
Designing dual-domain thermoplastic elastomers from ABA triblock copolymers: introducing bond-exchangeable subdomains into B-block strands†
Mikihiro
Hayashi
 *ab and
Tatsuya
Mizuno
a
*ab and
Tatsuya
Mizuno
a
aDepartment of Life Science and Applied Chemistry, Graduate School of Engineering, Nagoya Institute of Technology, Gokiso-cho Showa-ku, Nagoya-city, Aichi 466-8555, Japan
bPRESTO, Japan Science and Technology Agency, 4-1-8, Honcho, Kawaguchi, Saitama 332-0012, Japan. E-mail: evh70675@ict.nitech.ac.jp
First published on 3rd September 2024
Abstract
Here, we propose dual domain thermoplastic elastomers (TPEs) with bond exchangeable subdomains. A simple poly(styrene)-b-poly(butadiene)-b-poly(styrene) (SBS) triblock copolymer is chemically modified to attach pyridine groups in the middle block, followed by cross-linking via pyridine quaternization with diiodo molecules. In this design, the network structure comprises both main domains of glassy poly(styrene) blocks and subdomains of self-aggregated quaternized pyridines within the middle block matrix, according to the scattering data. The scattering measurement at high temperatures reveals that the presence of subdomains significantly enhances the thermal stability of the micro-phase separated network. This fact eventually results in a thermally stable rubbery plateau region in the temperature-ramp rheology even at temperatures higher than the glass transition temperature of poly(styrene). In addition, the formation of subdomains affects the tensile properties, especially the Young's modulus and the recovery ratio in cyclic load–unload tests. Since the quaternized pyridines in the subdomains enable trans-N-alkylation bond exchange at high temperatures, the stress relaxation and other useful functions of TPEs, such as reprocessability and recyclability, are exhibited. Thus, the present system can lead to a new methodology to create functionalized TPEs composed of triblock copolymers. We also discuss the important role of the fraction of quaternized pyridine units and the unique relaxation behaviors in these dual domain bond exchange materials.
Introduction
Thermoplastic elastomers (TPEs) have garnered attention due to their inherent sustainability, stemming from their ability to flow when heated.1 These materials typically feature self-aggregated molecular network structures composed of rigid cross-link domains connected by flexible polymer strands. In many cases, block copolymers containing glassy or crystalline blocks and molten blocks serve as the foundational components. A primary class of block copolymer-based TPEs consists of glassy-rubber-glassy ABA triblock copolymers.2–5 These materials exhibit stable rubber-like properties at room temperature but can flow at temperatures exceeding the glass transition temperature (Tg) of the glassy blocks or melting temperature (Tm) of the crystalline blocks. To date, researchers have amassed a wealth of fundamental knowledge, including insights into the effects of morphological characteristics and the loop/bridge ratio of the B block strands on macroscopic properties.6–9Additionally, efficient chemical techniques for versatile functionalization have been proposed.10–14 All the knowledge attained is crucial for expanding the application range of TPEs, which is increasingly important given the growing awareness of the need for sustainable societies. Along with the development of supramolecular polymers, the use of supramolecular interactions has been explored. Notably, integrating hydrogen bonds and metal–ligand coordination into the flexible B block strands has been recognized as a useful strategy for enhancing mechanical properties.15,16 Crucially, in such cases, the thermoplastic function remains intact due to the reversible nature of supramolecular bonds. Moreover, some reports suggest that this reversibility dissipates internal stress, thereby enhancing toughness.17,18 Similarly, incorporating covalently bonded cross-links into the B block strands can also improve mechanical properties.19 These systems are known to enhance mechanical properties, such as Young's modulus, maximum stress, and elastic recovery. The enhanced elastic recovery is because the stress propagation of A block domains is suppressed due to the presence of stable additional cross-links in the strands, which eventually improves the stability of phase separated network structures. However, there must be a limit to the incorporation fraction in order to preserve the thermoplastic nature of the material.
Hence, a new trend of combining block copolymer-based TPEs with the vitrimer concept has emerged more recently. In the vitrimer concept, the network topology undergoes alteration through associative bond exchange at elevated temperatures, maintaining polymer connectivity throughout the bond exchange process. This characteristic enables thermoplastic-like functions, where the relationship between temperature and viscosity or relaxation time is gentle, following Arrhenius dependence due to the dominance of reaction kinetics in relaxation. Various bond exchange mechanisms, including boronic ester exchange and disulfide exchange,20,21 have been incorporated into the B block strands of ABA triblock copolymer-based TPEs, resulting in enhanced mechanical properties while preserving the thermoplastic nature.
As an intriguing candidate for designing a new type of TPE, we have investigated vitrimer-like materials utilizing the trans-N-alkylation of quaternized pyridine groups.22–24 In our previous studies, poly(acrylate)s bearing pyridine side groups randomly along the chain were cross-linked by dihalogen molecules, forming cross-links through pyridine-halogen quaternization reactions. Although the resulting materials were insoluble in typical organic solvents due to their covalent nature, bond exchange occurred at sufficiently high temperatures (≫100 °C) without catalysts. Consequently, these materials exhibited remarkable creep and stress relaxation, along with advantageous features such as reprocessability, healability, and recyclability.25 Despite trans-N-alkylation occurring via a dissociative mechanism, the plots of relaxation time versus temperature followed Arrhenius dependence, indicating their vitrimer-like nature. Importantly, we revealed that the quaternized pyridine groups formed nano-sized aggregates due to the repulsive forces between the generated positive and negative charges against hydrophobic acrylate units.22–24 Thus, the resulting vitrimer-like material possessed unique self-aggregated bond-exchangeable nanodomains, setting it apart from other vitrimer-type materials.
In this study, we integrate the aforementioned quaternized pyridine cross-links into the B block strands of ABA triblock copolymer-based TPEs. The component polymer utilized is the poly(styrene)-b-poly(butadiene)-b-poly(styrene), SBS, triblock copolymer. To carry out cross-linking via the quaternization reaction, pyridine side groups are introduced into the middle block (Fig. 1a), followed by their reaction with diiodo cross-linkers at the mole ratio of iodo/pyridine = 1 and 0.5. The primary objective of this study is to propose a novel concept of functional TPEs with dual domains; the main glassy cross-link domains are formed through the self-assembly of the A blocks, while the subdomains are established via the self-aggregation of quaternized pyridines within the B block matrix (Fig. 1b). The subdomains worked as additional cross-links to enhance the thermal stability of the micro-phase separated network structure, which is investigated by temperature-ramp scattering measurements. We assess the effects of subdomain formation on the temperature-ramp rheology and tensile properties by a single elongation test and cyclic load–unload tests. The stress relaxation behaviors originating from the bond exchange nature of quaternized pyridines are then investigated (Fig. 1c), revealing some unique features due to the formation of a micro-phase separated structure. Reprocessability and recyclability are finally demonstrated, revealing that the present materials still exhibit the functions of TPEs, owing to the bond exchange nature in the subdomains. The realization of dual-domain TPEs combined with the bond exchange concept has not been attempted previously, and thus the present concept could offer valuable insights for the design of new types of functional materials.
 | ||
| Fig. 1 (a) Synthesis of the target triblock copolymer with pyridine side groups on the middle block. (b) Schematic representation of the dual domain structure after quaternization of pyridine groups with diiodo molecules. (c) Illustration of the trans-N-alkylation bond exchange of quaternized pyridine groups. | ||
Experimental section
Materials
Poly(styrene)-b-poly(butadiene)-b-poly(styrene) (SBS) was kindly provided by JSR Corporation. The product number was TR2827. 2-Mercaptoethanol, 4-methoxyphenol, diphenyl(2,4,6-trimethylbenzoyl)phosphine oxide (TPO), pyridine, isonicotinoyl chloride hydrochloride, 1,6-diiodohexane, and 1-iodohexane were purchased from Tokyo Chemical Industry Co., Ltd (Japan). These chemicals and other organic solvents were used as received.Synthesis of precursor polymers
The hydroxyl (OH) groups were incorporated into the poly(butadiene) block of SBS through a thiol–ene reaction as follows. SBS (3 g), 2-mercaptoethanol (ME, 12 mL), and TPO (radical initiator, 22 mg) were dissolved in chloroform (400 mL). The solution was degassed with argon gas, and UV light (365 nm) was irradiated for 1 h at 25 °C with stirring. The reaction was terminated by adding 4-methoxyphenol (3 g) to the solution. The resulting polymer, designated as SBS-OH, was purified by reprecipitation using THF as the good solvent (20 mL) and methanol (500 mL) as the poor solvent, 4 times. The yield after the purification was ca. 90%.The pyridine groups were then incorporated into SBS via an esterification reaction as follows: SBS-OH (2.5 g), pyridine (Py, 7.5 mL), and isonicotinoyl chloride hydrochloride (INCH, 8.3 g) were dissolved in dichloromethane (300 mL), and the solution was stirred at 25 °C for 24 h. The mole ratio of OH groups, Py, and INCH was set to be [OH]![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :[Py]:[INCH] = 1:30:60. After concentration of the reaction solution using a rotary evaporator, the solution was washed twice with distilled water (200 mL) and twice with a NaHCO3 saturated aqueous solution (200 mL). The polymer was further purified by reprecipitation using THF (20 mL) as the good solvent and water (500 mL) as the poor solvent, two times. The resulting polymer was designated as SBS-Py.
:[Py]:[INCH] = 1:30:60. After concentration of the reaction solution using a rotary evaporator, the solution was washed twice with distilled water (200 mL) and twice with a NaHCO3 saturated aqueous solution (200 mL). The polymer was further purified by reprecipitation using THF (20 mL) as the good solvent and water (500 mL) as the poor solvent, two times. The resulting polymer was designated as SBS-Py.
Cross-linking procedure
The following are the descriptions for the sample preparation at the mole ratio of iodo/pyridine = 1. SBS-Py (0.6 g) and 1,6-diiodohexane (28.6 μL) were dissolved in chloroform (total 10 mL), and the solutions were combined in a Teflon-made small box with a length, width and height of 40 mm, 25 mm and 20 mm, respectively. The solvent was slowly evaporated using a heating stage and a vacuum oven. The dried mixture was then heated at 110 °C for 24 h in an oven for cross-linking. The thickness of the obtained film was ca. 0.5 mm. In the case of the sample preparation at the mole ratio of iodo/pyridine = 0.5, the amount of 1,6-diiodohexane was half (=14.3 μL), and other procedures were the same.Polymer characterization
Proton nuclear magnetic resonance (1H-NMR) was performed using a Bruker Analytik DPX400 spectrometer (400 MHz). CDCl3 with an internal standard of tetramethylsilane (TMS) was used as the deuterated solvent. Size exclusion chromatography (SEC) was conducted using a JASCO PU4185 pump system, equipped with a JASCO RI-4035 differential refractometer and JASCO CO-2065 Plus Intelligent Column Oven [eluent, tetrahydrofuran (THF); flow rate = 0.35 mL min−1; temperature = 40 °C; Tosoh Inc.]. Poly(styrene)s were used as standards.Spectroscopy
Fourier transform infrared (FT-IR) spectroscopy was performed using an FT/IR-4700 spectrometer combined with an ATR attachment (JASCO Co.). The measurements were performed at 25 °C.Swelling test
The swelling test was carried out to estimate the swelling ratio (Rs) depending on the reaction time. The cross-linked sample was immersed in the solvent (THF, good solvent for SBS-Py), and Rs was estimated, according to Rs = (ms/mi) × 100, where mi and ms are the initial and swollen masses, respectively. The gel fraction (fgel) was also estimated by the swelling test. The solution was replaced with a new solvent at an interval of 24 h three times. The final swollen samples were completely dried in a vacuum, and the dried mass (md) was compared with mi. The fgel was defined by the relationship fgel = (md/mi) × 100.Morphological investigation
Small-angle X-ray scattering (SAXS) measurements were conducted at the BL-6A beamline of the Photon Factory of the High Energy Accelerator Research Organization (KEK) in Tsukuba, Japan. The X-ray wavelength was 1.5 Å and the distance of sample-to-detector was ca. 2000 mm, where the detector was a PILATUS 2M (Dectris Ltd). The measurements were first performed at room temperature (25 °C). The temperature dependence of the self-assembly state was then measured using a hot stage (Linkam Scientific Instruments Ltd). The temperature was raised from 20 to 180 °C with an interval of 20 °C, and the thermal equilibration time at each target temperature was set to 3 min. The measurements were carried out in an N2 gas environment. Transmission electron microscopy (TEM) was performed using a JEOL JEM-z2500 microscope at an accelerating voltage of 200 kV. Ultrathin films with a thickness of 70 nm were prepared with a Leica EM FC7 ultramicrotome (cryomicrotome) under dry conditions and the films were stained with osmium tetroxide vapor for clearer contrast, wherein osmium tetroxide preferentially stained the poly(butadiene) in the present system.Thermal decomposition
Thermogravimetric analysis (TGA) was performed using a TG/DTA7300 (Hitachi High-Tech). The sample was heated from 30 °C to 500 °C at a temperature-ramp rate of 10 °C min−1. The measurement was carried out using approximately 10 mg samples in an N2 gas environment.Temperature-ramp rheology
The temperature-ramp rheology was conducted, using a uniaxial rheometer Discovery DMA 850 (TA Instruments). A strain of 0.1% was applied and the frequency, f, was fixed to 1 Hz. The sample was heated from −90 °C to 200 °C at a ramp rate of 5 °C min−1. All measurements were performed under an N2 gas flow.Tensile test
The tensile test was performed using an AGS-500NX (Shimadzu). For the test, dogbone-shaped samples were prepared with a cutting die, where the gauge length and gauge width were 13 mm and 4 mm, respectively. The tests were carried out at an elongation rate of 10 mm min−1 at 25 °C. The stress–strain curve was represented with nominal stress and nominal strain. Cyclic loading–unloading tests were also performed using the same setup. The sample was elongated to the target strain at an elongation rate of 10 mm min−1, and the cross-head was then returned to the initial position at the same deformation rate. The residual strain was determined at the point at which the stress reached zero during the unloading.Stress relaxation
The stress relaxation test was performed using a shear-type rheometer MCR102e (Anton Paar) and 8 mm disposable plates. The measurements were carried out from 140 °C to 170 °C at an interval of 10 °C, with an application of 5% strain. All measurements were conducted in an N2 gas environment. The tests were started after a thermal equilibration time of 10 min at each target temperature.Results
Synthesis and cross-linking
For the chemical modification of the SBS triblock copolymer (see Fig. 1a), hydroxyl (OH) groups were initially introduced into the B block through a thiol–ene radical reaction using 2-mercaptoethanol. Subsequently, the attached OH groups underwent an esterification reaction with isonicotinoyl chloride hydrochloride (INCH), resulting in the incorporation of pyridine side groups randomly along the poly(butadiene) middle block of the SBS triblock copolymers. The resulting triblock copolymer was designated as SBS-Py. The synthesis was verified using 1H-NMR. By comparing the spectra of SBS and SBS-OH (black and green in Fig. 2), the appearance of new signals A and B is observed after the reaction with 2-mercaptoethanol, indicating the attachment of OH groups to the poly(butadiene) block. The comparison of the spectra of SBS-OH and SBS-Py (green and red in Fig. 2) then reveals the appearance of pyridine signals (signals C and D) after the reaction with isonicotinoyl chloride, where the signals of original A and B in the SBS-OH spectrum were shifted to signals A′ and B′ in the SBS-Py spectrum. These indicate the quantitative chemical modification of OH groups to attach pyridine groups to the poly(butadiene) block. The whole 1H-NMR spectra and integral ratio of signals are provided in ESI, Fig. S1–S3.† According to size exclusion chromatography (SEC), the main peak position remained unchanged after attachment of pyridine side groups (Fig. S4†), although there was some generation of smaller molecular weight species, probably due to the occurrence of unwanted side reactions during the chemical modifications. Due to the occurrence of side reactions, the apparent number average molecular weight and dispersity of SBS-Py changed from 80 kg mol−1 and 1.2 for SBS to 70 kg mol−1 and 1.7 for SBS-Py. Based on the 1H-NMR data, SBS-Py possesses a mass fraction of the poly(styrene) end block, ϕPS, of ca. 0.22 and a molecular weight between the pyridine groups (i.e., equivalent molecular weight of the pyridine group, MPy) in the middle block of ca. 1.4 kg mol−1 (see the estimation in the ESI†). | ||
| Fig. 2 Expanded 1H-NMR spectra of SBS, SBS-OH, and SBS-Py, focusing on the important signals to confirm the chemical modification of the B block. The signals with alphabets originate from the protons with the same alphabets in the chemical structures. | ||
For the quaternization reaction of the pyridine groups, a diiodo molecule, 1,6-diiodohexane, was blended with SBS-Py through solution casting using chloroform. The mole ratio of iodo/pyridine was varied to be 1 and 0.5 to assess the effects of the quaternized pyridine fraction on the physical properties. A thermal cross-linking reaction was then conducted on the dried sample at 110 °C for 24 h, resulting in the formation of a transparent elastomer film (Fig. 3a). The obtained samples are coded as SBS-CL-1 and SBS-CL-0.5, depending on the mole ratio of iodo/pyridine (=1 or 0.5).
 | ||
| Fig. 3 (a) Macroscopic appearance of the cross-linked sample, SBS-CL-1. (b) FT-IR spectra of SBS-Py, SBS-CL-0.5, and SBS-CL-1, focusing on the peaks from free and quaternized pyridines. The dotted line is a guide to the eye to see the change in relative intensity between the peaks at 1600 cm−1 and 1640 cm−1. (c) De-crosslinking behavior of SBS-CL-1 in 1-iodohexane. | ||
The resulting cross-linked samples did not dissolve but instead swelled in the good solvent, tetrahydrofuran. The swelling ratio and gel fraction were 348 and 96% for SBS-CL-1, and 796 and 92% for SBS-CL-0.5. These data indicate sufficient progress in cross-linking. The progress of the quaternization reaction was actually confirmed using FT-IR (Fig. 3b). According to our previous studies,22–24 the free pyridine and quaternized pyridine provide signals at 1600 cm−1 and 1640 cm−1, respectively (see also Fig. S5† for our previous data). Notably, in the present system, the SBS triblock copolymer possesses signals at 1600 cm−1 from the phenyl ring of poly(styrene) and 1640 cm−1 from the double bond of poly(butadiene) (Fig. S6†). Although the exact evaluation of the fraction of quaternized pyridines is difficult, the relative signal intensity at 1640 cm−1 was found to increase gradually with the addition of diiodo cross-linkers, by comparing the spectra of the three samples. The data thus revealed that the quaternization reaction of pyridine side groups in the B block progressed successfully. To assess the side reaction during cross-linking, investigation of chemical decross-linking using monohalogen molecules should be informative. In our previous study,25 a poly(acrylate) elastomer bearing quaternized pyridine cross-links was fully dissolved in monohalogen solvent at a high temperature, which was due to the bond exchange reaction of quaternized pyridine cross-links with the solvent molecules bearing halogen groups. Based on this knowledge, SBS-CL-1 was immersed in 1-iodohexane and heated to 160 °C (Fig. 3c). In the present case, SBS-CL-1 was completely dissolved in the heated 1-iodohexane, indicating that cross-linking indeed progressed through the pyridine quaternization reaction and that the remaining vinyl bonds in the poly(butadiene) block did not undergo side reactions to form covalently bonded cross-links.
Morphological analysis
In this design, we anticipated the formation of dual domain structures composed of poly(styrene) main domains and subdomains of aggregated quaternized pyridines. Structural characterization at a nanometer scale was thus conducted using small-angle X-ray scattering (SAXS). Fig. 4a presents the SAXS spectra of SBS-Py, SBS-CL-0.5, and SBS-CL-1. A strong scattering peak was commonly observed at q ∼ 0.23 nm−1 for the three samples, which reflected the correlation length between the poly(styrene) domains, since SBS-Py without quaternized pyridines exhibited this peak. The domain spacing D, calculated using D ∼ 2π/q1, was ca. 27 nm, where q1 represents the peak top position. Importantly, SBS-CL-1 exhibited another broad scattering peak at q ∼ 0.75 nm−1. The peak appearance was similar to that observed in our previous studies using random copolymers cross-linked via quaternized pyridines.22–24 Therefore, the additional peak in SBS-CL-1 could originate from the self-assembled quaternized pyridine units in the poly(butadiene) block. In the case of SBS-CL-0.5, a shoulder-like peak was observed, where the peak intensity in the q range of 0.5 nm−1 < q < 2 nm−1 was obviously larger than that of SBS-Py. These data indicate that, although the number of quaternized pyridines was not sufficient to form distinct aggregates, small aggregates were distributed in the poly(butadiene) matrix. We performed TEM observation for SBS-Py and SBS-CL-1 (Fig. S7†), where osmium staining was conducted to obtain the clear difference between the poly(styrene) domains and poly(butadiene) matrix. The TEM images were similar between the two samples, which is consistent with the similar peak top position of the poly(styrene) domains in the SAXS data. On the other hand, the aggregates of quaternized pyridines were not obvious in the TEM images probably due to the insufficient fraction and size, although we expected the color difference between the poly(butadiene) matrix and the quaternized pyridine aggregates, owing to the large scattering length density of the iodo units.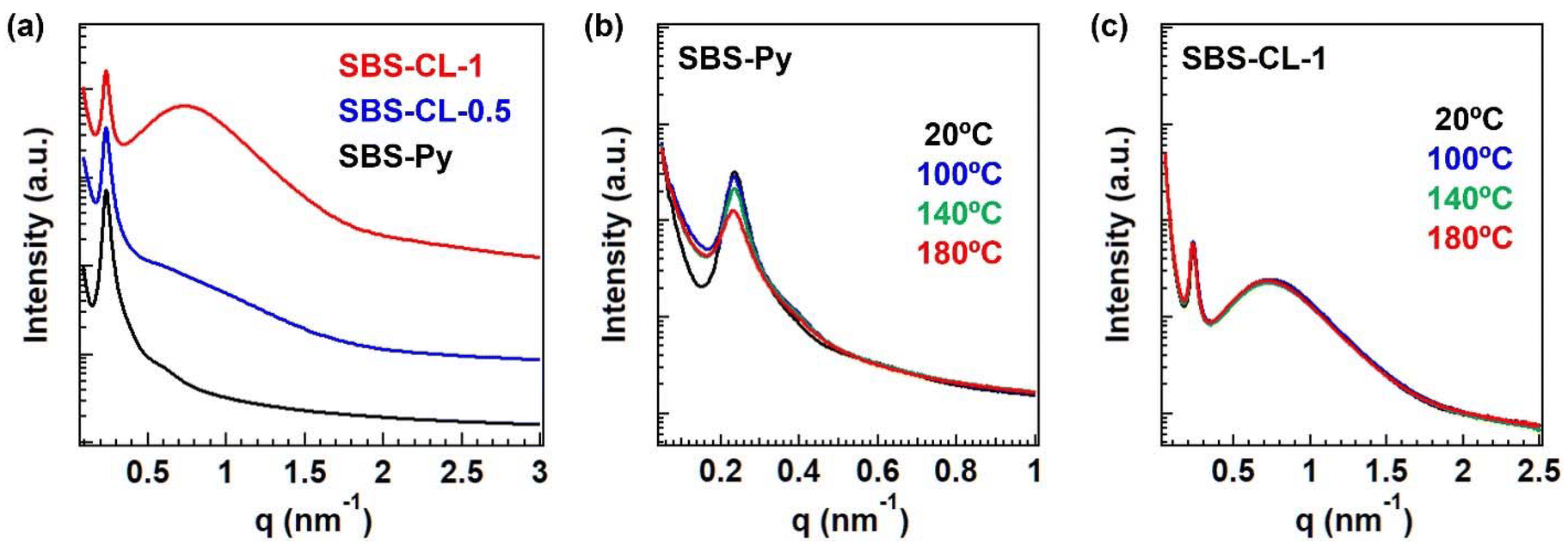 | ||
| Fig. 4 (a) SAXS spectra at 25 °C for the three samples and temperature-ramp SAXS spectra of (b) SBS-Py and (c) SBS-CL-1 (see also the whole spectra in the temperature region from 20 °C to 180 °C in the ESI†). | ||
Therefore, we conducted fitting analysis for SBS-CL-1 to estimate the possible size and correlation distance of quaternized pyridine aggregates, based on the Yarusso–Cooper (Y–C) model, in which spherical-shaped aggregates with radius R1 are assumed to be distributed randomly, while maintaining the radius of closest approach RCA.26 The entire function and fitting data are described in the ESI (Fig. S8†). The resulting value of the aggregate size, R1, was 2.4 nm and the distance between the aggregates, da, was estimated to be 5.2 nm. From these data, we confirmed that the nano-aggregates of quaternized pyridines were distributed in the poly(butadiene) matrix with a much shorter distance than the distance between the poly(styrene) domains, as designed.
To explore the role of subdomain formation, we conducted temperature-ramp SAXS measurements from 20 to 180 °C with an interval of 20 °C. Fig. 4b and c present the selected spectra recorded for SBS-Py and SBS-CL-1 at 20 °C, 100 °C, 140 °C, and 180 °C. The data for all measured temperatures are summarized in Fig. S9.† In the data for SBS-Py, broadening of the peak and lowering of the scattering intensity were observed with an increase in temperature, indicating the disordering of the morphology. On the other hand, for SBS-CL-1, the primary peak remained unchanged and the spectra were completely overlapped in the whole temperature range. The observed difference indicates that the thermal stability of the morphology was significantly enhanced due to the presence of subdomains. Importantly, the peak of the subdomains for SBS-CL-1 did not exhibit any changes upon heating. The result thus indicates that the subdomains of quaternized pyridine aggregates in the poly(butadiene) matrix worked to inhibit the disordering of the phase separated network structure.
Thermal and mechanical properties
First, thermogravimetric analysis (TGA) measurements for SBS-CL-1 revealed that the decomposition temperature, determined at the temperature of 5% weight loss, was ca. 300 °C (Fig. S10†), with decomposition initiating around 220 °C. Thus, all subsequent measurements were conducted below 200 °C. Fig. 5 illustrates the temperature-ramp rheology for SBS-Py, SBS-CL-0.5, and SBS-CL-1, plotting the storage modulus (E′) and loss tangent (tanδ) against temperatures. The prominent peak of tanδ at ca. −50 °C is derived from the glass transition of the middle poly(butadiene) block. Above this transition, a rubbery plateau was observed for all samples. A clear difference was found in the reduction behaviors of E′ in the high temperature region. For SBS-Py, a sharp reduction of E′ was observed at ca. 100 °C, above which the modulus reached the detection limit. This decrease in E′ is primarily attributed to softening of the poly(styrene) domains originating from the glass transition. Although the reduction of E′ at ca. 100 °C was also observed for SBS-CL-0.5, the reduction manner was much more gradual, compared with the data of SBS-Py, and the modulus finally reached the limit at ca. 170 °C. Notably, among the samples, the modulus was significantly stable for SBS-CL-1, and the plateau persisted up to 200 °C. In the previous section, we demonstrated the enhanced thermal stability of the phase separated network by forming subdomains in the poly(butadiene) matrix. Thus, in the case of SBS-CL-1, the softening of the poly(styrene) domain does not induce the flow of the entire network, which was the reason for the stable plateau region.
 | ||
| Fig. 5 Temperature-ramp rheology data for SBS-Py, SBS-CL-0.5, and SBS-CL-1. | ||
The plateau modulus was also different between samples. For example, the modulus at 25 °C was 2.1 MPa for SBS-Py, 3.0 MPa for SBS-CL-0.5, and 5.8 MPa for SBS-CL-1. The order of the modulus was always SBS-Py < SBS-CL-0.5 < SBS-CL-1, which was simply interpreted by the formation of additional cross-links in the poly(butadiene) strands. A difference in modulus was also observed in the tensile test performed at room temperature (i.e., 25 °C). Fig. 6a compares the stress–strain curves of SBS-Py, SBS-CL-0.5, and SBS-CL-1. Although the elongation at break was sacrificed, the Young's modulus increased with an increase in the fraction of quaternized pyridines; the value was 2.8 MPa for SBS-Py, 3.3 MPa for SBS-CL-0.5, and 6.1 MPa for SBS-CL-1. On the other hand, we observed an interesting phenomenon in the cyclic load–unload tests. For block copolymer-based elastomers with micro-phase separated structures, the elastic recovery is usually reduced by the increase in the Young's modulus.20,27–29 The main reason is the larger stress accumulation in the cross-link domains for the sample with a larger Young's modulus, which induces chain pullout from the domain and the unrecoverable deformation of the morphology. We then performed cyclic load–unload tests by changing the target strain (εt) from 25 to 100%. From these data, the recovery ratio (Rrex) was estimated, according to  , where εr is the residual strain. The values of Rrec were compared for the three samples as a function of εt in Fig. 6b, and the actual data are summarized in Fig. 6c; Rrec was the highest for SBS-CL-1 and the lowest for SBS-Py, and thus the recoverability was improved with an increase in the Young's modulus in the present sample series, unlike the usual trend. In fact, our group previously demonstrated19 that the partial incorporation of additional covalent cross-links into the middle block of the ABA triblock copolymer-based elastomer serves to inhibit the propagation of internal stress to the cross-link domains of the A blocks, thereby enhancing structural stability during cyclic deformation. A similar rationale should apply to the observed improvement in residual strain, once again emphasizing the importance of subdomains in enhancing mechanical properties.
, where εr is the residual strain. The values of Rrec were compared for the three samples as a function of εt in Fig. 6b, and the actual data are summarized in Fig. 6c; Rrec was the highest for SBS-CL-1 and the lowest for SBS-Py, and thus the recoverability was improved with an increase in the Young's modulus in the present sample series, unlike the usual trend. In fact, our group previously demonstrated19 that the partial incorporation of additional covalent cross-links into the middle block of the ABA triblock copolymer-based elastomer serves to inhibit the propagation of internal stress to the cross-link domains of the A blocks, thereby enhancing structural stability during cyclic deformation. A similar rationale should apply to the observed improvement in residual strain, once again emphasizing the importance of subdomains in enhancing mechanical properties.
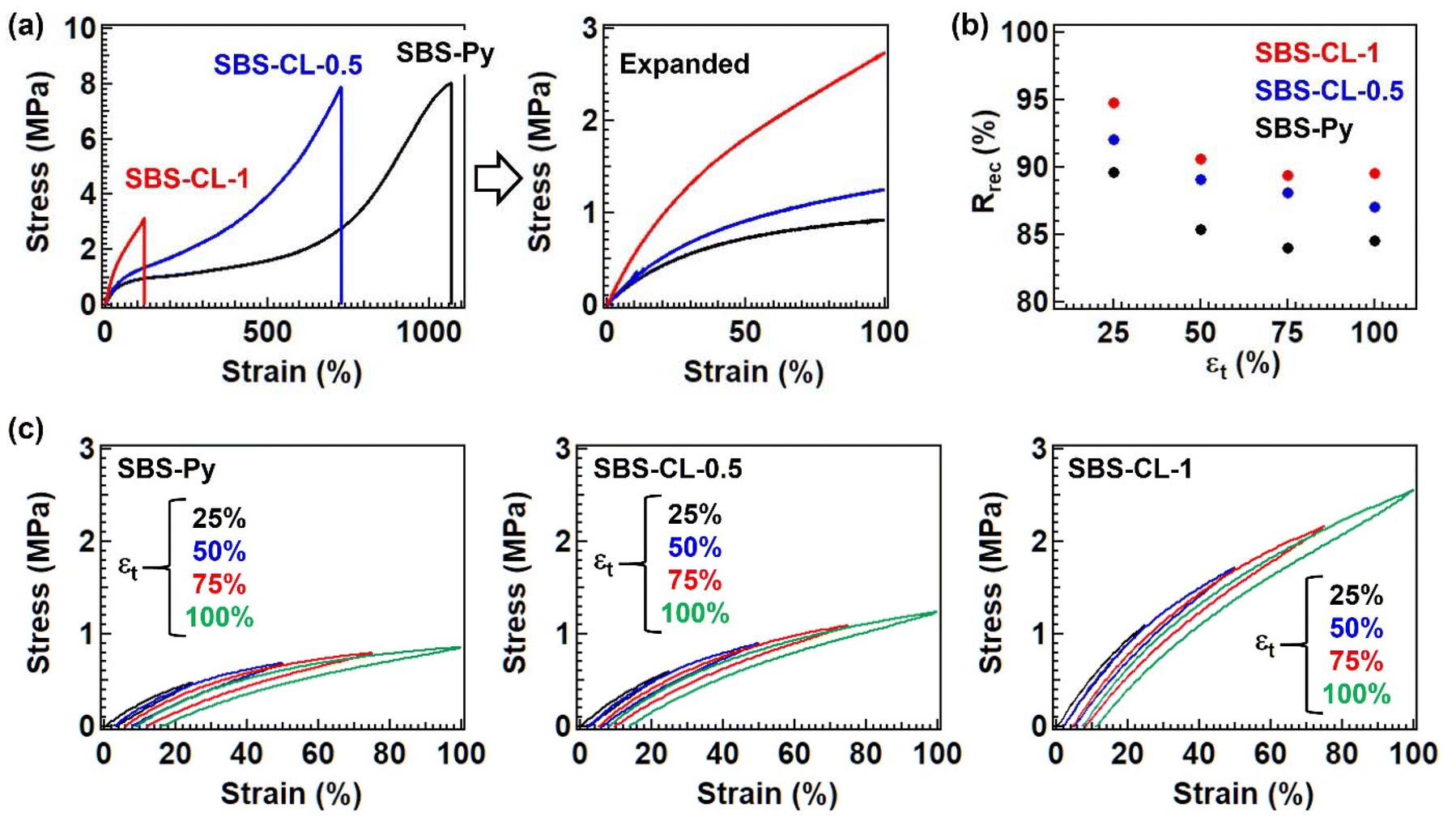 | ||
| Fig. 6 (a) Stress–strain curves and (b) plots of the recovery ratio (Rrec) at different target strains (εt) for SBS-Py, SBS-CL-0.5, and SBS-CL-1. (c) Raw data of the cyclic stress–strain curves. | ||
Stress relaxation and functions
In the preceding section, we elucidated the essential role of subdomains in influencing thermal and mechanical properties. Of greater importance is the assessment of how bond exchange via trans-N-alkylation operates within the micro-phase separated structure. For vitrimer materials, bond exchange properties are typically evaluated through stress relaxation tests.30–33 We here assess the stress relaxation properties mainly for the sample with a clear dual domain structure, that is, SBS-CL-1. Fig. 7a provides data for SBS-CL-1 measured at 140–170 °C, temperatures exceeding the Tg of the poly(styrene) block, where the stress (σ) was normalized by the initial stress (σ0). The non-normalized stress is shown in Fig. S11;† the initial stress decreased with an increase in temperatures, which is consistent with the data of temperature-ramp rheology. The progress of stress relaxation was confirmed, with relaxation rates increasing as temperatures rose. The result indicates the acceleration of bond exchange via trans-N-alkylation at higher temperatures. We also measured the stress relaxation for SBS-Py and SBS-0.5 (Fig. S12†), and the relaxation rate was in the order of SBS-Py ≫ SBS-CL-0.5 ≫ SBS-CL-1. This is understandable, considering that the formation of subdomains in the middle block should retard the chain diffusion. For SBS-CL-1, we tentatively estimated the relaxation time (τ), at which the stress was reduced to 1/e of the initial stress (Table S4†). From the plots of τ as a function of inverse temperature (Fig. S13†), the Arrhenius dependence was observed and the activation energy estimated from the slope was ca. 134 kJ mol−1. | ||
| Fig. 7 (a) Stress relaxation data at 140 °C, 150 °C, 160 °C, and 170 °C for SBS-CL-1, where the Y-axis indicates the relaxation modulus (G) normalized by the initial modulus (G0) at t = 0. (b) Data and (c) schematic explanation for three different relaxation regions, where the representative data at 170 °C is provided. | ||
Importantly, the relaxation curves were not monotonic for SBS-CL-1, unlike other reported data. For example, according to our previous studies for the samples with trans-N-alkylation bond exchange using quaternized pyridines in simple random copolymers, the relaxation curves were well fitted with a model function, i.e., the Kohlrausch–Williams–Watts (KWW) function.22–24 On the other hand, the present block copolymer sample exhibited complex relaxation curves that could not be fitted with the KWW function. Presumably, there are three kinds of relaxation mechanisms, depending on the time scale (Fig. 7b for the representative data at 170 °C and see also the schematic in Fig. 7c). On the short time scale (i.e., region A), the origin of relaxation should be the molten poly(styrene), since the temperature is much higher than the Tg. On the longer time scale (≫10 s), the relaxation should originate from the trans-N-alkylation of quaternized pyridines. It should be noted here that the micro-phase separated structure is maintained in the temperature range for stress relaxation, according to the temperature-ramp SAXS data. Considering this fact, on the middle time scale (i.e., region B), relaxation via bond exchange progressed in the limited space due to the constraint from the connection with poly(styrene) block domains. On the longer time scale (i.e., region C), some part of the middle block could be released from the constraint, which enables larger space diffusion. In fact, a similar relaxation behavior, that is, the tail of relaxation on the long time scale, was reported for block copolymer-based vitrimers, according to the report from Brent S. Sumerlin et al.34 Thus, unusual multi-step relaxation could be a special phenomenon originating from the block architecture with restricted diffusions to find exchange partners, although we need to accumulate more knowledge to confirm its universality.
Finally, several useful functions of SBS-CL-1 derived from its bond exchange nature are demonstrated as follows. The first is reprocessability (Fig. 8a); a strip-shaped film was twisted and fixed to paper using temperature-resistant tape (i.e., Kapton tape) and then subjected to heating in an oven at 160 °C for 1 h, followed by cooling to 25 °C. The resulting sample retained its twisted shape, demonstrating its reprocessability. This reprocessability arises from the loss of elastic recovery due to stress relaxation during heating. Secondly, mechanical recyclability is illustrated (Fig. 8b): the film sample was fractured into small pieces, which were then placed in a Teflon-made flat mold with a hole spacer and hot-pressed at 180 °C for 15 min. The pieces fused back into a film, indicating the progress of chain interpenetration between fractured pieces during hot-pressing. These functions are indeed crucial for TPEs, with the bond exchange nature in the subdomain of quaternized pyridines playing an essential role.
 | ||
| Fig. 8 (a) Reprocessability and (b) recyclability of SBS-CL-1 (see the explanation in the text). | ||
Conclusions
In conclusion, this study introduces a novel concept of block copolymer-based thermoplastic elastomers (TPEs), where the network structure comprises main glassy domains of the end blocks and subdomains along the middle block strands. Central to this concept is the incorporation of quaternized pyridines into the middle block, which was effective at enhancing the thermal stability of the phase separated networks and also some mechanical properties, such as the modulus and elastic recovery. Notably, bond exchange via trans-N-alkylation occurred at high temperatures, imparting the important functions of TPEs. The relaxation behavior differed from that of non-phase separated vitrimer systems with the same bond exchange mechanism, which was attributed to the relaxation of molten end blocks and the restriction of chain diffusion in the thermally stable phase separated structure. While the bond exchange or vitrimer concept has been applied to block copolymer frameworks previously, the dual domain concept with bond-exchangeable subdomains is novel. The insights gained from this study have potential implications not only for TPEs but also for a wide range of block copolymer-based materials. While this study has laid the groundwork for the concept, future research will focus on elucidating the benefits and impacts of the dual domain structure on physical properties, potentially expanding the application scope of sustainable polymeric materials.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Ms A. Mori for her assistance in performing TEM, which was supported by the Equipment Sharing Division, Organization for Co-Creation Research and Social Contributions, Nagoya Institute of Technology. We also thank Prof. A. Takasu for his assistance in performing SEC and Prof. K. Nagata for his assistance in performing TGA. This work was supported by JST, PRESTO Grant Number JPMJPR23N7 (M. H.), Japan and Foundation of Public Interest of Tatematsu, Japan.References
- G. Zanchin and G. Leone, Prog. Polym. Sci., 2021, 113, 101342 CrossRef CAS.
- W. Y. Wang, W. Lu, A. Goodwin, H. Q. Wang, P. C. Yin, N. G. Kang, K. L. Hong and J. W. Mays, Prog. Polym. Sci., 2019, 95, 1–31 CrossRef CAS.
- W. Wang, R. Schlegel, B. White, K. Williams, D. Voyloy, C. Steren, A. Goodwin, E. Coughlin, S. Gido, M. Beiner, K. Hong, N. Kang and J. Mays, Macromolecules, 2016, 49, 2646–2655 CrossRef CAS.
- M. Nasiri, D. Saxon and T. Reineke, Macromolecules, 2018, 51, 2456–2465 CrossRef CAS.
- S. Wang, S. Kesava, E. Gomez and M. Robertson, Macromolecules, 2013, 46, 7202–7212 CrossRef CAS.
- C. C. Honeker and E. L. Thomas, Chem. Mater., 1996, 8, 1702–1714 CrossRef CAS.
- A. Takano, I. Kamaya, Y. Takahashi and Y. Matsushita, Macromolecules, 2005, 38, 9718–9723 CrossRef CAS.
- Z. Huo, S. Arora, V. Kong, B. Myrga, A. Statt and J. Laaser, Macromolecules, 2023, 56, 1845–1854 CrossRef CAS PubMed.
- K. Madathil, B. Upadhyay, W. Ledford, S. Kilbey and G. Stein, Macromolecules, 2023, 56, 7102–7112 CrossRef CAS.
- K. J. Henderson, T. C. Zhou, K. J. Otim and K. R. Shull, Macromolecules, 2010, 43, 6193–6201 CrossRef CAS.
- Z. X. Shentu, Z. M. Zhang, J. Zhao, C. S. Chen, Q. Wu, L. Wang and X. Z. Yan, J. Mater. Chem. A, 2021, 9, 19619–19624 RSC.
- I. Kawarazaki, M. Hayashi, A. Shibata and M. Kaai, Polymer, 2021, 234, 124235 CrossRef CAS.
- I. Kawarazaki, M. Hayashi and A. Takasu, Polymer, 2020, 192, 122343 CrossRef CAS.
- M. Hayashi and L. Chen, Polym. Chem., 2020, 11, 1713–1719 RSC.
- M. Hayashi, A. Noro and Y. Matsushita, Macromol. Rapid Commun., 2016, 37, 678–684 CrossRef CAS PubMed.
- X. Guo, S. Nakagawa and N. Yoshie, Macromolecules, 2024, 57, 2351–2362 CrossRef CAS.
- M. Hayashi, S. Matsushima, A. Noro and Y. Matsushita, Macromolecules, 2015, 48, 421–431 CrossRef CAS.
- S. Kawana, S. Nakagawa, S. Nakai, M. Sakamoto, Y. Ishii and N. Yoshie, J. Mater. Chem. A, 2019, 7, 21195–21206 RSC.
- I. Kawarazaki and M. Hayashi, ACS Appl. Polym. Mater., 2021, 3, 1271–1275 CrossRef CAS.
- H. G. Fang, X. C. Gao, F. Zhang, W. J. Zhou, G. B. Qi, K. Song, S. Cheng, Y. S. Ding and H. H. Winter, Macromolecules, 2022, 55, 10900–10911 CrossRef CAS.
- L. Q. Zhu, L. Xu, S. Y. Jie and B. G. Li, Eur. Polym. J., 2022, 180, 111600 CrossRef CAS.
- Y. Oba, T. Kimura, M. Hayashi and K. Yamamoto, Macromolecules, 2022, 55, 1771–1782 CrossRef CAS.
- T. Kito and M. Hayashi, Soft Matter, 2024, 20, 2961–2968 RSC.
- T. Kito and M. Hayashi, Eur. Polym. J., 2024, 208, 112862 CrossRef CAS.
- M. Hayashi, Y. Oba, T. Kimura and A. Takasu, Polym. J., 2021, 53, 835–840 CrossRef CAS.
- D. J. Yarusso and S. L. Cooper, Macromolecules, 1983, 16, 1871–1880 CrossRef CAS.
- J. Kong, V. Lipik, M. Abadie, G. Deen and S. Venkatraman, Polym. Int., 2012, 61, 43–50 CrossRef CAS.
- G. Gregory, G. Sulley, L. Carrodeguas, T. Chen, A. Santmarti, N. Terrill, K. Lee and C. Williams, Chem. Sci., 2020, 11, 6567–6581 RSC.
- W. Shi, N. Lynd, D. Montarnal, Y. Luo, G. Fredrickson, E. Kramer, C. Ntaras, A. Avgeropoulos and A. Hexemer, Macromolecules, 2014, 47, 2037–2043 CrossRef CAS.
- R. G. Ricarte and S. Shanbhag, Polym. Chem., 2024, 15, 815–846 RSC.
- M. Hayashi, Nihon Reoroji Gakkaishi, 2022, 50, 15–20 CrossRef CAS.
- T. Isogai and M. Hayashi, Polym. Chem., 2024, 15, 269–275 RSC.
- M. Hayashi and T. Inaba, ACS Appl. Polym. Mater., 2021, 3, 4424–4429 CrossRef CAS.
- J. Lessard, G. Scheutz, S. Sung, K. Lantz, T. Epps and B. Sumerlin, J. Am. Chem. Soc., 2020, 142, 283–289 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: 1H-NMR, SEC data, FT-IR, TEM observation, fitting analysis of the SAXS data of SBS-CL-1, temperature-ramp SAXS data, TGA thermogram, non-normalized stress relaxation curves, comparison of stress relaxation of the three samples, and Arrhenius plots of relaxation time. See DOI: https://doi.org/10.1039/d4py00858h |
| This journal is © The Royal Society of Chemistry 2024 |