
Lewis acid-induced homo- and heterogeneous nickel catalysts for ethylene polymerization and copolymerization with polar monomers†
Wanlu
Tian
,
Chao
Li
*,
Chen
Tan
 * and
Min
Chen
*
* and
Min
Chen
*
Institute of Physical Science and Information Technology, Anhui University, Hefei, Anhui 230601, China. E-mail: misschen@ahu.edu.cn
First published on 4th January 2024
Abstract
Lewis acids have been widely investigated to tune the properties of olefin polymerization catalysts. However, the application of this strategy in heterogeneous olefin–polar monomer copolymerization has rarely been studied. Herein, a series of [N, O]-type nickel catalysts bearing Lewis base response moieties was designed and synthesized. These catalysts can be modulated by Lewis acids such as B(C6F5)3 and MAO, resulting in greatly enhanced catalytic performances. This is due to the tuning of Lewis acid to the electronic and steric hindrance effects of the catalysts. Moreover, this Lewis acid–base combination was used as an anchoring strategy for heterogeneous catalysis, leading to increased thermal stability, the formation of ultra-high molecular weight polyethylene (Mn up to 205.3 × 104 g mol−1), and excellent morphology control. The immobilized nickel systems also promoted the copolymerization of ethylene with polar monomers, generating copolymer with high molecular weight and high activity.
Introduction
The current global annual output of polyolefin is nearly 200 million tons. Polyolefin is widely used in every corner of life and has become one of the most important polymers in the history of materials.1 Since the discovery of the Ziegler–Natta catalyst, the iteration of transition metal catalysts has become the main driving force to promote the development of the polyolefin industry.2–7In recent years, there has been considerable use of olefin–polar monomer coordination copolymerization.8,9 This route provides a direct and economical method to synthesize new functionalized polyolefin materials with improved and designable properties.10–14 Among olefin–polar monomer copolymerization catalysts, low-cost nickel-based catalysts are highly anticipated. Historically, hundreds of nickel catalysts bearing various substitutes have been synthesized and their performances investigated in olefin–polar monomer copolymerization.15–32 This trial-and-error research strategy leads to more complicated catalyst synthesis.
Alternatively, with a tunable catalyst strategy, the catalytic performance can be switched during olefin polymerization.33–35 Lewis acid modulation is an interesting method for tuning the performances of transition-metal catalysts.36–38 The reactivity of nickel catalysts in olefin polymerization can be tuned by the addition of Lewis acids such as boranes and alumina. For instance, the Bazan, Lee, and Chen groups developed olefin polymerization catalysts based on [P, O]-, [N, N]-, and [N, O]-type nickel complexes (Scheme 1A–D).39–42 When B(C6F5)3 (BCF) was added, zwitterionic nickel catalytic species were generated by the formation of coordination interactions between the borane acceptor and oxygen donor. Compared to corresponding neutral nickel complexes, these zwitterionic species exhibited higher activity in ethylene polymerization but generated low molecular weight products, which was ascribed to the enhanced electrophilicity. This strategy can also be used in heterogeneous catalytic nickel systems through the introduction of solid-modified Lewis acid.
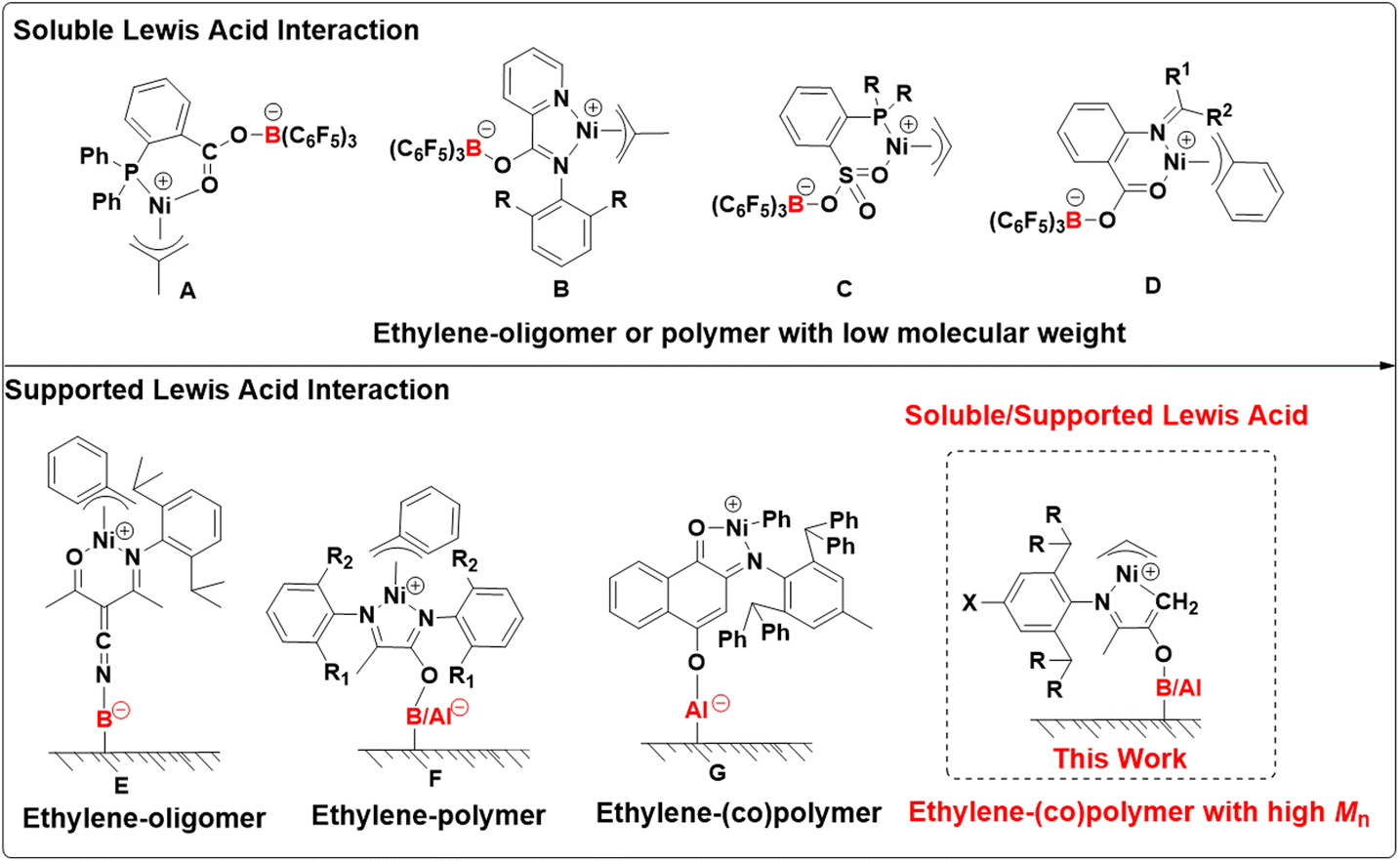 | ||
| Scheme 1 (A–D) Soluble Lewis acid-induced nickel catalysts for ethylene homogeneous polymerization generating low molecular weight oligomer and polyethylene. (E–G) Supported Lewis acid-induced nickel catalysts for heterogeneous ethylene (co)polymerization. (H) This work, a combination of soluble and supported Lewis acid-induced nickel catalysts for ethylene (co)polymerization producing (co)polymer with high molecular weight. | ||
Heterogeneous catalysts are predominantly for industrial polyolefin production because they offer many distinct advantages, such as controlling the morphology of the polymer and preventing reactor fouling.43–51 For instance, Rojas and Scott prepared heterogeneous [N, O]- and [N, N]-type nickel catalysts based on B(C6F5)3 or Al-modified supports for ethylene polymerization to improve catalytic activity (Scheme 1E and F).52–54 Recently, Shiono and Cai et al. prepared heterogeneous nickel catalysts via the reaction of an anilinonaphthoquinone ligand with methylaluminoxane-modified silica (MMAO/SiO2). This system can mediate ethylene copolymerization with 5-hexene-1-yl acetate and allyl acetate and lead to satisfactory polymer morphology control (Scheme 1G).55,56
As mentioned above, the Lewis acid-induced strategy is very practical in olefin polymerization. Developing unique nickel systems using this strategy for olefin (co)polymerization demonstrates exciting opportunities and will attract wide application interests. Inspired by the pioneering work by Bazan et al.,57 we designed and characterized a series of [N, O]-type nickel complexes bearing Lewis base response moieties (Scheme 1). With these nickel complexes, homogeneous and heterogeneous ethylene polymerization and copolymerization with polar monomers were studied by introducing soluble and supported B/Al Lewis acid, respectively. It is hypothesized that the introduction of Lewis acid units can significantly decrease the electron cloud density of the Ni center and increase the steric hindrance, thus leading to the simultaneous improvement of catalytic activity and molecular weight of the generated polyolefins.
Results and discussion
Ligands and catalyst synthesis
Ligands L1–L2 were easily prepared in high yield from 2,3-butanedione and amine through a condensation reaction. Deprotonation of ligands with lithium diisopropylamide (LDA) in THF provides lithium salt after evaporating the solvent. Subsequent reaction of the salt with allylnickel chloride dimer in dichloromethane affords complexes Ni1 and Ni2 (Scheme 2), which were characterized by 1H, 13C NMR, H–H COSY spectra, and single crystal diffraction. In examining Ni2, for example, the 1H NMR spectrum shows the proton signals adjacent to benzene ring (–CHPh2) at 5.9 and 5.6 ppm and terminal olefinic protons at 4.7 and 4.3 ppm. The values at 5.3, 3.1, 1.9, 1.6, and 0.9 ppm were attributed to the allyl group coordinated to the nickel metal center. | ||
| Scheme 2 Design of Lewis-acid-induced homogeneous and heterogeneous nickel complexes utilizing soluble and supported Lewis acids. | ||
The H–H COSY spectrum also showed the correlation of corresponding protons. A single crystal of Ni1 was obtained from a solution of toluene and pentane, as shown in Fig. 1, and the length of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond adjacent to the oxygen atom was shorter than that of the C–C bond adjacent to the nitrogen atom (1.32 Å vs. 1.50 Å). To further investigate the truly catalytical active species, the coordination reaction of Ni2 with 2 eq. of B(C6F5)3 was conducted. The 1H NMR, 19F NMR, and H–H COSY spectra indicated a successful Lewis acid-induced isomerization process to generate the Ni2-BCF complex (see the ESI†).
C bond adjacent to the oxygen atom was shorter than that of the C–C bond adjacent to the nitrogen atom (1.32 Å vs. 1.50 Å). To further investigate the truly catalytical active species, the coordination reaction of Ni2 with 2 eq. of B(C6F5)3 was conducted. The 1H NMR, 19F NMR, and H–H COSY spectra indicated a successful Lewis acid-induced isomerization process to generate the Ni2-BCF complex (see the ESI†).
 | ||
| Fig. 1 The molecular structure of Ni1 (CCDC2295534†). Hydrogen atoms were omitted for clarity, and ellipsoids were set at 30% probability. Selected bond lengths (Å) and angles (°): Ni1–O1 1.893 (4), Ni1–N1 1.897 (5), Ni1–C7 2.007 (7), Ni1–C8 1.989 (8), Ni1–C14 1.998 (13); N1–Ni1–O1 85.2 (2), O1–Ni1–C7 99.5 (3), N1–Ni1–C8 101.2 (3), N1–Ni1–C8 101.2 (3), C7–Ni1–C8 74.2 (3). | ||
Homogeneous ethylene polymerization
For ethylene polymerization, we explored the catalytic performance of these two nickel catalysts at different temperatures (30 °C, 50 °C, and 80 °C). Nickel complexes Ni1 and Ni2 were not active without the addition of Lewis acid cocatalyst (Table 1, entries 1 and 2). Even after increasing the polymerization temperature to 80 °C or ethylene pressure to 20 atm, polyethylene generation using Ni1 and Ni2 remained elusive in our work. In contrast, after using the BCF cocatalyst, these two catalysts exhibited activity in ethylene polymerization. The strongest catalytic activity for Ni1 was at 50 °C, with polymerization activity at 3.2 × 105 g mol−1 h−1 (Table 1, entries 3–5). Catalyst Ni2 bearing larger steric hindrance (Table 1, entries 6–8) also showed similar trends, with the highest catalytic activity of 6.4 × 105 g mol−1 h−1 at 50 °C, (Table 1, entry 7).| Ent. | Cat. | Lewis acid | T (°C) | Yieldb (g) | Act.b (105) | M n (104) | PDIc | B | T m (°C) |
|---|---|---|---|---|---|---|---|---|---|
| a Polymerization conditions: Ni catalyst = 5 μmol in 2 mL CH2Cl2, heptane = 28 mL, ethylene = 8 atm, 0.5 h. b The yields and activities are an average of at least two repetitive cycles. The activity is in units of 105 g mol−1 h−1. c Determined by gel permeation chromatography (GPC) in trichlorobenzene at 150 °C with polystyrene standards. The molecular weight is in units of 104 g mol−1. d Determined by 1H NMR in C2D2Cl4 at 120 °C. e Determined by differential scanning calorimetry (DSC, second heating). f 10 eq. of B(C6F5)3 was added. g 100 eq. of MAO was added. | |||||||||
| 1 | Ni1 | — | 30 | 0 | — | — | — | — | — |
| 2 | Ni2 | — | 30 | 0 | — | — | — | — | — |
| 3f | Ni1 | BCF | 30 | 0.5 | 2.0 | 32.0 | 1.80 | 10 | 127.5 |
| 4f | Ni1 | BCF | 50 | 0.8 | 3.2 | 18.9 | 1.56 | 13 | 122.6 |
| 5f | Ni1 | BCF | 80 | 0.6 | 2.4 | 8.7 | 1.45 | 22 | 116.4 |
| 6f | Ni2 | BCF | 30 | 1.2 | 4.8 | 130.8 | 2.36 | 6 | 130.9 |
| 7f | Ni2 | BCF | 50 | 1.6 | 6.4 | 115.5 | 2.54 | 11 | 129.5 |
| 8f | Ni2 | BCF | 80 | 1.3 | 5.2 | 96.0 | 1.88 | 13 | 125.9 |
| 9g | Ni2 | MAO | 30 | 0.5 | 2.0 | 95.8 | 1.44 | 12 | 126.5 |
| 10g | Ni2 | MAO | 50 | 0.9 | 3.6 | 75.4 | 1.43 | 20 | 120.3 |
| 11g | Ni2 | MAO | 80 | 0.6 | 2.4 | 56.8 | 1.98 | 23 | 118.5 |
Molecular weight (Mn) and molecular weight distribution (PDI) of the prepared polyethylene at different temperatures were tested by high-temperature gel permeation chromatography (GPC). The results showed that the molecular weight of polyethylene decreased with the increase in temperature for the same catalyst. This was ascribed to the increased chain transfer rate in the ethylene polymerization process when the temperature increased. For these two different catalysts, the molecular weight of polyethylene prepared by the Ni2 catalyst with the larger steric hindrance at 30 °C was as high as 130.8 × 104 g mol−1 (Table 1, entry 6), and thus, it would be classified as an ultra-high molecular weight polyethylene (UHMWP). At 50 °C and 80 °C, the molecular weights of polyethylene generated by catalyst Ni2 were 115.5 × 104 g mol−1 and 96.0 × 104 g mol−1, which were 6 times and 10 times higher than those of the polyethylene prepared by Ni1, respectively (Table 1, entries 7 and 8).
In addition, the branching density of polyethylene (the number of branches per 1000 carbon atoms in the polyethylene chain) can be adjusted by using different catalysts and polymerization temperatures. A lower branching density of the prepared polyethylene using Ni2 was observed, with the generation of additional linear polyethylene with a high melting point (Tm up to 130.9 °C). The steric hindrance of a catalyst has an important influence on the molecular weight and branching density of polyethylene due to the decreased β-H elimination rate for Ni2, with a larger steric hindrance.
Excluding the study of borane BCF, different Lewis acid cocatalysts such as methylaluminoxane (MAO) were also introduced to interact with the nickel complex in this system (Table 1, entries 9–11). Compared to BCF, Ni2 exhibited decreased activity in ethylene polymerization with cocatalyst MAO. Additionally, the generated polyethylene exhibited a lower molecular weight and melting point, and higher branching density (Table 1, entries 9–11 vs. 6–8). As such, the selection of different Lewis acids in this work was advantageous because they effectively modulated the topological structures of the produced polyethylene, which provided versatile catalytic performances for olefin polymerization.
DFT calculation
To further understand the structural differences and influence of Lewis acid modulation, density functional theory (DFT) calculations were carried out to study the electronic nature and steric effects of these catalysts. As depicted in Fig. 2, catalytic species 1A–1C and 2A–2C were classified as three types, starting from the same α-ketone-imine ligand structure. 1A and 2A are previously reported cationic nickel catalytic species with the ability to mediate olefin polymerization with high activity.58 | ||
| Fig. 2 DFT studies for nickel complex species 1A–1C and 2A–2C: the numbers in parentheses are frontier-orbital electron densities on the Ni centers; Vbur is the calculated topographic steric map around the nickel catalytic center. | ||
The results of frontier-orbital electron densities (FED) on the Ni centers indicated that the electron cloud densities of the Ni centers of 1B and 2B were significantly higher than those of the other four Ni complex species. This may explain the near complete absence of catalytic activity for these two complexes, because a Ni center with a high electron cloud density may be not beneficial for the coordination and electrophilic activation of monomer. Moreover, steric maps were generated using the SambVca 2.1 A tool, which provides the quantified steric hindrance around the catalytic species. It was indicated that the use of BCF modulation resulted in significantly increased steric hindrance (Vbur value: 1C > 1B ≈ 1A; 2C > 2B ≈ 2A). For 1C and 2C, this may lead to the simultaneous inhibition of ethylene coordination and chain transfer, resulting in lower catalytic activities and higher polymer molecular weights compared with 1A and 2A, respectively.
Supported Lewis acid-induced heterogeneous ethylene polymerization
Extensive academic research efforts have focused on homogeneous nickel catalysts for olefin polymerization.15–18,37 However, heterogeneous catalysts have been dominating industrial polyolefin production because they offer many distinct advantages, such as controlling the polymer morphology and preventing reactor fouling.43–51 From this perspective, heterogenization of soluble catalysts on solid supports represents an attractive strategy to bridge these two fields. The Lewis acid-induced heterogeneous strategy using a Lewis acid-modified support provides an efficient approach for this field.Supported Lewis acids (BCF/SiO2 or MAO/SiO2) in this work were prepared through BCF- and MAO-modified SiO2. The supported catalysts were accessed by mixing nickel complex with Lewis acid-modified SiO2 in toluene, stirring for 3 h, and then washing the solid precipitate with toluene. The maximum catalyst-supporting capacity was approximately 5 μmol Ni1/Ni2 per 100 mg of SiO2. In the solid-state19 F MAS NMR spectrum, the chemical shifts of peaks (−135, −155, and −166 ppm) were characteristic of BCF/SiO2, and newly generated peaks (−123, −142, and −158 ppm) were assigned to Ni2-BCF/SiO2. This indicated that Ni2 was successfully immobilized on –SiOB(C6F5)2 by binding to the carbonyl group of the ligand (see the ESI†).
Compared with the homogeneous system, catalytic activity of the heterogeneous catalyst Ni2-BCF/SiO2 was significantly increased at 30 °C, 50 °C, and 80 °C (Table 2, entries 1–3 vs.Table 1, entries 6–8, Fig. 3a and b). Excellent catalytic performance was observed for Ni2-BCF/SiO2, with an increase in activity up to 1.28 × 106 g mol−1 h−1 (Table 2, entry 2). The prepared polyethylene was UHMWPE with an increased molecular weight of 205.3 × 104 g mol−1 and 183.8 × 104 g mol−1, and a lower branch density and higher Tm at 30 °C and 50 °C, respectively (Table 2, entries 1 and 2). At the elevated temperature of 80 °C, the heterogeneous catalyst Ni2-BCF/SiO2 continued to produce polyethylene with a high molecular weight of 120.8 × 104 g mol−1 (Table 2, entry 3), which enabled a great advantage for the industrial application of UHMWPE preparation.
 | ||
| Fig. 3 Comparisons of the homogeneous and heterogeneous ethylene polymerization systems using Ni2: (a) molecular weight comparisons of generated polyethylene at 30 °C and 50 °C; (b) branching degree comparisons of generated polyethylene at 30 °C and 50 °C; (c and d) time-dependent studies (polymer yields vs. polymerization time) at 80 °C. | ||
| Ent. | Cat. | Supported Lewis acid | T (°C) | Yieldb (g) | Act.b (105) | M n (104) | PDIc | B | T m (°C) |
|---|---|---|---|---|---|---|---|---|---|
| a Polymerization conditions: Ni catalyst = 5 μmol, heptane = 30 mL, ethylene = 8 atm, 0.5 h. b The yields and activities are an average of at least two repetitive cycles. The activity is in units of 105 g mol−1 h−1. c Determined by gel permeation chromatography (GPC) in trichlorobenzene at 150 °C with polystyrene standards. The molecular weight is in units of 104 g mol−1. d Determined by 1H NMR in C2D2Cl4 at 120 °C. e Determined by differential scanning calorimetry (DSC, second heating). | |||||||||
| 1 | Ni2 | BCF/SiO2 | 30 | 1.9 | 7.6 | 205.3 | 1.75 | 3 | 134.0 |
| 2 | Ni2 | BCF/SiO2 | 50 | 3.2 | 12.8 | 183.8 | 2.28 | 4 | 131.9 |
| 3 | Ni2 | BCF/SiO2 | 80 | 2.1 | 8.4 | 120.8 | 2.21 | 11 | 129.2 |
| 4 | Ni2 | MAO/SiO2 | 30 | 1.0 | 4.0 | 132.5 | 2.37 | 6 | 130.1 |
| 5 | Ni2 | MAO/SiO2 | 50 | 1.2 | 4.8 | 121.8 | 1.51 | 14 | 127.5 |
| 6 | Ni2 | MAO/SiO2 | 80 | 0.7 | 2.8 | 85.5 | 1.64 | 15 | 123.7 |
Similarly, the Ni2-MAO/SiO2 system exhibited increased activity for ethylene polymerization compared to the homogeneous system, producing polyethylene with higher molecular weight and lower branching density (Table 2, entries 4–6 vs.Table 1, entries 9–11; Fig. 3a and b). To investigate the thermal stability of homogeneous and heterogeneous systems, time-dependent studies at 80 °C were performed. As depicted in Fig. 3, the heterogeneous catalysts Ni2-BCF/SiO2 and Ni2-MAO/SiO2 continued their ethylene polymerization activity within 60 minutes, while the homogeneous Ni2 catalyst lost nearly all activity within 30 minutes (Fig. 3c and d). The striking behavior difference between the homogeneous and heterogeneous nickel systems can be explained by the increased steric environment around the nickel center through the heterogenization step, which inhibited the chain transfer rate during the polymerization process.
Excluding the study of catalytic performance by nickel catalysts, polymer morphology control is also important for industrial polymerization procedures. Heterogeneous catalysts usually demonstrate distinct advantages compared with homogeneous systems. In our work, the homogeneous catalyst produced sticky polyethylene on the polymerization reactor (Fig. 4a and c). In contrast, the corresponding heterogeneous nickel complex generated free-flowing polyethylene with excellent morphology control at 50 °C (Fig. 4b and d), and no reactor fouling due to the leaching of catalyst into solution. This advantage offers the possibility to conduct a continuous polymerization process for industrial applications.
 | ||
| Fig. 4 (a) Polyethylene sample prepared from Ni2-BCF at 50 °C in solution. (b) Polyethylene sample prepared from Ni2-BCF/SiO2 at 50 °C in solution. (c) Polyethylene sample accessed from Ni2-BCF at 50 °C after drying. (d) Polyethylene sample prepared from Ni2-BCF/SiO2 at 50 °C after drying. (e) Stress–strain curves of polyethylene samples obtained using Ni2-BCF and Ni2-BCF/SiO2 at 50 °C. (f) Stress–strain curves of polyethylene samples obtained using Ni2-MAO and Ni2-MAO/SiO2 at 50 °C. | ||
Tensile strength analysis was performed for the prepared polyethylene samples. As Fig. 4e shows, the generated polyethylene possesses enhanced mechanical properties utilizing the Ni2-BCF/SiO2 supported catalyst compared to its homogeneous counterpart (Fig. 4e, tensile strength 36.6 vs. 18.9 MPa; elongation at break 542% vs. 415%). A similar trend was also observed for the supported Ni2-MAO/SiO2 and homogeneous systems (Fig. 4f, tensile strength 26.1 vs. 14.0 MPa; elongation at break 553% vs. 407%). It should be noted that the mechanical properties of the polyethylene produced by the heterogenization system may be the result of the combined performance of polyethylene with the left SiO2 support.
Ethylene-polar monomer copolymerization with Lewis acid-induced homogeneous and supported nickel catalysts
As the largest class of thermoplastic polymers, polyolefin materials have wide applications and a huge annual production. The introduction of even a small amount of polar functional groups into polyolefins could result in great control over important material properties. As the most direct and economic strategy, the coordination–insertion copolymerization of olefin with polar-functionalized monomers can enable molecular level control of the copolymer microstructures, which is one of the greatest challenges in this field. Thus, the copolymerization of ethylene and polar monomers (methyl 10-undecenoate and 6-chlorohex-1-ene) was investigated at 50 °C in this work.For the homogeneous system with BCF or MAO as the cocatalyst, Ni2 was active in the copolymerization of ethylene with methyl 10-undecenoate, generating copolymer with moderated molecular weight and comonomer incorporation ratio (Table 3, entries 1 and 3, respectively). In contrast, there was higher activity of the heterogeneous Ni2-BCF/SiO2 and Ni2-MAO/SiO2 systems as compared to the soluble system, and copolymer was generated with higher molecular weight, up to 22.8 × 104 g mol−1 and 14.1 × 104 g mol−1, respectively (Table 3, entries 2 and 4, respectively).
| Ent. | Cat. | Lewis acid | Co-monomer | Yieldb (g) | Act.b (104) | M n (104) | PDIc | X M (%) | T m (°C) |
|---|---|---|---|---|---|---|---|---|---|
| a Polymerization conditions: catalyst = 10 μmol, 50 °C, total volume of heptane and polar monomer (0.5 M L−1) = 18 mL, time = 1 h, 8.0 atm of ethylene. Lewis acid = 10 eq. of B(C6F5)3 or 100 eq. of MAO. b The activity is in units of 104 g mol−1 h−1. c Determined by gel permeation chromatography (GPC) in trichlorobenzene at 150 °C with polystyrene standards. The molecular weight is in units of 104 g mol−1. d Calculated by 1H NMR spectroscopy. e Determined by differential scanning calorimetry (DSC, second heating). | |||||||||
| 1 | Ni2 | BCF |

|
0.2 | 2.0 | 11.6 | 1.87 | 0.6 | 118.0 |
| 2 | Ni2 | BCF/SiO2 |

|
0.5 | 5.0 | 22.8 | 2.06 | 0.2 | 123.5 |
| 3 | Ni2 | MAO |
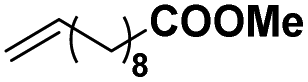
|
0.1 | 1.0 | 6.4 | 2.17 | 0.5 | 116.7 |
| 4 | Ni2 | MAO/SiO2 |

|
0.4 | 4.0 | 14.1 | 1.96 | 0.3 | 118.2 |
| 5 | Ni2 | BCF |

|
0.5 | 5.0 | 14.7 | 2.95 | 0.5 | 122.1 |
| 6 | Ni2 | BCF/SiO2 |

|
0.8 | 8.0 | 29.5 | 1.70 | 0.3 | 126.0 |
| 7 | Ni2 | MAO |

|
0.4 | 4.0 | 6.9 | 2.20 | 0.4 | 121.1 |
| 8 | Ni2 | MAO/SiO2 |

|
0.6 | 6.0 | 18.9 | 1.56 | 0.2 | 124.0 |
A similar trend was observed for the copolymerization of ethylene with 6-chlorohex-1-ene (Table 3, entries 5–8), affording copolymer with high molecular weight (29.5 × 104 g mol−1). The copolymers showed a slightly decreased comonomer incorporation ratio produced by the heterogenization system, which may be ascribed to the increased steric hindrance around the nickel catalytic center through the heterogenization process, which subsequently decreased the likelihood for the occurrence of polar monomer insertion (Table 3, entries 2, 4, 6, and 8). The heterogeneous strategy in this work provides a general method for additional Lewis acid-induced catalytic systems in the olefin copolymerization process.
Conclusions
We designed and characterized [N, O] nickel catalysts with different amounts of steric hindrance. These complexes mediated binding to Lewis acids such as B(C6F5)3 and MAO. The catalytic behaviors of nickel catalysts in homogeneous ethylene polymerization can be tuned by the addition of Lewis acids. To extend soluble Lewis acids to their supported counterparts, heterogeneous ethylene polymerization systems (Ni2-BCF/SiO2 and Ni2-MAO/SiO2) were designed and investigated. Compared to its homogeneous counterparts, the immobilized nickel catalyst behaved with high activity and thermal stability during ethylene polymerization, and was able to produce UHMWPE with excellent morphology control (molecular weight up to 205.3 × 104 g mol−1). Moreover, these immobilized nickel systems also promoted the copolymerization of ethylene with polar monomers, generating copolymer with high molecular weight and high activity. The ease of the Lewis acid-induced olefin (co)polymerization in this work demonstrates exciting opportunities for the synthesis of high-performance polyolefin materials and may inspire additional applications in other metal catalysis fields.Author contributions
Min Chen conceived the idea and designed the experiments. Wanlu Tian conducted the experiments and analysed the data. Chen Tan performed the DFT calculations. Min Chen and Chao Li wrote the manuscript together. Min Chen, Chao Li, and Chen Tan acquired the financial funding support for this project.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (NSFC, 21971230, U19B6001, 52373002, 22201003), Natural Science Foundation of Anhui Province (2308085Y35, 2023AH030002), and Hefei Natural Science Foundation (202304). We also thank the Anhui Province Key Laboratory of Environment-friendly Polymer Materials, Excellent Research and Innovation Team Project of Anhui Province (2022AH010001). We are also grateful for the mentorship and strong support from Professor Changle Chen (USTC).References
- M. Stürzel, S. Mihan and R. Mülhaupt, Chem. Rev., 2016, 116, 1398–1433 CrossRef PubMed.
- C. Tan and C. L. Chen, Angew. Chem., Int. Ed., 2019, 58, 7192–7200 CrossRef CAS PubMed.
- C. L. Chen, Nat. Rev. Chem., 2018, 2, 6–14 CrossRef CAS.
- H. B. Wang, Y. Yang, M. Nishiura, Y. Higaki, A. Takahara and Z. Hou, J. Am. Chem. Soc., 2019, 141, 3249–3257 CrossRef CAS PubMed.
- Y. Jiang, Z. Zhang, S. H. Li and D. M. Cui, Angew. Chem., Int. Ed., 2022, 61, e202112966 CrossRef CAS PubMed.
- Y. Wu, T. H. Nan, X. L. Ji, B. Liu and D. M. Cui, Angew. Chem., Int. Ed., 2022, 61, e202205894 Search PubMed.
- Y. X. Zhang, Y. X. Zhang, X. Q. Hu, C. Q. Wang and Z. B. Jian, ACS Catal., 2022, 12, 14304–14320 CrossRef CAS.
- L. K. Johnson, C. M. Killian and M. Brookhart, J. Am. Chem. Soc., 1995, 117, 6414–6415 CrossRef CAS.
- L. K. Johnson, S. Mecking and M. Brookhart, J. Am. Chem. Soc., 1996, 118, 267–268 CrossRef CAS.
- C. Tan, C. Zou and C. L. Chen, Macromolecules, 2022, 55, 1910–1922 CrossRef CAS.
- G. L. Zhou, L. Cui, H. L. Mu and Z. B. Jian, Polym. Chem., 2021, 12, 3878–3892 RSC.
- T. T. Wang, C. J. Wu, X. L. Ji and D. M. Cui, Angew. Chem., Int. Ed., 2021, 60, 25735–25740 Search PubMed.
- H. B. Wang, Y. Yang, M. Nishiura, Y. Higaki, A. Takahara and Z. M. Hou, J. Am. Chem. Soc., 2019, 141, 3249–3257 Search PubMed.
- H. B. Wang, Y. A. Zhao, M. Nishiura, Y. Yang, G. Luo, Y. Luo and Z. M. Hou, J. Am. Chem. Soc., 2019, 141, 12624–12633 CrossRef CAS PubMed.
- H. L. Mu, L. Pan, D. P. Song and Y. S. Li, Chem. Rev., 2015, 115, 12091–12137 CrossRef CAS PubMed.
- H. L. Mu, G. L. Zhou, X. Q. Hu and Z. B. Jian, Coord. Chem. Rev., 2021, 435, 213802 CrossRef CAS.
- Z. Chen and M. Brookhart, Acc. Chem. Res., 2018, 51, 1831–1839 CrossRef CAS PubMed.
- C. Tan, M. Chen and C. L. Chen, Trends Chem., 2023, 5, 147–159 Search PubMed.
- J. L. Rhinehart, L. A. Brown and B. K. Long, J. Am. Chem. Soc., 2013, 135, 16316–16319 CrossRef CAS PubMed.
- H. Zhang, C. Zou, H. P. Zhao, Z. G. Cai and C. L. Chen, Angew. Chem., Int. Ed., 2021, 60, 17446–17451 Search PubMed.
- G. W. K. Moore, S. E. L. Howell, M. Brady and X. Xu, Nat. Commun., 2021, 12, 1 CrossRef CAS PubMed.
- F. Lin and S. Mecking, Angew. Chem., Int. Ed., 2022, 61, e202203923 CrossRef CAS PubMed.
- B. S. Xin, N. Sato, A. Tanna, Y. Oishi, Y. Konishi and F. Shimizu, J. Am. Chem. Soc., 2017, 139, 3611–3614 CrossRef CAS PubMed.
- C. Tan, C. Zou and C. L. Chen, J. Am. Chem. Soc., 2022, 144, 2245–2254 CrossRef CAS PubMed.
- T. Vaidya, K. Klimovica, A. M. LaPointe, I. Keresztes, E. B. Lobkovsky, O. Daugulis and G. W. Coates, J. Am. Chem. Soc., 2014, 136, 7213–7216 CrossRef CAS PubMed.
- Z. Chen, M. D. Leatherman, O. Daugulis and M. Brookhart, J. Am. Chem. Soc., 2017, 139, 16013–16022 CrossRef CAS PubMed.
- D. Meinhard, M. Wegner, G. Kipiani, A. Hearley, P. Reuter, S. Fischer, O. Marti and B. Rieger, J. Am. Chem. Soc., 2007, 129, 9182–9191 CrossRef CAS PubMed.
- W. B. Du, H. D. Zheng, Y. W. Li, C. S. Cheung, D. H. Li, H. Gao, H. Y. Deng and H. Y. Gao, Macromolecules, 2022, 55, 3096–3105 CrossRef CAS.
- Y. P. Zhang, H. L. Mu, L. Pan, L. X. Wang and Y. S. Li, ACS Catal., 2018, 8, 5963–5976 Search PubMed.
- M. Baur, F. Lin, T. O. Morgen, L. Odenwald and S. Mecking, Science, 2021, 374, 604–607 Search PubMed.
- S. Xiong, M. M. Shoshani, X. Zhang, H. A. Spinney, A. J. Nett, B. S. Henderson, T. F. Miller and T. Agapie, J. Am. Chem. Soc., 2021, 143, 6516–6527 CrossRef CAS PubMed.
- X. L. Wang, Y. P. Zhang, F. Wang, L. Pan, B. Wang and Y. S. Li, ACS Catal., 2021, 11, 2902–2911 Search PubMed.
- C. L. Chen, ACS Catal., 2018, 8, 5506–5514 CrossRef CAS.
- J. S. Yang, X. Q. Hu and Z. B. Jian, Chin. J. Chem., 2022, 40, 2919–2926 Search PubMed.
- M. Chen, B. P. Yang and C. L. Chen, Angew. Chem., Int. Ed., 2015, 54, 15520–15524 CrossRef CAS PubMed.
- A. Maity and T. S. Teets, Chem. Rev., 2016, 116, 8873–8911 CrossRef CAS PubMed.
- T. V. Tran and L. H. Do, Eur. Polym. J., 2021, 142, 110100 CrossRef CAS.
- M. A. Escobar, T. O. Srofymchuk, B. E. Rodriguez, C. Lopez-Lira, R. Tapia, C. Daniliuc, H. Berke, F. M. Nachtigall, L. S. Santos and R. S. Rojas, ACS Catal., 2015, 5, 7338–7342 CrossRef CAS.
- Z. J. A. Komon, X. Bu and G. C. Bazan, J. Am. Chem. Soc., 2000, 122, 12379–12380 CrossRef CAS.
- B. M. Boardman and G. C. Bazan, Acc. Chem. Res., 2009, 42, 1597–1606 Search PubMed.
- B. Y. Lee, X. H. Bu and G. C. Bazan, Organometallics, 2001, 20, 5425–5431 CrossRef CAS.
- M. Chen, W. P. Zou;, Z. G. Cai and C. L. Chen, Polym. Chem., 2015, 6, 2669–2676 RSC.
- C. Zou, C. Tan and C. L. Chen, Acc. Mater. Res., 2023, 4, 496–506 Search PubMed.
- J. D. Pelletier and J. M. Basset, Acc. Chem. Res., 2016, 49, 664–677 CrossRef CAS PubMed.
- R. J. Witzke, A. Chapovetsky, M. P. Conley, D. M. Kaphan and M. Delferro, ACS Catal., 2020, 10, 11822–11840 CrossRef CAS.
- J. R. Severn, J. C. Chadwick, R. Duchateau and N. Friederichs, Chem. Rev., 2005, 105, 4073–4147 CrossRef CAS PubMed.
- J. Li, Y. Wang, W. Cai, G. Yang, Q. H. Tian, Y. S. Huang, D. Peng, C. Zou and C. Tan, Macromolecules, 2023, 56, 3015–3023 CrossRef CAS.
- M. M. Stalzer, M. Delferro and T. J. Marks, Catal. Lett., 2015, 145, 3–14 CrossRef CAS.
- M. H. Ji, G. F. Si, Y. Pan, C. Tan and M. Chen, J. Catal., 2022, 415, 51–57 CrossRef CAS.
- C. Zou, Q. Wang, G. F. Si and C. L. Chen, Nat. Commun., 2023, 14, 1442 CrossRef CAS PubMed.
- D. Peng, M. H. Xu, C. Tan and C. L. Chen, Macromolecules, 2023, 56, 2388–2396 CrossRef CAS.
- S. A. Correa, C. G. Daniliuc, H. S. Stark and R. S. Rojas, Organometallics, 2019, 38, 3327–3337 CrossRef CAS.
- Y. J. Wanglee, J. Hu, R. E. White, M. Y. Lee, S. M. Stewart, P. Perrotin and S. L. Scott, J. Am. Chem. Soc., 2012, 134, 355–366 CrossRef CAS PubMed.
- S. L. Scott, B. C. Peoples, C. Yung, R. S. Rojas, V. Khanna, H. Sano, T. Suzuki and F. Shimizu, Chem. Commun., 2008, 35, 4186–4188 Search PubMed.
- X. Fu, L. J. Zhang, R. Tanaka, T. Shiono and Z. G. Cai, Macromolecules, 2017, 50, 9216–9221 CrossRef CAS.
- H. Zhang, Z. Y. Zhang, Z. G. Cai, M. Y. Li and Z. Liu, ACS Catal., 2022, 12, 9646–9654 CrossRef CAS.
- Y. F. Chen, B. M. Boardman, G. Wu and G. C. Bazan, J. Organomet. Chem., 2007, 692, 4745–4749 CrossRef CAS.
- T. Liang, S. B. Goudari and C. L. Chen, Nat. Commun., 2020, 11, 372 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2295534. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3py01266b |
| This journal is © The Royal Society of Chemistry 2024 |