
Recent advances in organolead halide crystalline materials for photocatalytic H2 evolution and CO2 reduction applications
Xueling
Song
 a,
Xiaoman
Li
a,
Yuxuan
Song
a,
Jingyi
Bi
a,
Lei
Wang
a,
Jigao
Wang
c,
Junjie
Liu
b,
Yanyan
Li
b and
Hui
Wang
*b
a,
Xiaoman
Li
a,
Yuxuan
Song
a,
Jingyi
Bi
a,
Lei
Wang
a,
Jigao
Wang
c,
Junjie
Liu
b,
Yanyan
Li
b and
Hui
Wang
*b
aSchool of Materials and Chemistry, University of Shanghai for Science and Technology, Shanghai 200093, P. R. China
bSchool of Polymer Science and Engineering, College of Chemistry and Molecular Engineering, College of Economics and Management, Qingdao University of Science and Technology, Qingdao 266042, P. R. China
cChemical Engineering & Applied Chemistry, University of Toronto, Toronto, Canada
First published on 30th April 2024
Abstract
The photocatalytic technique has been widely recognized as a feasible technological route for sustainable energy conversion of solar energy into chemical energy. Photocatalysts play a vital role in the whole catalytic process. In particular, organolead halide perovskites have become emerging photocatalysts, owing to their precisely tunable light absorption range, high carrier diffusion mobility, and longer carrier lifetime and diffusion length. Nevertheless, their intrinsic structural instability and high carrier recombination rate are the major bottlenecks for further development in photocatalytic applications. This Frontier is focused on the recent research about the instability mechanism of organolead halide perovskites. Then, we summarize the recently developed strategies to improve the structural stability and photocatalytic activity of organolead halide materials, with an emphasis on the construction of organolead halide crystalline catalysts with high intrinsic structural stability. Finally, an outlook and challenges of organometal halide photocatalysts are presented, demonstrating the irreplaceable role of this class of emergent materials in the field of photo-energy conversion.
1. Introduction
With the rapid development of society and the economy, energy scarcity and the greenhouse effect have become two major global challenges.1,2 To satisfy the sustainable development of humanity, it is urgent to exploit efficient and environmentally friendly strategies for green energy synthesis (such as clean hydrogen energy and high value-added chemicals) and environmental purification. Although much attention has been paid to thermocatalysis and electrocatalysis for overcoming these two problems, solar energy-driven catalysis has been regarded as a more promising technique in recent years. As is well known, the design and development of photocatalysts is one of the prerequisites in photocatalytic applications. To date, various efficient photocatalysts have been reported, such as metal oxides,3,4 metal (oxygen) sulfides,5,6 metal (oxygen) nitrides,7,8 metal halide perovskites (all inorganic and hybrid organic–inorganic),9–11 metal–organic frameworks,12,13 covalent organic frameworks,14,15 and organometal halide coordination polymers.16 Taking the utilization of sunlight and the designability of the structure into consideration, hybrid organic–inorganic semiconductors have attracted much attention in contrast to all inorganic photocatalysts.Generally, the photocatalytic process of a semiconductor-photocatalyst system involves the following three sequential steps (Fig. 1): (i) light absorption to generate carriers, (ii) the separation and migration of photo-generated carriers, and (iii) free-carrier induced surface redox catalytic reactions. The above three steps directly depend on the intrinsic properties of semiconductor photocatalysts, together determining the photocatalytic efficiency.17,18 In the photocatalytic process, the photocatalyst is undoubtedly the key component to transform solar energy to chemical energy. The appropriate conduction band minimum (CBM) position to match the reduction potential of the target reaction is the prerequisite to drive photocatalytic reactions (such as H2 evolution and CO2 reduction). Apparently, an ideal photocatalyst should exhibit the following desirable characteristics: broad light absorption, efficient carrier separation and migration efficiency, good catalytic stability, and tunable energy band position. However, most of the existing photocatalysts are far from the desirable materials.
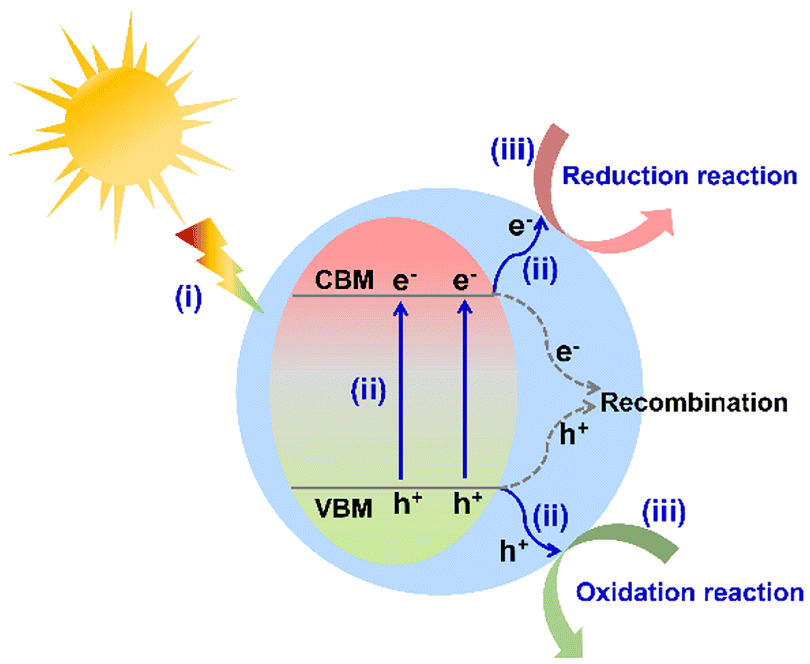 | ||
| Fig. 1 Basic photocatalytic mechanism of a semiconductor system (VBM represents the valence band maximum). | ||
Metal halide perovskites are a class of excellent crystalline optoelectronic materials with the formula ABX3 (A and B are cations, X is halide anions). Considering their excellent light-harvesting ability and suitable band structure, metal halide perovskites have been developed as attractive photocatalysts. Very recently, organolead halide perovskites with the general formula of APbX3 (A = organoammonium cations, such as methylammonium (MA+), formamidinium (FA+)) have been widely exploited for photocatalytic H2 evolution and CO2 reduction owing to their intriguing photochemical advantages, structural diversity, as well as low cost and easy availability.19,20 However, the moisture-sensitive nature of organolead halide perovskites largely limited their rapid development in the photocatalytic field.21,22 Since 2016, many breakthrough strategies have been gradually explored and employed to realize photocatalytic applications in the presence of water.
To date, most existing reviews have mainly focused on the enhancement strategies for photocatalytic applications (such as H2 generation, CO2 reduction, pollutant degradation, organic transformation, etc.) of metal halide perovskites (including all-inorganic and organic–inorganic hybrid materials).23–26 A comprehensive, systematic review about the developed approaches to maintain the structural stability while enhancing the photocatalytic efficiency of the most concerned organolead halide perovskites is scarce. In this Frontier, we briefly summarize the structural diversity and excellent optoelectronic properties of organolead halide perovskites in the first section. Subsequently, the moisture-sensitive nature of organolead halide perovskites was overviewed from the point of experimental results and theoretical calculation. The enhancement strategies for structural stability and photocatalytic performance were emphasized. Finally, we present our perspective on the future directional aspects of organometal halide crystalline materials and further improvement strategies in photocatalytic applications.
2. Diverse structure and photochemical properties of organolead halide perovskites
2.1 Diverse structure
Organolead halide perovskites are a class of ionic crystals. Normally, one Pb2+ cation coordinates six X− anions to form one [PbX6]4− octahedron, with Pb2+ at the center site and X− at the six vertex sites (Fig. 2a). Subsequently, the formed [PbX6]4− octahedra extended to chains, layers and networks by sharing vertices. The inorganic components of lead halide are surrounded by A-site cations via ionic interactions to construct diverse organolead halide structures. According to the dimensionality of the extended corner-sharing PbX6 octahedron at the molecular level, researchers have reported diverse crystal structures of organolead halide perovskites, including 0D, 1D, 2D and 3D topology networks (Fig. 2b).27 Generally, the low-dimensional structures were accessible due to the introduction of large-sized A-site cations.28 Obviously, the structure of organolead halide perovskites can be modulated flexibly just by altering the coordination modes between lead halide and organoammonium ligands in contrast to all-inorganic photocatalysts, leading to excellent photochemical properties.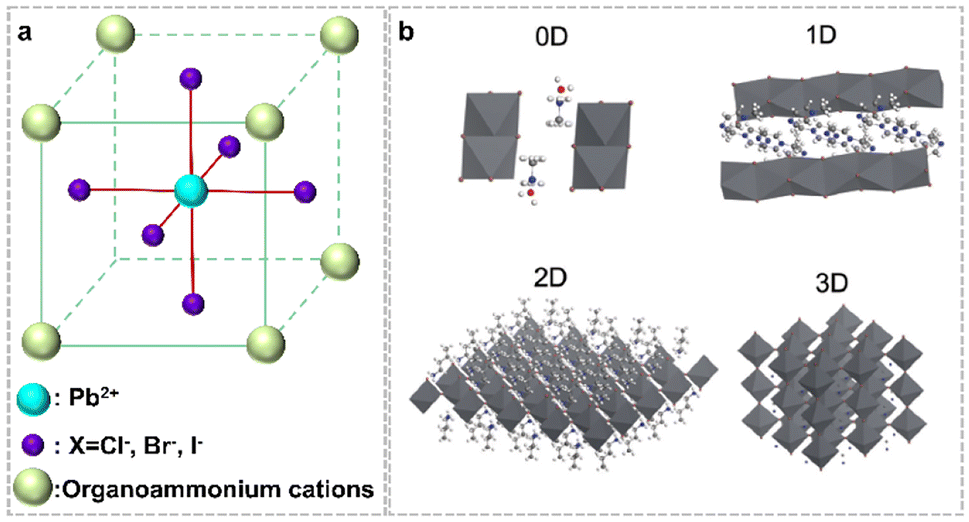 | ||
| Fig. 2 (a) Basic crystal structure of a traditional organolead halide perovskite. (b) Different dimensions of the reported organolead halide perovskite. Reproduced from ref. 27 with permission from the Royal Society of Chemistry, Copyright 2018. | ||
2.2 Photochemical properties
As a class of ionic crystal semiconductors, the band structure of organolead halide perovskites mainly includes CB, VB and bandgap (Eg). As illustrated in Fig. 3a, the energy band edge of common lead halide perovskites can be adjusted flexibly to modulate the photochemical properties.24 The VB of organolead halide perovskites is mainly composed of the antibonding hybrid state of the 6s orbitals of Pb2+ and np orbitals of X− (n = 3, 4, and 5 for Cl, Br, and I, respectively), with major contribution from X np. Taking CH3NH3PbX3 (MAPbX3) as an example, the VB position is 1.4, 1.2, and 1.0 eV vs. the reversible hydrogen electrode (RHE) for MAPbCl3, MAPbBr3, and MAPbI3, respectively. Thus, the position of VB shifts to a more negative potential while varying the halogen from Cl (3p) to Br (4p) to I (5p). However, the CB composition is somewhat disputed. One view demonstrated the CB only consisting of the 6p orbitals of Pb2+. It is hard to explain the reason of the variation of the CB position for APbX3 with the same A-site cation. Specifically, the CB position is −1.5, −1.0, and −0.5 eV vs. RHE for MAPbCl3, MAPbBr3, and MAPbI3, respectively. Obviously, the contribution from X− to CB cannot be neglected. A more mainstream view demonstrated the CB consisting of the antibonding states with dominant Pb2+ 6p orbitals and minor X−np orbitals (Fig. 3b) on the basis of theoretical and experimental results.29 Generally, the hybridization extent was enhanced from I to Br to Cl owing to the decreased bond lengths of Pb–X, leading to the shifting of the CB position to higher energies. Additionally, the influence of organic cations on the band structure is slight (Fig. 3c).30 On the basis of the above discussion, the X-site anions play a vital role in adjusting the energy band position, the band gap, and the light absorption range of organolead halide photocatalysts.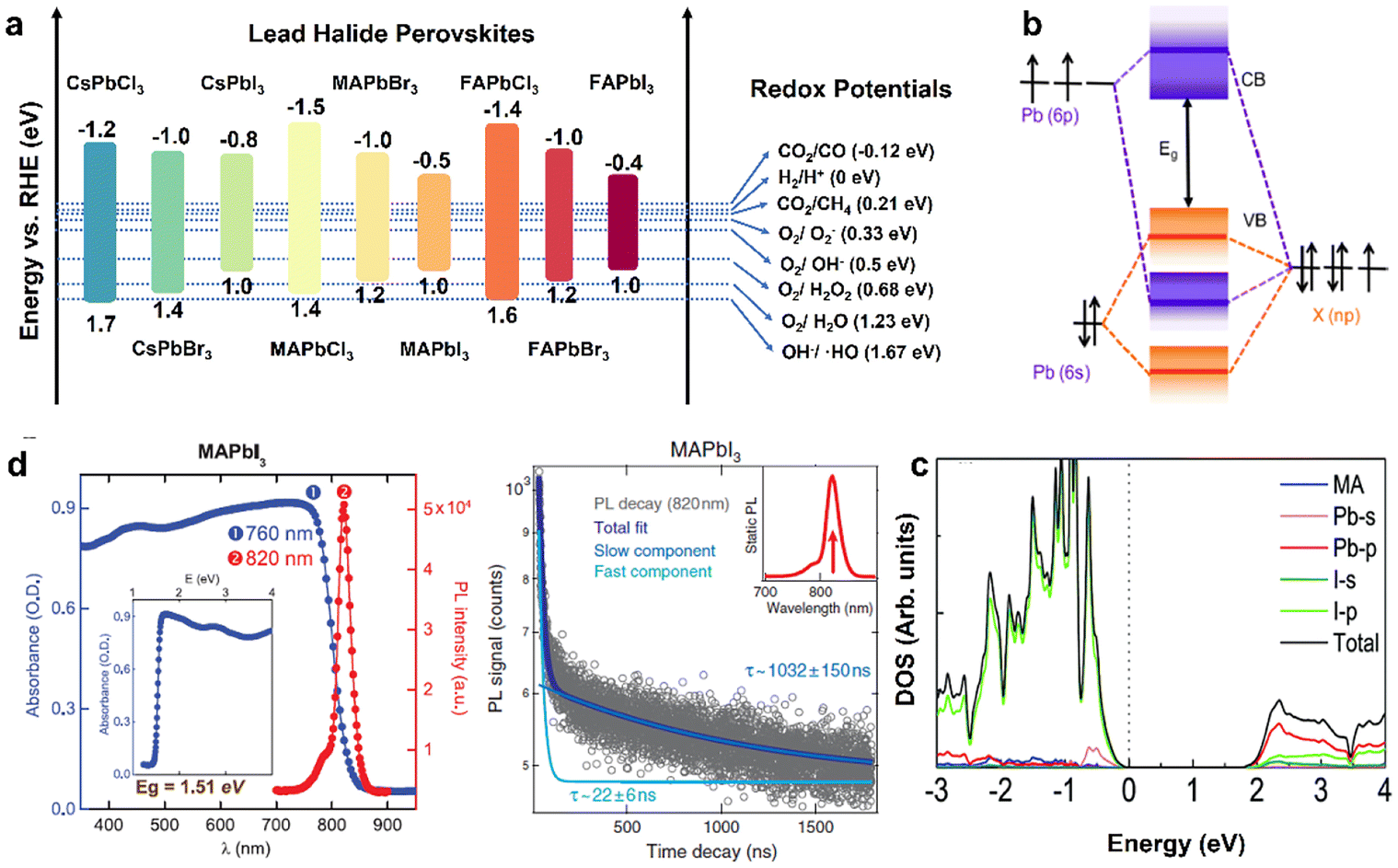 | ||
| Fig. 3 (a) Band energy positions of the common lead halide perovskites (vs. RHE) and the redox potential of photocatalytic reactions. Reproduced from ref. 24 with permission from the American Chemical Society, Copyright 2020. (b) Schematic illustration of bonding/antibonding orbitals in the CB and VB of APbX3. Reproduced from ref. 29 with permission from American Chemical Society, Copyright 2016. (c) Density of states (DOS) of the pure MAPbI3 without the spin–orbit coupling effect. Reproduced from ref. 30 with permission from the Royal Society of Chemistry, Copyright 2017. (d) Steady-state absorbance and photoluminescence spectra (left) and time-dependent photoluminescence spectra (right) of a MAPbI3 crystal. Reproduced from ref. 31 with permission from Science, Copyright 2015. | ||
According to the requirements for desirable photocatalysts, organolead halide perovskites have the potential to develop as a class of highly efficient photocatalysts owing to their attractive optoelectronic properties, including a precisely tunable direct bandgap, low excitonic binding energy, high light-absorption coefficient as well as carrier diffusion mobility. Taking MAPbX3 as an example, (a) the band gap was varied from 2.9 eV (MAPbCl3) to 2.2 eV (MAPbBr3) to 1.5 eV (MAPbI3); meanwhile, the value of the bandgap was also adjusted by changing the organoammonium cations (Fig. 3a);24 (b) the tunable bandgap is in favour of a broad light absorption range; MAPbI3 possesses a wide light absorption edge of above 800 nm, promoting the utilization of sunlight (Fig. 3d, left);31 (c) MAPbI3 also exhibited a microsecond carrier lifetime (τ ≈ 22 and1032 ns) and a micrometer carrier diffusion length (2–8 μm) (Fig. 3d, right),31 efficiently enhancing the separation efficiency of carriers and further improving the photocatalytic performance. The reaction potentials of the common photocatalytic reactions are also displayed in Fig. 3a. In general, the more negative CB potential than the target reduction reaction or the more positive VB potential than the target oxidation reaction is the basic energy band requirement for photocatalysts. On the basis of the reaction thermodynamics, the electronic band structure of organolead halide perovskites should be suitably matched with the target redox potentials to trigger the reactions. Impressively, the existing organolead halide perovskites can almost drive photocatalytic H2 evolution and CO2 reduction. Therefore, organolead halide perovskites are promising photocatalysts to synthesize high-valued chemical fuels and promote the conversion efficiency of solar energy.
3. Moisture-sensitive nature of organolead halide perovskites
Organolead halide perovskites have been considered as promising photoelectric semiconductor materials. However, the moisture-sensitive nature has developed as the major bottleneck due to: (1) the labile ionic crystalline structures, (2) the hydrophilic organoammonium cations in the lattice, and (3) the susceptible dissociation and migration of the corner halides in this class of perovskites, which severely limit their practical utilization and further development in photocatalytic applications.32,33 In most cases, this structural damage results in a negative effect on the photoelectric performance of perovskites and perovskite-based devices. As displayed in Fig. 4a, the resistance of the MAPbI3 film decreased in different relative humidity (RH) environments.34 Obviously, the resistance of the perovskite film decreased rapidly in the presence of ≥75% RH conditions. The composition of the MAPbI3 film during the degradation process was also verified by powder X-ray diffraction data. As shown in Fig. 4b, the as-synthesized (D0) MAPbI3 perovskite phase gradually vanished along with the appearance of a new PbI2 phase by prolonging the exposure time in air.35 And the noticeable colour change from the deep-brown colour of perovskites to the bright-yellow colour of PbI2 after storage in the lab for twelve days, also provide the first-hand experimental phenomenon for the moisture-sensitive feature of organolead halide perovskites. Therefore, it is valuable to figure out the detailed decomposition procedure of MAPbI3 to guide the research direction for enhancing the structural stability and photocatalytic performance.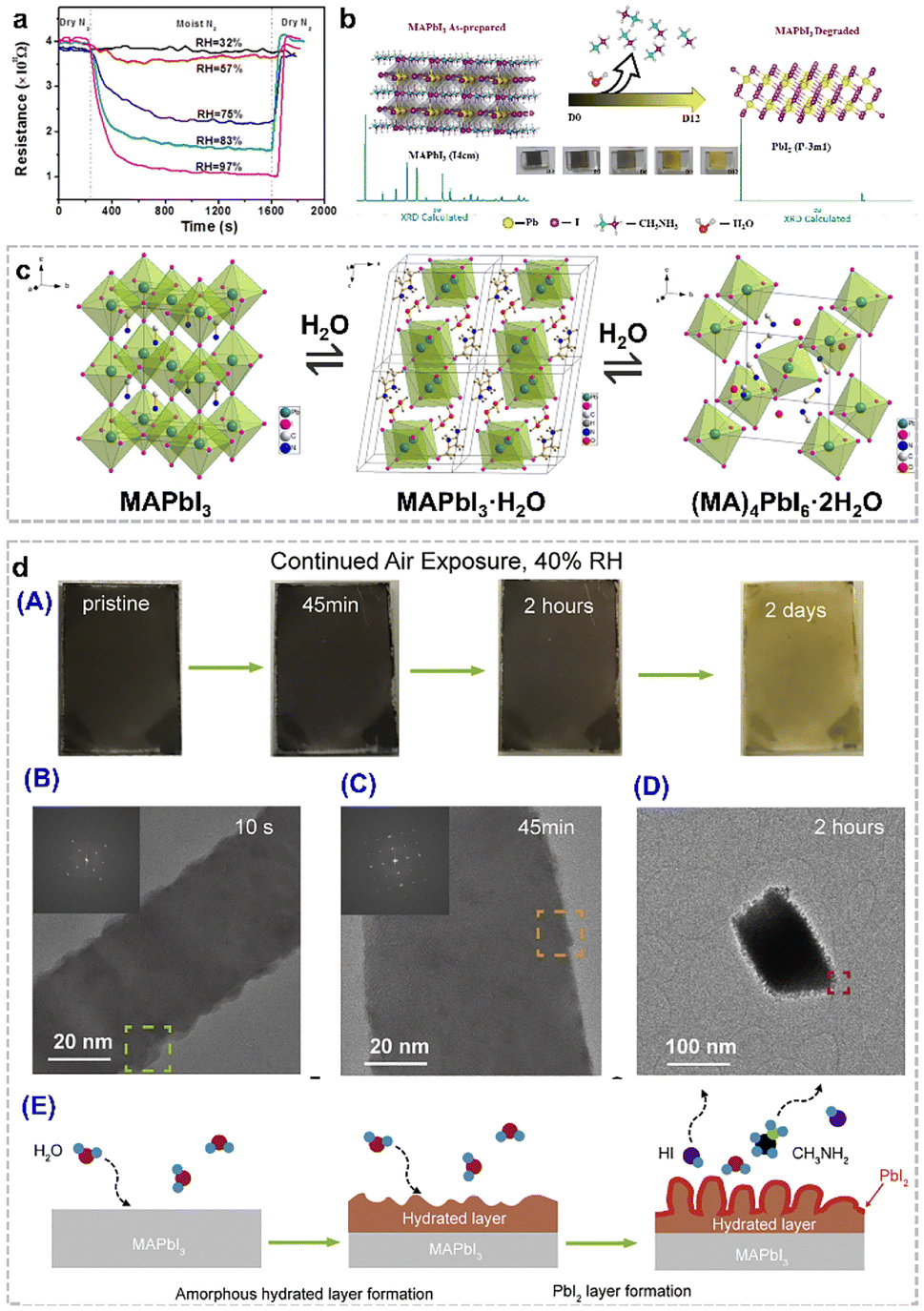 | ||
| Fig. 4 (a) Dynamic resistance curves of the perovskite film in various RH environments at room temperature. Reproduced from ref. 34 with permission from the American Chemical Society, Copyright 2015. (b) Structure transition from MAPbI3 to PbI2 under an ambient atmosphere for twelve days at three days interval. Reproduced from ref. 35 with permission from Elsevier, Copyright 2023. (c) Structure transition from MAPbI3 to the monohydrate phase (MAPbI3·H2O) and the dihydrate phase (MA4PbI6·2H2O). Reproduced from ref. 36 with permission from the American Chemical Society, Copyright 2015. (d) Colour change of MAPbI3 nanowires after moisture exposure under 40% RH air conditions for two days (A); cryo-electron microscopy images of MAPbI3 nanowires after 10 s (B), 45 min (C), and 2 h (D) moisture exposure; and proposed nano-scale decomposition mechanism (E). Reproduced from ref. 37 with permission from Elsevier, Copyright 2019. | ||
To date, much effort has been devoted to elucidate the degradation mechanism of organolead halide perovskites. The speculated humidity-induced decomposition mechanism in the early stage was gradually verified by more rigorous experimental control and advanced characterization methods. Kelly et al.38 and Leguy et al.36 revealed the existence of a dihydrate phase (MA4PbI6·2H2O) and a monohydrate phase (MAPbI3·H2O) during the degradation process by in situ absorption spectroscopy and grazing-incidence wide-angle X-ray scattering, respectively. The pale-yellow MAPbI3·H2O consists of an isolated [PbI3]− double chain architecture and ionic coordinated MA+. The H2O molecules are inserted into [PbI3]− double chains via the hydrogen bond interaction between H2O and MA+ (Fig. 4c).36 The 0D MA4PbI6·2H2O intermediate is built by isolated [PbI6]4− octahedra and MA+⋯H2O⋯MA+ (Fig. 4c).36 The transformation from MAPbI3·H2O to MA4PbI6·2H2O with PbI2 as the by-product is worth noting.39 The discovery of two intermediate phases demonstrated the moisture-driven degradation steps of organolead halide perovskites. Generally, the hydration reaction is likely to occur when the perovskites were placed in the moist environment due to the rapid formation of hydrogen bonds between water molecules and organoammonium cations and [PbX6]4− octahedra.40–42 The surface coordinated H2O molecules gradually penetrate into the perovskite crystals to form monohydrate and dihydrate intermediate phases. The whole moisture-sensitive reaction can be simply considered as the decomposition of organolead halide perovskites to organoammonium halide and PbX2.
The above degradation mechanism was proposed mainly based on the chemical reaction between organolead halide perovskites and H2O molecules. In general, this mechanism was confirmed by various in situ and ex situ characterization methods as well as theoretical calculations. However, the atomic-scale information was severely lacking due to the highly beam-sensitive nature of organolead halide perovskites.43,44 In recent years, high-resolution (scanning) transmission electron microscopy ((S)TEM) with ultra-low electron doses and equipped with advanced electron detectors and image-acquisition technology has been successfully applied to reveal the nature of boundaries, defects and decomposition pathways.45–47 Cui et al. have developed cryo-electron microscopy protocols to study the nanoscale moisture-sensitive decomposition process of MAPbI3 nanowires via capturing atomic-level information. Similarly, the colour change from dark-brown to light-yellow was optically observed after moisture exposure for two days under 40% RH air conditions, indicating the complete degradation of MAPbI3.37 The initial degradation state can be preserved well by plunge-freezing in liquid nitrogen, and further detected by high-resolution cryo-electron microscopy. As shown in Fig. 4d, an apparent rough surface was observed only after 10 s moisture exposure, indicating the extreme sensitivity of MAPbI3 without any protecting layers. After 45 min moisture exposure, a thin amorphous layer assigned to hydrated intermediates formed on the surface of MAPbI3. Prolonging the moisture exposure time to 2 h, nanoscale whiskers containing PbI2 nanograins dispersed in the amorphous layer were detected on the rougher perovskite surface. The observed nanoscale information indicated the rapid moisture-induced MAPbI3 degradation process. A nanoscale degradation pathway was also demonstrated: (1) an amorphous hydrated compound layer was firstly formed on the surface of MAPbI3 and (2) this layer then facilitated the further degradation of MAPbI3 to PbI2 and other gases. Obviously, the cryo-electron microscopy protocol plays a vital role in investigating the initial degradation process from a nano-scale view.
4. Enhancement strategies for structural stabilization and photocatalytic activity of organolead halide crystalline materials
The common hydration of organolead halide perovskites results in a significant change of the light-absorption and crystalline structures, further influencing their optical and electronic properties. For photocatalytic applications, most of the catalytic environments involve aqueous solution or humid conditions. Therefore, the structural instability of organolead halide catalysts must be resolved. Additionally, in contrast to photocatalytic H2 evolution, the CO2 photoreduction reaction is a multi-electron transfer process, resulting in the difficulty of control of products. The common reduction products contain CO (2e− reduction pathway), CH4 (8e− reduction pathway), and other polycarbonate chemicals (multielectron pathway). Therefore, except for the photocatalytic CO2 reduction rate, the selectivity of the reduction product is also an important criterion to evaluate the photocatalytic performance of organolead halide crystalline materials. In this section, we will review the enhancement strategies for the structural stability against H2O of organolead halide materials (including establishing suitable catalytic reaction media, encapsulation protection layers as well as efficient direct crystal engineering methods). The improved photocatalytic H2 evolution and CO2 reduction pathways are also simultaneously summarized in this section.4.1 Establishing dynamic equilibrium reaction media of organolead halide perovskites for photocatalytic H2 evolution
To overcome the hydrolysis-reaction-induced decomposition of organolead halide perovskites in aqueous solution or humid conditions, establishing a dynamic interfacial equilibrium or other suitable organic reaction media is an efficient strategy to improve structural stability.48–50 Until 2016, Park and co-workers pioneered the photocatalytic hydrogen generation applying organolead halide perovskites in hydriodic acid aqueous solution via building the dynamic equilibrium to maintain the relative stability of photocatalysts (Fig. 5a).48 This strategy is beneficial for keeping organolead halide perovskites well and driving photocatalytic HX splitting into H2. Under this circumstance, the oxidation of I− occurred along with the HX reduction reaction, leading to a deepened colour change. So, a reducing agent of H3PO2 is essential in this reaction system to reduce I3− to I− for maintaining the light absorption of the solution undisturbed. The initial STH value of MAPbI3 is reported as 0.81%.48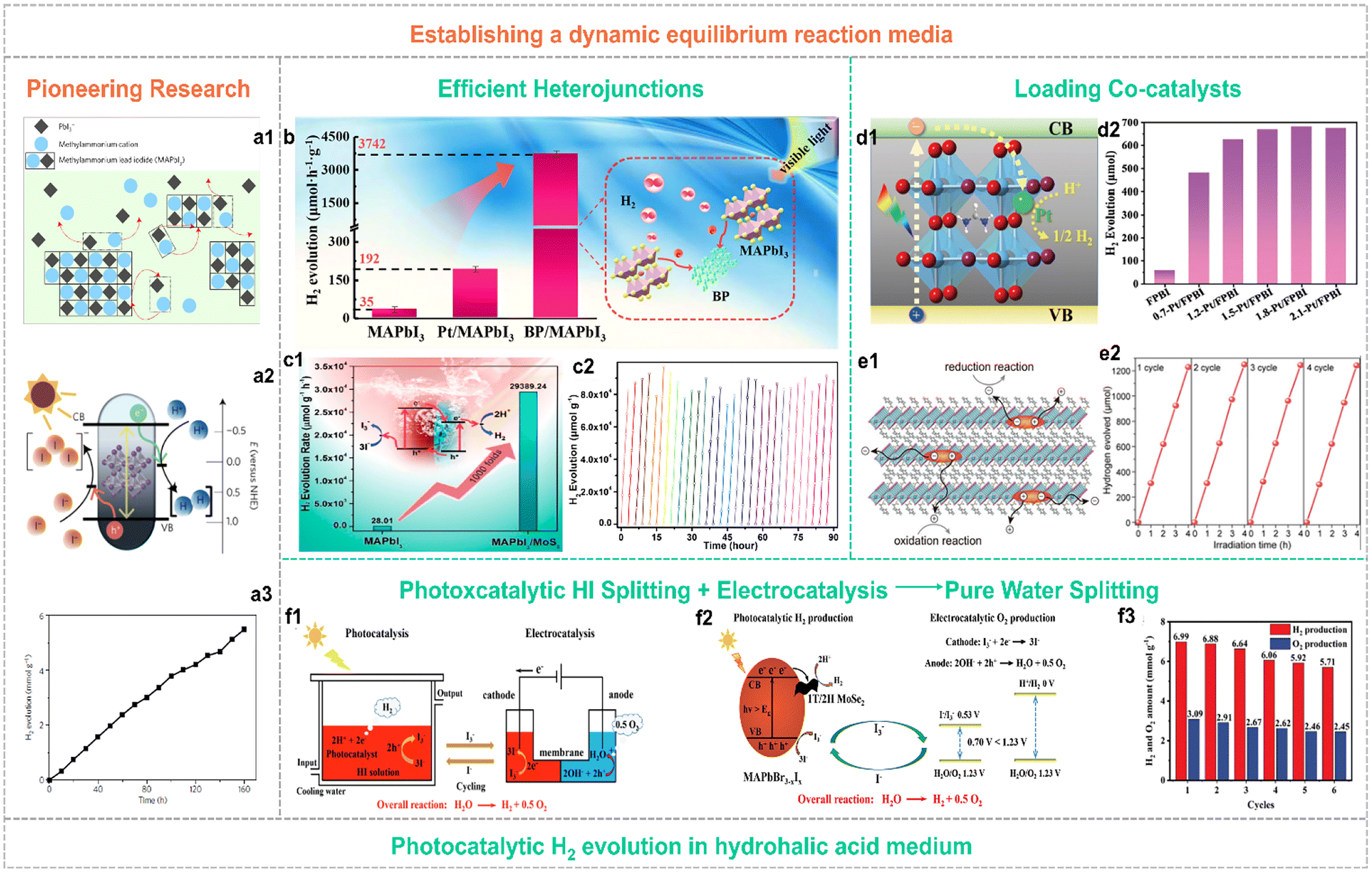 | ||
| Fig. 5 (a1) Schematic illustration of the dynamic dissolution–precipitation equilibrium for MAPbI3 perovskites in a saturated HI solution. (a2) Schematic energy band diagram of MAPbI3 perovskites for the photocatalytic HI splitting reaction. NHE is the abbreviation for normal hydrogen electrode. (a3) Stable photocatalytic H2 evolution performance of MAPbI3 over 160 h. Reproduced from ref. 48 with permission from the Nature Publishing Group, Copyright 2016. (b) Schematic illustration of the photocatalytic mechanism for the type I BP/MAPbI3 heterojunction. Reproduced from ref. 51 with permission from Elsevier, Copyright 2019. (c1) Schematic illustration of the photocatalytic mechanism and enhanced activity for the type II MAPbI3/MoS2 heterojunction. (c2) Stable photocatalytic H2 yield of the MAPbI3/MoS2 composite in 30 consecutive cycles. Reproduced from ref. 52 with permission from the American Chemical Society, Copyright 2021. (d1) Schematic illustration of electron transfer in the Pt/FAPbBr3−xIx composite. (d2) Photocatalytic H2 evolution activity of individual FAPbBr3−xIx and w-Pt/FAPbBr3−xIx composites (w represented the weight percent of Pt single-atom, wt%). Reproduced from ref. 50 with permission from the Royal Society of Chemistry, Copyright 2022. (e1) Schematic illustration of the efficient carrier separation and transfer pathway in 2D hybrid perovskites for the photocatalytic redox reaction. (e2) Cycling performance of photocatalytic H2 evolution applying Pt(2.0 wt%)/PMPI as the photocatalyst (PMPI = (C6H5CH2NH3)2PbI4). Reproduced from ref. 57 with permission from Wiley, Copyright 2021. (f1) Schematic illustration of the strategy for robust pure water splitting by tandem photocatalytic H2 production and electrocatalytic O2 evolution with I3−/I− as a redox shuttle mediator. (f2) Reaction mechanism of this pure water splitting strategy. (f3) Cycling performance of 3.0 wt% MoSe2/MAPbBr3−xIx for photocatalytic pure water spitting. Reproduced from ref. 59 with permission from Wiley, Copyright 2023. | ||
This strategy has been widely adopted by many research groups. To further improve the photocatalytic efficiency, various decoration methods have been explored to enhance the separation and transfer efficiency of photo-generated carriers. In particular, diverse organolead halide perovskite-based heterojunctions and co-catalysts modified composites have been reported. Tao et al.51 anchored black phosphorus (BP) on the surface of MAPbI3via electrostatic interaction to construct an efficient type I heterojunction for promoting the electron transfer (Fig. 5b). The optimized photocatalyst of 1.2% BP/MAPbI3 exhibited a high HI splitting rate of 3742 μmol h−1 g−1, a high AQE value of 23.2% at 420 nm, and an enhanced HI splitting efficiency of 0.93% under visible-light irradiation. More interestingly, Zhang et al.52 designed a type II heterostructure consisting MoS2 nanoflowers and MAPbI3 microcrystals, which efficiently promote the carrier separation and provide more H2 evolution reaction sites (Fig. 5c). The reported H2 evolution rate reached up to ∼30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 μmol h−1 g−1 with a high solar HI splitting efficiency of 7.35%, that outperforming the previous composites (such as MAPbI3/reduced graphene oxide (rGO),53 Ni3C/MAPbI3,54 MAPbI3/Pt/TiO2,55et al.). As is well known, Pt is considered as the most efficient electron-capturing agent to drive the H2 evolution reaction, originating from its larger work function than other noble metals. However, the photocatalytic H2 performance of most of the Pt modified organolead halide perovskites was far from the expected effect, probably due to the falling off of Pt nanoparticles in the built dynamic dissolution–precipitation equilibrium and the low utilization efficiency of the residual Pt nanoparticles on the surface of perovskites. Recently, the single-atom photocatalysts have received much more research attention.56 Wang et al.50 loaded the individual Pt single-atoms onto the FAPbBr3−xIx (FA = CH(NH2)2) substrate perovskite material via a facile self-adsorption and photoreduction method (Fig. 5d1). The reported single Pt atoms modified FAPbBr3−xIx exhibited a surprising photocatalytic H2 evolution rate of 6826 μmol h−1 g−1 (Fig. 5d2) and an extraordinarily high STH efficiency of 4.5%. This work further demonstrated the promotion effect of Pt single-atoms in capturing photo-generated electrons for enhancing photocatalytic H2 evolution performance. In contrast to 3D organolead halide perovskites, 2D layered counterparts showed significantly improved environmental stability and slightly inferior optoelectronic properties. Therefore, Zong et al. investigated the visible-light driven photocatalytic H2 evolution performance of a series of 2D organolead halide perovskites with different chain length organoammonium cations.57 The 2D hybrid perovskite showed superior carrier separation efficiency in favour of the photocatalytic redox reaction (Fig. 5e1). The perovskites with the shortest organoammonium cation chain loading with 2.0 wt% Pt cocatalysts exhibited an optimum H2 evolution rate of 333 μmol h−1 with a solar-to-chemical conversion efficiency (STC) of ∼1.57% (Fig. 5e2). Additionally, 2D halide perovskites exhibited enhanced humidity resistance and appropriate band edge position for photocatalytic HI splitting activity when exposed to an external electric field.58 Very recently, an efficient and feasible pure water splitting strategy was proposed by integrating MoSe2/MAPbBr3−xIx photocatalytic HI splitting and electrocatalytic I3− reduction-O2 evolution reaction into one loop (Fig. 5f1 and f2).59 The 3.0 wt% MoSe2/MAPbBr3−xIx composite showed pure water splitting into H2 and O2 (a stoichiometric rate of 1:2) with high total H2 production energy conversion efficiency of 2.67% and good cycling stability in the whole photocatalytic and electrocatalytic synergetic system (Fig. 5f3).
000 μmol h−1 g−1 with a high solar HI splitting efficiency of 7.35%, that outperforming the previous composites (such as MAPbI3/reduced graphene oxide (rGO),53 Ni3C/MAPbI3,54 MAPbI3/Pt/TiO2,55et al.). As is well known, Pt is considered as the most efficient electron-capturing agent to drive the H2 evolution reaction, originating from its larger work function than other noble metals. However, the photocatalytic H2 performance of most of the Pt modified organolead halide perovskites was far from the expected effect, probably due to the falling off of Pt nanoparticles in the built dynamic dissolution–precipitation equilibrium and the low utilization efficiency of the residual Pt nanoparticles on the surface of perovskites. Recently, the single-atom photocatalysts have received much more research attention.56 Wang et al.50 loaded the individual Pt single-atoms onto the FAPbBr3−xIx (FA = CH(NH2)2) substrate perovskite material via a facile self-adsorption and photoreduction method (Fig. 5d1). The reported single Pt atoms modified FAPbBr3−xIx exhibited a surprising photocatalytic H2 evolution rate of 6826 μmol h−1 g−1 (Fig. 5d2) and an extraordinarily high STH efficiency of 4.5%. This work further demonstrated the promotion effect of Pt single-atoms in capturing photo-generated electrons for enhancing photocatalytic H2 evolution performance. In contrast to 3D organolead halide perovskites, 2D layered counterparts showed significantly improved environmental stability and slightly inferior optoelectronic properties. Therefore, Zong et al. investigated the visible-light driven photocatalytic H2 evolution performance of a series of 2D organolead halide perovskites with different chain length organoammonium cations.57 The 2D hybrid perovskite showed superior carrier separation efficiency in favour of the photocatalytic redox reaction (Fig. 5e1). The perovskites with the shortest organoammonium cation chain loading with 2.0 wt% Pt cocatalysts exhibited an optimum H2 evolution rate of 333 μmol h−1 with a solar-to-chemical conversion efficiency (STC) of ∼1.57% (Fig. 5e2). Additionally, 2D halide perovskites exhibited enhanced humidity resistance and appropriate band edge position for photocatalytic HI splitting activity when exposed to an external electric field.58 Very recently, an efficient and feasible pure water splitting strategy was proposed by integrating MoSe2/MAPbBr3−xIx photocatalytic HI splitting and electrocatalytic I3− reduction-O2 evolution reaction into one loop (Fig. 5f1 and f2).59 The 3.0 wt% MoSe2/MAPbBr3−xIx composite showed pure water splitting into H2 and O2 (a stoichiometric rate of 1:2) with high total H2 production energy conversion efficiency of 2.67% and good cycling stability in the whole photocatalytic and electrocatalytic synergetic system (Fig. 5f3).
4.2 Encapsulating protection layers for organolead halide perovskites to enhance stability against moisture and improve photocatalytic performance
Over the past few years, great progress has been made to overcome the moisture-sensitivity of organolead halide perovskites. And various efficient enhanced methods are still needed to exploit. An encapsulation-based stability strategy has been developed as a feasible way to enhance stability against moisture of organolead halide perovskites.22,60,61 Notably, the crafted protection layers could not affect the original photochemical advantages (e.g., wide light absorption range, high carrier transfer efficiency, etc.) and the mass transfer capability of perovskites in practice. To realize this purpose, a few efficient encapsulation materials have been reported.Wang et al. firmly crafted a continuous organic passivating membrane on the surface of MAPbI3 crystals via a light-irradiation pathway in saturated HI aqueous solution.22 The formed passivating membrane was a homogeneous and dense MA+ cation capping layer with hydrophobic –CH3 moieties exposed on the surface (Fig. 6a1), resulting in enhanced stability and crystallinity against moisture conditions. More importantly, the perovskite catalysts with the MA+ passivating layer manifested a 2.24 times improvement of photocatalytic H2 generation rate due to the drastically increased carrier mobility rate and lifetime (Fig. 6a2).
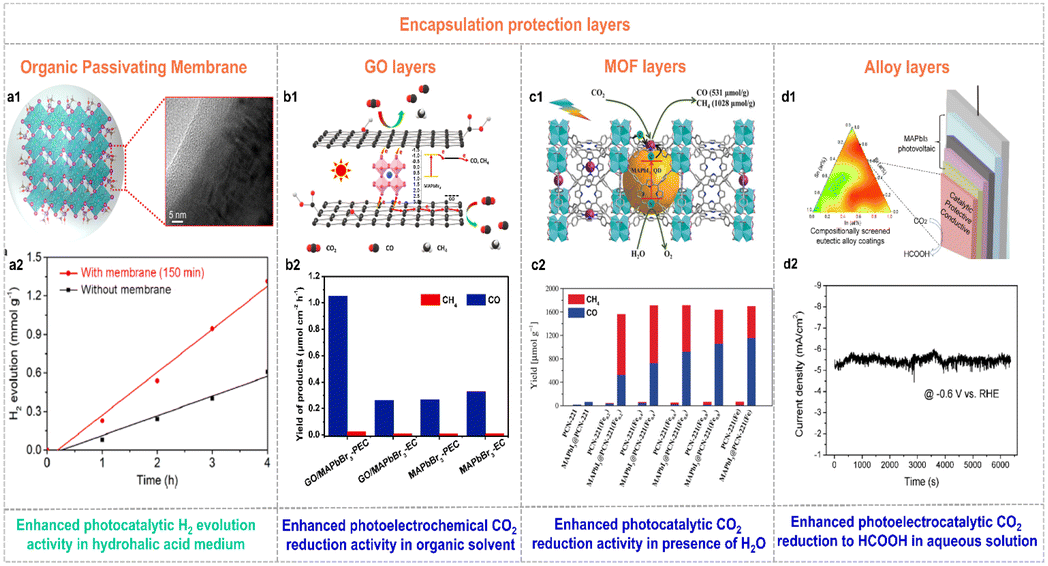 | ||
| Fig. 6 (a1) Schematic illustration of the growth of the organic passivating membrane on the surface of MAPbI3 crystals. (a2) Time-dependent photocatalytic H2 evolution of the individual MAPbI3 and MAPbI3 with the passivating layer. Reproduced from ref. 22 with permission from American Chemical Society, Copyright 2021. (b1) Possible photoelectron transfer pathway in GO/MAPbBr3 composites for photoelectrochemical CO2 reduction. (b2) Yield of CO and CH4 for electrochemical and photoelectrochemical CO2 reduction on MAPbBr3 and GO/MAPbBr3 composites (EC and PEC were the abbreviations of electrochemical and photoelectrochemical CO2 reduction, respectively). Reproduced from ref. 62 with permission from Elsevier, Copyright 2019. (c1) Schematic illustration of the synthesis and photocatalytic CO2 reduction mechanism of MAPbI3@PCN-221(Fe)x. (c2) Yields of CO and CH4 on the pristine PCN-221(Fe)x and MAPbI3@PCN-221(Fe)x in the CO2-satturated ethyl acetate/water solution over 80 h irradiation. Reproduced from ref. 60 with permission from Wiley, Copyright 2019. (d1) Schematic illustration of a halide perovskite photocathode coated with the catalytic, protective, and conductive alloy layer for CO2 conversion. (d2) Stability curve of the In0.4Bi0.6/perovskite photocathode under AM 1.5G irradiation at −0.6 V vs. RHE. Reproduced from ref. 63 with permission from the American Chemical Society, Copyright 2019. | ||
Similarly, the photocatalytic CO2 reduction performance of organolead halide perovskites was also considerably improved by introducing protection layers. For example, Shen et al. introduced GO to wrap MAPbBr3 quantum dots (QDs) to block out the solvent molecules and maintain the quantum size of perovskites in tetrabutylammonium hexafluorophosphate-propylene carbonate solution.62 The reported GO/MAPbBr3 composites showed higher photoelectrochemical CO2 reduction activity with a CO yield of 1.05 μmol cm−2 h−1 under solar irradiation (Fig. 6b2), benefiting from the protection as well as excellent photoelectrons extraction and transfer ability of GO (Fig. 6b1). Impressively, GO co-catalysts provided efficient catalytic sites for the accumulated photoexcited electrons and the absorbed CO2 molecules. Considering the well-known diversity and confinement effect of the pore structure in metal–organic frameworks (MOFs), Lu et al. encapsulated MAPbI3 QDs in the pores of Fe-porphyrin based MOFs (PCN-221(Fe)x, x represents the Fe contents) to improve the stability against H2O and photocatalytic CO2 reduction activity (Fig. 6c1).60 As shown in Fig. 6c2, the MAPbI3@PCN-221(Fe)x composites exhibited much enhanced photocatalytic stability and CO2 reduction activity applying H2O as a sacrificial reductant. Intriguingly, the optimal catalyst MAPbI3@PCN-221(Fe)0.2 possessed a record-high CO2 photoreduction total yield of 1559 μmol g−1 and high stability over 80 h illumination. The selectivity of CO and CH4 was 34% and 66%, respectively. The excellent photocatalytic performance was attributed to the outstanding photophysical properties and enhanced stability of organolead halide perovskites as well as the rich catalytic active sites in MOFs. Furthermore, Chen et al. picked out the In0.4Bi0.6 alloy from ternary In–Bi–Sn alloys as a catalytic, protective and conductive alloy layer to coat on the lead halide perovskite photovoltaic devices (Fig. 6d1).63 The designed photoelectrocatalytic system could be operated stably in aqueous solution for converting CO2 to value-added HCOOH with a nearly 100% faradaic efficiency at an applied potential of −0.6 V vs. RHE at least 1.5 h (Fig. 6d2). Additionally, Cheng et al. employed a microfluidic blow spinning technique to build MAPbBr3/polyacrylonitrile (PAN) composite photocatalysts.64 With the encapsulation of the outer PAN polymer, MAPbBr3 showed excellent structural stability in water. The optimal MAPbBr3/PAN nanofiber film with 7.40 wt% MAPbBr3 perovskites exhibited high CH4 photocatalytic rate of 63.25 μmol g−1 h−1 in deionized water under simulated sunlight.
4.3 Direct crystal engineering of organolead halide materials with high intrinsic stability for photocatalytic applications in the presence of water
Note that this section reviewed the direct crystal engineering for organolead halide crystalline materials with intrinsic stability, which was different from the improved strategies based on unstable perovskites. It is still challenging to construct high-stability organolead halide crystalline materials from the point of the crystalline structure. The efficient approaches are the modification and substitution of organoammonium cations.Generally, organic linkers could be applied to modulate the dimensionality of lead halide sublattices. Specifically, the aminoterephthalic acid coordinated with PbX2 (X = Br, I) to afford carboxylate-based organolead halide materials with 1D Pb–X–Pb secondary building units (Fig. 7a1), with chemical formula [Pb2X]3+(NH2-bdc)2 (TMOF-10-NH2(X)).70 Similarly, 2D layered (Fig. 7b1, TJU-16) and 3D skeleton-like (Fig. 7c1, TJU-32) carboxylate-based organolead halide materials were also obtained by applying adipate anions and iminodiacetate anions as template agents, respectively.71,72 TJU-16 and TJU-32 showed outstanding structural rigidity under aqueous conditions over a wide range of pH and even in boiling water, in contrast to TMOF-10-NH2(I) and traditional organolead halide perovskites (Fig. 7a2–c2). Taking TJU-16 as an example, the intrinsic stability was attributed to the following characteristics: (1) the halide anions were usually coordinated in the inner region in the inorganic component, reducing the direct contact with guest H2O molecules and (2) the hydrophobic and low-polar feature of organic carboxylates greatly suppressed the adsorption and penetration of guest H2O molecules. DFT calculations also demonstrated the higher energy cost and reaction carrier for the hydrolysis reaction on the surface of this layered organolead iodide crystalline material than the perovskites with a similar structure. This demonstrated that the organocarboxylate-oriented strategy is efficient to construct diverse and robust materials. As shown in Fig. 7a3 and b3, the edge of light-absorption was gradually shifted to the visible-light range from the chloride to the bromide to the iodide with the same crystalline structure. Taking TMOF-10-NH2(X) as an example, the optical bandgap is 2.82 eV and 2.77 eV for the bromine and the iodide, respectively. This trend is more obvious for layered organolead halide materials. And the displayed three organolead iodide crystalline materials all responded to visible light (Fig. 7a3–c3), indicating their great potential in solar-energy conversion. According to the calculated density of states (DOS) (Fig. 7a4–c4), the filled I 5p orbitals and the empty Pb 6p orbitals mainly contributed to the VBM and CBM, respectively, similar to the energy band composition of organolead iodide perovskites (Fig. 3c). More impressively, this kind of rigid organolead halide crystalline materials showed excellent carrier transfer properties, including high carrier mobility, microsecond-scaled carrier lifetime and micrometer-scaled carrier diffusion length, which is an essential factor for photocatalytic applications.
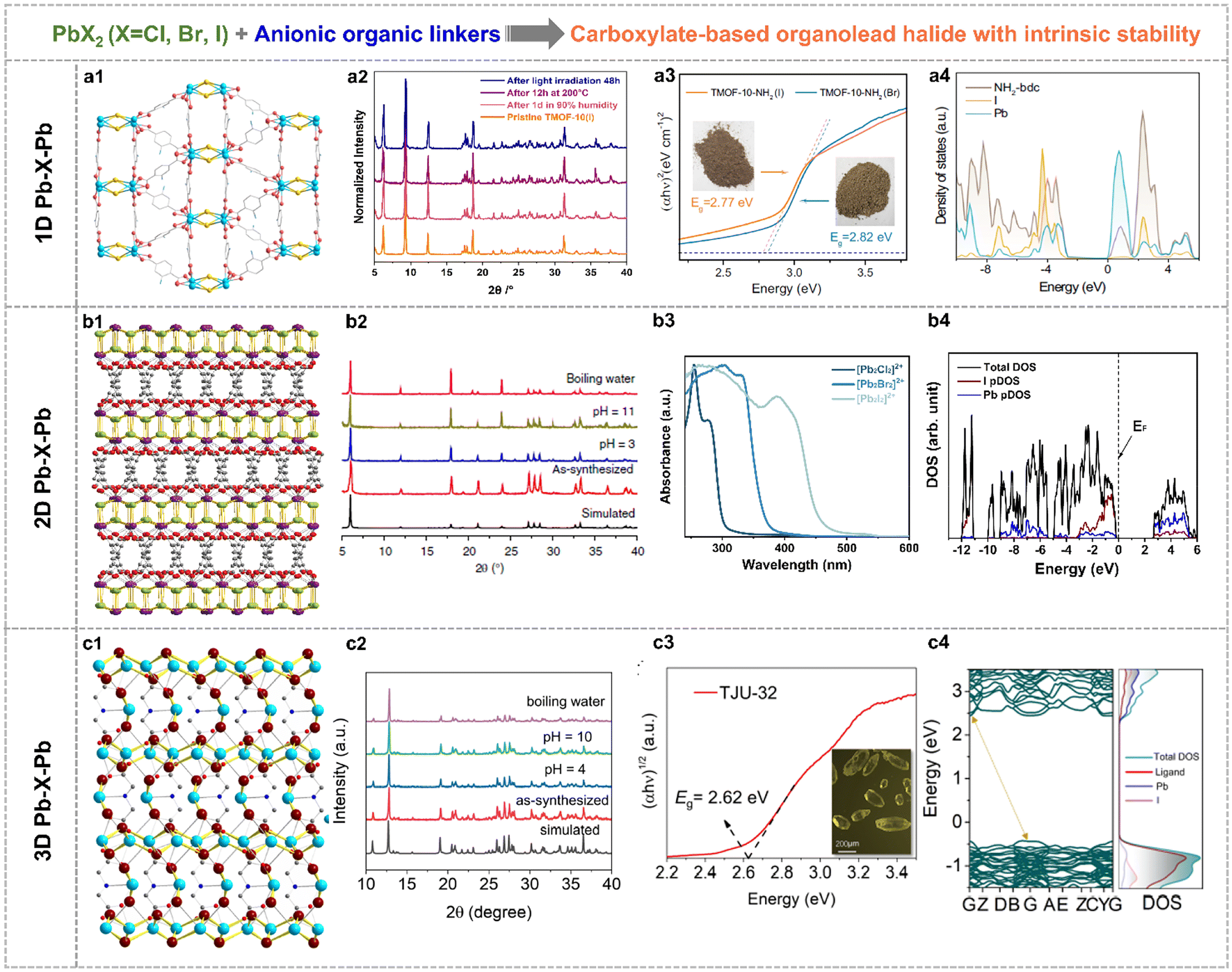 | ||
| Fig. 7 Crystallographic views of (a1) TMOF-10-NH2(I) with 1D Pb-X-Pb building units (Pb sky blue, I gold, O red and C grey), (b1) TJU-16 with 2D Pb–X–Pb building units (Pb purple, I lime, O red and C grey) and (c1) TJU-32 with 3D Pb–X–Pb building units (I dark red, N blue). PXRD patterns of (a2) TMOF-10-NH2(I), (b2) TJU-16 and (c2) TJU-32 before and after different treatments, indicating good structural stability. Photo-absorption ability of (a3) TMOF-10-NH2(X), (b3) the layered organolead halide materials and (c3) TJU-32. Calculated DOS of (a4) TMOF-10(I), (b4) TJU-16 and (c4) TJU-32. Reproduced from ref. 71 and 70, with permission from the Nature Publishing Group, Copyright 2020 and 2022, respectively, and reproduced from ref. 72, with permission from Wiley, Copyright 2024. | ||
The constructed organolead halide crystalline materials have emerged as promising photocatalysts to achieve efficient solar-energy transformation. The reported robust layered organolead iodide crystalline material of TJU-16 has been explored as a semiconductor photocatalyst to drive the photocatalytic water splitting reaction.71 It is worth mentioning that this layered semiconductor showed a visible-light absorption edge of ca. 480 nm, the appropriate energy band position straddling the water-splitting redox potential as well as excellent carrier diffusion and transfer characteristics. The overall water-splitting photocatalytic reaction was realized by applying TJU-16 as the photocatalyst without any additional photosensitizers and sacrificial agents. The optimal H2 evolution activity and solar-to-hydrogen energy conversion efficiency was up to 31 μmol g−1 h−1 and 0.014%, by loading trace Rh of 0.22 wt.‰ as the co-catalyst, respectively (Fig. 8a1). TJU-16-Rh0.22 also showed excellent photocatalytic sustainability over five cycles (Fig. 8a2). Much attention has also been paid to explore the CO2 photoreduction performance of highly robust organolead halide crystalline materials. Chen et al. modified TJU-16 with Au co-catalysts, promoting the spatial charge accumulation and the photocatalytic activity for CO2 reduction with a solar-to-fuel conversion efficiency of 0.034%.73 TMOF-10-NH2(I) consisting of lead iodide chains and coordinated carboxylate ligands was capable of driving CO2 photoreduction and water oxidation simultaneously (Fig. 8b1).70 The pristine TMOF-10-NH2(I) exhibited a high CO evolution rate of 78 μmol g−1 h−1 and high CO selectivity of ∼100% in the solid–gas photocatalytic system applying water vapor as the reductant (Fig. 8b2). In particular, the Ru1.58@TMOF-10-NH2(I) composite afforded a nearly 2-fold enhancement in the CO evolution rate of 154 μmol g−1 h−1 and an apparent quantum yield (AQY) of ∼1.36% at 400 nm. Very recently, TJU-32 with a 3D lead iodide skeleton achieved artificial photosynthesis of C2H5OH with an evolution rate of 17.6 μmol g−1 h−1 and a high selectivity of 80.4% in the presence of H2O (Fig. 8c1 and c2).72 Similarly, the photocatalytic performance of TJU-32 was further improved by loading the Rh co-catalyst. The optimal catalyst of Rh0.11@TMOF-10-NH2(I) with the Rh amount of 0.11 wt% exhibited a three-fold enhancement in C2H5OH evolution rate of 50.5 μmol g−1 h−1 and photocatalytic stability over consecutive five runs (Fig. 8c3), affording a high AQY of 1.4% at 400(±15) nm.
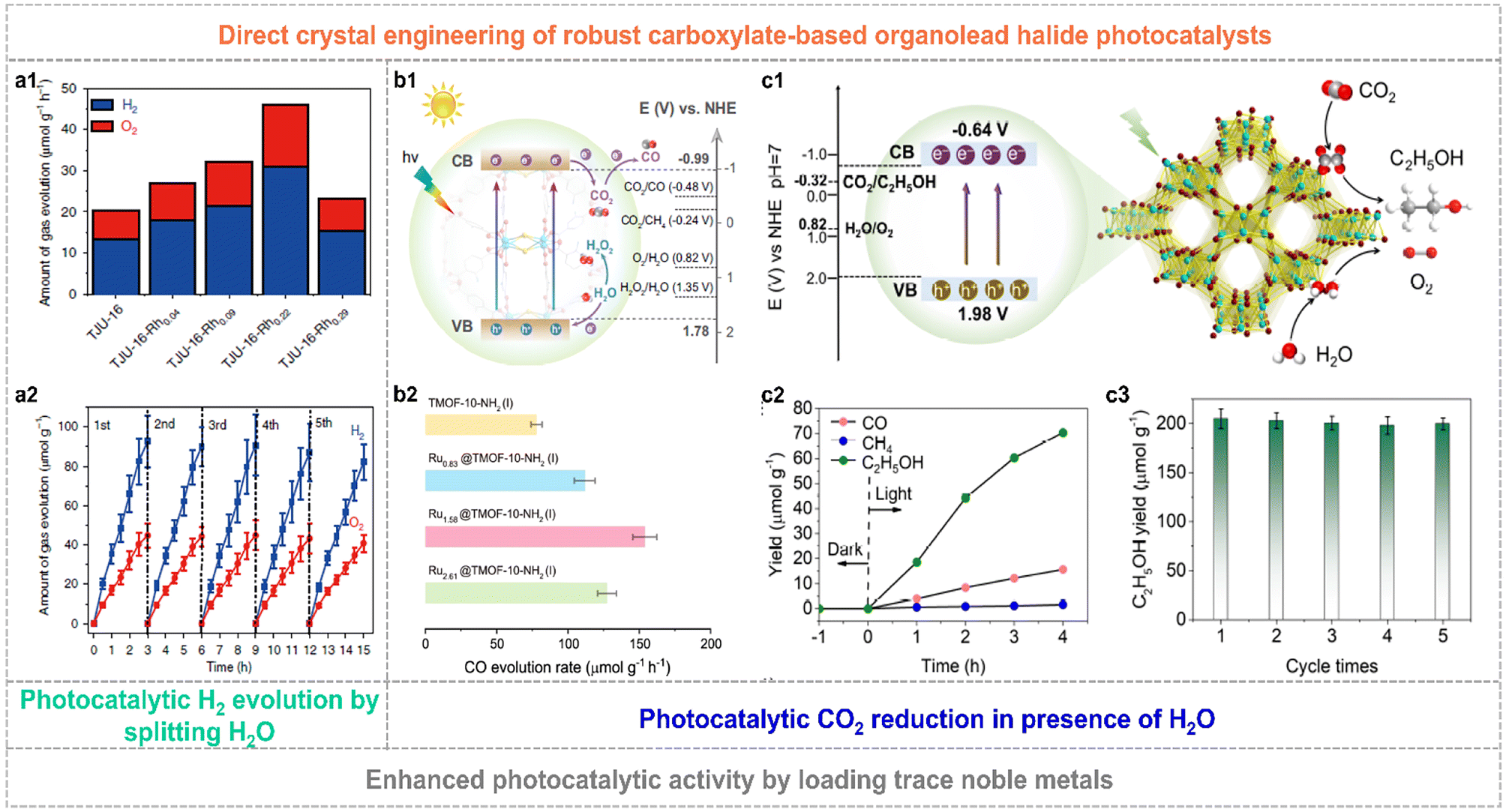 | ||
| Fig. 8 (a1) Photocatalytic activity of TJU-16 and Rh modified TJU-16. (a2) Photocatalytic stability of TJU-16-Rh0.22 for five repeated cycles. Reproduced from ref. 71, with permission from Nature Publishing Group, Copyright 2020. (b1) Schematic band structure diagram of TMOF-10-NH2(I). (b2) Photocatalytic CO evolution rate over pristine TMOF-10-NH2(I) and Ru-modified composites. Reproduced from ref. 70, with permission from Nature Publishing Group, Copyright 2022. (c1) Schematic illustration of photocatalytic CO2 reduction to C2H5OH for TJU-32. (c2) Gas yields of TJU-32 in photocatalytic CO2 reduction. (c3) Enhanced photocatalytic C2H5OH evolution rate and excellent stability over five cycles of the Rh0.11@TJU-32 catalyst. Reproduced from ref. 72, with permission from Wiley, Copyright 2024. | ||
In general, the development of high-stability organolead halide crystalline materials with excellent photocatalytic stability provides a critical advancement in applying H2O as the green catalytic environment instead of HX aqueous solution or water vapor as the green reductant. To date, the involved enhancement strategy for photocatalytic performance has been mainly focused on loading metallic co-catalysts (such as Rh, Ru, Au). It is still a key research topic to design and construct high-stability and high-efficiency organometallic halide catalysts to shorten the gap between the experimental study and commercial-scale requirements.
5. Summary and outlook
The Frontier mainly focuses on the diverse structure, excellent optoelectronic characteristics, and moisture-sensitive nature of organolead halide perovskites. Meanwhile, it emphasizes the diverse enhancement strategies for structural stability and photocatalytic activity of organolead halide crystalline materials. However, the photocatalytic research on the highly efficient organometallic halide catalysts is still in the initial stage. Several further research challenges were proposed as follows.I. Especially for organolead halide crystalline photocatalysts with high intrinsic stability, it is still urgent to explore enhancement strategies to widen the light absorption and solar-energy conversion efficiency. It is wise to choose or design organic carboxylates with visible-light absorption for enhancing the utilization of the solar spectrum. Moreover, the improved conductivity of organolead halide materials is also urgent to accelerate the photocarrier separation and transfer.
II. Considering the high toxicity of Pb to the environment and human beings, the development of organolead halide photocatalysts has been largely limited in practical industrial applications. To date, several lead-free (Sn2+, Bi3+, Cu+, etc.) halide crystalline materials have been explored. However, their stability against H2O and photocatalytic durability are still far from satisfactory in long-term photocatalysis. As a result, more efficient strategies should be further explored to enhance structural and photocatalytic stability in water or construct desired robust lead-free organometallic halide crystalline materials.
III. Photocatalytic CO2 reduction measurement was primarily carried out under a high-purity CO2 atmosphere. Nevertheless, the emitted CO2 concentration in the industrial exhaust gas is in the range of 5–20%, resulting in unsatisfactory photoreduction activity and/or selectivity. Organometallic halide crystalline materials exhibited the huge potential to achieve efficient low concentration CO2 photoreduction, benefiting from the precisely tunable structures and excellent photochemical properties.
IV. In contrast to C1 products, the research on C2/C2+ products has attracted much more attention due to their higher market price. Nonetheless, the main products of CO2 photoreduction are CO (the two-electron pathway) and CH4 (the eight-electron pathway) for our concerned organolead halide crystalline. It exhibited great potential to obtain C2/C2+ products via Cu-element doping or constructing organocopper halide catalysts for overcoming the high C–C coupling reaction energy barrier.
Author contributions
Hui Wang: conceiving the idea and supervision. Xueling Song: writing – original draft, visualization and funding acquisition. Xiaoman Li, Yuxuan Song, Jingyi Bi, Lei Wang, Jigao Wang, Junjie Liu, Yanyan Li and Hui Wang: reviewing and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (No. 22201180).References
- L. Gibson, E. N. Wilman and W. F. Laurance, Trends Ecol. Evol., 2017, 32, 922–935 CrossRef PubMed.
- J. Q. Luo, W. W. Zhang, H. B. Yang, Q. W. Fan, F. Q. Xiong, S. J. Liu, D. S. Li and B. Liu, EcoMat, 2021, 3, e12079 CrossRef CAS.
- C. Karthikeyan, P. Arunachalam, K. Ramachandran, A. M. Al-Mayouf and S. Karuppuchamy, J. Alloys Compd., 2020, 828, 154281 CrossRef CAS.
- W. B. Jiang, H. Y. Loh, B. Q. L. Low, H. J. Zhu, J. X. Low, J. Z. X. Heng, K. Y. Tang, Z. B. Li, X. J. Loh, E. Y. Ye and Y. J. Xiong, Appl. Catal., B, 2023, 321, 122079 CrossRef CAS.
- Q. Wang, M. Nakabayashi, T. Hisatomi, S. Sun, S. Akiyama, Z. Wang, Z. H. Pan, X. Xiao, T. Watanabe, T. Yamada, N. Shibata, T. Takata and K. Domen, Nat. Mater., 2019, 18, 827–832 CrossRef CAS PubMed.
- Q. H. Zhu, Q. Xu, M. M. Du, X. F. Zeng, G. F. Zhong, B. C. Qiu and J. L. Zhang, Adv. Mater., 2022, 34, 2202929 CrossRef CAS PubMed.
- Z. Wang, Y. Inoue, T. Hisatomi, R. Ishikawa, Q. Wang, T. Takata, S. S. Chen, N. Shibata, Y. Ikuhara and K. Domen, Nat. Catal., 2018, 1, 756–763 CrossRef CAS.
- J. D. Xiao, S. Nishimae, J. J. M. Vequizo, M. Nakabayashi, T. Hisatomi, H. H. Li, L. H. Lin, N. Shibata, A. Yamakata, Y. Inoue and K. Domen, Angew. Chem., Int. Ed., 2022, 61, e202116573 CrossRef CAS PubMed.
- H. W. Huang, B. Pradhan, J. Hofkens, M. B. J. Roeffaers and J. A. Steele, ACS Energy Lett., 2020, 5, 1107–1123 CrossRef CAS.
- H. W. Huang, D. Verhaeghe, B. Weng, B. Ghosh, H. W. Zhang, J. Hofkens, J. A. Steele and M. B. J. Roeffaers, Angew. Chem., Int. Ed., 2022, 61, e202203261 CrossRef CAS PubMed.
- Z. Y. Chen, N. Y. Huang and Q. Xu, Coord. Chem. Rev., 2023, 481, 215031 CrossRef CAS.
- Y. Chen, D. K. Wang, X. Y. Deng and Z. H. Li, Catal. Sci. Technol., 2017, 7, 4893–4904 RSC.
- S. Y. Wang, Z. W. Ai, X. W. Niu, W. J. Yang, R. Kang, Z. Y. Lin, A. Waseem, L. Jiao and H. L. Jiang, Adv. Mater., 2023, 35, 2302512 CrossRef CAS PubMed.
- H. Wang, H. Wang, Z. W. Wang, L. Tang, G. M. Zeng, P. Xu, M. Chen, T. Xiong, C. Y. Zhou, X. Y. Li, D. N. Huang, Y. Zhu, Z. X. Wang and J. W. Tang, Chem. Soc. Rev., 2020, 49, 4135–4165 RSC.
- G. B. Wang, S. Li, C. X. Yan, F. C. Zhu, Q. Q. Lin, K. H. Xie, Y. Geng and Y. B. Dong, J. Mater. Chem. A, 2020, 8, 6957–6983 RSC.
- C. Sun, R. A. Xi and H. H. Fei, Acc. Chem. Res., 2023, 56, 452–461 CrossRef CAS PubMed.
- C. B. Bie, L. X. Wang and J. G. Yu, Chem, 2022, 8, 1567–1574 CAS.
- J. Wang, J. L. Liu, Z. L. Du and Z. Q. Li, J. Energy Chem., 2021, 54, 770–785 CrossRef CAS.
- T. M. Brenner, D. A. Egger, L. Kronik, G. Hodes and D. Cahen, Nat. Rev. Mater., 2016, 1, 1–16 Search PubMed.
- S. Park, S. Choi, S. Kim and K. T. Nam, J. Phys. Chem. Lett., 2021, 12, 8292–8301 CrossRef CAS PubMed.
- J. L. Yang and T. L. Kelly, Inorg. Chem., 2017, 56, 92–101 CrossRef CAS PubMed.
- F. Y. Liu, M. Y. Wang, X. L. Liu, B. Wang, C. F. Li, C. N. Liu, Z. Lin and F. Huang, Nano Lett., 2021, 21, 1643–1650 CrossRef CAS PubMed.
- J. Chen, C. W. Dong, H. Idriss, O. F. Mohammed and O. M. Bakr, Adv. Energy Mater., 2020, 10, 1902433 CrossRef CAS.
- H. W. Huang, B. Pradhan, J. Hofkens, M. B. J. Roeffaers and J. A. Steele, ACS Energy Lett., 2020, 5, 1107–1123 CrossRef CAS.
- X. Wang, Y. Peng, S. Yang, H. G. Yang and Y. Hou, Mater. Chem. Front., 2023, 7, 4635–4657 RSC.
- H. Zhao, K. Kordas and S. Ojala, J. Mater. Chem. A, 2023, 11, 22656–22687 RSC.
- E. Z. Shi, Y. Gao, B. P. Finkenauer, Akriti, A. H. Coffey and L. T. Dou, Chem. Soc. Rev., 2018, 47, 6046–6072 RSC.
- C. K. Zhou, H. R. Lin, S. Lee, M. Chaaban and B. W. Ma, Mater. Res. Lett., 2018, 6, 552–569 CrossRef CAS.
- V. K. Ravi, G. B. Markad and A. Nag, ACS Energy Lett., 2016, 1, 665–671 CrossRef CAS.
- J. L. Chang, G. Z. Wang, Y. H. Huang, X. K. Luo and H. Chen, New J. Chem., 2017, 41, 11413–11421 RSC.
- D. Shi, V. Adinolfi, R. Comin, M. J. Yuan, E. Alarousu, A. Buin, Y. Chen, S. Hoogland, A. Rothenberger, K. Katsiev, Y. Losovyj, X. Zhang, P. A. Dowben, O. F. Mohammed, E. H. Sargent and O. M. Bakr, Science, 2015, 347, 519–522 CrossRef CAS PubMed.
- S. Chen, H. J. Yin, P. R. Liu, Y. Wang and H. J. Zhao, Adv. Mater., 2023, 35, 2203836 CrossRef CAS PubMed.
- K. K. Ren, S. Z. Yue, C. H. Li, Z. B. Fang, K. A. M. Gasem, J. Leszczynski, S. C. Qu, Z. J. Wang and M. H. Fan, J. Mater. Chem. A, 2022, 10, 407–429 RSC.
- L. Hu, G. Shao, T. Jiang, D. B. Li, X. L. Lv, H. Y. Wang, X. S. Liu, H. S. Song, J. Tang and H. Liu, ACS Appl. Mater. Interfaces, 2015, 7, 25113–25120 CrossRef CAS PubMed.
- K. Dhivyaprasath and M. Ashok, Sol. Energy, 2023, 255, 89–98 CrossRef CAS.
- A. M. A. Leguy, Y. Hu, M. Campoy-Quiles, M. I. Alonso, O. J. Weber, P. Azarhoosh, M. van Schilfgaarde, M. T. Weller, T. Bein, J. Nelson, P. Docampo and P. R. F. Barnes, Chem. Mater., 2015, 27, 3397–3407 CrossRef CAS.
- Y. B. Li, W. J. Zhou, Y. Z. Li, W. X. Huang, Z. W. Zhang, G. X. Chen, H. S. Wang, G. H. Wu, N. Rolston, R. Vila, W. Chiu and Y. Cui, Joule, 2019, 3, 2854–2866 CrossRef CAS PubMed.
- J. L. Yang, B. D. Siempelkamp, D. Y. Liu and T. L. Kelly, ACS Nano, 2015, 9, 1955–1963 CrossRef CAS PubMed.
- J. A. Christians, P. A. M. Herrera and P. V. Kamat, J. Am. Chem. Soc., 2015, 137, 1530–1538 CrossRef CAS PubMed.
- U. G. Jong, C. J. Yu, G. C. Ri, A. P. McMahon, N. M. Harrison, P. R. F. Barnes and A. Walsh, J. Mater. Chem. A, 2018, 6, 1067–1074 RSC.
- R. T. Wang, Y. Zhang, X. X. Wu, W. W. Zhang, L. X. Chi and F. Xu, Sustainable Energy Fuels, 2023, 7, 1974–1980 RSC.
- R. T. Wang, A. F. Xu, W. Q. Li, Y. N. Li and G. Xu, J. Phys. Chem. Lett., 2021, 12, 5332–5338 CrossRef CAS PubMed.
- J. H. Ran, O. O. Dyck, X. Z. Wang, B. Yang, D. B. Geohegan and K. Xiao, Adv. Energy Mater., 2020, 10, 1903191 CrossRef CAS.
- B. Yuan and Y. Yu, Chem, 2022, 8, 327–339 CAS.
- Y. H. Deng, Nature, 2021, 594, E6–E7 CrossRef CAS PubMed.
- M. U. Rothmann, J. S. Kim, J. Borchert, K. B. Lohmann, C. M. O'Leary, A. A. Sheader, L. Clark, H. J. Snaith, M. B. Johnston, P. D. Nellist and L. M. Herz, Science, 2020, 370, eabb5940 CrossRef CAS PubMed.
- J. Lv, H. Zhang, D. L. Zhang, L. M. Liu and Y. Han, Acc. Mater. Res., 2022, 3, 552–564 CrossRef CAS.
- S. Park, W. J. Chang, C. W. Lee, S. Park, H. Y. Ahn and K. T. Nam, Nat. Energy, 2017, 2, 1–8 Search PubMed.
- C. Cai, Y. Teng, J. H. Wu, J. Y. Li, H. Y. Chen, J. H. Chen and D. B. Kuang, Adv. Funct. Mater., 2020, 30, 2001478 CrossRef CAS.
- Y. Q. Wu, Q. Wu, Q. Q. Zhang, Z. Z. Lou, K. F. Liu, Y. D. Ma, Z. Y. Wang, Z. K. Zheng, H. F. Cheng, Y. Y. Liu, Y. Dai, B. B. A. Huang and P. Wang, Energy Environ. Sci., 2022, 15, 1271–1281 RSC.
- R. Li, X. T. Li, J. J. Wu, X. D. Lv, Y. Z. Zheng, Z. J. Zhao, X. Q. Ding, X. Tao and J. F. Chen, Appl. Catal., B, 2019, 259, 118075 CrossRef CAS.
- W. H. Guan, Y. Li, Q. X. Zhong, H. Y. Liu, J. N. Chen, H. C. Hu, K. X. Lv, J. Gong, Y. Xu, Z. H. Kang, M. H. Cao and Q. Zhang, Nano Lett., 2021, 21, 597–604 CrossRef CAS PubMed.
- Y. Q. Wu, P. Wang, X. L. Zhu, Q. Q. Zhang, Z. Y. Wang, Y. Y. Liu, G. Z. Zou, Y. Dai, M. H. Whangbo and B. B. Huang, Adv. Mater., 2018, 30, 1704342 CrossRef PubMed.
- Z. J. Zhao, J. J. Wu, Y. Z. Zheng, N. Li, X. T. Li and X. Tao, ACS Catal., 2019, 9, 8144–8152 CrossRef CAS.
- W. S. Han, Y. Z. Wei, J. W. Wan, N. Nakagawa and D. Wang, Inorg. Chem., 2022, 61, 5397–5404 CrossRef PubMed.
- Z. H. Xue, D. Y. Luan, H. B. Zhang and X. W. Lou, Joule, 2022, 6, 92–133 CrossRef CAS.
- H. Wang, H. F. Zhang, J. H. Wang, Y. Y. Gao, F. T. Fan, K. F. Wu, X. Zong and C. Li, Angew. Chem., Int. Ed., 2021, 60, 7452–7457 CrossRef.
- S. Park, S. Oh and J. Lee, J. Mater. Chem. A, 2023, 11, 6311–6320 RSC.
- X. L. Liu, Q. Q. Zhang, S. L. Zhao, Z. Y. Wang, Y. Y. Liu, Z. K. Zheng, H. F. Cheng, Y. Dai, B. B. Huang and P. Wang, Adv. Mater., 2023, 35, 2208915 CrossRef CAS PubMed.
- L. Y. Wu, Y. F. Mu, X. X. Guo, W. Zhang, Z. M. Zhang, M. Zhang and T. B. Lu, Angew. Chem., Int. Ed., 2019, 58, 9491–9495 CrossRef CAS PubMed.
- H. Y. Zhang, X. X. He, H. Wang, L. J. Chen, G. P. Xu, N. Zhang, K. Qu, Q. Q. He, Y. W. Peng and J. Pan, Nanotechnology, 2023, 34, 245601 CrossRef PubMed.
- Q. L. Wang, L. M. Tao, X. X. Jiang, M. K. Wang and Y. Shen, Appl. Surf. Sci., 2019, 465, 607–613 CrossRef CAS.
- J. Chen, J. Yin, X. P. Zheng, H. A. Ahsaine, Y. Zhou, C. W. Dong, O. F. Mohammed, K. Takanabe and O. M. Bakr, ACS Energy Lett., 2019, 4, 1279–1286 CrossRef CAS.
- R. Cheng, Z. B. Liang, L. L. Zhu, H. Li, Y. Zhang, C. F. Wang and S. Chen, Angew. Chem., Int. Ed., 2022, 61, e202204371 CrossRef CAS PubMed.
- Y. Zhao, H. Y. Zhong, L. Li, W. L. Lin, Y. E. Huang, B. Y. Su, X. H. Wu, X. Y. Huang and K. Z. Du, ACS Appl. Energy Mater., 2021, 4, 13550–13555 CrossRef CAS.
- T. Sheikh, S. Maqbool, P. Mandal and A. Nag, Angew. Chem., Int. Ed., 2021, 60, 18265–18271 CrossRef CAS PubMed.
- A. Leblanc, N. Mercier, M. Allain, J. Dittmer, T. Pauporté, V. Fernandez, F. Boucher, M. Kepenekian and C. Katan, ACS Appl. Mater. Interfaces, 2019, 11, 20743–20751 CrossRef PubMed.
- S. K. Yu, Z. R. Zhang, Z. H. Ren, H. L. Zhai, Q. Y. Zhu and J. Dai, Inorg. Chem., 2021, 60, 9132–9140 CrossRef CAS PubMed.
- W. J. Chen, Y. L. Shi, J. Chen, P. C. Ma, Z. B. Fang, D. Ye, Y. Y. Lu, Y. B. Yuan, J. Zhao and Z. G. Xiao, Adv. Mater., 2021, 33, 2104842 CrossRef CAS PubMed.
- X. F. Chen, C. D. Peng, W. Y. Dan, L. Yu, Y. N. Wu and H. H. Fei, Nat. Commun., 2022, 13, 4592 CrossRef CAS PubMed.
- X. L. Song, G. F. Wei, J. Sun, C. D. Peng, J. L. Yin, X. Zhang, Y. L. Jiang and H. H. Fei, Nat. Catal., 2020, 3, 1027–1033 CrossRef CAS.
- J. L. Yin, X. L. Song, C. Sun, Y. L. Jiang, Y. N. He and H. H. Fei, Angew. Chem., Int. Ed., 2024, e202316080 CAS.
- R. Chen, G. D. Gao and J. S. Luo, Nano Res., 2022, 15, 10084–10089 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2024 |