
Polymerization-induced self-assembly of (2-(4-vinylbenzyl)iso-indoline-1,3-dione) for the synthesis of hydrazine responsive block copolymer nanoparticles†
Shivshankar R.
Mane
 *ab and
Andrea S.
Carlini
*abc
*ab and
Andrea S.
Carlini
*abc
aDepartment of Chemistry and Biochemistry, University of California Santa Barbara, California 93106, USA. E-mail: srmane@ucsb.edu; acarlini@ucsb.edu
bCenter for Polymers and Organic Solids, University of California Santa Barbara, California 93106, USA
cDepartment of Biomolecular Science and Engineering, University of California Santa Barbara, California 93106, USA
First published on 20th February 2024
Abstract
Well-defined core–shell nanoparticles with a phthalimide core are synthesized using reversible addition–fragmentation chain transfer (RAFT)-mediated dispersion polymerization-induced self-assembly (PISA) of a novel vinyl benzyl phthalimide monomer, namely (2-(4-vinylbenzyl)iso-indoline-1,3-dione) (VBzPHT). Diblock copolymers consisting of poly(poly(ethylene glycol) methyl ether methacrylate)-block-(2-(4-vinylbenzyl)iso-indoline-1,3-dione) (PEGMA15-b-PVBzPHTn) self-assemble into nanoparticles during polymerization in methanol at 70 °C, when using poly(methyl ether methacrylate)-macro RAFT agent (macro-CTA, DP = 15, Mn = 7600 g mol−1, Đ = 1.14) and 2,2′-azobis(isobutyronitrile) (AIBN) as an initiator. While maintaining a constant chain length of the solvophilic macro-CTA agent, we show that varying the solvophobic PVBzPHT block length (DP = 15–200) under concentrated conditions (15 wt%) achieves self-assembled structures of increasing size. These nanoparticles, ranging from 95 to 389 nm in hydrodynamic diameter, are assessed using both dynamic light scattering and dry state transmission electron microscopy. Finally, we investigate hydrazine-responsive deprotection of phthalimide bearing amphiphilic (PEGMA15-b-PVBzPHTn) block copolymers, leading to the formation of poly(poly(ethylene glycol) methyl ether methacrylate)-block-vinyl benzyl amine (PEGMA15-b-(PVBzNH2)n). This increased solvophilicity leads to complex aggregated assemblies in situ. The insights of this study offer guidelines to the preparation of well-defined nanoparticles through PISA, with unique post-synthetic responsiveness amenable to applications in drug delivery and biocatalysis.
Introduction
Mother Nature sets a perfect example of monodisperse polymers that achieve well-defined sizes and shapes.1 This phenomenon inspires researchers to design amphiphilic homopolymers and block copolymers that self-assemble into nanoparticles such as micelles, vesicles, and worm-like structures via solution self-assembly. This can be done by interchanging single and mixed solvent systems or by slowly introducing new solvents to the solution under very dilute conditions (less than 1 wt% polymer).2–12 In other instances, post-polymerization modifications to the polymer are used to modulate self-assembly, but this can be difficult to implement on a large scale.13 Self-assembled nanoparticles have unique properties that are relevant to a wide range of applications, such as drug delivery, biocatalysis, bio-imaging, and protein immobilization.14–18 Traditionally, higher-order morphologies19,20 have been accessed by tuning the solvophobic–solvophilic balance (i.e. core and corona-forming blocks). However, precise control over size and shape of block copolymer assemblies at high solids content (10–50 wt%) in polar or non-polar solvents remains a key challenge.21–23 In contrast, one-pot methods such as polymerization induced self-assembly (PISA) can access well-defined polymeric nanoparticles at high solid content, without the need for any post-polymerization modifications.24,25In PISA block-copolymer formulations, a solvophilic precursor block is chain extended with another monomer to form a more solvophobic block. As this new block grows, the polymer chain becomes decreasingly soluble in the solvent system. Self-assembly of these polymer chains into nanoparticles then occurs when the growing block becomes insoluble at a critical degree of polymerization.26–28 Reports of PISA in the last decade primarily employ controlled/living polymerizations such as atom transfer radical polymerization (ATRP),29–31 nitroxide-mediated polymerization (NMP),32,33 ring-opening metathesis polymerization (ROMP),34–37 living anionic polymerization (LAP),38,39 ring-opening polymerization,40,41 and reversible addition–fragmentation chain transfer polymerization (RAFT).42–45
The most common strategy, RAFT-mediated PISA, generally involves chain extension of a soluble macromolecular chain transfer agent (CTA) in a suitable solvent, followed by addition of a second soluble monomer. Despite the initial solubility of this second monomer, its respective insoluble polymer acts as the driving force for in situ PISA.46–48 Most importantly, encoding these polymers with stimuli-responsive functional groups is gaining substantial interest in the context of PISA,49,50 to further manipulate their shapes and sizes. This is achieved by shifting the amphiphilicity of diblock copolymer assemblies through molecular deprotection, rearrangement, crosslinking, or altering solvent conditions. External stimuli to initiate these transformations have been reported with light,51,52 temperature,53–55 and pH.56 For example An and co-workers report temperature-sensitive poly(dimethylacrylamide)–poly(diacetone acrylamide) block copolymer nanoparticles, in which reversible morphological transitions from spheres to worms and lamellae are observed.53 In another example, poly(N,N-dimethylacrylamide) shows a lower critical solution temperature that can be controlled by varying solids content and degree of polymerization.54 Similarly, Zetterlund and co-workers employ RAFT dispersion PISA during the synthesis of P(N,N-diethylaminoethyl methacrylate)-stat-poly((ethylene glycol) methyl ether methacrylate) (PDEAEMA-stat-PEGMA), and observe that resulting particle morphologies can ranging from sphere to rods to vesicles through simple manipulation of pH and ionic strength.56 Integral to accessing specific self-assembled and stimuli-responsive morphologies is the choice of initial core-forming monomer used during PISA.57,58 Acrylates (MEA),59 methacrylates (HEMA, DEGMA),60,61 and acrylamides (NIPAM)62 represent the most commonly employed monomers for dictating initial polymer morphologies. Despite extensive PISA efforts with a limited library of known core-forming monomers,58 there remains a tremendous design scope for new monomers and those that bear stimuli-responsive functional groups.
Herein, we demonstrate for the first time a facile PISA synthesis of benzyl phthalimide-based block copolymer nanoparticles by using a newly identified hydrazine responsive core-forming monomer, vinyl benzyl phthalimide (VBzPHT) (Fig. 1). We use RAFT dispersion polymerization of VBzPHT with a block stabilizing poly((ethylene glycol) methyl ether methacrylate) (PEGMA) macro-chain transfer agent (macro-CTA). In situ, this produces poly(poly(ethylene glycol) methyl ether methacrylate)-block-(2-(4-vinylbenzyl)iso-indoline-1,3-dione) PEGMA15-b-PVBzPHTn diblock copolymer nanoparticles at high solids content (15 wt%) bearing hydrazine-responsive benzyl phthalimide group in their core. Tailoring the degree of polymerization prepares a series of PEGMA15-b-PVBzPHTn diblock copolymer nanoparticles with low dispersity. Hydrazine-induced deprotection of the resulting nanoparticles disrupts the balance of solvophobic core and solvophilic corona components, leading to particle disassembly and aggregation.
 | ||
| Fig. 1 Synthesis of responsive block copolymer nanoparticles by reversible addition–fragmentation chain transfer (RAFT)-mediated polymerization-induced self-assembly (PISA). | ||
Experimental
Materials
All the precursors as 4-vinyl benzyl chloride, potassium phthalimide and the RAFT-CTA agent 4-cyano-4-(phenylcarbonothioylthio)pentanoic acid were purchased from Sigma-Aldrich, and used as received. The 2,2′-azobis(isobutyronitrile) AIBN was also purchased from Sigma-Aldrich and recrystallized twice with methanol prior to use. The poly(ethylene glycol) methyl ether methacrylate (PEGMA) monomer (Mn = 500 g mol−1) was purchased from TCI and passed through alumina prior to use. All other reagents were used as received, unless otherwise noted.Monomer synthesis
In a round bottom flask, potassium phthalimide (15.77 g, 0.085 mol, 1.3 eq.) was dissolved in 100 mL DMF, to this 4-vinyl benzyl chloride (9.99 g, 0.0655 mol, 1 eq.) was added and the reaction mixture kept on heating at 110 °C for 6 h. After this reaction was cooled to room temperature, 400 ml water and 200 mL ethyl acetate were added to the solution. The aqueous layer was washed with ethyl acetate (3 × 100 mL) and dried over MgSO4. The solvent was evaporated under reduced pressure and the obtained solid was re-dissolved in chloroform at 40 °C and precipitated in cold pentane. The crude product was filtered and washed with cold pentane. Finally, it was dried using high vacuum and to obtain VBzPHT as a free white powder (13.54 g, yield 80%).631H NMR in DMSO-d6 (δ ppm): 7.75–7.95 (m, 4H), 7.45 (d, 2H), 7.25 (d, 2H), 6.72 (m, 1H), 5.77 (dd, 1H), 5.21 (dd, 1H), 4.79 (s, 2H). 13C NMR in DMSO-d6 (δ ppm) 168.04, 137.17, 136.3, 135.91, 134.02, 132.07, 128.86, 122.45, 122.32, 114.15, 41.31.PEGMA-CTA (macro-CTA) synthesis
In a Schlenk tube, the desired amount of poly(ethylene glycol) methyl ether methacrylate (PEGMA) (2.863 g, 5.7269 mmol, 16 eq.) was taken and to this, RAFT-CTA 4-cyano-4-(phenylcarbonothioylthio)pentanoic acid (100 mg, 0.3579 mmol, 1 eq.) was added, followed by the addition of AIBN (5.877 mg, 0.03579 mmol, 0.1 eq.). All the solids were dissolved in 1,4-dioxane solvent at an inert atmosphere. The reaction mixture was degassed 3 times by freeze–pump–thaw cycle and kept in a preheated bath at 70 °C for 6 h. After complete polymerization, the reaction was quenched by sudden cooling of the tube in an ice water bath and exposure to air. The macro-CTA was obtained by several precipitation in cold dry diethyl ether. The molecular weight measured by SEC is (Mn = 7600 g mol−1, Đ = 1.14). 1H NMR in DMSO-d6 (δ ppm): 7.82 (dd, 2H), 7.64 (dd, 1H), 7.47 (d, 2H), 4.25–3.15 (m, PEG proton), 2.1–0.81 (m, background aliphatic protons).DP and Mn,NMR calculation: degree of polymerization for macro-CTA was calculated by 1H NMR spectroscopy (Fig. S3†).

| Mn,NMR = (molecular weight of PEGMA500 × DPm) |
RAFT dispersion polymerization for PEGMA15-b-PVBzPHTn block copolymer synthesis
A typical RAFT dispersion polymerization for the synthesis of PEGMA15-b-PVBzPHT50 block copolymer at 15 wt% total solid content was performed as follows: in a Schlenk tube equipped with a magnetic spin bar, 100.0 mg (0.0131 mmol) of macro- (Mn = 7600 g mol−1, Đ = 1.14), 173.21 mg (0.6578 mmol) VBzPHT, and 1.08 mg (0.0065 mmol) AIBN were taken. The entire solid was dissolved in 1.125 g of methanol at inert atmosphere. The reaction mixture was degassed 3 times by freeze–pump–thaw cycle and kept in a preheated bath temperature at 70 °C for 24 h. The polymerization reaction was stopped by sudden cooling the tube in an ice water bath, and exposing the solution to air. 100 μL of the resultant mixture was taken out for 1H NMR analysis to determine the monomer conversion, and a portion of the mixture was diluted with methanol followed by filtering through a 0.45 μm PTFE filter for TEM and DLS analysis. Finally, the PEGMA15-b-PVBzPHT50 block copolymer was obtained by precipitation in cold dry diethyl ether and purified by several precipitations followed by vacuum drying at 40 °C for 12 h and measured SEC to obtain molecular weight (Mn = 17![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 300 g mol−1, Đ = 1.08) see Table 1, P3. By targeting desired PVBzPHT block lengths, similar polymerization reactions were carried out by adjusting the feed ratio, which allowed access to spherical morphology.
300 g mol−1, Đ = 1.08) see Table 1, P3. By targeting desired PVBzPHT block lengths, similar polymerization reactions were carried out by adjusting the feed ratio, which allowed access to spherical morphology.
| Polymer | Composition | [M]/[I]a | Conversiona (%) | M n (g mol−1) | M n (g mol−1) | Đ | Sized (nm) (PDI) |
|---|---|---|---|---|---|---|---|
| a Determined by 1H NMR spectroscopy. b Molecular weight (g mol−1) determined by SEC in DMF. c Polydispersity index (Mw/Mn) determined by SEC. d Size and dispersity measured by DLS. | |||||||
| Macro-CTA | PEGMA15 | 15 | 94 | 7720 | 7600 | 1.14 | — |
| P1 | PEGMA15-b-PVBzPHT15 | 15 | 93 | 11545 |
11300 |
1.12 | 95 (0.32) |
| P2 | PEGMA15-b-PVBzPHT25 | 25 | 96 | 14175 |
14000 |
1.09 | 118 (0.32) |
| P3 | PEGMA15-b-PVBzPHT50 | 50 | 94 | 20750 |
17300 |
1.08 | 151 (0.31) |
| P4 | PEGMA15-b-PVBzPHT100 | 100 | 90 | 33900 |
31400 |
1.13 | 380 (0.16) |
| P5 | PEGMA15-b-PVBzPHT200 | 200 | 94 | 60200 |
57000 |
1.11 | 389 (0.27) |
DP and Mn,NMR calculation: for the PEGMA15-b-PVBzPHTn (P1–P5) block copolymer, using 1H NMR spectroscopy (Fig. S6†).

| Mn,NMR = (molecular weight of PEGMA-CTA) + (molecular weight of VBzPHT × DPn). |
Stimuli-responsive study control experiment
In a two-neck round bottom flask, 10.5 g of VBzPHT monomer was dissolved in 60 mL ethanol solvent and kept for heating at 40 °C followed by drop-wise addition of hydrazine monohydrate (7.68 mL). Finally, the reaction mixture stirred for 1 h at 40 °C and RT for 19 h. After the complete reaction, 100 mL of 10% KOH in water was added to maintain a pH 10 solution. The product was extracted from 3 × 100 mL chloroform and the organic layer was dried over MgSO4 and solvent evaporated to yield VBzNH2. 1H NMR in DMSO-d6 (δ ppm): 7.23 (d, 2H), 7.15 (d, 2H), 6.51 (m, 1H), 5.58 (dd, 1H), 5.08 (dd, 1H), 3.61 (s, 2H). A similar protocol was employed for the PEGMA15-b-PVBzPHTn block copolymers P2 and P3 in water at ∼5 mg mL−1. At 24 h incubation time, samples were collected and diluted with water to ∼1 mg mL−1 for direct analysis by TEM and DLS. Samples for NMR analyses were precipitated and resuspended in DMSO-d6 solvent.Polymer characterization
Results and discussion
Monomer synthesis
Reacting vinyl benzyl chloride with potassium phthalimide at 110 °C (Scheme S1†) achieves synthesis of the core forming VBzPHT monomer. Confirmation by 1H and 13C NMR spectroscopy indicates appropriate monomer formation. The 1H NMR spectrum shows the appearance of characteristic signals at δ 7.75–7.95 ppm corresponding to the phthalimide proton, in addition to the vinylic backbone protons at δ 5.21–6.72 ppm (Fig. S1†). Similarly, in 13C NMR, the signal at δ 168.04 ppm denotes the phthalimide carbonyl group (amide), and δ 122.45, 132.07 ppm represent the aromatic carbons of the phthalimide ring (Fig. S2†).Synthesis of PEGMA15-CTA (macro-CTA)
The RAFT solution polymerization of PEGMA in 1,4-dioxane solvent synthesizes macro-CTA at 70 °C by using 4-cyano-4-(phenylcarbonothioylthio)pentanoic acid as a RAFT agent, and AIBN as a radical initiator. The polymerization reaction quenches after 6 h in ice cold water, leading to 94% monomer conversion. The molecular weight (Mn = 7600 g mol−1; Đ = 1.14) of the macro-CTA was obtained from SEC, and the degree of polymerization (DP) of 15 was calculated from the monomer conversion (Fig. S3† and Table 1). A 1H NMR spectrum of PEGMA15 macro-CTA and its corresponding SEC curve are shown in Fig. 2b, S3,† and Fig. 3a respectively. The signals at δ 4.25–3.15 ppm correspond to PEG protons, in addition to this, the parent RAFT-CTA protons at δ 7.82–7.47 ppm along with methylene protons at δ 2.1–0.81 ppm confirm the formation of macro-CTA.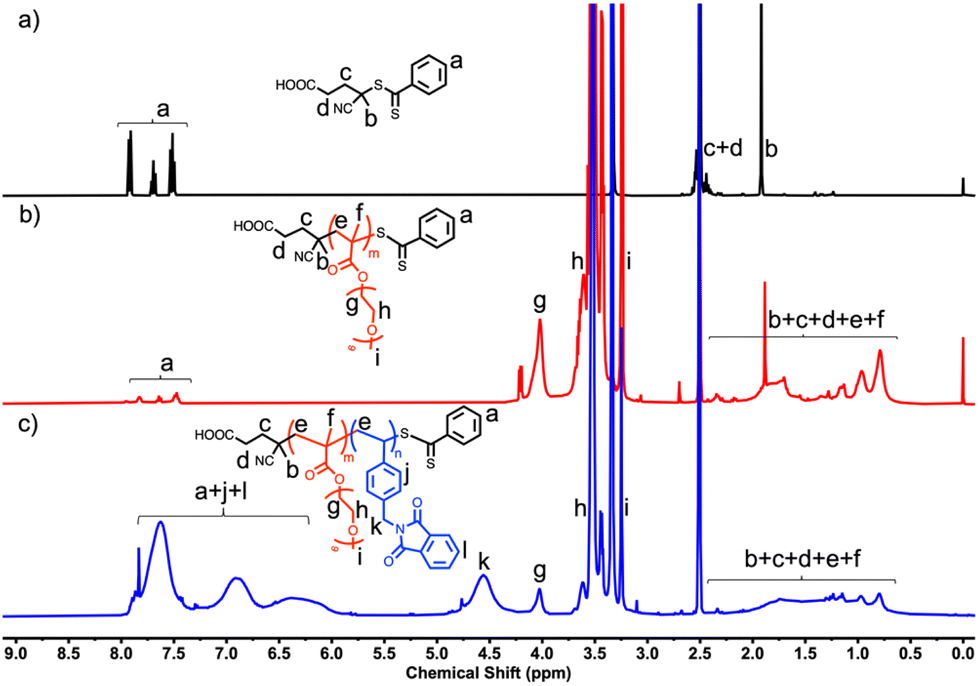 | ||
| Fig. 2 1H NMR overlay (a) RAFT agent, (b) macro-CTA, (c) PEGMAm-b-PVBzPHTn block copolymer in DMSO-d6. | ||
 | ||
| Fig. 3 (a) Representative SEC traces for macro-CTA, and PEGMAm-b-PVBzPHTn block copolymer: P1–P5; (b) [M]/[I] vs. Mn of P1–P5. | ||
RAFT dispersion polymerization for PEGMA15-b-PVBzPHTn block copolymer nanoparticles
Poly(ethylene glycol) methyl ether methacrylate PEGMA15-CTA, also known as macro-CTA, was used in the current study for RAFT dispersion polymerization of VBzPHT in methanol at 70 °C for 24 hours to in situ prepare nanoparticles of PEGMA15-b-PVBzPHTn block copolymer at 15 wt% total solids content (Scheme 1). Here we study how the size and shape of the nanoparticles evolve with the length of the solvophobic block, namely PVBzPHT, while keeping the chain length constant for macro-CTA. The target DP of the PVBzPHT chains sequentially increases from 15 to 200. The methanol solvent system is chosen for its ability to act as a good solvent for both the macro-CTA and VBzPHT monomer, but as a poor solvent for the PVBzPHT block, which is an essential basic condition for the PISA process to form in situ block copolymer nanoparticles. Thus, PVBzPHT block chains are insoluble in methanol, which leads to the formation of in situ self-assembly by varying the DP of the core-forming VBzPHT monomer. | ||
| Scheme 1 RAFT-mediated synthesis of poly(poly(ethylene glycol) methyl methacrylate) (macro-CTA), followed by the synthesis of PEGMAm-b-PVBzPHTn block copolymer (P1–P5). | ||
As the polymerization proceeds, the solution becomes turbid, indicating intra-strand nucleation as well as interstrand self-assembly of the PEGMA15-b-PVBzPHTn diblock copolymer. The polymerization reaction was analyzed by H1 NMR spectroscopy and calculated the conversion (Fig. S4–S8†). The VBzPHT phthalimide aromatic protons corresponding to the broad signals from δ 7.93–6.01 ppm, the PEG protons at δ 4.25–3.15 ppm and the backbone methylene protons at δ 2.1–0.81 ppm confirms the diblock copolymer PEGMA15-b-PVBzPHTn formation. 1H NMR analysis indicates ∼94% VBzPHT monomer conversion in each block copolymer case. Fig. 2 shows an overlay for the 1H NMR spectra of RAFT-agent, PEGMA15-CTA and PEGMA15-b-PVBzPHTn diblock copolymer. 2D HSQC NMR of P1, P4, and P5 provide additional structural characterization (Fig. S9–S11†).
The molecular weight (Mn = 7600 g mol−1; Đ = 1.14) of the macro-CTA followed by different block lengths (P1–P5; DP = 15–200) was measured by SEC. The observed molecular weight ranges from (Mn = 11300 g mol−1 to 57000 g mol−1) with narrow dispersity (Đ < 1.2). The normalized SEC traces for PEGMA15-b-PVBzPHTn along with macro-CTA were collected using a refractive index detector, as shown in Fig. 3 and Fig. S12.† The SEC curves shift towards a lower elution time as the degree of polymerization of the core-forming PVBzPHT block increases. This result suggests the complete initiation of macro-CTA, which results in the successful synthesis of a well-defined PEGMA15-b-PVBzPHTn diblock copolymer. Table 1 gives a summary of the results for PEGMA15-b-PVBzPHTn (P1–P5) diblock copolymer nanoparticles formed by RAFT dispersion polymerization in methanol.
Following the NMR and SEC analysis, all the PEGMA15-b-PVBzPHTn block copolymers were characterized using a combination of dynamic light scattering (DLS) and transmission electron microscopy (TEM) in methanol at 1 mg mL−1. DLS experiments of PEGMA15-b-PVBzPHTn diblock copolymers show their nanoparticle assemblies possessing low dispersity values (0.16–0.32) and increasing hydrodynamic sizes with respect to increasing solvophobicity (Fig. 4 and Table 1). In the case of solvophilic P1, and P2 low dispersity particles with diameters of 95 and 118 nm, respectively. These sizes are maintained even after incubation in water for 24 h (Fig. S13†). As PISA progresses with longer solvophobic PVBzPHT blocks, nanoparticle sizes increase significantly, with diameters for P3–P5 being 151, 380 and 389 nm, respectively. Additionally, autocorrelation functions reveal a large shift in delay times between P2 and P3. This transition can be explained as a consequence of increasing the solvophobic block fraction that ultimately increases the packing parameter of diblock copolymer resulting from the formation of higher order morphologies.64–66
 | ||
| Fig. 4 DLS analysis for PEGMAm-b-PVBzPHTn block copolymer (P1–P5) in methanol. Inset shows autocorrelation functions. | ||
Amphiphilic polymers generated in this study show a high morphological dependence on solvation. Specifically, NPs with large solvophobic VBzPHT blocks (P3–P5) show increasing divergence between observed diameters by TEM and DLS. Unstained dry state TEM images show the morphology of our polymeric nanostructures (Fig. 5 and Fig. S14†). We observe discrete spherical nanoparticles of P1 and P2 by TEM, with observed diameters that closely agree with DLS data. However, TEM of polymers bearing larger solvophobic VBzPHT blocks (P3–P5) show increasing divergence between TEM and DLS results. This is exemplified by the observation of smaller, non-discrete, and lower contrast particles by TEM. We suspect that nanoparticle sizes of our more solvophobic polymers are more strongly influenced by drying effects during TEM sample preparation.67 Specifically, polymeric core–shell nanoparticles bearing hydrated coronas, which contribute to hydrodynamic drag in DLS, are often collapsed in the desiccated and high-vacuum environment of TEM. To further investigate solvent effects, we precipitate our nanoparticles from methanol and resuspend them in water (pH ∼ 6.0, for 24 h) as a control (Fig. S13†). Hydrophilic P1 and P2 display low sensitivity to these altered environmental conditions by DLS. In contrast, more hydrophobic P4 and P5 show a significant deswelling effect (>50%) from and 380/389 nm to 152/190 nm diameters, respectively as they exclude the more polar solvent. Therefore, unstained TEM morphologies are unlikely to be representative of solvated nanoparticles.
 | ||
| Fig. 5 Representative TEM images for PEGMA15-b-PVBzPHTn copolymer: P1 (a & f), P2 (b & g), P3 (c & h), P4 (d & i), and P5 (e & j). | ||
Hydrazine-responsive study for PEGMA15-b-PVBzPHTn
The polymers presented in this manuscript were designed with a sufficiently high molecular weight corona-forming block to generate kinetically-trapped spheres. We suspect that reducing the hydrophobic core content in situ would enable the formation of higher-order morphologies. To do this, phthalimide protected amines can undergo deprotection by using hydrazine hydrate.68,69 We demonstrate the responsive functionality of our VBzPHT monomer and resulting PEGMA15-b-PVBzPHTn block copolymers by subjecting them to hydrazine treatment for phthalimide deprotection. Upon addition of hydrazine hydrate, monomer deprotection of the phthalimide amine group occurs, therefore converting vinyl benzyl phthalimide (VBzPHT) to vinyl benzyl amine (VBzNH2). Confirmation by 1H NMR spectroscopy after 20 h indicates the complete removal of phthalimide ring, as there is no signal at δ 7.75–7.95 ppm that corresponds to the phthalimide aromatic protons (Fig. S15†). Similarly, addition of hydrazine hydrate to particle solutions converts the solvophobic phthalimide block (PVBzPHT) of PEGMA15-b-PVBzPHTn polymer into solvophilic PEGMA15-b-(PVBzNH2)n. For these experiments, we used P2 and P3 suspended in water, given their known stability in an aqueous environment (Fig. S13†). 1H NMR at 20 h shows a complete loss of signal for phthalimide at δ 7.25–7.95 ppm (Fig. S16 and S17†), indicating ∼100% deprotection. The morphology of both deprotected nanoparticles by TEM (Fig. 6c and Fig. S19†) shows unique morphologies. For instance, treated P2 yields low contrast aggregated assemblies approximately 757 nm in size by DLS (Fig. S18a†). Conversely, treated P3 yields larger lamellae-like assemblies approximately 967 nm in size (Fig. S18b†). Fig. 6 depicts a schematic representation of the proposed disassembly and aggregation process for PEGMA15-b-PVBzPHTn to PEGMA15-b-(PVBzNH2)n, with accompanying TEM images (Fig. S19†). We suspect that decreasing amphiphilicity, increasing solvation in water, and intermolecular hydrogen bonding between polymers with newly exposed amines are likely driving forces towards this molecular rearrangement.70–72 The reduced crystallinity and increased sizes observed in treated P3 are likely attributable to the presence of twice the amount of hydrogen bonding and aromatic π–π stacking interactions between benzyl amines, in comparison to that of treated P2. | ||
| Fig. 6 Schematic representation for disassembly of PEGMA15-b-VBzPHTn to PEGMA15-b-(VBzNH2)n. (a) Chemical structure, (b) schematic disassembly and aggregation process, (c) representative TEM images for P2 before hydrazine treatment, (d & e) P2 after deprotection and aggregation after hydrazine treatment for 20 h. | ||
Conclusions
In summary, the focus of this study explores RAFT-mediated dispersion polymerization-induced self-assembly of a newly identified benzyl phthalimide (VBzPHT) monomer. In combination with PEGMA15 macro-CTA, VBzPHT acting as the core-forming monomer for a variety of polymeric self-assembled nanoparticles. Variations in the chain length of the solvophobic block (DP 15–200), while maintaining a fixed length solvophilic macro-CTA, produces spherical morphologies ranging from 95 nm to 389 nm in diameter by DLS and TEM. Stimuli-responsive deprotection of the phthalimide bearing PVBzPHT core block results in structurally dynamic rearrangements into aggregated assemblies.This PISA method represents a facile approach for the preparation of well-defined nanoparticles in which their unique responsive behavior poses a wide range of applications in drug delivery. Specifically, future studies aim to utilize the hydrazine-deprotected benzyl amine as a versatile linker for pH-responsive therapeutic cargos (e.g. doxorubicin conjugation via an imine linker).
Author contributions
S. R. M. conceived the idea, and designed and performed the experiments. S. R. M. and A. S. C. analysed the data and wrote the paper.Conflicts of interest
The author declares no conflict of interest.Acknowledgements
S. R. M. would like to thank the CSIR-SRA (Scientist's Pool Scheme, Award No. 13(9139-A)/2020-Pool) for the funding. We thank the University of California and a UCSB Faculty Research Grant for financial support. Thanks to Natasha Cao for manuscript revisions.Notes and references
- J. Jennings, G. He, S. M. Howdle and P. B. Zetterlund, Chem. Soc. Rev., 2016, 45, 5055–5084 RSC.
- D. E. Discher and A. Eisenberg, Science, 2002, 297, 967–973 CrossRef CAS PubMed.
- Y. Mai and A. Eisenberg, Chem. Soc. Rev., 2012, 41, 5969 RSC.
- R. J. Williams, A. P. Dove and R. K. O'Reilly, Polym. Chem., 2015, 6, 2998–3008 RSC.
- B. M. Discher, Y. Y. Won, D. S. Ege, J. C. M. Lee, F. S. Bates, D. E. Discher and D. A. Hammer, Science, 1999, 284, 1143–1146 CrossRef CAS PubMed.
- S. R. Mane, A. Sathyan and R. Shunmugam, ACS Appl. Nano Mater., 2020, 3, 2104–2117 CrossRef CAS.
- S. R. Mane, A. Sathyan and R. Shunmugam, Sci. Rep., 2017, 7, 44857 CrossRef CAS PubMed.
- G. Mellot, P. Beaunier, J. M. Guigner, L. Bouteiller, J. Rieger and F. Stoffelbach, Macromol. Rapid Commun., 2019, 40, 1–7 CrossRef PubMed.
- S. R. Mane, K. Chatterjee, H. Dinda, J. Das Sarma and R. Shunmugam, Polym. Chem., 2014, 5, 2725–2735 RSC.
- S. R. Mane and R. Shunmugam, ACS Macro Lett., 2014, 3, 44–50 CrossRef CAS PubMed.
- D. Kumar, S. A. Mohammad, A. Kumar, S. R. Mane and S. Banerjee, Polym. Chem., 2022, 13, 1960–1969 RSC.
- T. I. Löbling, O. Ikkala, A. H. Gröschel and A. H. E. Müller, ACS Macro Lett., 2016, 5, 1044–1048 CrossRef PubMed.
- K. Kita-Tokarczyk, J. Grumelard, T. Haefele and W. Meier, Polymer, 2005, 46, 3540–3563 CrossRef CAS.
- D. Keller, A. Beloqui, M. Martínez-Martínez, M. Ferrer and G. Delaittre, Biomacromolecules, 2017, 18, 2777–2788 CrossRef CAS PubMed.
- L. C. S. Huang, D. Le, I. L. Hsiao, S. Fritsch-Decker, C. Hald, S. C. Huang, J. K. Chen, J. R. Hwu, C. Weiss, M. H. Hsu and G. Delaittre, Polym. Chem., 2021, 12, 50–56 RSC.
- S. R. Mane, I.-L. Hsiao, M. Takamiya, D. Le, U. Straehle, C. Barner-Kowollik, C. Weiss and G. Delaittre, Small, 2018, 14, 1801571 CrossRef PubMed.
- A. Beloqui, S. R. Mane, M. Langer, M. Glassner, D. M. Bauer, L. Fruk, C. Barner-Kowollik and G. Delaittre, Angew. Chem., Int. Ed., 2020, 59, 19951–19955 CrossRef CAS PubMed.
- Z. Wang, M. C. M. Van Oers, F. P. J. T. Rutjes and J. C. M. Van Hest, Angew. Chem., Int. Ed., 2012, 51, 10746–10750 CrossRef CAS PubMed.
- B. Charleux, G. Delaittre, J. Rieger and F. D'Agosto, Macromolecules, 2012, 45, 6753–6765 CrossRef CAS.
- L. P. D. Ratcliffe, A. Blanazs, C. N. Williams, S. L. Brown and S. P. Armes, Polym. Chem., 2014, 5, 3643–3655 RSC.
- M. Semsarilar, V. Ladmiral, A. Blanazs and S. P. Armes, Langmuir, 2013, 29, 7416–7424 CrossRef CAS PubMed.
- Y. Li and S. P. Armes, Angew. Chem., Int. Ed., 2010, 49, 4042–4046 CrossRef CAS PubMed.
- X. Wang and Z. An, Macromol. Rapid Commun., 2019, 40, 1800325 CrossRef PubMed.
- X. Zhang, S. Boissé, W. Zhang, P. Beaunier, F. D'Agosto, J. Rieger and B. Charleux, Macromolecules, 2011, 44, 4149–4158 CrossRef CAS.
- S. Boissé, J. Rieger, K. Belal, A. Di-Cicco, P. Beaunier, M. H. Li and B. Charleux, Chem. Commun., 2010, 46, 1950–1952 RSC.
- C. Liu, C. Y. Hong and C. Y. Pan, Polym. Chem., 2020, 11, 3673–3689 RSC.
- N. J. W. Penfold, J. Yeow, C. Boyer and S. P. Armes, ACS Macro Lett., 2019, 8, 1029–1054 CrossRef CAS PubMed.
- S. L. Canning, G. N. Smith and S. P. Armes, Macromolecules, 2016, 49, 1985–2001 CrossRef CAS PubMed.
- G. Wang, M. Schmitt, Z. Wang, B. Lee, X. Pan, L. Fu, J. Yan, S. Li, G. Xie, M. R. Bockstaller and K. Matyjaszewski, Macromolecules, 2016, 49, 8605–8615 CrossRef CAS.
- Y. Wang, G. Han, W. Duan and W. Zhang, Macromol. Rapid Commun., 2019, 40, 1800140 CrossRef PubMed.
- J. Wang, L. Yuan, Z. Wang, M. A. Rahman, Y. Huang, T. Zhu, R. Wang, J. Cheng, C. Wang, F. Chu and C. Tang, Macromolecules, 2016, 49, 7709–7717 CrossRef CAS.
- X. G. Qiao, M. Lansalot, E. Bourgeat-Lami and B. Charleux, Macromolecules, 2013, 46, 4285–4295 CrossRef CAS.
- X. G. Qiao, P. Y. Dugas, B. Charleux, M. Lansalot and E. Bourgeat-Lami, Polym. Chem., 2017, 8, 4014–4029 RSC.
- D. B. Wright, M. A. Touve, L. Adamiak and N. C. Gianneschi, ACS Macro Lett., 2017, 6, 925–929 CrossRef CAS PubMed.
- J. C. Foster, S. Varlas, L. D. Blackman, L. A. Arkinstall and R. K. O'Reilly, Angew. Chem., Int. Ed., 2018, 57, 10672–10676 CrossRef CAS PubMed.
- D. B. Wright, M. A. Touve, M. P. Thompson and N. C. Gianneschi, ACS Macro Lett., 2018, 7, 401–405 CrossRef CAS PubMed.
- D. Le, M. Dilger, V. Pertici, S. Diabaté, D. Gigmes, C. Weiss and G. Delaittre, Angew. Chem., Int. Ed., 2019, 58, 4725–4731 CrossRef CAS PubMed.
- J. Wang, M. Cao, P. Zhou and G. Wang, Macromolecules, 2020, 53, 3157–3165 CrossRef CAS.
- D. M. Day and L. R. Hutchings, Eur. Polym. J., 2021, 156, 110631 CrossRef CAS.
- E. Guégain, C. Zhu, E. Giovanardi and J. Nicolas, Macromolecules, 2019, 52, 3612–3624 CrossRef.
- J. Jiang, X. Zhang, Z. Fan and J. Du, ACS Macro Lett., 2019, 8, 1216–1221 CrossRef CAS PubMed.
- S. L. Canning, G. N. Smith and S. P. Armes, Macromolecules, 2016, 49, 1985–2001 CrossRef CAS PubMed.
- M. Nardi, T. Scherer, L. Yang, C. Kübel, C. Barner-Kowollik and E. Blasco, Polym. Chem., 2021, 12, 1627–1634 RSC.
- F. D'Agosto, J. Rieger and M. Lansalot, Angew. Chem., Int. Ed., 2020, 59, 8368–8392 CrossRef PubMed.
- S. Piogé, T. N. Tran, T. G. McKenzie, S. Pascual, M. Ashokkumar, L. Fontaine and G. Qiao, Macromolecules, 2018, 51, 8862–8869 CrossRef.
- N. J. Warren and S. P. Armes, J. Am. Chem. Soc., 2014, 136, 10174–10185 CrossRef CAS PubMed.
- J. Cao, Y. Tan, Y. Chen, L. Zhang and J. Tan, Macromol. Rapid Commun., 2021, 42, 1–12 Search PubMed.
- S. J. Hunter, J. R. Lovett, O. O. Mykhaylyk, E. R. Jones and S. P. Armes, Polym. Chem., 2021, 12, 3629–3639 RSC.
- F. D. Jochum and P. Theato, Chem. Soc. Rev., 2013, 42, 7468–7483 RSC.
- F. D. Jochum, L. Z. Borg, P. J. Roth and P. Theato, Macromolecules, 2009, 42, 7854–7862 CrossRef CAS.
- A. Bagheri, C. Boyer and M. Lim, Macromol. Rapid Commun., 2019, 40, 1800510 CrossRef PubMed.
- C. A. Boyer and G. M. Miyake, Macromol. Rapid Commun., 2017, 48, 1700327 CrossRef PubMed.
- X. Wang, J. Zhou, X. Lv, B. Zhang and Z. An, Macromolecules, 2017, 50, 7222–7232 CrossRef CAS.
- C. A. Figg, A. Simula, K. A. Gebre, B. S. Tucker, D. M. Haddleton and B. S. Sumerlin, Chem. Sci., 2015, 6, 1230–1236 RSC.
- S. Kessel, N. P. Truong, Z. Jia and M. J. Monteiro, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 4879–4887 CrossRef CAS.
- P. B. Zetterlund, D. Zhou, S. Perrier, R. P. Kuchel and S. Dong, Polym. Chem., 2017, 8, 3082–3089 RSC.
- J. C. Foster, S. Varlas, B. Couturaud, J. R. Jones, R. Keogh, R. T. Mathers and R. K. O'Reilly, Angew. Chem., Int. Ed., 2018, 57, 15733–15737 CrossRef CAS PubMed.
- S. R. Mane, New J. Chem., 2020, 44, 6690–6698 RSC.
- S. Sugihara, A. H. Ma'Radzi, S. Ida, S. Irie, T. Kikukawa and Y. Maeda, Polymer, 2015, 76, 17–24 CrossRef CAS.
- K. Bauri, A. Narayanan, U. Haldar and P. De, Polym. Chem., 2015, 6, 6152–6162 RSC.
- K. Parkatzidis, N. P. Truong, M. Rolland, V. Lutz-Bueno, E. H. Pilkington, R. Mezzenga and A. Anastasaki, Angew. Chem., Int. Ed., 2022, 61, e2021134 CrossRef PubMed.
- X. Wang, C. A. Figg, X. Lv, Y. Yang, B. S. Sumerlin and Z. An, ACS Macro Lett., 2017, 6, 337–342 CrossRef CAS PubMed.
- S. Arias, E. Maron and H. G. Börner, Biomacromolecules, 2021, 22, 213–221 CrossRef CAS PubMed.
- A. Blanazs, A. J. Ryan and S. P. Armes, Macromolecules, 2012, 45, 5099–5107 CrossRef CAS.
- X. Dai, Y. Zhang, L. Yu, X. Li, L. Zhang and J. Tan, ACS Macro Lett., 2019, 8, 955–961 CrossRef CAS PubMed.
- Y. Mai and A. Eisenberg, Chem. Soc. Rev., 2012, 41, 5969–5985 RSC.
- B. K. Wilson and R. K. Prudhomme, J. Colloid Interface Sci., 2021, 604, 208–220 CrossRef CAS PubMed.
- S. Srichan, H. Mutlu and J. Lutz, Eur. Polym. J., 2015, 62, 338–346 CrossRef CAS.
- J. W. Cleveland, J. Il Choi, R. S. Sekiya, J. Cho, H. J. Moon, S. S. Jang and C. W. Jones, ACS Appl. Mater. Interfaces, 2022, 14, 11235–11247 CrossRef CAS PubMed.
- F. K. Wolf, A. M. Hofmann and H. Frey, Macromolecules, 2010, 43, 3314–3324 CrossRef CAS.
- W. Qi, Y. Zhang, J. Wang, G. Tao, L. Wu, Z. Kochovski, H. Gao, G. Chen and M. Jiang, J. Am. Chem. Soc., 2018, 140, 8851–8857 CrossRef CAS PubMed.
- Y. Song, Y. Chen, P. Li and C.-M. Dong, Biomacromolecules, 2020, 21, 5345–5357 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Monomer synthesis scheme and NMR data. See DOI: https://doi.org/10.1039/d4py00073k |
| This journal is © The Royal Society of Chemistry 2024 |