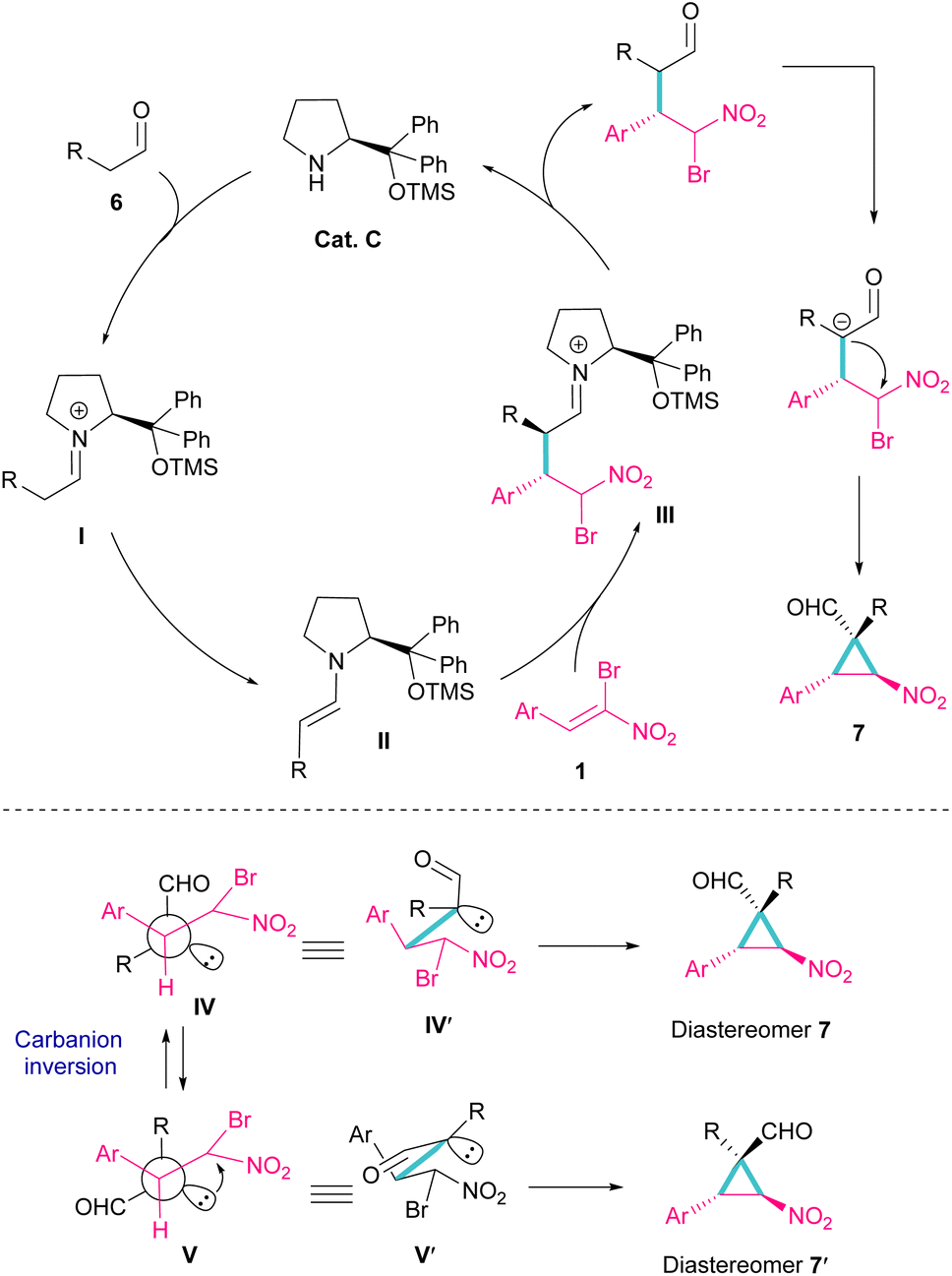
Another organocatalyst was used in the reaction of bromonitroalkenes 1 and alkyl aldehydes 6 (Scheme 4).34 A series of enantioenriched cyclopropanes 7 with quaternary carbon centers were prepared in the presence of a chiral pyrrolidine catalyst C. As illustrated in Scheme 5, the reaction started with the condensation of pyrrolidine with aldehyde to form imine I, followed by a 1,3-H shift to enamine II. The attack of bromonitrostyrene to the alkene moiety produced intermediate III. By release of the catalyst and subsequent intramolecular SN2 reaction, cyclopropane 7 was provided along with the elimination of bromide. It seems that the formation of two diastereomers 7 and 7′ from two configurations IV and V is possible. The configuration at C2 is generated from the intramolecular attack of the anion with DABCO, while the configuration at C4 is from the acidic proton at this C-atom, providing the thermodynamically stable product. In this case, the NO2 group is at the trans position to the aryl ring at C3.
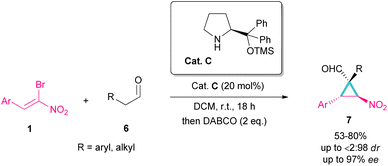 |
| | Scheme 4 Reaction of bromonitroalkenes and alkyl aldehydes catalyzed by chiral organocatalyst. | |
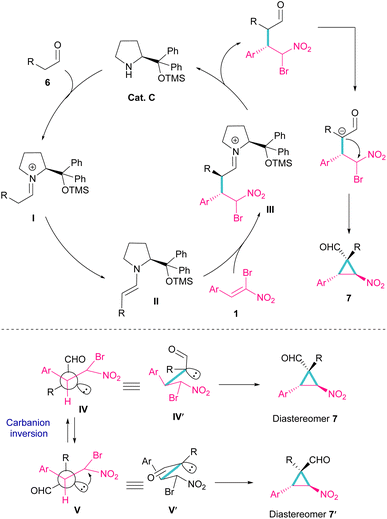 |
| | Scheme 5 Putative mechanism for reaction of bromonitroalkenes and alkyl aldehydes. | |
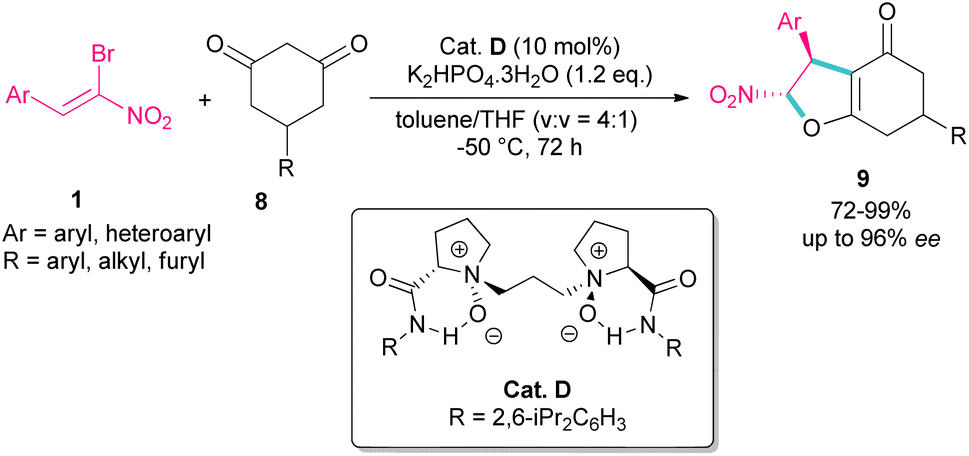
A domino Michael-alkylation reaction was reported by Feng and co-workers for the synthesis of dihydrofurans 9 from bromonitrostyrene 1 (Scheme 6).35 For this purpose, they treated bromonitrostyrene 1 and cyclohexane-1,3-diones 8 in the presence of the organocatalyst D. For dimedone, two stereocenters of the bicyclic 2,3-dihydrofurans were obtained with high diastereo- and enantioselectivity, while three stereocenters of the bicyclic 2,3-dihydrofurans were prepared from prochiral 5-monosubstituted cyclohexane-1,3-dione products via the desymmetrization reaction.
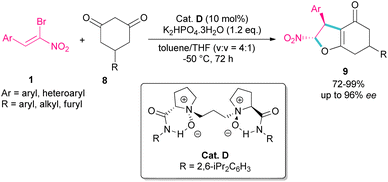 |
| | Scheme 6 Reaction of cyclohexane-1,3-dione and bromonitrostyrenes catalyzed by organocatalyst. | |
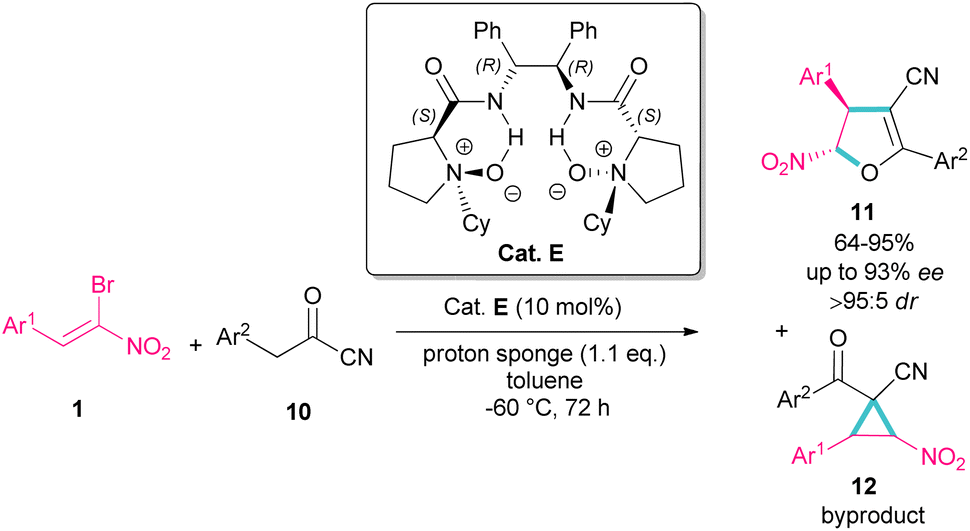
In 2016, Feng et al. introduced a chiral organocatalyst E for the asymmetric synthesis of dihydrofurans 11 from (Z)-bromonitrostyrenes 1 and α-substituted cyano ketones 10 (Scheme 7).36 In this regard, a domino Michael addition-alkylation between bromonitrostyrene and α-substituted cyano ketone in the presence of chiral N,N′-dioxide E was conducted under very mild reaction conditions. The authors found that the use of proton sponge, added in six equal parts, could enhance enantioselectivity and also the performance of the reaction in a very low temperature prevent the formation of the cyclopropane compound as a byproduct and thus increasing the yield of dihydrofuran 11. In addition to excellent enantioselectivity, high diastereo- and regioselectivity were also observed in this reaction.
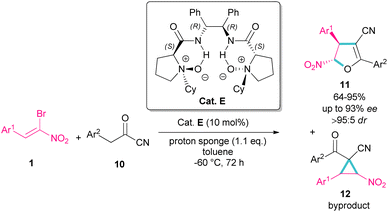 |
| | Scheme 7 Organocatalysis reaction of (Z)-bromonitrostyrenes and α-substituted cyano ketones. | |
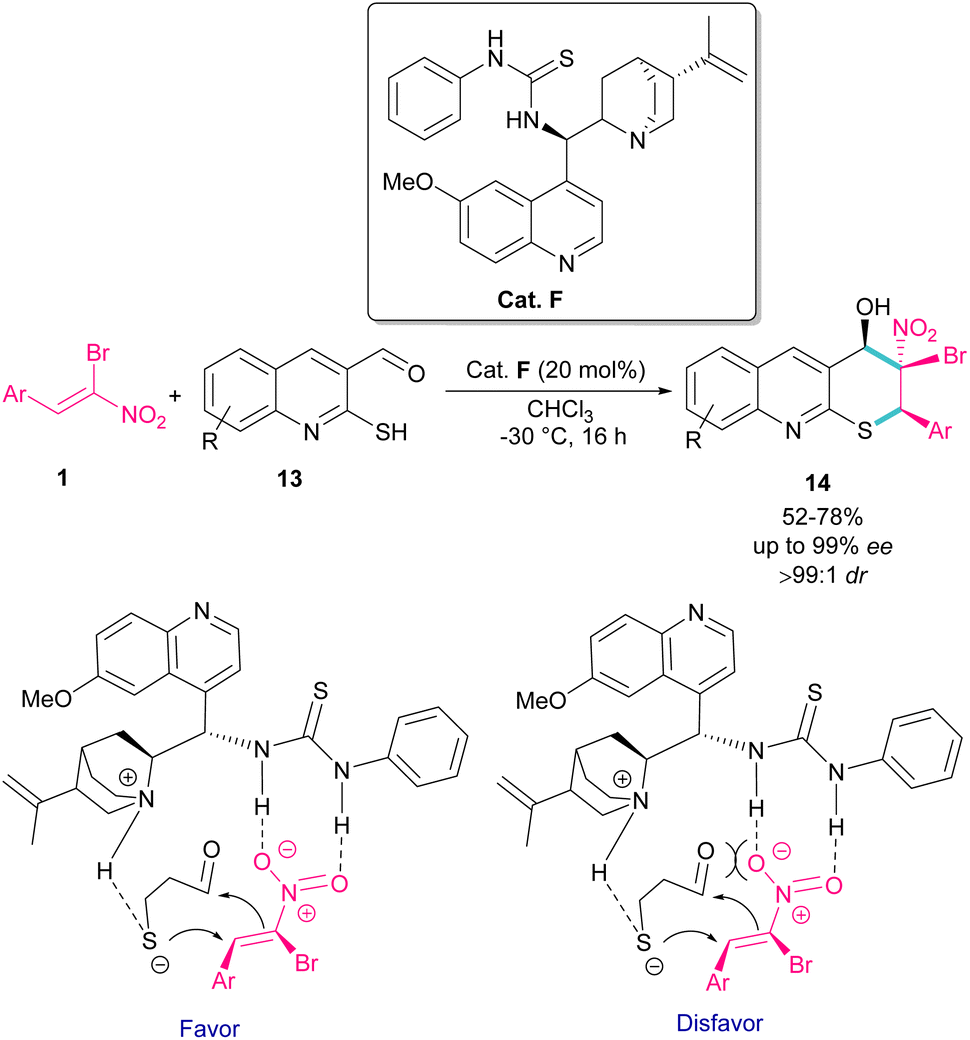
The use of α-bromonitroalkenes 1 in the synthesis of 3,4-dihydro-2H-thiopyrano[2,3-b]quinolines 14 was reported by Xie and co-workers in 2018 (Scheme 8).37 An organocatalysis system was proposed for this synthetic method, which started with the deprotonation of 2-mercaptoquinoline-3-carbaldehyde 13 by tertiary amine of catalyst F, and the activation of α-bromonitroalkene 1 by the thiourea moiety of F through the formation of two hydrogen bonds. The subsequent domino Michael/Henry reaction resulted in product 14. This method had the advantages of excellent enantio- and dieastereoselectivity, mild reaction conditions, and the ability of 3,4-dihydro-2H-thiopyrano[2,3-b]quinolines to undergo further organic transformations. In addition, salicylaldehyde can also act as a coupling partner in the cyclization with bromonitroalkene.
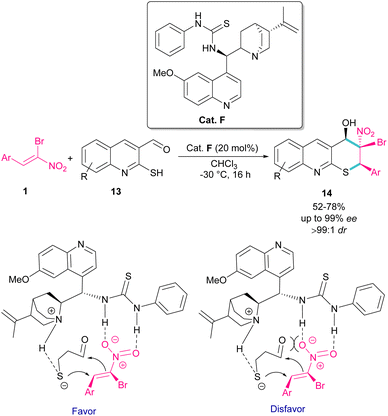 |
| | Scheme 8 Organocatalyst-catalyzed reaction of 2-mercaptoquinoline-3-carbaldehyde and α-bromonitroalkene. | |
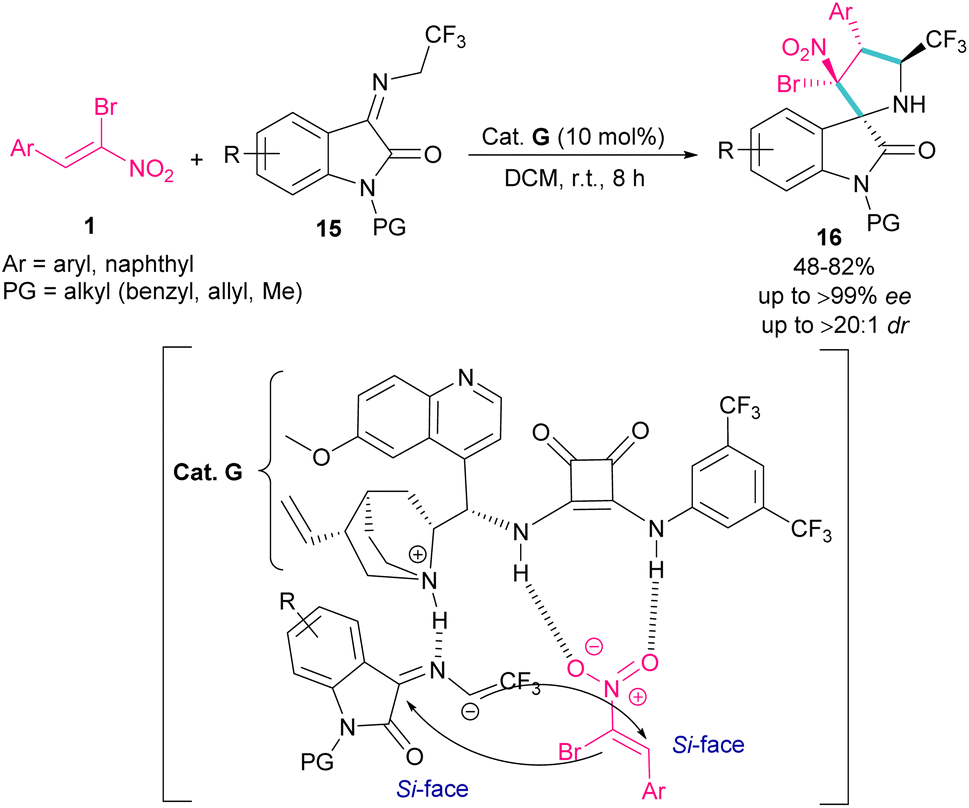
In 2020, an organocatalysis system was developed to prepare pyrrolidinyl spiro-oxindoles bearing quaternary carbon centers 16 (Scheme 9).38 Han and co-workers designed a (3 + 2)-cycloaddition reaction between isatin-derived ketimine 15 and (Z)-α-bromonitroalkene 1 in the presence of cinchonidine-derived squaramide G. This bifunctional organocatalyst can control the stereoselectivity by the hydrogen bonding interactions with both substrates blocking one side of each substrate, allowing nucleophilic attack from the Si-face.
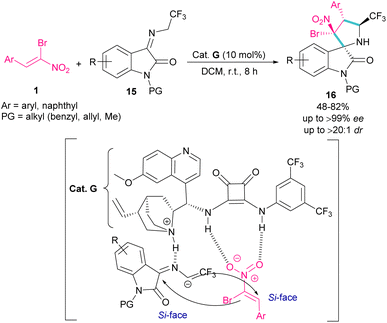 |
| | Scheme 9 Reaction of isatin-derived ketimine and (Z)-α-bromonitroalkene catalyzed by cinchonidine-derived squaramide. | |
In 2022, Tanyeli and co-workers extended a new organocatalysis system for the synthesis of chiral dihydrofurans 18 from bromonitroalkenes 1 and 1,3-dicarbonyl compounds 17 (Scheme 10).39 Their reaction proceeded through domino type Michael-SN2 reactions in the presence of a quinine-derived sterically encumbered squaramide H. The main advantages of this method that differentiates it from previous methods was the performance of the reaction in room temperature at short reaction times, while other similar reactions required cryogenic conditions and longer times to proceed. As shown in transition state, the quinoline moiety of the catalyst H activates the nucleophile 17 by hydrogen bonding. At the same time, the electrophile 1 is also activated by double hydrogen bonding with the squaramide group of H. Consequently, the nucleophilic attack of 17 from the Si-face of 1 afforded product with excellent enantioselectivity.
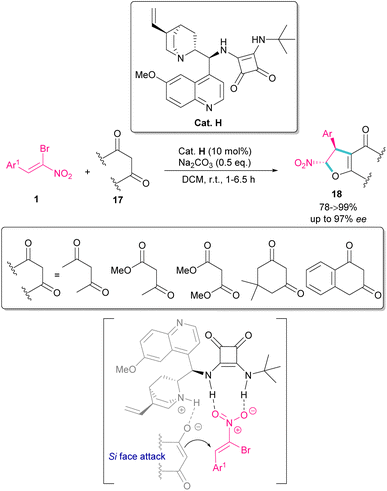 |
| | Scheme 10 Squaramide-catalyzed reaction of bromonitroalkenes and 1,3-dicarbonyl compounds. | |
2.2. Metal-catalyzed transformations of bromonitrostyrenes
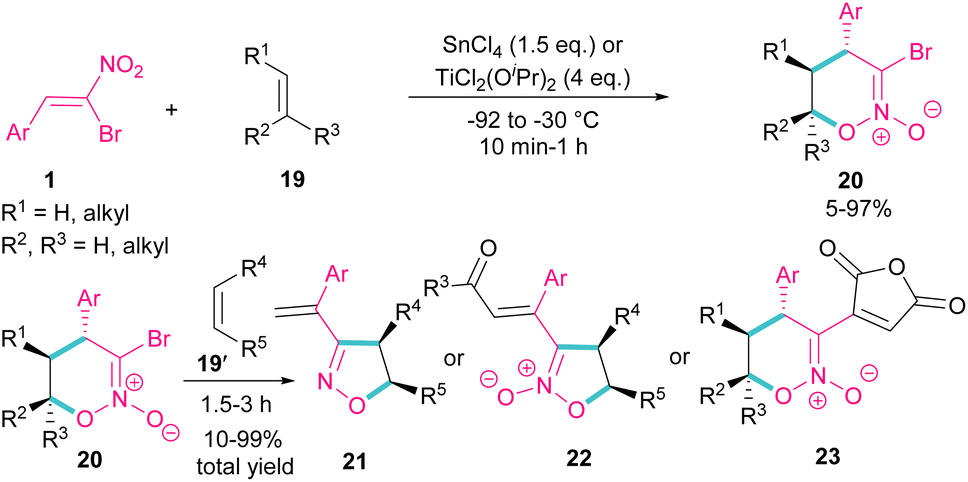
Bromonitrostyrenes can be applied for the construction of 3-bromo-5,6-dihydro-4H-1,2-oxazine N-oxides (Scheme 11).40 An alkene was used as a coupling partner and the cyclization reaction was performed in the presence of SnCl4 or TiCl2(OiPr)2 as a Lewis acid catalyst. (4 + 2)-Cycloaddition of alkene to bromonitrostyrene was sensitive to the reaction conditions, where an increase in temperature or time led to 3-chloro-1,2-oxazine N-oxide as byproduct. In the next stage, (3 + 2)-cycloaddition of the obtained 3-bromo-5,6-dihydro-4H-1,2-oxazine N-oxides 20 with another alkene reactant 19′ was performed and three products, including 3-vinyloxazole 21, isoxazole N-oxide 22, and 3-functionalized 1,2-oxazine N-oxide 23 were obtained in different ratios.
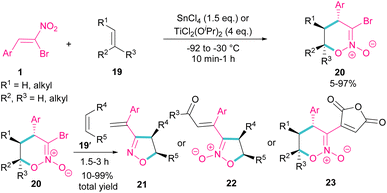 |
| | Scheme 11 Reaction of bromonitrostyrenes with alkenes in the presence of SnCl4. | |
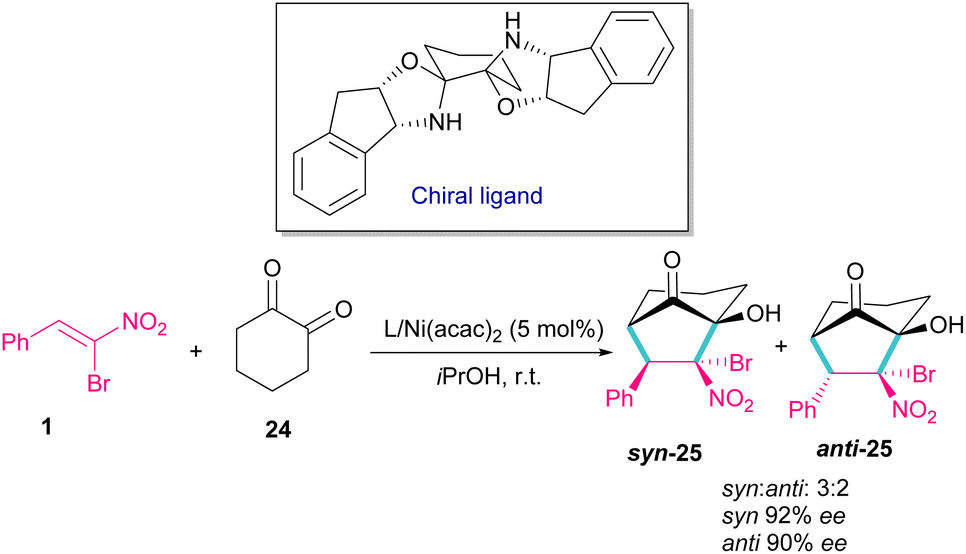
In 2014, a chiral bisoxazolidine in the combination with Ni(acac)2 catalyst was used for the preparation of optically active bicycle[3,2,1] octane of 1,2-diones 25 from nitroalkenes and 1 1,2-cyclohexadione 24 (Scheme 12).41 α-Bromonitroalkene 1 in a domino Michael–Henry reaction with diketone 24 can furnish product 25 with two syn and anti isomers. In this reaction, excellent enantioselectivity was observed (98% ee), but diastereoselectivity was relatively lower (2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 dr) in comparison with other nitroalkenes as coupling reactants (up to 99% ee, up to 7:1 dr).
:1 dr) in comparison with other nitroalkenes as coupling reactants (up to 99% ee, up to 7:1 dr).
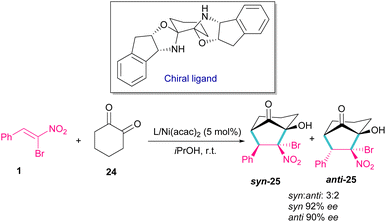 |
| | Scheme 12 Ni-catalyzed reaction of bromonitrostyrenes with 1,2-cyclohexadione. | |
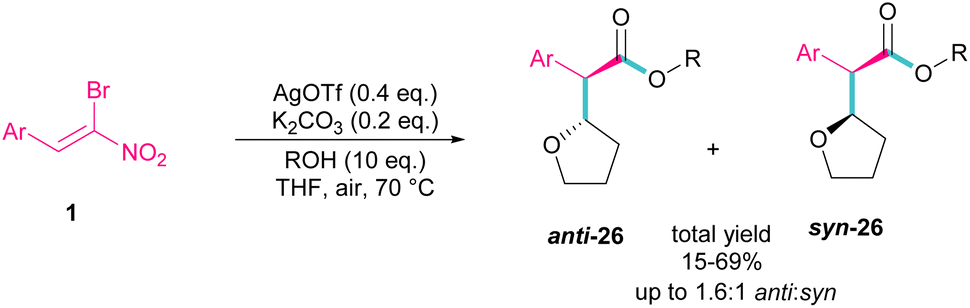
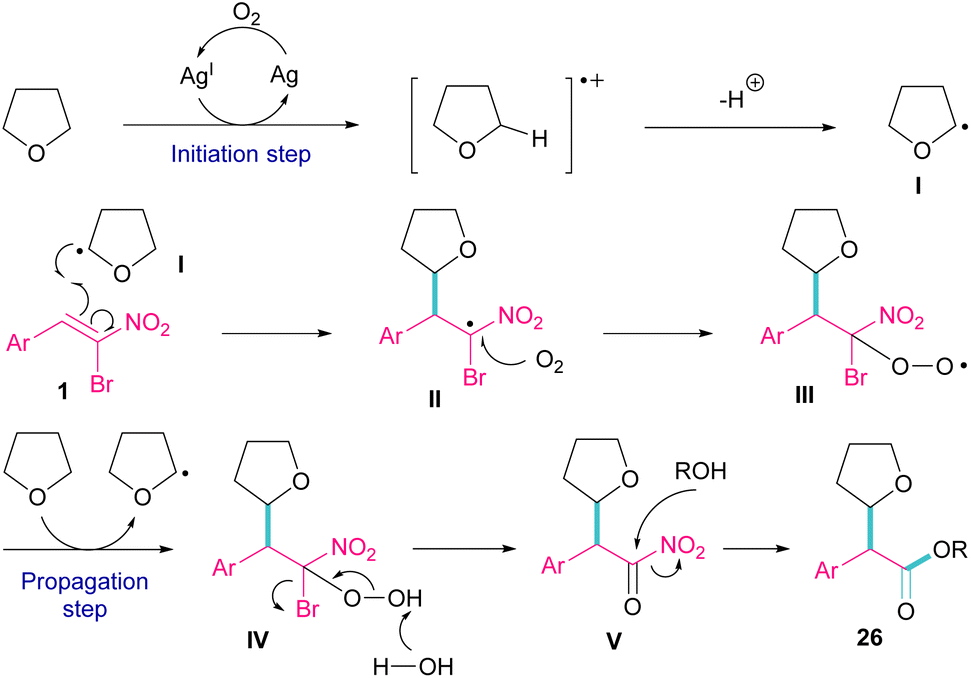
Silver triflate can catalyze the three-component reaction of tetrahydrofuran, β-bromonitrostyrene and alcohol via a radical pathway (Scheme 13).42 The reaction was initiated by the formation of radical I from the interaction of Ag(I) with THF, which attacked β-bromonitrostyrene 1 to form intermediate II. Further attack of O2 on II led to the peroxy radical III. Then, III abstracted a hydrogen from THF to give hydroperoxide IV. The cleavage of the O–O bond by H2O resulted in intermediate V with the release of bromide. In this step, the nucleophilic attack of alcohol to the carbonyl group along with the removal of the second leaving group (NO2) led to product 26. It is noteworthy that only less hindered alcohols can attack intermediate V (Scheme 14).
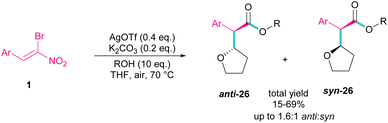 |
| | Scheme 13 Ag-catalyzed addition of THF radicals to β-bromonitrostyrenes. | |
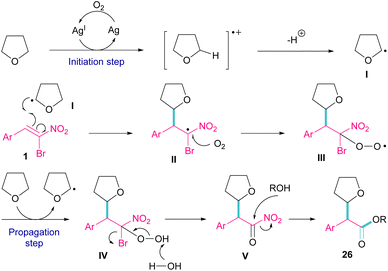 |
| | Scheme 14 Plausible mechanism for Ag-catalyzed addition of THF radicals to β-bromonitrostyrenes. | |
2.3. Base-mediated transformations of bromonitrostyrenes
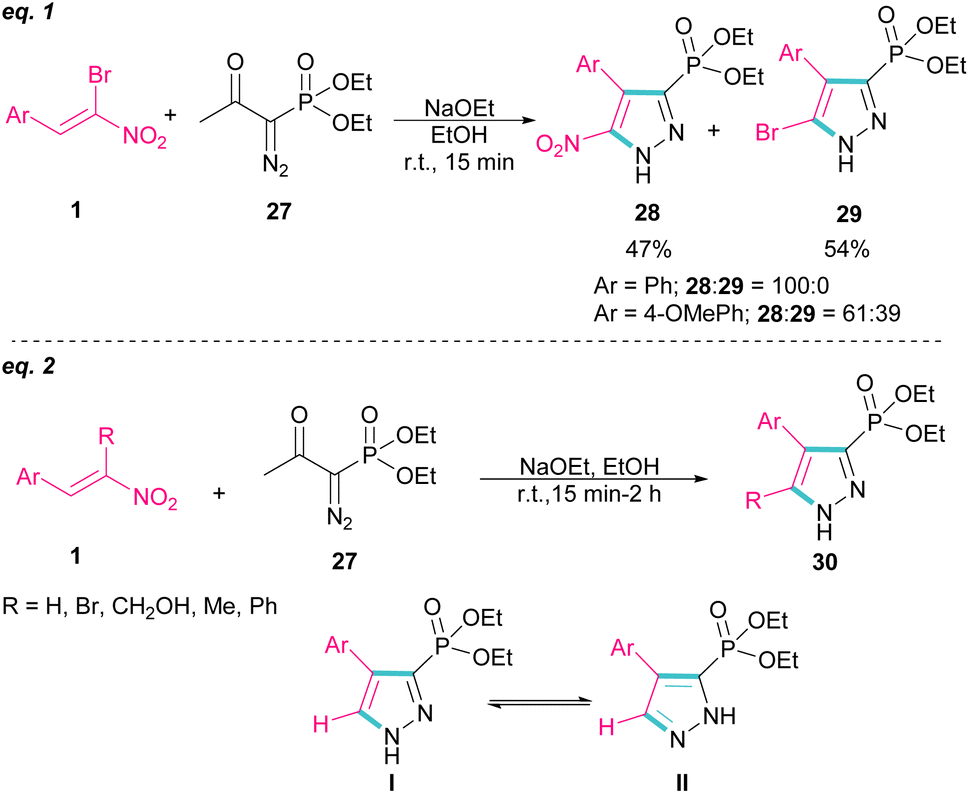
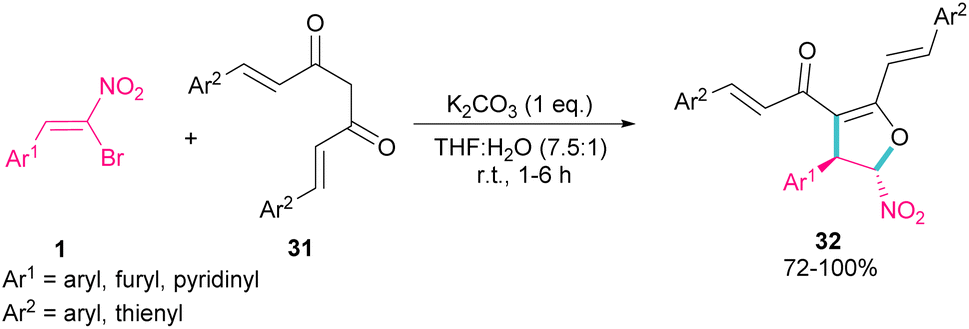
In 2007, Namboothiri and co-workers reported the synthesis of phosphonylpyrazoles 28 and 29 from diethyl 1-diazo-2-oxopropylphosphonate 27 with α-bromonitroalkenes 1 under basic conditions (Scheme 15, eqn (1)).43 The elimination of the leaving group in the final product depended on the substituent at the aryl ring of bromonitroalkene substrate. When Ar = Ph, product 28 was the major product, while products 28 and 29 were obtained in a ratio of 61:39 if bromonitrostyrene contained a 4-OMePh moiety. In a similar work, by dynamic NMR investigation, the Namboothiri group showed that two tautomers I and II are presented in such a reaction mixture that have a small energy difference but a high barrier to interconversion (Scheme 15, eqn (2)).44 Also, they could extend the substrate scope of bromonitrostyrenes in this work. Namboothiri et al. also reported the reaction of α-bromonitroalkenes 1 with curcumins 31 in the presence of a base (Scheme 16).45 Single diastereomer of dihydrofurans 32 was obtained in a Michael addition–alkenylation cascade reaction.
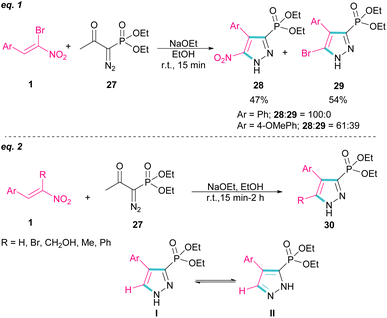 |
| | Scheme 15 Reaction of diethyl 1-diazo-2-oxopropylphosphonate and α-bromonitroalkenes. | |
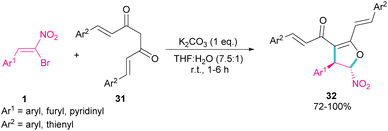 |
| | Scheme 16 Base-mediated reaction of α-bromonitroalkenes with curcumins. | |
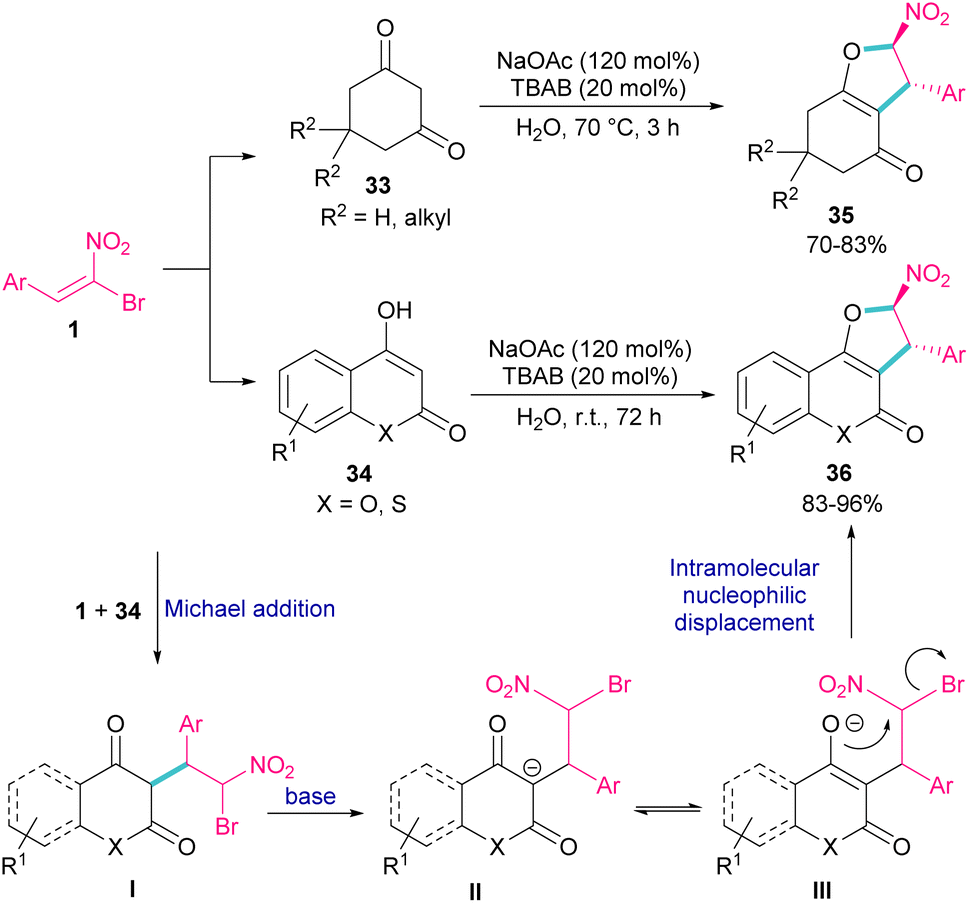
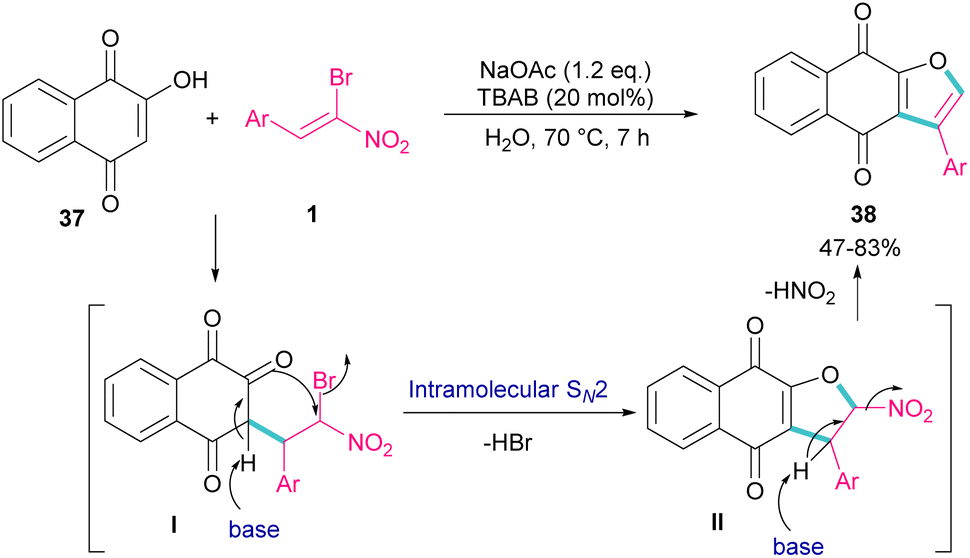
In 2011, Xie et al. developed a method for the assembly of dihydrofurans from domino reaction of bromonitrostyrenes with 4-hydroxycoumarins or 1,3-carbonyl compounds (Scheme 17).46 In this work, tricyclic 2,3-dihydrofurans 36 and bicyclic 2,3-dihydrofurans 35 were obtained in high yields under basic conditions in a aqueous solution. In all cases, only the trans isomer of products was isolated, showing excellent diastereoselectivity of this method. Regarding the mechanism, Michael addition of bromonitroalkene 1 to 1,3-dicarbonyl compounds 33 gave intermediate I, which was converted to the enolate II under basic conditions. Finally, product 36 was formed via an intramolecular nucleophilic displacement in III. Similar reaction conditions were used by Jianwu and co-workers for the synthesis of furan structures (Scheme 18).47 In their reaction, α-bromonitroalkenes 1 and 2-hydroxynaphthalene-1,4-diones 37 were applied as starting materials to form 3-phenylnaphtho[2,3-b]furan-4,9-diones 38 via Michael addition and subsequent intramolecular SN2 reaction. At last, the absorption properties of the obtained products were determined by UV-Vis spectra and fluorescence spectroscopy.
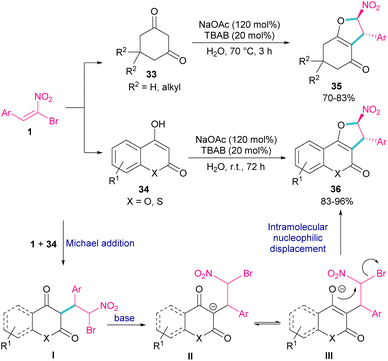 |
| | Scheme 17 Reaction of bromonitrostyrenes with 4-hydroxycoumarins mediated by base. | |
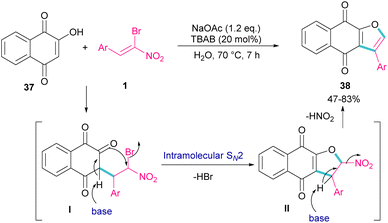 |
| | Scheme 18 Reaction of α-bromonitroalkenes and 2-hydroxynaphthalene-1,4-diones under basic conditions. | |
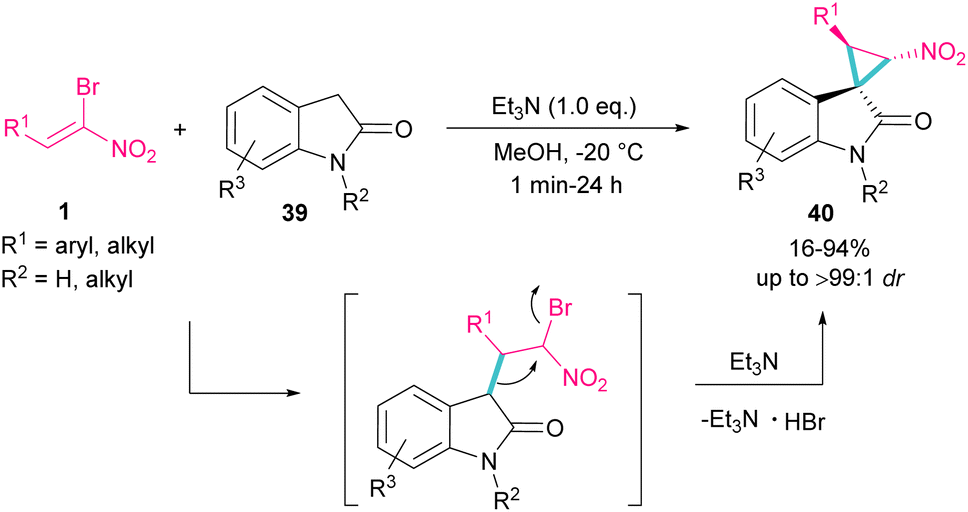
Spirocyclopropyl oxindole frameworks 40 were constructed in high yield with excellent diastereoselectivity from the reaction of bromonitroalkene 1 and N-protected indolin-2-ones 39 (Scheme 19).48 Due to high reactivity of indolin-2-one under ambient temperature and consequently low diastereoselectivity of the obtained product, a very low temperature was necessary for this reaction. The formation of cyclopropane was accomplished by the abstraction of a proton by a base and the liberation of bromide. Not only N-protected indolin-2-one but also N-H indolin-2-one was included in this transformation.
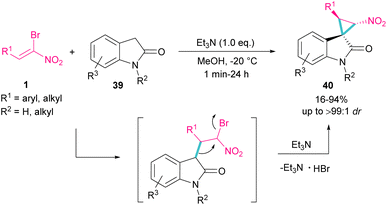 |
| | Scheme 19 Reaction of bromonitroalkene with N-protected indolin-2-ones. | |
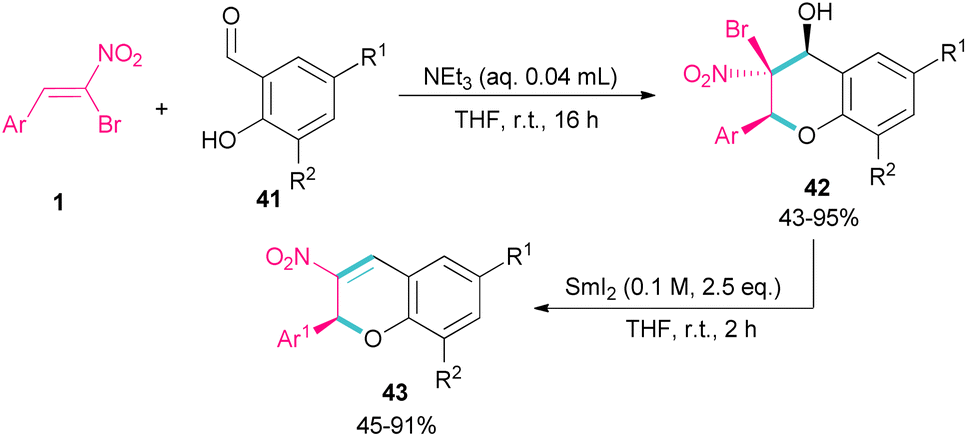
Soengas et al. established a two-step procedure for the preparation of 2-C-glycosyl-3-nitrochromenes 43 starting from bromonitrostyrenes 1 and ortho-hydroxybenzaldehydes 41 (Scheme 20).49 For this purpose, first, they treated bromonitrostyrenes 1 with ortho-hydroxybenzaldehydes 41 under basic conditions to achieve (2S,3S,4S)-3-bromo-3,4-dihydro-4-hydroxy-3-nitro-2H-1-benzopyrans 42 in moderate to excellent yields. In the next stage, the reaction was conducted in the presence of 0.1 M solution of SmI2 in THF for couple hours to form 3-nitrochromenes 43. SmI2 can promote the β-elimination in benzopyran 42 with complete stereoselectivity. All steps were performed under mild conditions and all products were obtained with enantiomeric purity.
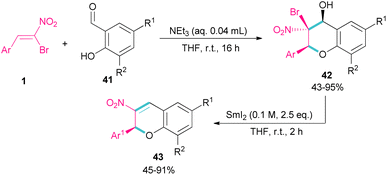 |
| | Scheme 20 NEt3-catalyzed reaction of bromonitroalkene with ortho-hydroxybenzaldehydes. | |
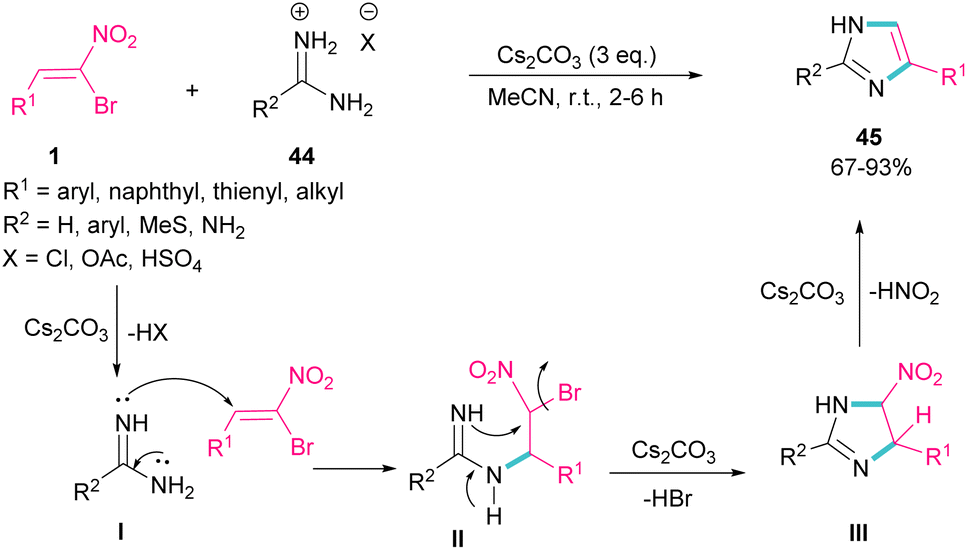
In 2015, again the Namboothiri research team synthesized imidazole scaffolds 45 through the reaction of amidine hydrochloride 44 with α-bromonitroalkenes 1 (Scheme 21).50 For the assembly of 2,5-disubstituted imidazoles from α-bromonitroalkenes, 3.0 equivalents of Cs2CO3 were required to promote all steps of this imidazole synthesis. First, Cs2CO3 promoted the formation of amidine I by the elimination of HX form 44. Michael addition of 1 to I gave intermediate II, which underwent an intramolecular SN2 process via a 5-exo-tet fashion to provide nitroimidazoline III along with the removal of HBr in the presence of Cs2CO3. Subsequent elimination of HNO2 by Cs2CO3 led to product 45. Moreover, the authors reported potential anti-parasitic activity of the resulting imidazoles.
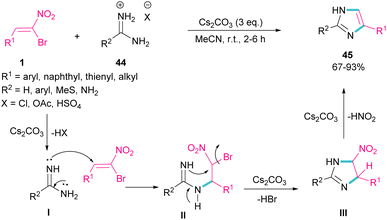 |
| | Scheme 21 Cs2CO3-promoted reaction of bromonitroalkene with amidines. | |
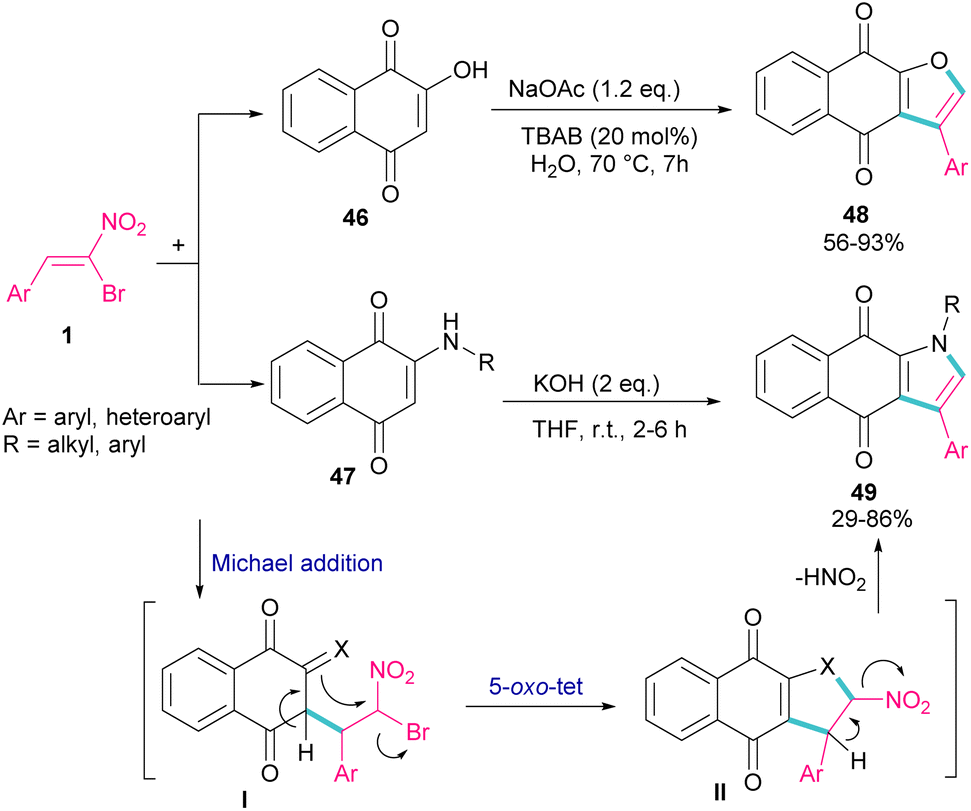
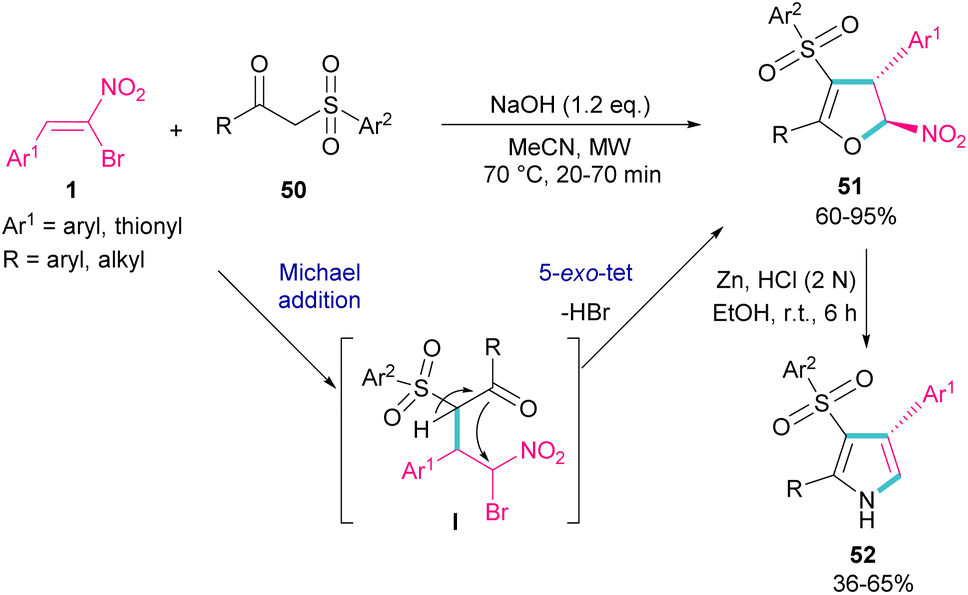
In 2018, Namboothiri and co-workers used α-bromonitroalkene 1 in the reaction with lawsone 46 and 2-aminonaphthoquinone 47 to produce furan and pyrrole fused quinonoid compounds 48 and 49 using two different basic conditions (Scheme 22).51 The transformations were performed via Michael addition of 1 to 47 to form intermediate I, followed by SN2 reaction in a 5-exo-tet manner to obtain intermediate II. Finally, pyrrole or furan frameworks were furnished after HNO2 removal. The authors also studied the anticancer activity of the obtained products. After a while, Namboothiri proposed a similar mechanism involving Michael addition/5-exo-tet reaction for the construction of dihydrofuran derivatives 51 from α-bromonitroalkenes 1 and β-ketosulfones 50 (Scheme 23).52 They could provide a series of pyrroles 52 by the reduction of dihydrofuran 51 in the presence of zinc powder under acidic conditions.
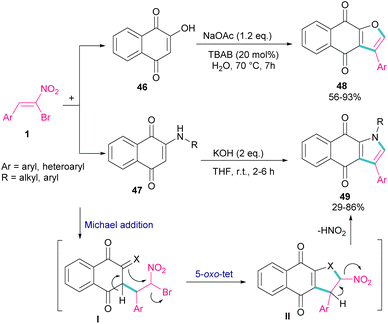 |
| | Scheme 22 Reaction of bromonitroalkene with lawsone and 2-aminonaphthoquinone under basic conditions. | |
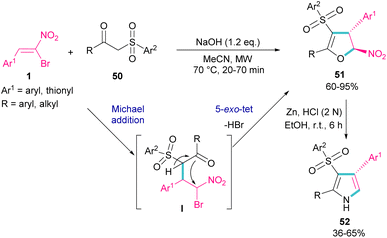 |
| | Scheme 23 Reaction of bromonitroalkene with β-ketosulfones. | |
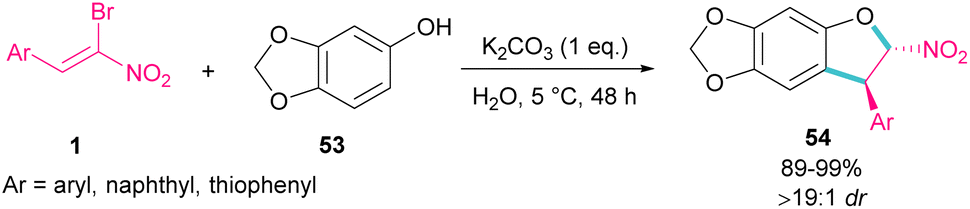
In 2020, Feng et al. presented a protocol for the construction of diastereoselective trans-3-aryl-2-nitro-2,3-dihydrobenzofurans 54 under basic conditions (Scheme 24).53 A diverse range of (Z)-bromonitrostyrenes 1, containing aryl, naphthyl and thiophenyl moieties reacted smoothly with sesamol 53 in water as a green solvent to form dihydrofurans in excellent yields. All products were purified only by a simple filtration procedure and could also be synthesized in the Gram-scale (1.20 g, 94%). In the same year, the synthesis of the oxazole rings from α-halo-β-naphthol and nitroalkenes was carried out.54 Bromonitroalkene as a coupling partner gave the desired product in 82% with >20:1 diastereoselectivity.
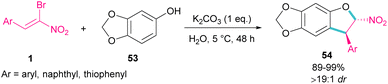 |
| | Scheme 24 Reaction of (Z)-bromonitrostyrenes and sesamol in the presence of K2CO3. | |
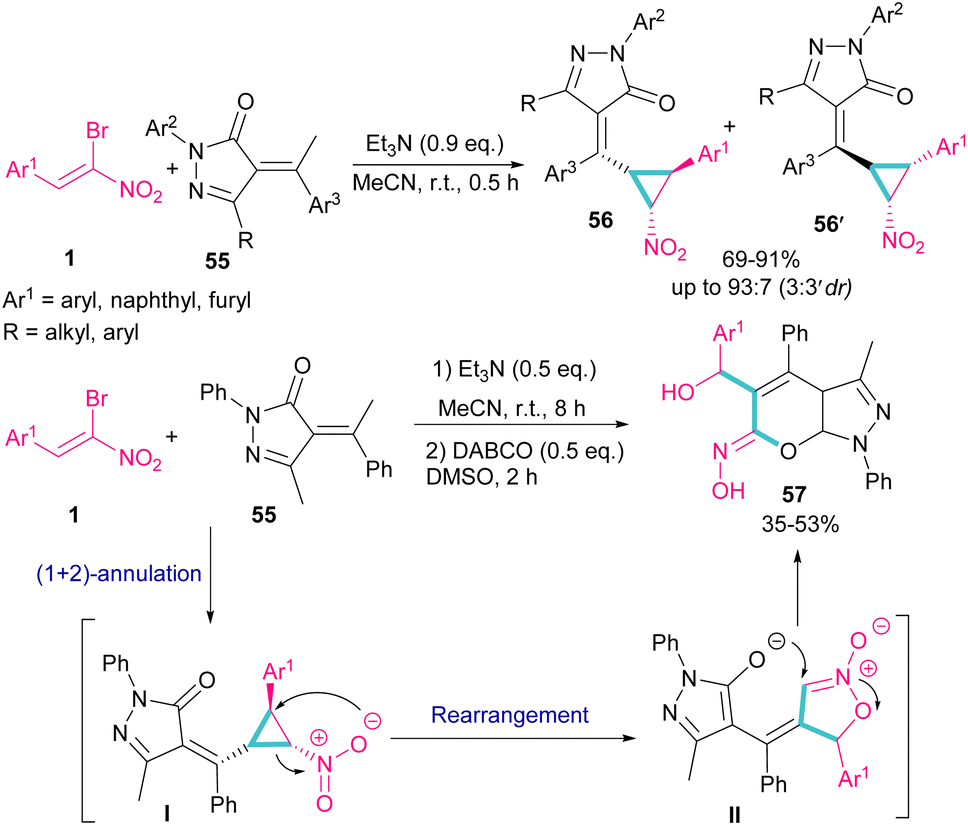
A chemoselective annulation of bromonitrostyrenes with α-alkylidene pyrazolones was established by Han and co-workers in 2021 (Scheme 25).55 A novel library of pyrazole-fused pyranone oximes were obtained, where α-alkylidene pyrazolone 55 acted as C1 synthon and react in (2 + 1)-cycloaddition with bromonitrostyrene 1 to provide vinylcyclopropane-based pyrazolone 56 and 56′ in the presence of a base. The formation of diastereoisomer 56 was favored due to the less steric hindrance between the aryl and nitro groups. In another stage, the authors conducted this reaction in two steps, in which Et3N was served as a base to obtain the cyclopropane product. Then, the addition of second base resulted in an intramolecular rearrangement towards the synthesis of pyrazole-fused pyranone oximes 57.
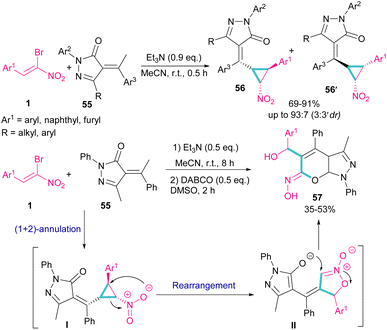 |
| | Scheme 25 Reaction of bromonitrostyrenes with α-alkylidene pyrazolones mediate by Et3N. | |
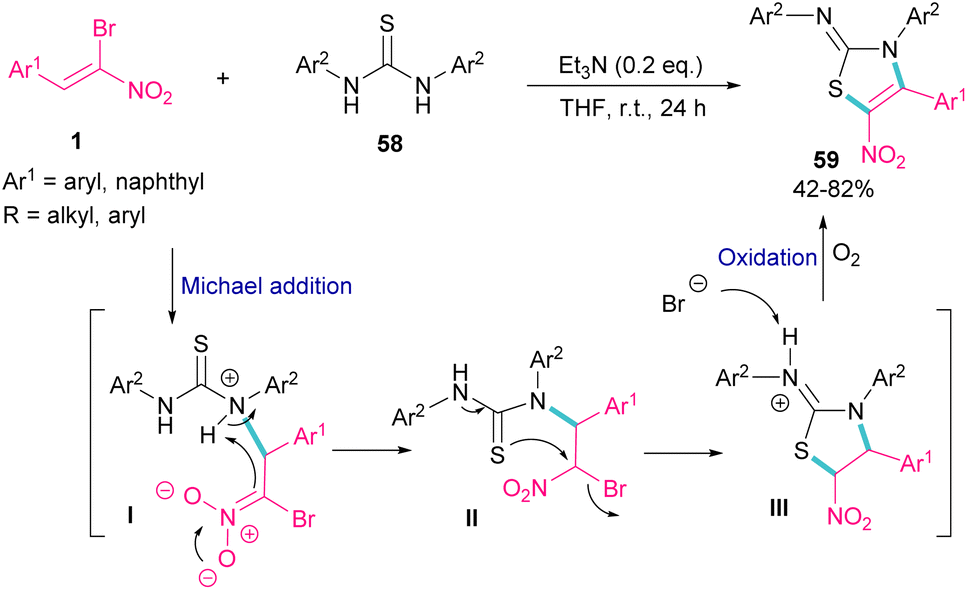
The synthesis of 2-iminothiazolines 59 can be obtained from the cyclization of 1,3-disubstituted thioureas 58 with 1-bromo-1-nitroalkenes 1 (Scheme 26).56 At first, Michael addition between 1-bromo-1-nitroalkenes 1 and 1,3-diphenylthiourea 58 produced intermediate II, which then underwent a tautomerism and nucleophilic substitution to afford the five-membered ring intermediate III. The deprotonation of III by bromide anion, followed by the oxidation yielded product 59. It should be noted that using the suitable amount of the base and the performance of the reaction under air is crucial for this reaction to proceed.
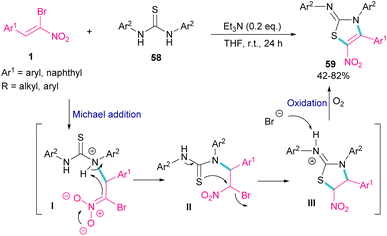 |
| | Scheme 26 Et3N-promoted reaction of 1,3-disubstituted thioureas with 1-bromo-1-nitroalkenes. | |
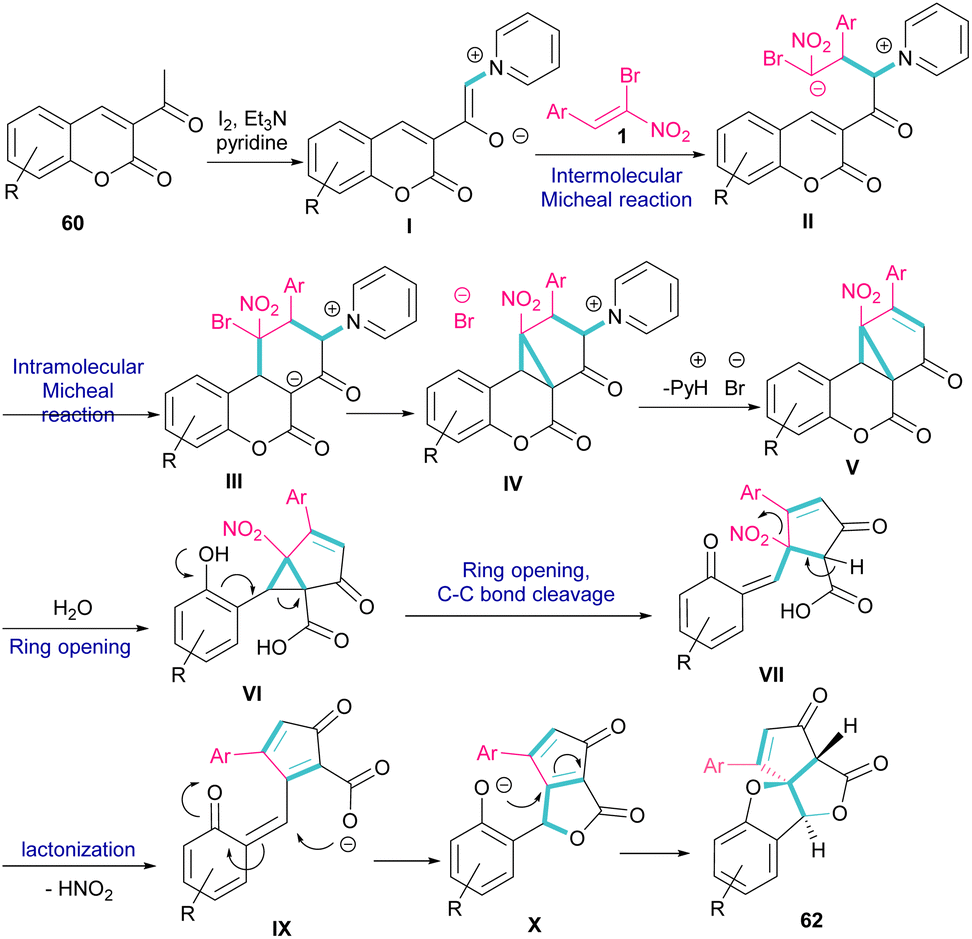
A new class of cyclopenta[c]furo[3,2-b]furan-5,6-diones, containing three carbon stereocenters were constructed by Yavari et al. in 2022 (Scheme 27).57 A three-component reaction, including β-bromo-β-nitrostyrenes 1, 3-acetyl-2H-chromen-2-ones 60, and pyridine 61 was carried out through three steps intermolecular and intramolecular Michael additions. Initially, the C–H bond functionalization of 3-acetyl-2H-chromen-2-one 60 with pyridine 61 in the presence of I2 and Et3N furnished pyridinium ylide I. Then, sequentially intermolecular Michael addition of 1 with I, and intramolecular Michael reaction gave intermediate II, which was converted to cyclopropane III. Intermediate IV was obtained via the elimination of pyridinium bromide in III, followed by two steps cyclopropane ring-opening and rearrangement to produce VI. The release of HNO2 gave VII, which underwent an intramolecular lactonization to render IX. Another intramolecular Michael addition of the phenoxide ion with the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond gave product 62 (Scheme 28).
C bond gave product 62 (Scheme 28).
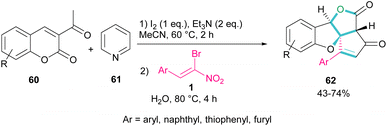 |
| | Scheme 27 Three-component reaction of β-bromo-β-nitrostyrenes, 3-acetyl-2H-chromen-2-ones, and pyridine. | |
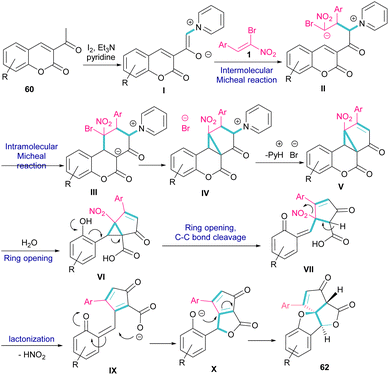 |
| | Scheme 28 Possible mechanism for reaction of β-bromo-β-nitrostyrenes, 3-acetyl-2H-chromen-2-ones, and pyridine. | |
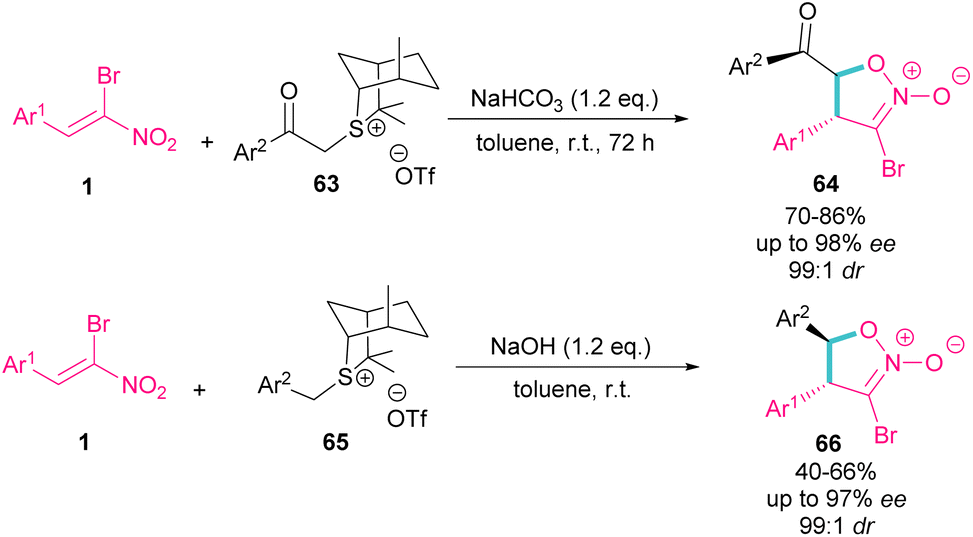
In the same year, the Xie group designed a (4 + 1) ylide annulation between chiral sulfonium salts 63 and 65 with α-bromonitroalkenes 1 access to enantioenriched isoxazoline N-oxides 64 and 66 (Scheme 29).58 Two types of chiral sulfonium salts, such as α-benzoyl sulfonium triflate salts 63 and α-benzyl sulfonium triflate salts 65 could smoothly generate the sulfonium ylide intermediates, which served as C1 synthon in (4 + 1)-annulation with bromonitroalkene as a 4-atomic synthon. All products were obtained in high yields with excellent enantio- and diastereoselectivity. Also, changing the anions of sulfonium salts from OTf− to Br−, ClO4− and BF4−, showed that the type of anion has a great effect on the activity, and enantioselectivity in this asymmetric annulation.
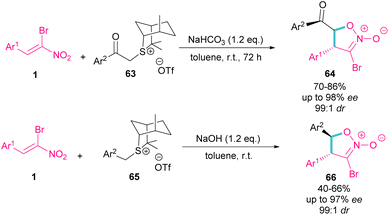 |
| | Scheme 29 Reaction of sulfonium salts and substituted α-bromonitroalkenes. | |
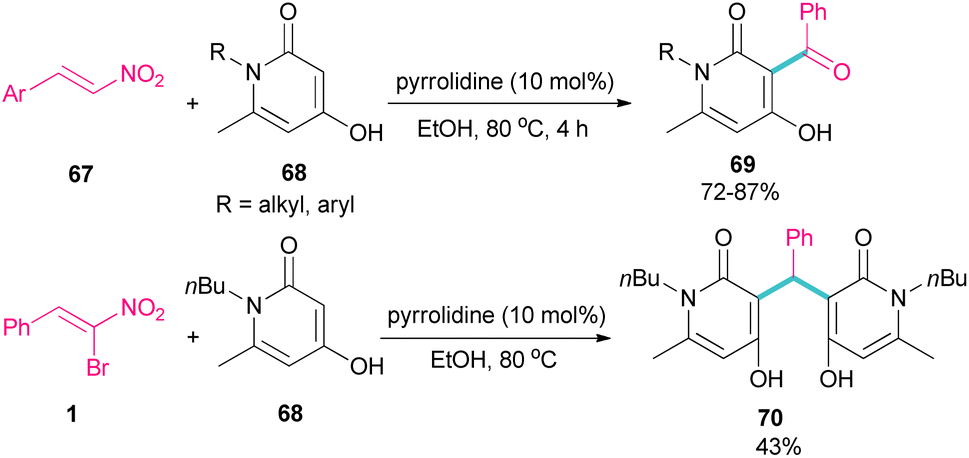
In 2023, Li et al. investigated the application of pyrrolidine as a catalyst in the reaction of 1-butyl-4-hydroxy-6-methylpyridin-2(1H)-one 68 and nitroalkenes 67 (Scheme 30).59 They achieved an open ring product 69, when α-bromonitrostyrene 1 was used as a coupling reactant, while the reaction of other nitroalkenes led to 4-hydroxy-3-benzoylpyridin-2(1H)-ones 70 as the main product. This may be because of the high steric hindrance in α-bromonitrostyrene, which inhibits the formation of product 69.
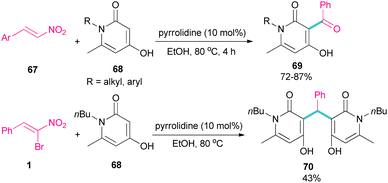 |
| | Scheme 30 Pyrrolidine-catalyzed reaction of 1-butyl-4-hydroxy-6-methylpyridin-2(1H)-one and nitroalkenes. | |
2.4. Catalyst-free transformations of bromonitrostyrenes
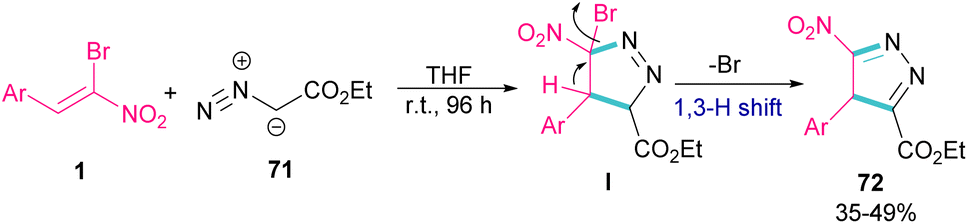
In 2009, Xie and co-workers reported the synthesis of pyrazoles 72 from 1,3-cycloaddition of α-bromo-α-nitroalkenes 1 with ethyl diazoacetate 71 under catalyst-free conditions (Scheme 31).60 In general, the reaction involved the nucleophilic addition of 71 to 1 to form intermediate I, followed by the elimination of bromide along with the 1,3-H shift to furnish the final pyrazole product 72.
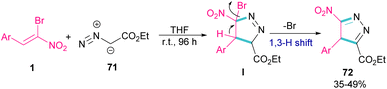 |
| | Scheme 31 Reaction of α-bromo-α-nitroalkenes with ethyl diazoacetate under catalyst-free conditions. | |
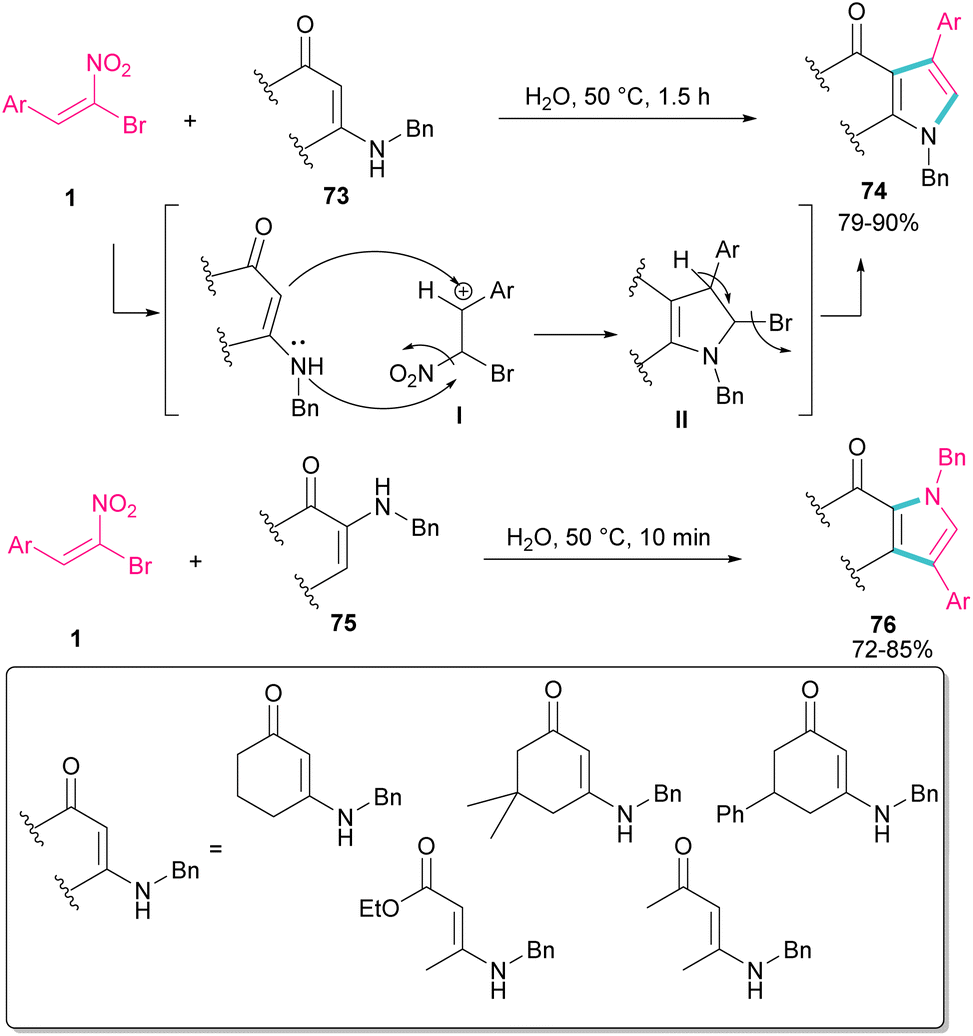
In 2010, Rueping and Parra treated (E)-β-bromonitrostyrenes 1 with enaminones 73 to synthesize pyrrole derivatives 74 (Scheme 32).61 The main advantages of their reaction were the performance of the reaction in water as a green solvent, very short reaction time, and excellent product yields. In this reaction, bromonitrostyrenes 1 acted as a trifunctional synthon and reacted well with both enaminone 73 and N-benzylenaminone 75 as binucleophilic synthons. First, the nucleophilic attack of nitrogen to the carbon atom in I concomitant with removal of a leaving group produced II. Next, the deprotonation and the release of another leaving group afforded the desired product 74.
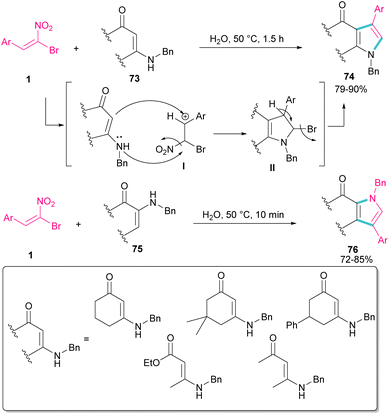 |
| | Scheme 32 Catalyst-free reaction of (E)-β-bromonitrostyrenes with enaminones. | |
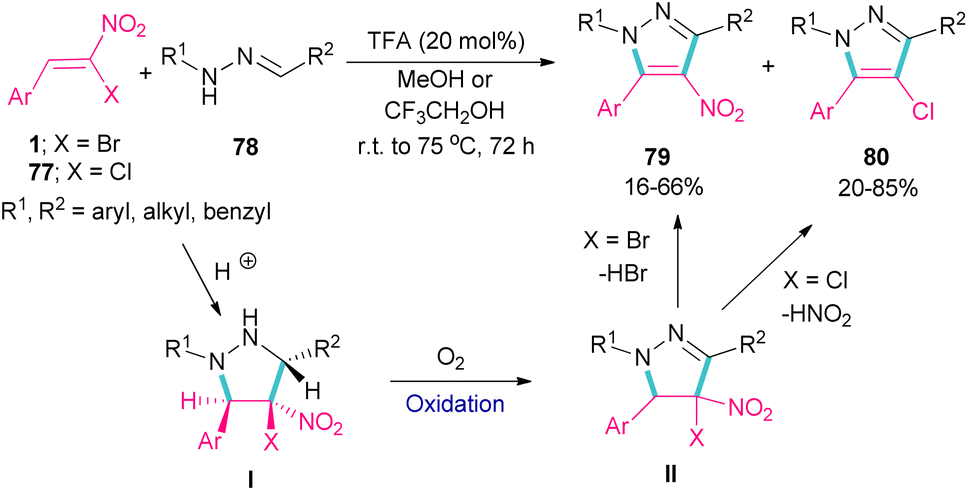
Deng et al. showed that depending on the type of nitrostyrenes and the reaction solvent, two different pyrazole products could be obtained under acidic conditions (Scheme 33).62 When β-chloro-β-nitrostyrenes 77 were used as reactants in the reaction with hydrazones 78, the elimination of HNO2 occurred to produce 4-chloro-tetrasubstituted pyrazoles 80 in MeOH as a solvent. Whereas, 4-nitro-tetrasubstituted pyrazoles 79 were obtained from β-bromo-β-nitrostyrenes 1 and hydrazones 78 as starting materials in MeOH. In this case, the removal of HBr was favored over HNO2, which could be due to the easy cleavage of the C–Br bond compared to the C–Cl bond. In fact, the leaving-group abilities of functional groups can be classified as Br > NO2 > Cl. In general, the reactions involved pyrazolidine intermediate I, which was subjected to oxidation and elimination steps. It should be noted that when a more acidic alcoholic solvent like CF3CH2OH was utilized, the formation of 4-bromo-tetrasubstituted pyrazoles was also observed in the reaction mixture.
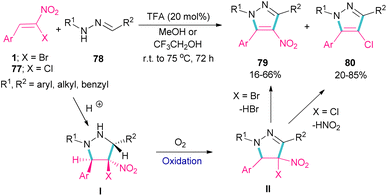 |
| | Scheme 33 TFA-catalyzed reaction of bromonitrostyrenes with hydrazones. | |
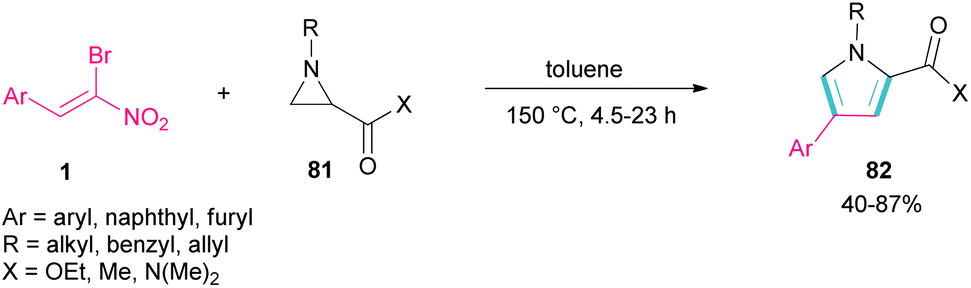
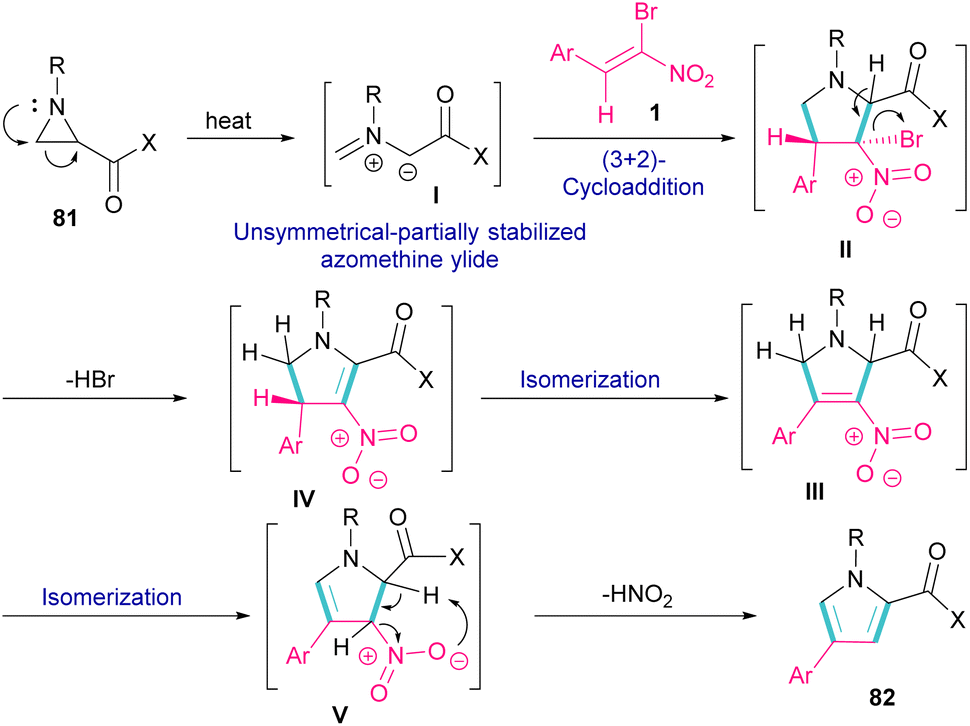
Another domino reaction was carried out by the Khan research team to construct 1,2,4-trisubstituted pyrrole derivatives 82 from 1,3-dipolar cycloaddition of unactivated aziridines 81 with β-bromo-β-nitrostyrenes 1 (Scheme 34).63 This reaction involved in situ generated unsymmetrical azomethine ylide from aziridine, followed by a cascade elimination and aromatization step. As shown in Scheme 35, the mechanism started with simultaneous cleavage of the C–C bond of aziridine 81 under heat to afford azomethine ylide I. The interaction of I with β-bromo-β-nitrostyrene 1 resulted in the unstable cycloadduct II, which readily underwent E2 elimination of HBr to render III. In this step, III was isomerized to IV and then V, followed by the elimination of HNO2 to form product 82.
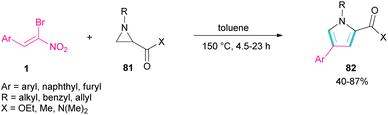 |
| | Scheme 34 Catalyst-free reaction of unactivated aziridines with β-bromo-β-nitrostyrenes. | |
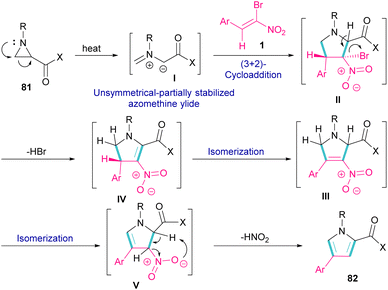 |
| | Scheme 35 Plausible mechanism for reaction of unactivated aziridines with β-bromo-β-nitrostyrenes. | |
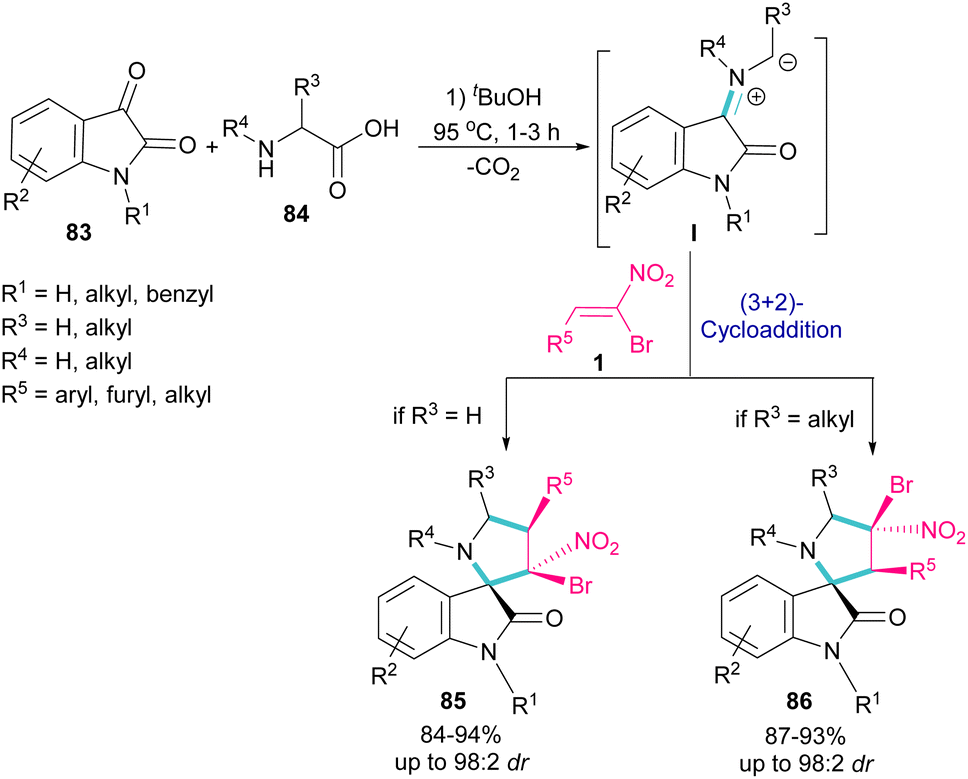
In 2019, Ganesh and co-workers developed a multi-component reaction, including oxindoles 83, bromonitrostyrenes 1 and α-amino acids 84 to generate tetra-substituted α-spiropyrrolidine structures 85 and 86 (Scheme 36).64 In the first step, azomethine ylide I was formed from the condensation of oxindole and α-amino acid. Next, (3 + 2)-cycloaddition of bromonitrostyrene with I provided spiropyrrolidine. Two diastereomers could be obtained depending on the substituent at the α-position of amino acid. If R3 = H, diastereomer 85 was the major product, while spiropyrrolidine 86 was obtained as a major diastereomer when R3 = alkyl. The diastereoselectivity may be due to the steric effects in transition states of azometine ylides.
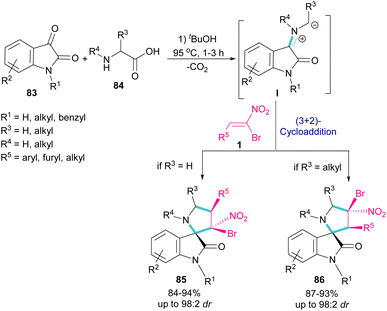 |
| | Scheme 36 Reaction of oxindole, bromonitrostyrenes and α-amino acids. | |
3. Conclusions
In this review, we have described the advances in the synthesis of carbocyclic and heterocyclic compounds from the reaction of α-bromonitrostyrenes with various coupling reactants in straightforward and atom economical manners. Organocatalytic domino Michael reactions offer a direct and sustainable route for the synthesis of diastereo- and enantioselective bioactive products under mild reaction conditions. Although it seems that the use of Lewis acid metal catalysts and bases as promoters in the reactions of α-bromonitrostyrenes could be studied further. Considering the great potential of α-bromonitrostyrene as a reactive dipolarophile with good leaving groups (bromo and nitro), the use of this synthon in the cross-coupling reactions, as well as regio- and stereoselective syntheses can provide useful insights for further researches in this field. In our opinion, α-bromonitrostyrenes can be considered as a simple and accessible building block for the construction of more complex organic molecules.
Conflicts of interest
There are no conflicts to declare.
Notes and references
- R. Jacques, R. Pal, N. A. Parker, C. E. Sear, P. W. Smith, A. Ribaucourt and D. M. Hodgson, Org. Biomol. Chem., 2016, 14, 5875–5893 RSC.
- Z. Chen, M. Pitchakuntla and Y. Jia, Nat. Prod. Rep., 2019, 36, 666–690 RSC.
- E. Mateev, M. Georgieva and A. Zlatkov, J. Pharm. Pharm. Sci., 2022, 25, 24–40 CAS.
- T. Shi, G. Yin, X. Wang, Y. Xiong, Y. Peng, S. Li, Y. Zeng and Z. Wang, Green Synth. Catal., 2023, 4, 20–34 CrossRef CAS.
- R. H. Mir, P. A. Mir, R. Mohi-Ud-Din, S. Sabreen, M. Maqbool, A. J. Shah, K. Shenmar, S. N. Raza and F. H. Pottoo, Anti-Cancer Agents Med. Chem., 2022, 22, 3291–3303 CrossRef CAS PubMed.
- H. Rostami and L. Shiri, ChemistrySelect, 2020, 5, 11197–11220 CrossRef CAS.
- Ş. G. Küçükgüzel and S. Şenkardeş, Eur. J. Med. Chem., 2015, 97, 786–815 CrossRef PubMed.
- K. Karrouchi, S. Radi, Y. Ramli, J. Taoufik, Y. N. Mabkhot, F. A. Al-Aizari and M. h. Ansar, Molecules, 2018, 23, 134 CrossRef PubMed.
- M. Lusardi, A. Spallarossa and C. Brullo, Int. J. Mol. Sci., 2023, 24, 7834–7859 CrossRef CAS PubMed.
- S. Hoz and D. Speizman, Tetrahedron Lett., 1979, 20, 4855–4856 CrossRef.
- M. M. Campbell, N. Cosford, L. Zongli and M. Sainsbury, Tetrahedron, 1987, 43, 1117–1122 CrossRef CAS.
- E. Sheremet, R. Tomanov, E. Trukhin and V. Berestovitskaya, Russ. J. Org. Chem., 2004, 40, 594–595 CrossRef CAS.
- W. E. Parham and J. L. Bleasdale, J. Am. Chem. Soc., 1951, 73, 4664–4666 CrossRef CAS.
- L.-P. Fan, P. Li, X.-S. Li, D.-C. Xu, M.-M. Ge, W.-D. Zhu and J.-W. Xie, J. Org. Chem., 2010, 75, 8716–8719 CrossRef CAS PubMed.
- R. G. Soengas, R. C. Acurcio and A. M. Silva, Eur. J. Org. Chem., 2014, 2014, 6339–6359 CrossRef CAS.
- S. D. Pasuparthy and B. Maiti, ChemistrySelect, 2022, 7, e202104261 CrossRef CAS.
- T. Das, S. Mohapatra, N. P. Mishra, S. Nayak and B. P. Raiguru, ChemistrySelect, 2021, 6, 3745–3781 CrossRef CAS.
- S. Nayak, P. Panda, S. Bhakta, S. K. Mishra and S. Mohapatra, RSC Adv., 2016, 6, 96154–96175 RSC.
- P. Sharma, R. Gupta and R. K. Bansal, Beilstein J. Org. Chem., 2021, 17, 2585–2610 CrossRef CAS PubMed.
- H. Luo, X. Yan, L. Chen, Y. Li, N. Liu and G. Yin, Eur. J. Org. Chem., 2016, 2016, 1702–1707 CrossRef CAS.
- Q. Liang, Asymmetric Domino Michael-Henry Reaction of 1,2-Diones with Nitroolefins Catalyzed by Chiral Ni (II) Complexes, Texas A&M University-Commerce, 2015 Search PubMed.
- X. Xie, L. Xiang, C. Peng and B. Han, Chem. Rec., 2019, 19, 2209–2235 CrossRef CAS PubMed.
- T. Chanda and J. C. G. Zhao, Adv. Synth. Catal., 2018, 360, 2–79 CrossRef CAS.
- Y. Hussain, M. Sharma, A. Kumar and P. Chauhan, Org. Chem. Front., 2022, 9, 572–592 RSC.
- M. Marčeková, B. Ferko, K. R. Detková and P. Jakubec, Molecules, 2020, 25, 3390–3413 CrossRef PubMed.
- Y. Gao, S. Yang, Y. Huo and X. Q. Hu, Adv. Synth. Catal., 2020, 362, 3971–3986 CrossRef CAS.
- H. D. Khanal, M. Perumal and Y. R. Lee, Org. Biomol. Chem., 2022, 20, 7675–7693 RSC.
- A. Z. Halimehjani and Z. Ghaffari, Org. Biomol. Chem., 2023, 21, 2653–2688 RSC.
- C. Li, K. Yin, X. Zhou, F. Zhang and Z. Shen, Tetrahedron, 2023, 149, 133717 CrossRef CAS.
- C. Li, X. Zhou, F. Zhang and Z. Shen, Curr. Org. Chem., 2023, 27, 108–118 CrossRef CAS.
- M. Rueping, A. Parra, U. Uria, F. Besselievre and E. Merino, Org. Lett., 2010, 12, 5680–5683 CrossRef CAS PubMed.
- U. Das, Y.-L. Tsai and W. Lin, Org. Biomol. Chem., 2013, 11, 44–47 RSC.
- C. Jarava-Barrera, F. Esteban, C. Navarro-Ranninger, A. Parra and J. Alemán, Chem. Commun., 2013, 49, 2001–2003 RSC.
- J. Luis-Barrera, R. Mas-Ballesté and J. Aleman, ChemPlusChem, 2015, 80, 1595–1600 CrossRef CAS PubMed.
- J. Feng, L. Lin, K. Yu, X. Liu and X. Feng, Adv. Synth. Catal., 2015, 357, 1305–1310 CrossRef CAS.
- J. Feng, X. Yuan, W. Luo, L. Lin, X. Liu and X. Feng, Chem.–Eur. J., 2016, 22, 15650–15653 CrossRef CAS PubMed.
- J.-W. Zhang, J.-L. Dong, Q.-C. Sun and J.-W. Xie, Synlett, 2018, 29, 603–608 CrossRef CAS.
- F.-Y. Chen, L. Xiang, G. Zhan, H. Liu, B. Kang, S.-C. Zhang, C. Peng and B. Han, Tetrahedron Lett., 2020, 61, 151806–151813 CrossRef CAS.
- Z. D. Susam, M. Bozdemir, G. Gündoğdu and C. Tanyeli, New J. Chem., 2022, 46, 599–606 RSC.
- L. V. Romashov, Y. A. Khomutova, V. M. Danilenko, S. L. Ioffe and A. V. Lesiv, Synthesis, 2010, 2010, 407–414 CrossRef.
- Q. Liang, J. He and B. Ni, Tetrahedron: Asymmetry, 2014, 25, 1146–1149 CrossRef CAS.
- X. Ge, K. L. M. Hoang, M. L. Leow and X.-W. Liu, RSC Adv., 2014, 4, 45191–45197 RSC.
- R. Muruganantham, S. M. Mobin and I. N. Namboothiri, Org. Lett., 2007, 9, 1125–1128 CrossRef CAS PubMed.
- R. Muruganantham and I. Namboothiri, J. Org. Chem., 2010, 75, 2197–2205 CrossRef CAS PubMed.
- N. Ayyagari, D. Jose, S. M. Mobin and I. N. Namboothiri, Tetrahedron Lett., 2011, 52, 258–262 CrossRef CAS.
- J.-W. Xie, P. Li, T. Wang and F.-T. Zhou, Tetrahedron Lett., 2011, 52, 2379–2382 CrossRef CAS.
- R. Zhang, D. Xu and J. Xie, Chin. J. Chem., 2012, 30, 1690–1694 CrossRef CAS.
- S. Roy and K. Chen, J. Chin. Chem. Soc., 2013, 60, 597–604 CrossRef CAS.
- R. G. Soengas, H. Rodriguez-Solla, A. M. Silva, R. Llavona and F. A. A. Paz, J. Org. Chem., 2013, 78, 12831–12836 CrossRef CAS PubMed.
- E. Gopi, T. Kumar, R. F. Menna-Barreto, W. O. Valença, E. N. da Silva Júnior and I. N. Namboothiri, Org. Biomol. Chem., 2015, 13, 9862–9871 RSC.
- T. V. Baiju, R. G. Almeida, S. T. Sivanandan, C. A. de Simone, L. M. Brito, B. C. Cavalcanti, C. Pessoa, I. N. Namboothiri and E. N. da Silva Junior, Eur. J. Med. Chem., 2018, 151, 686–704 CrossRef CAS PubMed.
- V. Mane, S. T. Sivanandan, R. G. Santana, A. Beatriz, E. N. da Silva Junior and I. N. Namboothiri, J. Org. Chem., 2020, 85, 8825–8843 CrossRef CAS PubMed.
- J. Feng, S. Wang, J. Feng, Q. Li, J. Yue, G. Yue, P. Zou and G. Wang, New J. Chem., 2020, 44, 11937–11940 RSC.
- M. Qi, M. Li, L. Bai, J. Liu and X. Luan, J. Org. Chem., 2023, 88, 5997–6007 CrossRef CAS PubMed.
- Y.-J. Zhong, T. Qi, Y.-L. Ji, H. Huang, X. Zhang, H.-J. Leng, C. Peng, J.-L. Li and B. Han, J. Org. Chem., 2021, 86, 2582–2592 CrossRef CAS PubMed.
- Y. Xu, X. Ge, Y. Zhang, H. Zhang and X.-W. Liu, RSC Adv., 2021, 11, 2221–2225 RSC.
- I. Yavari, R. Mohsenzadeh and P. Ravaghi, J. Org. Chem., 2022, 87, 2616–2623 CrossRef CAS PubMed.
- P. Gong, J. Wang, W. B. Yao, X. S. Xie and J. W. Xie, Adv. Synth. Catal., 2022, 364, 1185–1199 CrossRef CAS.
- L. Li, Y. Liu, S. Zhou, J. Li, C. Qi and F. Zhang, Synth. Commun., 2023, 53, 492–502 CrossRef CAS.
- J.-W. Xie, Z. Wang, W.-J. Yang, L.-C. Kong and D.-C. Xu, Org. Biomol. Chem., 2009, 7, 4352–4354 RSC.
- M. Rueping and A. Parra, Org. Lett., 2010, 12, 5281–5283 CrossRef CAS PubMed.
- X. Deng, J. T. Liang and N. S. Mani, Eur. J. Org. Chem., 2014, 2014, 410–417 CrossRef CAS.
- V. Kumar, A. Awasthi, A. Metya and T. Khan, J. Org. Chem., 2019, 84, 11581–11595 CrossRef CAS PubMed.
- M. P. Rao, S. S. Gunaga, J. Zuegg, R. Pamarthi and M. Ganesh, Org. Biomol. Chem., 2019, 17, 9390–9402 RSC.
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  ,
Mohammad Mahdi Aghanour Ashtiani
,
Mohammad Mahdi Aghanour Ashtiani