
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Mimicking the extracellular world: from natural to fully synthetic matrices utilizing supramolecular biomaterials
Laura
Rijns
 ab,
Martin G. T. A.
Rutten
ab,
Annika F.
Vrehen
ab,
Ana A.
Aldana
c,
Matthew B.
Baker
cd and
Patricia Y. W.
Dankers
*abe
ab,
Martin G. T. A.
Rutten
ab,
Annika F.
Vrehen
ab,
Ana A.
Aldana
c,
Matthew B.
Baker
cd and
Patricia Y. W.
Dankers
*abe
aInstitute for Complex Molecular Systems, Eindhoven University of Technology, 5600 MB Eindhoven, The Netherlands. E-mail: p.y.w.dankers@tue.nl
bDepartment of Biomedical Engineering, Laboratory of Chemical Biology, Eindhoven University of Technology, 5600 MB Eindhoven, The Netherlands
cDepartment of Complex Tissue Regeneration, MERLN Institute for Technology Inspired Regenerative Medicine, Maastricht University, 6200 MD Maastricht, The Netherlands
dDepartment of Instructive Biomaterials Engineering, MERLN Institute for Technology Inspired Regenerative Medicine, Maastricht University, 6200 MD Maastricht, The Netherlands
eDepartment of Chemical Engineering and Chemistry, Eindhoven University of Technology, 5600 MB, Eindhoven, The Netherlands
First published on 25th July 2024
Abstract
The extracellular matrix (ECM) has evolved around complex covalent and non-covalent interactions to create impressive function—from cellular signaling to constant remodeling. A major challenge in the biomedical field is the de novo design and control of synthetic ECMs for applications ranging from tissue engineering to neuromodulation to bioelectronics. As we move towards recreating the ECM's complexity in hydrogels, the field has taken several approaches to recapitulate the main important features of the native ECM (i.e. mechanical, bioactive and dynamic properties). In this review, we first describe the wide variety of hydrogel systems that are currently used, ranging from fully natural to completely synthetic to hybrid versions, highlighting the advantages and limitations of each class. Then, we shift towards supramolecular hydrogels that show great potential for their use as ECM mimics due to their biomimetic hierarchical structure, inherent (controllable) dynamic properties and their modular design, allowing for precise control over their mechanical and biochemical properties. In order to make the next step in the complexity of synthetic ECM-mimetic hydrogels, we must leverage the supramolecular self-assembly seen in the native ECM; we therefore propose to use supramolecular monomers to create larger, hierarchical, co-assembled hydrogels with complex and synergistic mechanical, bioactive and dynamic features.
 Laura Rijns | Laura Rijns (1996) is a postdoc at Stanford University with Professor Zhenan Bao, focused on improving the communication between electronic materials and living tissue. Her research is funded by a Niels Stensen and Rubicon Fellowship. She was selected as MIT ChemE Rising Star 2024. She obtained her PhD (2023) “cum laude” from Eindhoven University of Technology (TU/e) with Professor Patricia Dankers and Professor E.W. (Bert) Meijer on supramolecular hydrogels. She was awarded the Materials-Driven Regeneration Young Talent Award 2021, Best PhD Thesis Award of BME TU/e 2024, and the ACS Global Outstanding Graduate Student & Mentor Award in Polymer Science & Engineering 2024. |
 Martin G.T.A. Rutten | Martin Rutten is postdoctoral researcher in the group of Professor Patricia Dankers on materials for translational medicine at Eindhoven University of Technology (TU/e). He received his MSc at Radboud University Nijmegen (the Netherlands) in the group of Dr P.H.J. Kouwer. In 2019 he started as a PhD student in the group of Professor Patricia Dankers at TU/e, The Netherlands. Here he focused on the design of multi-component hydrogel networks that are able to mimic the complex time and length scales of natural tissues. |
 Annika F. Vrehen | Annika Vrehen is a preclinical scientist at VivArt-X, investigating biomaterial designs for regeneration therapies. She received her bachelor's degree in Medical Sciences & Engineering and her masters Biomedical Engineering at Eindhoven University of Technology (TU/e). In this master she focused on the development of a renal cellular read-out for biomaterial screening in the group of Professor Patricia Dankers. During her masters, she performed an internship at King's College London in the group of Dr Georgina Ellison-Hughes. In 2023, Annika received her PhD in biomedical engineering at the TU/e, by focusing on designing corneal stromal microenvironments based on supramolecular hydrogels. |
 Ana A. Aldana | Ana Agustina Aldana is a dedicated researcher specializing in materials chemistry and biomaterials. Dr Aldana has worked at several renowned research centers, including the MERLN Institute at Maastricht University, the Institute for Biomaterials at Erlangen-Nuremberg University, the Harry Perkins Institute of Medical Research, and INTEMA Institute of Materials Sciences and Technology. With over 20 peer-reviewed publications, her research merges chemistry and engineering to design advanced biomaterials for biomedical applications. She has received prestigious awards, including the Marie Curie Individual Fellowship and the Endeavour Individual Fellowship, highlighting her significant contributions to biomedical science. |
 Matthew B. Baker | Matthew B. Baker is an Assistant Professor at Maastricht University, and group leader of the BioMatt group. He is a chemist by training, and the group takes a molecular view of materials design. He received his BSc in chemistry (2006) at Clemson University and worked shortly for Tetramer Technologies, LLC. He obtained his PhD (2012) in Physical Organic Chemistry with Professor Ronald K. Castellano at the University of Florida. Afterwards, he performed his postdoc under the guidance of Professor E.W. (Bert) Meijer on the design and characterization of supramolecular hydrogels as extracellular matrix mimics. |
 Patricia Y.W. Dankers | Patricia Y.W. Dankers is Full Professor in Biomedical Materials and Chemistry at Eindhoven University of Technology (TU/e). She obtained her first PhD (2006) at TU/e under the supervision of Professor E.W. (Bert) Meijer on supramolecular bioactive biomaterials. She obtained her second PhD (2013) in medical sciences at Groningen University on kidney regenerative medicine under the supervision of Professor Marja J.A. van Luyn. In 2010 she was a visiting professor with Professor Samuel I. Stupp at Northwestern University, Chicago (USA). She is a VENI (2008), VIDI (2017) and VICI (2023) laureate and received an ERC starting grant (2012) and has been awarded various awards. |
Introduction: the native ECM and its mechanical, bioactive and dynamic properties
The extracellular matrix (ECM) – the environment around cells – dictates cell differentiation, proliferation, fate, and enables communication within living tissue. This ECM is composed of both proteoglycans and fibrous proteins, yielding a network with both elastic and viscous properties. The ECM has a bidirectional communication with the cell; the cell remodels the matrix and the matrix provides information to control cellular behavior. Understanding, reprogramming and mimicking this environment around cells is important to many fields – from regenerative medicine to cancer research to wearable electronics.1–5Recreating the native ECM in a simplistic, controlled manner to grow cells into complex living tissue will have many far-reaching applications useful for personalized medicine,6 (local) immunomodulation7 and non-invasive neuromodulation.4,8,9 Many labs world-wide focus on mimicking the ECM using sophisticated biomaterials, with pioneering contributions by the Bhattacharya,10–12 Varghese,13,14 Vemula,15,16 Pal,17,18 Heilshorn,19,20 Anseth,21,22 Burdick,23,24 DeForest,25 Tirrell,26,27 Lutolf,28,29 Clevers,30,31 Dankers,32,33 Mata,34,35 Mano,36,37 Chaudhuri38,39 and Mooney7,40 labs. Prof. Bhattacharya and his team beautifully leverage synthetic molecular and supramolecular systems to recreate self-assembled structures seen in biology, elegantly utilizing a wide mix of building blocks, ranging from natural moieties, like sugar-derived low molecular weight gelators (LMWGs)12 to two-component hydrogels based on fatty acids and amines,11 all the way towards organogels.10
In this review, we first dissect the complex features of the ECM into simpler pieces, classified as its mechanical, bioactive and dynamic properties=. After this, we discuss the current state-of-the-art ECM mimicking biomaterials, ranging from completely natural to intermediate hybrid to fully synthetic matrices (Fig. 1). We end by taking a chemistry approach and provide a forward-thinking perspective on how to recreate the complexity of our ECM in a fully synthetic, controlled fashion. Synthetic supramolecular materials allow for orthogonal control over their properties, such that the influence of only one matrix property on cellular outcome can be evaluated – making these types of materials ideal for future applications as artificial ECM.
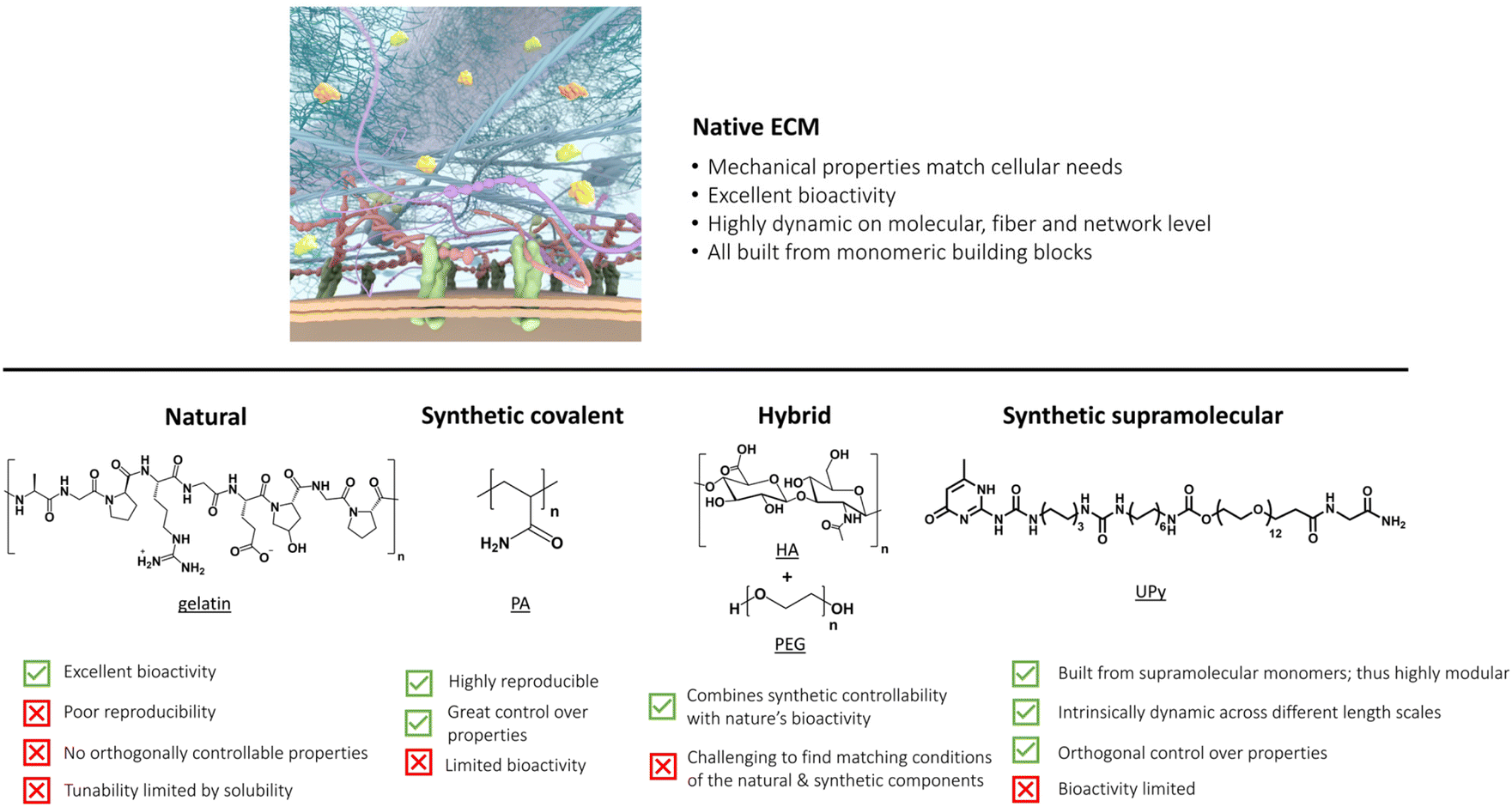 | ||
| Fig. 1 Overview of important properties of our native ECM, which is the benchmark (top). Bottom part shows the current state-of-the-art ECM mimicking hydrogels with their properties. Part of the figure is created by the ICMS Animation Studio. | ||
The ECM's mechanical properties
Starting with the mechanical properties, the ECM is a viscoelastic material due to combinations of different classes of macromolecules, along with the covalent and supramolecular interactions from which it is assembled (Fig. 1). In general, the elastic properties are covered by the fibrous proteins such as collagens, elastins, fibronectins and laminins, which provide tensile strength, and serve as adhesive sites for cells.41 The viscous proteoglycans on the other hand cover the majority of interstitial space within the tissue in the form of a hydrated gel, serving a variety of functions owing to their unique buffering, hydration, binding and force bearing proteins.42 As the concentration, variety, and hierarchical order of proteins can differ greatly between tissues, the mechanical properties of the ECM also show a large variation, ranging from 1.9 kPa in the lungs43 to 20 GPa in cortical bone.44As numerous studies show that the mechanical properties of the ECM play a crucial role in cellular behavior, one must realize that the cell is also a viscoelastic material.45 At short timescales (<1 s), the mechanical response is dominated by individual fibers and the cell behaves as an elastic solid. In contrast, at longer timescales, processes like remodeling are dominant and cause the cell to relax. This mechanical behavior is mainly due to the properties of the cytoskeleton, a complex network of self-assembled protein filaments in the cytoplasm of cells. This network behaves as semi-flexible filaments, with specific relaxation mechanisms and an increase in stiffness upon an applied stress, i.e. stress stiffening, most likely induced by molecular motors as myosin.46,47 The interplay between the mechanical and dynamic parameters of the cell and ECM is proposed to play an important role in cellular function and behavior.
Biochemical properties of the ECM
Next to the ECM's complex mechanical properties, the biochemical information of the ECM also greatly impacts cellular behavior (Fig. 1). An abundant and important receptor that regulates many ECM–cell interactions is the integrin receptor.48,49 Integrins are heterodimeric transmembrane proteins that consist of an α and β subunit, which are non-covalently attached. Structurally, they consist of an extracellular ligand-binding domain, a transmembrane domain and a cytoplasmic tail. These integrins are often the first components in a signaling cascade; by tuning integrin–ECM interactions, e.g. via different integrin subunits, the integrin activation state, the binding site of the substrate and even the presence of divalent cations, a large extent of specific cell–matrix signaling dynamics are possible.50 Integrins interact with the extracellular world by binding to ECM glycoproteins, like laminin, collagen and fibronectin, and transmitting this information into the cytoplasm of the cell. Once bound, the intracellular parts of the integrin receptors are linked to the cytoskeleton via multi-molecule complexes known as focal adhesions, again in a large variety of different states. Due to this, forces in the ECM are directly transferred via integrin towards the cytoskeleton, changing assembly and organization of cytoskeletal fibers, inducing signaling pathways, migration and gene expression (both directly and indirectly).51–53 While there are over 24 integrin types, with different specificities and binding strength towards the various ligand proteins, synthetic extracellular matrices moved towards simplicity early in their development, thus trying to unravel general common signals: in the 1980s, it was discovered that integrins only need the tripeptide arginine–glycine–aspartic acid (RGD) sequence to obtain successful binding and cellular adhesion.49,54,55 In addition, other markers have been identified via significant work in the cancer therapeutics space for specific integrin binders, which provided potential power to synthetic biomaterials. Growth factors (e.g. fibroblast growth factors (FGFs), hepatocyte growth factors (HGFs), and vascular endothelial growth factors (VEGFs)) can be bound to ECM proteins, like fibronectin, vitronectin, collagens and proteoglycans themselves, or in combination with heparin and heparan sulphate.56–58 This results in the ability of the ECM to induce a rapid and localized signaling cascade by regulating the bioavailability of growth factors; their distribution, activation, and presentation to cells.Yet integrin binding is not the whole story, the cell–ECM interaction is a complex and dynamic micro-environment that involves many more receptors (e.g. syndecans59) and operates in both directions. The cell and the matrix determine each other's function and course, that is the cell via constant remodeling and pulling of surrounding fibers and the matrix via its specific properties in terms of mechanical stiffness, bioactivity and dynamics. In addition, the cells can also transduce intercellular generated forces via the ECM through other cells as a way of cell–cell communication (in addition to direct cell–cell interactions).
Dynamic properties of the ECM
Next to to the macroscopic bulk stiffness of tissues and materials, fundamental insights into dynamics is also highly important as the ECM itself is a dynamic, multicomponent network, primarily held together by non-covalent interactions.60 To illustrate, the proteoglycans and glycoproteins can both store and dissipate deformation energy, while displaying a time-dependent mechanical response. This results in stress relaxation behavior when being deformed, or creep when a mechanical stress is applied.61 Stress relaxation tests quantify this dynamic behavior, which is measured by applying a constant deformation and measuring the stress over time.62,63 In contrast to the wide variety in elastic moduli, the relaxation half times of most tissues cover only a few orders of magnitude, i.e. seconds to hour,64,65 except for bone.66 To compare, most conventional synthetic polymeric hydrogels have relaxation half times of hours or show almost no stress relaxation at all, such as for example covalently crosslinked polyacrylamide.64 The ECM also responds to externally applied stresses, which is often quantified via a creep test, where a constant stress is applied and the responding deformation is measured over time.62,63 In addition, viscoelastic materials will show a certain recovery of the deformation, as they tend to return to their original conformation. However, owing to their ability to flow, they will only partially return to their initial state, which is a measure for the plasticity of the system. In order to translate this knowledge on the dynamics into the design of emergent function found in the ECM, one could think of using dynamic (e.g. supramolecular) biomaterials for dissipating stress, while more static (e.g. covalent) bonds could be introduced to mimic the elastic behavior found in the ECM.In addition, it is important to note that the abovementioned properties interfere and overlap with each other, as time-dependent behavior for both the mechanical and biochemical side of the ECM is often observed. To illustrate, the ECM's ‘dynamic’ and mechanical properties manifest themselves in stress relaxation or stress stiffening behavior, which are both fiber rearrangements leading to relaxation or stiffening of the matrix, respectively. And, depending on cellular needs, a tight, stiff mechanical environment might be preferred for proliferation. However, later in time, a more loose, dynamic environment with increased mesh size, decreased stiffness and increased mobility of fibers in the network might support and favor tissue growth and differentiation.67,68 Such changes in network properties could be achieved through inclusion of light- or enzyme sensitive groups in the hydrogel.68–70 Additionally, the dynamic or robust incorporation of different bioactive signals (e.g. ‘dynamic’ delivery of large growth factors with many possible ligand-receptor interactions or ‘robust’ incorporation of cell adhesion motifs to withstand cell pulling forces) is important for cells. Lastly, it is important to realize that the optimal extracellular environment depends on cell type. For example, neural cells have different requirements for growth and differentiation (‘soft’ environment) as compared to bone cells, which need more stiff surroundings. This means that design criteria to create the optimal ECM mimicking hydrogel for healthy cell growth vary and should be tailored towards cellular needs.
The summation of all these properties can lead to increasingly complex behavior from simple ECM components via the culmination of finely tuned molecular interactions.63,71 Over the years, various materials ranging from natural to fully synthetic have been created to transfer specific ECM properties to smart materials.40,72 In this review, we discuss the advantages and limitations of all these materials as platform for cell culture and compare their mechanical, bioactive and dynamic properties. For ease of discussion, we classify the materials in natural, synthetic covalent, hybrid and synthetic supramolecular hydrogels according to the nature of the polymers (Fig. 1). Finally, we provide a forward thinking perspective for the challenges in the field, highlighting the emergence of complex function that arises from using simple supramolecular building blocks and the importance of dynamics.
Natural extracellular matrices
Matrices built from from natural sources are suitable as ECM mimetics or replacements. These materials, in general, are characterized by their powerful natural biochemical composition (which is good for cells), but they are limited in tailorability and reproducibility. We briefly discuss these materials below, highlighting how the complexity of ECM derivates has led to the use of simple hydrogel design, allowing for more insight into pathways and mechanisms underlying the cell-ECM communication.A conceptually appealing approach to mimicking the natural ECM is to utilize ECMs derived from living tissue (Table 1). So-called decellularized ECMs (dECMs) are biomaterials in which all cells are removed, leaving only the tissue's native microenvironment with tissue specific proteins and microstructure.73,74 The main advantage of dECM lies in the high resemblance to the natural ECM in terms of bioactivity. However, the ECM's microstructure is almost never fully preserved in dECMs. Besides the extensive practical steps needed (and ethical obligations) in obtaining this ECM, the harsh processing steps often alter the final mechanical strength. In addition, the viscoelastic properties can very easily from batch to batch, while the material is difficult to process into new shapes or forms.75–78 This leads to a very delicate balance between removing as much cellular material while maintaining enough bioactivity, and structural integrity. Other limitations of dECMs include limited scalability, donor variation and inflammatory reactions to the material.79,80
| Type of ECM mimic | Used material | Selected features why to use/not use the material | Group | Ref. |
|---|---|---|---|---|
| Natural | dECMs (e.g. Matrigel) | ✓ Highly bioactive | Many labs | 73–83 |
| ✗ Poor reproducibility | ||||
| ✗ Poor control over properties | ||||
| Collagen | ✓ Dynamic mechanical properties, like stress relaxation | Weaver, Koenderink, Chaudhuri, Shenoy, Weitz, MacKintosh | 84–92 | |
| ✗ No orthogonally controllable properties | ||||
| Gelatin | ✓ Good bioactivity | Isaksson | 93 | |
| ✓ Easy to handle | ||||
| ✗ Limited mechanical strength | ||||
| ✗ Scalability limited | ||||
| Fibrin | ✓ Dynamic mechanical properties, like stress stiffening | Koenderink | 93 and 94 | |
| ✓ Associated with wound healing | ||||
| ✗ No orthogonally controllable properties | ||||
| Hyaluronic acid (HA) | ✓ Therapeutic effects in wound healing, inflammation | Engler, Vemula | 95–104 | |
| ✓ Hydrogelation tunable through many parameters | ||||
| ✗ Many purification steps required | ||||
| ✗ Limited mechanical strength | ||||
| Chondroitin sulfate (CS) | ✓ Useful for growing cartilage tissue and chondrogenic differentiation | Varghese | 105–107 | |
| ✗ Limited mechanical strength | ||||
| Alginate | ✓ Highly tunable and controllable mechanics, dynamics and pore size | Chaudhuri, Mooney | 108–113 | |
| ✓ Easy to obtain | ||||
| ✗ Degradation difficult to control | ||||
| Fatty-acid derived | ✓ Good control over self-assembling and mechanical properties | Bhattacharya | 17 | |
| ✗ Gelation might occur in organic solvents only; making cell culture impossible | ||||
| Synthetic covalent | Poly(ethylene glycol) (PEG) | ✓ Relatively biocompatible and non-toxic | Anseth, Bowman | 119–127 |
| ✓ Easy to functionalize | ||||
| ✓ Tunable elastic moduli over a large range (G′: 100 Pa–100 kPa) | ||||
| ✗ Lacks dynamic properties | ||||
| Polyisocyanide (PIC) | ✓ Dynamic mechanical properties, like stress stiffening | Nolte, Rowen, Kouwer | 129–133 | |
| ✓ Orthogonally controllable properties | ||||
| ✗ Difficult to introduce bioactivity | ||||
| Poly(N-isopropylacrylamide) (PNIPAM) | ✓ Temperature-sensitive, enabling to change network properties | Matsumi, Okano | 134 and 135 | |
| ✗ Difficult to introduce bioactivity | ||||
| Polyvinyl alcohol (PVA) | ✓ Biocompatible | Lee, Peppas, Lauprêtre, Al-ayah | 136–139 | |
| ✓ Tunable mechanics | ||||
| ✗ Not degradable | ||||
| ✗ Very slow to no dynamic behavior | ||||
| Polyacrylamide (PA) | ✓ Cheap | Pruitt, Jacobson | 140–143 | |
| ✓ Tunable mechanics | ||||
| ✗ Bioactivity and dynamics are limited | ||||
| ✗ Monomeric precursors are toxic, which can be problematic for cell culture | ||||
| Hybrid | Alginate + PEG | ✓ Processability | Chaudhuri, Baker | 148 and 149 |
| ✓ Dynamic mechanical properties | ||||
| ✓ High level of control | ||||
| ✗ Possibilities for cell adhesion limited | ||||
| ✗ Challenging to find matching concentrations/conditions of individual components | ||||
| Fibronectin + PEG | ✓ Ability to bind and retain growth factors; highly bioactive | Salmeron-Sanchez | 150 | |
| ✗ Challenging to find matching concentrations/conditions of individual components | ||||
| Collagen + PEG | ✓ Tunable mechanical properties | Myung, Gu | 153 and 154 | |
| ✓ Transparent | ||||
| ✗ Challenging to find matching concentrations/conditions of individual components | ||||
| Synthetic supramolecular | Peptide amphiphiles (PAs) | ✓ Control over dynamics | Stupp, Mata | 185–192 |
| ✓ Responsive to external stimuli (pH, salt etc.) | ||||
| ✓ Tunable morphology, mechanics and dynamics | ||||
| ✗ Bioactivity limited to short peptide mimics | ||||
| Self-assembling peptide hydrogels (SAPHs) | ✓ Precise control over material properties by tuning pH and ionic strength | Saiani | 193 and 194 | |
| ✗ Limited mechanical strength | ||||
| Engineered proteins | ✓ Highly tunable | Tirrell | 26 and 27 | |
| ✗ Structural information of assembled proteins often unknown | ||||
| Elastin-like proteins (ELPs) | ✓ Highly tunable properties by tuning assembly conditions, lower critical solution temperature (LCST) or by changing amino acid sequence | Heilshorn | 184, 203–205 | |
| ✗ Synthesis can be challenging | ||||
| Bis-urea (BU) | ✓ Tunable fiber morphology | Sijbesma, Palmans | 207–210 | |
| ✓ Complex mechanical properties, like stress stiffening | ||||
| ✗ Bioactivity limited to short sequences | ||||
| Benzene-1,3,5-tricarboxamide (BTA) | ✓ Fiber morphology as well as gel mechanics, dynamics and bioactivity orthogonally tunable | Meijer, Baker, Dankers | 213–225 | |
| ✓ Highly dynamic on molecular, fiber and network level | ||||
| ✗ Bioactivity limited to short sequences | ||||
| Ureido-pyrimidinone (UPy) | ✓ Dynamics across different length scales - on molecular, fiber and network level | Dankers, Meijer | 227–231 | |
| ✓ Orthogonal control over gel properties | ||||
| ✓ Highly modular | ||||
| ✗ Synthesis of supramolecular monomers can be challenging and expensive | ||||
| ✗ Hands-on lab experience required |
The dECM, which is used most often for cell culture is Matrigel. Matrigel is a gelatinous mixture originated from murine Englebreth–Holm–Swarm tumors that primarily consists of laminin, collagen IV and enactin.81,82 Besides these fiber-like structures that mainly provide physical support to cells, Matrigel also consists of a variety of growth factors and enzymes responsible for sending chemical information to cells. Despite Matrigel's excellent biological properties, Matrigel's ill-defined composition, tumor sourcing, complexity and high batch-to-batch variability often leads to poor reproducibility in experiments and prohibits a clean path to clinical use, while it also lacks the mechanical tunability that synthetic matrices offer.82,83
Other natural polymers can be categorized into protein-based, like collagen, carbohydrate-based, like alginate, or fatty-acid based, like fatty-acid amides derived from naturally occurring amino acids (although in organic solvents).
For the protein-based polymers, collagen restitutes the main component of the natural ECM, consisting of amino acids that are linked via peptide bonds and form a triple helix via supramolecular interactions (Table 1 and Fig. 2A). Reconstituted collagens normally come from animal sources, with collagen I the most abundant form.84 The exact mechanical aspects of the hydrogel depend on the concentration, ionic strength, temperature, pH, introduction of chemical crosslinks, dehydrothermal or UV-treatment.85–87 Besides its bulk mechanical stiffness, collagen possesses dynamic mechanical properties in the form of strain-stiffening behavior,88 caused by fibers that align in the direction of the applied strain, causing a transition from bending to streching.89 In addition to strain-stiffening, collagen shows strain enhanced stress relaxation; higher strains do not only increase the stiffness of the network, but also increase the relaxation speed of internal stress dissipation, a mechanism proposed to be important for cellular interactions (Fig. 2A).90 When subjected to compression and tension tests, collagen also shows plastic behavior, i.e. unrecoverable deformation, caused via stretch depended deformation of the weak crosslinks in the network.91,92 Plasticity gives cells the ability to generate permanent channels in the network through pulling forces and therefore increase the ability for cell migration.92
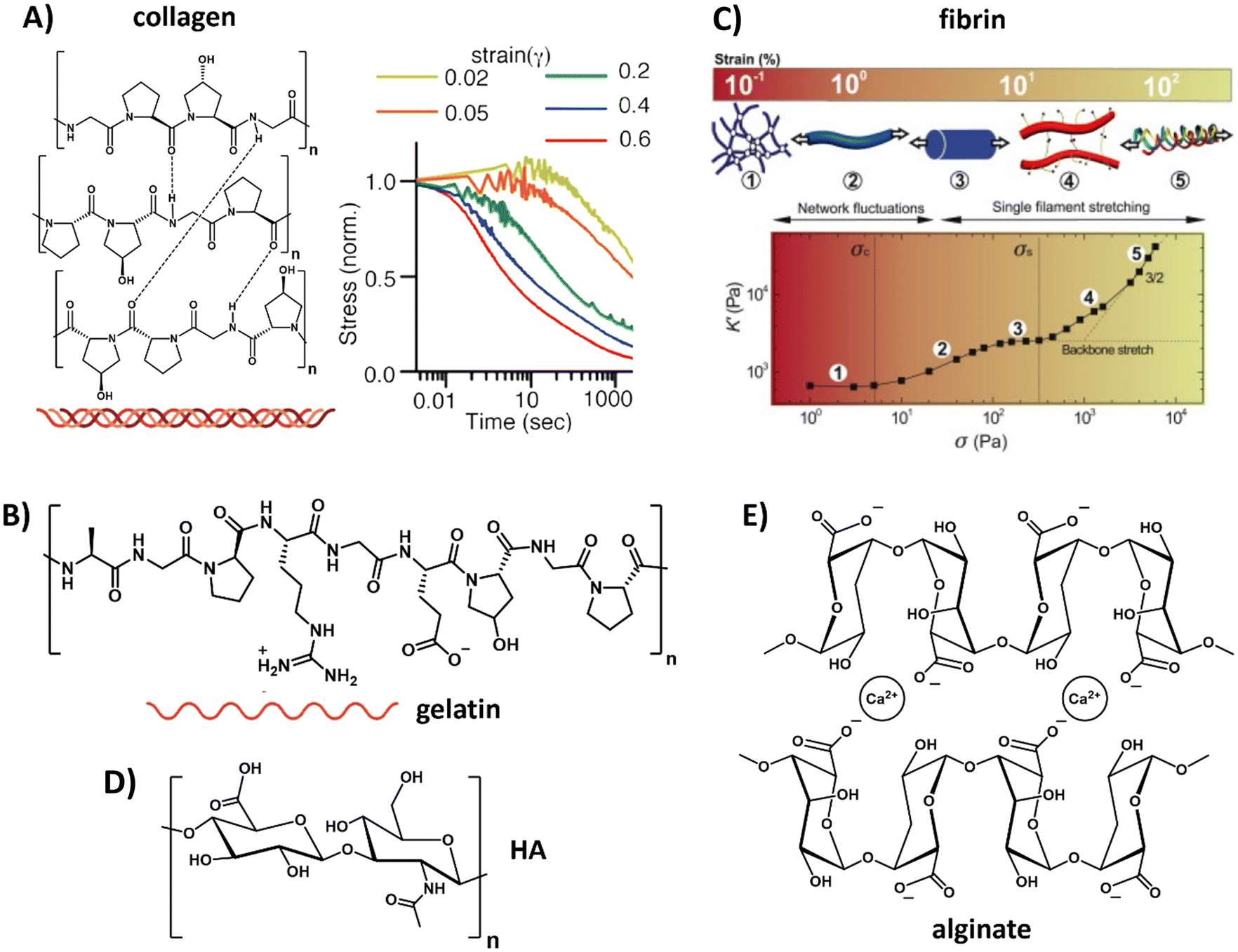 | ||
| Fig. 2 Natural derived hydrogels. (A) Chemical structure of collagen, composed of three fibers that bundle into a helix. Collagen exhibits strain-enhanced stress relaxation, i.e. at increasing strains, dissipation of the internal stress proceeds faster. Left part, adapted with permission from ref. 93, right part, adapted with permission from ref. 90. Available under a CC-BY 4.0. Copyright 2021 ACS. (B) Chemical structure of gelatin, obtained when the triple helix of collagen is broken into single fibers. Adapted with permission from ref. 93. Available under a CC-BY 4.0. Copyright 2021 ACS. (C) Composition and architecture of fibrin, which is composed of bundles of protofibrils, which in turn are assembled out of two strands of fibrin monomers. This architecture gives fibrin a unique stress stiffening response: first, thermal energy fluctuations between crosslinks are dissipated, after which the fibers themselves are stretched and crosslinks might get broken. Adapted with permission from ref. 94. Copyright 2010 Elsevier. (D) Chemical structure of hyaluronic acid (HA). (E) Chemical structure of alginate. | ||
A variation to collagen is gelatin, which can be obtained out of collagen by breaking the triple helix structure into single fibers (Table 1 and Fig. 2B).93 Similar to collagen, gelatin possesses great biocompatibility and, in addition, can easily form a gel by lowering the temperature. However, gelatin suffers from batch-to-batch variations and scalability. The main drawback of gelatin as compared to collagen is that the disruption of the helical structure limits the physical strength of the gel as well as its mechanical performance.
Another class of natural biopolymers often used in ECM mimics are polymers associated with wound healing, e.g. fibrin. Fibrin shows a unique hierarchical supramolecular protein structure (Table 1 and Fig. 2C).94 As with other natural ECM polymers, fibrin hydrogels possess stress relaxation and stress stiffening.90,94 Unique about its stress stiffening is the presence of various stress-stiffening regimes. At small stresses, the crosslinks in the network (that bend due to thermal fluctuations) are stretched out, leading to a linear stress–strain correlation.94 At increasing stress the network stiffens non-linearly due to an entropic stiffening response of the semiflexible protofibrils and flexible linker chains in the network. At even higher stresses, fibers are stretched axially and crosslinks can be broken under increasing force without disrupting the overall network architecture (Fig. 2C).94
Belonging to the class of carbohydrate-based natural polymers, hyaluronic acid (HA) is one of the proteoglycans in the ECM, i.e. a linear polymer made from repeating disaccharides glucuronic acid and glucosamine (Table 1 and Fig. 2D). A very comprehensive review on HA, its properties and applications was recently written by the Vemula lab.95 HA can be used in its native form, but is more recently often used as HA-conjugate and in HA-modified systems. HA is growing as a very interesting ECM mimicking material and biomaterial because of its therapeutic features, having an influential role on ECM organization and remodeling, cell growth and differentiation, promoting wound healing and modulating immunological processes like inflammation. Changes in molecular mass of HA are known to influence molecular and cellular signaling mechanisms and ECM remodeling processes. In addition, variations in HA's molecular sequence cause changes in the structure, distribution and density and allows for the binding of specific ligands or growth factors.96 Furthermore, the negative charge of proteoglycans attracts sodium ions and thereby water via osmose. This keeps the cells hydrated but the water also provides resistance against compression.97 Hydrogelation can be induced via various pathways, i.e. crosslinking,98,99 light radical polymerization,100 esterification101 or annealing as water is removed.102 Although HA shows very good compatibility, it has limited mechanical strength and requires multiple purifications steps before it can be used safely without immunogenic side reactions.103,104
Another carbohydrate-based polymer that is found in the human body in cartilage is chondroitin sulfate (CS), being a glycosaminoglycan consisting of alternating repeating units of N-acetyl galactosamine and glucuronic acid. The Varghese lab has ample expertise with CS as biomaterial to study fundamental cell–material interactions and especially for growing cartilage tissue and inducing chondrogenic differentiation.105,106 Because of CS's hydrophilic nature and low mechanical strength, it has frequently been functionalized and mixed with other polymers to improve its properties for regenerative medicine applications.107
Other carbohydrate-based natural polymers, but not found in our native ECM, include alginate, agarose and chitosan. Alginate, for example, is a linear copolymer consisting of mannuronic acid and guluronic acid. The two sugar monomers can be either present as repeating or alternating blocks,108,109 which allows for varying mechanical properties as well as varying pore size via tuning the ratio between the two monomers (Table 1 and Fig. 2E).110 In addition, control over the mechanical and dynamic properties is possible by using different concentrations of calcium together with different molecular weight alginate, as shown by Chaudhuri et al.64 Alginate can easily be obtained, has limited toxicity and forms gels conveniently via the use of divalent cations (e.g. calcium or barium).109 However, alginate's degradation cannot be fully controlled and happens unpredictably over time due to ion exchange of calcium with surrounding media, causing chains to dissolute.111 In addition, cell adhesion to alginate is limited and requires functionalization for protein binding and attachment of cells.112,113
The Pal and Bhattacharya labs have great expertise with fatty-acid and sugar-derived hydrogels, where they systemically studied the physical gelation mechanisms of these natural components (Table 1). To illustrate, different fatty-acid amides derived from naturally occurring amino acids were designed and synthesized and it was found that especially the alanine-derived moieties were efficient gelators (in organic solvents), because of their ability to self-assemble into layered structures.17 Increasing fatty acid chain lengths improved gel mechanical strength by increasing the contribution of van der Waals interactions. In addition, they created various aqueous hydrogels based on sugar derivatives as natural components, being more compatible for future cell culture applications. For example, they reported the gelation mechanisms and behavior of sugar-based azobenzene gels as influence of pH and salts.114
To increase the complexity of natural ECM mimics, there is a movement towards recreating the multi-component nature of the native ECM. Therefore, hybrid gels are being developed, where two gels with different properties (i.e. one labile and one slow degrading gel) are mixed.115 For example, Koenderink et al. showed the importance of an increase in complexity by combining two natural ECM components, i.e. collagen and hyaluronan.71 The interaction of hyaluronan inside and around the collagen fibers created a soft hydrated matrix, interacting and stabilizing collagen. Additionally, the presence of hyaluronan shifted the stress stiffening response: it (1) lowered the stress sensitivity as the onset strain increased upon inclusion of hyaluronan (i.e. the strain at which the non-linear stiffening starts) and it (2) resulted in a great increase of the linear elastic modulus (G°) well above the sum of moduli of the individual collagen and hyaluronan gels.71 Another example is the combination of alginate and reconstituted basement membrane in presence of calcium.116,117 By increasing the calcium concentration, (1) stiffer networks and (2) hydrogels with increased plasticity could be formed, while maintaining the network's architecture. We will elaborate on the design strategies and properties of hybrid hydrogels later, but completely synthetic covalent hydrogels are first discussed in full detail.
Synthetic covalent hydrogels
To overcome the limitations of natural-derived matrices, including batch-to-batch variability and poor reproducibility, a growing interest arose towards fully synthetic hydrogels. Synthetic gels possess tunable mechanical properties and degradation rates, while bioactivity can be introduced by incorporating functional moieties and small peptides.118 Furthermore, they allow for full control over their input parameters (e.g. type of fibers, ligand, concentrations etc.), such that the influence of a single element on cellular behavior can be assessed. In fact, many of the seminal contributions to cell–matrix interactions come from synthetic systems, and they remain both a useful tool for scientists and a promising option for clinical translation.The most important synthetic hydrogels for biomedical applications have been designed around poly(ethylene glycol) (PEG), a hydrophilic polymer which can retain large amounts of water,119 and is relatively biocompatible and non-toxic (Table 1).120 To form hydrogels, PEG polymers are usually functionalized with reactive end groups, e.g. azides, alkynes, maleimides, norbornenes, thiols, alkenes or acrylates.121,122 To promote cell adhesion, cell binding groups like RGD, GFOGER and catechol can easily be introduced to the artificial matrix through the aforementioned click chemistry strategies or functional groups.123–127 Further control over the morphology of these hydrogels can be achieved by using star or branched derivatives.128 Mechanical advantages of PEG lay mainly in the possibility to tune a large range of elastic moduli (from ∼100 Pa to >100 kPa).65,120 This scaling ability depends however largely on the crosslink density, thereby simultaneously affecting the diffusion and the structure of the network. While good control over mechanical properties is possible, PEG based hydrogels usually lack dynamic properties in terms of stress relaxation, stress stiffening or self-healing, mainly due to the covalent structure of the network mesh. Adding matrix metalloproteinase (MMP)-degradable crosslinks to PEG networks can however deliver cell-responsive degradability into this synthetic matrix.127
An oligo(ethylene glycol) OEG based polymer with more control over dynamic properties is PIC, synthesized from isocyanopeptides grafted with OEG side chains (Table 1 and Fig. 3A).129,155 The resulting polymer possesses tunable lower critical solution temperature (LCST) behavior, as well as a tunable bulk stiffness, which can both be controlled by the length of the polymer chains.130 (Fig. 3B). Next to that, once above the LCST, the hydrogel shows stress-stiffening behavior in the same mechanical regime as natural fibers (e.g. collagen), making PIC very suited for mimicking the complex dynamic mechanical aspects of the ECM.130 Control over the stress stiffening properties can be achieved by changing the length of the polymer chains, concentration, temperature or by incorporating supramolecular particles (Fig. 3B and C).131 Cell spreading behavior can be controlled by varying PIC contour length (Lc), i.e. the length of the polymer chain, which determines the stress stiffening properties (Fig. 3D).132 A higher degree of cell spreading with lower circularity (which is desired) was observed for human adipose derived stem cells in hydrogels that contained smaller critical stresses (i.e. the stress required before stiffening of the PIC polymers starts). Controlled dynamics in terms of stress relaxation and self-healing are however lacking and although introduction of bioactive signals can be achieved,133 the possibilities are limited due to solubility issues.
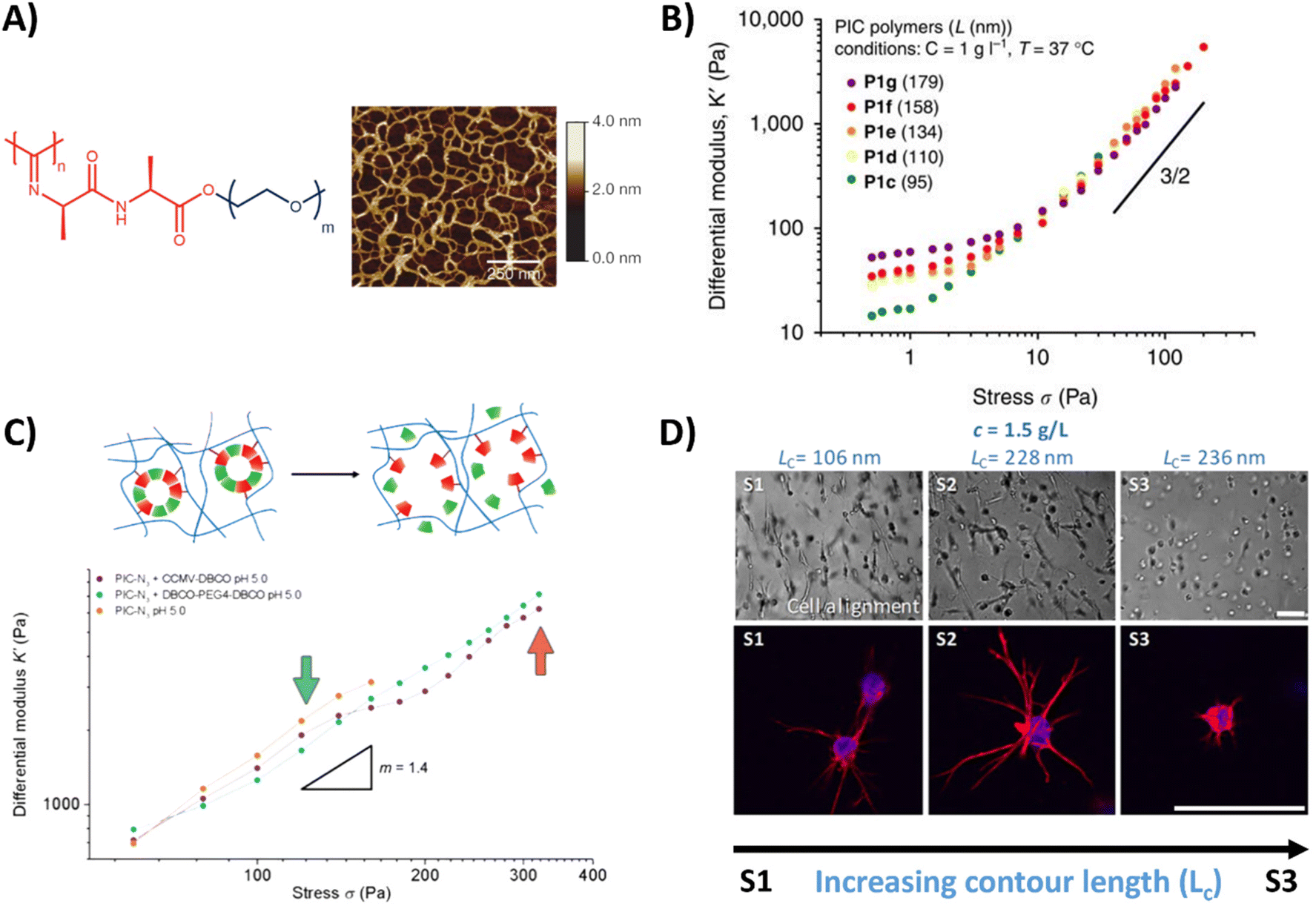 | ||
| Fig. 3 PIC hydrogel and its mechanical properties. (A) Chemical structure of the PIC hydrogel and AFM image showing the bundle like meshes, formed once heated above the LCST. Adapted with permission from ref. 155. Copyright 2013 Springer Nature. (B) Stress stiffening properties of PIC hydrogels with different polymer length, showing that the bulk plateau modulus and the critical stress can be increased by increasing the polymer length. Adapted with permission from ref. 130. Available under a CC-BY 4.0. Copyright 2014 Springer Nature. (C) Stress stiffening properties of PIC hydrogels crosslinked with supramolecular particles. Adapted with permission from ref. 131. Copyright 2018 Royal Society of Chemistry (RSC). (D) Brightfield and fluorescence images of hASCs, showing that cell spreading depends on PIC contour length (Lc) and thus the stress stiffening properties. Scale bar is 70 μm. Adapted with permission from ref. 132. Available under a CC BY-NC-ND 4.0. Copyright 2019 ACS. | ||
Poly(N-isopropylacrylamide) (PNIPAM) is a widely used polymer for hydrogels due to its thermodynamic properties. Similar to PIC, PNIPAM possess a LCST, above which the polymer network collapses and undergoes a transition from a hydrated coil to a dehydrated globule (Table 1 and Fig. 4A).134 As the LCST is around 32 °C, cells can easily be detached from the gel by lowering the temperature.135 Direct incorporation of bioactive molecules in PNIPAM is however challenging and is often achieved by copolymerization with other polymeric systems. As the polymer simply collapses above its LCST, precise control over the mechanical properties is often difficult.
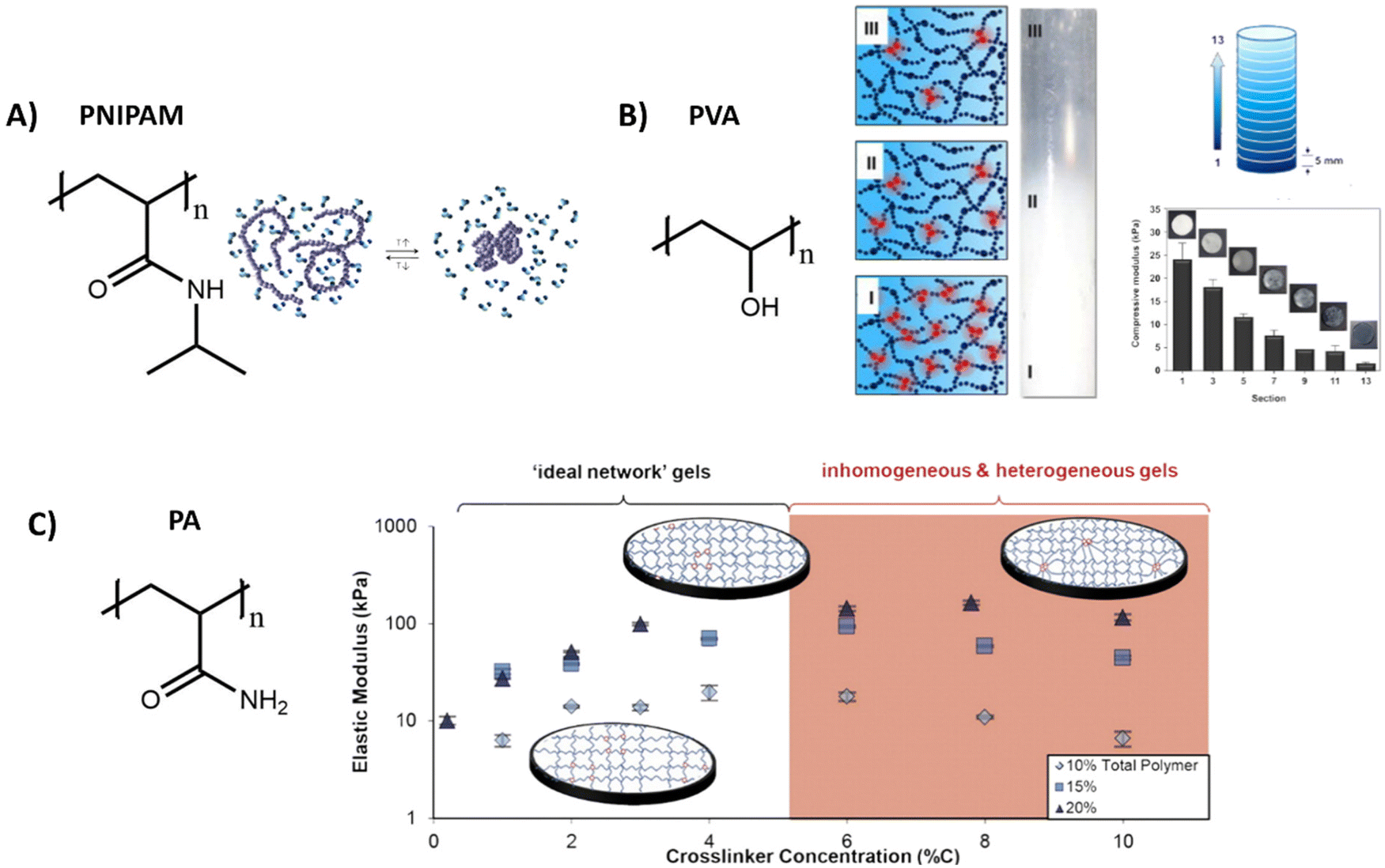 | ||
| Fig. 4 Various structures and applications of synthetic hydrogels. (A) Chemical structure of PNIPAM, which can undergo a reversible coil to globule transition as influence of temperature. Adapted with permission from ref. 144. Copyright 2012 Elsevier. (B) Chemical structure of polyvinyl alcohol (PVA), which forms hydrogels with various stiffnesses by tuning freezing and thawing cycles, allowing for the creation of hydrogels with a stiffness gradient. Adapted with permission from ref. 136. Copyright 2015 Elsevier. (C) Chemical structure of polyacrylamide (PA). Varying the crosslinker concentration gives control over the gel stiffness, which has an optimum after which the stiffness decreases due to inhomogeneities. Adapted with permission from ref. 141. Copyright 2016 ACS. | ||
Another widely used synthetic polymer is polyvinyl alcohol (PVA), due to its biocompatibility and tunable mechanical properties (Table 1). PVA is a simple polymer carbon chain with hydroxyl side groups and can be obtained via hydrolysis out of polyvinyl acetate (Fig. 4B). A big advantage of PVA is that it can be crosslinked without using chemical crosslinkers, but by simply using freeze/thaw cycles to induce PVA crystallite formation.136 The mechanical properties of PVA hydrogels can be tuned through changing the polymer concentration, freezing temperature and by varying the number of freeze–thaw cycles.137,138 By gradual freezing and cooling, Kim et al. even created a stiffness gradient in the polymer network, (Fig. 4B)136 yielding an ideal material to study cell migration behavior. PVA however is not degradable and lacks molecular dynamics (movement of monomers), since the monomers are covalently bound to each other,104 but does exhibit slow bulk dynamics (stress relaxation, fiber rearrangements) with <5% relaxation in 5 minutes – likely due to OH hydrogen bonding rearrangements.139
Another class of synthetic gels are polyacrylamide (PA) gels, which are inexpensive and often used to study the influence of stiffness on cell behavior (Table 1).140 PA gels are fabricated by reacting acrylamide monomer and bis-acrylamide crosslinker, in presence of ammonium persulfate and tetramethyl ethylenediamine as initiator for redox radical polymerization (Fig. 4C). By increasing the crosslinker concentration, an increase in mechanical stiffness can be achieved, until an optimum is reached after which the stiffnesses decreases (Fig. 4C).141 Polyacrylamide itself does not show any bioactivity, but it can be introduced by using a bifunctional crosslinker, like sulfo-SANPAH, via hydrazine modification,142 or by simple RGD coupling.143 However, as with the previous synthetic systems, bioactivity and dynamics of these systems are limited and 3D cell culture might be problematic as the monomeric precursors are toxic before polymerization.
The best of both worlds – hybrid hydrogels systems
Although naturally-derived hydrogels are substantially biocompatible, their lowest possible stiffness is limited by the minimum concentration needed for complete hydrogel network formation and at the higher end by the solubility of the natural-derived component.145 Furthermore, the rational design, tailorable bioactivity, and processability is often limited to narrow ranges. On the other hand, synthetic materials often lack in bioactivity. Therefore, the field has started to design hybrid hydrogel systems, which combine the biological and mechanical power of naturally-derived hydrogels with the tailorability of synthetic methodology. This can be as simple as mixing a natural and synthetic network together, or the introduction of smart synthetically modified natural polymers into the hydrogel.146,147 However, due to the limited range of solvents, temperatures, and concentrations it is challenging to combine natural and synthetic polymers within one hybrid hydrogel.145 Examples of some of the most used designs and properties of hybrid hydrogels are presented in Fig. 5 and discussed below.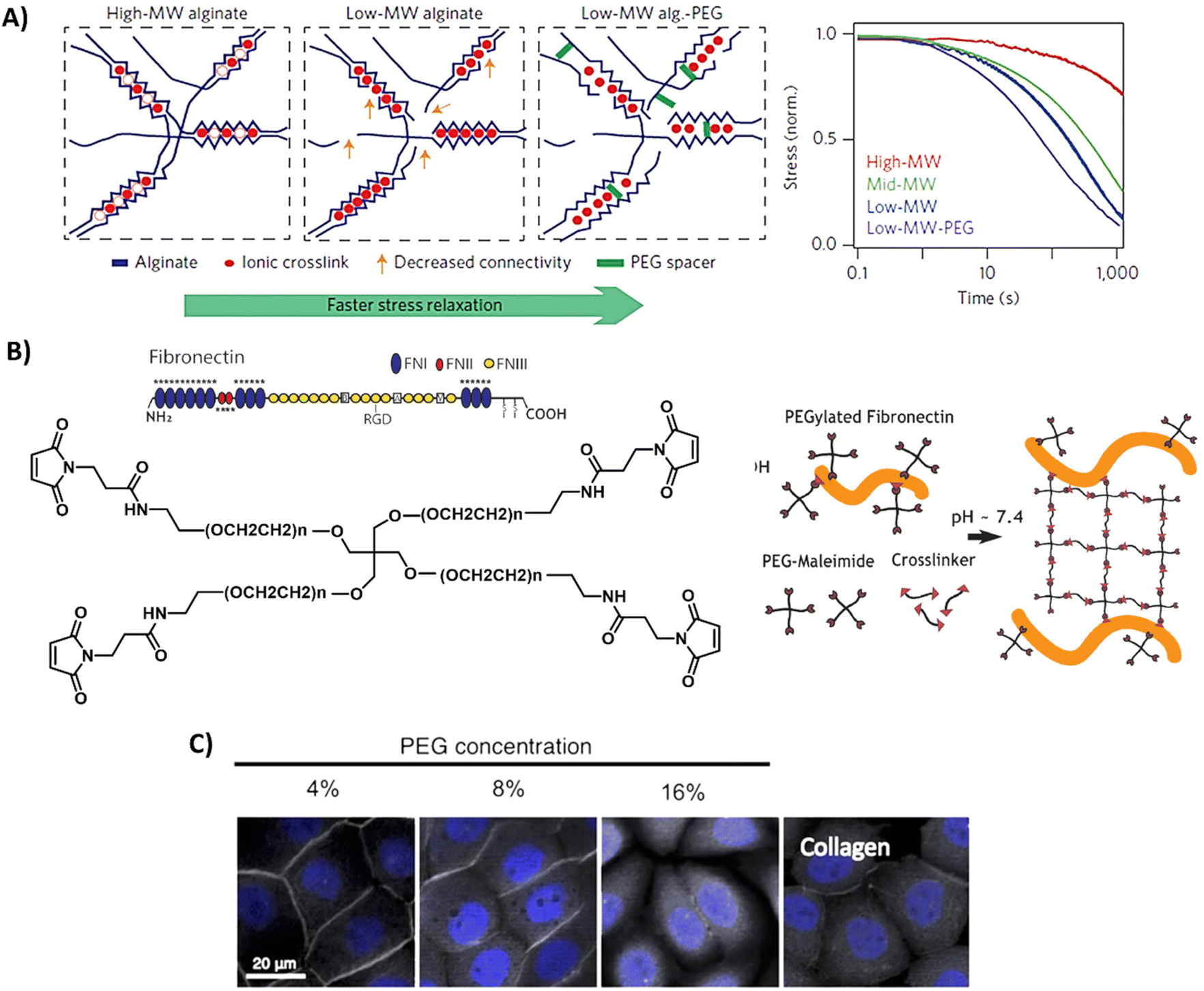 | ||
| Fig. 5 Design and properties of hybrid hydrogels. (A) Gels based on alginate and PEG, with tunable stress relaxation independent of the bulk stiffness. Adapted with permission from ref. 64. Copyright 2015 Springer Nature. (B) Gels based on fibronectin and PEG. Adapted with permission from ref. 150. Available under a CC-BY 4.0. Copyright 2020 Elsevier. (C) Gels based on collagen of which the primary amines are crosslinked via NHS moieties on multi-arm PEG polymers, showing successful protein expression of ZO-1 (grey) of immortalized corneal epithelial cells and aSMA (yellow) in corneal stromal stem cells after 2-day cell culture on PEG-collagen hydrogels. Adapted with permission from ref. 153. Available under a CC-BY 4.0. Copyright 2020 Springer Nature. | ||
Alginate is often used in hybrid hydrogels to impart processability and control the dynamic mechanical properties. By covalently coupling PEG to alginate, Chaudhuri et al. formed hydrogels with similar mechanical stiffness, but varying stress relaxation and creep (Table 1 and Fig. 5A).64,148 High stress relaxation enhanced cell spreading owing to the cells remodeling their substrate. Double network alginate-PEG systems have also been developed, enabling the formation of a tough injectable matrix. This approach allowed the investigation of cell adhesion kinetics on an identical hydrogel composition, with different molecular attachment of adhesive ligands—highlighting the importance of the molecular network on cell adhesion.149 Other hybrid gels based on PEG include mixes of a fibronectin network consisting of 4-arm-PEG-maleimide, crosslinked with PEG-dithiol and thiolated protease-degradable peptides as developed by Trujillo et al. (Table 1 and Fig. 5B).150 Owing to the ability of ECM proteins, like fibronectin, to bind growth factors (see previous section), this hybrid hydrogel possesses the ability to recruit and retain VEGF and bone morphogenic protein 2 (BMP2), outperforming plain PEG in growth factor retention, and herewith providing a highly controllable 3D environment to promote bone regeneration and vascularization (Table 1).150 The Seliktar lab made mixes of PEG with albumin, fibrinogen and gelatin, revealing that the PEG-proteins mixes contain different degradation rates and mechanisms as compared to PEG only.151 PEG could also be mixed with fibrin to benefit from fibrin's bioactivity.152 In addition, these hybrid fibrin/PEG hydrogels showed slower degradation as compared to pure fibrin gels, revealing that PEG prevents fast degradation of fibrin. As collagen is one of the most abundant ECM proteins, Fernandes-Cunha et al. designed in situ forming hybrid hydrogels crosslinked with various concentrations of collagen (4, 8, and 16% v/v PEG to collagen) with 4-arm and an 8-arm PEG-NHS (Table 1 and Fig. 5C), providing hybrid hydrogels with different mechanical properties and transparency.153 All hydrogels supported corneal epithelial and stromal cells to proliferate, to adhere and to express desirable cell morphology, but best results were obtained using a 4-arm crosslinker (independent of stiffness), showing the influence of structural morphology on cellular behavior.153 This was also previously observed and hypothesized to be caused by differences in collagen fiber alignment after PEG crosslinking.154
Other hybrid hydrogels consisting of a covalently crosslinked bovine serum albumin (BSA) network reinforced with non-covalently adsorbed polyelectrolytes were studied by Khoury et al. Besides the reinforcement and large stiffening effect, the non-covalently attached polyelectrolytes can create and break local bonds, allowing the gels to heal any structural damage and function as shape memory.145 The ability of these gels to self-heal can be beneficial for cells: for example in a scenario where gel rupture occurred as a result of cell pulling forces, and after self-healing, the gel is able to provide mechanical support again to the cells.
A special synthetic polymer is the polyisocyanide (PIC),155 one of the few synthetic polymers which exhibit stress stiffening: when subjected to mechanical stress, the polymer becomes stiffer and more resistant to deformation. However, the solubility and limited bioactivity of PIC limits its wide use. Therefore, hybrid versions are being developed, for example by combining PIC with Matrigel, as done by the Kouwer lab,156 exhibiting both stress stiffening properties and good bioactivity, while possessing great control over the gel through carefully tuning the ratio of the two materials.
And lastly, MMP cleavable peptides are interesting crosslinkers in synthetic materials, imparting a biomimetic degradation mechanism. Jha and coworkers studied the influence of degradation kinetics of MMP crosslinkers on biological outcomes of a cell laden hybrid hydrogel.157 RGD-functionalized HA, heparin and different MMP cleavable peptides were used to tune hydrogel degradation rate. Interestingly, the hydrogels crosslinked with the slow MMP-degradable peptides supported the cardiac progenitor cell (CPC) survival, proliferation, and endothelial cell differentiation.157
Grafting on natural or synthetic polymers – combining both worlds within the same macromolecule
While the combination of natural and synthetic polymers within one hybrid hydrogel can be limited due to solubility issues, chemical modification of natural polymers can overcome this issue and broaden the tools for designing hybrid hydrogels. Synthetic and natural polymers modified with biomolecules are also included in this category. Among the variety of chemical strategies, the synthesis of photo-polymerizable derivate (e.g. via methacrylation) is the most common tool for obtaining tunable mechanical properties and spatial and temporal control of crosslinking. Other common strategies to prepare photo-polymerizable polymers is the addition of norbornene, allowing photo-initiated thiol–ene reactions.158,159 The modular design of this strategy allows for the tailorability of mechanical performance via the structure of the thiol cross-linker and cross-linking density,160 while still allowing for bioactivity through for example RGD-cysteine.While irreversible covalent cross-linking enables tunable hydrogel mechanics and degradation, the use of reversible interactions, both dynamic covalent and non-covalent ones, results in viscoelastic properties. Dynamic covalent cross-linked hydrogels can tailor viscoelasticity and stiffness independently, allowing to decouple the effects of stress relaxation and stiffness on cellular behavior.
Complex hybrid hydrogels can be designed through combining cross-linking chemistries and polymers. For example, Li et al.161 developed double dynamically cross-linked hydrogels based on PEG, poly(vinyl alcohol) (PVA), and glycol chitosan (GC). PEG was functionalized with benzaldehyde and phenylboronic acid as end groups, thus, this macromolecule reacts with both PVA and GC via borate ester and imine formation, respectively. The dynamic nature of both cross-linking renders self-healing and shear-thinning behavior, promising features for injection applications.
Synthetic supramolecular biomaterials
A recent and growing interest lies in mimicking the dynamic properties of the ECM, as the ECM itself is highly dynamic and predominantly held together via non-covalent interactions.60 Among these dynamic features, stress relaxation properties have recently come to the forefront, as a rising number of studies show the important effect of stress relaxation on cell shape and more complex biological function.148,162–167 Next to these macroscopic dynamic properties, attention for the importance of molecular dynamics is also growing.168–171 Current synthetic covalent matrices however have long stress relaxation times, of hours.64 Yet, the relaxation half times of most of our tissue cover only the range of seconds to hours.64,65Supramolecular assemblies based on non-covalent interactions, are arising as very promising candidates as ECM mimics owing to their inherent controllable dynamics, adaptability, tunability and control over mechanical and bioactive properties.172,173 Supramolecular interactions are directional, and examples include hydrogen bonds, π–π interactions and hydrophobic forces to form fiber-like structures, similar to nature. To introduce function into these gels, bioactive cues can be coupled to the monomeric building blocks and mixed into the supramolecular fibers.174–176 By co-assembling different supramolecular monomers, it is possible to tune ligand presentation177 as well as morphology of the supramolecular structure.178 Additionally, the supramolecular formulation procedure used greatly impacts the type of supramolecular structure that is formed, ranging from fibers to hydrogels, elastomers and solid meshes.179,180 For ease of discussion, we here discuss promising classes of synthetic supramolecular biomaterials, starting with (1) peptide amphiphiles (PAs), peptides and proteins, followed by (2) structures based on self-assembling supramolecular monomers.
Firstly, PAs, peptides and engineered proteins are a class of aqueous, self-assembling structures, on which the Stupp, Tirrell, Heilshorn and Besenius and other labs have made great effort in their design, synthesis and characterization (Table 1 and Fig. 6).26,181–184
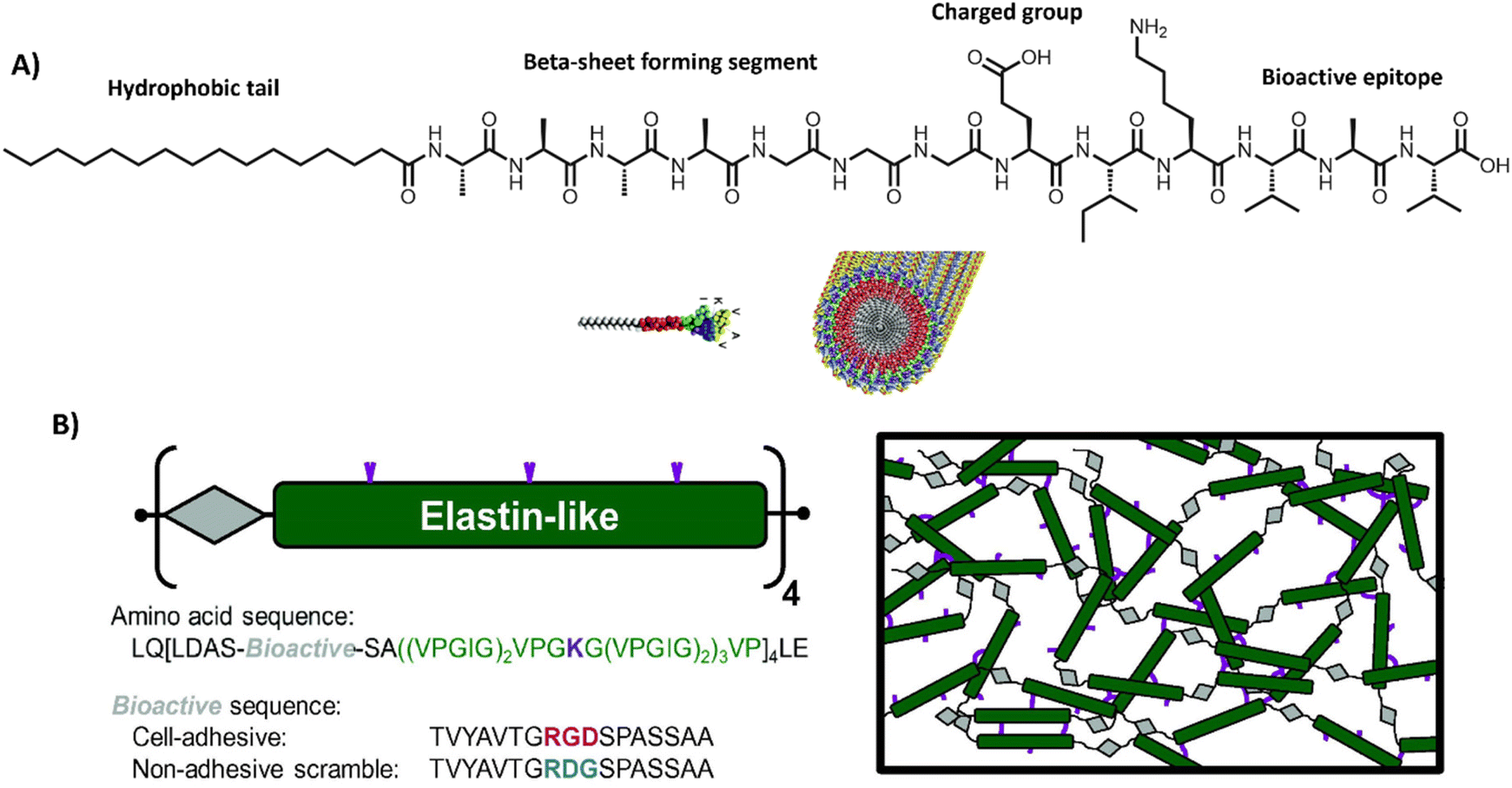 | ||
| Fig. 6 Peptide amphiphiles and engineered proteins. (A) PAs based containing the RADA sequence, which assemble into stable β-sheets. Adapted with permission from ref. 206. Available under a CC-BY 3.0. Copyright 2012 Elsevier. (B) ELPs designed by repeating units of a bioactive sequence (grey), which can be cell adhesive or non-adhesive and an elastin-like sequence (green), which can assemble into hydrogels, often via additional crosslinks, e.g. lysine (purple). Adapted with permission from ref. 184. Available under a CC BY-NC-ND 3.0. Copyright 2013 Elsevier. | ||
PAs are amphiphilic building blocks that usually consist of a hydrophobic domain, functionalized with a more polar peptide sequence (Table 1 and Fig. 6A). The hydrophobic domain causes a hydrophobic collapse as well as aids in shielding water, whereas the more hydrophilic amino acids provide more directional hydrogen bonds.185 Charged amino acids could be installed to introduce responsiveness towards external stimuli, such as pH186 or salt concentration.183,187 Additionally, short bioactive moieties, e.g. IKVAV,188 could be used to achieve cellular binding.187,189 By varying the molecular design of the different regions within the PA (i.e. only tuning the hydrophilic part, or only the hydrophobic region, or both simultaneously),190 it is possible to control the assembly's morphology, structure and mechanical properties.191 One of the most well-known PA-based, fibrous hydrogels is PuraMatrix, forming stable β-sheets through a combination of electrostatic and hydrophobic interactions.20 More recently, multicomponent, PA-based supramolecular hydrogels are being developed for applications like regenerative medicine – as pioneered by the Mata lab.34,35,192
Self-assembling peptide hydrogels (SAPHs) are variations on PAs, consisting of shorter sequences, typically only a few amino acids, such as RADA (Table 1).193 Another well-known sequence, created by Saiani lab, is FEFEFKFK, forming anti-parallel β sheets in dilute state and self-holding hydrogels at higher concentrations above its critical gelation concentration. Precise control over material properties is possible by tuning pH or ionic strength of the solution.194 Recently, a growing attention and focus has arisen on the modulation of dynamics within PA structures – investigating the potential of this elusive property to control cellular behavior.168,195,196 For example, the Stupp lab demonstrated that their bioactive PAs with greater supramolecular motion (i.e. movement of monomeric peptide amphiphile within peptide fibril) outperform the more static counterparts in promoting vascular growth, axonal regeneration, survival of motor neurons and functional recovery of spinal cord injury in mice. They hypothesize that the greater internal motion of the bioactive moieties results in polyvalency, with that promoting receptor clustering to achieve effective signaling.168
Interestingly, other groups, including our own laboratory, recently also discovered the importance of molecular dynamics in supramolecular hydrogels impacting cell spreading behavior32,197 – where bioactive, supramolecular monomers must be robustly incorporated inside the supramolecular stacks, or else cells might pull out loosely bound monomers. In addition, many groups, including the Adams, Ulijn, Xu and Tuttle groups, started focusing on high-throughput options to screen different combinations of short peptide sequences (i.e. tripeptides) for their ability to self-assemble.34,198–202
To introduce more structural organization in biomaterials, elastin-like proteins (ELPs) can be utilized (Table 1 and Fig. 6B), well-studied by the Heilshorn lab.184 ELPs consist of short repetitive amino acid sequences that provide elasticity and mechanical stability. A big advantage of ELPs is that they are highly controllable and tunable, by varying amino acid sequence, LCST, assembly conditions or by introducing additional chemical203,204 or enzymatic crosslinks.205 Bioactive ELPs can be engineered by incorporating cell adhesion moieties, like RGD.184 Overall, protein-based materials are very promising candidates for studying fundamental principles underlying cell–material interactions because of their high modularity and biocompatible nature.
Lastly, self-assembling monomeric building blocks can be used to create bioactive, fibrous networks. Herein, bola-amphiphiles are frequently utilized, which are comprised of a hydrophobic core, shielded by two hydrophilic end groups (Table 1 and Fig. 7A). Similar to PAs and peptide-based materials, the shape, structure and properties of bola-amphiphilic materials is highly tunable through engineering its molecular design. The Sijbesma and Palmans laboratories designed a bola-amphiphilic monomer that consists of complementary bis-urea (BU) motifs,207–209 separated through a hydrophobic spacer and shielded with OEG blocks, yielding well-defined micellar rods once assembled in aqueous solutions (Fig. 7A). The mechanical properties including yield strain of these BU-based supramolecular structures are tunable by varying PEG length and aliphatic linker length (Fig. 7B).210
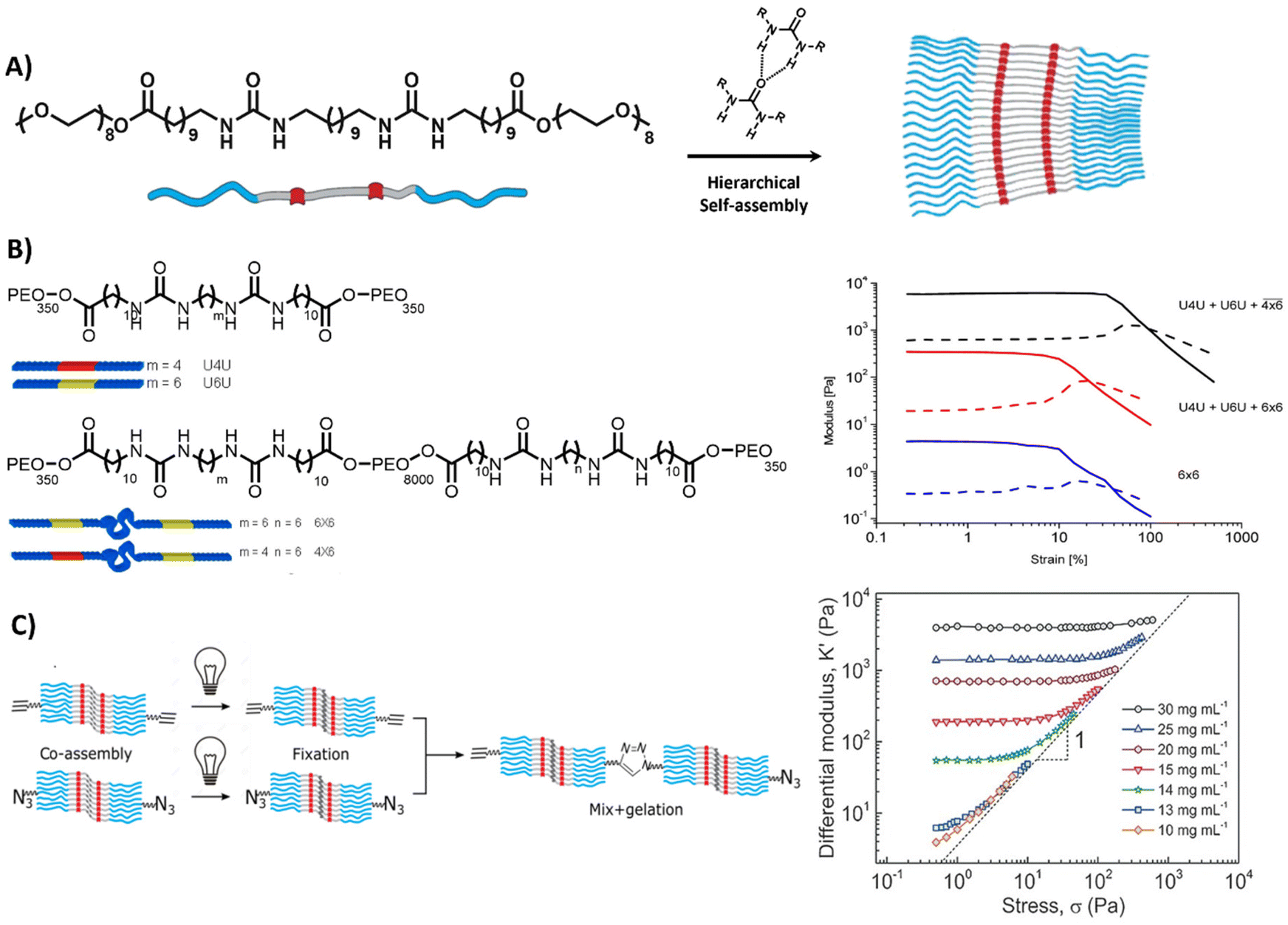 | ||
| Fig. 7 Bis-urea based hydrogel. (A) Chemical structure and morphology of bis-urea (BU) (red) building blocks, separated by an alkyl spacer (grey) and flanked by OEG chains, assembling into rod like structures via hydrogen bonds and hydrophobic interactions. Adapted with permission from ref. 211. Available under a CC-BY-NC 4.0. Copyright 2017 Wiley-VCH. (B) Chemical structure of BU molecules with varying length of OEG chains and alkyl spacer. Variations in length can be used to form different crosslinks, i.e. loops inside the fiber (intra) and links between the fibers (inter), resulting in different mechanical properties. Adapted with permission from ref. 210. Copyright 2014 ACS. (C) Stress stiffening hydrogels based on azide- and ethyne-functionalized BU building blocks to induce covalent crosslinks between the assembled rods. Adapted with permission from ref. 211. Available under a CC-BY-NC 4.0. Copyright 2017 Wiley-VCH. | ||
Complex mechanical properties could be introduced into the material through the incorporation of azide- and ethyne-functionalized BU monomers, leading to covalent crosslinks between the self-assembled rods,211 and surprisingly yielding stress-stiffening behavior (Fig. 7C). The stress stiffening behavior is hypothesized to occur through the presence of soft bending modes in bundles of BU fibers.211 In addition, the modular nature of the supramolecular system allowed for the easy incorporation of bioactive monomers by mixing it with the non-functionalized BU material.212 The ability to tune functionality as well as mechanical features (i.e. stress stiffening) in these supramolecular materials makes them interesting candidates to mimic crucial aspects of the ECM.
Another class of synthetic supramolecular assemblies are C3-symmetrical discotic molecules, among those the benzene-1,3,5-tricarboxamide (BTA) unit – well studied by the Meijer lab and Baker laboratories (Table 1).213–215 BTA monomers self-assemble through triple amide hydrogen bonding combined with π–π stacking of the aromatic cores to form supramolecular, double-helical polymers.216 To ensure self-assembly of this supramolecular motif in water, the BTA is most frequently equipped with C12 hydrophobic arms followed by tetra(ethylene glycol) (EG4) hydrophilic chains on the outer ends (Fig. 8A).217 The hydrophobic spacer has a dual function, as it shields water from the core such that it cannot compete for hydrogen bonding and the hydrophobic effect further stimulates supramolecular polymerization, while the EG4 domains provide water-solubility. The morphology of the supramolecular stacks can be tuned by varying hydrophobic chain length, as the core amides become more or less shielded from water, respectively increasing or decreasing the tendency of the monomers to hydrogen bond and assemble into longer aggregates.218 BTA fibers are highly dynamic, with a homogeneous exchange profile of BTA monomers over total fiber length.213 More recently, the BTA has been functionalized with peptides and carbohydrates, leaving the BTA a versatile platform for biomedical applications.177,219–221
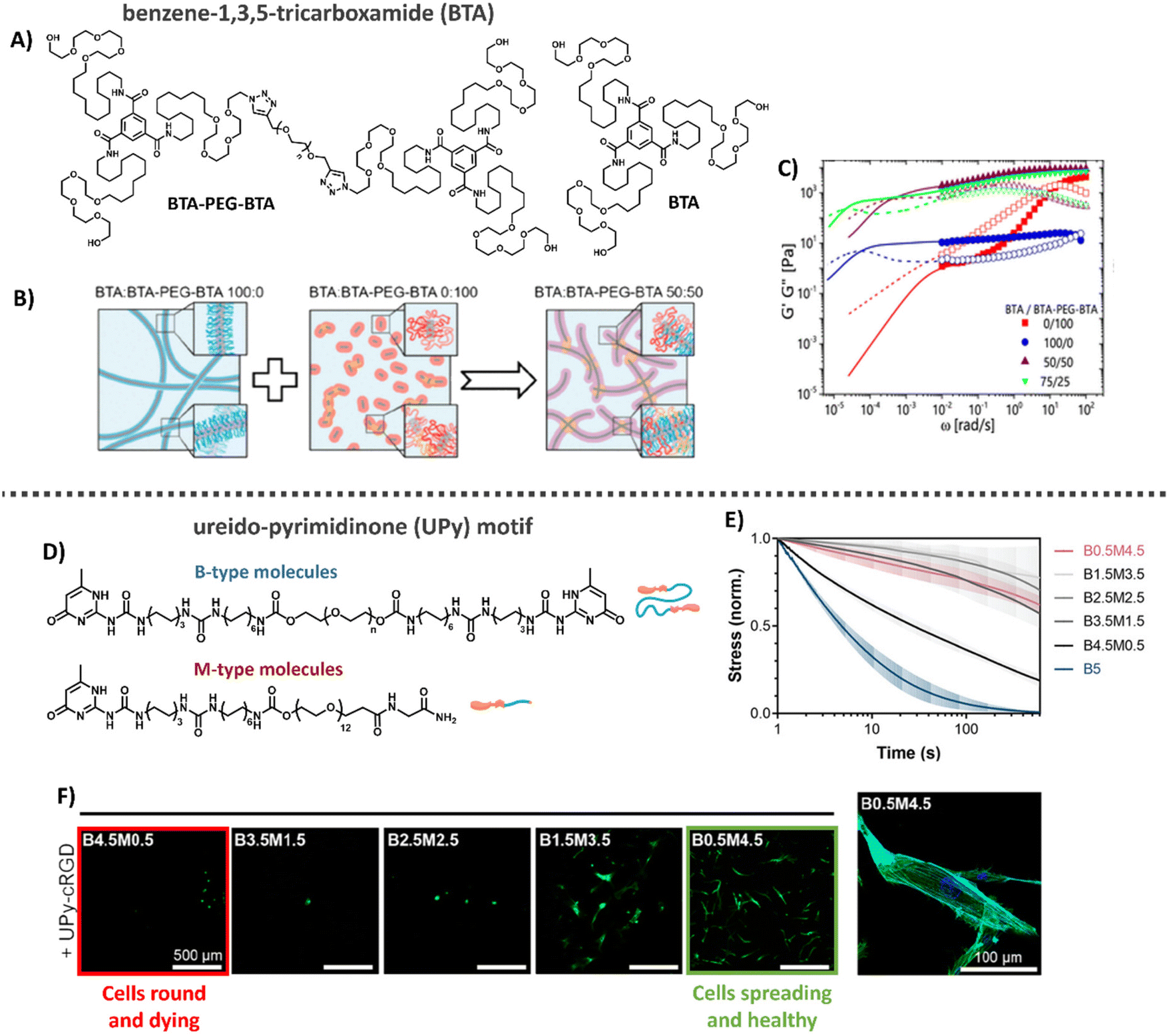 | ||
| Fig. 8 Benzene-1,3,5-tricarboxime (BTA) and Ureido-pyrimidinone (UPy)-based hydrogels. (A) Chemical structure of BTA and BTA-PEG-BTA. (B) Cartoons showing the supramolecular self-assembly, with BTA forming long entangled fibers, and the bifunctional BTA-PEG-BTA, forming micelles. Upon mixing BTA-PEG-BTA can function as a crosslink between BTA fibers. Adapted with permission from ref. 222. Available under a CC BY-NC 4.0. Copyright 2020 ACS. (C) Mechanical analysis of BTA based hydrogels, where mixing of the two different BTA molecules results in gels with tunable mechanical properties that are stable over a long range of time scales and show two different modes of relaxation. Adapted with permission from ref. 222. Available under a CC BY-NC 4.0. Copyright 2020 ACS. (D) Different UPy molecules used to form hydrogels, M-type molecules stack into long static 1D fibers and can be functionalized with a large variety of different biochemical end groups. UPy-PEG-UPy form short dynamic fibers and function as crosslinkers between M-type molecules. (E) Dynamic properties of UPy-based hydrogels. By changing the ratio between B-and M-type molecules the gel's dynamic properties can be tuned. (F) Cell adhesion behavior on UPy hydrogels. Changes in dynamics are used to tune cellular adhesion inside the hydrogels, with cells round and dying in case of fast dynamics (left, red) and cells spreading and healthy in case of slow dynamics (right, green). Images D–F: adapted with permission from ref. 32. Available under a CC-BY 3.0. Copyright 2021 Wiley-VCH. | ||
Supramolecular hydrogels based on BTAs have been engineered via both molecular tuning and modular mixing. Recently, BTA was mixed with PNIPAM-functionalized BTA (BTA-PNIPAM), enabling a temperature-sensitive sol-gel transition to allow facile cell encapsulation.232 In addition, by combining the monomeric small BTA moleculewith a bifunctional, telechelic BTA-PEG-BTA, i.e. two BTA moieties separated by a PEG spacer (Fig. 8A), the monofunctional BTA fibers are cross-linked non-covalently to create 3D hydrogel networks: the BTA-PEG-BTA forms small micelles itself, but acts as supramolecular crosslinker upon mixing with BTA (Fig. 8B).197,215,222
In this latter cross-linked system, hydrogel stiffnesses and dynamic properties could be tuned by changing either the total hydrogel concentration or the ratio between the two supramolecular building blocks (Fig. 8C) formed within the material.197,215,223 Recent developments focused on facilitating BTA synthetic route showed big improvements in production speed and in the ability to modify the structures of these complex supramolecular hydrogelators.215 For example, careful tuning of the hydrophobics on the exterior of the BTA-PEG-BTA has shown the ability to tune the viscoelasticity and stress relaxation of these materials over 5 orders of magnitude, unlocking applications like 3D bioprinting with living cells.223 Further expansion of the BTA-PEG-BTA architecture with a synthetically addressable norbornene pendent group has allowed the in situ covalent reinforcement of the BTA hydrogels in cell compatible conditions. This has led to the creation of tough (strain energy ∼180 kJ m−3) and fibrous hydrogels that can be utilized as bioinks with spatiotemporal modification of properties.224 Moreover, the BTA supramolecular hydrogels exhibit excellent biocompatibility for cellular encapsulation, where cellular morphology and aggregates size could be controlled by the dynamic properties of the gels, and have been able to create a 3D bioprinted meniscus model.197,225
Overall, the supramolecular nature of the system allows great control over the dynamics and mechanical properties of the system and shows therefore great potential for mimicking these aspects of the ECM. The ability to install synthetically addressable handles into the BTA architecture unlocks new possibilities to covalently modify these materials during the course of cell culture or during processing. Of note, it is important that the dynamics in this system matches with cellular time scales to support cellular adhesion on bioactive ligands, and is an active area of study.
Another supramolecular building block, well-studied by our own laboratory, is the ureido-pyrimidinone (UPy) motif (Table 1).226,227 UPy monomers undergo fourfold hydrogen bonding in a DDAA (donor–donor–acceptor–acceptor) fashion to form dimers, while urea or urethane groups support supramolecular polymerization in the lateral direction (Fig. 8D).227–230 To ensure fiber formation of this supramolecular motif in water, hydrophobic spacers are attached onto urea moieties, while PEG chains are utilized on the chains-ends as water-compatible units.171,231 UPy fibers are less dynamic as compared to BTA supramolecular fibers, with barely any monomer exchange occurring over total fiber length in a 24 hours timeframe.197 To translate the supramolecular polymers into a supramolecular hydrogel network, a bifunctional (B) UPy supramolecular crosslinker is required,32 similar to the BTA supramolecular hydrogel system. The mechanical and dynamic properties of the UPy supramolecular hydrogel are also tunable by tuning the M/B UPy ratio or changing the hydrogel's concentration (Fig. 8D and E).
Bioactive function can be introduced into the hydrogel system by including bio-functionalized UPys as integrin-binding ligands which promote cell adhesion (Fig. 8F). Through recent developments, it is possible to control the effective ligand concentration (i.e. mol% bioactive monomers in supramolecular fiber) inside supramolecular polymers, which showed to regulate epithelial cell polarity.33 High effective ligand concentrations (>5 mol%) led to multivalent effects through dynamic recruitment of the bioactive fibers, with that promoting efficient receptor clustering. Importantly, the effective ligand concentration can be tuned independent from other properties, like stiffness and stress relaxation. Recently, temperature-responsiveness was also introduced into the UPy system.233 Currently, other UPy ligands (i.e. other small peptides towards large proteins and carbohydrates) are being engineered and explored as well.
Overall, the tunable and highly modular design of the supramolecular UPy hydrogel system allows for excellent and orthogonal control over its mechanical, bioactive and dynamic properties (Fig. 8), making it an ideal platform to fundamentally study cell–material interactions.
Conclusion and future directions
The cellular environment is critical to the function and homeostasis from tissue to organ to organism. Mimicking and recreating this environment is a critical endeavor to many future fields, from regenerative medicine, to advanced soft matter, drug delivery, medical devices, and wearables. Along this review, we recap the long journey on hydrogel design to mimic our native ECM. Naturally sourced hydrogels claim powerful biological activity, but the lack of reproducibility and the complexity itself makes it difficult to understand the underlying mechanisms of cell–material interactions. Thus, designing simpler hydrogels became useful for understanding the effect of individual features (e.g. stiffness, RGD peptide sequence) on cell–matrix interactions and cellular response. The drawbacks and limitations of using either synthetic or natural polymers lead scientists towards developing hybrid hydrogels, not only improving tailorability and reproducibility, but also capturing ECM complexity.Overall, many different strategies have been investigated to engineer specific facets of the ECM. However, nature has evolved all its chemical processes around only a limited number of molecules that can assemble into a large library of materials with different stiffnesses, bioactivity and dynamics, and special processes as stress stiffening and stress relaxation. Even so, part of the tunability of the ECM arises from the complex and synergistic interplay between the various components, giving rise to tunable properties and strong reinforced assemblies. The current synthetic covalent materials can only represent one specific property of the ECM, for other properties, a completely different material is needed. Supramolecular assemblies are arising as the next-generation biomaterials to their inherent controllable dynamics, adaptability, tunability and great control over their structural and physical properties. At the same time, a supramolecular copolymerization approach can elegantly be used to control the structure's morphology,178 dynamics170,178 and ligand presentation. Like in the natural ECM, these systems can assemble into larger, hierarchical complexes with tunable mechanical, bioactive and dynamic properties. As nature designed all cellular processes with a limited number of molecules, we propose that reducing the complexity of hydrogels towards minimal co-assembling supramolecular components is the way towards fully synthetic ECM mimics (Fig. 9).
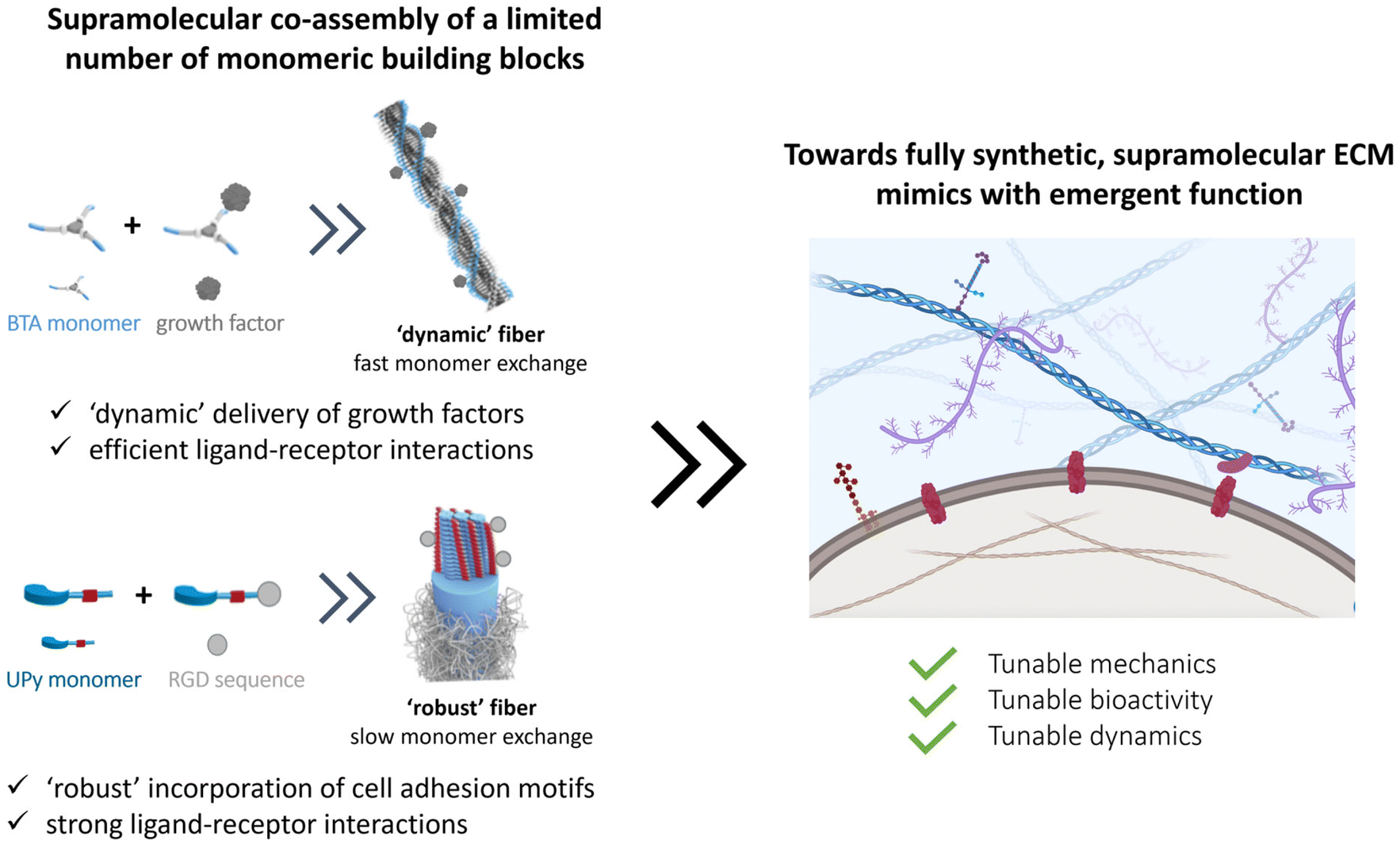 | ||
| Fig. 9 Towards the design of more complex ECM mimics with minimal components using supramolecular interactions. The co-assembly of a limited number of monomeric building blocks should allow for the assembly of higher assembled structures with tunable mechanical, bioactive and dynamic properties to fabricate fully functional ECM mimics. Figure created using Biorender software. | ||
Data availability
No primary research results, software or code have been included and no new data were generated or analyzed as part of this review.Conflicts of interest
Patricia Dankers is co-founder and share holder of spin-off company VivArt-X that focusses on women's health using supramolecular materials; especially targeting breast regeneration.Acknowledgements
We thank the ICMS Animation Studio for support with the artwork. The researchers are financially supported by the Ministry of Education, Culture and Science (Gravity Programs 024.003.013 and 024.005.020), the Research Program of Chemelot InSciTe, project EyeSciTe and the European Union's Horizon Research and Innovation Program under grant agreement 101079482 (‘SUPRALIFE’). A. A. A. would like to thank the support of Marie Skłodowska-Curie Individual Fellowships under grant agreement 101028471.References
- K. Liu, Y. Jiang, Z. Bao and X. Yan, CCS Chem., 2019, 1, 431–447 CrossRef CAS.
- V. R. Feig, H. Tran, M. Lee and Z. Bao, Nat. Commun., 2018, 9, 2740 CrossRef.
- Y. Wang, C. Zhu, R. Pfattner, H. Yan, L. Jin, S. Chen, F. Molina-Lopez, F. Lissel, J. Liu, N. I. Rabiah, Z. Chen, J. W. Chung, C. Linder, M. F. Toney, B. Murmann and Z. Bao, Sci. Adv., 2017, 3, e1602076 CrossRef.
- B. C. K. Tee, A. Chortos, A. Berndt, A. K. Nguyen, A. Tom, A. McGuire, Z. C. Lin, K. Tien, W. G. Bae, H. Wang, P. Mei, H. H. Chou, B. Cui, K. Deisseroth, T. N. Ng and Z. Bao, Science, 2015, 350, 313–316 CrossRef CAS.
- Z. Yuan, Y. Li, S. Zhang, X. Wang, H. Dou, X. Yu, Z. Zhang, S. Yang and M. Xiao, Mol. Cancer, 2023, 22, 48 CrossRef CAS PubMed.
- F. Schutgens, M. B. Rookmaaker, T. Margaritis, A. Rios, C. Ammerlaan, J. Jansen, L. Gijzen, M. Vormann, A. Vonk, M. Viveen, F. Y. Yengej, S. Derakhshan, K. M. de Winter-de Groot, B. Artegiani, R. van Boxtel, E. Cuppen, A. P. A. Hendrickx, M. M. van den Heuvel-Eibrink, E. Heitzer, H. Lanz, J. Beekman, J. L. Murk, R. Masereeuw, F. Holstege, J. Drost, M. C. Verhaar and H. Clevers, Nat. Biotechnol., 2019, 37, 303–313 CrossRef CAS.
- M. O. Dellacherie, B. R. Seo and D. J. Mooney, Nat. Rev. Mater., 2019, 4, 379–397 CrossRef.
- S. Santhanam, V. R. Feig, K. W. McConnell, S. Song, E. E. Gardner, J. J. Patel, D. Shan, Z. Bao and P. M. George, Adv. Mater. Technol., 2023, 8, 2201724 CrossRef CAS.
- Y. Liu, J. Li, S. Song, J. Kang, Y. Tsao, S. Chen, V. Mottini, K. McConnell, W. Xu, Y. Q. Zheng, J. B. H. Tok, P. M. George and Z. Bao, Nat. Biotechnol., 2020, 38, 1031 CrossRef CAS PubMed.
- S. Sahoo, N. Kumar, C. Bhattacharya, S. S. Sagiri, K. Jain, K. Pal, S. S. Ray and B. Nayak, Des. Monomers Polym., 2011, 14, 95–108 CrossRef CAS.
- H. Basit, A. Pal, S. Sen and S. Bhattacharya, Chem. – Eur. J., 2008, 14, 6534–6545 CrossRef CAS.
- S. Datta and S. Bhattacharya, Chem. Soc. Rev., 2015, 44, 5596–5637 RSC.
- H. L. Lim, Y. Hwang, M. Kar and S. Varghese, Biomater. Sci., 2014, 2, 603–618 RSC.
- N. S. Hwang, S. Varghese, Z. Zhang and J. Elisseeff, Tissue Eng., 2006, 12, 2695–2706 CrossRef CAS.
- S. Zhang, J. Ermann, M. D. Succi, A. Zhou, M. J. Hamilton, B. Cao, J. R. Korzenik, J. N. Glickman, P. K. Vemula, L. H. Glimcher, G. Traverso, R. Langer and J. M. Karp, Sci. Transl. Med., 2015, 7, 300ra128 Search PubMed.
- A. Jain, S. Dhiman, A. Dhayani, P. K. Vemula and S. J. George, Nat. Commun., 2019, 10, 450 CrossRef CAS PubMed.
- A. Pal, Y. K. Ghosh and S. Bhattacharya, Tetrahedron, 2007, 63, 7334–7348 CrossRef CAS.
- S. Bhattacharya, A. Srivastava and A. Pal, Angew. Chem., Int. Ed., 2006, 45, 2934–2937 CrossRef CAS PubMed.
- D. Sengupta and S. C. Heilshorn, Tissue Eng., Part B, 2010, 16, 285–293 CrossRef CAS.
- K. J. Lampe and S. C. Heilshorn, Neurosci. Lett., 2012, 519, 138–146 CrossRef CAS.
- D. S. W. Benoit and K. S. Anseth, Acta Biomater., 2005, 1, 461–470 CrossRef.
- A. M. Rosales and K. S. Anseth, Nat. Rev. Mater., 2016, 1, 15012 CrossRef CAS PubMed.
- S. R. Caliari and J. A. Burdick, Nat. Methods, 2016, 13, 405–414 CrossRef CAS PubMed.
- S. Uman, A. Dhand and J. A. Burdick, J. Appl. Polym. Sci., 2020, 137, 48668 CrossRef CAS.
- C. A. DeForest, B. E. Kirkpatrick and K. S. Anseth, Nat. Chem. Eng., 2024, 1, 2–5 CrossRef.
- F. Sun, W. Bin Zhang, A. Mahdavi, F. H. Arnold and D. A. Tirrell, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 11269–11274 CrossRef CAS PubMed.
- M. T. Kozlowski, H. N. Zook, D. N. Chigumba, C. P. Johnstone, L. F. Caldera, H. P. Shih, D. A. Tirrell and H. T. Ku, Front. Bioeng. Biotechnol., 2023, 11, 1144209 CrossRef PubMed.
- N. Gjorevski and M. P. Lutolf, Nat. Protoc., 2017, 12, 2263–2274 CrossRef CAS PubMed.
- S. Rezakhani, N. Gjorevski and M. P. Lutolf, Biomaterials, 2021, 276, 121020 CrossRef CAS PubMed.
- N. Sachs, Y. Tsukamoto, P. Kujala, P. J. Peters and H. Clevers, Development, 2017, 144, 1107–1112 CrossRef CAS.
- N. Gjorevski, N. Sachs, A. Manfrin, S. Giger, M. E. Bragina, P. Ordóñez-Morán, H. Clevers and M. P. Lutolf, Nature, 2016, 539, 560–564 CrossRef CAS PubMed.
- M. Diba, S. Spaans, S. I. S. Hendrikse, M. M. C. Bastings, M. J. G. Schotman, J. F. Van Sprang, D. J. Wu, F. J. M. Hoeben, H. M. Janssen and P. Y. W. Dankers, Adv. Mater., 2021, 33, 2008111 CrossRef CAS PubMed.
- L. Rijns, M. J. Hagelaars, J. J. B. Van Der Tol, S. Loerakker, C. V. C. Bouten and P. Y. W. Dankers, Adv. Mater., 2023, 2300873 CrossRef PubMed.
- B. O. Okesola, Y. Wu, B. Derkus, S. Gani, D. Wu, D. Knani, D. K. Smith, D. J. Adams and A. Mata, Chem. Mater., 2019, 31, 7883–7897 CrossRef CAS.
- A. Majkowska, K. E. Inostroza-Brito, M. Gonzalez, C. Redondo-Gómez, A. Rice, J. C. Rodriguez-Cabello, A. E. Del Rio Hernandez and A. Mata, Biomacromolecules, 2023, 24, 4419–4429 CrossRef CAS PubMed.
- N. M. Alves and J. F. Mano, Int. J. Biol. Macromol., 2008, 43, 401–414 CrossRef CAS PubMed.
- J. Mota, N. Yu, S. G. Caridade, G. M. Luz, M. E. Gomes, R. L. Reis, J. A. Jansen, X. Frank Walboomers and J. F. Mano, Acta Biomater., 2012, 8, 4173–4180 CrossRef CAS PubMed.
- D. Indana, P. Agarwal, N. Bhutani and O. Chaudhuri, Adv. Mater., 2021, 33, 2101966 CrossRef CAS PubMed.
- J. Lou, R. Stowers, S. Nam, Y. Xia and O. Chaudhuri, Biomaterials, 2018, 154, 213–222 CrossRef CAS.
- J. Lou and D. J. Mooney, Nat. Rev. Chem., 2022, 6, 726–744 CrossRef CAS.
- T. Rozario and D. W. DeSimone, Dev. Biol., 2010, 341, 126–140 CrossRef CAS PubMed.
- C. Frantz, K. M. Stewart and V. M. Weaver, J. Cell Sci., 2010, 123, 4195–4200 CrossRef CAS PubMed.
- A. J. Booth, R. Hadley, A. M. Cornett, A. A. Dreffs, S. A. Matthes, J. L. Tsui, K. Weiss, J. C. Horowitz, V. F. Fiore, T. H. Barker, B. B. Moore, F. J. Martinez, L. E. Niklason and E. S. White, Am. J. Respir. Crit. Care Med., 2012, 186, 866–876 CrossRef CAS PubMed.
- P. J. Thurner, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2009, 1, 624–649 Search PubMed.
- K. E. Kasza, A. C. Rowat, J. Liu, T. E. Angelini, C. P. Brangwynne, G. H. Koenderink and D. A. Weitz, Curr. Opin. Cell Biol., 2007, 19, 101–107 CrossRef CAS PubMed.
- P. Chen and V. B. Shenoy, Soft Matter, 2011, 7, 355–358 RSC.
- G. H. Koenderink, Z. Dogic, F. Nakamura, P. M. Bendix, F. C. MacKintosh, J. H. Hartwig, T. P. Stossel and D. A. Weitz, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 15192–15197 CrossRef CAS.
- S. Sani, M. Messe, Q. Fuchs, M. Pierrevelcin, P. Laquerriere, N. Entz-Werle, D. Reita, N. Etienne-Selloum, V. Bruban, L. Choulier, S. Martin and M. Dontenwill, ChemBioChem, 2021, 22, 1151–1160 CrossRef CAS.
- T. G. Kapp, F. Rechenmacher, S. Neubauer, O. v. Maltsev, E. A. Cavalcanti-Adam, R. Zarka, U. Reuning, J. Notni, H. J. Wester, C. Mas-Moruno, J. Spatz, B. Geiger and H. Kessler, Sci. Rep., 2017, 7, 39805 CrossRef CAS.
- M. A. Arnaout, S. L. Goodman and J.-P. Xiong, Curr. Opin. Cell Biol., 2002, 14, 641–651 CrossRef CAS.
- C. H. Damsky and Z. Werb, Curr. Opin. Cell Biol., 1992, 4, 772–781 CrossRef CAS PubMed.
- S. Mathew, X. Chen, A. Pozzi and R. Zent, Pediatr. Nephrol., 2012, 27, 891–900 CrossRef.
- A. Pozzi and R. Zent, J. Am. Soc. Nephrol., 2013, 24, 1034–1039 CrossRef CAS.
- B. Felding-Habermann and D. A. Cheresh, Curr. Opin. Cell Biol., 1993, 5, 864–868 CrossRef CAS PubMed.
- M. Aumailley, M. Gerl, A. Sonnenberg, R. Deutzmann and R. Timpl, FEBS Lett., 1990, 262, 82–86 CrossRef CAS.
- R. O. Hynes, Science, 2009, 326, 1216–1219 CrossRef CAS PubMed.
- M. F. Brizzi, G. Tarone and P. Defilippi, Curr. Opin. Cell Biol., 2012, 24, 645–651 CrossRef CAS PubMed.
- O. Saksela and M. Laiho, FASEB J., 1997, 11, 51–59 CrossRef.
- M. J. Kwon, B. Jang, J. Y. Yi, I. O. Han and E. S. Oh, FEBS Lett., 2012, 586, 2207–2211 CrossRef CAS PubMed.
- S. H. Kim, J. Turnbull and S. Guimond, J. Endocrinol., 2011, 209, 139–151 CAS.
- Y. Fung, Biomechanics: Mechanical Properties of Living Tissues, Springer, New York, 1993 Search PubMed.
- T. G. Mezger, Applied Rheology: With Joe Flow on Rheology Road, Anton Paar GmbH, 2018 Search PubMed.
- O. Chaudhuri, Biomater. Sci., 2017, 5, 1480–1490 RSC.
- O. Chaudhuri, L. Gu, D. Klumpers, M. Darnell, S. A. Bencherif, J. C. Weaver, N. Huebsch, H.-P. Lee, E. Lippens, G. N. Duda and D. J. Mooney, Nat. Mater., 2016, 15, 326–334 CrossRef CAS.
- K. H. Vining and D. J. Mooney, Nat. Rev. Mol. Cell Biol., 2017, 18, 728–742 CrossRef CAS PubMed.
- N. Sasaki, Y. Nakayama, M. Yoshikawa and A. Enyo, J. Biomech., 1993, 26, 1369–1376 CrossRef CAS.
- M. J. Hagelaars, L. Rijns, P. Y. W. Dankers, S. Loerakker and C. V. C. Bouten, Tissue Eng., Part B, 2022, 29, 203–216 CrossRef.
- L. Rijns, M. B. Baker and P. Y. W. Dankers, J. Am. Chem. Soc., 2024, 146(26), 17539–17558 CrossRef CAS PubMed.
- A. S. Adhikari, J. Chai and A. R. Dunn, J. Am. Chem. Soc., 2011, 133, 1686–1689 CrossRef CAS PubMed.
- D. Xiang, X. Wu, W. Cao, B. Xue, M. Qin, Y. Cao and W. Wang, Front. Chem., 2020, 8, 7 CrossRef CAS.
- F. Burla, J. Tauber, S. Dussi, J. van der Gucht and G. H. Koenderink, Nat. Phys., 2019, 15, 549–553 Search PubMed.
- U. Blache, E. M. Ford, B. Ha, L. Rijns, O. Chaudhuri, P. Y. W. Dankers, A. M. Kloxin, J. G. Snedeker and E. Gentleman, Nat. Rev. Methods Primers, 2022, 2, 98 Search PubMed.
- T. Hoshiba, H. Lu, N. Kawazoe and G. Chen, Expert Opin. Biol. Ther., 2010, 10, 1717–1728 Search PubMed.
- J. J. Song and H. C. Ott, Trends Mol. Med., 2011, 17, 424–432 CrossRef CAS PubMed.
- J. S. Cartmell and M. G. Dunn, Tissue Eng., 2004, 10, 1065–1075 CrossRef CAS PubMed.
- T. Woods and P. F. Gratzer, Biomaterials, 2005, 26, 7339–7349 CrossRef CAS.
- J. J. Lovekamp, D. T. Simionescu, J. J. Mercuri, B. Zubiate, M. S. Sacks and N. R. Vyavahare, Biomaterials, 2006, 27, 1507–1518 CrossRef CAS PubMed.
- G. Mattei, V. di Patria, A. Tirella, A. Alaimo, G. Elia, A. Corti, A. Paolicchi and A. Ahluwalia, Acta Biomater., 2014, 10, 875–882 CrossRef CAS.
- K. H. Hussein, K.-M. Park, K.-S. Kang and H.-M. Woo, Mater. Sci. Eng., C, 2016, 67, 766–778 CrossRef CAS PubMed.
- Y. S. Kim, M. Majid, A. J. Melchiorri and A. G. Mikos, Bioeng. Transl. Med., 2019, 4, 83–95 CrossRef PubMed.
- C. S. Hughes, L. M. Postovit and G. A. Lajoie, Proteomics, 2010, 10, 1886–1890 CrossRef CAS.
- H. K. Kleinman and G. R. Martin, Semin. Cancer Biol., 2005, 15, 378–386 CrossRef CAS.
- M. Bao, J. Xie, W. T. S. Huck, M. Bao, J. Xie and W. T. S. Huck, Adv. Sci., 2018, 5, 1800448 CrossRef PubMed.
- J. K. Mouw, G. Ou and V. M. Weaver, Nat. Rev. Mol. Cell Biol., 2014, 15, 771–785 CrossRef CAS.
- K. S. Weadock, E. J. Miller, L. D. Bellincampi, J. P. Zawadsky and M. G. Dunn, J. Biomed. Mater. Res., 1995, 29, 1373–1379 CrossRef CAS.
- M. G. Haugh, M. J. Jaasma and F. J. O'Brien, J. Biomed. Mater. Res., Part A, 2009, 89A, 363–369 CrossRef CAS.
- E. E. Antoine, P. P. Vlachos and M. N. Rylander, Tissue Eng., Part B, 2014, 20, 683–696 CrossRef CAS.
- A. J. Licup, S. Münster, A. Sharma, M. Sheinman, L. M. Jawerth, B. Fabry, D. A. Weitz and F. C. MacKintosh, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 9573–9578 CrossRef CAS PubMed.
- F. Burla, S. Dussi, C. Martinez-Torres, J. Tauber, J. van der Gucht and G. H. Koenderink, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 8326–8334 CrossRef CAS.
- S. Nam, K. H. Hu, M. J. Butte and O. Chaudhuri, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 5492–5497 CrossRef CAS.
- P. L. Chandran and V. H. Barocas, J. Biomech. Eng., 2004, 126, 152–166 CrossRef PubMed.
- E. Ban, J. M. Franklin, S. Nam, L. R. Smith, H. Wang, R. G. Wells, O. Chaudhuri, J. T. Liphardt and V. B. Shenoy, Biophys. J., 2018, 114, 450–461 CrossRef CAS PubMed.
- X. Zhao, X. Chen, H. Yuk, S. Lin, X. Liu and G. Parada, Chem. Rev., 2021, 121, 4309–4372 CrossRef CAS.
- I. K. Piechocka, R. G. Bacabac, M. Potters, F. C. Mackintosh and G. H. Koenderink, Biophys. J., 2010, 98, 2281–2289 CrossRef CAS PubMed.
- N. G. Kotla, I. L. Mohd Isa, A. Larrañaga, B. Maddiboyina, S. K. Swamy, G. Sivaraman and P. K. Vemula, Adv. Healthcare Mater., 2023, 12, 2203104 CrossRef CAS.
- H. Cui, C. Freeman, G. A. Jacobson and D. H. Small, IUBMB Life, 2013, 65, 108–120 CrossRef CAS.
- E. Han, S. S. Chen, S. M. Klisch and R. L. Sah, Biophys. J., 2011, 101, 916 CrossRef CAS.
- K. P. Vercruysse, D. M. Marecak, J. F. Marecek and G. D. Prestwich, Bioconjugate Chem., 1997, 8, 686–694 CrossRef CAS.
- G. D. Prestwich, D. M. Marecak, J. F. Marecek, K. P. Vercruysse and M. R. Ziebell, JCR, 1998, 53, 93–103 CrossRef CAS.
- M. G. Ondeck and A. J. Engler, J. Biomech. Eng., 2016, 138, 21003 CrossRef.
- M. Mensitieri, L. Ambrosio, L. Nicolais, D. Bellini and M. O'Regan, J. Mater. Sci. Mater. Med., 1996, 7, 695–698 CrossRef CAS.
- J. Fujiwara, M. Takahashi, T. Hatakeyama and H. Hatakeyama, Polym. Int., 2000, 49, 1604–1608 CrossRef CAS.
- L.-S. Liu, A. Y. Thompson, M. A. Heidaran, J. W. Poser and R. C. Spiro, Biomaterials, 1999, 20, 1097–1108 CrossRef CAS PubMed.
- K. Y. Lee and D. J. Mooney, Chem. Rev., 2001, 101, 1869–1880 CrossRef CAS PubMed.
- D. A. Wang, S. Varghese, B. Sharma, I. Strehin, S. Fermanian, J. Gorham, D. H. Fairbrother, B. Cascio and J. H. Elisseeff, Nat. Mater., 2007, 6, 385–392 CrossRef CAS.
- S. Varghese, N. S. Hwang, A. C. Canver, P. Theprungsirikul, D. W. Lin and J. Elisseeff, Matrix Biol., 2008, 27, 12–21 CrossRef CAS PubMed.
- N. A. Moghadam, F. Bagheri and M. B. Eslaminejad, Colloids Surf., B, 2022, 219, 112786 CrossRef CAS.
- F. A. Johnson, D. Q. Craig and A. D. Mercer, J. Pharm. Pharmacol., 1997, 49, 639–643 CrossRef CAS.
- O. Smidsrød and G. Skjåk-Bræk, Trends Biotechnol., 1990, 8, 71–78 CrossRef PubMed.
- J. L. Drury and D. J. Mooney, Biomaterials, 2003, 24, 4337–4351 CrossRef CAS PubMed.
- M. A. LeRoux, F. Guilak and L. A. Setton, J. Biomed. Mater. Res., 1999, 47, 46–53 CrossRef CAS.
- E. Alsberg, K. W. Anderson, A. Albeiruti, R. T. Franceschi and D. J. Mooney, J. Dent. Res., 2001, 80, 2025–2029 CrossRef CAS PubMed.
- K. Y. Lee, H. J. Kong and D. J. Mooney, Macromol. Biosci., 2008, 8, 140–145 CrossRef CAS.
- A. Srivastava, S. Ghorai, A. Bhattacharjya and S. Bhattacharya, J. Org. Chem., 2005, 70, 6574–6582 CrossRef CAS PubMed.
- N. Huebsch, E. Lippens, K. Lee, M. Mehta, S. T. Koshy, M. C. Darnell, R. M. Desai, C. M. Madl, M. Xu, X. Zhao, O. Chaudhuri, C. Verbeke, W. S. Kim, K. Alim, A. Mammoto, D. E. Ingber, G. N. Duda and D. J. Mooney, Nat. Mater., 2015, 14, 1269–1277 CrossRef CAS.
- K. M. Wisdom, K. Adebowale, J. Chang, J. Y. Lee, S. Nam, R. Desai, N. S. Rossen, M. Rafat, R. B. West, L. Hodgson and O. Chaudhuri, Nat. Commun., 2018, 9, 4144 CrossRef.
- O. Chaudhuri, S. T. Koshy, C. Branco da Cunha, J.-W. Shin, C. S. Verbeke, K. H. Allison and D. J. Mooney, Nat. Mater., 2014, 13, 970–978 CrossRef CAS.
- E. A. Phelps, N. O. Enemchukwu, V. F. Fiore, J. C. Sy, N. Murthy, T. A. Sulchek, T. H. Barker and A. J. García, Adv. Mater., 2012, 24, 64–70 CrossRef CAS PubMed.
- B. V. Slaughter, S. S. Khurshid, O. Z. Fisher, A. Khademhosseini and N. A. Peppas, Adv. Mater., 2009, 21, 3307–3329 CrossRef CAS.
- K. A. Kyburz and K. S. Anseth, Ann. Biomed. Eng., 2015, 43, 489–500 CrossRef.
- C. E. Hoyle and C. N. Bowman, Angew. Chem., Int. Ed., 2010, 49, 1540–1573 CrossRef CAS.
- M. A. Azagarsamy and K. S. Anseth, ACS Macro Lett., 2013, 2, 5–9 CrossRef CAS PubMed.
- C. A. DeForest and K. S. Anseth, Nat. Chem., 2011, 3, 925–931 CrossRef CAS PubMed.
- J. L. West and J. A. Hubbell, Macromolecules, 1999, 32, 241–244 CrossRef CAS.
- S. Bhutani, A. L. Y. Nachlas, M. E. Brown, T. Pete, C. T. Johnson, A. J. García and M. E. Davis, ACS Biomater. Sci. Eng., 2018, 4, 200–210 CrossRef CAS PubMed.
- M. Fujita, G. M. Policastro, A. Burdick, H. T. Lam, J. L. Ungerleider, R. L. Braden, D. Huang, K. G. Osborn, J. H. Omens, M. M. Madani and K. L. Christman, Nat. Commun., 2021, 12, 3764 CrossRef CAS.
- M. P. Lutolf, J. L. Lauer-Fields, H. G. Schmoekel, A. T. Metters, F. E. Weber, G. B. Fields and J. A. Hubbell, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 5413–5418 CrossRef CAS PubMed.
- Z. Zhang, A. Loebus, G. de Vicente, F. Ren, M. Arafeh, Z. Ouyang and M. C. Lensen, Chem. Mater., 2014, 26, 3624–3630 CrossRef CAS.
- P. H. J. Kouwer, P. de Almeida, O. ven den Boomen, Z. H. Eksteen-Akeroyd, R. Hammink, M. Jaspers, S. Kragt, M. F. J. Mabesoone, R. J. M. Nolte, A. E. Rowan, M. G. T. A. Rutten, V. A. A. Le Sage, D. C. Schoenmakers, C. Xing and J. Xu, Chin. Chem. Lett., 2018, 29, 281–284 CrossRef CAS.
- M. Jaspers, M. Dennison, M. F. J. Mabesoone, F. C. MacKintosh, A. E. Rowan and P. H. J. Kouwer, Nat. Commun., 2014, 5, 5808 CrossRef.
- D. C. Schoenmakers, L. Schoonen, M. G. T. A. Rutten, R. J. M. Nolte, A. E. Rowan, J. C. M. van Hest and P. H. J. Kouwer, Soft Matter, 2018, 14, 1442–1448 RSC.
- K. Liu, S. M. Mihaila, A. Rowan, E. Oosterwijk and P. H. J. Kouwer, Biomacromolecules, 2019, 20, 826–834 CrossRef CAS.
- J. Rožanc, C. Zhang, B. Vihar, U. Maver, C. Ma, K. Liu, Q. Li, Y. Xiong, C. Xu, W. Zhang, C. Ruan, X. Li and X. Lei, Bioengineering, 2022, 9, 453 CrossRef.
- K. Jain, R. Vedarajan, M. Watanabe, M. Ishikiriyama and N. Matsumi, Polym. Chem., 2015, 6, 6819–6825 RSC.
- O. H. Kwon, A. Kikuchi, M. Yamato, Y. Sakurai and T. Okano, J. Biomed. Mater. Res., 2000, 50, 82–89 CrossRef CAS.
- T. H. Kim, D. B. An, S. H. Oh, M. K. Kang, H. H. Song and J. H. Lee, Biomaterials, 2015, 40, 51–60 CrossRef CAS.
- C. M. Hassan, N. A. Peppas and Springer Berlin Heidelberg, Biopolymers, PVA Hydrogels, Anionic Polymerisation Nanocomposites, 2000, vol. 153, pp. 37–65 Search PubMed.
- R. Ricciardi, F. Auriemma, C. De Rosa and F. Lauprêtre, Macromolecules, 2004, 37, 1921–1927 CrossRef CAS.
- W. Nafo and A. Al-Mayah, PLoS One, 2020, 15, e0233021 CrossRef CAS PubMed.
- S. Syed, A. Karadaghy and S. Zustiak, J. Visualized Exp., 2015, 97, e52643 Search PubMed.
- A. K. Denisin and B. L. Pruitt, ACS Appl. Mater. Interfaces, 2016, 8, 21893–21902 CrossRef CAS PubMed.
- V. Damljanović, B. C. Lagerholm and K. Jacobson, BioTechniques, 2005, 39, 847–851 CrossRef.
- B. K. Brandley and R. L. Schnaar, Anal. Biochem., 1988, 172, 270–278 CrossRef CAS PubMed.
- C. Weber, R. Hoogenboom and U. S. Schubert, Prog. Polym. Sci., 2012, 37, 686–714 CrossRef CAS.
- L. R. Khoury and I. Popa, Nat. Commun., 2019, 10, 5439 CrossRef PubMed.
- R. Goyal, M. E. Vega, A. K. Pastino, S. Singh, M. Guvendiren, J. Kohn, N. S. Murthy and J. E. Schwarzbauer, J. Biomed. Mater. Res., Part A, 2017, 105, 2162–2170 CrossRef CAS PubMed.
- M. Setayeshmehr, E. Esfandiari, M. Rafieinia, B. Hashemibeni, A. Taheri-Kafrani, A. Samadikuchaksaraei, D. L. Kaplan, L. Moroni and M. T. Joghataei, Tissue Eng., Part B, 2019, 25, 202–224 CrossRef CAS PubMed.
- S. Nam, R. Stowers, J. Lou, Y. Xia and O. Chaudhuri, Biomaterials, 2019, 200, 15–24 CrossRef CAS PubMed.
- A. A. Aldana, F. L. C. Morgan, S. Houben, L. M. Pitet, L. Moroni and M. B. Baker, J. Polym. Sci., 2021, 59, 2832–2843 CrossRef CAS.
- S. Trujillo, C. Gonzalez-Garcia, P. Rico, A. Reid, J. Windmill, M. J. Dalby and M. Salmeron-Sanchez, Biomaterials, 2020, 252, 120104 CrossRef CAS.
- A. Berdichevski, Y. Shachaf, R. Wechsler and D. Seliktar, Biomaterials, 2015, 42, 1–10 CrossRef CAS PubMed.
- O. M. Benavides, J. P. Quinn, S. Pok, J. Petsche Connell, R. Ruano and J. G. Jacot, Tissue Eng., Part A, 2015, 21, 1185–1194 CrossRef CAS.
- G. M. Fernandes-Cunha, K. M. Chen, F. Chen, P. Le, J. H. Han, L. A. Mahajan, H. J. Lee, K. S. Na and D. Myung, Sci. Rep., 2020, 10, 16671 CrossRef CAS.
- S. Lin and L. Gu, Materials, 2015, 8, 551–560 CrossRef.
- P. H. J. Kouwer, M. Koepf, V. A. A. le Sage, M. Jaspers, A. M. van Buul, Z. H. Eksteen-Akeroyd, T. Woltinge, E. Schwartz, H. J. Kitto, R. Hoogenboom, S. J. Picken, R. J. M. Nolte, E. Mendes and A. E. Rowan, Nature, 2013, 493, 651–655 CrossRef CAS.
- Y. Zhang, M. M. P. Zegers, A. Nagelkerke, A. E. Rowan, P. N. Span and P. H. J. Kouwer, Adv. Sci., 2021, 8, 2003380 CrossRef CAS.
- A. K. Jha, K. M. Tharp, S. Browne, J. Ye, A. Stahl, Y. Yeghiazarians and K. E. Healy, Biomaterials, 2016, 89, 136–147 CrossRef CAS PubMed.
- N. B. Cramer, S. K. Reddy, M. Cole, C. Hoyle and C. N. Bowman, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 5817–5826 CrossRef CAS.
- J. van Hoorick, A. Dobos, M. Markovic, T. Gheysens, L. van Damme, P. Gruber, L. Tytgat, J. van Erps, H. Thienpont, P. Dubruel, A. Ovsianikov and S. van Vlierberghe, Biofabrication, 2020, 13, 015017 CrossRef PubMed.
- H. W. Ooi, C. Mota, A. Tessa Ten Cate, A. Calore, L. Moroni and M. B. Baker, Biomacromolecules, 2018, 19, 3390–3400 CrossRef CAS PubMed.
- Y. Li, L. Yang, Y. Zeng, Y. Wu, Y. Wei and L. Tao, Chem. Mater., 2019, 31, 5576–5583 CrossRef CAS.
- F. L. C. Morgan, J. Fernández-Pérez, L. Moroni and M. B. Baker, Adv. Healthcare Mater., 2022, 11, 2101576 CrossRef CAS PubMed.
- L. Zou, A. S. Braegelman and M. J. Webber, ACS Appl. Mater. Interfaces, 2019, 11(6), 5695–5700 CrossRef CAS PubMed.
- V. Yesilyurt, A. M. Ayoob, E. A. Appel, J. T. Borenstein, R. Langer and D. G. Anderson, Adv. Mater., 2017, 29, 1605947 CrossRef PubMed.
- A. C. Yu, H. Lian, X. Kong, H. Lopez Hernandez, J. Qin and E. A. Appel, Nat. Commun., 2021, 12, 746 CrossRef CAS.
- S. Tang, H. Ma, H.-C. Tu, H.-R. Wang, P.-C. Lin, K. S. Anseth, S. Tang, H. Ma, K. S. Anseth, H. Tu, H. Wang and P. Lin, Adv. Sci., 2018, 5, 1800638 CrossRef.
- A. Chrisnandy, D. Blondel, S. Rezakhani, N. Broguiere and M. P. Lutolf, Nat. Mater., 2022, 21, 479–487 CrossRef CAS PubMed.
- Z. Álvarez, A. N. Kolberg-Edelbrock, I. R. Sasselli, J. A. Ortega, R. Qiu, Z. Syrgiannis, P. A. Mirau, F. Chen, S. M. Chin, S. Weigand, E. Kiskinis and S. I. Stupp, Science, 2021, 374, 848–856 CrossRef PubMed.
- N. M. Matsumoto, R. P. M. Lafleur, X. Lou, K. C. Shih, S. P. W. Wijnands, C. Guibert, J. W. A. M. Van Rosendaal, I. K. Voets, A. R. A. Palmans, Y. Lin and E. W. Meijer, J. Am. Chem. Soc., 2018, 140, 13308–13316 CrossRef CAS PubMed.
- B. N. S. Thota, X. Lou, D. Bochicchio, T. F. E. Paffen, R. P. M. Lafleur, J. L. J. van Dongen, S. Ehrmann, R. Haag, G. M. Pavan, A. R. A. Palmans and E. W. Meijer, Angew. Chem., Int. Ed., 2018, 57, 6843–6847 CrossRef CAS PubMed.
- S. I. S. Hendrikse, S. P. W. Wijnands, R. P. M. Lafleur, M. J. Pouderoijen, H. M. Janssen, P. Y. W. Dankers and E. W. Meijer, Chem. Commun., 2017, 53, 2279–2282 RSC.
- X. Lou, R. P. M. Lafleur, C. M. A. Leenders, S. M. C. Schoenmakers, N. M. Matsumoto, M. B. Baker, J. L. J. van Dongen, A. R. A. Palmans and E. W. Meijer, Nat. Commun., 2017, 8, 15420 CrossRef PubMed.
- T. Aida, E. W. Meijer and S. I. Stupp, Science, 2012, 335, 813–817 CrossRef CAS.
- M. B. Baker, R. P. J. Gosens, L. Albertazzi, N. M. Matsumoto, A. R. A. Palmans and E. W. Meijer, ChemBioChem, 2016, 17, 207–213 CrossRef CAS PubMed.
- P. Y. W. Dankers, M. C. Harmsen, L. A. Brouwer, M. J. A. Van Luyn and E. W. Meijer, Nat. Mater., 2005, 4, 568–574 CrossRef CAS.
- S. Spaans, P. P. K. H. Fransen, B. D. Ippel, D. F. A. De Bont, H. M. Keizer, N. A. M. Bax, C. V. C. Bouten and P. Y. W. Dankers, Biomater. Sci., 2017, 5, 1541–1548 RSC.
- L. Rijns, L. Su, K. Maxeiner, G. Morgese, D. Y. W. Ng, T. Weil and P. Y. W. Dankers, Chem. Commun., 2023, 59, 2090–2093 RSC.
- S. I. S. Hendrikse, L. Su, T. P. Hogervorst, R. P. M. Lafleur, X. Lou, G. A. van der Marel, J. D. C. Codee and E. W. Meijer, J. Am. Chem. Soc., 2019, 141, 13877–13886 CrossRef CAS.
- M. H. Bakker and P. Y. W. Dankers, in Self-assembling Biomaterials, Elsevier, 2018, pp. 177–204 Search PubMed.
- M. G. T. A. Rutten, L. Rijns and P. Y. W. Dankers, J. Polym. Sci., 2024, 62(1), 155 CrossRef CAS.
- M. von Gröning, I. de Feijter, M. C. A. Stuart, I. K. Voets and P. Besenius, J. Mater. Chem. B, 2013, 1, 2008–2012 RSC.
- C. M. Madl, B. L. Lesavage, R. E. Dewi, C. B. Dinh, R. S. Stowers, M. Khariton, K. J. Lampe, D. Nguyen, O. Chaudhuri, A. Enejder and S. C. Heilshorn, Nat. Mater., 2017, 16, 1233–1242 CrossRef CAS PubMed.
- J. D. Hartgerink, E. Beniash and S. I. Stupp, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 5133–5138 CrossRef CAS.
- K. J. Lampe, A. L. Antaris and S. C. Heilshorn, Acta Biomater., 2013, 9, 5590–5599 CrossRef CAS PubMed.
- H. Cui, M. J. Webber and S. I. Stupp, Biopolymers, 2010, 94, 1–18 CrossRef CAS PubMed.
- R. Appel, S. Tacke, J. Klingauf and P. Besenius, Org. Biomol. Chem., 2015, 13, 1030–1039 RSC.
- E. Beniash, J. D. Hartgerink, H. Storrie, J. C. Stendahl and S. I. Stupp, Acta Biomater., 2005, 1, 387–397 CrossRef PubMed.
- K. Tashiro, G. C. Sephel, B. Weeks, M. Sasaki, G. R. Martin, H. K. Kleinman and Y. Yamada, J. Biol. Chem., 1989, 264, 16174–16182 CrossRef CAS.
- H. Storrie, M. O. Guler, S. N. Abu-Amara, T. Volberg, M. Rao, B. Geiger and S. I. Stupp, Biomaterials, 2007, 28, 4608–4618 CrossRef CAS PubMed.
- D. Lombardo, M. Kiselev, S. Magazù and P. Calandra, Adv. Condens. Matter Phys., 2015, 151683 Search PubMed.
- A. P. Nowak, V. Breedveld, L. Pakstis, B. Ozbas, D. J. Pine, D. Pochan and T. J. Deming, Nature, 2002, 417, 424–428 CrossRef CAS.
- C. Redondo-Gómez, Y. Abdouni, C. R. Becer and A. Mata, Biomacromolecules, 2019, 20, 2276–2285 CrossRef PubMed.
- C. Ligorio, J. A. Hoyland and A. Saiani, Gels, 2022, 8, 211 CrossRef CAS PubMed.
- S. Wan, S. Borland, S. M. Richardson, C. L. R. Merry, A. Saiani and J. E. Gough, Acta Biomater., 2016, 46, 29–40 CrossRef CAS PubMed.
- R. Z. Pavlović, S. A. Egner, L. C. Palmer and S. I. Stupp, J. Polym. Sci., 2023, 61, 870–880 CrossRef.
- Z. Álvarez, J. A. Ortega, K. Sato, I. R. Sasselli, A. N. Kolberg-Edelbrock, R. Qiu, K. A. Marshall, T. P. Nguyen, C. S. Smith, K. A. Quinlan, V. Papakis, Z. Syrgiannis, N. A. Sather, C. Musumeci, E. Engel, S. I. Stupp and E. Kiskinis, Cell Stem Cell, 2023, 30, 219–238 CrossRef PubMed.
- L. Rijns, J. W. Peeters, S. I. S. Hendrikse, M. E. J. Vleugels, X. Lou, H. M. Janssen, E. W. Meijer and P. Y. W. Dankers, Chem. Mater., 2023, 35, 8203–8217 CrossRef CAS.
- J. K. Gupta, D. J. Adams and N. G. Berry, Chem. Sci., 2016, 7, 4713–4719 RSC.
- N. Singh, M. Kumar, J. F. Miravet, R. V. Ulijn and B. Escuder, Chem. – Eur. J., 2017, 23, 981–993 CrossRef CAS PubMed.
- P. W. J. M. Frederix, G. G. Scott, Y. M. Abul-Haija, D. Kalafatovic, C. G. Pappas, N. Javid, N. T. Hunt, R. V. Ulijn and T. Tuttle, Nat. Chem., 2014, 7, 30–37 CrossRef PubMed.
- R. Orbach, L. Adler-Abramovich, S. Zigerson, I. Mironi-Harpaz, D. Seliktar and E. Gazit, Biomacromolecules, 2009, 10, 2646–2651 CrossRef CAS.
- J. Shi, Y. Gao, Z. Yang and B. Xu, Beilstein J. Org. Chem., 2011, 7, 167 CrossRef CAS PubMed.
- K. S. Straley and S. C. Heilshorn, Soft Matter, 2009, 5, 114–124 RSC.
- D. W. Lim, D. L. Nettles, L. A. Setton and A. Chilkoti, Biomacromolecules, 2007, 8, 1463–1470 CrossRef CAS.
- M. K. McHale, L. A. Setton and A. Chilkoti, Tissue Eng., 2005, 11, 1768–1779 CrossRef CAS PubMed.
- A. Tan, J. Rajadas and A. M. Seifalian, J. Controlled Release, 2012, 163, 342–352 CrossRef CAS.
- N. Chebotareva, P. H. H. Bomans, P. M. Frederik, N. A. J. M. Sommerdijk and R. P. Sijbesma, Chem. Commun., 2005, 39, 4967–4969 RSC.
- A. Pal, S. Karthikeyan and R. P. Sijbesma, J. Am. Chem. Soc., 2010, 132, 7842–7843 CrossRef CAS PubMed.
- M. Vleugels, R. Bosman, S. Wijker, B. Fehér, J. Spiering, L. Rijns, R. Bellan, P. Y. W. Dankers and A. R. A. Palmans, Chem. – Eur. J., 2023, e202303361 Search PubMed.
- M. M. E. Koenigs, A. Pal, H. Mortazavi, G. M. Pawar, C. Storm and R. P. Sijbesma, Macromolecules, 2014, 47, 2712–2717 CrossRef CAS.
- M. Fernandez-Castano Romera, R. P. M. Lafleur, C. Guibert, I. K. Voets, C. Storm and R. P. Sijbesma, Angew. Chem., Int. Ed., 2017, 56, 8771–8775 CrossRef CAS PubMed.
- R. C. van Gaal, A. B. C. Buskermolen, B. D. Ippel, P. P. K. H. Fransen, S. Zaccaria, C. V. C. Bouten and P. Y. W. Dankers, Biomaterials, 2019, 224, 119466 CrossRef CAS PubMed.
- L. Albertazzi, D. van der Zwaag, C. M. A. Leenders, R. Fitzner, R. W. van der Hofstad and E. W. Meijer, Science, 2014, 344, 491–495 CrossRef CAS PubMed.
- C. M. A. Leenders, L. Albertazzi, T. Mes, M. M. E. Koenigs, A. R. A. Palmans and E. W. Meijer, Chem. Commun., 2013, 49, 1963–1965 RSC.
- S. Hafeez, H. W. Ooi, D. Suylen, H. Duimel, T. M. Hackeng, C. van Blitterswijk and M. B. Baker, J. Am. Chem. Soc., 2022, 144, 4057–4070 CrossRef CAS PubMed.
- R. P. M. Lafleur, S. Herziger, S. M. C. Schoenmakers, A. D. A. Keizer, J. Jahzerah, B. N. S. Thota, L. Su, P. H. H. Bomans, N. A. J. M. Sommerdijk, A. R. A. Palmans, R. Haag, H. Friedrich, C. Böttcher and E. W. Meijer, J. Am. Chem. Soc., 2020, 142, 17644–17652 CrossRef CAS PubMed.
- C. M. A. Leenders, M. B. Baker, I. A. B. Pijpers, R. P. M. Lafleur, L. Albertazzi, A. R. A. Palmans and E. W. Meijer, Soft Matter, 2016, 12, 2887–2893 RSC.
- P. Besenius, K. P. van den Hout, H. M. H. G. Albers, T. F. A. de Greef, L. L. C. Olijve, T. M. Hermans, B. F. M. de Waal, P. H. H. Bomans, N. A. J. M. Sommerdijk, G. Portale, A. R. A. Palmans, M. H. P. van Genderen, J. A. J. M. Vekemans and E. W. Meijer, Chem.- Eur. J., 2011, 17, 5193–5203 CrossRef CAS PubMed.
- K. Matsuura, H. Hayashi, K. Murasato and N. Kimizuka, Chem. Commun., 2011, 47, 265–267 RSC.
- K. Murasato, K. Matsuura and N. Kimizuka, Biomacromolecules, 2008, 9, 913–918 CrossRef CAS PubMed.
- M. K. Müller and L. Brunsveld, Angew. Chem., Int. Ed., 2009, 48, 2921–2924 CrossRef PubMed.
- E. Vereroudakis, M. Bantawa, R. P. M. Lafleur, D. Parisi, N. M. Matsumoto, J. W. Peeters, E. del Gado, E. W. Meijer and D. Vlassopoulos, ACS Cent. Sci., 2020, 6, 1401–1411 CrossRef CAS PubMed.
- S. Hafeez, F. R. Passanha, A. J. Feliciano, F. A. A. Ruiter, A. Malheiro, R. P. M. Lafleur, N. M. Matsumoto, C. van Blitterswijk, L. Moroni, P. Wieringa, V. L. S. LaPointe and M. B. Baker, Biomater. Sci., 2022, 10, 4740–4755 RSC.
- S. Hafeez, M. C. Decarli, A. Aldana, M. Ebrahimi, F. A. A. Ruiter, H. Duimel, C. van Blitterswijk, L. M. Pitet, L. Moroni and M. B. Baker, Adv. Mater., 2023, 35, 2301242 CrossRef CAS PubMed.
- S. Hafeez, A. Aldana, H. Duimel, F. A. A. Ruiter, M. C. Decarli, V. Lapointe, C. van Blitterswijk, L. Moroni and M. B. Baker, Adv. Mater., 2023, 2207053 CrossRef CAS PubMed.
- H. Kautz, D. J. M. Van Beek, R. P. Sijbesma and E. W. Meijer, Macromolecules, 2006, 39, 4265–4267 CrossRef CAS.
- F. H. Beijer, R. P. Sijbesma, H. Kooijman, A. L. Spek and E. W. Meijer, J. Am. Chem. Soc., 1998, 120, 6761–6769 CrossRef CAS.
- R. P. Sijbesma, F. H. Beijer, L. Brunsveld, B. J. Folmer, J. H. Hirschberg, R. F. Lange, J. K. Lowe and E. W. Meijer, Science, 1997, 278, 1601–1604 CrossRef CAS PubMed.
- J. H. K. K. Hirschberg, L. Brunsveld, A. Ramzi, J. A. J. M. Vekemans, R. P. Sijbesma and E. W. Meijer, Nature, 2000, 407, 167–170 CrossRef CAS.
- M. M. L. Nieuwenhuizen, T. F. A. de Greef, R. L. J. van der Bruggen, J. M. J. Paulusse, W. P. J. Appel, M. M. J. Smulders, R. P. Sijbesma and E. W. Meijer, Chem. – Eur. J., 2010, 16, 1601–1612 CrossRef CAS PubMed.
- R. E. Kieltyka, A. C. H. Pape, L. Albertazzi, Y. Nakano, M. M. C. Bastings, I. K. Voets, P. Y. W. Dankers and E. W. Meijer, J. Am. Chem. Soc., 2013, 135, 11159–11164 CrossRef CAS PubMed.
- L. Rijns, H. Duijs, R. P. M. Lafleur, R. Cardinaels, A. R. A. Palmans, P. Y. W. Dankers and L. Su, Biomacromolecules, 2024, 25(8), 4686–4696 CrossRef CAS PubMed.
- D. J. Wu, M. G. T. A. Rutten, J. Huang, M. J. G. Schotman, J. F. van Sprang, B. M. Tiemeijer, G. M. ter Huurne, S. P. W. Wijnands, M. Diba and P. Y. W. Dankers, Macromolecules, 2024, 57(14), 6606–6615 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2024 |