
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Crystal facet engineering of spinel NiCo2O4 with enhanced activity and water resistance for tuneable catalytic methane oxidation†
Yash
Boyjoo
abc,
Yonggang
Jin
*a,
Xin
Mao
d,
Guangyu
Zhao
a,
Thomas
Gengenbach
e,
Aijun
Du
 d,
Hua
Guo
*a and
Jian
Liu
*bf
d,
Hua
Guo
*a and
Jian
Liu
*bf
aCSIRO Mineral Resources, 1 Technology Court, Pullenvale, QLD 4069, Australia. E-mail: yonggang.jin@csiro.au; hua.guo@csiro.au
bState Key Laboratory of Catalysis, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, 457 Zhongshan Road, Dalian 116023, China. E-mail: jian.liu@surrey.ac.uk
cCurtin Mauritius, Telfair, Moka, Mauritius
dSchool of Chemistry, Physics and Mechanical Engineering, Queensland University of Technology, Brisbane, QLD 4001, Australia
eCSIRO Manufacturing, Bayview Avenue, Clayton, VIC 3168, Australia
fDICP-Surrey Joint Centre for Future Materials, Department of Chemical and Process Engineering and Advanced Technology Institute, University of Surrey, Guildford, Surrey GU2 7XH, UK
First published on 26th December 2023
Abstract
Spinel NiCo2O4 are excellent catalysts for complete methane oxidation. Nevertheless, the spinel structure is thermally unstable and its activity is negatively affected by humidity. Herein, we report crystal facet engineering synthesis of spinel NiCo2O4 hexagonal nanosheets with different exposed facets. Density functional theory (DFT) simulations predict that a more viable reaction mechanism for methane oxidation occurs on 112-NiCo2O4 with {112} exposed facets compared with 111-NiCo2O4 with {111} exposed facets. Detailed material characterization and catalytic oxidation testing verified the DFT results showing that 112-NiCo2O4 has better thermal stability as well as higher catalytic activity towards methane oxidation than 111-NiCo2O4. Conversely, 111-NiCo2O4 has the enhanced water resistance of the two catalysts. DFT calculations suggest that OH groups tend to preferentially adsorb onto metal sites, which (1) reduces the number of active sites available and (2) makes CH4 adsorption and activation a more arduous process. This study offers insights on the behavior of spinel oxide catalysts towards methane combustion in dry and humid conditions, further demonstrating that crystal facet engineering can be a practical strategy to tune the activity and water resistance of metal-oxide catalysts.
Broader contextCatalytic oxidation is ideal for the treatment of low concentration effluents. In this context, spinel NiCo2O4 have been found to be excellent catalysts for complete CH4 oxidation. Nevertheless, the spinel structure is thermally unstable and its activity is negatively affected by humidity. Herein, we report crystal facet engineering synthesis of spinel NiCo2O4 hexagonal nanosheets with different exposed facets. Experimental and density functional theory (DFT) studies show that 112-NiCo2O4 with {112} exposed facets has better thermal stability as well as higher catalytic activity towards methane oxidation than 111-NiCo2O4 with {111} exposed facets. Conversely, 111-NiCo2O4 has the enhanced water resistance of the two catalysts. Modelling studies suggest that OH groups tend to preferentially adsorb onto metal sites. Nevertheless it may be possible that in humid conditions, CH4 oxidation follows an alternative pathway on 111-NiCo2O4. This study offers insights on the behavior of spinel oxide catalysts towards CH4 combustion in dry and humid conditions, further demonstrating that crystal facet engineering can be a practical strategy to tune the activity and water resistance of metal-oxide catalysts. These low-cost and high-performance catalysts are highly demanded for mitigating fugitive CH4 emissions in resources and energy sectors such as coal mining, natural gas industry, natural gas engines, etc. |
Introduction
Approximately 70% of the methane (CH4) emitted from coal mines is released as ventilation air methane (VAM).1 Being a much more potent greenhouse gas than CO2,1 efficient remediation pathways are required to be considered for the removal of CH4 from VAM. Since thermal oxidation of dilute methane in the ventilation air requires reaction temperatures usually above 1000 °C, catalytic oxidation of lean CH4 has been investigated extensively as a promising way to lower this temperature via the use of a catalytic process.2 Catalytic oxidation is ideal for the treatment of low concentration effluents and has been successfully used for the treatment of CH43–5 but also for the combustion of other hydrocarbons such as carbon monoxide,6 formaldehyde,7 toluene,8,9 propane10 and chlorinated volatile organic compounds (VOCs).11It is generally agreed that CH4 total oxidation proceeds through a Mars–Van Krevelen (M–vK) mechanism, during which the hydrocarbon molecules get adsorbed on the metal active sites followed by stepwise oxidation using lattice oxygen.2 The oxygen vacancies created as a result can be filled by adsorption and dissociation of oxygen from the air during reaction. While supported PdOx nanoparticles are the state-of-the-art catalytic materials for CH4 complete oxidation,12 non-noble metal oxides such as Co3O4 hexagonal nanosheets,13,14 Co3O4 nanoparticles,5,15 Co3O4 nanotubes,16 CuO nanobelts,17 bowtie-shaped spinel NiCo2O4 catalysts,18 and composites such as Co3O4/CeO2,19 MnOx–NiO20 and NiO/CeO221 have also proved to be appealing candidates, with the added benefit of being much cheaper than noble metal catalysts. Spinel Co3O4 or NiCo2O4 type materials are among the most attractive as they can deliver complete CH4 oxidation at temperatures in the neighbourhood of 500 °C. This is attributed to the spinel structure that allows the formation of oxygen vacancies rather easily, due to the uneven metal–oxygen bond lengths within the material's matrix. Nevertheless, the main issue with the spinel structure is that it is thermally unstable and decomposes at elevated temperatures into a much less active material.22 For example, NiO tends to segregate from the spinel NiCo2O4 structure at elevated temperatures,22,23 which deactivates the catalyst.
PdOx and metal oxides nanoparticles are generally deactivated by the presence of water vapour that is normally present in dilute methane emissions.12,24,25 This is because water molecules tend to compete for active sites or facilitate the coalescence of PdOx nanoparticles. Consequently, the development of efficient, active and water-resistant catalysts presents a major challenge in this current field of research. Some of the strategies that have been used so far for metal oxides are compositing with other metal oxides25 or, in the case of PdOx, the use of supports or the addition of Pt as a promoter.12
Crystal facet engineering (CFE) of nanocrystals strategy has been widely reported for the enhanced catalytic activity of catalysts via the design and creation of specific exposed crystal planes on its surface. The unique atomic arrangements that occur as a consequence of CFE allows the possibility to stabilise high index crystal planes that can: expose abundant unsaturated active sites to adsorb and activate reactants, facilitate formation of oxygen vacancies and create structural defects such as steps and edges.26 Over the past decade or so, CFE has been successfully applied in batteries,27,28 gas-sensing,29 water-splitting,30,31 photocatalysis,32 CO2 conversion33,34 and oxygen reduction/evolution reactions (ORR/OER).35,36 Notably, CFE has been successfully applied to the catalytic oxidation of hydrocarbons. High energy surface facets in MnO2 catalysts could facilitate the creation of oxygen vacancies compared with the low-index facets such as {110} or {100}, resulting in enhanced activities for VOC oxidation.7,8 The exposed crystal facet {112} in Co3O4 was found to be responsible for its higher activity during the combustion of methane when compared with the {001} and {011} planes.13 Unsurprisingly, the same exposed facet on Co3O4 hexagonal nanosheets was active for CO and toluene oxidation due to the stabilisation of oxygen vacancies, which allows the unhampered removal and replenishment of lattice oxygen during reaction.14 Oxygen vacancies created during CFE have also shown good ability to trap metal cations within the lattice37 and favours single-atom doping38 for applications in catalytic oxidation reactions. CFE can be relatively easily achieved through modifying synthesis procedures (e.g., use of different capping agents, solvent ratios and hydrothermal synthesis temperatures) and is therefore an attractive and straightforward approach to be employed to tune the catalytic properties of crystal surfaces.
Herein, we applied CFE to synthesise two NiCo2O4 catalysts with similar morphologies and textural parameters but with different exposed crystal facets. As a result, we created 111-NiCo2O4 and 112-NiCo2O4 having a hexagonal nanoplate morphology and with {111} and {112} exposed crystal facets, respectively. Combined experimental and theoretical studies were performed to investigate the effect of the exposed crystal facets on the materials’ catalytic activities and stabilities for CH4 oxidation reaction under both dry and humid conditions. We found that the high index {112} exposed facet afforded better activity and stability to NiCo2O4 hexagonal nanosheets while the {111} facet offered enhanced water resistance. Through theoretical calculations, we then acquired fundamental understandings on how the presence of water vapour in the feed decreases the catalysts’ activities. The impressive stability of 112-NiCo2O4 is demonstrated through long-term stability testing under humid conditions for 230 h reaction time. This study offers some insights into the behaviour of different exposed facets of spinel NiCo2O4 for CH4 catalytic oxidation under dry and humid conditions. It also shows that CFE can be a useful strategy to tune the activity and water resistance of metal-oxide materials by offering alternative reaction pathways in catalytic oxidation reactions for the treatment of lean effluents.
Results and discussion
Characterization
The procedure for the synthesis of 111-NiCo2O4 and 112-NiCo2O4 is given in the ESI.† The X-ray diffraction (XRD) patterns of the 111-NiCo2O4 and 112-NiCo2O4 samples calcined at different temperatures are shown in Fig. S1 and S2 (ESI†), respectively. Both 111-NiCo2O4 and 112-NiCo2O4 samples exhibit spinel NiCo2O4 structure at 400 °C calcination temperature. However, the spinel structure of 111-NiCo2O4 decomposes to show obvious NiO peaks (at 2θ = 51° and 74°) at a calcination temperature of 450 °C while these peaks appear in 112-NiCo2O4 at a higher calcination temperature of 500 °C. The spinel structure is made up of AO4 and BO6 tetrahedral and octahedral cells, respectively, where A and B are metal atoms. When Ni atoms are doped into the spinel structure, they tend to occupy the octahedral sites of NiCo2O4.39,40 Due to the difference in the atomic size of Ni relative to Co in NiCo2O4, the metal–oxygen bond lengths are uneven within the structure making it thermal unstable. Consequently, spinel NiCo2O4 has been reported to decompose at around 400 °C resulting in the segregation of NiO from the structure.3,23 The results obtained here attest for the higher thermal stability of 112-NiCo2O4 compared with 111-NiCo2O4. This could be attributed to the surface atomic arrangement in the high-index-faceted 112-NiCo2O4 that enhances its structural stability even at high temperatures.26Scanning electron microscope (SEM) images in Fig. 1a and d show thin hexagonal nanoplate morphology of size ca. 200–350 nm for 111-NiCo2O4-400 and 300–400 nm for 112-NiCo2O4-400. Some small differences exist between the two samples such as the presence of small nanoparticles in 111-NiCo2O4-400 and the presence of a few through holes on the surfaces of 112-NiCo2O4-400 but we do not expect these minor variances to affect the overall CH4 oxidation rates. Transmission electron microscopy (TEM) images in Fig. 1b and e show that both 111-NiCo2O4-400 and 112-NiCo2O4-400 have thin hexagonal nanoplate morphology. From the high-resolution Scanning transmission electron microscope (STEM) image of 111-NiCo2O4-400 in Fig. 1c, the visible planes are (2−20) and (20−2), both with lattice distance of 0.28 nm as confirmed by the Fast Fourier Transform (FFT) pattern (inset of Fig. 1c) and perpendicular to the {111} exposed facet according to the Weiss zone law.41 Similarly in Fig. 1f, the {112} exposed facet is dominant in 112-NiCo2O4-400 which is normal to the (11−1) and (1−31) planes with lattice distances of 0.46 nm and 0.24 nm, respectively and in accordance with FFT pattern (inset of Fig. 1f). These results confirm that {111} and {112} are the dominant exposed facets on the surface of 111-NiCo2O4-400 and 112-NiCo2O4-400, respectively. From N2 adsorption isotherms (Fig. S3, ESI†), it was evaluated that both samples had a similar Brunauer, Emmett and Teller (BET) specific surface areas with 38 m2 g−1 for 111-NiCo2O4-400 and 37 m2 g−1 for 112-NiCo2O4-400. These initial results confirm that the basic textural parameters of both samples (particles sizes, morphologies and surface areas) are comparable and therefore ideal for studying the effect of crystal exposed facet as a predominant variable for the catalytic oxidation of CH4.
 | ||
| Fig. 1 (a) SEM, (b) STEM and (c) high-resolution STEM for 111-NiCo2O4-400 (inset: corresponding FFT pattern), (d) SEM, (e) STEM and (f) high-resolution STEM for 112-NiCo2O4-400 (inset: corresponding FFT pattern). | ||
Hydrogen temperature-programmed-reduction (H2-TPR) results in Fig. 2a reveal two reduction peaks for both samples. The first smaller peak is attributed to the reduction of M3+ cations to M2+ (M being the sum of Ni and Co atoms) while the second larger peak is due to the complete reduction of M2+ to M0.3 It was theoretically shown that M3+ species adsorb and activate the hydrocarbon molecules to be subsequently reduced to M2+ species during the oxidation reaction due to the donation of adjacent lattice oxygen atoms.5,6 Hence, comparing the first peak temperatures for both samples, M3+ species in 112-NiCo2O4-400 can be more easily reduced (228.6 °C) than in 111-NiCo2O4-400 (261.2 °C). Good reducibility or the ability to give away lattice oxygen atoms is a vital characteristic for metal oxide catalysts used in oxidation reactions. Furthermore, by calculating and comparing the areas under the peaks, it can be determined that 112-NiCo2O4 had slightly higher amount of M3+ at 58% compared to 53% for 111-NiCo2O4-400. The CH4 consumption and CO2 evolved during methane temperature-programmed-reduction (CH4-TPR) are shown in Fig. 2b. The CO2 produced is a result of the reaction of the adsorbed CH4 with lattice oxygen from the NiCo2O4 surface. The reduction peaks are attributed to the reduction process of M3+ to M2+. 13 Water was also detected while negligible CO or H2 occurred during this time (see Fig. S4 and S5 in the ESI† for more details). The CH4 consumption peaks centred at 452 °C and 469 °C, closely correspond to the CO2 evolution peaks centred at 446 °C and 466 °C for 112-NiCo2O4-400 and 111-NiCo2O4-400, respectively. Additionally, the ratio of the areas under the CO2 formation peaks divulge that 20% more CO2 was formed on 112-NiCo2O4-400 relative to 111-NiCo2O4-400. The lower reduction temperature of the 112-NiCo2O4 sample from the CH4-TPR results may be due to the atomic arrangement on the 112 surface which facilitates CH4 adsorption, activation and oxidation (as confirmed by DFT later). It is noticed that the amount of CO2 generated (20% more for 112-NiCo2O4-400 compared with 111-NiCo2O4-400) does not correspond to the amounts of M3+ as suggested by the H2-TPR profiles in the respective samples. This could be due to: (1) not all the M3+ species act as active sites or (2) different atomic arrangement on the differently exposed crystal facet, which creates a more active pathway for CH4 oxidation reaction (as revealed later in the DFT calculation section) on the 112-NiCo2O4 sample. Nevertheless, the CH4-TPR findings are in line with the H2-TPR results indicating 112-NiCo2O4-400 as a more active catalyst than 111-NiCo2O4-400.
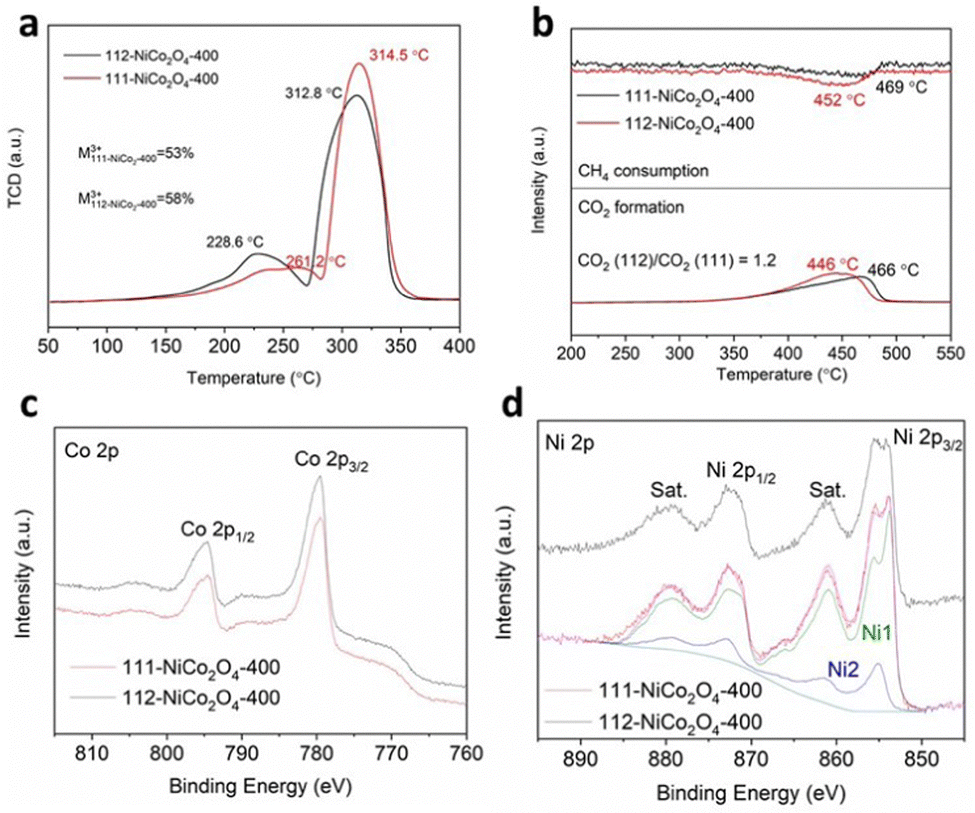 | ||
| Fig. 2 (a) H2-TPR, (b) CH4-TPR, high-resolution XPS of (c) Co 2p and (d) Ni 2p for 111-NiCo2O4-400 and 112-NiCo2O4-400. A representative Ni 2p peak-fit using NiO (Ni1) and Ni(OH)2 (Ni2) model spectra is shown for 111-NiCo2O4-400. See text for details. | ||
X-ray photoelectron spectroscopy (XPS) analysis of transition metal oxides is not a trivial exercise because the relevant metal 2p photoelectron signals are very complex, and different oxidation states cannot easily be determined, let alone quantified. Herein we mainly rely on the important studies published over the last 10–15 years to extract reliable and meaningful information about the nature of the Co and Ni species in the NiCo2O4 samples.42–45 The Co 2p spectra for both 111-NiCo2O4-400 and 112-NiCo2O4-400 in Fig. 2c are similar and exhibit relatively narrow peak widths with flat, weak satellite structures. These features are characteristic to the presence of both Co3+ and Co2+, with predominantly the higher oxidation state specie, in a spinel lattice.42 Nevertheless, the quantification of all species is very difficult, not only because of the complex peak shape of the Co 2p doublet but also because of its significant overlap with both Co and Ni Auger peaks. An attempt at identifying and quantifying the two different oxidation states would be associated with large uncertainties and, therefore, be unreliable. The Ni 2p spectra for both samples are presented in Fig. 2d. They clearly display the characteristics of NiO, i.e., a sharp intense peak at around 853.7 eV, a broader, weaker peak about 2 eV above the first peak, and a strong loss/satellite structure between 860 and 862 eV.43–45 However, compared to the sharp low binding energy (BE) peak, the second peak at 855–856 eV is more intense as what would be observed in a pure NiO sample. This points to the possible presence of some Ni(OH)2 which has been shown to be inherently present as an impurity in the form of a thin layer over synthesised NiO samples following exposure to ambient atmosphere.43,44 Furthermore, the intense loss/satellites features indicate that the oxidation state of Ni is mainly 2+, i.e., it is most probably present as NiO and Ni(OH)2 rather than NiOOH.43,44 Previous studies39,40 have shown that the Ni is mostly present inside NiO6 octahedral cells in the spinel structure of NiCo2O4 and this is depicted in Fig. S6a (ESI†). Similarly, the NiO crystal structure is an arrangement of NiO6 octahedral cells, as shown in Fig. S6b (ESI†). Therefore, the Ni 2p spectra of both 111-NiCo2O4-400 and 112-NiCo2O4-400 samples are consistent with the NiO6 octahedral cells present in the spinel NiCo2O4 structure. The O 1s spectra of both samples are similar and presented in Fig. S7 (ESI†). The C 1s spectra of the samples were used to determine the contribution to the total O 1s signal by organic carbon (see Fig. S8 for more details, ESI†). The compositions (atomic concentrations) of the samples are shown in Table S1 (ESI†). The measured Ni/Co atomic ratios are 1.0 and 0.7 for 111-NiCo2O4-400 and 112-NiCo2O4-400, respectively, deviating significantly from the stoichiometric value of 0.5. This can be attributed to the surface chemical arrangement being quite different from the bulk, noting that the XPS sampling depth is of the order of just a few nm. The Ni 2p spectra for both samples could be fitted reasonably well using model spectra derived from NiO and Ni(OH)2 reference compounds (components Ni1 and Ni2, Fig. 2d). The ratio of NiO/Ni(OH)2 for 111-NiCo2O4-400 was higher at 5.5 compared to 3.2 for 112-NiCo2O4-400, implying that more NiO6 cells are present on the surface of 111-NiCo2O4-400. Due to the thermal instability of the spinel NiCo2O4 structure, at high temperatures, we surmise that the NiO6 octahedral cells tend to detach from the structure and migrate towards the surface of the catalyst where they can assemble into NiO fine nanocrystals, which then get coated with a thin Ni(OH)2 layer once exposed to ambient air.22,23 This is consistent with the observed variation of the NiO/Ni(OH)2 ratio in Table S1 (ESI†). Indeed, it was found that the samples calcined at a higher temperature of 450 °C present a surface enriched in NiO relative to the samples calcined at 400 °C. Furthermore, this ratio is higher for 111-NiCo2O4-450 compared to 112-NiCo2O4-450, which is in line with our XRD observations (Fig. S1 and S2, ESI†) that suggest that the sample with {112} exposed facets have a better thermal stability than the one with {111} exposed facets.
Comparison of 111 vs. 112 – DFT and experimental validation
An earlier density functional theory (DFT) study on NiCo2O4 with {110} exposed facets revealed that the most active site for CH4 adsorption and activation was Ni3+ situated in octahedral cells within the spinel lattice and that CH4 oxidation could follow two reaction pathways.5 In pathway 1, only Ni3+ acted as the active site while in pathway 2, both Ni3+ and Co3+ were considered as active sites. Both pathways shared steps in common for CH4 adsorption and its dissociation into *CH3 and *H. In our work according to DFT modelling, 2 analogous pathways were energetically feasible on the surfaces of 111-NiCo2O4 and 112-NiCo2O4 for complete CH4 oxidation. Firstly, it needs to be specified that the {111} facet exposes only Ni atoms on its surface while the {112} facet exposes both Ni and Co atoms (see Fig. S9 for DFT generated model representations, ESI†). The types of exposed metal atoms as well as their geometrical arrangement will clearly affect the adsorption of CH4 as well as its reaction mechanism towards complete oxidation. For pathway 1 on 111-NiCo2O4, only the Ni atom in the octahedral cell was the active site while in pathway 2 an O atom associated with a neighbouring Co atom also participated in the reaction mechanism. Regarding the 112-NiCo2O4 surface, pathway 1 involved one Ni and one Co atom in the reaction route while pathway 2 entailed two Ni atoms and one Co atom as participatory active sites. Fig. 3 displays the Gibbs free energy profiles of both pathways on the 112-NiCo2O4 surface as well as some representative molecular conformations derived from DFT modelling. The corresponding results for 111-NiCo2O4 are given in Fig. S10 (ESI†). Simplified diagrams of the detailed interactions of CH4 and intermediates on the catalysts’ surfaces are presented in Fig. S11–S14 (ESI†). The complete oxidation of CH4 on both NiCo2O4 surfaces consisted of 11 elementary steps. The first step involving the adsorption of CH4 on Ni active sites, exhibited a stronger binding energy of −0.30 eV on 112-NiCo2O4 compared to −0.19 eV on 111-NiCo2O4. This means that the 112-NiCo2O4 surface had a relatively better affinity for CH4 capture and adsorption. Following the initial adsorption step, CH4 had to overcome an activation barrier to dissociate into adsorbed *CH3 and *H species. Thereafter sequential oxidation of *CH3 to, *CH2, *CHO, *CO and finally *CO2 occurred by reaction with lattice surface oxygen. In detail, taking the 112-surface and pathway 1 as example, a H atom migrates from the *CH3 to produce *CH2 species with one water molecule released to the atmosphere. DFT calculated results suggest that an uphill free energy change is required for this step. However, for pathway 2, there is interaction between the *CH3 and the closest Co atom, which facilitates the migration of a H atom from *CH3 to the lattice O in the vicinity of the Co atom. As a result, the free energy change strongly decreases to −0.87 eV. Hence pathway 2 is much more favourable for CH4 oxidation on the 112-surface. After the formation of *CH2 + *OH + *OH, the *CH2 can dissociate into *CH, releasing one H atom to the lattice O of the nearby Co atom to form a water molecule, with the free energy change slightly increasing for this step. Then the first O2 molecule can be adsorbed into the generated oxygen vacancy with a significantly downhill free energy change. The adsorbed oxygen reacts with the *CH species to produce *CHO and *OH intermediates. The next step is the formation of CO molecule on the Ni active site. After that, another O2 molecule that gets adsorbed into an oxygen vacancy site reacts with the *CO to produce *CO2 which eventually desorbs from the active site and ends the complete oxidation cycle on 112-NiCo2O4. From Fig. S10a (ESI†), pathway 1 is the favoured route for CH4 oxidation on 111-NiCo2O4 since two uphill free energy changes are involved with a total energy requirement of 2.78 eV compared with three uphill free energy steps in pathway 2 having a total energy demand of 3.37 eV.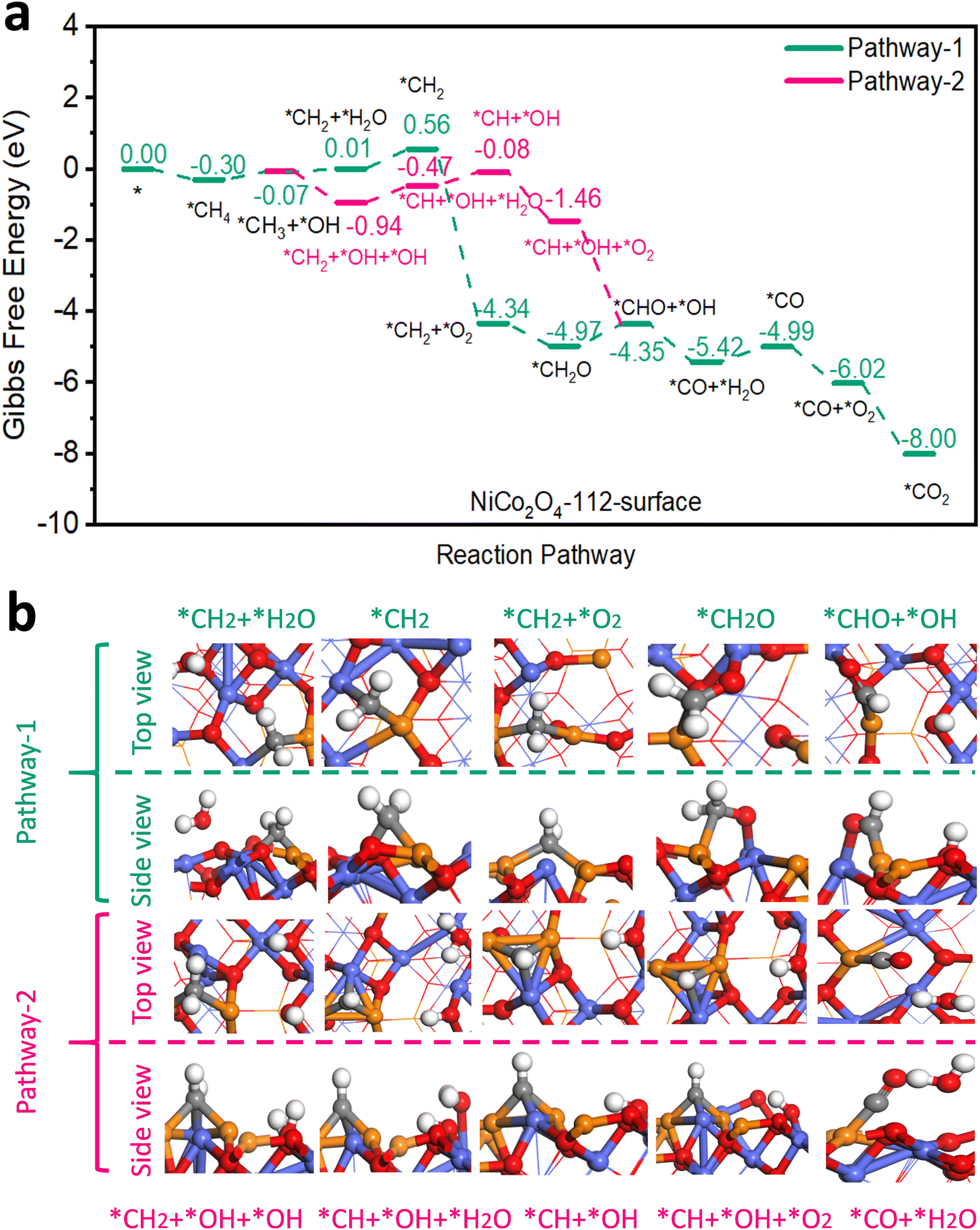 | ||
| Fig. 3 (a) Gibbs free energy diagram showing pathways and (b) selected molecular conformations for CH4 oxidation on the 112-NiCo2O4 surface. | ||
For 112-NiCo2O4 the potential limiting step for pathway 1 is the formation of *CHO with an energy requirement of 0.62 eV while for pathway 2, the potential limiting step is the formation of *CH with an energy requirement of 0.47 eV. In the case of 111-NiCo2O4, the potential limiting step for pathway 1 is the formation of *CH2 with an energy barrier of 1.61 eV while for pathway 2, the potential limiting step is the formation of *CH with an energy requirement of 1.38 eV. For both pathways, these intermediate reaction energy barriers were much larger on the 111-surface compared to the 112-surface. Consequently, CH4 oxidation should theoretically be favoured on the 112-surface compared with the 111-surface.
We performed CH4 catalytic oxidation light-off experiments to verify this. We used the samples calcined at 400 °C due to their comparable physical and textural parameters (see XRD, SEM, TEM and N2 adsorption results discussed earlier). The CH4 catalytic oxidation experiments were carried out up to a temperature of 500 °C for the sake of completion of the light-off curves (Fig. 4a). However due to the thermal instability of the samples at temperatures higher than their calcination temperatures, we compared the data up to a catalytic oxidation temperature of 400 °C (Fig. S15, ESI†). From Fig. S15 (ESI†), 112-NiCo2O4 outperforms 111-NiCo2O4 under both dry and humid conditions over the whole temperature range investigated, achieving 80% and 65% methane conversion at 400 °C, respectively. These findings are in line with our DFT simulation results.
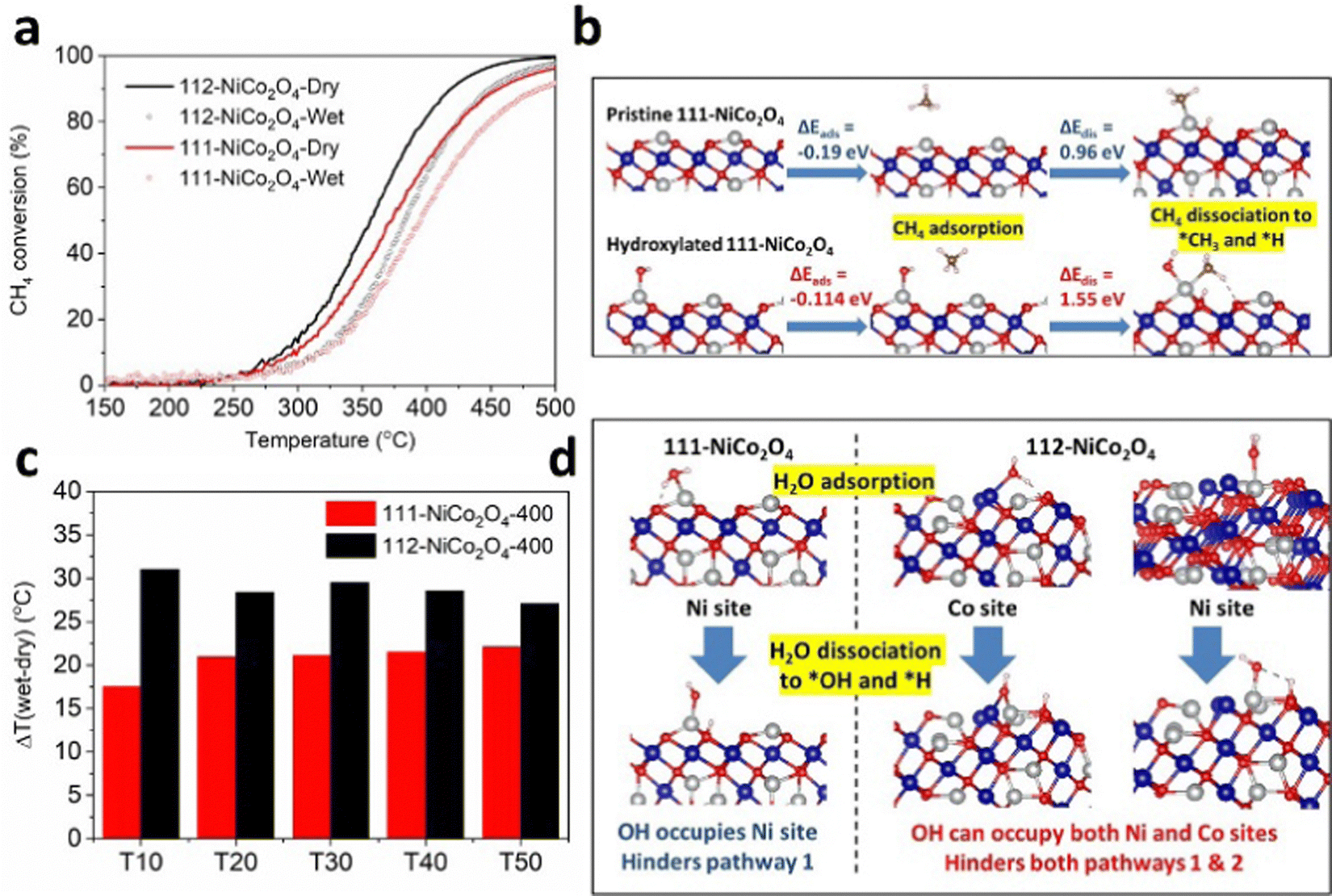 | ||
| Fig. 4 (a) Light-off curves for CH4 oxidation (3.2% water vapor concentration under humid conditions), (b) DFT simulations comparing the adsorption and dissociation of methane on pristine and hydroxylated 111-NiCo2O4, (c) the temperature difference between wet and dry conditions at several Tx points for 111-NiCo2O4-400 and 112-NiCo2O4-400 and (d) water adsorption and dissociation on 111-NiCo2O4 and 112-NiCo2O4 active sites. | ||
Effect of humidity
The presence of water vapour in the feed reduces the catalytic activity of both samples. To explain this, DFT simulations were carried out to calculate the adsorption energies and activation barriers of water on both surfaces (Table S2, ESI†). Interestingly, computer modelling indicated that water could be adsorbed on both Co and Ni atoms on the 112-surface, with the Co sites strongly preferred with an adsorption energy of −1.59 eV. Interaction of water molecules with the Ni site is also feasible due to an exothermic adsorption energy of −0.52 eV. Conversely, water could only adsorb on the Ni sites on the 111-surface with an adsorption energy of −0.94 eV. These adsorption energies are much more exothermic than those for CH4, meaning that water molecules will preferentially adsorb on the active sites instead of CH4 when the feed is humid. Furthermore, the energy required to dissociate a water molecule into adsorbed *OH (on the active metal sites) and *H (on the neighbouring lattice O sites) is 0.84 eV on the 112-surface and 0.66 eV on the 111-surface, resulting in heavily hydroxylated NiCo2O4 surfaces. These energy requirements are less positive than the corresponding energies required for CH4 (Table S3, ESI†) and suggest that water adsorption and dissociation on the spinel NiCo2O4 active sites are feasible and indeed thermodynamically preferred to CH4 adsorption and activation. We performed further DFT calculations to determine how CH4 is adsorbed and activated on hydroxylated 112-NiCo2O4 and 111-NiCo2O4 surfaces. From molecular modelling, a stable conformation of the hydroxyl molecule could be maintained on the 112-NiCo2O4 surface when the O from OH is bonded to one Ni and one Co atoms. Methane can then adsorb onto the hydroxylated Ni atoms with an adsorption energy of −0.14 eV and then dissociate into *CH3 and *H species with a high energy requirement of 1.38 eV. On the 111-NiCo2O4 surface, the O atom from the hydroxyl is bonded to one Ni atom. The corresponding adsorption and dissociation energies of CH4 on the hydroxylated Ni atoms is calculated as −0.11 eV and 1.55 eV, respectively. Fig. 4b and Fig. S16 (ESI†) compares the adsorption and dissociation of CH4 on both pristine and hydroxylated surfaces of 111-NiCo2O4 and 112-NiCo2O4, respectively. The results show that CH4 adsorption and dissociation is much harder on hydroxylated NiCo2O4 surfaces compared to the pristine surfaces. Hence, the presence of water vapour in the feed has a dual negative effect upon the activity of the catalyst. Firstly, H2O adsorption and activation are thermodynamically preferred to CH4 onto the active sites. This creates a hydroxylated surface which reduces the number of active sites available for CH4 adsorption and activation. Secondly, CH4 adsorption and activation is energetically harder on the hydroxylated surfaces relative to the pristine surfaces. These findings explain our experimental results as to how the presence of humidity in the feed negatively impacts the active sites on the spinel surface and reduces the catalytic activity towards methane oxidation.Although the 112-NiCo2O4 sample performs better than 111-NiCo2O4 under humid conditions, it appears that relative to its activity under dry conditions the 111-NiCo2O4 sample is more resistant to water vapour than 112-NiCo2O4. This is evident in Fig. 4c, where we compare the temperature difference between wet and dry conditions at different Tx (x = 10, 20, 30, 40, 50 and 60, where for example T10 is the temperature at which 10% methane conversion occurs). This temperature difference is consistently larger for the 112-NiCo2O4 sample over the temperature range of interest. Fig. 4d shows the molecular arrangements for H2O adsorption and dissociation on both 111-NiCo2O4 and 112-NiCo2O4, as determined from DFT simulations. The favoured adsorption and dissociation of water on both Ni and Co sites on 112-NiCo2O4 could hinder both pathways 1 and 2 for the methane oxidation reaction (since both Ni and Co sites are involved in the reaction mechanism, as mentioned in the DFT modelling section). However, since water adsorbs on the Ni sites on 111-NiCo2O4 (as only Ni sites are exposed to the reactants, see DFT section), this may hinder pathway 1 of the reaction but it may be possible that the methane oxidation reaction shifts to the alternative pathway 2 on this surface during reaction under humid conditions. On the 111-NiCo2O4 surface, some H atoms from dissociated H2O molecules will occupy the oxygen sites bonded to the Ni active sites, which may thwart the oxidation reaction. Therefore, the lattice O associated with a Co atom could alleviate this burden by participating in the oxidation reaction via pathway 2 (Fig. S12, ESI†). These reasons may explain why 111-NiCo2O4 is more resistant to water vapour than 112-NiCo2O4.
Interestingly, the results in this study reveal that different exposed crystal facets provide different sought-after properties for the catalytic oxidation of methane, i.e., high activity and thermal stability on the {112} exposed facet of 112-NiCo2O4 and good water resistance/relative hydrophobicity on the {111} exposed facet of 111-NiCo2O4.
Stability tests
Preliminary stability tests were performed at 500 °C under humid conditions (3.2% water) over ca. 35 hours on 111-NiCo2O4 and 112-NiCo2O4 samples calcined at 450 °C (Fig. 5a). As expected, 112-NiCo2O4-450 shows better activity and stability with initial methane conversion of 97.6% and a final conversion of 90.4%, hence a 7.2% activity loss after 35 h reaction. Comparatively, 111-NiCo2O4-450 starts at 89.9% methane conversion but loses 11.2% activity after 35 h reaction, reaching only 78.8% conversion. To estimate the relative degree of NiO segregation from the spent samples, we calculated the ratio R of the peak heights at 50.756° with Miller index (012) for NiO to the peak height at 52.332° with Miller index (004) for NiCo2O4 from the XRD patterns of the fresh and spent samples in Fig. S17 and S18 (ESI†). The value of R is 2.2 for spent 111-NiCo2O4-450 compared with 1.6 for spent 112-NiCo2O4-450. This means that more NiO was segregated from the bulk spinel structure in the spent 111-NiCo2O4-450. This is expected as we earlier showed that 112-NiCo2O4 was more thermally stable than 111-NiCo2O4. XPS provides the relevant surface data of the tested samples. Table S1 (ESI†) presents a comparison of the NiO/Ni(OH)2 ratios based on XPS analysis of the fresh and spent samples. The appropriately peak-fitted Ni 2p spectra for fresh and spent 111-NiCo2O4-450 and 112-NiCo2O4-450 are shown in Fig. 5b. Please note that we observed hardly any difference between the Co 2p spectra for fresh and spent samples, indicating that Co did not undergo any significant changes during testing (see Fig. S19, ESI†). However, a large increase in the NiO/Ni(OH)2 ratios for both after-test samples was determined when compared to the fresh samples (Table S1, ESI†), consistent with the detachment of the NiO6 cells from the spinel structure, followed by their migration towards the catalyst surface and assembling into NiO nanocrystals. Nevertheless, from Table S1 (ESI†), while the Ni/Co ratios for both spent samples are similar, it is interesting to see that the NiO/Ni(OH)2 values for both spent samples are also comparable at ca. 14. This may be attributed to the high reaction temperature of 500 °C as well as long reaction time under humid conditions which are severe conditions for the integrity of the catalyst structure to be maintained. Hence, XPS results indicate similar degrees of atomic rearrangements occurring on the surfaces of spent 111-NiCo2O4-450 and 112-NiCo2O4-450 although XRD results showed more NiO being segregated within the bulk of 111-NiCo2O4-450, which can be attributed to the lower thermal stability of NiCo2O4 with {111} exposed facets. Nevertheless, the comparatively similar surface NiO/Ni(OH)2 as well as Ni/Co ratios on the spent samples further confirm our findings from Fig. 4a, i.e., the {112} exposed facet is more active than the {111} exposed facet in NiCo2O4 for methane oxidation reaction, (since despite the harsh conditions used, it can be assumed that some of the exposed crystal facets initially present in the pristine samples are retained during stability testing). The SEM image in Fig. 5c show the thin hexagonal morphology of fresh 112-NiCo2O4-450. The structure is still somehow preserved in the spent sample (Fig. 5d) although a certain degree of sintering as well as edge-etching are observed on the particles. On the other hand, spent 111-NiCo2O4-450 shows a relatively higher level of particles sintering (Fig. S20, ESI†). Hence an overall better thermal structural stability is evident with 112-NiCo2O4-450 when compared with 111-NiCo2O4-450.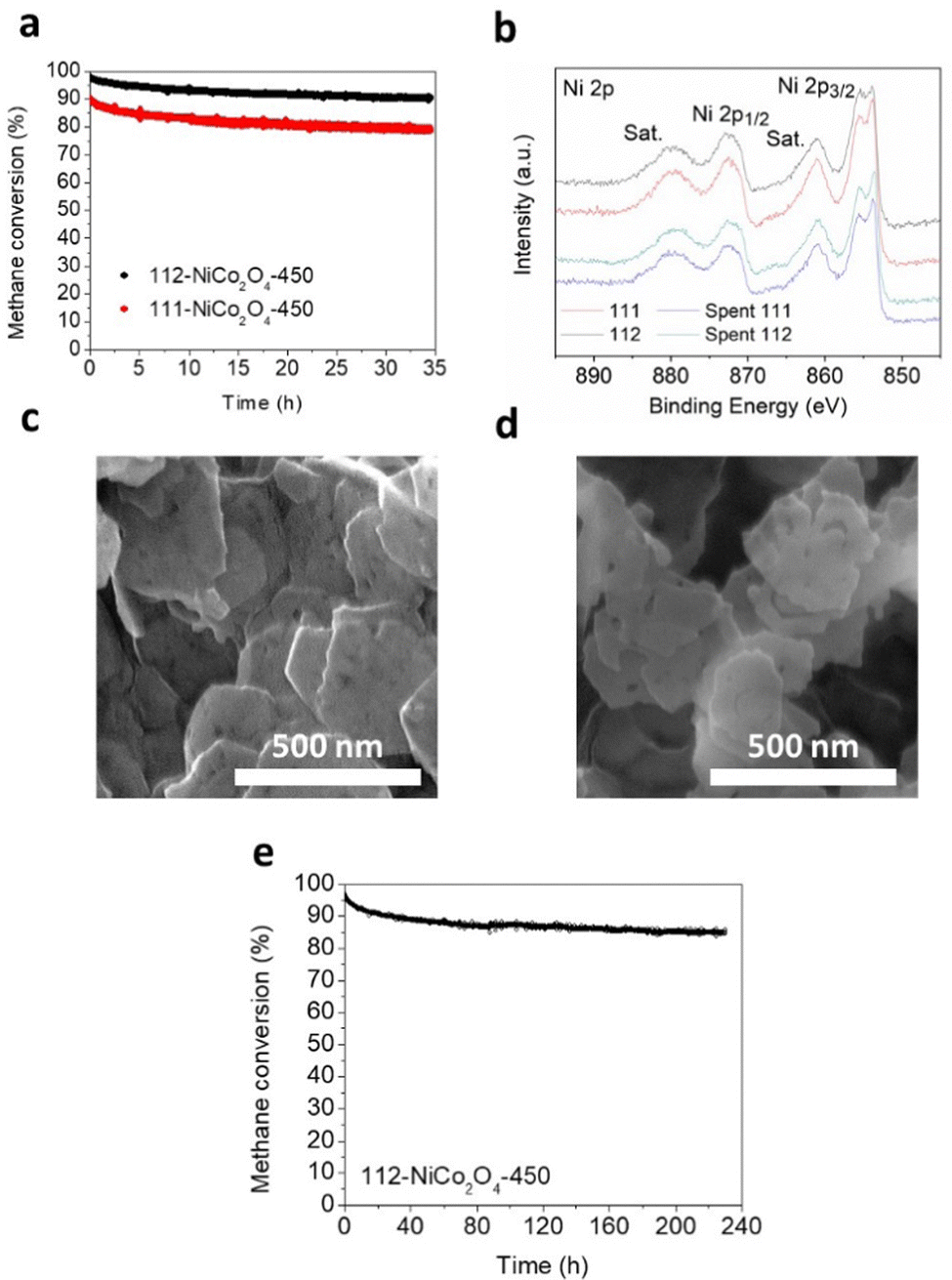 | ||
| Fig. 5 (a) Preliminary stability rest comparing 111-NiCo2O4-450 and 112-NiCo2O4-450 at 3.2% water vapor concentration and a reaction temperature of 500 °C, (b) high-resolution XPS Ni 2p spectra for fresh and spent 111-NiCo2O4-450 and 112-NiCo2O4-450, SEM image of (c) 112-NiCo2O4-450 and (d) spent 112-NiCo2O4-450 and (e) long-term stability over 230 h for the catalytic oxidation of CH4 using 112-NiCo2O4-450 at 3.2% water vapor concentration and a reaction temperature of 500 °C. | ||
Long term stability test was performed on 112-NiCo2O4-450 for ca. 230 h (Fig. 5d) at 500 °C under humid conditions (3.2% water). It can be seen that after the relatively large activity loss from 97.1% to 88.3% in the first 50 h of reaction time, good stability is maintained such that the catalyst still retains 85.7% activity after 230 h reaction. Table S4 (ESI†) compares the activity of the catalyst in this work with other similar types of catalysts from previous publications. The impressive maintenance in activity can be credited to the high structural stability and activity of the NiCo2O4 spinel catalyst with {112} exposed facets.
Conclusions
The use of spinel NiCo2O4 materials for catalytic oxidation reaction at high temperatures has been impaired mostly due to its low thermal stability. The stability and activity of NiCo2O4 for methane oxidation is largely dependent on the atomic arrangement at the catalysts’ surface. We demonstrate that NiCo2O4 with {112} exposed facets is more stable and active than its counterpart with {111} exposed facets. DFT simulations indicate that the reaction mechanism for methane oxidation is less challenging on 112-NiCo2O4 as it offers less energetically intensive alternate pathways for the oxidation of reaction intermediates as opposed to 111-NiCo2O4. In addition, 111-NiCo2O4 had the better water resistance of the two samples. This is because different exposed facets could also affect the way that water molecules present in the feed interact with the surface-active sites and therefore affect the activity of the material under humid conditions. The experimental and theoretical findings in this work would pave the way for crystal facet engineering (in conjunction with other strategies such as doping, compositing and hierarchical structure design) as a powerful, elegant and facile method towards the development of abundant metal oxide catalyst materials with tuneable activity, stability and hydrophobicity for the complete oxidation of dilute methane. These low-cost and high-performance catalysts are highly demanded for mitigating fugitive methane emissions in resources and energy sectors such as coal mining, natural gas industry, natural gas engines, etc.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was funded by CSIRO and the Australian Coal Industry's Research Program (ACARP). Partially supported by the National Key R&D Program of China (no. 2022YFA1504500, 2021YFA1502804).References
- I. Karakurt, G. Aydin and K. Aydiner, Renewable Sustainable Energy Rev., 2011, 15, 1042–1049 CrossRef CAS.
- L. He, Y. Fan, J. Bellettre, J. Yue and L. Luo, Renewable Sustainable Energy Rev., 2020, 119, 109589 CrossRef CAS.
- T. Lim, S. Cho, H. Yang, M. Engelhard and D. Kim, Appl. Catal., A, 2015, 505, 62–69 CrossRef CAS.
- L. Hu, Q. Peng and Y. Li, J. Am. Chem. Soc., 2008, 130, 16136–16137 CrossRef CAS PubMed.
- F. Tao, J. Shan, L. Nguyen, Z. Wang, S. Zhang, L. Zhang, Z. Wu, W. Huang, S. Zeng and P. Hu, Nat. Commun., 2015, 6, 7798 CrossRef CAS PubMed.
- X. Xie, Y. Li, Z. Liu, M. Haruta and W. Shen, Nature, 2009, 458, 746–749 CrossRef CAS PubMed.
- S. Rong, P. Zhang, F. Liu and Y. Yang, ACS Catal., 2018, 8, 3435–3446 CrossRef CAS.
- J. Huang, R. Fang, Y. Sun, J. Li and F. Dong, Chemosphere, 2021, 263, 128103 CrossRef CAS PubMed.
- J. Niu, H. Liu, Y. Zhang, X. Wang, J. Han, Z. Yue and E. Duan, Chemosphere, 2020, 259, 127427 CrossRef CAS PubMed.
- Y. Jian, M. Tian, C. He, J. Xiong, Z. Jiang, H. Jin, L. Zheng, R. Albilali and J. Shi, Appl. Catal., B, 2021, 283, 119657 CrossRef CAS.
- H. Liu, X. Li, Q. Dai, H. Zhao, G. Chai, Y. Guo, Y. Guo, L. Wang and W. Zhan, Appl. Catal., B, 2021, 282, 119577 CrossRef CAS.
- D. Jiang, K. Khivantsev and Y. Wang, ACS Catal., 2020, 10, 14304–14314 CrossRef CAS.
- L. Hu, Q. Peng and Y. Li, J. Am. Chem. Soc., 2008, 130, 16136–16137 CrossRef CAS PubMed.
- S. Zhao, T. Li, J. Lin, P. Wu, Y. Li, A. Li, T. Chen, Y. Zhao, G. Chen, L. Yang, Y. Meng, X. Jin, Y. Qiu and D. Ye, Chem. Eng. J., 2021, 420, 130448 CrossRef CAS.
- Y. Zheng, Y. Liu, H. Zhou, W. Huang and Z. Pu, J. Alloys Compd., 2018, 734, 112–120 CrossRef CAS.
- Z. Fei, S. He, L. Li, W. Ji and C. Au, Chem. Commun., 2012, 48, 853–855 RSC.
- Q. Kong, Y. Yin, B. Xue, Y. Jin, W. Feng, Z.-G. Chen, S. Su and C. Sun, Beilstein J. Nanotechnol., 2018, 9, 2526–2532 CrossRef CAS PubMed.
- Y. Dai, V. P. Kumar, C. Zhu, H. Wang, K. J. Smith, M. O. Wolf and M. J. MacLachlan, Adv. Funct. Mater., 2019, 29, 1807519 CrossRef.
- J. Dou, Y. Tang, L. Nie, C. Andolina, X. Zhang, S. House, Y. Li, J. Yang and F. Tao, Catal. Today, 2018, 311, 48–55 CrossRef CAS.
- Y. Zhang, Z. Qin, G. Wang, H. Zhu, M. Dong, S. Li, Z. Wu, Z. Li, Z. Wu, J. Zhang, T. Hu, W. Fan and J. Wang, Appl. Catal., B, 2013, 129, 172–181 CrossRef CAS.
- X. Huang, J. Li, J. Wang, Z. Li and J. Xu, Front. Chem. Sci. Eng., 2020, 14, 534–545 CrossRef CAS.
- M. Cabo, E. Pellicer, E. Rossinyol, O. Castel, S. Surinach and M. Baro, Cryst. Growth Des., 2009, 9, 4814–4821 CrossRef CAS.
- S. Verma, H. Joshi, T. Jagadale, A. Chawla, R. Chandra and S. Ogale, J. Phys. Chem. C, 2008, 112, 15106–15112 CrossRef CAS.
- A. Datye and M. Votsmeier, Nat. Mater., 2021, 20, 1049–1059 CrossRef CAS PubMed.
- D. Qiao, G. Lu, Y. Guo, Y. Wang and Y. Guo, J. Rare Earths, 2010, 28, 742–746 CrossRef CAS.
- C. Xiao, B. Lu, P. Xue, N. Tian, Z. Zhou, X. Lin, W. Lin and S. Sun, Joule, 2020, 4, 2562–2598 CrossRef CAS.
- X. Han, G. He, Y. He, J. Zhang, X. Zheng, L. Li, C. Zhong, W. Hu, Y. Deng and T. Ma, Adv. Energy Mater., 2018, 8, 1702222 CrossRef.
- C. Chang, K. Chen, Y. Hsieh, C. Chang and H. Tuan, ACS Nano, 2022, 16, 1486–1501 CrossRef CAS PubMed.
- X. Gao and T. Zhang, Sens. Actuators, B, 2018, 277, 604–633 CrossRef CAS.
- S. Wang, G. Liu and L. Wang, Chem. Rev., 2019, 119, 5192–5247 CrossRef CAS PubMed.
- L. Fang, Z. Jiang, H. Xu, L. Liu, Y. Guan, X. Gu and Y. Wang, J. Catal., 2018, 357, 238–246 CrossRef.
- G. Liu, J. Yu, G. Lu and H. Cheng, Chem. Commun., 2011, 47, 6763–6783 RSC.
- L. Ma, R. Ye, Y. Huang, T. Reina, X. Wang, C. Li, X. Li Zhang, M. Fan, R. Zhang and J. Liu, Chem. Eng. J., 2022, 446, 137031 CrossRef CAS.
- C. Yang, C. Pei, R. Luo, S. Liu, Y. Wang, Z. Wang, Z. Zhao and J. Gong, J. Am. Chem. Soc., 2020, 142, 19523–19531 CrossRef CAS PubMed.
- Q. Kuang, X. Wang, Z. Jiang, Z. Xie and L. Zheng, Acc. Chem. Res., 2014, 47, 308–318 CrossRef CAS PubMed.
- S. Devaguptapu, S. Hwang, S. Karakalos, S. Zhao, S. Gupta, D. Su, H. Xu and G. Wu, ACS Appl. Mater. Interfaces, 2017, 9, 44567–44578 CrossRef CAS PubMed.
- J. Bae, D. Shin, H. Jeong, C. Choe, Y. Choi, J. Han and H. Lee, ACS Catal., 2021, 11, 11066–11074 CrossRef CAS.
- Z. Xu, Y. Zhang, L. Qin, Q. Meng, Z. Xue, L. Qiu, G. Zhang, X. Guo and Q. Li, Small, 2020, 16, 2002071 CrossRef CAS PubMed.
- M. Iliev, P. Silwal, B. Loukya, R. Datta, D. Kim, N. Todorov, N. Pachauri and A. Gupta, J. Appl. Phys., 2013, 114, 033514 CrossRef.
- J. Marco, J. Gancedo, M. Gracia, J. Gautier, E. Rios, H. Palmer, C. Greaves and F. Berry, J. Mater. Chem., 2001, 11, 3087–3093 RSC.
- S. Sun, X. Zhang, J. Cui and S. Liang, Nanoscale, 2020, 12, 16657–16677 RSC.
- M. Biesinger, B. Payne, A. Grosvenor, L. Lau, A. Gerson and R. Smart, Appl. Surf. Sci., 2011, 257, 2717–2730 CrossRef CAS.
- M. Biesinger, B. Payne, L. Lau, A. Gerson and R. Smart, Surf. Interface Anal., 2009, 41, 324–332 CrossRef CAS.
- A. Grosvenor, M. Biesinger, R. Smart and N. McIntyre, Surf. Sci., 2006, 600, 1771–1779 CrossRef CAS.
- M. Biesinger, L. Lau, A. Gerson and R. Smart, Phys. Chem. Chem. Phys., 2012, 14, 2434–2442 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ey00281k |
| This journal is © The Royal Society of Chemistry 2024 |