
BODIPY directed one-dimensional self-assembly of gold nanorods†
Hemant
 ab,
Atikur
Rahman
a,
Priyanka
Sharma
a,
Asifkhan
Shanavas
a and
Prakash P.
Neelakandan
*ab
ab,
Atikur
Rahman
a,
Priyanka
Sharma
a,
Asifkhan
Shanavas
a and
Prakash P.
Neelakandan
*ab
aInstitute of Nano Science and Technology, Knowledge City, Sector 81, Mohali 140306, India. E-mail: ppn@inst.ac.in
bAcademy of Scientific and Innovative Research (AcSIR), Ghaziabad 201002, India
First published on 29th May 2024
Abstract
The assembly of anisotropic nanomaterials into ordered structures is challenging. Nevertheless, such self-assembled systems are known to have novel physicochemical properties and the presence of a chromophore within the nanoparticle ensemble can enhance the optical properties through plasmon–molecule electronic coupling. Here, we report the end-to-end assembly of gold nanorods into micrometer-long chains using a linear diamino BODIPY derivative. The preferential binding affinity of the amino group and the steric bulkiness of BODIPY directed the longitudinal assembly of gold nanorods. As a result of the linear assembly, the BODIPY chromophores positioned themselves in the plasmonic hotspots, which resulted in efficient plasmon–molecule coupling, thereby imparting photothermal properties to the assembled nanorods. This work thus demonstrates a new approach for the linear assembly of gold nanorods resulting in a plasmon–molecule coupled system, and the synergy between self-assembly and electronic coupling resulted in an efficient system having potential biomedical applications.
Introduction
Self-assembled ordered structures of metal nanospecies, particularly those of gold nanomaterials, have attracted tremendous attention in various research fields.1–8 Nanomaterial assemblies hold significant appeal for fundamental scientific exploration and practical applications, as they facilitate the direct connection between minute nanoscale entities and the broader macroscopic world. These organized nanostructures have immense potential in therapeutic sensors,9–11 catalysis,12–14 and optoelectronic device applications including photonic wave-guiding15 and optical switching.16 The characteristics of these nanoparticle assemblies are intricately intertwined not only with the size and geometry of the constituent nanoparticles but also with the spatial organization of these building blocks. As a result, the field of nanoscience is on a quest to develop innovative methods for assembling nanomaterials into functional devices.17 Besides applications, such investigations aid in understanding the fundamental concepts of self-assembly. Further, electronic coupling between the plasmons of adjacent nanoparticles results in a strong enhancement of the local electric field in the gap between nanoparticle assemblies, known as plasmonic hotspots. This enhancement in turn enables the amplification of phenomena such as Raman scattering, multiphoton excitation, and non-linear spectroscopies of the adsorbed molecules.18–20Gold nanorods are extensively studied on account of their shape anisotropy, controllable aspect ratio, and unique optical properties in both visible and near-infrared regions.21–23 The anisotropic shape allows nanorods to be assembled via two orientational modes: “side-by-side” and “end-to-end”. End-to-end assembly of gold nanorods has been accomplished through different protocols such as usage of bridging motifs,24,25 change in the physical properties of the medium,26–29 coordination bond formation,30,31 non-covalent interactions,32–37 and protein–ligand interactions.38,39 These techniques typically require extensive nanoparticle functionalization, employ non-aqueous media, and are associated with high expenses.34,36,38 Moreover, one must be cautious as uncontrolled aggregation is often observed during the self-assembly and such systems have limited practical applications.
As metal nanoparticles are placed in close proximity, their plasmonic resonances couple with each other resulting in significant changes in their optical properties.40–43 The shape, size, and inter-particle gap are the decisive factors determining the coupling and the resultant variation in the optical properties of the plasmon-coupled systems. Furthermore, integrating organic chromophores with metal nanomaterials can lead to functional nanomaterials with versatile properties.44,45 The chromophores can influence the physicochemical properties of nanomaterials through plasmon–molecule electronic coupling wherein the plasmonic resonance degenerates with molecular resonance. In plasmon–molecule coupled systems, the properties of the nanomaterials and the chromophores are mutually modified, resulting in new materials with unique characteristics distinct from the nanoparticle or the chromophore.46,47
Herein, we demonstrate a hitherto unknown approach to assembling gold nanorods into end-to-end linked, micrometer-long chains using a rigid, linear organic molecule belonging to the BODIPY family. The linear chains were observed to be robust under a variety of conditions. Our results show that the preferential binding affinity of the amino group to the ends of gold nanorods aided the assembly, and the bulkiness of the organic ligand and the electrostatic repulsion between the positively charged surfactants on the lateral surface were decisive in driving the assembly in one dimension. Further, it was observed that the longitudinal plasmon band of gold nanorods coupled with the electronic transitions in BODIPY thereby imparting efficient photothermal properties to the assembled nanorods. Thus, by employing a linear chromophore having complementary optical properties of gold nanorods, we demonstrate a novel methodology for the self-assembly of gold nanorods. The uniqueness of this approach is the synergy between self-assembly process and the ensuing electronic interaction which promoted each other leading to the remarkable photophysical properties that were absent in the individual components.
Results and discussion
Self-assembly of gold nanorods
Cetyltrimethylammonium bromide (CTAB) protected gold nanorods (AuNRs) were synthesized using a seed-mediated growth method28,48 and characterized using high-resolution transmission electron microscopy (HR-TEM) which revealed dimensions of 9.8 × 42.5 nm with an aspect ratio of 4.3 ± 0.5 (Fig. S1, ESI†). The UV-Vis absorption spectrum of AuNRs showed two peaks at 514 and 679 nm, corresponding to the transverse and longitudinal localized surface plasmon resonance bands, respectively. It is well-known that surfactants have a strong affinity towards the {100} and {110} crystal planes on the lateral sides of the gold nanorods.25 As {111} facets have the closest packed structures of gold atoms and low surface energy compared to the lateral sides, the ends are more active for binding and conjugating external moieties.39 It is also known that the amino group has a high affinity to gold nanostructures through soft–soft interactions or dative covalent bond formation.49 On the basis of these facts, we hypothesized that a rigid, linear molecule functionalized with amino groups on both ends would serve as a bridge aiding the linear assembly of gold nanorods (Fig. 1a). To test this hypothesis, we synthesized the BODIPY molecules 1–3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 50–52 (Fig. 1b) and studied their interaction with AuNRs. As the electronic states of the BODIPY chromophore are known to strongly couple with the LSPR of gold nanoparticles,53–55 apart from inducing a linear assembly of AuNRs, the diaminoBODIPYs could be expected to couple electronically with AuNRs thereby leading to unique photophysical properties. The absorption spectra of BODIPY molecules 1–3, their aggregates and AuNRs are shown in Fig. S2.† The overlap between the longitudinal surface plasmon resonance band of AuNRs and the absorption of 1 indicates the plausibility of plasmon–molecule coupling between them.
50–52 (Fig. 1b) and studied their interaction with AuNRs. As the electronic states of the BODIPY chromophore are known to strongly couple with the LSPR of gold nanoparticles,53–55 apart from inducing a linear assembly of AuNRs, the diaminoBODIPYs could be expected to couple electronically with AuNRs thereby leading to unique photophysical properties. The absorption spectra of BODIPY molecules 1–3, their aggregates and AuNRs are shown in Fig. S2.† The overlap between the longitudinal surface plasmon resonance band of AuNRs and the absorption of 1 indicates the plausibility of plasmon–molecule coupling between them.
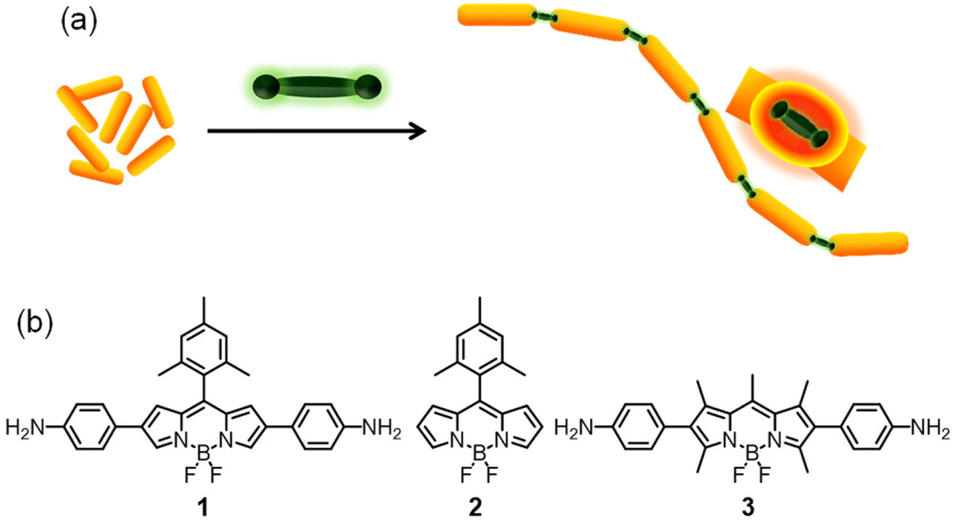 | ||
| Fig. 1 (a) Schematic representation of the BODIPY-induced end-to-end assembly of gold nanorods (AuNRs) and (b) the chemical structure of the BODIPY molecules used in the study. | ||
To study the self-assembly of AuNRs in the presence of molecules 1–3, AuNRs were suspended in water and treated with 1–3. As shown in Fig. 2a, the consecutive addition of 1 in acetone to a solution of AuNRs in water resulted in a gradual red shift in the UV-Vis absorption peak at 710 nm corresponding to the longitudinal plasmon band of AuNRs. Moreover, adding 1 led to a decrease in the absorbance and broadening of the peak. Upon adding 10 μM of 1, we observed a red shift of ∼50 nm along with an 11% decrease in absorption maximum. On the other hand, adding 2 or 3 resulted in minor changes in the absorbance of AuNRs (Fig. 2b and c). Although an increase in absorbance in the region of the transverse plasmon resonance band was observed upon adding 2, this has been attributed to the absorption of 2.
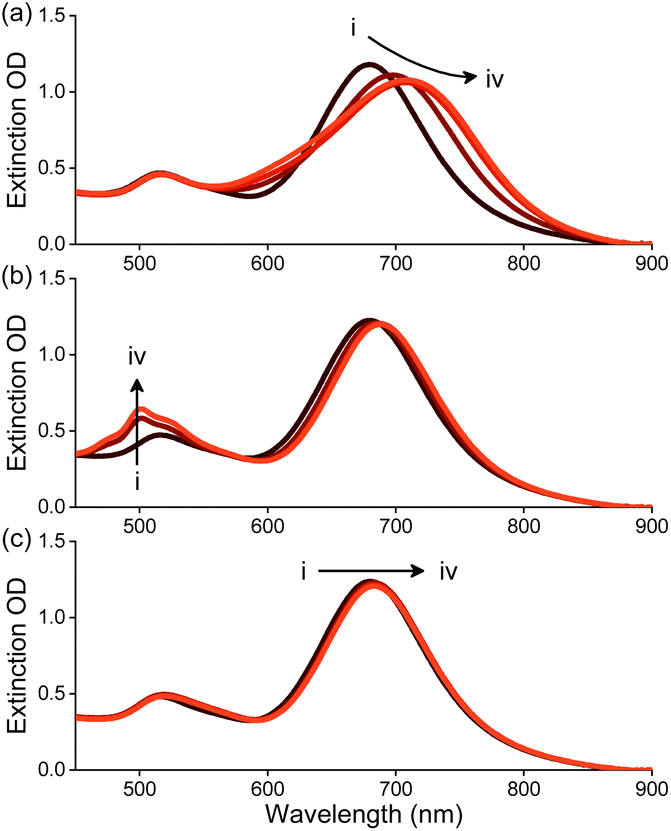 | ||
| Fig. 2 Changes in the extinction spectrum of AuNRs (20 μg mL−1) with the addition of (a–c) 1–3, respectively in water. [1–3], (i–iv) 0–10 μM, respectively. | ||
The red shift of the longitudinal band of AuNRs in the presence of 1 has been attributed to an end-to-end assembly of AuNRs. This is in accordance with prior reports wherein bathochromic shifts were observed for assembled nanorods through plasmon–plasmon coupling.56 The linear assembly of AuNRs was then visualized using high-resolution transmission electron microscopy (HR-TEM), which showed the formation of micrometer-long chains of end-to-end assembled AuNRs in the presence of 1 (Fig. 3a–c). On the other hand, in the presence of 2 and 3, HR-TEM revealed random and lateral aggregates of AuNRs, respectively (Fig. 3d–f). It is inferred that the amine groups in BODIPY preferentially bind to the terminal ends of AuNRs as compared to the lateral sides, thereby inducing the end-to-end assembly.
 | ||
| Fig. 3 High-resolution transmission electron micrographs of the (a–c) end-to-end, (d) random and (e and f) lateral aggregates of AuNRs induced by (a–c) 1, (d) 2 and (e and f) 3. | ||
The gap between two nanorods in the end-to-end assembled AuNRs was measured as ∼2.6 nm (Fig. S3†), which matches the sum of the theoretical length of 1 and two amine–gold bonds.57 Subsequently, the atomic composition within the gap of the end-to-end assembled AuNRs was determined through EDX analysis. While we observed a notable decrease in the gold composition, all elements present in molecule 1 were detected within the gap (Fig. S4 and S5†). These experiments, thus, establised the presence of 1 within the gap. Further, we estimated the number of molecules present in the gap between two nanorods using the dimensions of the gold nanorods and the BODIPY molecule. Our calculations showed that approximately 196 BODIPY molecules were present at each junction between two AuNRs.
Mechanism of self-assembly
The driving force for the linear self-assembly of AuNRs could be one or a combination of the following: (i) the preferential binding affinity of the amino groups to the terminal ends of the nanorods, or (ii) the steric features of the BODIPYs that favours their binding at the terminal or lateral ends of the nanorods, or (iii) plasmon–molecule interactions between BODIPY and gold. It was intriguing that among the two diamino BODIPYs (1 and 3), only 1 induced the linear self-assembly whereas adding 3 resulted in lateral assembly. To understand the difference, we looked into the geometry of these compounds. It has been reported that sterically bulky surfactants induce linear assembly of gold nanorods.33 On a similar note, we assume that molecule 1 having the sterically bulky mesityl group at the meso-position of the BODIPY preferentially binds to the terminal ends of AuNRs resulting in the end-to-end assembly. On the other hand, molecule 3, which is devoid of steric hindrance, binds to the lateral surface of the AuNRs wherein they could be stabilized through π–π stacking interactions.Next, we studied the interaction between AuNRs and the protonated form of molecule 1 (denoted as 1H). It is well established that surfactants and protonated amines having cationic groups bind to the gold surfaces through electrostatic interactions between the cationic groups and the anionic sites on the gold surface.58,59 When a solution of 1H was added to an aqueous solution of AuNRs, we observed a bathochromic shift of 30 nm in the longitudinal LSPR peak of AuNRs (Fig. S6†). Further, TEM analysis of a mixture of AuNRs and 1H revealed linear chains of end-to-end assembled AuNRs (Fig. S7†). These observations were in tune with literature reports of binding interactions between cationic groups and gold and thus, we infer that the protonated BODIPY 1H is capable of binding to gold nanorods and induce the assembly of AuNRs. We further observed that the fluorescence of 1H was completely quenched in the presence of AuNRs (Fig. S8†). The quenching of fluorescence of a chromophore in the proximity of a metal surface often serves as a primary indicator of plasmon–molecule coupling through surface plasmon-induced resonance energy transfer.45,54 On the basis of these experiments, it was concluded that the rigid, linear and sterically bulky geometry of 1 was crucial for the linear self-assembly of AuNRs. Further, the placement of the BODIPY chromophore in the gap between nanorods resulted in plasmon–molecule coupling which, as discussed in the later sections, has a significant role in deciding the photophysical properties of the self-assembled AuNRs.
Stability of self-assembled nanorods
Owing to the plasmonic interactions and the presence of BODIPY, the end-to-end self-assembled AuNRs exhibited absorption in the NIR region. However, the stability of the end-to-end assembled AuNRs could be detrimental to their applications. Thus, we evaluated the stability of the end-to-end assembled AuNRs using a competitive ligand. Thiol groups are known to have greater affinity towards gold surfaces and 1,6-hexane-dithiol (HDT) has been reported to induce longitudinal assembly of gold nanorods.25 We thus titrated a solution of HDT in acetone with the end-to-end assembled AuNRs. As shown in Fig. S9,† no significant changes were observed in the longitudinal LSPR peak of the end-to-end assembled AuNRs, thereby indicating the stability of the linear AuNRs arrays. Further, TEM showed that the end-to-end assembled AuNRs remained intact in the presence of HDT (Fig. S10†), thereby highlighting the strong interactions between 1 and AuNRs. This experiment further proved that the hydrophobic interactions between the linear aromatic molecules of 1 played a key role in stabilizing the end-to-end assembled AuNRs as compared to the HDT molecules having a higher affinity thiol group.We also studied the stability of the self-assembled AuNRs in the presence of an external stimulus such as HCl. It was observed that adding HCl to a solution of the end-to-end assembled AuNRs resulted in a minor blue shift in the longitudinal LSPR peak maximum (Δλ, 10 nm) along with the formation of a new peak at ∼580 nm (Fig. S11†). Further, TEM analysis showed that the end-to-end assembled AuNRs remained fairly undisturbed after adding HCl. It is known that the removal of capping agents from the gold surface can result in changes in the absorption maximum owing to the alterations in the refractive index of the medium surrounding the gold surface.60 We infer that the addition of HCl resulted in the protonation of CTAB which led to their displacement from the surface of gold thereby leading to a blue shift in the LSPR peak maximum. This inference was supported by TEM images, which revealed an aggregated mass in the background which could be attributed to the displaced CTAB (Fig. S12†). It is thus inferred that the end-to-end assembly of AuNRs is a stable process and the assembled nanorods showed remarkable stability even in the presence of a competitive ligand and external stimuli. Further, the new peak at ∼580 nm could be assigned to the formation of protonated BODIPY 1H upon the addition of HCl (Fig. S13†).
The close proximity of gold nanorods generates a strong electromagnetic field in the gap between the nanorods, commonly referred to as plasmonic hot spots. The presence of a chromophore in the hotspots results in a significant enhancement in the intensity of the Raman scattering of the chromophore and this phenomenon, known as the surface-enhanced Raman scattering (SERS), has emerged as a highly sensitive methodology in sensing.61 We performed Raman scattering measurements of 1 in the absence and presence of AuNRs (Fig. 4) as a confirmation for the placement of 1 in the hotspots generated as a result of the end-to-end assembly of AuNRs. The Raman scattering profile for molecule 1 at a concentration of 1 mM exhibited peaks at 940, 1580, and 1620 cm−1 corresponding to the symmetric C–C (C–CH3), aromatic C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C and C–N (C–NH2) stretching, respectively.62 However, at a concentration of 10 μM, which was employed for the self-assembly of AuNRs, we did not observe any Raman signal for molecule 1. Next, we recorded the Raman scattering of the end-to-end assembled AuNRs (containing molecule 1 (10 μM) and AuNRs) wherein peaks were observed at 940, 1580 and 1620 cm−1 thereby substantiating the presence of molecule 1 in the hot spots generated as a result of the end-to-end assembly of AuNRs. Further, the SERS enhancement factor of molecule 1 in the presence of AuNRs was found to be 1.09 × 105 which is similar to the enhancement factors reported for chromophores in the vicinity of gold nanostructures.63 We further studied changes in the Raman scattering of molecules 2 and 3 in the presence of AuNRs. We did not observe any significant enhancement in the Raman signals (Fig. S14†), thereby supporting our hypothesis that these compounds do not bind in the hot spots.
C and C–N (C–NH2) stretching, respectively.62 However, at a concentration of 10 μM, which was employed for the self-assembly of AuNRs, we did not observe any Raman signal for molecule 1. Next, we recorded the Raman scattering of the end-to-end assembled AuNRs (containing molecule 1 (10 μM) and AuNRs) wherein peaks were observed at 940, 1580 and 1620 cm−1 thereby substantiating the presence of molecule 1 in the hot spots generated as a result of the end-to-end assembly of AuNRs. Further, the SERS enhancement factor of molecule 1 in the presence of AuNRs was found to be 1.09 × 105 which is similar to the enhancement factors reported for chromophores in the vicinity of gold nanostructures.63 We further studied changes in the Raman scattering of molecules 2 and 3 in the presence of AuNRs. We did not observe any significant enhancement in the Raman signals (Fig. S14†), thereby supporting our hypothesis that these compounds do not bind in the hot spots.
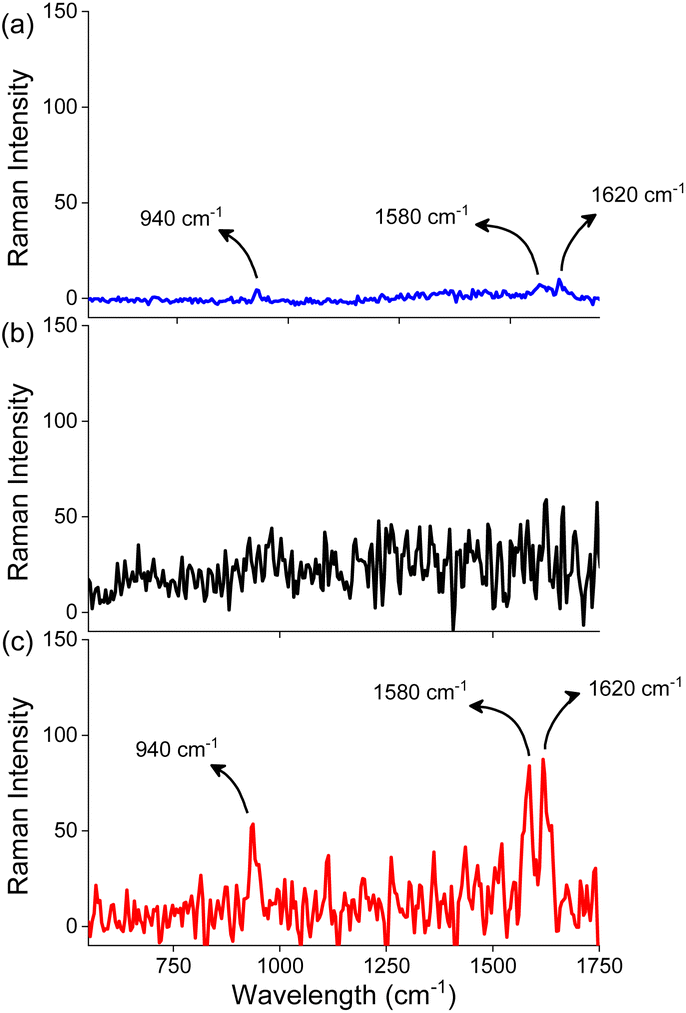 | ||
| Fig. 4 Raman spectra of (a) 1 (1 mM) in the absence of AuNRs, (b) 1 (10 μM) in the absence of AuNRs, and (c) 1 (10 μM) in the presence of AuNRs in water. | ||
Investigation of photothermal properties
Photothermal conversion relies on the generation of heat by a photosensitizer upon irradiation using light.64–67 Owing to efficient absorption of NIR light, AuNRs have been recognized as potential photothermal agents.68 As the end-to-end assembled AuNRs exhibited NIR absorption, we were interested in investigating their photothermal properties. An aqueous solution of the assembled AuNRs was irradiated with a 750 nm laser and the change in temperature was monitored at different time intervals using a thermal imaging camera. As presented in Fig. 5a, we observed an increase of 4.2 °C per minute of irradiation and after 6 minutes the temperature of the solution increased from 28 to 53 °C. The increase in temperature as a result of irradiation was linearly fitted to derive the thermal time constant (τ) and was used to determine the photothermal conversion efficiency (η) (Fig. S15†). A maximum photothermal conversion efficiency of 70% was obtained at a laser power of 0.526 W which is better than that reported for surfactant functionalized gold nanorods and other metal nanocrystals.69 Under similar experimental conditions, we then tested the photothermal effect for the individual components, i.e., AuNRs and 1. While the rise in temperature with molecule 1 was negligible, AuNRs showed a temperature rise of 5 °C for the same duration of irradiation (Fig. S16†) and the corresponding photothermal conversion efficiency was obtained as 20%.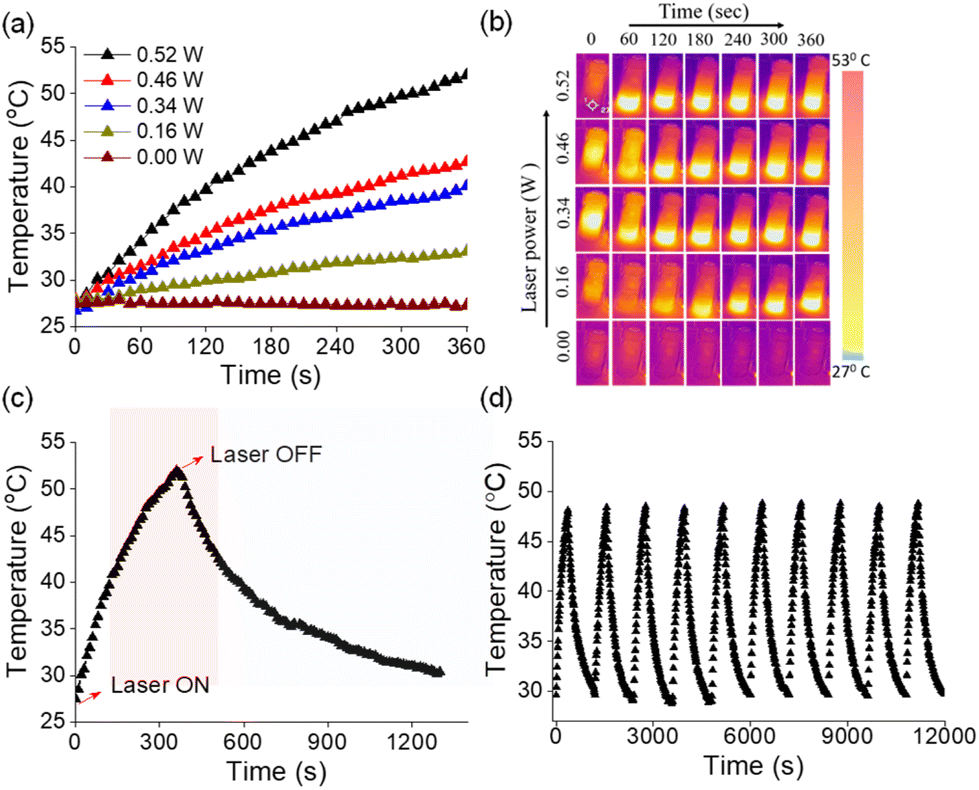 | ||
| Fig. 5 (a) Increase in temperature of an aqueous solution of end-to-end assembled AuNRs (20 μg mL−1) upon irradiation using a 750 nm laser at different laser powers and (b) the corresponding photographs showing the increase in temperature obtained using a FLIR pro-thermal imaging camera. (c) The increase in temperature of the end-to-end assembled AuNRs upon laser irradiation (0.52 W) followed by a decrease in temperature upon turning off the laser. (d) Photothermal heating and cooling cycles of the end-to-end assembled AuNRs upon laser irradiation (0.49 W). | ||
An ideal photothermal agent should be stable at elevated temperatures and the process should be reversible so as to allow its repeated usage. To test this, we monitored the absorbance of the end-to-end assembled AuNRs over a range of temperatures. We did not observe significant changes in absorption maxima of assembled AuNRs as the temperature increased from 25 to 55 °C (Fig. S17†), thereby confirming their structural integrity and stability at higher temperatures. Next, we investigated the photothermal efficiency of the end-to-end assembled AuNRs at different laser power intensities. We observed that the increase in temperature was proportional to the laser power (Fig. 5b), thereby implying the regulation of the generated heat by modulating the laser power. Furthermore, the photothermal effect was observed to be a reversible process upon turning off the irradiation source (Fig. 5c). The photothermal heating and cooling curves of end-to-end assembled AuNRs were monitored over ten cycles to assess the stability and durability of their photothermal conversion properties (Fig. 5d). The results showed that the AuNRs maintained their photothermal heating and cooling capabilities consistently across multiple cycles, indicating a high degree of stability. These experiments indicate that the end-to-end assembled AuNRs could function as a controlled photothermal agent for multiple cycles.
Conclusions
In summary, we demonstrated a simple one-step method to produce a linear chain of end-to-end assembled gold nanorods. Our results show that the diamino-functionalized organic chromophore, 1, effectively interacted with the longitudinal ends of gold nanorods and drove their linear assembly. The steric bulkiness of the functionalized BODIPY chromophore was crucial in promoting the longitudinal assembly. The placement of the chromophore in the plasmonic hot spots – generated as a result of the linear assembly – led to strong electronic coupling between AuNRs and 1, and endowed photothermal properties to the self-assembled system. This work thus establishes a unique design strategy wherein the synergy between self-assembly and electronic coupling resulted in superior physicochemical properties, and could serve as a platform for designing novel inorganic–organic nanohybrids for diverse photonic and biomedical applications. This work, thus, not only emphasizes the importance of nanomaterial design and optimization but also underscores the promising role of gold nanorods in advancing targeted cancer treatments with improved efficacy and reduced side effects.Author contributions
Hemant, A. Rahman, and P. P. Neelakandan designed the experiments. Hemant performed the experiments and conducted the data analysis. P. Sharma and A. Shanavas assisted with photothermal conversions. Each author supported the manuscript's writing and talked about the results.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
Hemant (201920-191620089158) and Atikur Rahman thank the University Grants Commission and INST Mohali, respectively, for their research fellowships.References
- S. Jambhulkar, D. Ravichandran, Y. Zhu, V. Thippanna, A. Ramanathan, D. Patil, N. Fonseca, S. V. Thummalapalli, B. Sundaravadivelan, A. Sun, W. Xu, S. Yang, A. M. Kannan, Y. Golan, J. Lancaster, L. Chen, E. B. Joyee and K. Song, Small, 2024, 20, 2306394 CrossRef CAS PubMed.
- A. Rao, S. Roy, V. Jain and P. P. Pillai, ACS Appl. Mater. Interfaces, 2023, 15, 25248–25274 CrossRef CAS PubMed.
- D. Gentili and G. Ori, Nanoscale, 2022, 14, 14385–14432 RSC.
- M. S. Lee, D. W. Yee, M. Ye and R. J. Macfarlane, J. Am. Chem. Soc., 2022, 144, 3330–3346 CrossRef CAS PubMed.
- M. A. Boles, M. Engel and D. V. Talapin, Chem. Rev., 2016, 116, 11220–11289 CrossRef CAS PubMed.
- C. Xi, P. F. Marina, H. Xia and D. Wang, Soft Matter, 2015, 11, 4562–4571 RSC.
- M. Grzelczak, J. Vermant, E. M. Furst and L. M. Liz-Marzán, ACS Nano, 2010, 4, 3591–3605 CrossRef CAS.
- Z. Nie, A. Petukhova and E. Kumacheva, Nat. Nanotechnol., 2010, 5, 15–25 CrossRef CAS.
- A. Amestoy, A. Rangra, V. Mansard, D. Saya, E. Pouget, E. Mazaleyrat, F. Severac, C. Bergaud, R. Oda and M.-H. Delville, ACS Appl. Mater. Interfaces, 2023, 15, 39480–39493 CrossRef CAS PubMed.
- S. R. Ahmed, J. Kim, V. T. Tran, T. Suzuki, S. Neethirajan, J. Lee and E. Y. Park, Sci. Rep., 2017, 7, 44495 CrossRef CAS.
- Y. Zhao, Y. Huang, H. Zhu, Q. Zhu and Y. Xia, J. Am. Chem. Soc., 2016, 138, 16645–16654 CrossRef CAS PubMed.
- J. Zhang, Z. Huang, Y. Xie and X. Jiang, Chem. Sci., 2022, 13, 1080–1087 RSC.
- S. Agrawal, R. A. Mysko, M. M. Nigra, S. K. Mohanty and M. P. Hoepfner, Langmuir, 2021, 37, 3281–3287 CrossRef CAS PubMed.
- A. La Torre, M. del C. Giménez-López, M. W. Fay, G. A. Rance, W. A. Solomonsz, T. W. Chamberlain, P. D. Brown and A. N. Khlobystov, ACS Nano, 2012, 6, 2000–2007 CrossRef CAS PubMed.
- H. Wei, D. Pan, S. Zhang, Z. Li, Q. Li, N. Liu, W. Wang and H. Xu, Chem. Rev., 2018, 118, 2882–2926 CrossRef CAS.
- N. O. Junker, A. Lindenau, M. Rütten, M. Lach, A. Nedilko, D. N. Chigrin, G. von Plessen and T. Beck, Adv. Funct. Mater., 2023, 33, 2303260 CrossRef CAS.
- Z. Chai, A. Childress and A. A. Busnaina, ACS Nano, 2022, 16, 17641–17686 CrossRef CAS PubMed.
- Y. Wy, H. Jung, J. W. Hong and S. W. Han, Acc. Chem. Res., 2022, 55, 831–843 CrossRef CAS PubMed.
- X. Lu, D. Punj and M. Orrit, Nano Lett., 2022, 22, 4215–4222 CrossRef CAS PubMed.
- H. Wei and H. Xu, Nanoscale, 2013, 5, 10794–10805 RSC.
- J. Zheng, X. Cheng, H. Zhang, X. Bai, R. Ai, L. Shao and J. Wang, Chem. Rev., 2021, 121, 13342–13453 CrossRef CAS PubMed.
- H. Chen, L. Shao, Q. Li and J. Wang, Chem. Soc. Rev., 2013, 42, 2679–2724 RSC.
- H. Chen, L. Shao, Q. Li and J. Wang, Chem. Soc. Rev., 2013, 42, 2679–2724 RSC.
- T. Jain, F. Westerlund, E. Johnson, K. Moth-Poulsen and T. Bjørnholm, ACS Nano, 2009, 3, 828–834 CrossRef CAS PubMed.
- S. T. Shibu Joseph, B. I. Ipe, P. Pramod and K. G. Thomas, J. Phys. Chem. B, 2006, 110, 150–157 CrossRef CAS PubMed.
- H. Duan, Z. Jia, M. Liaqat, M. D. Mellor, H. Tan, M.-P. Nieh, Y. Lin, S. Link, C. F. Landes and J. He, ACS Nano, 2023, 17, 12788–12797 CrossRef CAS PubMed.
- H. J. Kim, W. Wang, W. Bu, M. M. Hossen, A. Londoño-Calderon, A. C. Hillier, T. Prozorov, S. Mallapragada and D. Vaknin, Sci. Rep., 2019, 9, 20349 CrossRef CAS PubMed.
- S. M. H. Abtahi, N. D. Burrows, F. A. Idesis, C. J. Murphy, N. B. Saleh and P. J. Vikesland, Langmuir, 2017, 33, 1486–1495 CrossRef CAS PubMed.
- Y. Wang, A. E. I. DePrince, S. K. Gray, X.-M. Lin and M. Pelton, J. Phys. Chem. Lett., 2010, 1, 2692–2698 CrossRef CAS PubMed.
- F. C.-M. Leung, S. Y.-L. Leung, C. Y.-S. Chung and V. W.-W. Yam, J. Am. Chem. Soc., 2016, 138, 2989–2992 CrossRef CAS PubMed.
- Y.-T. Chan, S. Li, C. N. Moorefield, P. Wang, C. D. Shreiner and G. R. Newkome, Chem. – Eur. J., 2010, 16, 4164–4168 CrossRef CAS PubMed.
- S. Vazirieh Lenjani, M. Mayer, R. Wang, Y. Dong, A. Fery, J.-U. Sommer and C. Rossner, J. Phys. Chem. C, 2022, 126, 14017–14025 CrossRef CAS.
- E. Severoni, S. Maniappan, L. M. Liz-Marzán, J. Kumar, F. J. García de Abajo and L. Galantini, ACS Nano, 2020, 14, 16712–16722 CrossRef CAS PubMed.
- J. Wu, Y. Xu, D. Li, X. Ma and H. Tian, Chem. Commun., 2017, 53, 4577–4580 RSC.
- Y. Xu, X. Wang and X. Ma, Dyes Pigm., 2017, 145, 385–390 CrossRef CAS.
- W. Ni, R. A. Mosquera, J. Pérez-Juste and L. M. Liz-Marzán, J. Phys. Chem. Lett., 2010, 1, 1181–1185 CrossRef CAS.
- K. G. Thomas, S. Barazzouk, B. I. Ipe, S. T. S. Joseph and P. V. Kamat, J. Phys. Chem. B, 2004, 108, 13066–13068 CrossRef CAS.
- M. Kincanon and C. J. Murphy, ACS Nano, 2023, 17, 24090–24103 CrossRef CAS PubMed.
- K. K. Caswell, J. N. Wilson, U. H. F. Bunz and C. J. Murphy, J. Am. Chem. Soc., 2003, 125, 13914–13915 CrossRef CAS PubMed.
- D. Liu and C. Xue, Adv. Mater., 2021, 33, 2005738 CrossRef CAS PubMed.
- N. Jiang, X. Zhuo and J. Wang, Chem. Rev., 2018, 118, 3054–3099 CrossRef CAS PubMed.
- J.-F. Li, C.-Y. Li and R. F. Aroca, Chem. Soc. Rev., 2017, 46, 3962–3979 RSC.
- A. Klinkova, R. M. Choueiri and E. Kumacheva, Chem. Soc. Rev., 2014, 43, 3976–3991 RSC.
- H. Chen, T. Ming, L. Zhao, F. Wang, L.-D. Sun, J. Wang and C.-H. Yan, Nano Today, 2010, 5, 494–505 CrossRef CAS.
- K. G. Thomas and P. V. Kamat, Acc. Chem. Res., 2003, 36, 888–898 CrossRef CAS PubMed.
- S. Elhani, H. Ishitobi, Y. Inouye, A. Ono, S. Hayashi and Z. Sekkat, Sci. Rep., 2020, 10, 1–11 CrossRef PubMed.
- T. Zhao, D. Meng, Z. Hu, W. Sun, Y. Ji, J. Han, X. Jin, X. Wu and P. Duan, Nat. Commun., 2023, 14, 81 CrossRef CAS PubMed.
- A. Kar, V. Thambi, D. Paital, G. Joshi and S. Khatua, Langmuir, 2020, 36, 9894–9899 CrossRef CAS PubMed.
- J. Lee and J. W. Ha, Anal. Chem., 2022, 94, 7100–7106 CrossRef CAS PubMed.
- P. P. Neelakandan, A. Jiménez and J. R. Nitschke, Chem. Sci., 2014, 5, 908–915 RSC.
- Y. Hayashi, S. Yamaguchi, W. Y. Cha, D. Kim and H. Shinokubo, Org. Lett., 2011, 13, 2992–2995 CrossRef CAS PubMed.
- A. J. Musser, P. P. Neelakandan, J. M. Richter, H. Mori, R. H. Friend and J. R. Nitschke, J. Am. Chem. Soc., 2017, 139, 12050–12059 CrossRef CAS PubMed.
- A. Rahman, P. P. Praveen Kumar, P. Yadav, T. Goswami, A. Shanavas, H. N. Ghosh and P. P. Neelakandan, ACS Appl. Nano Mater., 2022, 5, 6532–6542 CrossRef CAS.
- P. P. P. Kumar, A. Rahman, T. Goswami, H. N. Ghosh and P. P. Neelakandan, ChemPlusChem, 2021, 86, 87–94 CrossRef CAS PubMed.
- P. P. P. Kumar, P. Yadav, A. Shanavas, S. Thurakkal, J. Joseph and P. P. Neelakandan, Chem. Commun., 2019, 55, 5623–5626 RSC.
- A. F. Stewart, B. P. Gagnon and G. C. Walker, Langmuir, 2015, 31, 6902–6908 CrossRef CAS PubMed.
- S. Y. Quek, L. Venkataraman, H. J. Choi, S. G. Louie, M. S. Hybertsen and J. B. Neaton, Nano Lett., 2007, 7, 3477–3482 CrossRef CAS PubMed.
- C. J. Murphy, L. B. Thompson, A. M. Alkilany, P. N. Sisco, S. P. Boulos, S. T. Sivapalan, J. A. Yang, D. J. Chernak and J. Huang, J. Phys. Chem. Lett., 2010, 1, 2867–2875 CrossRef CAS.
- C. J. Orendorff, T. M. Alam, D. Y. Sasaki, B. C. Bunker and J. A. Voigt, ACS Nano, 2009, 3, 971–983 CrossRef CAS PubMed.
- R. del Caño, J. M. Gisbert-González, J. González-Rodríguez, G. Sánchez-Obrero, R. Madueño, M. Blázquez and T. Pineda, Nanoscale, 2020, 12, 658–668 RSC.
- Y. Bao and A. Oluwafemi, Chem. Commun., 2023, 60, 469–481 RSC.
- J. S. Sandoval and D. W. McCamant, J. Phys. Chem. A, 2023, 127, 8238–8251 CrossRef CAS PubMed.
- T. Köker, N. Tang, C. Tian, W. Zhang, X. Wang, R. Martel and F. Pinaud, Nat. Commun., 2018, 9, 607 CrossRef PubMed.
- X. Cui, Q. Ruan, X. Zhuo, X. Xia, J. Hu, R. Fu, Y. Li, J. Wang and H. Xu, Chem. Rev., 2023, 123, 6891–6952 CrossRef CAS PubMed.
- J. Li, W. Zhang, W. Ji, J. Wang, N. Wang, W. Wu, Q. Wu, X. Hou, W. Hu and L. Li, J. Mater. Chem. B, 2021, 9, 7909–7926 RSC.
- S. Luo, X. Ren, H. Lin, H. Song and J. Ye, Chem. Sci., 2021, 12, 5701–5719 RSC.
- M. Kim, J.-H. Lee and J.-M. Nam, Adv. Sci., 2019, 6, 1900471 CrossRef PubMed.
- R. Zhou, M. Zhang, J. Xi, J. Li, R. Ma, L. Ren, Z. Bai, K. Qi and X. Li, Nanoscale Res. Lett., 2022, 17, 68 CrossRef CAS PubMed.
- X. Liu, B. Li, F. Fu, K. Xu, R. Zou, Q. Wang, B. Zhang, Z. Chen and J. Hu, Dalton Trans., 2014, 43, 11709–11715 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental section and supporting figures. See DOI: https://doi.org/10.1039/d4nr02161d |
| This journal is © The Royal Society of Chemistry 2024 |