DOI:
10.1039/D4RA02206H
(Review Article)
RSC Adv., 2024,
14, 15167-15177
Catalytic asymmetric carbenoid α-C–H insertion of ether
Received
22nd March 2024
, Accepted 2nd May 2024
First published on 13th May 2024
Abstract
Significant advancements have been made in catalytic asymmetric α-C–H bond functionalization of ethers via carbenoid insertion over the past decade. Effective asymmetric catalytic systems, featuring a range of chiral metal catalysts, have been established for the enantioselective synthesis of diverse ether substrates. This has led to the generation of various enantioenriched, highly functionalized oxygen-containing structural motifs, facilitating their application in the asymmetric synthesis of bioactive natural products.
1. Introduction
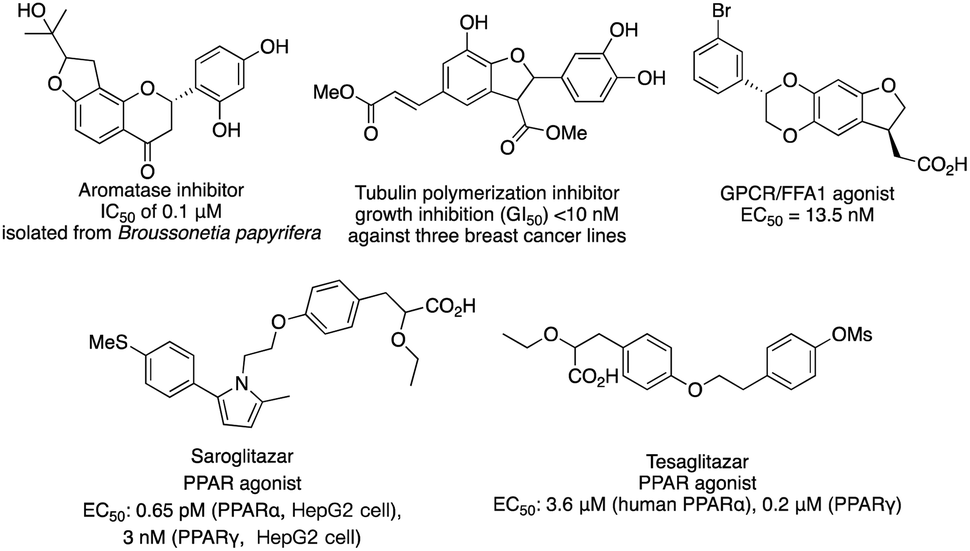
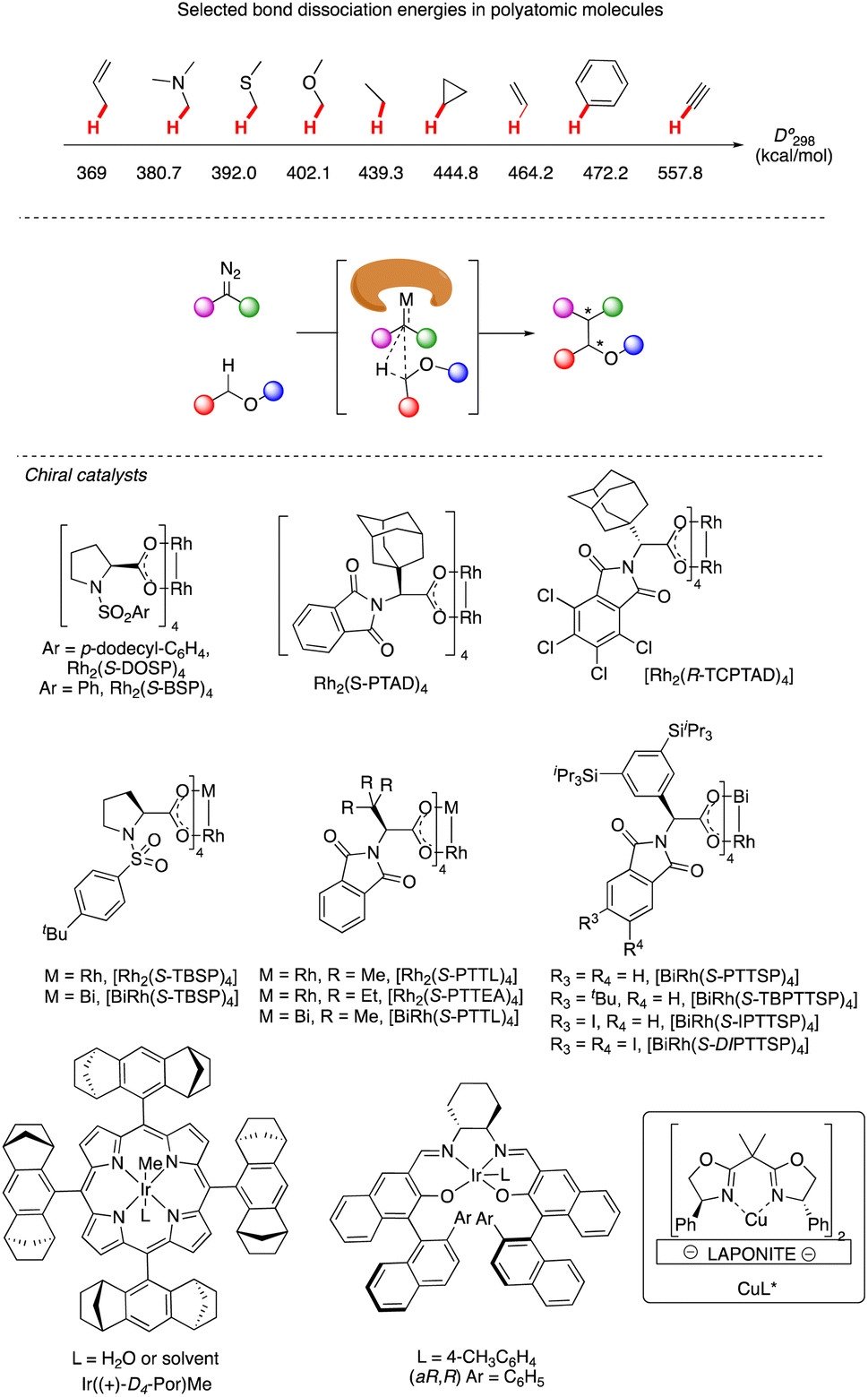
C–H functionalization continues to be one of the forefronts of organic chemistry.1 The resurgence of carbenoid transformations from diazo compounds has garnered significant attention due to their effectiveness in transition-metal and enzyme-catalyzed C–H functionalization.2 While the spontaneous decomposition of ethyl diazoacetate was first studied in 1906 by Silberrad and Roy,3 it was not until Yates suggested the participation of transition metals in diazo decompositions that the true potential of diazo chemistry began to emerge.4 Particularly noteworthy is the ability of in situ formed highly active metal carbenoids to insert into C–H bonds of aromatics and alkanes.5 Especially with direct C–H bond functionalization with different transition metals, such as Cu,6 Ir,7 Rh8 and others,9 the in situ formed highly active metal carbenoids are ideal to insert into C–H bonds of aromatics and alkanes. Despite extensive research on alkyl amines and alcohols, catalytic asymmetric α-C–H functionalization of ethers remains challenging. Early studies by Adams, Frenette, and coworkers in 1989 demonstrated that the C–H bond adjacent to an ether is a preferred site for insertion catalyzed by rhodium acetate.10 Subsequent breakthroughs by Doyle and Davies and their coworkers showcased the remarkable efficiency and enantioselectivity achievable using chiral rhodium carboxamides (Rh2(5S-MEPY)4) and newly designed dirhodium complexes, respectively.11 Since then, the chiral dirhodium complex has dominated the asymmetric C–H insertion of diazo compounds.12 Fig. 1 showcases highly active ether natural products and drug candidates, emphasizing the significance of ether compound synthesis methodology in drug discovery and natural product synthesis (Fig. 1). Ether compounds possess higher α-C–H bond dissociation energy and limited metal coordination capability compared to compounds such as amines, sulfur, and olefins. This characteristic presents a challenge for the functionalization of α-C–H bonds in ethers (Fig. 2). Herein, we present a comprehensive review of recent advancements in α-C–H bond carbenoid insertion of ethers, underscoring the importance of this methodology in the total synthesis of ether natural products.13
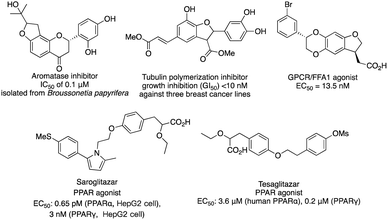 |
| | Fig. 1 Representative bioactive nature products. | |
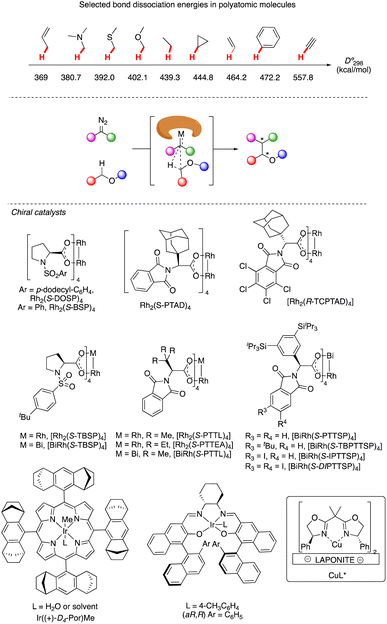 |
| | Fig. 2 Representative catalysts for α-C–H bond carbenoid insertion of ether. | |
2. Intramolecular α-C–H bond carbenoid insertion of ether
2.1 Synthesis of dihydrobenzofurans
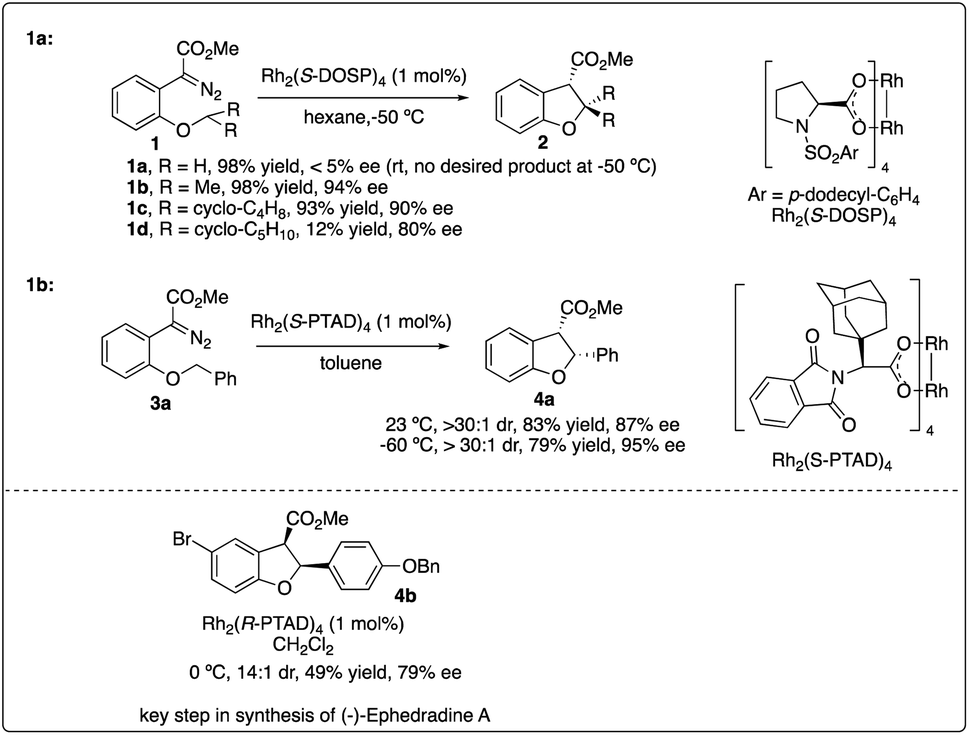
In 1996, Davies and his colleagues reported the development of a newly designed chiral catalyst, Rh2(S-DOSP)4, commonly known as Davies's catalyst, and successfully applied it to intermolecular enantioselective C–H insertion reactions.11b Subsequently, they achieved the intramolecular asymmetric synthesis of dihydrobenzofuran 2 with high enantioselectivity using the same robust catalyst (Scheme 1a).14 Through the exploration of intramolecular substrates, they investigated the selectivity between tertiary and secondary sites, highlighting the importance of both the site of C–H insertion and the catalyst in facilitating effective asymmetric intramolecular C–H insertion reactions.
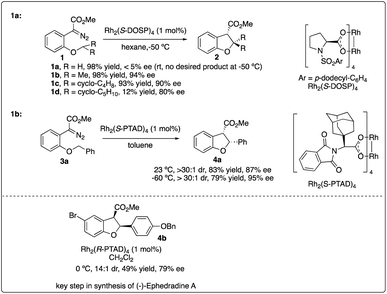 |
| | Scheme 1 Enantioselectivity intramolecular C–H insertion reactions reported by Davises group. | |
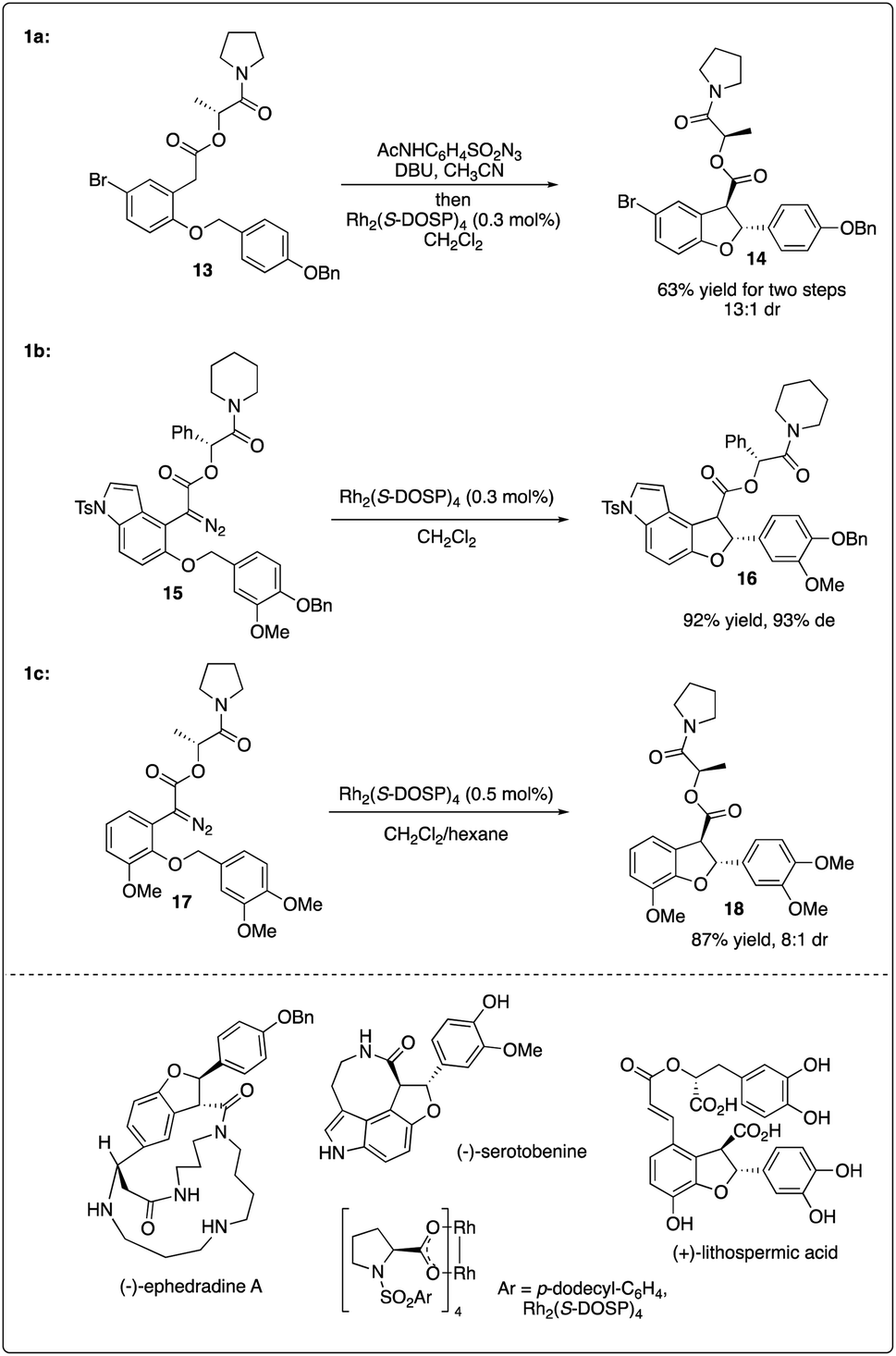
In 2006, Davies and colleagues15a introduced a novel adamantly variant, Rh2(S-PTAD)4, as a backup chiral catalyst to address instances where Rh2(S-DOSP)4 exhibited low asymmetric induction. They successfully demonstrated the enhanced ability of Rh2(S-PTAD)4 in promoting enantioselective intramolecular C–H insertion of α-phenyldiazoacetate 3a, serving as a pivotal step in the synthesis of (−)-ephedradine without the need for a chiral auxiliary (Scheme 1b). Furthermore, Davies and Walji developed an immobilization strategy using Argopore resin to heterogenize chiral rhodium catalysts, enabling their application in asymmetric intramolecular insertion reactions.15b This strategy induced similar reactivity and stereoselectivity as their homogeneous counterparts, facilitating the recycling of rhodium catalysis without compromising catalytic efficiency or stereoselectivity.
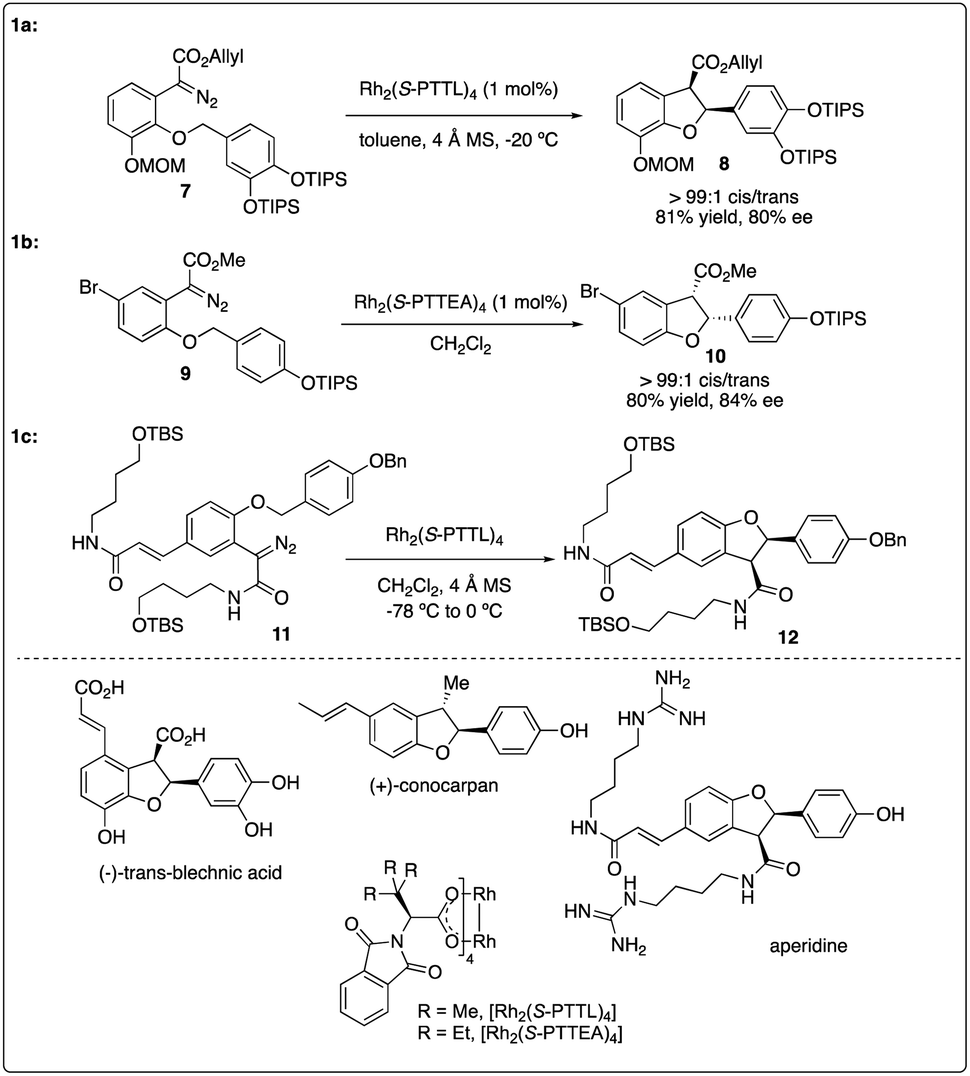
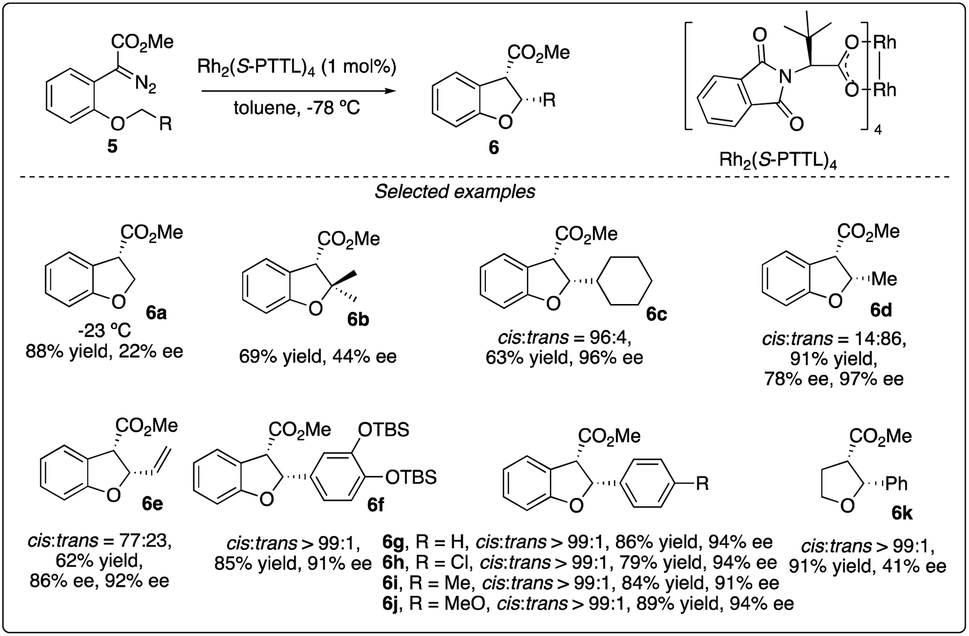
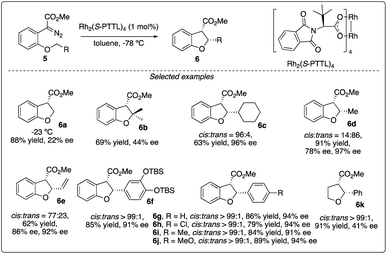
Chiral dirhodium carboxamide (e.g., Rh2(5S-MEPY)4) and carboxylate (such as Rh2(S-DOSP)4) catalysts have been demonstrated as efficient catalysts to construct optically active molecules by the enantioselective intramolecular C–H insertion reactions. Subsequently, Hashimoto and colleagues16 identified Rh2(S-PTTL)4 (referred to as Hashimoto's catalyst) as the optimal catalysis for the enantioselective C–H insertion reaction of α-phenyldiazoacetate 5 (Scheme 2). Substrates bearing with a benzene ring consistently yielded excellent cis/trans ratios and high ee values (6f–6g), while those with simple methine or methyl groups resulted in disappointment outcomes (6a–d). Notably, the product containing a 3,4-(TBSO)2C6H3 group (6f) features an interesting motif found in dihydrobenzofuran-type neolignan blechnic acid. This motif was exploited in their following work on the asymmetric total synthesis of (−)-trans-blechnic acid, wherein they modified the rhodium catalyzed insertion reaction reported earlier (Scheme 3a).17 Additionally, aliphatic substrates were tested, yielding excellent diastereoselectivity, high yields and moderate enantioselectivity under standard reaction conditions (6k).
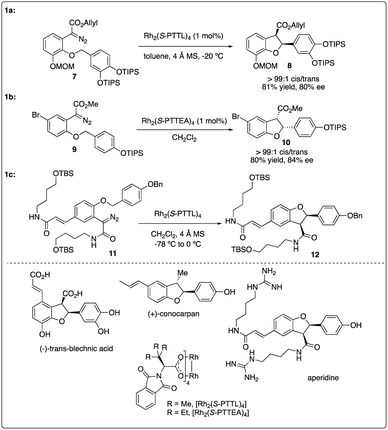 |
| | Scheme 2 Enantioselectivity intramolecular C–H insertion reactions reported by Hashimoto group. | |
 |
| | Scheme 3 Key step in natural product total synthesis reported by Hashimoto group and Wakimoto group. | |
In 2009, Hashimoto and colleagues achieved the asymmetric synthesis of (+)-conocarpan with excellent diastereoselectivity and good enantioselectivity by employing Rh2(S-PTTEA)4-catalyzed intramolecular C–H insertion reaction of diazo ester 9 as the key step (Scheme 3b).18 2011, Wakimoto and coworkers reported the total synthesis of aperidine, wherein the key carbenoid insertion step was catalyzed Hashimoto's catalyst Rh2(S-PTTL)4, affording the product in 75% yield and completely stereoselective manner (Scheme 3c).19
Sometimes, the use of a chiral auxiliary alone may not suffice to achieve the necessary asymmetric induction in rhodium-catalyzed C–H carbenoid insertion reactions. To achieve good enantioselectivity in the synthesis of nature products, double asymmetric induction is frequently employed. Fukuyama and colleagues reported the Rh2(S-DOSP)4-catalyzed key enantioselective synthesis step of (−)-ephedradine A by utilizing intramolecular C–H insertion of diazoacetate bearing a chiral auxiliary, resulting in the formation of a trans-dihydrobenzofuran with excellent diastereoselectivity (Scheme 4a).20 Subsequently, they employed a similar strategy combining a chiral rhodium catalyst and chiral auxiliary to access the asymmetric synthesis of (−)-serotobenine (Scheme 4b).21
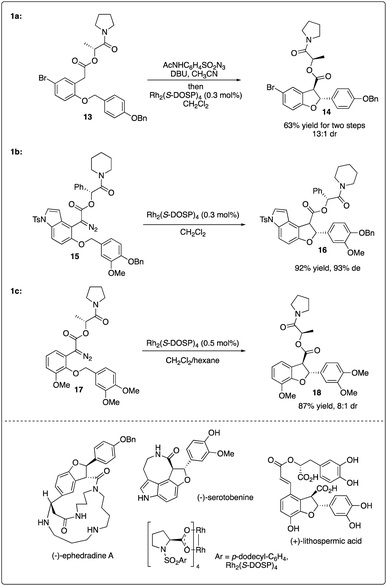 |
| | Scheme 4 Key steps in natural product total synthesis combining with chiral auxiliary and chiral rhodium strategy. | |
Recently, Yu and Wang realized the intramolecular C–H insertion step of the enantioselective synthesis of (+)-lithospermic acid by combining Davies's catalyst Rh2(S-DOSP)4 and Fukuyama's chiral auxiliary, achieving good yield and diastereoselectivity (Scheme 4c).22
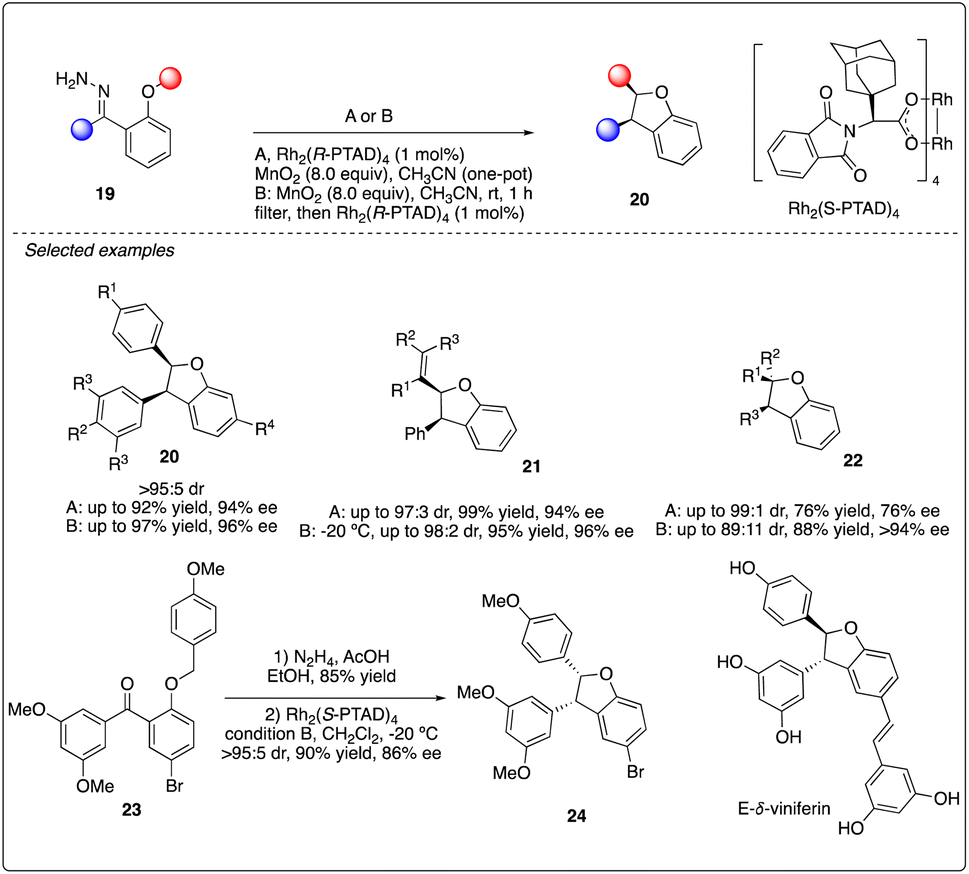
Additionally, Shaw reported Rh2(R-PTAD)4-catalyzed enantioselective intramolecular C–H insertion reactions of donor–donor carbenoid with very high stereoselectivity and good to excellent enantioselectivity (Scheme 5). They developed two unique reaction conditions to achieve the oxidation and insertion reactions step by step or in one-pot from hydrazone 19. This methodology was successfully applied to the asymmetric synthesis of E-δ-viniferin.23
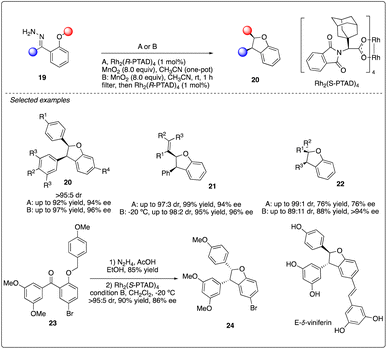 |
| | Scheme 5 Enantioselective intramolecular C–H insertion reactions of donor–donor carbenoid and key step in total synthesis of E-δ-viniferin. | |
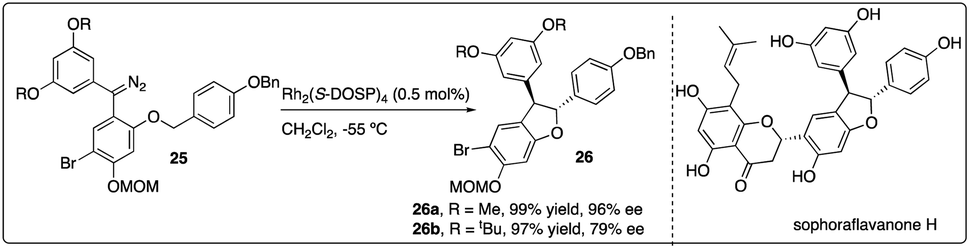
Very recently, Kan and colleagues constructed the dihydrobenzofuran ring 26 of natural product sophoraflavanone H via Rh2(S-DOSP)4-catalyzed asymmetric C–H insertion reaction of donor–donor carbinoid 25, achieving excellent yield and moderate enantioselectivity. Interestingly, the enantioselectivity of the dihydrobenzofuran 26 could be enhanced by introducing a methyl ether, albeit the removal of which posed challenges (Scheme 6).24
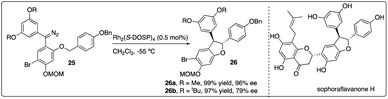 |
| | Scheme 6 Key step in total synthesis of sophoraflavanone H by Kan group. | |
2.2 Synthesis of chromanones and pyran
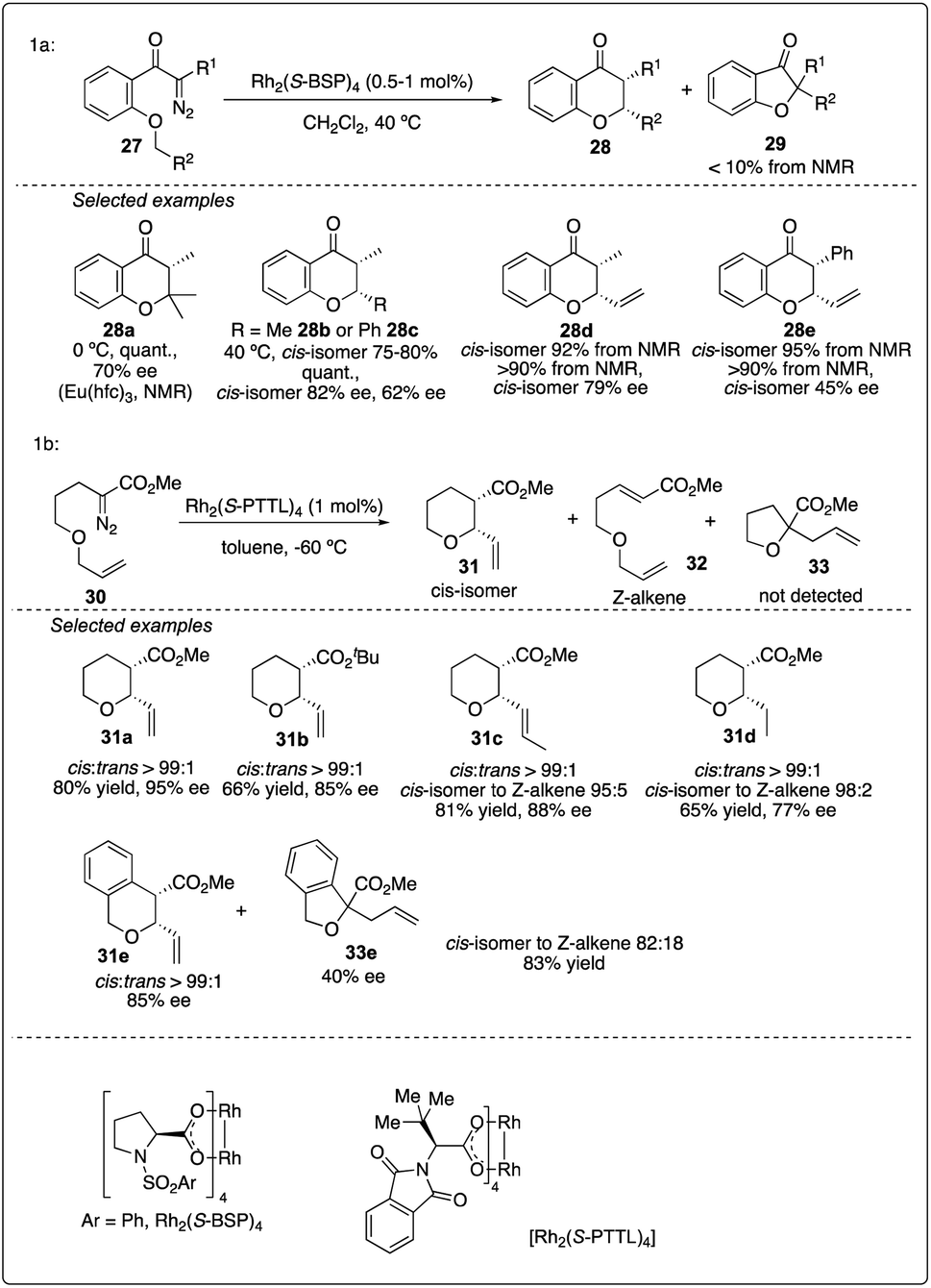
The efficiency of asymmetric intramolecular carbenoid C–H insertion has been demonstrated to preferentially produce five and six-membered rings. In 1992, McKervey and Ye reported Rh2(S-BSP)4-catalyzed asymmetric C–H insertion reaction of ketocarbenoids, leading to the formation of chromanones 28 in excellent yields and good ee values (Scheme 7a)25 In 2015, Hashimoto and colleagues achieved the Rh2(S-PTTL)4-catalyzed asymmetric 1,6-C–H insertion reaction of α-diazo esters 30 by slightly modifying the substrate structure. This method was preferred over Z-alkene formation or tandem ylide formation-rearrangement and 1,2-hydride shift product (Scheme 7b).26
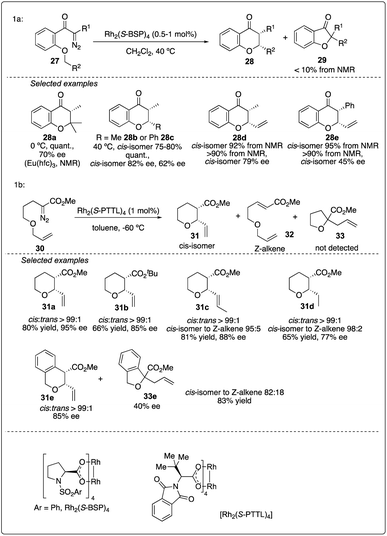 |
| | Scheme 7 Chromanones and pyran synthesis via enantioselectivity intramolecular C–H insertion reactions. | |
2.3 Synthesis of β-lactones
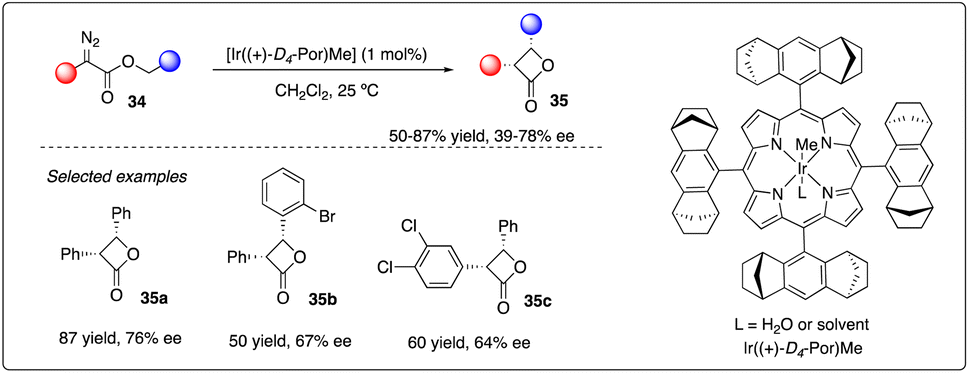
Four-membered rings like β-lactones are common yet unique structural motifs in natural products and pharmaceuticals, posing a challenge but accessible via intramolecular C–H carbene insertion. In 2013, Che and coworkers reported the use of [Ir((+)-D4-Por)Me], supported by D4-Halterman porphyrin ligand, as iridium catalysis. They successfully achieved enantioselective intramolecular carbene insertion into C–H bonds of α-diazoesters 34, yielding a series of aromatic substituted cis-β-lactones 35 with good stereoselectivity and enantioselectivities (Scheme 8).27
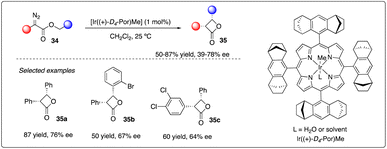 |
| | Scheme 8 Synthesis of β-lactones via iridium catalyzed asymmetric C–H insertion reactions by Che group. | |
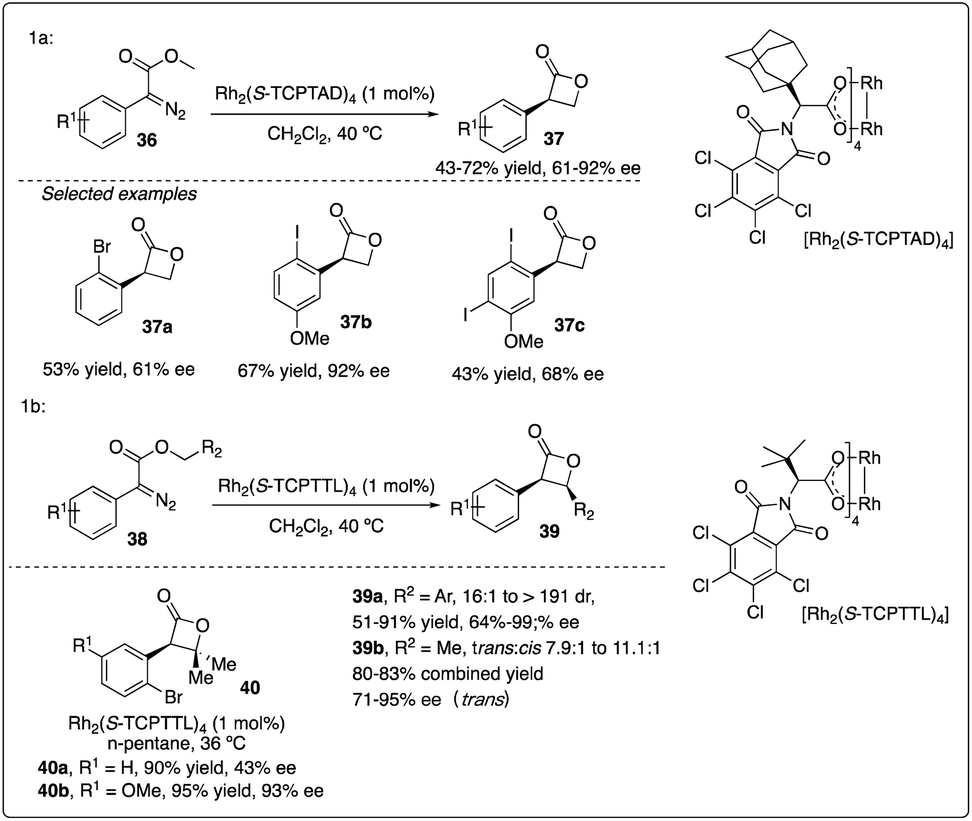
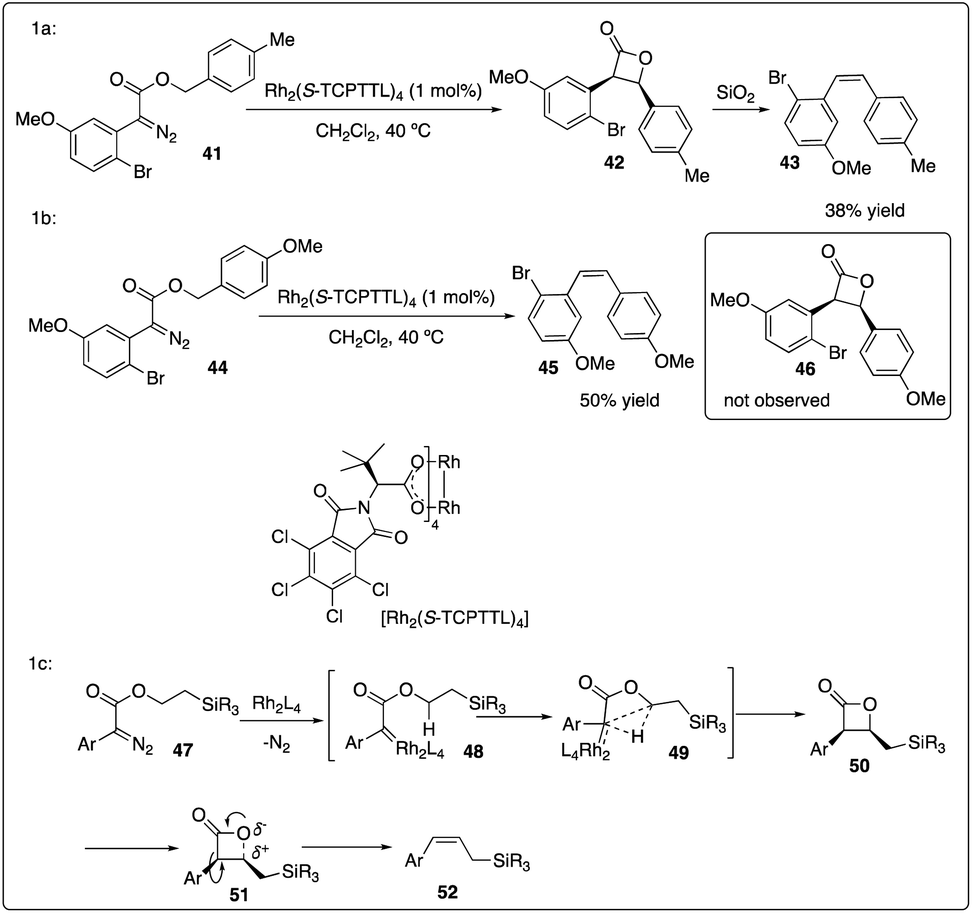
Davies and colleagues discovered that the ortho-substituent (such as halo and trifluoromethyl) on aryldiazoacetate 36 could interfere with intermolecular C–H insertion, thereby enhancing the formation of β-lactone 37. Firstly, they developed asymmetric methyl C–H insertion catalyzed by Rh2(S-TCPTAD)4, achieving good to excellent enantioselectivities. Secondly, they switched to Rh2(S-TCPTTL)4 to modify the cis/trans selectivity of methylene C–H insertion (Scheme 9).28 Interestingly, when ethyl aryldiazoacetate 38b was employed as substrates, trans-formed product 39b was obtained with good to excellent enantioselectivity. However, when electron-donating methyl or methoxyl group (Scheme 10a and b) were introduced to the para-position of the benzyl ester, the desired C–H insertion product 42 was unstable to undergo CO2 extrusion, similar to previous findings reported by the same group (Scheme 10c). They proposed the mechanism for Z-product formation, suggesting that the silicon could direct the C–H insertion away from the more sterically hindered α-position and facilitate the polarization of the lactone C–O bond.29
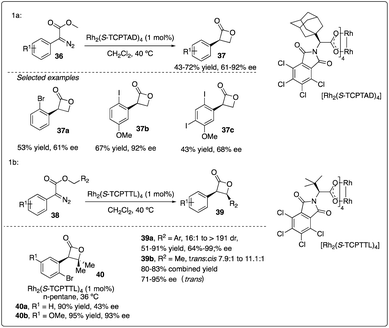 |
| | Scheme 9 Synthesis of β-lactones via rhodium catalyzed asymmetric C–H insertion reactions by Davies group. | |
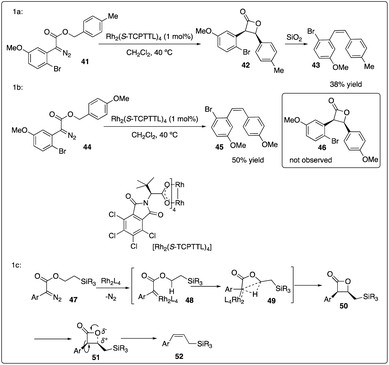 |
| | Scheme 10 Control reactions and proposal mechanism for Z-product formation by Davies group. | |
3. Intermolecular α-C–H bond carbenoid insertion of ether
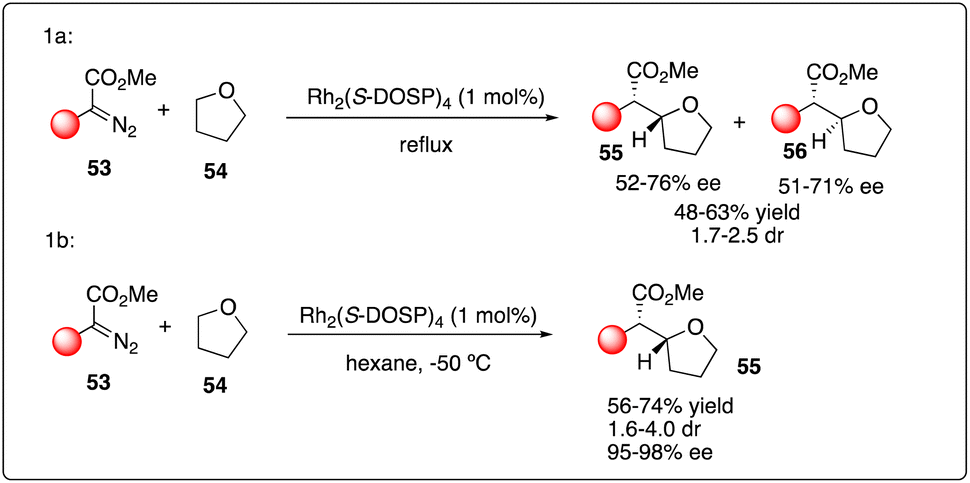
The challenge of achieving asymmetric intermolecular carbenoid C–H insertion persisted until the emergence of a robust catalyst system system capable of overcoming these major obstacles in organic synthesis. Based upon their previous study on Rh2(S-DOSP)4-catalyzed asymmetric cyclopropanations with diazoacetates, Davies and Hansen proposed utilizing the decomposition of aryldiazoacetates 53 by Rh2(S-DOSP)4 to enable asymmetric intermolecular C–H insertions. Employing this strategy, they successfully realized the C–H insertion of aryldiazoacetates 53 with tetrahydrofuran 54 as a carbenoid trap, yielding a series of products with moderate dr values and good enantioselectivity (Scheme 11a).30 Davies and Hansen also proposed the asymmetric induction model by Rh2(S-DOSP)4 to predict the configuration in the product. Subsequently, they optimized the reaction conditions,31 discovering that the degassing of the solvent significantly increased the reactivity and the enantioselectivity. Furthermore, they achieved further improvement in enantioselectivity by employing nonpolar solvents such as THF (2 equiv.) in hexane, highlighting the remarkable selectivity of the carbenoid in the presence of hexane (Scheme 11b). Additionally, they investigated the kinetic isotope effect for the C–H insertion process through competition experiments between d8-tetrahydrofuran and tetrahydrofuran.
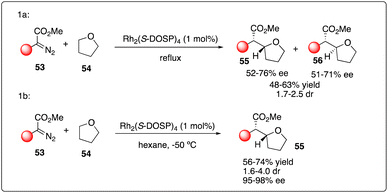 |
| | Scheme 11 Enantioselective intermolecular C–H insertion reactions of donor–accepter carbenoid and THF by Davies group. | |
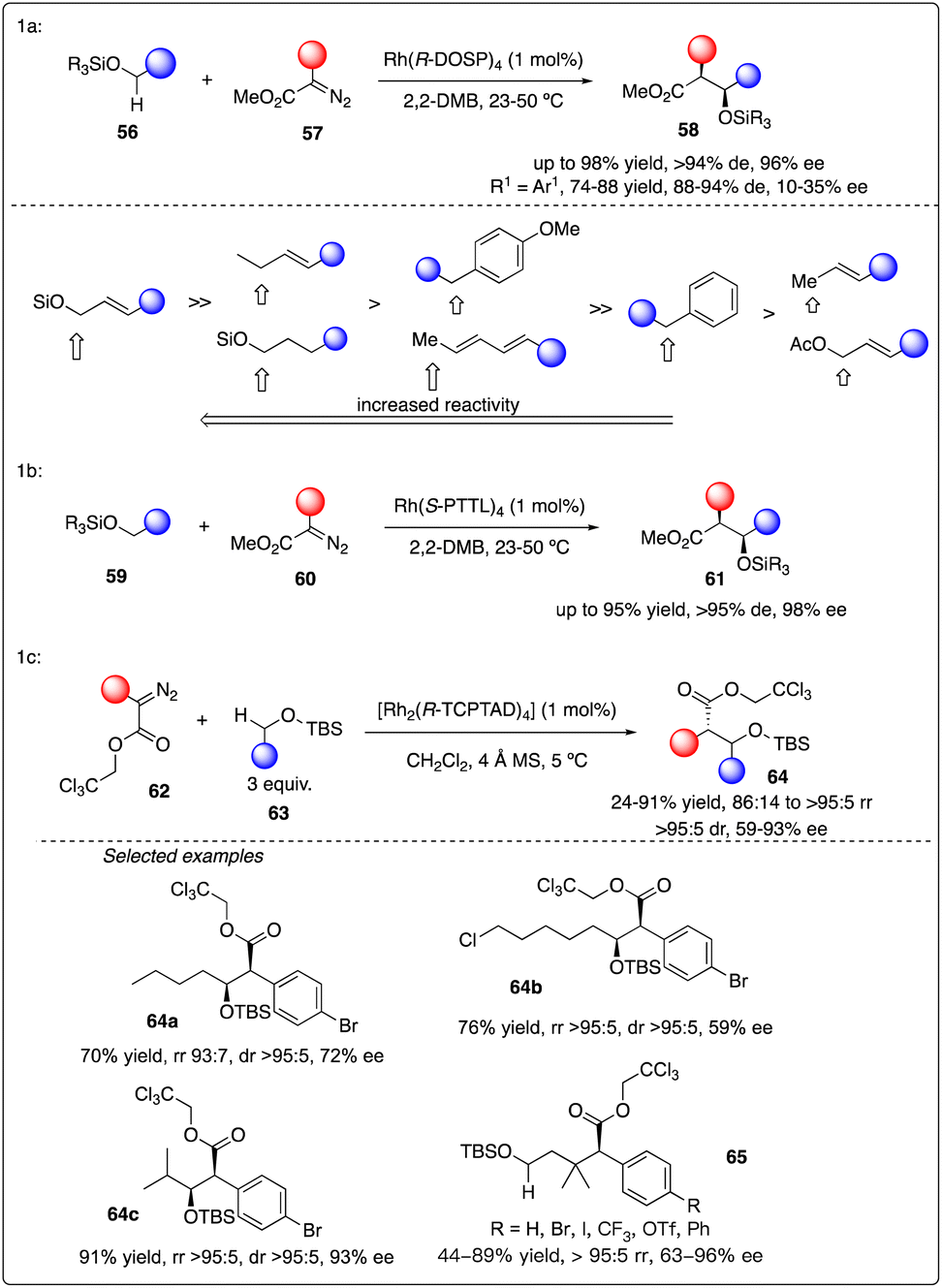
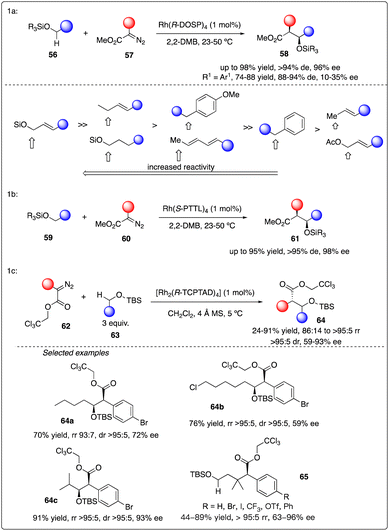
Subsequently, they delved into the comprehensive chemistry of rhodium carbenoid-induced C–H insertion of silyl ethers 56 to synthesize silyl-protected β-hydroxy esters 58 with excellent regio-, diastereo-, and enantioselectivity (Scheme 12a).32 They particular emphasized the critical requirement for selectivity by utilizing donor/acceptor-substituted carbenoids 57 and catalytic rhodium prolinate complex Rh2(S-DOSP)4. Additionally, they discovered that the C–H insertion reaction is facilitated with the trans-allyl silyl ether and summarized the general reactivity trends (Scheme 12b). They elucidated that a silyl allyl ether is highly activated for C–H insertion, while acetoxyl group exerts a deactivating effect presumably due to its electron-withdrawing nature. Despite consistently low enantioselectivity observed in the C–H insertion of benzyl silyl ethers 59 by Rh2(S-DOSP)4 (10–35% ee), and moderately high enantioselectivity achieved through chiral auxiliary mediated C–H functionalization reactions using ethyl (S)-lactate-based aryldiazoacetate (79–88% de, 68–85% ee), they ultimately attained the best results (91–95% de, 95–98% ee) by employing Hashimoto's Rh2(S-PTTL)4 catalyst.33
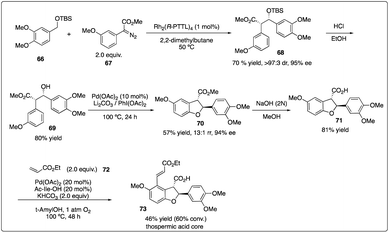 |
| | Scheme 12 Enantioselective intermolecular C–H insertion reaction for the synthesis of thospermic acid core by Davies and Yu group. | |
Recently, Davies and Sigman and their coworkers discovered that the site- and stereoselective C–H functionalization at α, γ, δ and even more distal positions to the siloxy ethers could be achieved by leveraging the preferences of Rh2(R-TCPTAD)4 and Rh2(S-2-Cl-5-BrTPCP)4 catalyst, yielding products with good to excellent enantioselectivity (Scheme 12c).34 Additionally, they developed a machine learning classification model to predict the major C–H functionalization site based on catalyst propensity and substrate electronics.
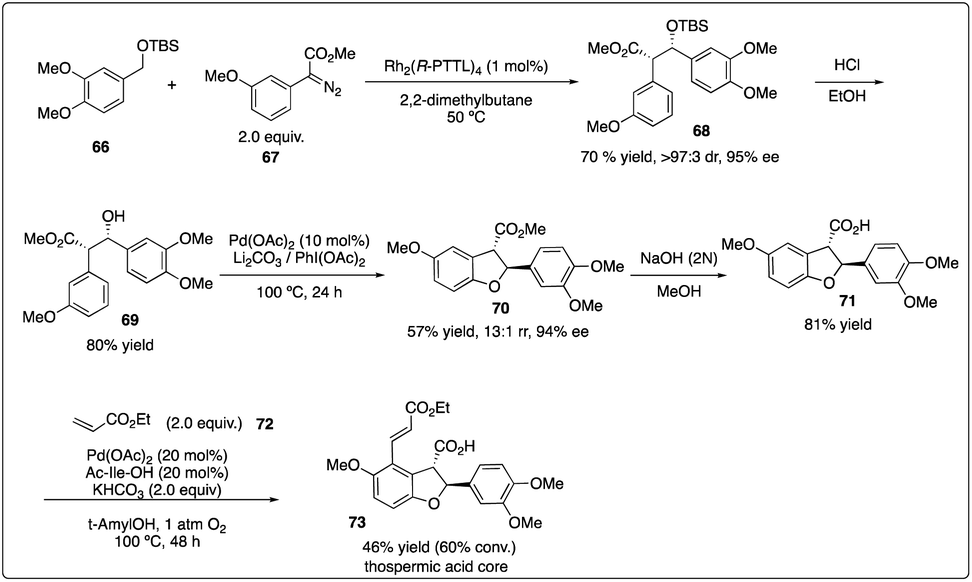
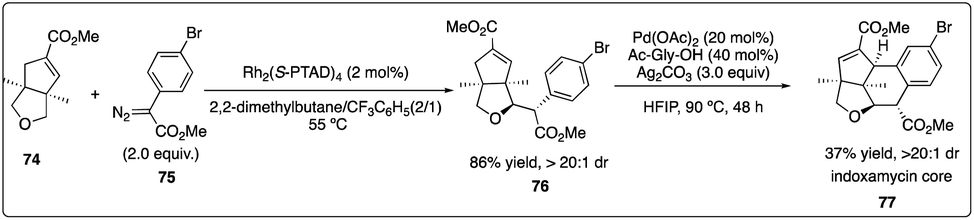
In 2013, Davies and Yu and their coworkers achieved a highly regio-, diastereo-, and enantioselective synthesis of dihydrobenzofurans 73 (the core of the thospermic acid family) involving three distinct C–H functionalization steps. Particularly noteworthy was a rhodium-catalyzed enantioselective intermolecular C–H insertion reaction (Scheme 13).35a Later, in a collaboration between the Sorensen and Yu groups with the Davies groups, another two sequential C–H functionalization reactions were described for the synthesis of the indoxamycin family core 77 with good yield and highly regio- and di stereoselectivity (Scheme 14).35b
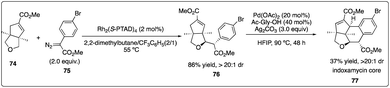 |
| | Scheme 13 Enantioselective intermolecular C–H insertion reaction for the synthesis of indoxamycin core by Sorensen group. | |
 |
| | Scheme 14 Enantioselective intermolecular C–H insertion reactions of donor–accepter carbenoid and silyl ether by Davies group. | |
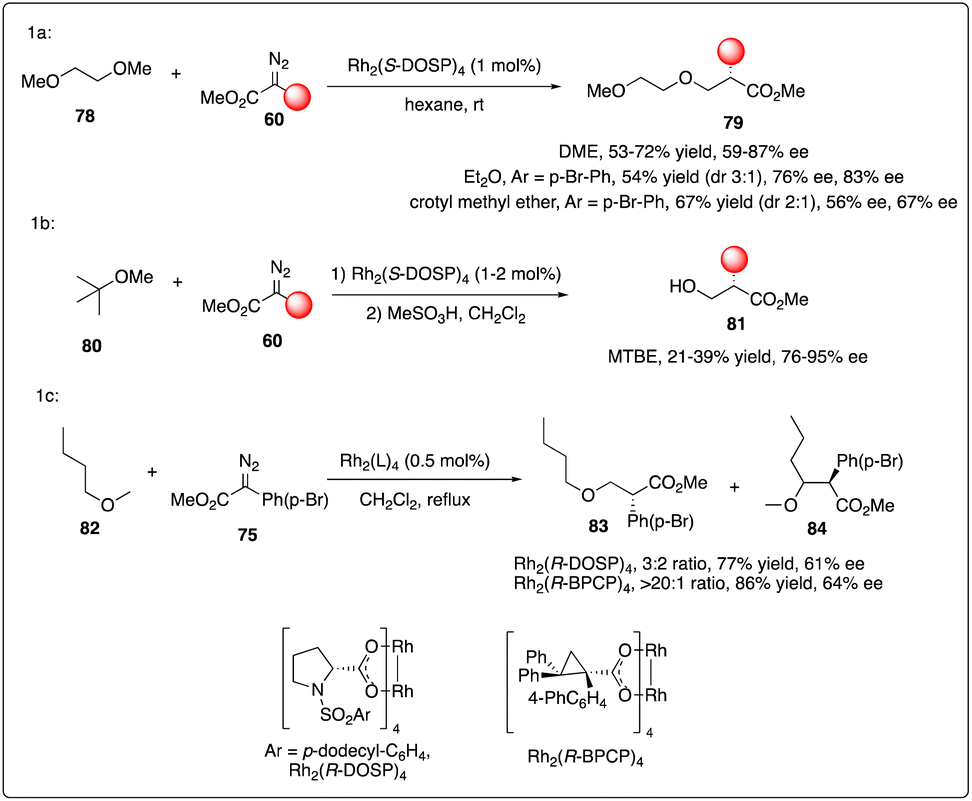
The electronic stabilization of the transition state during C–H activation favored α to alkoxy or silyloxy groups in a concerted non-synchronous manner, as demonstrated with excellent enantioselectivity and regioselectivity results. However, the Davies group observed a dramatical site selectivity when alkyl ethers like DME 78, Et2O, and unsymmetric ethers were used.36a In comparison to a mixture obtained with Rh2(R-DOSP)4 catalyst, the Rh2(R-BPCP)4 catalyzed C–H carbenoid insertion reaction selectively targeted the primary C–H bond with moderate enantioselectivity (Scheme 15).36b
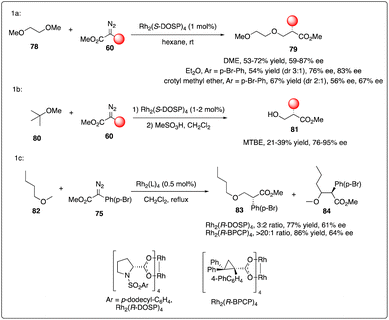 |
| | Scheme 15 Enantioselective intermolecular C–H insertion reactions of donor–accepter carbenoid and primary methine by Davies group. | |
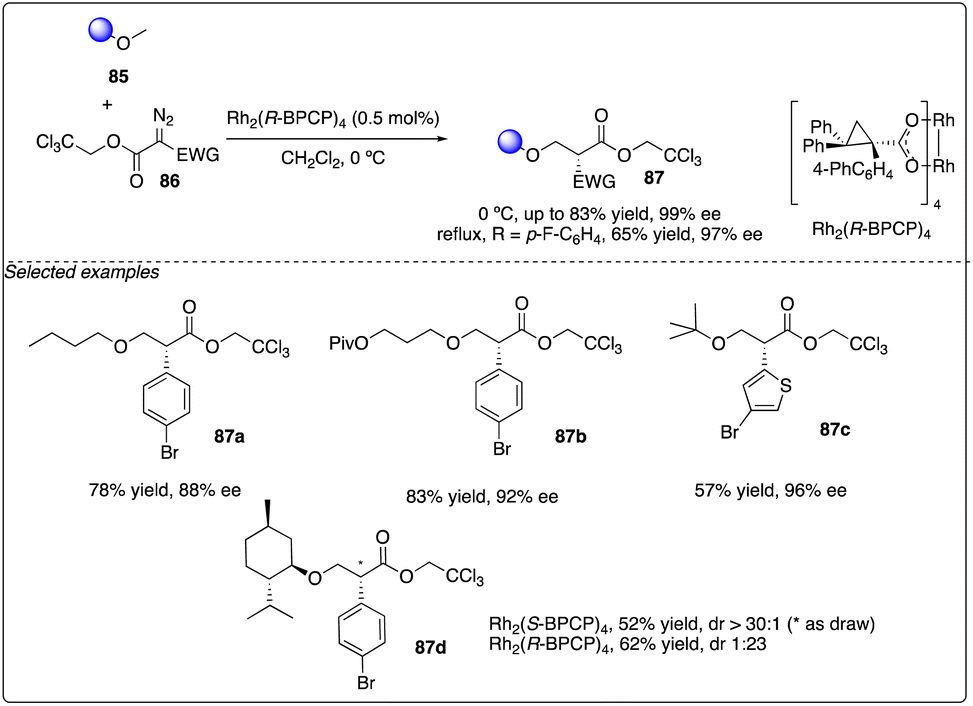
Later, Davies and Guptill optimized the aforementioned C–H insertion reaction of diazo compounds 86 with a 2,2,2-trichloroethyl (TCE) ester as the acceptor group. They discovered that the TCE ester could enhance reactivity and improve the levels of regioselectivity and enantioselectivity between primary and secondary C–H bonds (Scheme 16).37
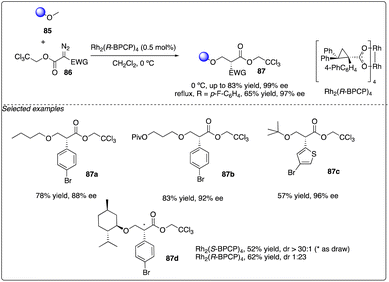 |
| | Scheme 16 Enantioselective intermolecular C–H insertion reactions of donor–accepter (TCE) carbenoid and primary methine by Davies group. | |
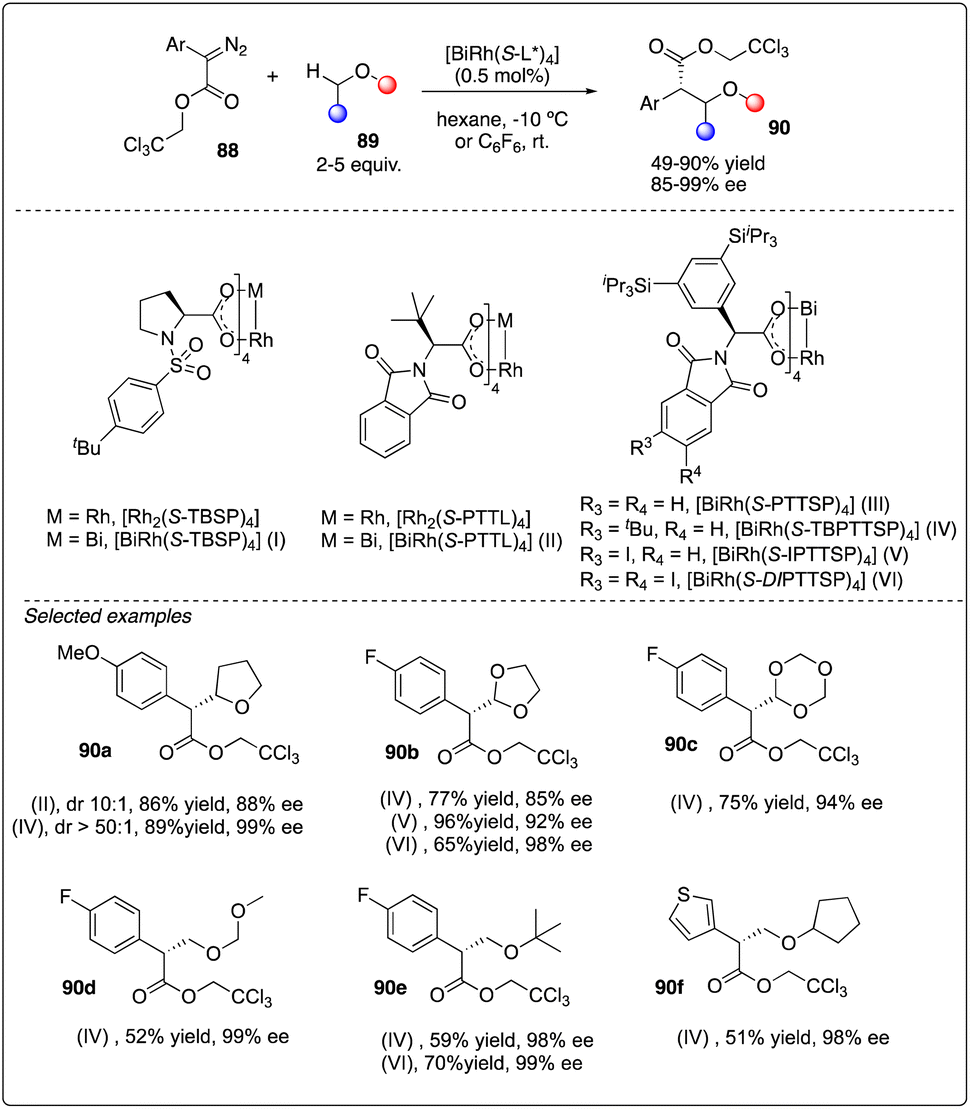
Recently, Davies, Musaev and Berry and their coworkers reported the first chiral [RhBi] paddlewheel catalyst, [BiRh(S-TBSP)4], and applied them to asymmetric cyclopropanation and C–H functionalization.38 Later, Fürstner and his coworkers designed a new type of [RhBi] paddlewheel catalysts39 and found it to be exceedingly effective in site-selective C–H functionalization with ethers (Scheme 17).40 Their rationale catalyst design involved using phenyl rings carrying lateral –Si(iPr)3 substituents to replace the tert-butyl residues, facilitating a large number of attractive interligand London dispersion interactions.41
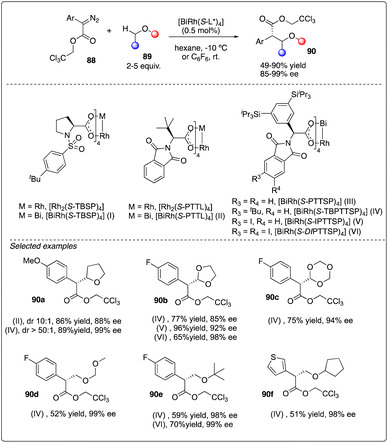 |
| | Scheme 17 Enantioselective intermolecular C–H insertion reactions of donor–accepter carbenoid with chiral [RhBi] catalyst by Fürstner group. | |
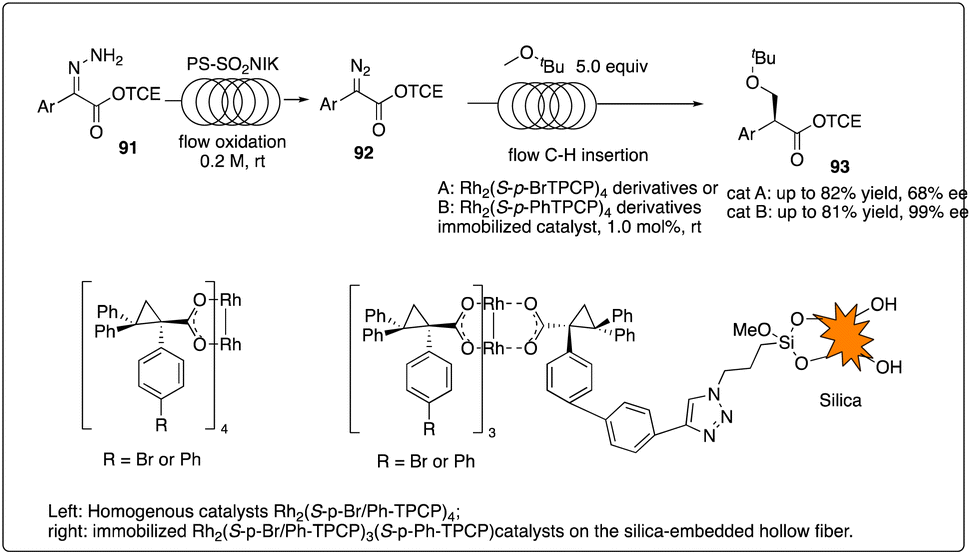
Davies and Jones and their coworkers developed a scalable flow reactor for enantioselective and regioselective rhodium C–H carbene insertion reactions (Scheme 18).42 Comparing to conventional batch reactors, the yield, site-selectivity and enantioselectivity of the product in this immobilized dirhodium hollow-fiber flow reactor were similar but more effective, scalable, recyclable.
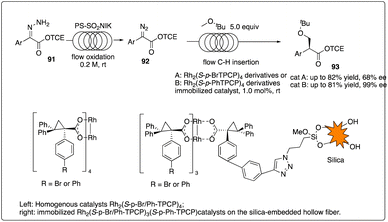 |
| | Scheme 18 Enantioselective intermolecular C–H insertion reactions with a scalable flow reactor by Davies and Jones group. | |
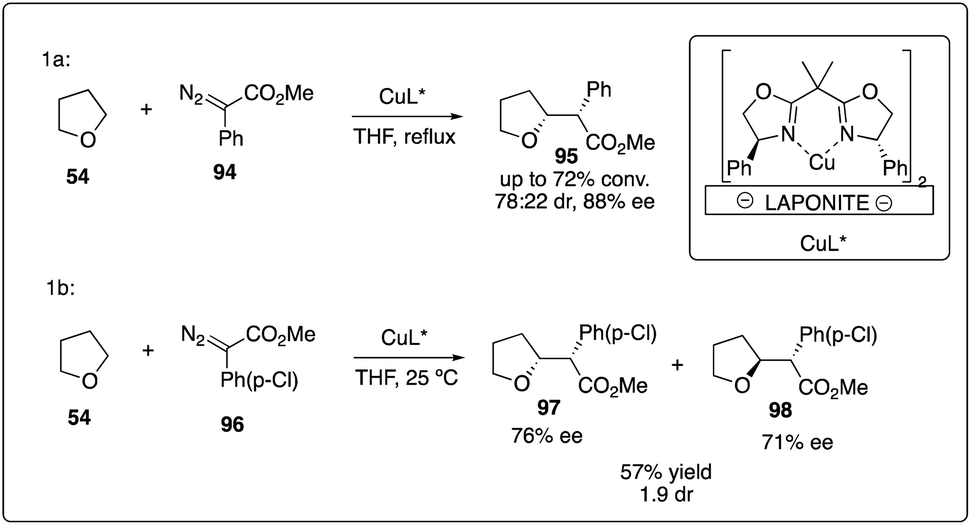
Copper complex was the first transition metal catalyst proposed to promote the decompositions of carbenoids.4 However, it was only recently that the asymmetric version for the α-C–H activation/carbenoid insertion of ethers was realized. In 2007, Fraile, Mayoral and coworkers reported an immobilized box–Cu complex catalyzed asymmetric insertion of a carbene into C–H bonds of THF with good enantioselectivity (Scheme 19a).43 This bis(oxazoline)-copper complex supported by Laponite could be recovered and reused with good reactivity and enantioselectivity control, demonstrating catalytic stability over three more cycles. Comparing with homogeneous box–copper complexes for carbene insertion reactions, the immobilized copper complexes could achieve higher chemoselectivities and stereoselectivities than normally used rhodium catalyst (Scheme 19b).44
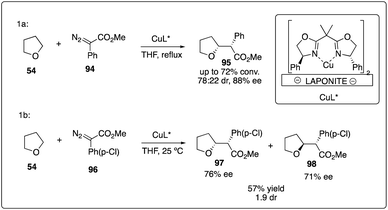 |
| | Scheme 19 Enantioselective intermolecular C–H insertion reactions of donor–accepter carbenoid and THF catalyzed by copper complex. | |
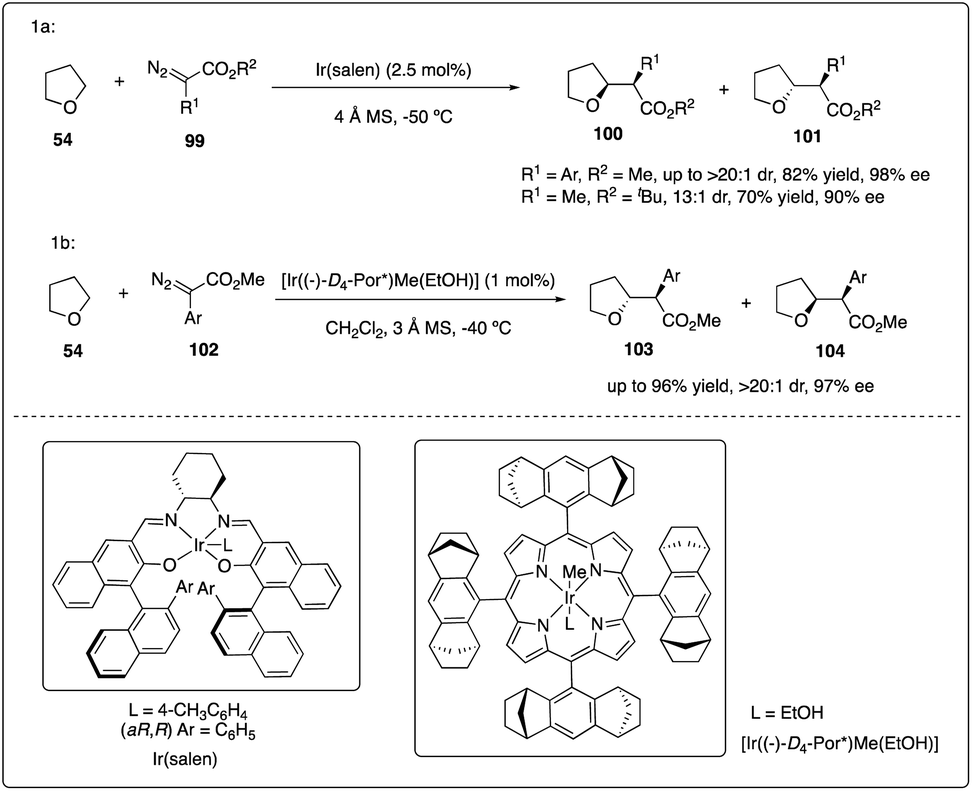
Katsuki and Suematsu discovered the iridium(III)-salen complexes show promising results in asymmetric carbenoid C–H insertion reactions (Scheme 20a). All the aryldiazoacetates 99 were well tolerated with good to excellent diastereoselectivity and enantioselectivity. They also achieved the first example of asymmetric intermolecular insertion with alkyl-substituted diazoacetate.45 Later, Che and coworkers reported chiral iridium porphyrin catalyzed asymmetric C–H carbene C–H insertion reactions with good to excellent diastereoselectivity, enantioselectivity and product turnovers (Scheme 20b).46
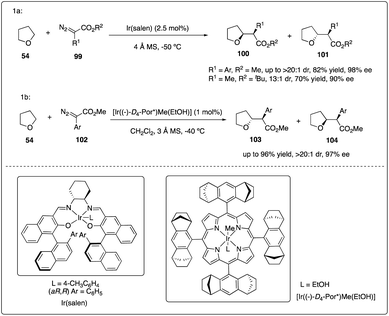 |
| | Scheme 20 Enantioselective intermolecular C–H insertion reactions of donor–accepter carbenoid and THF catalysed by iridium complex. | |
4. Summary and outlook
Catalytic asymmetric α-C–H bond functionalization of ether via carbenoid insertion has made enormous progress over the past decade. Effective asymmetric catalytic systems, including various chiral metal catalysts, have already been established for the enantioselective carbenoid insertion into the C–H bond adjacent to the oxygen atom in a variety of structurally diverse ether substrates. This has led to the generation of various enantioenriched highly-functionalized oxygen-containing structural motifs, enabling corresponding applications in asymmetric synthesis of bioactive natural products.
Despite these elegant advancements, challenges still remain and further exploration of catalytic enantioselective insertion of carbenoids into α-C–H bond functionalization of ethers is highly in demand. Currently, rhodium based asymmetric catalytic systems are the most commonly employed and well-studied, however, asymmetric catalytic systems based on more readily available transition metals, such as copper, iron, and cobalt, which might enhance the value of asymmetric C–H functionalization reactions, are underexplored and should draw more attention from the chemistry community. In addition, researches in this area is likely to focus on novel ligand families design and development to explore new type of substrates and transformations with a high level of chemo-, regio- and/or stereo-selectivities. Moreover, the discovery and further development of carbenoid insertion transformations with the in situ generated diazo substrates from more accessible precursors, like hydrazones, would greatly promote the practicality and versatility of current asymmetric C–H functionalization methodologies. Despite these challenges, we believe that catalytic asymmetric carbenoid insertion into α-C–H bond of ether will undoubtedly witness improvement and meet the requirements for more practical applications in the synthesis of pharmaceuticals or natural products in the future.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We gratefully acknowledge the National Natural Science Foundation of China (22201308) and China Postdoctoral Science Foundation funded project (2022M713510) is acknowledged. We appreciated the advice from Dr Shou-Guo Wang, Shenzhen Institutes of Advanced Technology, Chinese Academy of Sciences.
References
-
(a) J. H. Docherty, T. M. Lister, G. Mcarthur, M. T. Findlay, P. Domingo-Legarda, J. Kenyon, S. Choudhary and I. Larrosa, Transition-metal-catalyzed C–H bond activation for the formation of C–C bonds in complex molecules, Chem. Rev., 2023, 123, 7692–7760 CrossRef CAS PubMed;
(b) M. I. Lapuh, S. Mazeh and T. Besset, Chiral transient directing groups in transition-metal-catalyzed enantioselective C–H bond functionalization, ACS Catal., 2020, 10, 12898–12919 CrossRef CAS.
-
(a) J. V. Santiago and A. H. L. Machado, Enantioselective carbenoid insertion into C(sp3)–H bonds, Beilstein J. Org. Chem., 2016, 12, 882–902 CrossRef CAS PubMed;
(b) S. Gandhi, Catalytic enantioselective cross dehydrogenative coupling of sp3 C–H of heterocycles, Org. Biomol. Chem., 2019, 17, 9683 RSC;
(c) S.-Y. Zhang, F.-M. Zhang and Y.-Q. Tu, Direct Sp3 α-C–H activation and functionalization of alcohol and ether, Chem. Soc. Rev., 2011, 40, 1937–1949 RSC.
- O. Silberrad and C. S. Roy, XXIV.—Gradual decomposition of ethyl diazoacetate, J. Chem. Soc., Trans., 1906, 89, 179–182 RSC.
-
(a) P. Yates, The copper-catalyzed decomposition of diazoketones, J. Am. Chem. Soc., 1952, 74, 5376–5381 CrossRef CAS;
(b) L. T. Scott and G. J. DeCicco, Intermolecular carbon-hydrogen insertion of copper carbenoids, J. Am. Chem. Soc., 1974, 96, 322–323 CrossRef CAS.
-
(a) M. P. Doyle, R. Duffy, M. Ratnikov and L. Zhou, Catalytic carbene insertion into C–H bonds, Chem. Rev., 2010, 110, 704–724 CrossRef CAS PubMed;
(b) B. D. Bergstrom, L. A. Nickerson, J. T. Shaw and L. W. Souza, Transition metal catalyzed insertion reactions with donor/donor carbenes, Angew. Chem., Int. Ed., 2021, 60, 6864–6878 CrossRef CAS PubMed.
-
(a) T. Aneeja, M. Neetha, C. M. A. Afsina and G. Anilkumar, Progress and prospects in copper-catalyzed C–H functionalization, RSC Adv., 2020, 10, 34429–34458 RSC;
(b) O. Daugulis, H. Do and D. Shabashov, Palladium- and copper-catalyzed arylation of carbon–hydrogen bonds, Acc. Chem. Res., 2009, 42, 1074–1086 CrossRef CAS PubMed.
- S. Kumar, S. Nunewar, S. Oluguttula, S. Nanduri and V. Kanchupalli, Recent advances in Rh(III)/Ir(III)-catalyzed C–H functionalization/annulation via carbene migratory insertion, Org. Biomol. Chem., 2021, 19, 1438–1458 RSC.
-
(a) C.-X. Liu, S.-Y. Yin, F. Zhao, H. Yang, Z. Feng, Q. Gu and S.-L. You, Rhodium-catalyzed asymmetric C–H functionalization reactions, Chem. Rev., 2023, 123, 10079–10134 CrossRef CAS PubMed;
(b) D. A. Colby, R. G. Bergman and J. A. Ellman, Rhodium-catalyzed C–C bond formation via heteroatom-directed C–H bond activation, Chem. Rev., 2010, 110, 624–655 CrossRef CAS PubMed.
-
(a) S. Barranco, J. Zhang, S. López-Resano, A. Casnati and M. H. Pérez-Temprano, Transition metal-catalysed directed C–H functionalization with nucleophiles, Nat. Synth., 2022, 1, 841–853 CrossRef;
(b) Y. He, Z. Huang, K. Wu, J. Ma, Y.-G. Zhou and Z. Yu, Recent advances in transition-metal-catalyzed carbene insertion to C–H bonds, Chem. Soc. Rev., 2022, 51, 2759–2852 RSC;
(c) F. Kakiuchi and N. Chatani, Catalytic methods for C–H bond functionalization: application in organic synthesis, Adv. Synth. Catal., 2003, 345, 1077–1101 CrossRef CAS;
(d) T. Lyons and M. Sanford, Palladium-catalyzed ligand-directed C–H functionalization reactions, Chem. Rev., 2010, 110, 1147–1169 CrossRef CAS PubMed;
(e) X. Chen, K. M. Engle, D. H. Wang and J.-Q. Yu, Palladium(II)-catalyzed C–H activation/C–C cross-coupling reactions: versatility and practicality, Angew. Chem., Int. Ed., 2009, 48, 5094–5115 CrossRef CAS PubMed;
(f) A. A. O. Sarhan and C. Bolm, Iron(III) chloride in oxidative C–C coupling reactions, Chem. Soc. Rev., 2009, 38, 2730–2744 RSC;
(g) C. L. Sun, B. J. Li and Z. J. Shi, Direct C–H transformation via iron catalysis, Chem. Rev., 2011, 111, 1293–1314 CrossRef CAS PubMed;
(h) T. Kitamura, Transition-metal-catalyzed hydroarylation reactions of alkynes through direct functionalization of C–H bonds: a convenient tool for organic synthesis, Eur. J. Org Chem., 2009, 1111–1125 CrossRef CAS;
(i) Y. Qian, J. Tang, X. Zhou, J. Luo, X. Yang, Z. Ke and W. Hu, Enantioselective multifunctionalization with Rh carbynoids, J. Am. Chem. Soc., 2023, 145, 26403–26411 CrossRef CAS PubMed.
-
(a) J. Adams, M.-A. Poupart, L. Grenier, C. Schaller, N. Ouimet and R. Frenette, Rhodium acetate catalyzes the addition of carbenoids α- to ether oxygens, Tetrahedron Lett., 1989, 30, 1749–1752 CrossRef CAS;
(b) P. Wang and J. Adams, Model studies of the stereoelectronic effect in Rh(II) mediated carbenoid C–H insertion reactions, J. Am. Chem. Soc., 1994, 116, 3296–3305 CrossRef CAS.
-
(a) M. P. Doyle, A. van Oeveren, L. J. Westrum, M. N. Protopopova and T. W. Clayton Jr, Asymmetric synthesis of lactones with high enantioselectivity by intramolecular carbon-hydrogen insertion reactions of alkyl diazoacetates catalyzed by chiral rhodium(II) carboxamides, J. Am. Chem. Soc., 1991, 113, 8982–8984 CrossRef CAS;
(b) H. M. L. Davies, P. R. Bruzinski, D. H. Lake, N. Kong and M. J. Fall, Asymmetric cyclopropanations by rhodium(II) N-(arylsulfonyl)prolinate catalyzed decomposition of vinyldiazomethanes in the presence of alkenes. Practical enantioselective synthesis of the four stereoisomers of 2-phenylcyclopropan-1-amino acid, J. Am. Chem. Soc., 1996, 118, 6897–6907 CrossRef CAS.
-
(a) H. M. L. Davies and K. Liao, Dirhodium tetracarboxylates as catalysts for selective intermolecular C–H functionalization, Nat. Rev. Chem., 2019, 3, 347–360 CrossRef CAS PubMed;
(b) R. Wu, D. Zhu and S. Zhu, Dirhodium: carbene transformations and beyond, Org. Chem. Front., 2023, 10, 2849–2878 RSC;
(c) H. Yu and F. Xu, Non-noble metal-catalyzed cross-dehydrogenation coupling (CDC) involving ether α-C(sp3)–H to construct C–C bonds, Beilstein J. Org. Chem., 2023, 19, 1259–1288 CrossRef CAS PubMed.
-
(a) Y.-R. Luo, Comprehensive Handbook of Chemical Bond Energies, CRC Press, 2007 CrossRef;
(b) Y.-H. Miao, Y.-H. Hu, J. Yang, T. Liu, J. Sun and X.-J. Wang, Natural source, bioactivity and synthesis of benzofuran derivatives, RSC Adv., 2019, 9, 27510–27540 RSC;
(c) A. Radadiya and A. Shah, Bioactive benzofuran derivatives: an insight on lead developments, radioligands and advances of the last decade, Eur. J. Med. Chem., 2015, 97, 356–376 CrossRef CAS PubMed.
- H. M. L. Davies, M. V. A. Grazini and E. Aouad, Asymmetric intramolecular C–H insertions of aryldiazoacetates, Org. Lett., 2001, 3, 1475–1477 CrossRef CAS PubMed.
-
(a) R. P. Reddy, G. H. Lee and H. M. L. Davies, Dirhodium tetracarboxylate derived from adamantylglycine as a chiral catalyst for carbenoid reactions, Org. Lett., 2006, 8, 3437–3440 CrossRef CAS PubMed;
(b) H. M. L. Davies and A. M. Walji, Universal strategy for the immobilization of chiral dirhodium catalysts, Org. Lett., 2005, 7, 2941–2944 CrossRef CAS PubMed.
- H. Saito, H. Oishi, S. Kitagaki, S. Nakamura, M. Anada and S. Hashimoto, Enantio- and diastereoselective synthesis of cis-2-aryl-3-methoxycarbonyl-2,3-dihydrobenzofurans via the Rh(II)-catalyzed C–H insertion process, Org. Lett., 2002, 4, 3887–3890 CrossRef CAS PubMed.
- M. Ito, R. Namie, J. Krishnamurthi, H. Miyamae, K. Takeda, H. Nambu and S. Hashimoto, Asymmetric total synthesis of (–)-trans-blechnic acid via rhodium(II)-catalyzed C–H insertion and palladium(II)-catalyzed C–H olefination reactions, Synlett, 2014, 25, 288–292 CAS.
- Y. Natori, H. Tsutsui, N. Sato, S. Nakamura, H. Nambu, M. Shiro and S. Hashimoto, Asymmetric synthesis of neolignans (−)-epi-conocarpan and (+)-conocarpan via Rh(II)-catalyzed C–H insertion process and revision of the absolute configuration of (−)-epi-conocarpan, J. Org. Chem., 2009, 74, 4418–4421 CrossRef CAS PubMed.
- T. Wakimoto, K. Miyata, H. Ohuchi, T. Asakawa, H. Nukaya, Y. Suwa and T. Kan, Enantioselective total synthesis of aperidine, Org. Lett., 2011, 13, 2789–2791 CrossRef CAS PubMed.
- W. Kurosawa, T. Kan and T. Fukuyama, Stereocontrolled total synthesis of (−)-ephedradine A (orantine), J. Am. Chem. Soc., 2003, 125, 8112–8113 CrossRef CAS PubMed.
- Y. Koizumi, H. Kobayashi, T. Wakimoto, T. Furuta, T. Fukuyama and T. Kan, Total synthesis of (−)-serotobenine, J. Am. Chem. Soc., 2008, 130, 16854–16855 CrossRef CAS PubMed.
- D. H. Wang and J. Q. Yu, Highly convergent total synthesis of (+)-lithospermic acid via a late-stage intermolecular C–H olefination, J. Am. Chem. Soc., 2011, 133, 5767–5769 CrossRef CAS PubMed.
-
(a) C. Soldi, K. N. Lamb, R. A. Squitieri, M. González-López, M. J. Di Maso and J. T. Shaw, Enantioselective intramolecular C–H insertion reactions of donor–donor metal carbenoids, J. Am. Chem. Soc., 2014, 136, 15142–15145 CrossRef CAS PubMed;
(b) J. T. Shaw, C–H insertion reactions of donor/donor carbenes: inception, investigation, and insights, Synlett, 2020, 31, 838–844 CrossRef CAS PubMed.
- H. Murakami, T. Asakawa, Y. Muramatsu, R. Ishikawa, A. Hiza, Y. Tsukaguchi, Y. Tokumaru, M. Egi, M. Inai, H. Ouchi, F. Yoshimura, T. Taniguchi, Y. Ishikawa, M. Kondo and T. Kan, Total synthesis of sophoraflavanone h and confirmation of its absolute configuration, Org. Lett., 2020, 22, 3820–3824 CrossRef CAS PubMed.
-
(a) M. A. McKervey and T. Ye, Asymmetric synthesis of substituted chromanones via C–H insertion reactions of α-diazoketones catalysed by homochiral rhodium(II) carboxylates, J. Chem. Soc., Chem. Commun., 1992, 823–824 RSC;
(b) T. Ye, C. Fernández García and M. A. McKervey, Chemoselectivity and stereoselectivity of cyclisation of α-diazocarbonyls leading to oxygen and sulfur heterocycles catalysed by chiral rhodium and copper catalysts, J. Chem. Soc., Perkin Trans., 1995, 1, 1373–1379 RSC;
(c) A. Rosales, I. Rodríguez-García, C. López-Sánchez, M. Álvarez-Corral and M. Muñoz-Dorado, Solvent influence in the Rh-catalyzed intramolecular 1,6 C–H insertions: a general approach to the chromane and flavanone skeletons, Tetrahedron, 2011, 67, 3071–3075 CrossRef CAS.
- M. Ito, Y. Kondo, H. Nambu, M. Anada, K. Takeda and S. Hashimoto, Diastereo- and enantioselective intramolecular 1,6-C–H insertion reactions of α-diazo esters catalyzed by chiral dirhodium(II) carboxylates, Tetrahedron Lett., 2015, 56, 1397–1400 CrossRef CAS.
- J.-C. Wang, Y. Zhang, Z.-J. Xu, V. K.-Y. Lo and C.-M. Che, Enantioselective intramolecular carbene C–H insertion catalyzed by a chiral iridium(III) complex of D4-symmetric porphyrin ligand, ACS Catal., 2013, 3, 1144–1148 CrossRef CAS.
- L. Fu, H. Wang and H. M. L. Davies, Role of ortho-substituents on rhodium-catalyzed asymmetric synthesis of β-lactones by intramolecular C–H insertions of aryldiazoacetates, Org. Lett., 2014, 16, 3036–3039 CrossRef CAS PubMed.
- D. M. Guptill, C. M. Cohen and H. M. L. Davies, Rhodium(II)-catalyzed stereoselective synthesis of allylsilanes, Org. Lett., 2013, 15, 6120–6123 CrossRef CAS PubMed.
- H. M. L. Davies and T. Hansen, Asymmetric intermolecular carbenoid C–H insertions catalyzed by rhodium(II) (S)-N-(p-dodecylphenyl)sulfonylprolinate, J. Am. Chem. Soc., 1997, 119, 9075 CrossRef CAS.
- H. M. L. Davies, T. Hansen and M. R. Churchill, Catalytic asymmetric C–H activation of alkanes and tetrahydrofuran, J. Am. Chem. Soc., 2000, 122, 3063–3070 CrossRef CAS.
-
(a) H. M. L. Davies, E. G. Antoulinakis and T. Hansen, Catalytic asymmetric synthesis of syn-aldol products from intermolecular C–H insertions between allyl silyl ethers and methyl aryldiazoacetates, Org. Lett., 1999, 1, 383–386 CrossRef CAS;
(b) H. M. L. Davies and E. G. Antoulinakis, EXT asymmetric catalytic C–H activation applied to the synthesis of syn-aldol products, Org. Lett., 2000, 2, 4153–4156 CrossRef CAS PubMed;
(c) H. M. L. Davies, R. E. J. Beckwith, E. G. Antoulinakis and Q. Jin, New strategic reactions for organic synthesis: catalytic asymmetric C–H activation α to oxygen as a surrogate to the aldol reaction, J. Org. Chem., 2003, 68, 6126 CrossRef CAS PubMed.
-
(a) H. M. L. Davies and J. R. Manning, Catalytic C–H functionalization by metal carbenoid and nitrenoid insertion, Nature, 2008, 451, 417 CrossRef CAS PubMed;
(b) H. M. L. Davies, Recent advances in catalytic enantioselective intermolecular C–H functionalization, Angew. Chem., Int. Ed., 2006, 45, 6422–6425 CrossRef CAS PubMed;
(c) H. M. L. Davies, Catalytic enantioselective C–H activation by means of metal–carbenoid-induced C–H insertion, Chem. Rev., 2003, 103, 2861 CrossRef CAS PubMed;
(d) H. M. L. Davies, J. Yang and J. Nikolai, Asymmetric C–H insertion of Rh(II) stabilized carbenoids into acetals: A C–H activation protocol as a claisen condensation equivalent, J. Organomet. Chem., 2005, 690, 6111–6124 CrossRef CAS;
(e) H. M. L. Davies, S. J. Hedley and B. R. Bohall, Asymmetric intermolecular C–H functionalization of benzyl silyl ethers mediated by chiral auxiliary-based aryldiazoacetates and chiral dirhodium catalysts, J. Org. Chem., 2005, 70, 10737–10742 CrossRef CAS PubMed.
- Y. T. Boni, R. C. Cammarota, K. Liao, M. S. Sigman and H. M. L. Davies, Leveraging regio- and stereoselective C(sp3)–H functionalization of silyl ethers to train a logistic regression classification model for predicting site-selectivity bias, J. Am. Chem. Soc., 2022, 144, 15549–15561 CrossRef CAS PubMed.
-
(a) H. Wang, G. Li, K. M. Engle, J. Q. Yu and H. M. L. Davies, Sequential C–H functionalization reactions for the enantioselective synthesis of highly functionalized 2,3-dihydrobenzofurans, J. Am. Chem. Soc., 2013, 135, 6774–6777 CrossRef CAS PubMed;
(b) T. A. Bedell, G. A. B. Hone, D. Valette, J.-Q. Yu, H. M. L. Davies and E. J. Sorensen, Rapid construction of a benzo-fused indoxamycin core enabled by site-selective C–H functionalizations, Angew. Chem., Int. Ed., 2016, 55, 8270–8274 CrossRef CAS PubMed.
-
(a) H. M. L. Davies and J. Yang, Influence of a β-alkoxy substituent on the C–H activation chemistry of alkyl ethers, Adv. Synth. Catal., 2003, 345, 1133–1138 CrossRef CAS;
(b) C. Qin and H. M. L. Davies, Role of sterically demanding chiral dirhodium catalysts in site-selective C–H functionalization of activated primary C–H bonds, J. Am. Chem. Soc., 2014, 136, 9792–9796 CrossRef CAS PubMed.
- D. M. Guptill and H. M. L. Davies, 2,2,2-Trichloroethyl aryldiazoacetates as robust reagents for the enantioselective C–H functionalization of methyl ethers, J. Am. Chem. Soc., 2014, 136, 17718–17721 CrossRef CAS PubMed.
- Z. Ren, T. L. Sunderland, C. Tortoreto, T. Yang, J. F. Berry, D. G. Musaev and H. M. L. Davies, Comparison of reactivity and enantioselectivity between chiral bimetallic catalysts: bismuth–rhodium- and dirhodium-catalyzed carbene chemistry, ACS Catal., 2018, 8, 10676–10682 CrossRef CAS.
- S. Singha, M. Buchsteiner, G. Bistoni, R. Goddard and A. Fürstner, A new ligand design based on london dispersion empowers chiral bismuth–rhodium paddlewheel catalysts, J. Am. Chem. Soc., 2021, 143, 5666–5673 CrossRef CAS PubMed.
- M. Buchsteiner, S. Singha, J. Decaens and A. Fürstner, Chiral bismuth-rhodium paddlewheel complexes empowered by london dispersion: the C–H functionalization nexus, Angew. Chem., Int. Ed., 2022, 61, e202212546 CrossRef CAS PubMed.
-
(a) J. P. Wagner and P. R. Schreiner, London dispersion in molecular chemistry—reconsidering steric effects, Angew. Chem., Int. Ed., 2015, 54, 12274–12296 CrossRef CAS PubMed;
(b) E. Solel, M. Ruth and P. R. Schreiner, London dispersion helps refine steric A–values: dispersion energy donor scales, J. Am. Chem. Soc., 2021, 143, 20837–20848 CrossRef CAS PubMed ; For reviews, see:;
(c) D. J. Liptrot and P. P. Power, London dispersion forces in sterically crowded inorganic and organometallic molecules, Nat. Rev. Chem., 2017, 1, 0004 CrossRef;
(d) K. L. Mears and P. P. Power, Beyond steric crowding: dispersion energy donor effects in large hydrocarbon ligands, Acc. Chem. Res., 2022, 55, 1337–1348 CrossRef CAS PubMed.
- C.-J. Yoo, D. Rackl, W. Liu, C. B. Hoyt, B. Pimentel, R. P. Lively, H. M. L. Davies and C. W. Jones, An immobilized-dirhodium hollow-fiber flow reactor for scalable and sustainable C–H functionalization in continuous flow, Angew. Chem., Int. Ed., 2018, 57, 10923–10927 CrossRef CAS PubMed.
- J. M. Fraile, J. I. García, J. A. Mayoral and M. Roldán, Simple and efficient heterogeneous copper catalysts for enantioselective C–H carbene insertion, Org. Lett., 2007, 9, 731–733 CrossRef CAS PubMed.
- J. M. Fraile, P. López-Ram-de-Viu, J. A. Mayoral, M. Roldán and J. Santafé-Valero, Enantioselective C–H carbene insertions with homogeneous and immobilized copper complexes, Org. Biomol. Chem., 2011, 9, 6075–6081 RSC.
- H. Suematsu and T. Katsuki, Iridium(III) catalyzed diastereo- and enantioselective C–H bond functionalization, J. Am. Chem. Soc., 2009, 131, 14218–14219 CrossRef CAS PubMed.
- J.-C. Wang, Z.-J. Xu, Z. Guo, Q.-H. Deng, C.-Y. Zhou, X.-L. Wan and C.-M. Che, Highly enantioselective intermolecular carbene insertion to C–H and Si–H bonds catalyzed by a chiral iridium(III) complex of a D4-symmetric halterman porphyrin ligand, Chem. Commun., 2012, 48, 4299–4301 RSC.
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *b
*b