
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Multiscale effects in tandem CO2 electrolysis to C2+ products
Lewis S.
Cousins
 and
Charles E.
Creissen
*
and
Charles E.
Creissen
*
School of Chemical and Physical Sciences, Keele University, Staffordshire, ST5 5BG, UK. E-mail: c.e.creissen@keele.ac.uk
First published on 12th December 2023
Abstract
CO2 electrolysis is a sustainable technology capable of accelerating global decarbonisation through the production of high-value alternatives to fossil-derived products. CO2 conversion can generate critical multicarbon (C2+) products such as drop-in chemicals ethylene and ethanol, however achieving high selectivity from single-component catalysts is often limited by the competitive formation of C1 products. Tandem catalysis can overcome C2+ selectivity limitations through the incorporation of a component that generates a high concentration of CO, the primary reactant involved in the C–C coupling step to form C2+ products. A wide range of approaches to promote tandem CO2 electrolysis have been presented in recent literature that span atomic-scale manipulation to device-scale engineering. Therefore, an understanding of multiscale effects that contribute to selectivity alterations are required to develop effective tandem systems. In this review, we use relevant examples to highlight the complex and interlinked contributions to selectivity and provide an outlook for future development of tandem CO2 electrolysis systems.
 Charles E. Creissen | Charles E. Creissen is a Lecturer in Physical Chemistry at Keele University, UK. He received his PhD in 2019 from the University of Cambridge, before moving to the Collège de France, Paris, as a Postdoctoral Research Associate and subsequently Research Engineer. In 2022, Charles moved to Keele University to establish his research group, working on the development of electrocatalysts for the sustainable production of commodity chemicals from CO2 and waste materials. |
Introduction
Electrochemical CO2 reduction (CO2R) is a sustainable technology capable of generating high-value precursors to commodity chemicals and fuels using renewable electricity. Recent efforts in computational and experimental research have been aimed at increasing selectivity for multicarbon (C2+) products with high rates of reaction.1–3 These targets represent high-value drop-in chemicals such as ethylene, ethanol, and propanol, which are heavily used in industrial processes and can therefore reduce carbon emissions in existing infrastructures.4,5 However, achieving high selectivity for multicarbon products requires catalysts that promote C–C coupling whilst avoiding kinetically favoured C1 products and H2 evolution. An effective method to increase C2+ selectivity is through tandem catalysis, in which an initial reactant generated at one catalytically active site can be further reacted at a secondary site. With Cu-based catalysts, this can be achieved through the promotion of CO generation at a specific site (COcat), which subsequently undergoes further reduction to C2+ products at a Cu site (C–Ccat).Tandem effects span multiple length scales with approaches extending from atomic scale modification through to macroscale device engineering. Consequently, in promoting tandem effects, a range of additional contributions can also alter selectivity which must also be considered to decipher routes for improved C2+ selectivity. In this review, we use recent examples of Cu-based bimetallic and hybrid (molecular or single-atom) tandem catalysts to highlight developments in methods to construct catalytic systems and understand the factors that contribute to enhanced C2+ selectivity. We first provide a mechanistic basis for tandem CO2R before looking more specifically at effects that occur on different length scales between atomic and macroscale, to provide an outlook and future perspectives on this research area.
Tandem CO2 conversion mechanisms
The mechanisms of CO2R are key to understanding how tandem catalysis can promote multicarbon product selectivity. Seminal work by Hori demonstrated that different metals give rise to distinct product distributions in aqueous conditions, but only Cu is capable of generating significant amounts of C2+ products.6 This divergence in product distributions is closely linked with the surface binding energy of intermediates involved in the reaction. If the binding energy is too strong, the surface is poisoned, while if it is too weak, the coverage and/or residence time is reduced.7,8 The relative binding energies for CO2R intermediates arising from the first electron and proton transfer steps are therefore good indicators of the favoured products (Fig. 1).8 A moderate binding energy for *COOH tends to generate CO, while a moderate binding energy for *OCOH tends to favour formic acid (‘*’ denotes a surface-bound species). Cu not only facilitates both of these pathways, but also displays a moderate binding energy for *CO, enabling C–C coupling between proximal *CO species to generate C2+ products (Fig. 1).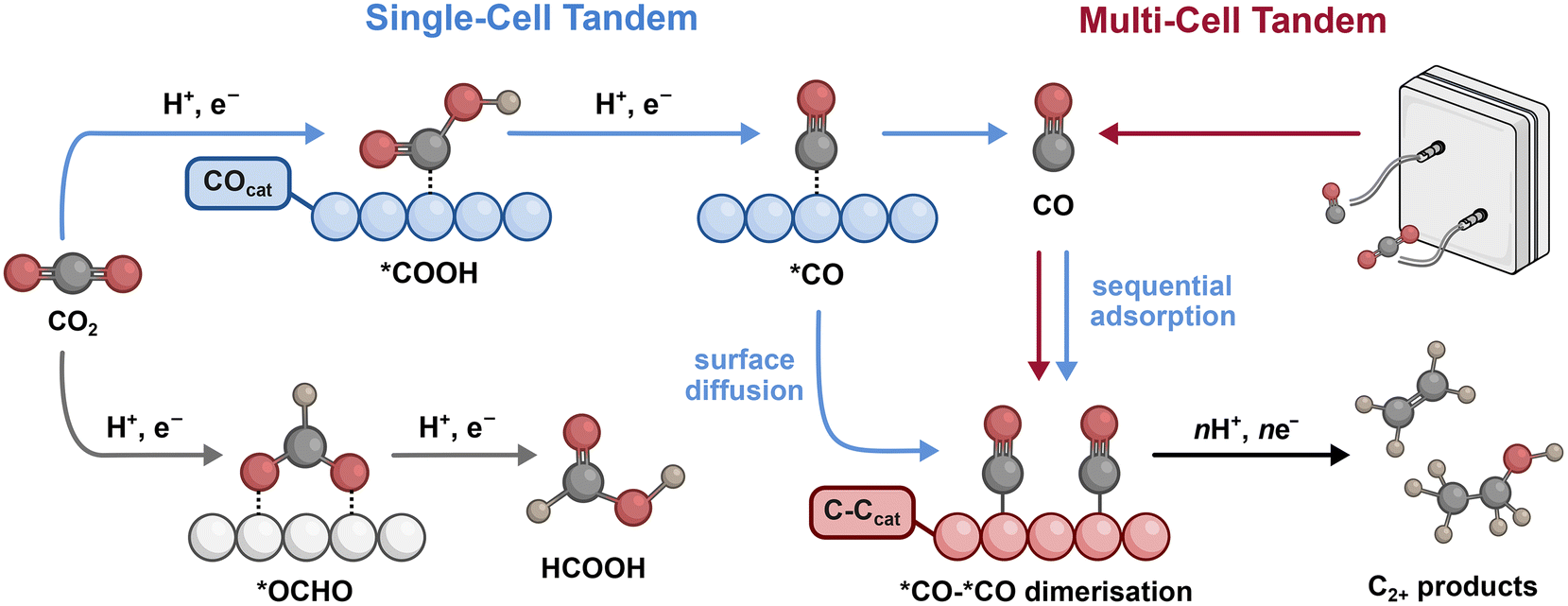 | ||
| Fig. 1 Reaction pathways for CO2 reduction showing the different surface-bound intermediates. The grey path is formate production, the blue path is the single-cell tandem pathway where CO generation occurs on a COcat (blue catalyst) and C–C coupling occurs at a C–Ccat (red catalyst). The red path is a multi-cell tandem where the CO is sourced from a separate electrochemical cell before being transferred to a C–Ccat. Note that the initial electron transfer to form *CO2−, as well as hydrogenated derivatives *COH and *CHO have been omitted for simplification in this figure. | ||
An overarching limitation in promoting C–C coupling is the inclination of Cu to generate kinetically favoured C1 products and H2. However, in tandem approaches, competitive C1 production is limited as CO becomes the major reactant due to continuous internal (single-cell) or external supply (multi-cell) (Fig. 1). In multi-cell tandem systems, the CO is sourced from electrolysis and requires sequential adsorption, in which CO is transported to a C–Ccat for subsequent reduction. In single-cell tandems the COcat and C–Ccat sites are combined internally and can rely on either CO transport and sequential adsorption or surface diffusion, in which the surface-bound *CO intermediate is transported across the surface. Both pathways inevitably result in *CO adsorption at the Cu site as is required for C–C coupling. Therefore, irrespective of the transport mechanism or configuration, tandem catalysis can be achieved through the effective coupling of a C–Ccat with a COcat. Here, we evaluate different combinations of Cu-based C–Ccats with metal COcats (bimetallic) or molecular and single-atom COcats (hybrid) in a variety of different configurations to highlight effects that can alter selectivity.
Atomic scale
COcat considerations
The COcat is a vital component of tandem CO2R seeing as the supply of CO is required for subsequent C–C coupling. Although a range of heteroatoms have been used in bimetallic Cu catalysts, the most common secondary elements for tandem systems are Ag, Au, and Zn, due to their inherent ability to selectively produce CO, however examples using Pd have also demonstrated tandem effects for specific cases which are discussed within.9,10 Aside from element variation, the onset potentials for metals are not highly tuneable, but can vary due to combination with other metals (see below for electronic effects). For hybrid catalysts however, the onset potential is highly sensitive to molecular structure and can be tuned by modification of functional groups or metal centre.11 For example, a hybrid tandem using Fe porphyrin showed that an early onset for the catalyst led to high C2+ selectivity at low overpotentials, while a catalyst using a Ni porphyrin with a more cathodic onset potential required larger overpotentials to enhance selectivity due to the reduced ability to generate CO.12 In another hybrid system, increased π-delocalisation on Fe porphyrins was shown to anodically shift the onset potential for CO evolution, giving rise to the highest multicarbon product selectivity.13 Similarly, SACs are highly effective COcats that exhibit onset potentials specific to the support and metal.14 In one example, a SAC based on N-coordinated Ni sites in a porphyrinic framework showed CO production across a wide potential range, which proved effective for C2+ production when coupled with Cu nanoparticles.15 Although the aim of the COcat is to increase the CO concentration, there are other contributions arising from the nature of the catalyst and the choice of metal, which are discussed within different subsections.Concentration, coverage, and binding configurations
Tandem catalysts aim to increase the local concentration of CO to promote C–C coupling. However, the enhancement of C2+ selectivity can involve several different contributions at the atomic level, including coverage of intermediates, binding configurations, concentration and availability of reactants (CO2 or CO), and the activity of distinct active sites.The increased coverage of *CO on the C–Ccat surface is often regarded as a beneficial contributor to multicarbon selectivity (Fig. 2a).16 This is not only because a higher presence of *CO can promote dimerisation between proximal species, but also because the coverage can alter the binding configurations of adsorbed intermediates. On Cu, the coverage has been correlated with the ratio of *COatop to *CObridge sites (Fig. 2b), and a favourable ratio will lead to the kinetically and thermodynamically preferred coupling pathways (COatop–CObridge and COatop–COatop) thereby enhancing C2+ selectivity.17 A good example of this used a molecular cobalt phthalocyanine (CoPc) catalyst deposited on a Cu surface to show that the increased coverage of *CO led to an enhancement in COatop, resulting in a reduced energy barrier for C–C coupling.18 The concentration of CO2 in solution can also play a role in altering coverage and binding configurations. A CuAg alloy tested in CO2 supersaturated solutions demonstrated that the coverage of *CO and ratio of *CObridge to *COatop increased with CO2 concentration resulting in a high selectivity towards propanol.19
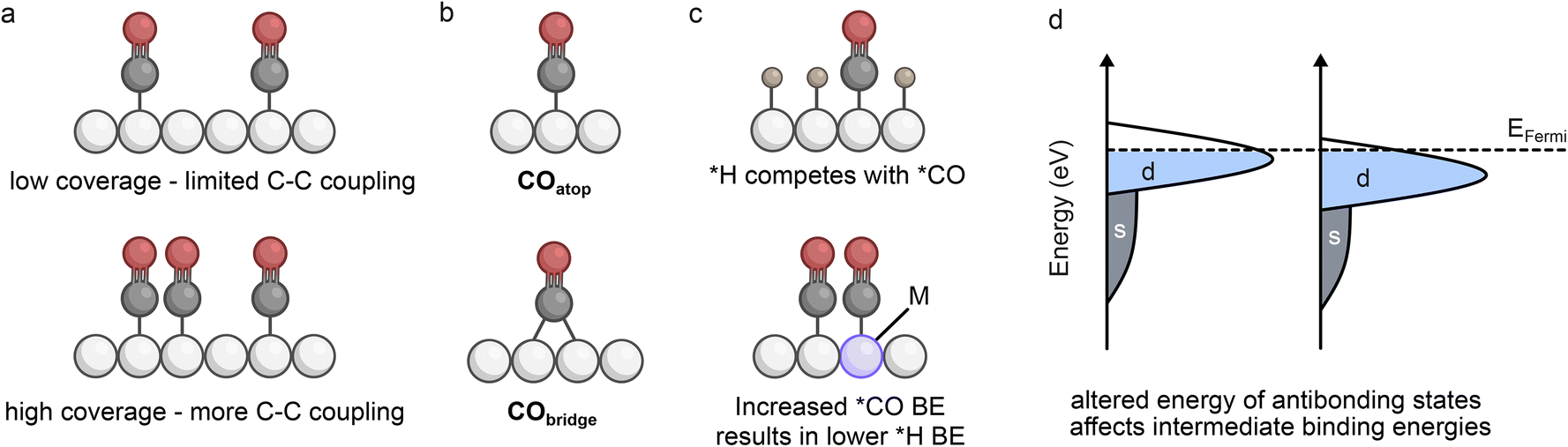 | ||
| Fig. 2 (a) Illustration demonstrating the influence of coverage on selectivity, (b) the two major binding configurations of *CO, (c) illustration of how competition between *H and *CO in acidic conditions can be overcome with addition of a metal (M) that increases the binding energy (BE) of *CO, (d) a simplified illustration of the degree of band-filling with respect to the position of the d-band centre relative to the Fermi level (EFermi). | ||
However, studies have demonstrated that *CO coverage is typically very low on electrochemically active surfaces and it is expected that high monolayer coverages would generally weaken surface binding due to lateral interactions between adjacent *CO species.22,23 Therefore coverage modifications are likely coupled with additional contributions to tandem catalysis including the local concentration of CO. The presence of an internal CO reservoir has been identified as a contributor to enhanced C2+ selectivity, which has been experimentally observed on Cu catalysts.24 This ‘pooling’ effect creates a high local CO concentration close to the catalyst and ensures that the availability of CO is high for sequential reaction, even under CO2-depleted conditions. Continuous reduction of CO2 to CO in single-cell tandems therefore feeds the growth of the reservoir to boost C2+ production. Observations of higher C2+ selectivity at lower CO2 flow rates for gas-fed flow cells also support the idea that an increased CO coverage and/or availability relative to CO2 can lead to more effective C–C coupling.20,21 An additional consideration is that higher C2+ selectivity has been observed with specific CO2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :CO ratios in mixed feeds.25 The effects can in part be attributed to CO reservoirs but also to the presence of distinct sites for CO reduction and CO2R on Cu that have recently been observed.26–28 As the activity is a combination of both CO and CO2 reduction, it is more favourable to provide the catalyst surface with both reactants to enhance activity. Therefore, the availability of both CO and CO2 can be essential to enhancing C2+ products at the C–Ccat.
:CO ratios in mixed feeds.25 The effects can in part be attributed to CO reservoirs but also to the presence of distinct sites for CO reduction and CO2R on Cu that have recently been observed.26–28 As the activity is a combination of both CO and CO2 reduction, it is more favourable to provide the catalyst surface with both reactants to enhance activity. Therefore, the availability of both CO and CO2 can be essential to enhancing C2+ products at the C–Ccat.
The presence of different heteroatoms can also influence the types of products generated due to mechanistic alterations.29,30 For example, the hydrogenation of *CO to form *CHO can be promoted by specific metals, as demonstrated by a CuPd catalyst that promoted *CO–*CHO coupling in direct CO reduction giving rise to higher selectivity for acetate.31 For low *CO coverage in CO2R, protonation and subsequent formation of CH4 can be enhanced. This was observed for a Ni-SAC system which followed the *CO dimerisation pathway when Ni-SAC was present as opposed to the CH4 pathway in the absence of a COcat.15 This effect was also evidenced by a study employing a Cu catalyst with low amounts of Au, which used CO2 dilution experiments to verify the coverage-dependent selectivity trends.32 Additionally, the promotion of asymmetric binding can enhance C2+ selectivity, as shown by a series of CuZn catalysts with varied compositions.33 Alternative pathways involving coupling between *CHx and *CO species to form oxygenates have been demonstrated on CuAg, showing that asymmetric coupling can also be influenced by tandem catalysts.34 Therefore, although increased *CO coverage tends to promote multicarbon product selectivity, specific intermediate binding configurations and alternative pathways should also be considered when developing tandem catalysts.
Recent efforts to maximise CO2 utilisation have explored acidic conditions which can avoid parasitic CO2 conversion to (bi)carbonate species observed in neutral and alkaline conditions.20,35,36 However, the pH can play a role in selectivity, as identified by a study using a CuAg catalyst with different compositions under neutral and acidic conditions.37 In this example, the presence of H3O+ was suggested to weaken the binding energy of *CO, which in turn lowers the coverage and limits C2+ selectivity. As increased *CO binding affinity has been correlated with a weaker *H binding affinity, incorporation of a strong *CO-binding component should prevent competition for *H adsorption (Fig. 2c).38 A study building on this principle explored CuPd catalysts in acidic conditions and showed that the high binding affinity of *CO promoted higher *CO coverage and limited *H binding (and H2 evolution) to enhance C–C coupling.39 The resulting selectivity increases for tandem systems can therefore be due to binding configuration, mechanistic pathways, and correlated binding energies, which can vary depending on the type of catalyst and the specific conditions employed.
Electronic effects
Bimetallic catalysts are highly susceptible to electronic structure modification, as the proximity of metals with different electronegativity results in shifting of the d-band maximum relative to the Fermi level (Fig. 2d). Normally when a d-band is shifted further from the Fermi level (lower in energy), the binding strengths of specific intermediates are decreased due to the lower energy of antibonding states – this is the case for *CO binding, as the d–π* back-bonding is less effective with a lower lying d-band.40,41 Alterations to electronic structure can therefore impact selectivity due to the product pathways being highly sensitive to intermediate binding energies.An interesting example aiming to isolate electronic effects from morphology used a magnetron sputtering co-deposition synthesis method to form a series of AuxCuy alloys.42 The results indicated that increasing Au content shifted the d-band away from the Fermi level, resulting in a reduced desorption energy (weaker binding energy) for *CO, as well as a lower binding affinity for oxygen. Consequently, although the reduced oxophilicity limited HCOO− formation, the weakened binding energy of *CO resulted in increased CO production as opposed to C2+ products. This suggests that electronic effects due to Au doping do not play a major role in promoting C2+ selectivity, but rather hinder it in this case due to favoured CO desorption. However, subtle shifts in the d-band position did give rise to selectivity alterations, highlighting the strong dependence of intermediate binding energies on electronic modifications.
In cases where both constituents have equally filled valence bands, and therefore similar electronegativity, electron transfer is limited. However, electronic modulation can still occur due to effects of strain.43,44 Although this is a structural property, the major consequential effects are electronic in nature. This is well-demonstrated by a CuAg catalyst, for which surface alloying induced compressive strain, giving rise to a shifted valence band which reduced the binding energy for adsorbed H and O intermediates relative to *CO.45 The outcome was that H2 evolution was suppressed while the formation of oxygenated C2+ products was promoted. Strain has also been suggested to impact selectivity in CuAu catalysts with fewer Cu layers on Au providing the highest degree of lattice strain, resulting in a greater selectivity towards ethylene.46 A recent report of an AgCu single atom alloy C–Ccat coupled with an Ag COcat suggested that compressive strain around the Cu due to the presence of Ag was responsible for a shift in the d-band compared with Cu, which promoted C–C coupling.47 Therefore structural effects can give rise to electronic modification and should also be considered when assessing the major selectivity-altering pathways.
Electronic effects can alter selectivity towards specific C2+ products in direct CO reduction and generally enhance multicarbon selectivity of C–Ccats. For example, although a bimetallic CuAg derived from electrochemical reduction of a mixed metal oxide was ineffective for CO2R, it achieved a high C2+ selectivity for direct CO reduction.48 Examples of multimetallic catalysts have proven effective for CO conversion to propanol, holding promise for the integration of such C–Ccats in segmented tandem electrodes (see section on gas diffusion electrodes).49,50 Additionally, recent work exploring intermetallic alloys for CO reduction identified the ability to independently modify d-band positions and local electric fields, which can alter the binding energies of intermediates.51 Although the examples underwent segregation in air, the development of novel methods for intermetallic catalyst synthesis may prove effective for alternative C–Ccats in tandem systems.52
Nanoscale to mesoscale
Morphology and interface
The composition and structural motifs in bimetallic catalysts can play a large role in directing selectivity, seeing as the diffusion of *CO or CO is highly dependent on the nature and distribution of COcat and C–Ccat components. The mixing patterns of nanoparticulate bimetallic catalysts can influence the spatial separation as well as electronic effects, giving rise to alterations in selectivity for CO2R. Some examples of common nanoparticle mixing patterns that have been employed for CO2R are shown in Fig. 3a.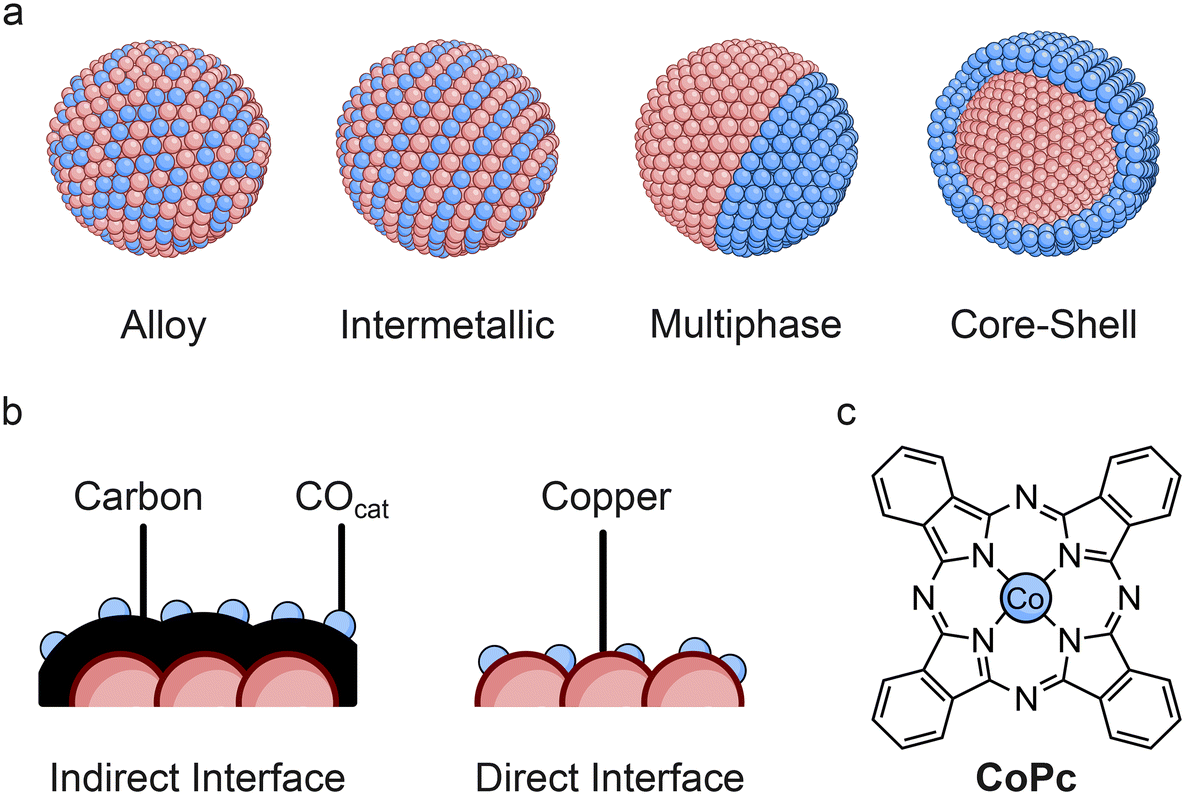 | ||
| Fig. 3 (a) Different types of mixing patterns that have been used in bimetallic catalysts (note, only spherical nanoparticles are shown for simplicity), (b) illustration of indirect (carbon interlayer) and direct interface between copper and COcat, (c) molecular structure of cobalt phthalocyanine. | ||
A study using CuPd catalysts with different structural arrangements, revealed that multiphase catalysts with distinct COcat and C–Ccat components were found to be more effective for C–C coupling than ordered and disordered catalysts, suggesting that tandem effects promote C2+ selectivity rather than electronic effects in this example.53 In another study, Ag–Cu mutliphase nanoparticles with predominantly (100) facets exposed showed a high ability to generate ethylene, while physically mixed Ag and Cu nanocubes predominantly formed CO.54 The benefits of separated COcat and C–Ccat sites was also highlighted for a series of Ag–Cu nanodimers, synthesised with a high degree of control over size and shape.55 This allowed for differentiation between effects of sequential catalysis, which was dominant for segregated structures, as opposed to electronic effects, which were dominant in surface alloyed catalysts.
Completely separating morphological effects from electronic effects in tandem catalysts is not straightforward due to the common presence of a direct interface between the different metals. However, in one case it was possible to exploit solely tandem effects through the separation of Ag and Cu components with a carbon interlayer in core–shell nanoparticles (Fig. 3b, indirect interface).56 The results demonstrated that C2+ selectivity was enhanced due to sequential catalysis and that the thickness of the Cu shell (and therefore the ratio of Ag to Cu) could be used to tune the ethanol/ethylene ratio. Nanoscale control over CO mass transport appears to have clear benefits for sequential catalysis with bimetallic catalysts and therefore an understanding of how to promote transport at such length scales could have a large impact on selectivity.10
Conversely, the presence of a direct interface has proven beneficial for hybrid systems featuring molecular catalysts. This was well demonstrated in a study using CoPc (Fig. 3c), which was either directly immobilised on Cu nanocubes (direct) or first immobilised on a carbon support (indirect).57 Direct deposition showed higher C2+ selectivity than the indirect interface, suggesting that surface *CO transport takes place and that sequential adsorption may be a limiting factor in hybrid systems. It is worth noting that such macrocycles are highly susceptible to aggregation and support interactions, which can alter activity depending on the immobilisation method and support used.58,59 Therefore, there are additional considerations for molecular species that may not be present for bimetallic catalysts.
Catalyst restructuring
Unintentional nanoscale effects can also contribute to altered selectivity for CO2R. One example is the dynamic restructuration that can occur under applied potentials.60–63 In bimetallic catalysts, this can alter shape, nano/mesostructure, and distributions of metals, all of which can impact the selectivity, CO transport, and stability.Recent work demonstrated that for alloyed and multiphase AgCu catalysts, Cu becomes mobile and can detach from the catalyst surface before redepositing as particles, which drastically alters the morphology.64 In other studies, the impact of high CO concentrations on instability has been identified, which is of particular significance for tandem systems.65,66 This was well-highlighted in a study employing a Ag-core–Cu-shell catalyst by tracking the structural changes under reaction conditions.66 It was shown that although the applied potential does play a role in restructuration, the CO concentration played a more dominant role. This is also supported by other work showing that in the presence of CO, intermetallic Cu–Cu bonds are weakened by CO binding which can lead to cluster formation.67 Similar observations have been noted for Cu-based single-atom catalysts and molecular catalysts.63 In most cases however, the formation of clusters under applied potentials can be reversed by removing the potential, which can be attributed to a high affinity of Cu atoms for nitrogen co-ordination in such networks. These effects account for C2+ production through *CO dimerisation, which would otherwise not be possible on isolated Cu sites.
The prevalence of restructuring and instability of electrocatalysts under a range of conditions necessitates full consideration of the active catalyst and also preventative measures. Recent examples have demonstrated that specific heteroatoms can prevent destabilisation, and therefore exploration of compositions that can simultaneously retain structural integrity and tandem properties is encouraged.68,69 Additionally, due to the reversible nature of common dynamic alterations, operando spectroscopic techniques should be used when possible to elucidate the active structure.70,71
Microscale to macroscale
Spatial separation
The trends for different nanoparticle mixing patterns generally show that nanoscale spatial separation of COcat and C–Ccat components promotes tandem catalysis. This also holds true for cascade systems in which the components are separated on larger length scales. For example, an alloyed Au0.01Cu0.99 catalyst was shown to exhibit minimal improvements over pure Cu, while a Cu surface featuring Au nanoparticles displayed a higher C2+ selectivity.72 This suggested that CO generation at a separate site was key to boosting C–C coupling. Separation at microscale also presents an opportunity to control CO diffusion pathways. In one case, a Cu nanoneedle array featuring Ag nanoparticles was used to increase the diffusion path for CO, resulting in higher coverage and enhanced C2+ selectivity.73 A notable example using well-defined and uniformly dispersed Cu dot or line arrays on Ag surfaces provided a deeper understanding of diffusional transport, revealing that sequential catalysis could be controlled through microstructure tuning.74 In this case, a 2D diffusion transport model was used to confirm that the flux of CO from the Ag catalyst was sufficient to facilitate effective C–C coupling at the Cu sites.Spatial separation of metals has also proven effective in convective flow systems. An example of this used a flowing electrolyte solution with an Ag COcat placed downstream from a Cu C–Ccat.75 Computational simulations were used to elucidate CO convection and diffusion routes to guide experimental operating conditions. A 1 mm gap between the plates enabled the separated components to individually drive CO2R and CO reduction, resulting in an increased overall C2+ selectivity for the setup. These results have expanded the scope for micro-to-macroscale separation and different arrangements to promote purely tandem catalysis have been extended to gas diffusion electrodes in high-rate electrolysis cells.
Gas diffusion electrodes
Recent approaches to spatially separate COcat and C–Ccat components in devices for high-rate conversion have taken advantage of gas-fed flow cells (GFFCs) and membrane electrode assemblies (MEAs), which bypass mass transport limitations for CO and CO2. The key difference between these devices is that GFFCs employ a flowing liquid catholyte solution while MEAs use a humidified gas feed (Fig. 4a). In both cases, the gaseous CO2 feed overcomes solubility issues with conventional H-cells to promote high current density operation. Importantly, in these configurations, gas diffusion electrodes (GDEs) are employed to enable the gaseous CO2 to pass directly to the catalyst/solution interface, however there are a number of CO diffusion pathways that must be considered in these systems which can vary depending on the type of device employed (Fig. 4b). For example, while GFFCs can potentially suffer from CO loss in both solution (3) and through the gas outlet (1), MEAs do not have the possibility of CO loss in solution as there is no separate liquid stream. Consequently, a number of different GDE configurations have been tested for tandem CO2R, including layered, mixed, and segmented electrodes (Fig. 4c).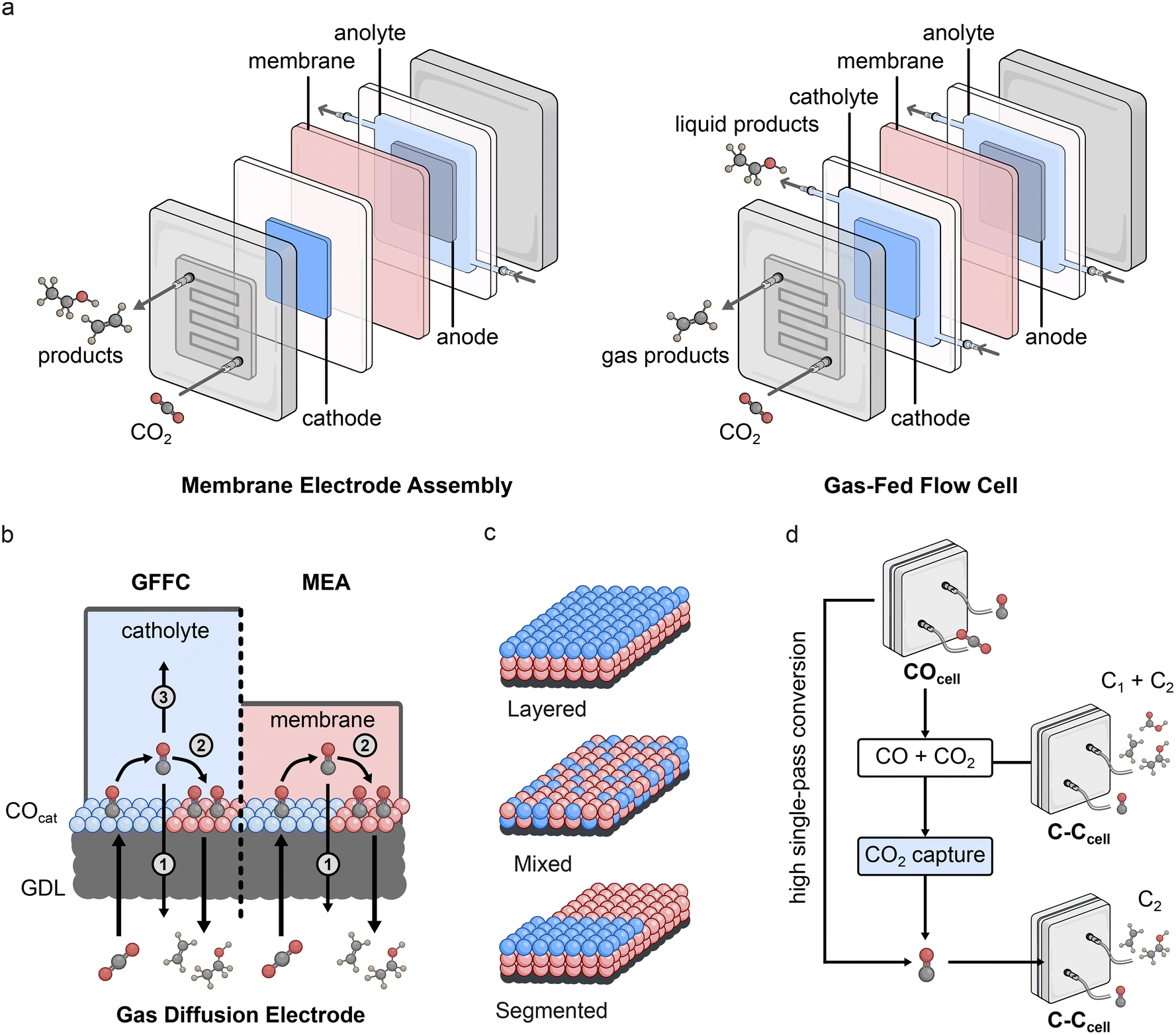 | ||
| Fig. 4 (a) Schematics showing membrane electrode assembly (MEA) and gas-fed flow cell (GFFC) configurations, (b) different interfaces for GFFC and MEA configurations with the different possible pathways for CO diffusion, in which (1) is through the gas outlet, (2) is towards the C–Ccat for further reduction, and (3) is CO loss in the solution stream (GDL = gas diffusion layer), (c) types of gas diffusion electrode arrangements that have been tested for tandem catalysis, (d) methods to combat CO purity issues in multi-cell tandems. | ||
A study using ZnO with Cu compared the selectivity for mixed and layered GDEs in a GFFC configuration.76 Results showed that although the mixed electrodes showed higher C2+ selectivity than pure Cu, layered electrodes gave the best performance, which was attributed to back-diffusion of CO away from the solution and towards the gas stream. The benefits of layered configurations over mixed were also highlighted for Cu/Ag and Cu/Ni-SAC GDEs, in which the Ni-SAC system resulted in higher C2+ selectivity due to the higher activity of the Ni catalyst for CO evolution.77 In addition, the study showed that the layered electrode was also effective in an MEA, albeit with lower selectivity than in the GFFC. Another Ni-SAC tandem arrangement with Cu nanocubes showed that a mixed configuration displayed a greater C2+ selectivity enhancement in an MEA than in a GFFC, which was attributed to the different transport properties of CO in the different configurations.78
The transport of CO has also been considered when forming segmented electrodes in MEAs. Such arrangements place the COcat at the gas inlet to increase the CO concentration at the C–Ccat placed downstream. An effective example of this employed an Ag catalyst as the COcat and varied the loading to enhance CO utilisation at the Cu C–Ccat.79 Segmented electrodes outperformed layered electrodes and were applicable to other COcats including Fe-SAC and ZnO. An important aspect of this study was the development of a 2D continuum model to rationalise the high selectivity of the segmented electrodes. The results demonstrated that spatial separation and consideration of CO mass transport and resulting *CO coverage accounted for the higher C2+ selectivity observed with such configurations.79 It is anticipated that exploration of similar configurations using a range of different catalysts will result in high-performing tandem GDEs.
Multi-cell tandems
Multi-cell tandem systems work by physically separating the COcat and C–Ccat processes into distinct cells – COcell and C–Ccell respectively (Fig. 4d). Gas products from the COcell are used as the reactant gas in the C–Ccell, and application of this system has shown increased C2+ selectivity compared to Cu single-cell systems. An important consideration in multi-cell setups is the CO:CO2 ratio in the reactant feed for the C–Ccell, as maximising CO purity avoids the formation of C1 products.80,81 One strategy is to increase the CO2R-to-CO single-pass conversion of the COcell, which would maximise CO purity, however this relies on catalysts and devices that do not generate (bi)carbonate under electrocatalytic conditions. An alternative approach to increase the purity is through subsequent capture of unconverted CO2, as has been demonstrated using either ethanolamine solution or 5 M NaOH (Fig. 4d).81,82 This method underscores the potential cooperation between carbon capture and utilisation.
A notable advantage of multi-cell systems is the ability to individually optimise catalysts for each reaction. While in single-cell systems, activities of COcat and C–Ccat components must be matched due to the application of a mutual potential, whereas in multi-cell systems they can operate at different potentials and rates can be balanced through alteration of relative geometric areas. Similarly, the separation eliminates contributions from additional factors such as electronic modification that can impact the ability of a tandem system to operate. This enables greater freedom for tuning the catalysts, potentially resulting in more selective C2+ production and consequently reduced energy requirements for product separation. However, the benefits must be balanced by the additional cost of construction and continual operation of a second cell.
Outlook
In this review, we evaluated the range of effects that can impact C2+ selectivity for tandem CO2 electrolysis. At the atomic scale, the nature of the COcat can influence the CO supply at different applied potentials and therefore can alter the observed enhancement in C2+ selectivity. This is particularly relevant to molecular catalysts and SACs which have onset potentials that are highly sensitive to structure. The increased CO concentration can enhance *CO coverage, but this can also affect the nature and binding configurations of the intermediates involved in the reaction as well as the specific surface binding energies, resulting in alterations to product distributions. The ‘pooling’ of CO in reservoirs can also play a role in increasing the availability of CO for sequential reduction to improve C2+ selectivity. In addition to these atomic-scale effects, the electronic modulation of the C–Ccat d-band by secondary metals can influence the binding energies of intermediates on the surface to alter selectivity.At the nanoscale, different nanoparticle mixing patterns have demonstrated that separating COcat and C–Ccat components is beneficial to C2+ selectivity and an indirect interface (carbon interlayer) can remove any influence of electronic alterations, promoting solely tandem effects. Conversely, for hybrid catalysts, the presence of a direct interface can enhance C2+ selectivity, reiterating the sensitivity of tandem systems to the types of catalysts used. Therefore, the optimal interface between COcat and C–Ccat can vary for different types of tandem system, which warrants further exploration. Catalyst restructuring can also play a role in selectivity alteration and this process can be dynamic under applied potentials and different conditions. However, because of the specific mechanisms and alterations, this can either have a positive or a negative effect on C2+ selectivity. Due to the difficulty in precisely controlling restructuration, prevention is instead preferable.
Spatial separation has also proven effective at larger length scales. At the microscale, consideration of CO transport has enabled design of catalysts that promote conversion of CO through understanding of diffusion or convection pathways. This has been extended to gas diffusion electrodes for MEAs and GFFCs to identify optimal pathways and consequently architectures for high-rate tandem electrolysis. Finally, at the macroscale, the use of additional electrolysers to supply CO has taken advantage of complete separation and innovative techniques to increase CO purity for sequential reduction to C2+ products.
The future development of tandem CO2R systems relies on a combination of experimental and theoretical contributions. Throughout this review, we have presented a range of mechanisms and designs that have been supported by computational methods. DFT calculations to determine types of intermediates and their surface binding energies are highly useful to outline processes at the atomic scale, but possibly the most important computational tools for tandem systems is the use of continuum modelling to determine local species concentrations and flux of primary (CO2) and secondary (CO) reactants.83 Progress in this domain offers an elegant route to design, optimise, and understand tandem processes at a range of length scales.
From a catalyst perspective there is a wide range of routes for development. Although currently most single-cell tandem systems are limited to Cu C–Ccats, the use of more selective C–Ccats could give rise to higher C2+ product specificity to target high-value chemicals. Furthermore, recent examples of C2+ products stemming from molecular catalysts opens routes to tune multicarbon selectivity through ligand and metal modifications.84 Aside from CO-based tandem pathways common to Cu catalysts, other metals have recently demonstrated C2–C4 production in cascade pathways stemming from formate-pathway intermediates rather than those that generate CO, and could therefore be utilised in tandem catalysis based on alternative primary products.85–87
When comparing multi-cell with single-cell tandems, there are a number of different considerations. Firstly, the selectivity of single-cell systems is typically dependent on the operating conditions including flow rate and current density, while multi-cell tandems are less restricted by these conditions as the input feed for the C–Ccell can be easily altered. This provides greater adaptability when used for industrial processes. However, in terms of cost, the additional electrolyser unit required for multi-cell configurations adds to the capital expenditure of the full system, therefore this must sufficiently counteract cost benefits of reduced separation if operating with higher selectivity, and possible associated costs such as (bi)carbonate regeneration. Finally, the energy efficiency of the C–Ccell in multi-cell tandems should be sufficiently high to minimise the energy demand for a full system, while in a single-cell tandem, the cooperation between COcat and C–Ccat sites can reduce the required energy input. Nonetheless, continued development of both types of system is necessary to identify additional benefits that each configuration could offer.
This review highlights the multitude of methods to promote C2+ selectivity using tandem systems, in which individual systems can display specific advantages at different length scales. However, the interdependent nature of many catalyst and device modifications means that consideration of multiple effects is often required to determine the dominant mechanisms for promotion of C2+ selectivity. As our understanding of tandem effects continues to grow, there is promise for selective CO2 electrolysis to C2+ products. Consideration of atomic to macroscale effects provides an opportunity to design and develop effective tandem systems that can sustainably generate high-value products at competitive rates of reaction.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by funding from a Royal Society Research Grant (RGS\R1\231022) and a Royal Society of Chemistry Research Enablement Grant (E22-9248199019).References
- I. E. L. Stephens, K. Chan, A. Bagger, S. W. Boettcher, J. Bonin, E. Boutin, A. K. Buckley, R. Buonsanti, E. R. Cave, X. Chang, S. W. Chee, A. H. M. da Silva, P. de Luna, O. Einsle, B. Endrődi, M. Escudero-Escribano, J. V. F. de Araujo, M. C. Figueiredo, C. Hahn, K. U. Hansen, S. Haussener, S. Hunegnaw, Z. Huo, Y. J. Hwang, C. Janáky, B. S. Jayathilake, F. Jiao, Z. P. Jovanov, P. Karimi, M. T. M. Koper, K. P. Kuhl, W. H. Lee, Z. Liang, X. Liu, S. Ma, M. Ma, H.-S. Oh, M. Robert, B. R. Cuenya, J. Rossmeisl, C. Roy, M. P. Ryan, E. H. Sargent, P. Sebastián-Pascual, B. Seger, L. Steier, P. Strasser, A. S. Varela, R. E. Vos, X. Wang, B. Xu, H. Yadegari and Y. Zhou, J. Phys.: Energy, 2022, 4, 042003 Search PubMed.
- D. Wakerley, S. Lamaison, J. Wicks, A. Clemens, J. Feaster, D. Corral, S. A. Jaffer, A. Sarkar, M. Fontecave, E. B. Duoss, S. Baker, E. H. Sargent, T. F. Jaramillo and C. Hahn, Nat. Energy, 2022, 7, 130–143 CrossRef CAS.
- Z. W. Seh, J. Kibsgaard, C. F. Dickens, I. Chorkendorff, J. K. Nørskov and T. F. Jaramillo, Science, 2017, 355, eaad4998 CrossRef PubMed.
- D. Karapinar, C. E. Creissen, J. G. Rivera de la Cruz, M. W. Schreiber and M. Fontecave, ACS Energy Lett., 2021, 6, 694–706 CrossRef CAS.
- P. De Luna, C. Hahn, D. Higgins, S. A. Jaffer, T. F. Jaramillo and E. H. Sargent, Science, 2019, 364, eaav3506 CrossRef CAS PubMed.
- Y. Hori, Modern Aspects of Electrochemistry, Springer, New York, 2008, pp. 89–189 Search PubMed.
- R. Kortlever, J. Shen, K. J. P. Schouten, F. Calle-Vallejo and M. T. M. Koper, J. Phys. Chem. Lett., 2015, 6, 4073–4082 CrossRef CAS PubMed.
- J. T. Feaster, C. Shi, E. R. Cave, T. Hatsukade, D. N. Abram, K. P. Kuhl, C. Hahn, J. K. Nørskov and T. F. Jaramillo, ACS Catal., 2017, 7, 4822–4827 CrossRef CAS.
- W. Zhu, B. M. Tackett, J. G. Chen and F. Jiao, Top. Curr. Chem., 2018, 376, 41 CrossRef PubMed.
- B. Cao, F.-Z. Li and J. Gu, ACS Catal., 2022, 12, 9735–9752 CrossRef CAS.
- C. E. Creissen, in Recent Developments in Functional Materials for Artificial Photosynthesis, ed. S. Ghosh and Q. Wang, The Royal Society of Chemistry, 2023, ch 6, pp. 120–156.
- F. Li, Y. C. Li, Z. Wang, J. Li, D. H. Nam, Y. Lum, M. Luo, X. Wang, A. Ozden, S. F. Hung, B. Chen, Y. Wang, J. Wicks, Y. Xu, Y. Li, C. M. Gabardo, C. T. Dinh, Y. Wang, T. T. Zhuang, D. Sinton and E. H. Sargent, Nat. Catal., 2020, 3, 75–82 CrossRef CAS.
- M. Wang, V. Nikolaou, A. Loiudice, I. D. Sharp, A. Llobet and R. Buonsanti, Chem. Sci., 2022, 13, 12673–12680 RSC.
- A. S. Varela, W. Ju, A. Bagger, P. Franco, J. Rossmeisl and P. Strasser, ACS Catal., 2019, 9, 7270–7284 CrossRef CAS.
- D.-L. Meng, M.-D. Zhang, D.-H. Si, M.-J. Mao, Y. Hou, Y.-B. Huang and R. Cao, Angew. Chem., 2021, 133, 25689–25696 CrossRef.
- Y. Huang, A. D. Handoko, P. Hirunsit and B. S. Yeo, ACS Catal., 2017, 7, 1749–1756 CrossRef CAS.
- C. Zhan, F. Dattila, C. Rettenmaier, A. Bergmann, S. Kühl, R. García-Muelas, N. López and B. R. Cuenya, ACS Catal., 2021, 11, 7694–7701 CrossRef CAS PubMed.
- X. Kong, J. Zhao, J. Ke, C. Wang, S. Li, R. Si, B. Liu, J. Zeng and Z. Geng, Nano Lett., 2022, 22, 3801–3808 CrossRef CAS PubMed.
- K. Qi, Y. Zhang, N. Onofrio, E. Petit, X. Cui, J. Ma, J. Fan, H. Wu, W. Wang, J. Li, J. Liu, Y. Zhang, Y. Wang, G. Jia, J. Wu, L. Lajaunie, C. Salameh and D. Voiry, Nat. Catal., 2023, 6, 319–331 CrossRef CAS.
- A. Perazio, C. E. Creissen, J. G. Rivera de la Cruz, M. W. Schreiber and M. Fontecave, ACS Energy Lett., 2023, 8, 2979–2985 CrossRef CAS.
- Y. C. Tan, K. B. Lee, H. Song and J. Oh, Joule, 2020, 4, 1104–1120 CrossRef CAS.
- X. Chang, H. Xiong, Q. Lu and B. Xu, JACS Au, 2023, 3, 2948–2963 CrossRef CAS PubMed.
- J. Hou, X. Chang, J. Li, B. Xu and Q. Lu, J. Am. Chem. Soc., 2022, 144, 22202–22211 CrossRef CAS PubMed.
- S. Louisia, D. Kim, Y. Li, M. Gao, S. Yu, I. Roh and P. Yang, Proc. Natl. Acad. Sci. U. S. A., 2022, 119, e2201922119 CrossRef CAS PubMed.
- X. Wang, J. F. de Araújo, W. Ju, A. Bagger, H. Schmies, S. Kühl, J. Rossmeisl and P. Strasser, Nat. Nanotechnol., 2019, 14, 1063–1070 CrossRef CAS PubMed.
- W. Gao, Y. Xu, H. Xiong, X. Chang, Q. Lu and B. Xu, Angew. Chem., Int. Ed., 2023, 62, e202313798 CrossRef CAS PubMed.
- W. Gao, Y. Xu, L. Fu, X. Chang and B. Xu, Nat. Catal., 2023, 6, 885–894 CrossRef CAS.
- Y. Lum and J. W. Ager, Nat. Catal., 2019, 2, 86–93 CrossRef CAS.
- P. Wei, D. Gao, T. Liu, H. Li, J. Sang, C. Wang, R. Cai, G. Wang and X. Bao, Nat. Nanotechnol., 2023, 18, 299–306 CrossRef CAS PubMed.
- J. Li, Z. Wang, C. McCallum, Y. Xu, F. Li, Y. Wang, C. M. Gabardo, C.-T. Dinh, T.-T. Zhuang, L. Wang, J. Y. Howe, Y. Ren, E. H. Sargent and D. Sinton, Nat. Catal., 2019, 2, 1124–1131 CrossRef CAS.
- H. Shen, Y. Wang, T. Chakraborty, G. Zhou, C. Wang, X. Fu, Y. Wang, J. Zhang, C. Li, F. Xu, L. Cao, T. Mueller and C. Wang, ACS Catal., 2022, 12, 5275–5283 CrossRef CAS.
- X. Wang, P. Ou, J. Wicks, Y. Xie, Y. Wang, J. Li, J. Tam, D. Ren, J. Y. Howe, Z. Wang, A. Ozden, Y. Z. Finfrock, Y. Xu, Y. Li, A. S. Rasouli, K. Bertens, A. H. Ip, M. Graetzel, D. Sinton and E. H. Sargent, Nat. Commun., 2021, 12, 3387 CrossRef CAS PubMed.
- J. Zhang, C. Guo, S. Fang, X. Zhao, L. Li, H. Jiang, Z. Liu, Z. Fan, W. Xu, J. Xiao and M. Zhong, Nat. Commun., 2023, 14, 1298 CrossRef CAS PubMed.
- C. Chen, S. Yu, Y. Yang, S. Louisia, I. Roh, J. Jin, S. Chen, P.-C. Chen, Y. Shan and P. Yang, Nat. Catal., 2022, 5, 878–887 CrossRef CAS.
- J. A. Rabinowitz and M. W. Kanan, Nat. Commun., 2020, 11, 5231 CrossRef CAS PubMed.
- J. E. Huang, F. Li, A. Ozden, A. S. Rasouli, F. P. G. de Arquer, S. Liu, S. Zhang, M. Luo, X. Wang, Y. Lum, Y. Xu, K. Bertens, R. K. Miao, C. T. Dinh, D. Sinton and E. H. Sargent, Science, 2021, 372, 1074–1078 CrossRef CAS PubMed.
- N. Ling, J. Zhang, M. Wang, Z. Wang, Z. Mi, S. Bin Dolmanan, M. Zhang, B. Wang, W. Ru Leow, J. Zhang and Y. Lum, Angew. Chem., Int. Ed., 2023, 62, e202308782 CrossRef CAS PubMed.
- Y.-J. Zhang, V. Sethuraman, R. Michalsky and A. A. Peterson, ACS Catal., 2014, 4, 3742–3748 CrossRef CAS.
- Y. Xie, P. Ou, X. Wang, Z. Xu, Y. C. Li, Z. Wang, J. E. Huang, J. Wicks, C. McCallum, N. Wang, Y. Wang, T. Chen, B. T. W. Lo, D. Sinton, J. C. Yu, Y. Wang and E. H. Sargent, Nat. Catal., 2022, 5, 564–570 CrossRef CAS.
- J. K. Nørskov, T. Bligaard, J. Rossmeisl and C. H. Christensen, Nat. Chem., 2009, 1, 37–46 CrossRef PubMed.
- Z.-J. Zhao, S. Liu, S. Zha, D. Cheng, F. Studt, G. Henkelman and J. Gong, Nat. Rev. Mater., 2019, 4, 792–804 CrossRef.
- K. Liu, M. Ma, L. Wu, M. Valenti, D. Cardenas-Morcoso, J. P. Hofmann, J. Bisquert, S. Gimenez and W. A. Smith, ACS Appl. Mater. Interfaces, 2019, 11, 16546–16555 CrossRef CAS PubMed.
- T. Kim, R. E. Kumar, J. A. Brock, E. E. Fullerton and D. P. Fenning, ACS Catal., 2021, 11, 6662–6671 CrossRef CAS.
- R. P. Jansonius, L. M. Reid, C. N. Virca and C. P. Berlinguette, ACS Energy Lett., 2019, 4, 980–986 CrossRef CAS.
- E. L. Clark, C. Hahn, T. F. Jaramillo and A. T. Bell, J. Am. Chem. Soc., 2017, 139, 15848–15857 CrossRef CAS PubMed.
- J. Monzó, Y. Malewski, R. Kortlever, F. J. Vidal-Iglesias, J. Solla-Gullón, M. T. M. Koper and P. Rodriguez, J. Mater. Chem. A, 2015, 3, 23690–23698 RSC.
- C. Du, J. P. Mills, A. G. Yohannes, W. Wei, L. Wang, S. Lu, J.-X. Lian, M. Wang, T. Guo, X. Wang, H. Zhou, C.-J. Sun, J. Z. Wen, B. Kendall, M. Couillard, H. Guo, Z. Tan, S. Siahrostami and Y. A. Wu, Nat. Commun., 2023, 14, 6142 CrossRef CAS PubMed.
- N. Martić, C. Reller, C. Macauley, M. Löffler, A. M. Reichert, T. Reichbauer, K. M. Vetter, B. Schmid, D. McLaughlin, P. Leidinger, D. Reinisch, C. Vogl, K. J. J. Mayrhofer, I. Katsounaros and G. Schmid, Energy Environ. Sci., 2020, 13, 2993–3006 RSC.
- X. Wang, P. Ou, A. Ozden, S.-F. Hung, J. Tam, C. M. Gabardo, J. Y. Howe, J. Sisler, K. Bertens, F. P. García de Arquer, R. K. Miao, C. P. O'Brien, Z. Wang, J. Abed, A. S. Rasouli, M. Sun, A. H. Ip, D. Sinton and E. H. Sargent, Nat. Energy, 2022, 7, 170–176 CrossRef CAS.
- W. Niu, Z. Chen, W. Guo, W. Mao, Y. Liu, Y. Guo, J. Chen, R. Huang, L. Kang, Y. Ma, Q. Yan, J. Ye, C. Cui, L. Zhang, P. Wang, X. Xu and B. Zhang, Nat. Commun., 2023, 14, 4882 CrossRef CAS PubMed.
- E. L. Clark, R. Nielsen, J. E. Sørensen, J. L. Needham, B. Seger and I. Chorkendorff, ACS Energy Lett., 2023, 8, 4414–4420 CrossRef CAS PubMed.
- T. Gong, K. K. Rudman, B. Guo and A. S. Hall, Acc. Chem. Res., 2023, 56, 1373–1383 CrossRef CAS PubMed.
- S. Ma, M. Sadakiyo, M. Heim, R. Luo, R. T. Haasch, J. I. Gold, M. Yamauchi and P. J. A. Kenis, J. Am. Chem. Soc., 2017, 139, 47–50 CrossRef CAS PubMed.
- Y. Ma, J. Yu, M. Sun, B. Chen, X. Zhou, C. Ye, Z. Guan, W. Guo, G. Wang, S. Lu, D. Xia, Y. Wang, Z. He, L. Zheng, Q. Yun, L. Wang, J. Zhou, P. Lu, J. Yin, Y. Zhao, Z. Luo, L. Zhai, L. Liao, Z. Zhu, R. Ye, Y. Chen, Y. Lu, S. Xi, B. Huang, C.-S. Lee and Z. Fan, Adv. Mater., 2022, 34, 2110607 CrossRef CAS PubMed.
- J. Huang, M. Mensi, E. Oveisi, V. Mantella and R. Buonsanti, J. Am. Chem. Soc., 2019, 141, 2490–2499 CrossRef CAS PubMed.
- J. Zhang, T. H. M. Pham, Y. Ko, M. Li, S. Yang, C. D. Koolen, L. Zhong, W. Luo and A. Züttel, Cell Rep. Phys. Sci., 2022, 3, 100949 CrossRef CAS.
- M. Wang, A. Loiudice, V. Okatenko, I. D. Sharp and R. Buonsanti, Chem. Sci., 2023, 14, 1097–1104 RSC.
- S. Ren, E. W. Lees, C. Hunt, A. Jewlal, Y. Kim, Z. Zhang, B. A. W. Mowbray, A. G. Fink, L. Melo, E. R. Grant and C. P. Berlinguette, J. Am. Chem. Soc., 2023, 145, 4414–4420 CrossRef CAS PubMed.
- D. Grammatico, A. J. Bagnall, L. Riccardi, M. Fontecave, B.-L. Su and L. Billon, Angew. Chem., Int. Ed., 2022, 61, e202206399 CrossRef CAS PubMed.
- R. Amirbeigiarab, J. Tian, A. Herzog, C. Qiu, A. Bergmann, B. Roldan Cuenya and O. M. Magnussen, Nat. Catal., 2023, 6, 1–10 CrossRef.
- J. Huang, N. Hörmann, E. Oveisi, A. Loiudice, G. L. De Gregorio, O. Andreussi, N. Marzari and R. Buonsanti, Nat. Commun., 2018, 9, 3117 CrossRef PubMed.
- R. M. Arán-Ais, R. Rizo, P. Grosse, G. Algara-Siller, K. Dembélé, M. Plodinec, T. Lunkenbein, S. W. Chee and B. R. Cuenya, Nat. Commun., 2020, 11, 3489 CrossRef PubMed.
- C. E. Creissen and M. Fontecave, Nat. Commun., 2022, 13, 2280 CrossRef CAS PubMed.
- P.-C. Chen, C. Chen, Y. Yang, A. L. Maulana, J. Jin, J. Feijoo and P. Yang, J. Am. Chem. Soc., 2023, 145, 10116–10125 CrossRef CAS PubMed.
- S. V. Somerville, P. B. O'Mara, T. M. Benedetti, S. Cheong, W. Schuhmann, R. D. Tilley and J. J. Gooding, J. Phys. Chem. C, 2023, 127, 289–299 CrossRef CAS PubMed.
- P. Wilde, P. B. O'Mara, J. R. C. Junqueira, T. Tarnev, T. M. Benedetti, C. Andronescu, Y. T. Chen, R. D. Tilley, W. Schuhmann and J. J. Gooding, Chem. Sci., 2021, 12, 4028–4033 RSC.
- B. Eren, D. Zherebetskyy, L. L. Patera, C. H. Wu, H. Bluhm, C. Africh, L.-W. Wang, G. A. Somorjai and M. Salmeron, Science, 2016, 351, 475–478 CrossRef CAS PubMed.
- X. K. Lu, B. Lu, H. Li, K. Lim and L. C. Seitz, ACS Catal., 2022, 12, 6663–6671 CrossRef CAS.
- V. Okatenko, A. Loiudice, M. A. Newton, D. C. Stoian, A. Blokhina, A. N. Chen, K. Rossi and R. Buonsanti, J. Am. Chem. Soc., 2023, 145, 5370–5383 CrossRef CAS PubMed.
- J. Timoshenko and B. Roldan Cuenya, Chem. Rev., 2021, 121, 882–961 CrossRef CAS PubMed.
- D. Mendoza, S.-T. Dong and B. Lassalle-Kaiser, Curr. Opin. Colloid Interface Sci., 2022, 61, 101635 CrossRef CAS.
- C. G. Morales-Guio, E. R. Cave, S. A. Nitopi, J. T. Feaster, L. Wang, K. P. Kuhl, A. Jackson, N. C. Johnson, D. N. Abram, T. Hatsukade, C. Hahn and T. F. Jaramillo, Nat. Catal., 2018, 1, 764–771 CrossRef CAS.
- C. Wei, Y. Yang, H. Ma, G. Sun, X. Wang, Y. Cheng, C. Zhang, B. S. Yeo, C. He and A. B. Wong, Adv. Funct. Mater., 2023, 33, 2214992 CrossRef CAS.
- Y. Lum and J. W. Ager, Energy Environ. Sci., 2018, 11, 2935–2944 RSC.
- Gurudayal, D. Perone, S. Malani, Y. Lum, S. Haussener and J. W. Ager, ACS Appl. Energy Mater., 2019, 2, 4551–4559 CrossRef CAS.
- T. Zhang, Z. Li, J. Zhang and J. Wu, J. Catal., 2020, 387, 163–169 CrossRef CAS.
- X. She, T. Zhang, Z. Li, H. Li, H. Xu and J. Wu, Cell Rep. Phys. Sci., 2020, 1, 100051 CrossRef.
- Y.-R. Lin, D. U. Lee, S. Tan, D. M. Koshy, T. Y. Lin, L. Wang, D. Corral, J. E. Avilés Acosta, J. A. Zamora Zeledon, V. A. Beck, S. E. Baker, E. B. Duoss, C. Hahn and T. F. Jaramillo, Adv. Funct. Mater., 2022, 32, 2113252 CrossRef CAS.
- T. Zhang, J. C. Bui, Z. Li, A. T. Bell, A. Z. Weber and J. Wu, Nat. Catal., 2022, 5, 202–211 CrossRef CAS.
- T. Möller, M. Filippi, S. Brückner, W. Ju and P. Strasser, Nat. Commun., 2023, 14, 5680 CrossRef PubMed.
- N. S. Romero Cuellar, C. Scherer, B. Kaçkar, W. Eisenreich, C. Huber, K. Wiesner-Fleischer, M. Fleischer and O. Hinrichsen, J. CO2 Util., 2020, 36, 263–275 CrossRef CAS.
- A. Ozden, Y. Wang, F. Li, M. Luo, J. Sisler, A. Thevenon, A. Rosas-Hernández, T. Burdyny, Y. Lum, H. Yadegari, T. Agapie, J. C. Peters, E. H. Sargent and D. Sinton, Joule, 2021, 5, 706–719 CrossRef CAS.
- J. C. Bui, E. W. Lees, L. M. Pant, I. V. Zenyuk, A. T. Bell and A. Z. Weber, Chem. Rev., 2022, 122, 11022–11084 CrossRef CAS PubMed.
- S.-T. Dong, C. Xu and B. Lassalle-Kaiser, Chem. Sci., 2023, 14, 550–556 RSC.
- S. P. Cronin, S. Dulovic, J. A. Lawrence, K. A. Filsinger, A. P. Hernandez-Gonzalez, R. Evans, J. W. Stiles, J. Morris, I. Pelczer and A. B. Bocarsly, J. Am. Chem. Soc., 2023, 145, 6762–6772 CrossRef CAS PubMed.
- M. Choi, S. Bong, J. W. Kim and J. Lee, ACS Energy Lett., 2021, 6, 2090–2095 CrossRef CAS.
- K. U. D. Calvinho, A. B. Laursen, K. M. K. Yap, T. A. Goetjen, S. Hwang, N. Murali, B. Mejia-Sosa, A. Lubarski, K. M. Teeluck, E. S. Hall, E. Garfunkel, M. Greenblatt and G. C. Dismukes, Energy Environ. Sci., 2018, 11, 2550–2559 RSC.
| This journal is © The Royal Society of Chemistry 2024 |