
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOxidative chemical vapor deposition for synthesis and processing of conjugated polymers: a critical review
Afshin
Dianatdar
and
Ranjita K.
Bose
 *
*
Department of Chemical Engineering, Engineering and Technology Institute Groningen (ENTEG), University of Groningen, 9747AG Groningen, The Netherlands. E-mail: r.k.bose@rug.nl; Tel: +31 50 3634486
First published on 16th August 2023
Abstract
Oxidative chemical vapor deposition (oCVD) has developed progressively in the last two decades as a solvent-free (or dry) methodology for synthesis and thin film deposition of conjugated polymers. This method has offered new opportunities beyond traditional solution processing methods in the research of these materials. It is crucial to have a clear understanding of the differences between the solvent-free vs. solvent-based methodologies for synthesis and thin film deposition of conjugated polymers. Herein, the strengths and limitations of each procedure are compared in order to provide guidelines for future research and development. This review systematically approaches this comparison by first characterizing the thin films in terms of their chemical and physical properties. Then, the interfacial properties of a conjugated polymer thin film with the underlying substrate are critically compared when two different processing methods are exploited. Finally, the effect of the substrate on the coating properties and performance is reviewed.
 Afshin Dianatdar | Afshin Dianatdar attended University of Groningen (The Netherlands) in 2017 to follow his PhD degree of chemical engineering. His research interests have been conjugated polymers, chemical vapor deposition, and their application in electrochemical energy storage devices. Upon graduation in 2022, he joined Johnson & Johnson (Belgium) as Formulation Scientist and currently involved in innovation projects around antifouling coatings for marine applications. |
 Ranjita K. Bose | Ranjita K. Bose obtained a bachelors in chemical engineering from Gujarat University (India) in 2006, followed by a PhD in chemical vapor deposition of polymers in 2011 from Drexel University (USA). From 2012 to 2017 she did postdoctoral research on self-healing polymers in the Delft University of Technology (The Netherlands). In 2017 she joined the University of Groningen (The Netherlands), where she is currently an associate professor of polymer engineering. She leads a team of 7 PhD students, 1 postdoctoral researcher and 3 MSc students working on chemical vapor deposition of polymer films and coatings, rheology, and structure–property relationships in polymers for self-healing applications. |
1. Introduction
Polymers have long been considered as insulating materials, and it was only about 35 years ago that numerous reports of polymers showing conductivity emerged in the literature.1 This was a breakthrough as until then, scientists had been dependent on inorganic materials such as metals and metal oxides for conductive properties. Therefore, the concept of a material with the flexibility of traditional polymers and conductivity of metals opened a wide scope of interest in academia and later in industry as well. The term “synthetic metals” was coined to describe these new materials, which are not metals but possess free electrons to conduct electricity.2,3The general term of “conductive polymers” encompasses a broad area of ion-conducting and electron-conducting polymers based on their mode of charge propagation.4 For ionic conductivity, conductive polymers should be able to be easily oxidized and reduced, subsequently allowing the counter ions to move freely in the polymer matrix. This ion is either an anion or a cation. Anion movement is possible in the case of oxidized conductive polymers, resulting in a polycationic backbone, where small counter ions such as Cl− could move along due to an anionic potential. On the other hand, when conductive polymers are reduced, cationic conductivity becomes possible in the resulting polyanionic structure.5 However, the current review focuses only on electron conducting polymers. These polymers are also called redox polymers. Redox polymers could again be classified into different groups based on the following (see Fig. 1(a)–(d)):6
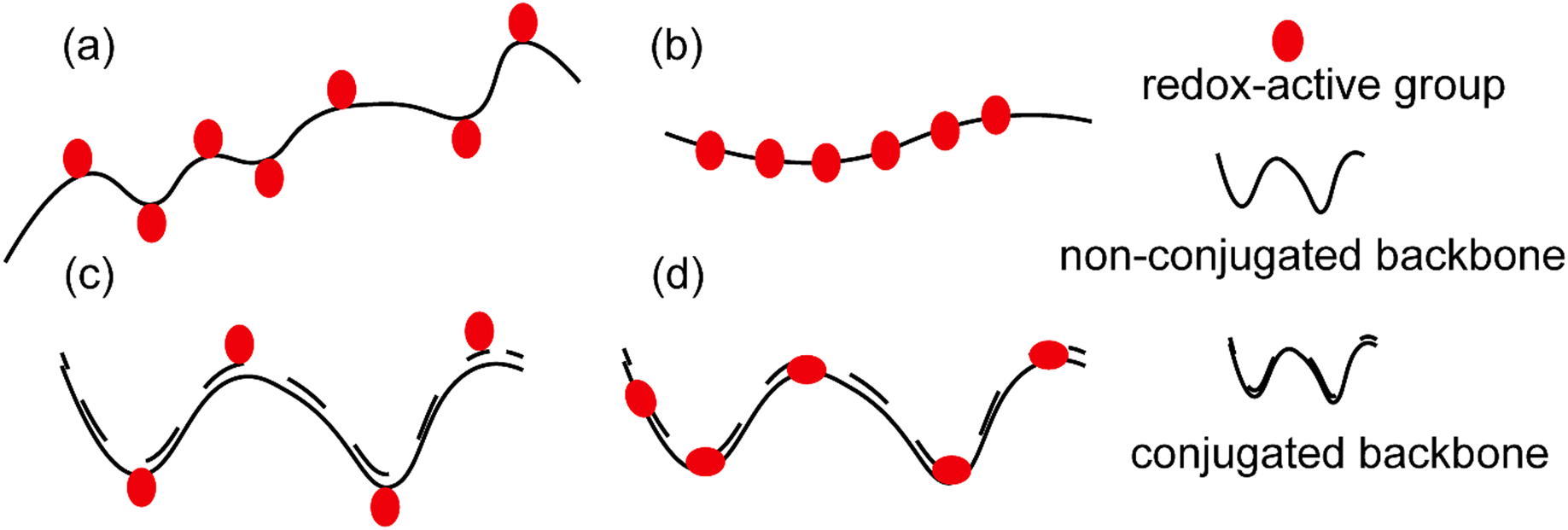 | ||
| Fig. 1 A schematic representation of (a) a polymer with non-conjugated backbone bearing redox active groups (red balls) in its side chains; (b) a polymer with non-conjugated backbone containing the redox active sites along its backbone; (c) a polymer with conjugated backbone bearing redox sites in its side chains; (d) a polymer with conjugated backbone including its redox groups along its backbone. | ||
(a) Location of redox-active sites: (i) polymers that embed the redox-active site(s) along their backbone; and (ii) polymers that bear the redox-active site(s) in their side chains.
(b) The chemical structure of their backbone: (i) conjugated and (ii) non-conjugated.
(c) Nature of the redox center: (i) organic and (ii) inorganic.
Clearly, polymers as a result of hybrid forms of the above have also been developed during the last few decades in the attempt for new materials development with modified properties.7 Additionally, complications resulting from the fact that the redox-active site(s) is associated with the polymer either through covalent bond(s) or via supramolecular interactions should also be considered for a precise distinction.8 The following discussion only includes organic materials, in which redox groups are covalently bonded within the polymer architecture.
a. Types of electroactive polymers
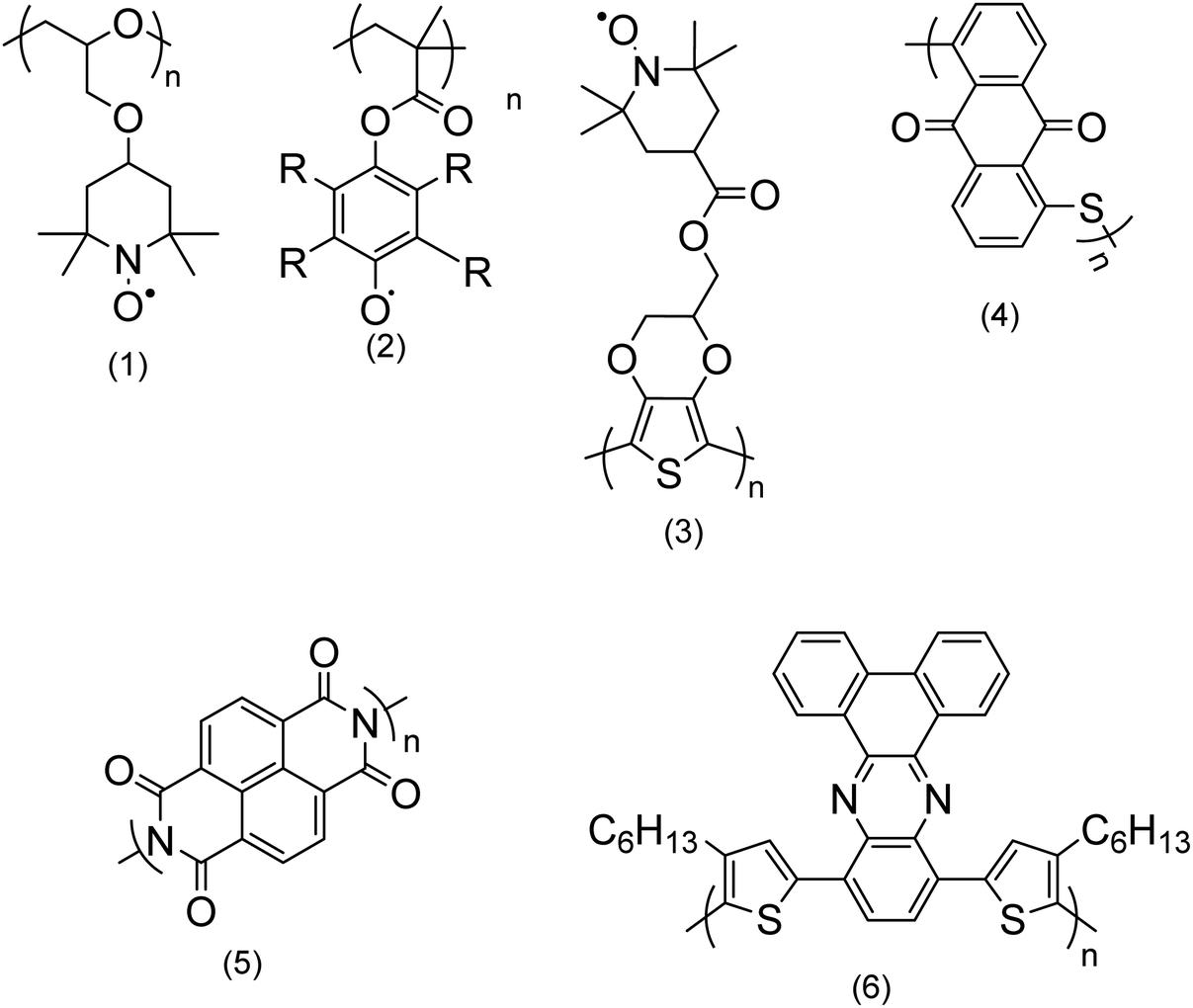 | ||
| Fig. 2 Chemical structures of a few redox polymers: structures (1) and (2) are examples of non-conjugated polymers with redox sites in their side chains; structure (3) shows an example of a conjugated polymer which bears redox active sites in its side chain; structures (4) and (5) are examples of non-conjugated polymers with redox centers along their backbone; and structure (6) is an example of a conjugated polymer with redox sites along its backbone.9 | ||
Non-conjugated backbone. The polymers which embed the electroactive group along their backbone, although their backbone does not have a regular alternating single and double bond, entirely fall within this group. Examples are structures (4) and (5) in Fig. 2.
Conjugated polymers (CPs). Redox-active polymers with conjugated backbones are the polymers giving fame to the field of electrically active macromolecules. In their simple definition, CPs (also called intrinsically conductive polymers) are macromolecules that have a conjugated sequence of single and double carbon bonds along their chains.15 The history began in 1976, when Alan MacDiarmid, Hideki Shirakawa and Alan J. Heeger together reported that the conductivity of ‘trans polyacetylene’ could be enhanced up to seven orders of magnitude upon exposure to halogens.16 This breakthrough opened a new era in the physics and chemistry of polymeric materials, and won them the Nobel Prize for Chemistry in 2000.17 Polyacetylene itself was not a stable polymer and never found practical applications.18 It stimulated the research into polymers with similar properties, which resulted in introduction of a wide range of conjugated polymers, some of the most important of which are presented in Fig. 3.
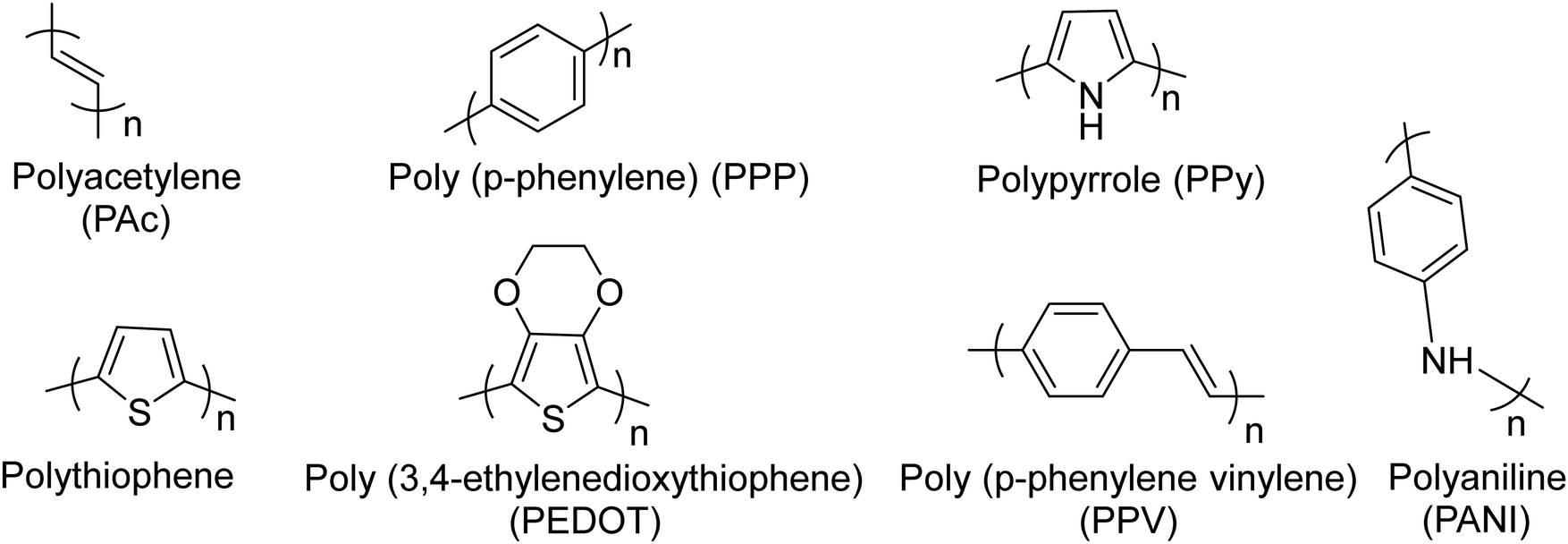 | ||
| Fig. 3 Chemical structures of the most common conjugated polymers, which have been exploited readily or as the building block for the synthesis of more sophisticated redox active polymers. | ||
From a comparison point of view, these polymers show a range of conductivities from insulator to metallic, with most reported conductivity in the range of 10−5–103 S cm−1, mostly lying in the semiconductive range (Fig. 4).19 Additionally, they could combine the conductivity of a metal with flexibility of a polymer (in a rough sense). These all gave rise to a large body of research from the 1980s up to now.
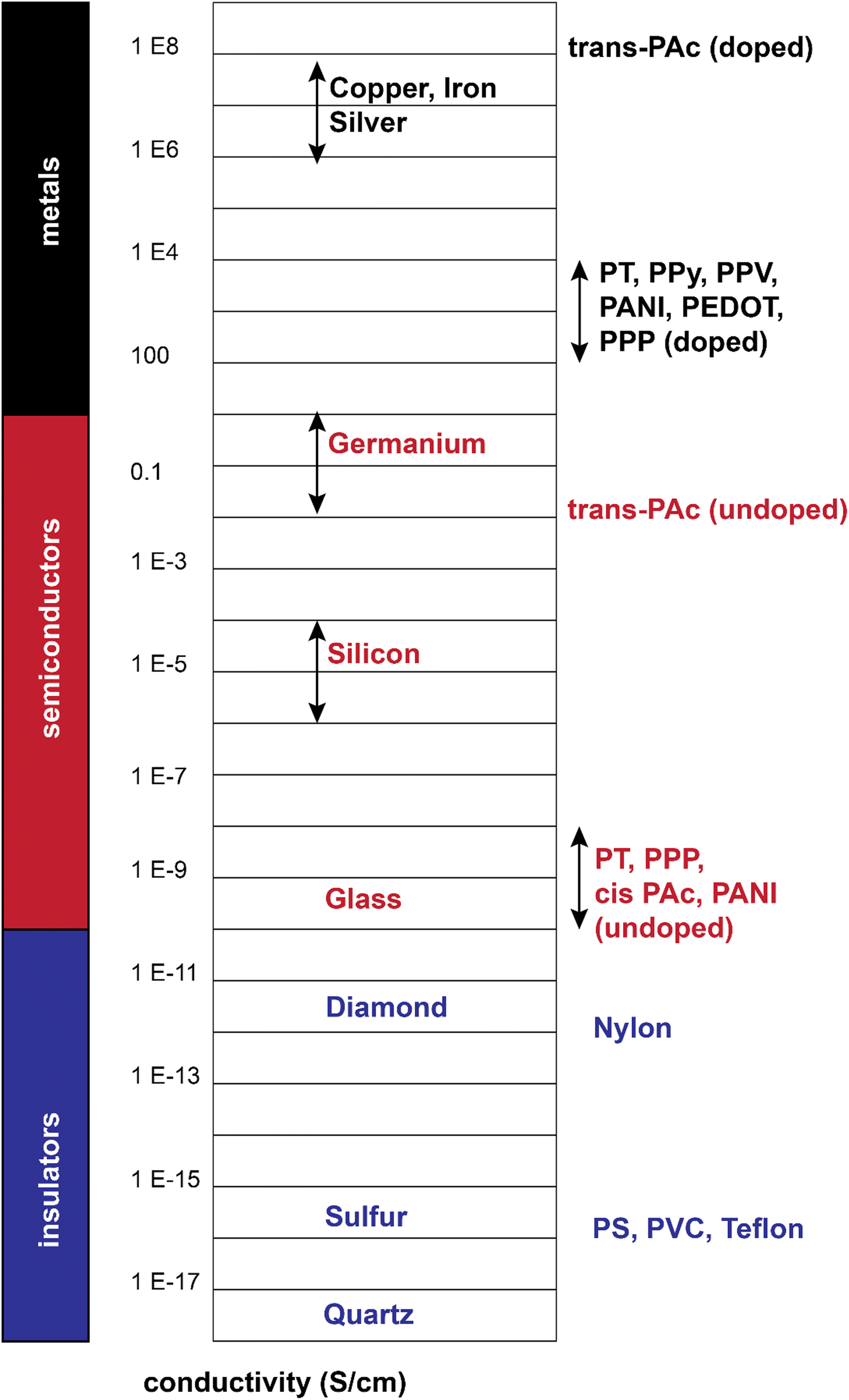 | ||
| Fig. 4 Electrical conductivity of doped/undoped conjugated polymers relative to selective inorganic materials.22–24 | ||
The conjugated structures in Fig. 3 consist of adjacent carbon atoms associated together with σ-bonds that hold the structure together and orthogonal π-bonds that are delocalized and allow electron mobility within the polymer chains (intramolecular charge transfer).20 Additionally, inter-chain electron hopping is another factor determining the overall conductivity of a CP. From a physical point of view, materials could be divided into three categories according to the electron orbital structure, namely insulators, semiconductors, and conductors (Fig. 5).19,21
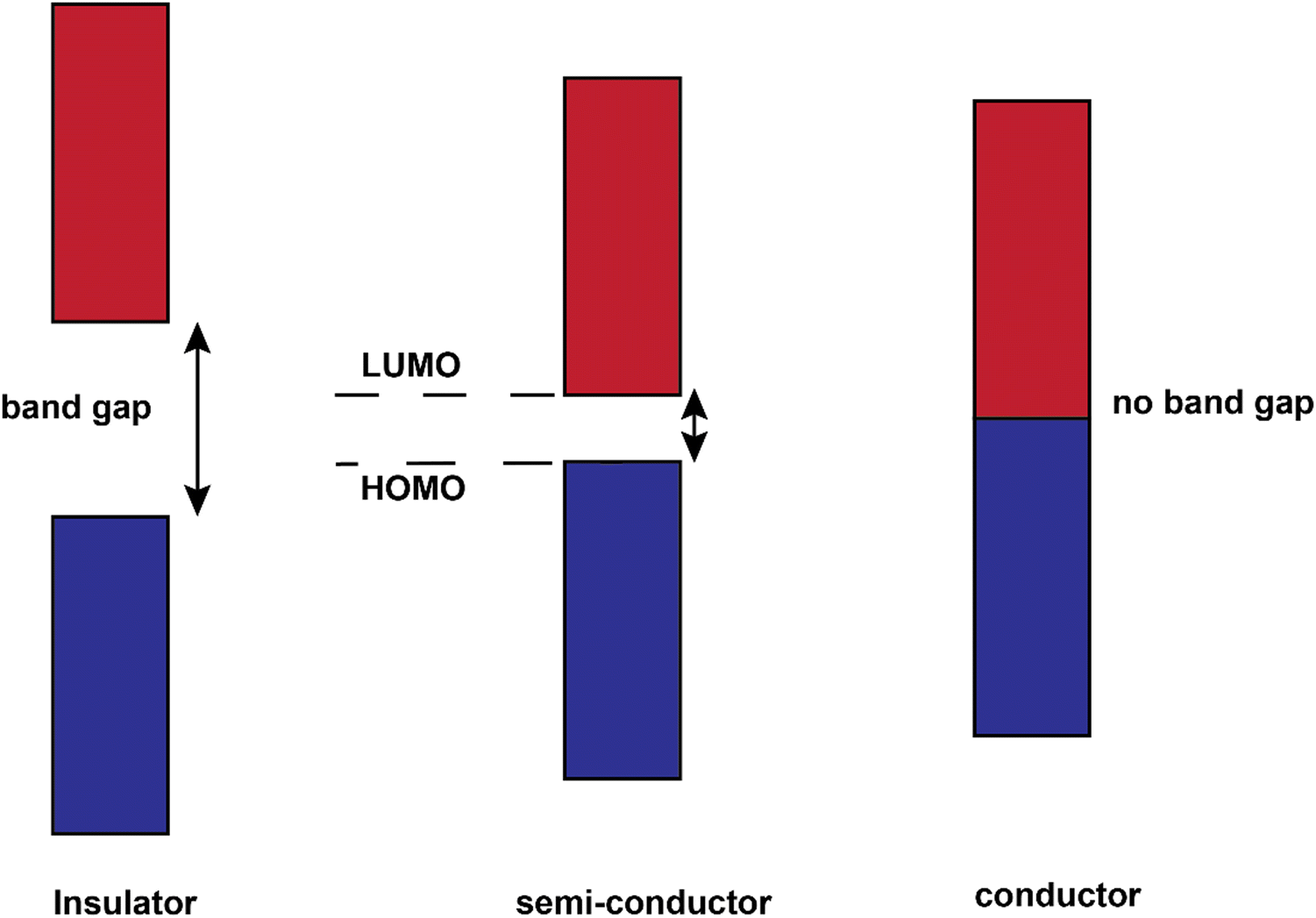 | ||
| Fig. 5 Band gap structures for insulators, semiconductors, and conductors. | ||
The determining criterion is the energy gap between the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO). For a material to show conductivity, it is required that electrons to be transferred from the HOMO (also called the valence band) to the LUMO (also called the conduction band). If such a transfer occurs, the electrons might potentially be able to freely transport within the material. The energy difference between these two “states” is called the band gap.23 In conductors (e.g., copper or gold), this gap does not exist. That is why electrons could freely move between the bands and the material. For insulators such as wood or rubber, the required energy for the transfer of an electron from the valence band to the conduction band is so large (>10 eV) that makes the process very difficult or impossible.25 For semiconductors, a certain amount of energy is required for an electron to be transferred between “valence band” and “conduction band” and for the sake of “neutral” conjugated polymers, this energy is between 3–6 eV which reduces to 1–4 eV upon doping.19 The synthesis and processing of CPs is the main scope of the current review.
b. Synthesis (polymerization)
There are different methodologies for the synthesis of CPs including solvent-based methods of chemical polymerization, electrochemical polymerization, emulsion polymerization, and photo-induced polymerization. Selected chemical polymerization routes could also be progressed in solvent-free mode of vapor-phase polymerization, oxidative chemical vapor deposition, and plasma polymerization.9,26–30The above categorization based on solvent-use is particularly relevant as CPs in their unmodified form are not soluble in common organic solvents and this complicates their initial polymerization and their further processing with the aid of solvents.31 This should be understood within the context of common CP applications in the devices (mentioned above), in which they are often in the form of a thin film of a few nanometers to a few micrometers.32 In other words, when CPs are synthesized (in bulk), they need to be further processed into a thin film by different coating methods such as spin coating or casting, which gives rise to challenges if the initial polymer is not soluble.33 On the other hand, vapor-phase deposition exploits a gas-phase reactor to polymerize a thin film of CPs on a surface from reactant vapors. Although the vapor-phase synthesis of PPy dates back to 1986,34 a systematic protocol was pioneered by K.K. Gleason35 of MIT in 2006. In this method, which is basically equal to oxidation polymerization without use of solvent, the monomer and the oxidant are separately metered into a vacuum chamber in the initial step and react directly onto the surface of a substrate resulting in a thin film.
c. Properties and applications
As mentioned above, CPs combine the conductivity of a metal with the flexibility of a polymer. This gave rise to a large body of research from 1980s up to now for a wide range of applications including but not limited to organic electronics: organic photovoltaics, field-effect transistors, light-emitting diodes; energy storage devices: batteries, supercapacitors, fuel cells;36–40 medical devices and procedures: sensor, drug delivery, tissue engineering, actuator;41–44 and other fields like corrosion,45–47 textiles,48 gas/liquid separation,49,50 and catalysts.51Electrical conductivity in CPs is a combination of electron and ion mobility. The ionic conductivity of CPs is poorly understood due to the lack of comprehensive studies.52 For ionic conductivity in solid-state polymers, the polymeric host should act as a solvent for the ions.53 However, being solids, they are inherently poor solvents, and therefore, their ionic conductivity only finds relevance in certain applications where liquid-based solvents are also present such as electrochemical energy storage devices. In this regard, although a common resistivity measurement method via a four-probe technique does not distinguish between ionic and electronic conductivity of a solid CP,54 it is reasonable to think that the dominant mechanism of charge transfer in these materials stems from electrical conductivity.
d. Aim and scope
As could be expected, each of the above methods offers their own advantages over the other, which makes either of them preferred under certain circumstances. Considering the importance of CPs in fundamental and applied research as well as potential industrial applications, there are numerous review papers, books (chapters) on CPs and their properties,55–60 an overview of different synthesis routes61,62 such as catalytic polymerization,33 electropolymerization63,64 or vapor-phase polymerization,65–68 along with their applications in different fields such as electrochemical energy storage devices or medical sensors.69 For the case of vapor-phase polymerization and oxidative chemical vapor deposition, previous reviews periodically detailed and updated the procedure of conjugated polymer synthesis, materials chemistry, the reaction mechanism, the properties of the polymers synthesized, and the structure–property relationships. These reviews significantly contributed to advancing the state-of-the-art. However, there is no systematic review outlining how oCVD (or other vapor-phase synthesis techniques) compares to the other solvent-based in different aspects of CP thin films including synthesis routes, polymer properties and processing aspects. The clarity in this case is of utmost importance as the synthesis method not only determines the possibility of using different synthesis routes but also vastly affects the properties of the final polymer.The remainder of this review critically addresses how these methods differ. First, a brief overview of each of the polymerization routes listed above is provided. Then, the most developed methodology in each category i.e., chemical synthesis (as a solvent-based method) and oxidative chemical vapor deposition (as a solvent-free method) is chosen for more comprehensive discussion and comparison. We will discuss in detail three sections: thin film, thin film–substrate interface, and substrate. It should be noted that each of these components is not independent of each other. The advantages and limitations of oCVD compared to solution processing are outlined following the reports in each field. It is worth mentioning that there are certain aspects of thin film deposition (on a substrate) that have not been directly addressed in oCVD research reports given that it is a relatively new field. For these aspects, a critical discussion of how oCVD may differ from solution processing is provided, analyzing the basic physics and chemistry of vapor-phase vs. solution-phase processing of polymers from the literature.
2. Solvent-based methods
a. Chemical polymerization
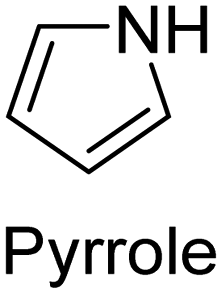
Chemical polymerization is the most developed technique for polymerization of conjugated polymers. It also serves as a platform for most of the other methods. Chemical polymerization broadly includes oxidation polymerization and organometallic polycondensation. In the former method, an oxidant is used to form a radical cation of the monomer. Then, two such cations form a C–C bond, resulting in a radical dication. This coupling continues till the formation of a conjugated polymer. An example for oxidation polymerization of polypyrrole is provided in Scheme 1.70,71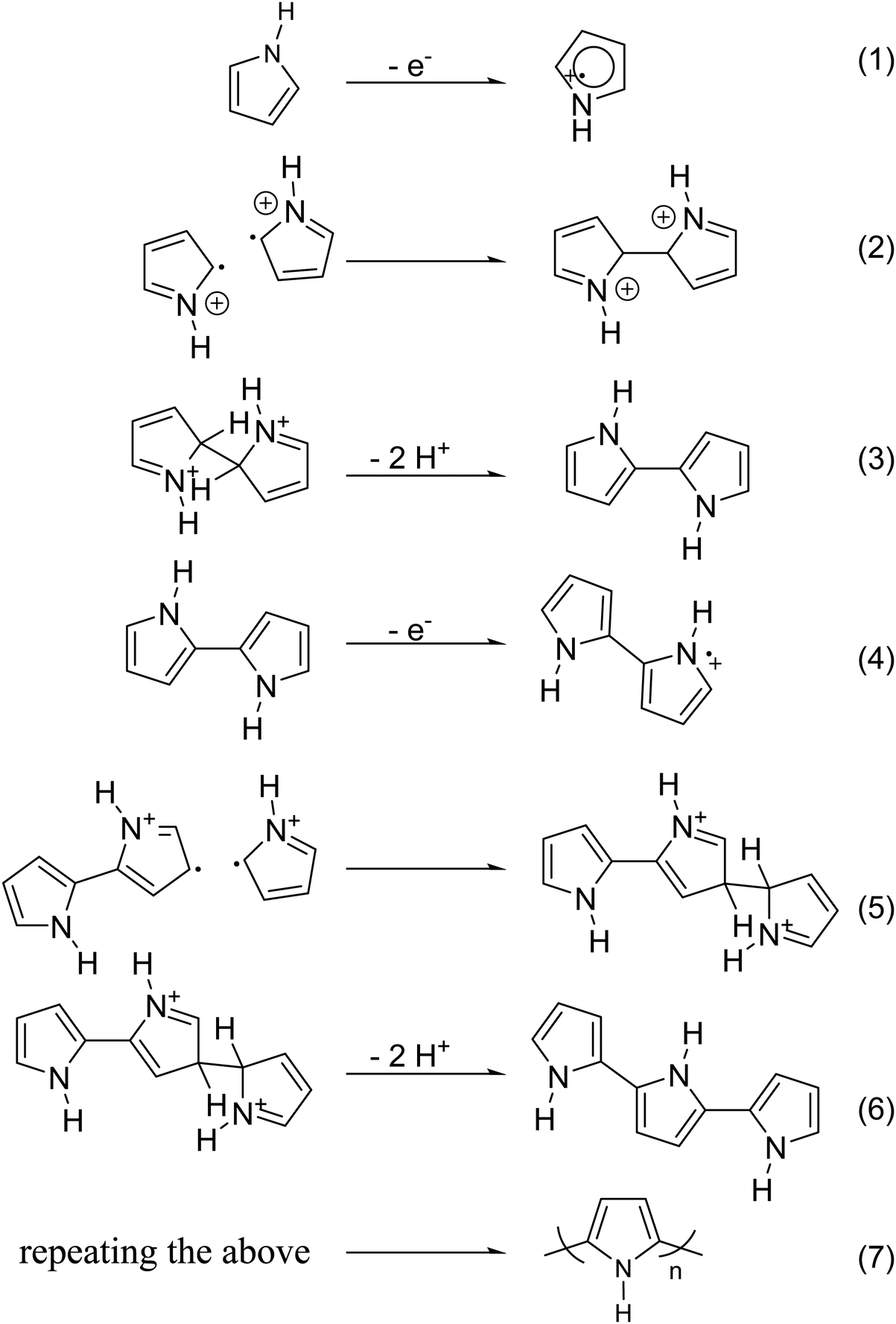 | ||
| Scheme 1 Oxidation polymerization of PPy, based on the reaction mechanism proposed by Diaz et al.79 | ||
Organometallic polycondensation proceeds where a bis-halogenated monomer undergoes C–C bonding via different coupling methods including Kumada, Suzuki–Miyaura, Stille, and Negishi polycondensation mechanisms.33,72,73Scheme 2 provides the general route of these reactions, details of which are beyond the scope of the current review and could be followed in the cited documents. For simplicity, from now on, we will use organometallic polycondensation as an umbrella term for all these methods.
 | ||
| Scheme 2 The general scheme for organometallic polycondensation for polymerization of CPs.33 | ||
b. Other synthesis routes
Electrochemical polymerization is another popular method for the synthesis of CPs. It uses an electrochemical cell consisting of a working electrode, a counter electrode, and a reference electrode dipped into a flask containing the monomer-dissolved in a solvent. The mechanism is similar to the oxidation polymerization explained above, albeit the oxidation is provided by the electrical current rather than an oxidant. In this method, a thin film of polymer is directly synthesized and formed on the surface of the working electrode, which has to be a conductive surface.74The next step is emulsion polymerization, wherein an organic monomer is first dispersed in an aqueous medium with the aid of a surfactant. An oxidant is then added to the solution, triggering the oxidation polymerization and resulting in (nano)particles of the CP dispersion.75
Finally, photo-induced polymerization is a comparatively newer method. Photons are used to initiate and proceed polymerization of a number of CPs. It is categorized into three different mechanistic pathways:28 (i) Oxidative radical polymerization, which was pioneered by Shimidzu et al., has been used for a range of electron-rich monomers such as thiophene. In this method, a solution containing a monomer, an electron acceptor (such as CCl4), and a salt (e.g. omnium salt) is irradiated with photons (of a certain wavelength), which triggers electron transfer from the monomer to the electron acceptor. This is followed by monomer–monomer coupling, deprotonation, and so forth to result in CP thin films.76–78 (ii) Cationic oxidative polymerization has been reported as a solvent-free polymerization method for a thienothiophene derivative. As the authors proposed, the C–Br bond of the bromine-modified thiophene derivative is broken as a result of photolysis. This releases elemental Br2 which initiates the polymerization by monomer addition to the cationic chain and elimination of HBr.80 (iii) Reductive polymerization that has been exploited for the polymerization of electron-deficient monomers such as benzotriazole and thienopyrazine. The synthesis mechanism was believed to proceed via chain growth-free radical polymerization. It was a solvent-based reaction using LiCl as a reductant and the reaction was initiated with light having a wavelength in the range of 400–626 nm.81
3. Solvent-free method: oxidative chemical vapor deposition (oCVD)
The general mechanism of all solvent-free methods is based on an initial oxidation of reactive monomers: the same as oxidation polymerization in the solution phase. For a long time, a common practice was to first oxidize the substrate (to be coated) with an oxidizing agent and then to expose the pre-treated substrate to vapors of monomers. In this way, the monomers should be first adsorbed on the surface of the substrate under reduced pressure and then activated by interactions with the oxidant present on the surface of the substrate. The method was named vapor-phase polymerization (VPP). It may be noted that although this method is frequently referred to as a solvent-free method in the literature, a solution of the oxidant is used for pre-treating the substrate.82 Another solvent-free method is plasma polymerization, which is expected to proceed similarly to oxidation polymerization. The difference is that instead of an oxidant, plasma is used for the initial activation of the monomer and subsequent cross-couplings where chemical structure and stoichiometry are not reliably controlled.83–94Finally, in the most developed solvent-free methodology, both the oxidant and the monomer are vaporized and metered into a gas-phase reactor under reduced pressure. They are then adsorbed on the surface of a substrate and initiate polymerization and film formation at the same time.35 This method was named oxidative chemical vapor deposition (oCVD) and is the subject of the current review.
Reactor design for oCVD
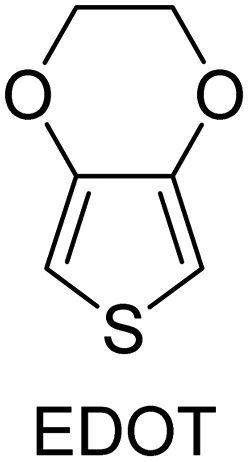
The early design for the oCVD reactor was inspired by the preceding works in the field of vapor phase material syntheses such as initiated chemical vapor deposition, metal–organic chemical vapor deposition, plasma-enhanced chemical vapor deposition, and atomic layer deposition.95–98 Building upon them as well as based on one pioneering work reported by Mohammadi et al.,34 a modified version of the oCVD reactor was developed in the group of K.K. Gleason at MIT and reported in 2006.35 The reactor consisted of a vacuum chamber with a stage at the bottom, the temperature of which could be controlled by water coolant. A crucible was placed under the chamber for vaporizing FeCl3, which could then be introduced into the reactor with argon (acting as a diluent) in the vertical direction and parallel to the EDOT flow. The monomer and the oxidant would be adsorbed on the reactor surface and oxidative polymerization would commence (Fig. 6(a)). Shortly after this primary design, the reactor configuration was changed by inversing the stage position under the ceiling and crucible at the bottom, in order to avoid substrate contamination with FeCl3 particulates.99 Also, the EDOT monomer flow was introduced into the chamber at a 90° angle relative to the oxidant flow direction (Fig. 6(b)). This reactor configuration is still in use up to today. However, other research groups again came up with reactor modifications. For example, the group of K.K. Lau changed the oxidant from FeCl3 (a solid) to SbCl5 (a liquid) and placed the substrate back at the bottom of the chamber (Fig. 6(c)) for deposition of polyaniline or polythiophene.100,101 An alternative reactor configuration exploited two crucibles inside the chamber allowing the vaporization of relatively high Mw monomer in addition to the oxidant crucible (Fig. 6(d)).102 However, this configuration used a smaller surface area of the substrate compared to the original one (26–52 cm2).30 Finally, in a more recent configuration and with inspiration from molecular layer deposition reactors, Andrew et al. adopted a hot-wall tubular shape reactor, in which the monomer and the oxidant would flow in the opposite direction and in parallel to the long-axis tubular direction (Fig. 6(e)).103 In this way, the monomer and oxidant mix in a controlled manner during their residence time above the substrate. This configuration has been used for both solid and liquid phase precursors, giving flexibility in reaction design.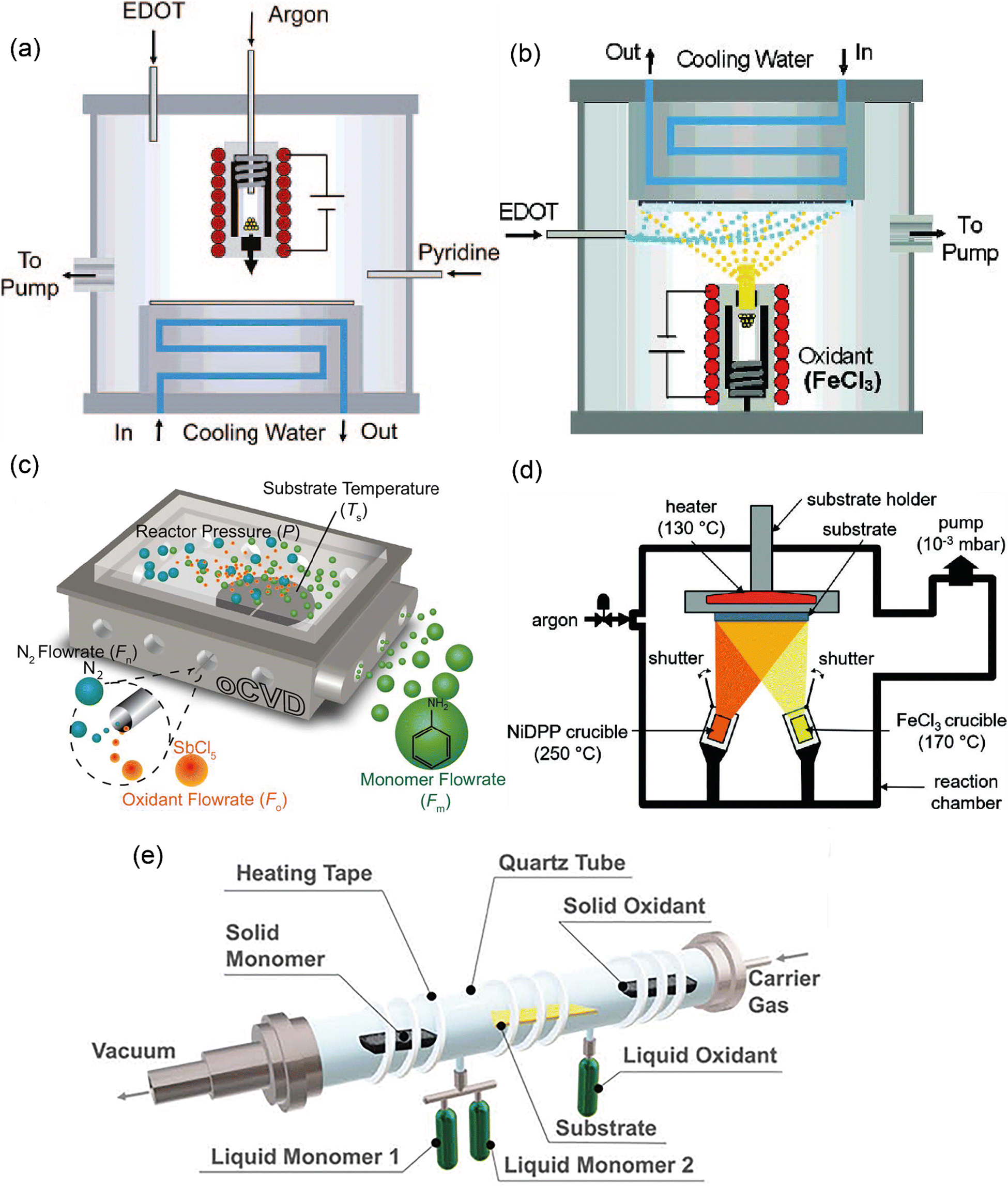 | ||
| Fig. 6 Different reactor configurations with (a) stage on the ceiling, with oxidant vaporizing by crucible at the bottom, and oxidant and monomer flow with 0° relatively. Reprinted with permission from.35 Copyright (2006) American Chemical Society. (b) Stage on the bottom, with oxidant vaporizing by crucible on the ceiling, and oxidant to monomer flow with 90°. Reprinted with permission from.99 Copyright (2007) American Chemical Society. (c) Stage on the bottom, with liquid oxidant and monomer flowing into the reactor at 90°;101 (d) with a substrate on the ceiling, and solid/oxidant vaporizing by crucible flowing at an angle <90°;106 (e) tubular reactor configuration with monomer/oxidant (solid/liquid) flowing into the reactor at 180°. Reproduced from ref. 30 with permission from The Royal Society of Chemistry. | ||
4. CP thin films
The first (and in a sense the most) important aspect of a thin CP film on a substrate is the properties of the film itself. Using the electronic property as the evaluation index, there are mainly five critical aspects that contribute to the thin film performance, namely (i) chemical structure, (ii) conjugation length, (iii) crystallinity, (iv) packing, and (v) doping. These aspects are not mutually exclusive as mentioned before. Below, each factor is discussed in more detail.a. Chemical structure
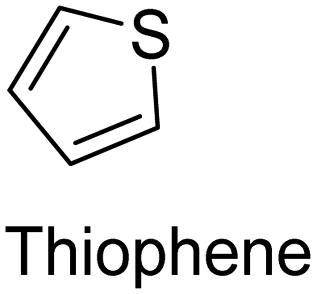
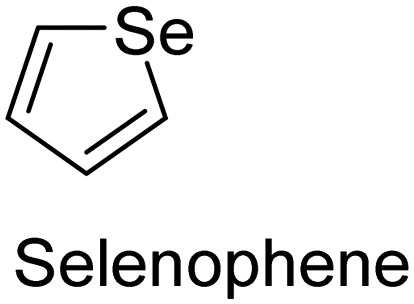
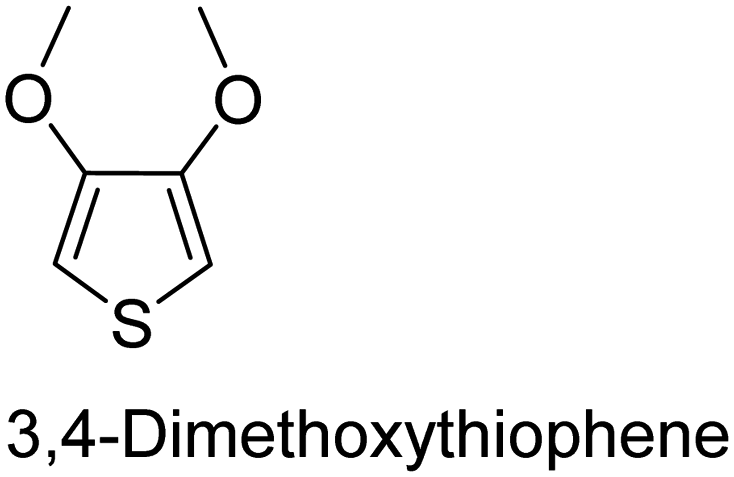
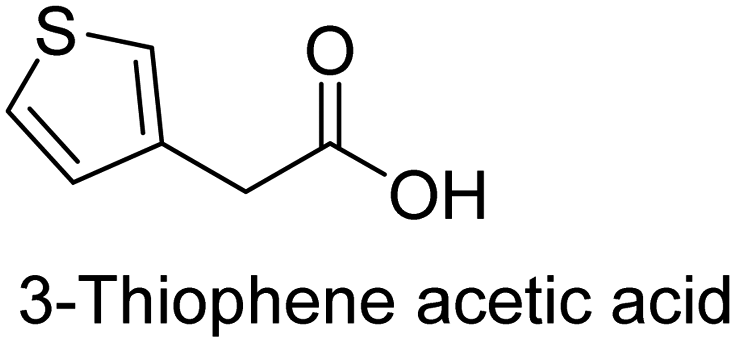
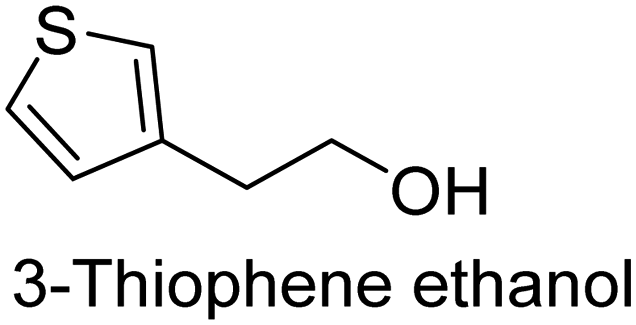
The nature of a CP itself primarily determines the electronic properties of the thin film. For example, polythiophenes and their derivatives usually show higher conductivities than polyaniline.104,105 Therefore, the first aspect to consider is to compare the range of monomers that can be polymerized by each method. Table 1 provides an overview of the monomers polymerized by oCVD reported until now along with the reported values (if applicable) of their electrical conductivities. In principle, oCVD replicates oxidation polymerization and should therefore be applicable to the various conjugated monomers polymerized by oxidative polymerization. It was also possible to polymerize 3-thiopheneethanol, a direct polymerization of which does not proceed either chemically or electrochemically.| Monomer | Oxidant used | Conductivity (S cm−1) | Ref. |
|---|---|---|---|
|
FeCl3, VOCl3, SbCl5 | 2.7 × 10−5–70 | 125–128 |
|
FeCl3, SbCl5 | 2–180 | 21,129,130 |
|
SbCl5 | NA | 101 |
|
FeCl3, Br2, CuCl2, SbCl5, VOCl3 | 9.1 × 10−4–6259 | 99,131–140 |
|
FeCl3 | 2–35 | 141,142 |
|
FeCl3 | 10 | 143 |
|
FeCl3, Br2 | 10−4–10 | 129,144,145 |
|
FeCl3 | Non-conductive | 107,146 |
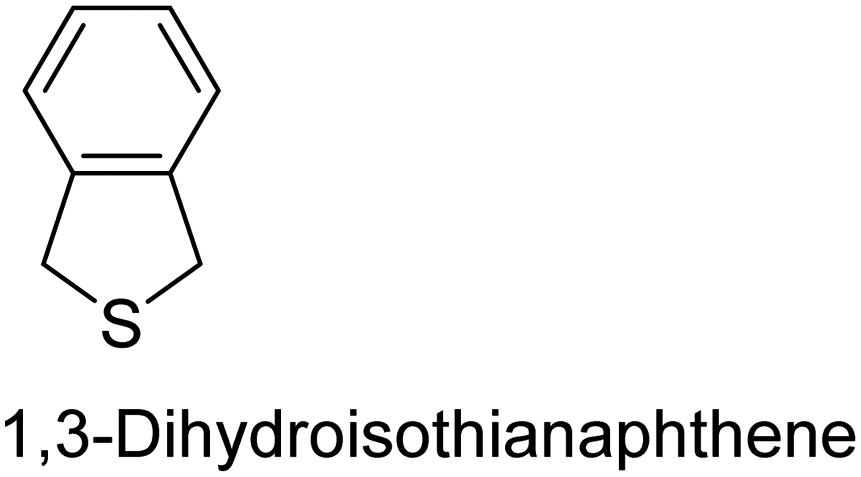
|
FeCl3 | 10−3–1 | 147 |
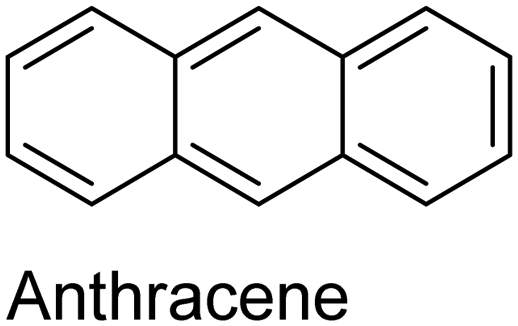
|
FeCl3 | NA | 148 |
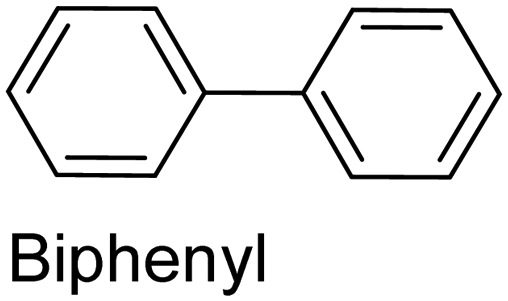
|
FeCl3 | NA | 148 |
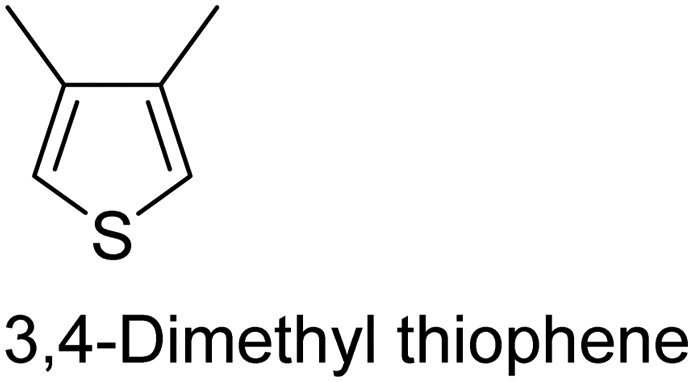
|
FeCl3 | NA | 103,149 |
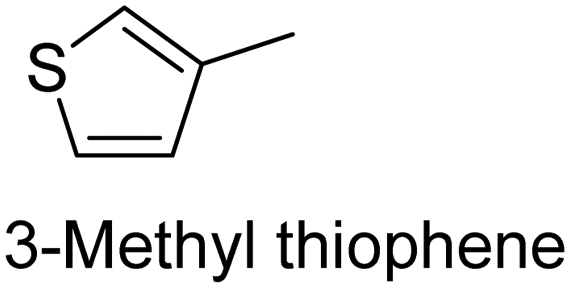
|
FeCl3 | NA | 103,149 |
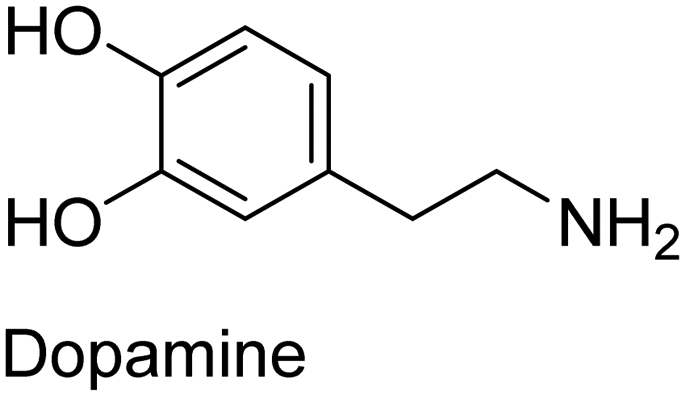
|
H2SO4/Na2SO4 | 0.4 | 150,151 |
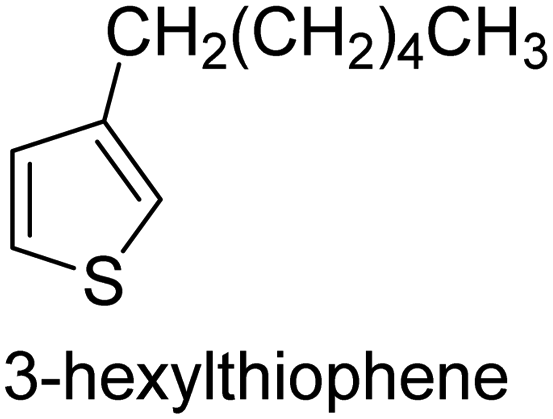
|
FeCl3 | 4 × 10−4–4 | 152 |
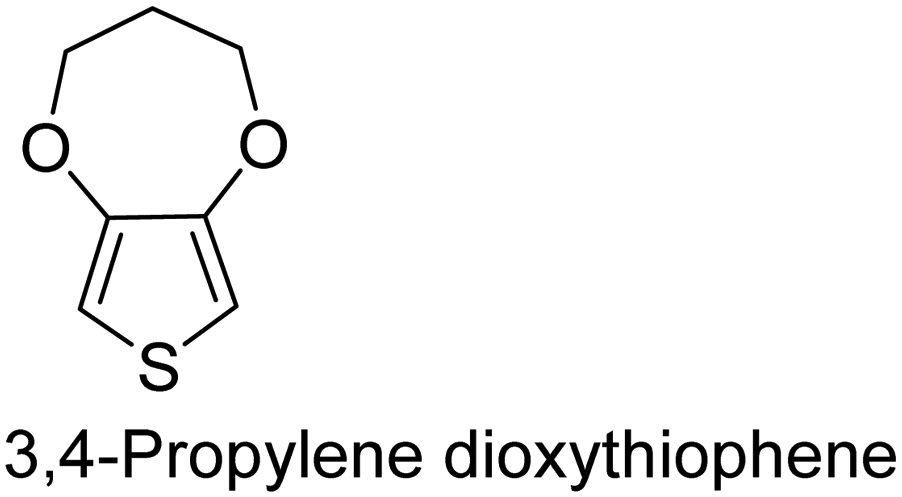
|
FeCl3 | 106 | 103,149 |
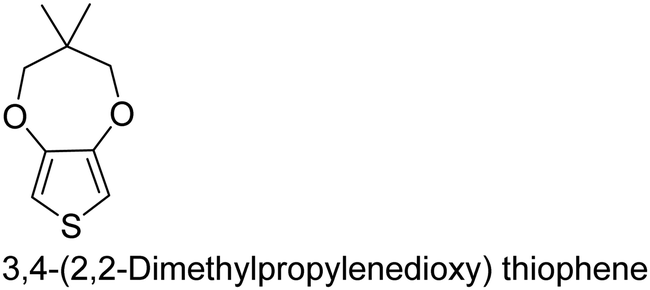
|
FeCl3 | 31 | 103,149 |
|
FeCl3 | 1.6 | 103,149 |
|
FeCl3 | NA | 103,149 |
|
FeCl3 | NA | 103,149 |
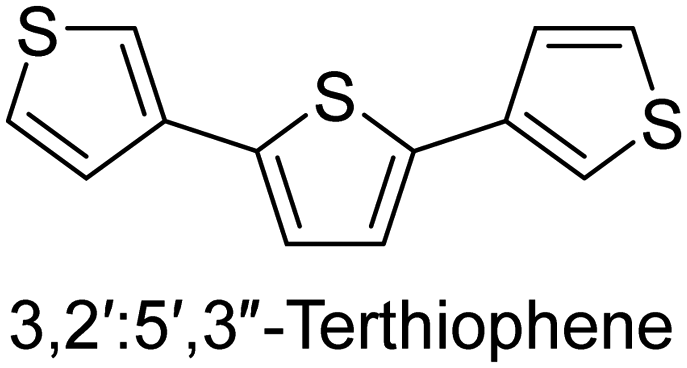
|
FeCl3 | NA | 103,149 |
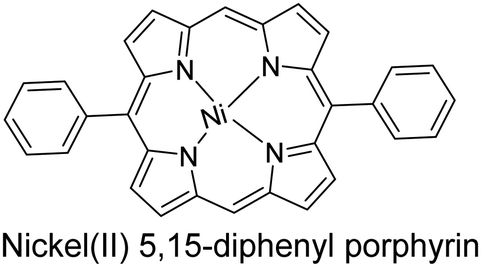
|
FeCl3, CuCl2, (Cu(ClO4)2·6H2O). FeCl3 | 3.6 × 10−2 | 102,106 |
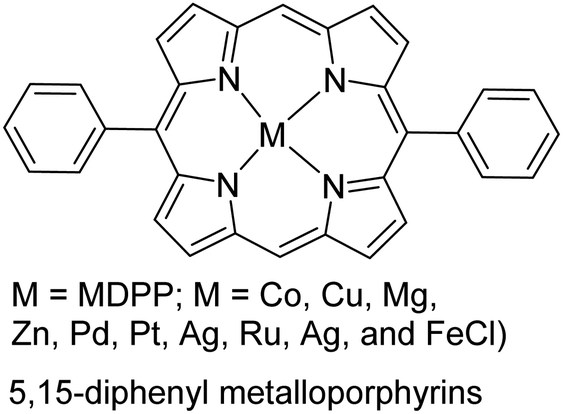
|
FeCl3 | NA | 114 |
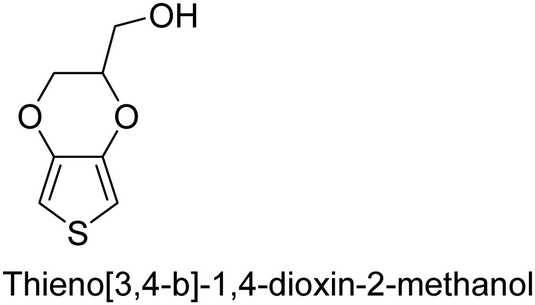
There is a need for an initial modification step to protect its hydroxyl group to avoid its nucleophilic attack in a polymerization environment. Although with oCVD, it was possible to directly copolymerize it with EDOT in a one-step oCVD without the need for protection of hydroxyl groups.107 Yet, important CPs like poly(p-phenylene) (PPP) or poly phenylene vinylene (PPV) are excluded from oxidative polymerization (therefore oCVD as well). However, it is worth mentioning that for the case of PPV, alternative CVD routes taking advantage of precursor pyrolysis have been done before, which could be a basis for other non-catalytic routes for the synthesis of CPs.108–111
With chemical methods and in particular with organometallic polycondensation, a wide range of monomers can be synthesized that is beyond the scope of oCVD. An immediate reason for this is the use of catalysts (in their solid form) which is not possible to implement in the current state-of-the-art vapor-based oCVD method. Another aspect is that organometallic polycondensation often involves multi-step reactions, while oCVD is a one-step process. Therefore, oCVD is not able to provide comparable control over the structure of CPs as it is a one-step, non-catalytic method.112,113
In addition to the different properties resulting from CPs synthesized via different polymerization methods, oxidation polymerization is more limited in terms of the variety of monomers that are possible to polymerize as compared to organometallic polycondensation.
One primary consideration of oCVD is the vapor pressure of monomers under deposition conditions. A monomer should have sufficiently high vapor pressure under the deposition temperature and pressure to be introduced into the reactor in the gas phase. This often becomes challenging as the monomer gets bulkier, which is associated with higher Mw, melting point, and boiling point. These non-volatile monomers tend to rapidly lose their kinetic energy upon entrance into the CVD chamber and condense, which restricts their further interaction with oxidants and polymerization.103 Despite this general challenge, the case for the deposition of porphyrin derivatives (with Mw > 400 g mol−1, melting point >350 °C) proved that with an accurate experimental design and the deposition conditions, it was possible to expand oCVD to heavier monomers.106 Cardenas-Morcoso et al. took porphyrin-based CP research to the next level by synthesizing a wide range of 5,15-diphenyl metalloporphyrins (MDPP; M = Co, Cu, Mg, Zn, Pd, Pt, Ag, Ru, Ag, and FeCl; see Table 1).114 This was particularly appealing due to the MDPP application for solar-based energy generation devices, but difficulty in its processing. Another strategy could be using a nitrogen bubbler to facilitate monomer vaporization and flow into the CVD reactor.115 One concern still remains that even with successful deposition of such monomers, the deposition rate could be possibly very low, the same bottleneck that atomic layer deposition faces.116
On the other hand, chemical synthesis and solution polymerization depend on the persistent solubility of the reactants as well as the growing polymeric chains in the reaction medium (solvent). However, alternating single and double bonds in conjugated chains of CPs are highly rigid, causing the chains to aggregate in the solvent rather than being dissolved. As a result, with the addition of each monomeric unit to a growing chain in a reaction medium, the tendency of the chain to precipitate out of the solvent becomes higher.117 This puts a limit on how large the chains could propagate before precipitation. As a solution to this challenge, conjugated monomers are modified with alkyl groups at the 3rd and/or 4th position. Such a limitation however is not applicable to oCVD, simply because it is a solvent-free method. Therefore, oCVD offers the possibility for direct synthesis of common conjugated monomers. This specific solvation issue becomes more relevant in processing and the final film properties are discussed further in the following sections.
In principle, a conjugated polymer could be oxidized or reduced by a dopant. As a result, the polymer turns into a polycation or polyanion, whose charges would be compensated by the counter ions from the dopant. The former is called p-type doping while the latter is called n-type doping of CPs. With this consideration, it could be hypothesized that any CP can be potentially p-doped or n-doped if strong enough oxidizing or reducing agents are used and/or by application of electrochemical methods. However, in practice, the most common CPs are more prone to oxidation. Their reduction, while studied theoretically and shown also in practice for a few of them, is challenging and might need very strong reducing agents like alkali metals e.g., sodium naphthanilide, sodium/potassium alloys, and lithium iodide.19,119,120 The main reason for this challenge lies in the electron-rich conjugated backbone of CPs in general. Nevertheless, due to the desired properties that electron-conducting CPs could offer, there has been enormous research on the modification of conjugated monomers to alter their orbital structure and make their backbone electron deficient, easing the process of n-doping. In this context, the CPS which readily tend to be oxidized rather than reduced have been frequently termed as p-type CPs, while the other modified CPs with electron gaining tendency have been called n-type CPs.121–124
Returning to the discussion on comparing the synthesis routes of CPs, as polymerization of n-type CPs often involves catalysts, it has been excluded from oCVD practice.121,153–155 Recently, Woods et al.156 reported a catalyst-free protocol for the synthesis of n-type CPs, although their precursors (e.g. lithium salt) are beyond the current scope of polymer CVD in general due to its extremely low vapor pressure at room temperature. Generally speaking, the electron-rich structure of common CPs (Fig. 3) needs to be modified with an electron withdrawing group in order to be able to accommodate an excess electron upon reduction. However, in oCVD, these-electron-rich structure is exactly the reason for the sp2–sp2 carbon coupling, making the process challenging. Nevertheless, Bilger et al.157 attempted to polymerize a thiophene–cyclopentadienone–thiophene triad in which the thiophenes were electronically decoupled from cyclopentadienone and were able to engage in C–C couplings upon exposure to another monomer in the presence of FeCl3 gas. This attempt of course opens new directions in the field of oCVD. However, there are some concerns that should be considered about this protocol in parallel. First, although ultraviolet photoelectron spectroscopy results suggested a low-lying valence band level (which is essential for n-type behavior in CPs), a donor–acceptor–donor (D–A–D) structure has not been considered among the best strategies for n-type monomer design (common strategies donor–acceptor (D–A) with strong A, D–A with weak D, A–D–A and A–A121). Second, if the donor has to be electronically decoupled from the acceptor in order to preserve its ability for C–C bonding, the acceptor can no longer apply its electron-withdrawing effect on the donor in order to provide the backbone with space for electron injection. Therefore, more investigation into this strategy is necessary to understand its potential.
b. Conjugation length
A charge carrier has a limited path to travel along the chain of a CP. Such a path is called the (effective) conjugation length, which is affected by Mw and the microstructure of the CP.158 When it comes to electron mobility along a polymeric chain as well as intermolecular charge transfer, with a more extended chain there is more space for the carrier to travel along the chain.159 On the other hand, the position at which monomers link to each other also impacts the electron mobility in the chain. This is called structural regularity and it could have three configurations as discussed later. Finally, there is the conformational regularity that impacts electron transfer in a single polymeric chain. For example, a chain twist could compromise carrier mobility as well.160,161 The combination of all these factors determines the effective conjugation length of CP polymeric chains.M w of a polymer determines how long the chains are and therefore directly influences the conjugation length.162,163 In solution synthesis, the growing polymeric chains need to continuously maintain their solvation in their reaction medium as additional units are added. However, as mentioned in the previous section, the rigid backbone of CPs in their basic chemical structure makes their solubility a challenge in the common organic solvents. Indeed, they tend to result in fine aggregates at best when so-called “good” solvents are used.27,164–166 Therefore, a growing chain of CP is highly susceptible to precipitate at oligomeric length when polymerizing in solution.167–171 To address this issue, researchers have came up with the idea to modify the monomer structure (e.g. adding an alkyl chain to the 3rd position) in order to enhance the solubility of the growing polymers in the reaction medium. This of course gives rise to partial distortion in the conjugation of the resultant CPs, negatively impacting their electronic properties.160,172,173 Another parameter influencing Mw is the CP crystallinity which would be discussed in the microstructure section.
As oCVD is a solvent-free method, the above concerns about solvation of unmodified conjugated monomer are not applicable. Therefore, oCVD might have the potential to grow CPs with higher Mw. Although via an indirect assessment, there have been reports revealing that oCVD provides control of conjugation length, because of the difficulty in estimating Mw of CPs in general, it was not possible to test the above hypothesis on oCVD potential to grow high Mw CPs. Yet, indirect methods showed how fine-tuning deposition conditions could increase the conjugation length.
In an early report, Im et al.99 observed that increasing the deposition temperature of PEDOT from 15 °C to 100 °C resulted in a continual decrease in the π–π transition energy measured by UV-vis spectroscopy. In another report, Goktas et al.107 copolymerized EDOT and 3-thiophene ethanol by oCVD and observed that the monomer feed ratio impacts the number of each unit in the final polymeric chains analyzed by AP-MALDI-HRMS spectroscopy; they attributed this observation to the conjugation length in part. In another research, PITN147 was synthesized by oCVD and it was observed that by increasing the deposition temperature from 70 °C to 130 °C, the band gap decreased from 1.14 eV to 1.05 eV. Additionally, the Raman and FTIR peaks downshifted continuously with increasing deposition temperature. These all were attributed to a higher conjugation length. Cheng et al.103 observed a higher oxidation potential (deeper HOMO) with reduced conjugation length. Smolin et al.174 synthesized PANI with oCVD and attributed the peak of 825 cm−1 to higher Mw PANI. However, in this case, it was observed that 1590 cm−1 and 1580 cm−1 peaks of quinonoid and benzenoid of aniline polymer shift to 1580 cm−1 and 1505 cm−1 for aniline oligomer.175
Borrelli et al.126 analyzed oCVD-grown unsubstituted PTs using Raman spectroscopy. As had been suggested before, the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C antisymmetric stretching vibration would lose intensity and downshift upon a higher conjugation length. Using this as the index, the research showed that the PT film deposited at 1 mTorr resulted in the shortest conjugation length. However, the other deposition pressures (75, 150, and 300 mTorr) all showed somewhat similar Raman (as well as UV-vis) spectra to each other. This suggests that pressure could be used to modulate the conjugation length to some degree.
C antisymmetric stretching vibration would lose intensity and downshift upon a higher conjugation length. Using this as the index, the research showed that the PT film deposited at 1 mTorr resulted in the shortest conjugation length. However, the other deposition pressures (75, 150, and 300 mTorr) all showed somewhat similar Raman (as well as UV-vis) spectra to each other. This suggests that pressure could be used to modulate the conjugation length to some degree.
Another parameter is the type of oxidant. Deposition of PEDOT with two different oxidants Br2 and FeCl3 revealed that bromine was able to result in a higher conjugation length, which was inferred from spectroscopy. This was attributed to a higher oxidizing strength of bromine compared to iron chloride.176
When it comes to structural regularity, organometallic polycondensation has been the method of choice for the synthesis of CPs, allowing 2,5 coupled polythiophenes.33 There could be three types of C–C linkages, which are named as tail–tail (TT) for 5,5 C–C coupling, head–tail (HT) for 2,5 C–C coupling, and head–head (HH) for 2,2 C–C coupling (Fig. 7(e)). HT coupling represents a regularity, control of which has not been possible by oxidation polymerization. The results showed that oxidation polymerization forms random 2,4 linkages, which undermines the effective conjugation length.177–182 Therefore, oCVD may not inherently be able to compete with chemical methods in forming regioregular CPs. However, there is an early report of the synthesis of regioregular polythiophenes with the controlled introduction of oxidants into the medium in multiple time spans, which may give a hint in oCVD processing as well.183 The microstructure of CPs is more extensively discussed in the next section.
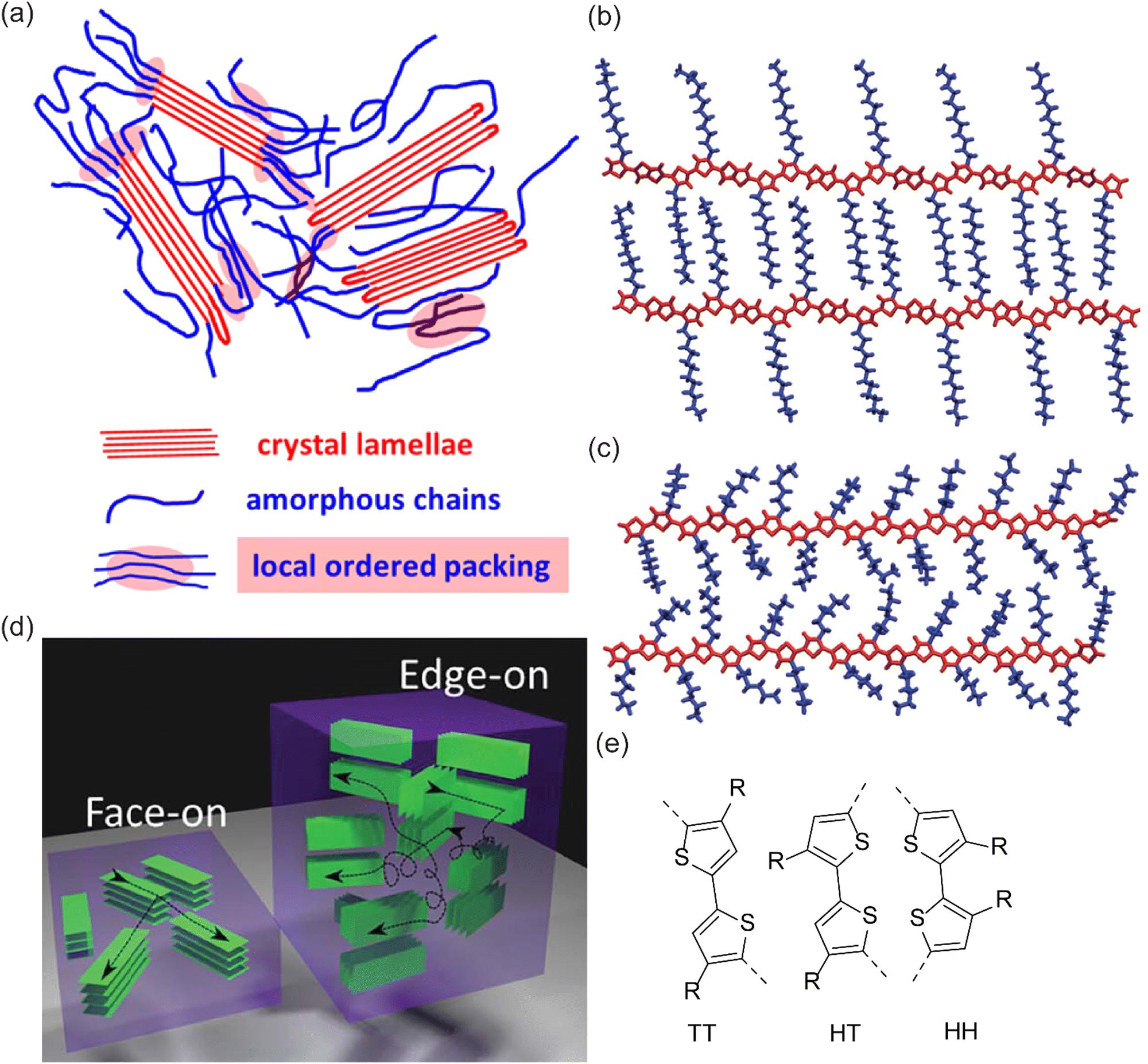 | ||
| Fig. 7 (a) An illustration of P3HT morphology comprising crystalline and amorphous phases. Reprinted with permission from.184 Copyright (2016) American Chemical Society; (b) and (c) thiophene-based CP with alkyl side chains that are interdigitated and randomly oriented, respectively. Reproduced from ref. 185 with permission from The Royal Society of Chemistry. (d) Illustration showing face-on vs. edge-on PEDOT orientation;186 (e) possible C–C bonding of two thiophene groups during polymerization of poly(thiophene) namely TT, HT, and HH. | ||
Regarding Mw of CPs synthesized by oCVD, one concern could be reactants “accessing” a polymerization spot of a growing chain, which might be undermined by the lower diffusion rate of the reactants to the potential chains embedded in the already-formed film. This phenomenon of diffusion into the polymer film is not yet understood.
c. Microstructure
As a polymeric chain has limited length, a carrier moving along the chain needs to hop between them at some point. This is called inter-molecular charge transfer. In this regard, how the polymeric chains are arranged relative to each other determines the polymer microstructure and electron mobility. This topic could be discussed in terms of crystallinity and thin film packing.From a structural perspective, Mw and the structure of the side chains can influence the tendency of the CPs for crystal nucleation and growth.192,194 For unmodified polymers of poly(arylene)s, crystallinity seemed to be associated with a lower Mw. As established before, poly(arylene)s – and so other CPs – are modified by side chain alkylation for better solubility. An enhanced solubility also allows for obtaining a higher Mw polymer during synthesis, which resulted in a less crystalline polymer.33 On the other hand, comparing alkyl-modified polymers within themselves, Zen et al. reported that the crystallinity of P3HT decreased significantly with lower Mw.194 Yet, an additional aspect regarding Mw would be the folding behavior. Again, in the case of P3HT, when Mn surpasses a critical value of 10 kDa, the chains will transit from the extended to the folded conformation, which requires the balancing of an energy barrier to unfold and form ordered crystals.59 Additionally, the structure of the alkyl-side chain impacts the crystallization tendency as well. If the alkyl structure is linear, resulting in a symmetrical CP structure, an improved crystallinity could be expected; while for branched side chains usually desired for better solubility, the crystallinity is undermined.192
After the synthesis of CPs, their solution is coated on the surface of a substrate, followed by solvent evaporation and film formation. Film morphology highly depends on the processing method and history. There are reports that the crystallinity could be controlled by the aid of solvent–polymer interaction60,191,195,196 and film annealing after deposition.197,198 The type of solvent and substrate surface features are the two main parameters affecting the crystallization of CPs. Sanda et al. used four different solvents for the processing 2,7-dioctyl[1]benzothieno[3,2-b][1]benzothiophene, and observed that a higher boiling point solvent resulted in larger polycrystalline domains.199 This is attributed to a lower evaporation rate of solvents with higher boiling points, allowing more time for crystals to grow. The same results have been noticed in other reports, suggesting a universal trend.191,195
Next, the substrate surface properties influence crystal nucleation and crystal growth. Physical and chemical properties of the surface including surface microstructure, roughness, and surface energy impact the interfacial interaction between substrate and solution. This controls the wetting behavior, the evaporation rate and the morphology development. In an interesting report, Zhang et al. showed that lowering the surface energy of the substrate by different functionalization groups, higher crystalline CP films were obtained. This was attributed to a lower penalty for heterogeneous nucleation of crystals.193 An additional aspect is the substrate's viscoelastic properties. In a report, Kim et al. vapor deposited pentacene film on the surface of a number of polymeric substrates at different temperatures. They observed that in case the temperature surpasses the glass transition temperature of the substrates, nucleation density is prompted yet its growth into larger grains is disrupted. However for rigid substrates (below Tg), an increase in temperature favors the formation of larger crystal grains.200 This could be understood in a broader context of interfacial dynamics between CPs solution and the substrate. In this regard, Diao et al. used a diketopyrrolopyrrole-based CP as a model compound and worked toward a general design rule for substrate-directed CP crystallization. They experimented with a template substrate modified with ionic liquids and studied how enthalpy of adsorption could be used as an index to quantify template–CP interaction. It was concluded that an increase in interaction at the interface is associated with a higher degree of crystallinity and larger domain size by the facilitation of CP nucleation via dynamic interaction.201
After solvent evaporation, an additional step of annealing has been shown to induce crystallization in certain cases.191,197,198 In another report, Jo et al. showed that exposing P3HT solution to a non-solvent, the π–π interaction of P3HT chains is encouraged by the unfavorable interaction of the polymer and its non-solvent, facilitating the crystallization of P3HT nanofibers.202
Another strategy has been using a template, which could assist the formation of crystalline domains during thin film processing.203 Through the so-called template-assisted crystallization, features of a surface including its roughness could stimulate crystallization by using a template, which favors the formation of a crystalline domain aligned with a pre-defined pattern.203 A well-known template in anodic aluminum oxide (AAO) with a channel size of 10 nm to 10 μm, which directs the CPs to be coated normally to the AAO pores.59,203
In oCVD, the deposition temperature has been used for crystallinity control.140 It was also observed that acid rinsing aids the broadening of the crystallographic signature and also intensifies it, which was attributed to a higher degree of crystallinity.136 The relationship between deposition temperature and post-processing to crystallinity remains to be understood. The crystallinity also depends on the regioregularity which could not be controlled by oCVD as mentioned before.
It was mentioned before that for the sake of solubility, long side chains – often alkyl – are introduced in the polymer structure. Generally speaking, such a modification undermines a close π–π stacking between planes of CP chains as a result of possible plane torsion. In this regard, for alkylated CPs, the best packing is obtained when these side chains are so-called interdigitated (Fig. 7(b)vs.Fig. 7(c)).205,206 The interdigitization tendency is favored for the case of linear alkyl chains rather than branched structures.160,192,207
Also, a related but different aspect of thin-film conductivity is their spatial conformation relative to an underlying substrate. This is due to the anisotropic nature of charge transfer in randomly oriented CP chains. An isotropic charge transfer of either in-plane or out-of-plane charge transfer might be preferred in different applications. For example, in the case of a field-effect transistor, since the direction of charge mobility is horizontal, an edge-on to the substrate (in-plane π stacking) is desired; while for OPVs with vertical charge mobility, a face-on CPs orientation (out-of-plane π stacking) is preferred.208 In this regard, research was done to shed light on the morphology control of CPs in order to stimulate the face-on and edge-on orientation of polymeric chains relative to the substrate (Fig. 7(d)).209
A relevant but different aspect is the coplanarity of CP chains: Chen et al. reported a systematic study of isoindigo-based polymers with increased polymer planarity by substituting thiophenes for phenyl rings and investigated how this structural modification impact polymer orientation toward silicon-based substrates. They concluded that in case the obtained polymer is soluble, interpolymer interaction is minimized and the van der Waals interaction between the polymer and the substrate is encouraged, favoring a face-on orientation of the synthesized CPs. However, for a less planar polymer (more chain twisting), the chains are more aggregated, resulting in favored edge-on orientation upon deposition.208 Mullen et al. copolymerized five different benzodithiophene isomers with alkylated dithiophene and observed that an increased degree of curvature (monomer angle between 106° and 180°) decreased ordering of chains and packing in their associated solid films.210 Consequently, although smaller curvature angles could prompt packing, the authors advise an intermediate level of curvature to preserve solubility as it was observed that molecules with the lowest degree of curvature were difficult to process. This indeed could be a potential opportunity for oCVD.210
The attempts for orientation control of CPs from the solution phase on a substrate have been followed in three different strategies:211
(i) By using shear forces including frictional transfer, mechanical rubbing, roller transfer, and strain alignment.
(ii) Fine-tuning of solution properties to prompt certain orientation: methods include the use of certain coating techniques such as dip coating, blade coating, solution shearing,212 wire-bar coating which have shown the ability to “guide” polymer chains for a certain orientation. Also, off-center spin coating, floating film-transfer method, and directional solvent evaporation could be included in this topic.
(iii) Substrate-directed CP ordering: for which epitaxial crystallization and dynamic template-directed orientation.
The details for each of these strategies and techniques are available in the literature59,60,211–213 and we do not discuss them here. The important point is except for the second strategy which exploits the possibilities with different solution processing methods to obtain the desired orientation, both the first and third strategies could be benefited by oCVD methodology. Yet, it is worth mentioning that not all of the above methods are easily translatable to scale-up and large-area processing. In addition, most of them are not applicable in multi-layer and device-relevant manufacturing as they will result in either chemical or mechanical damage to the other layers.211
There is less information in oCVD on CP and their packing control. A well-studied case is PEDOT deposition using FeCl3, which showed a transition from edge-on to face-on orientation when the deposition temperature was increased from a lower temperature to 300 °C. The oCVD-grown PEDOT with face-on orientation in this case was associated with a record conductivity above 6000 S cm−1. With the motivation of lowering the deposition temperature, the same research group reported that by using VOCl3 as an oxidant, it was possible to obtain pure face-on orientation at a deposition temperature as low as 140 °C. However, in the latter case, the conductivity was lower (2800 S cm−1).140,214 From a post-processing point of view, PEDOT film rinsing with HBr showed to improve packing.136
d. Doping (and oxidants)
Doping is the process that offers the conjugated polymers the conductivity they are known for. It is achieved by the incorporation of a small quantity of a chemical that lowers the resistivity of a CP. There could be three types of dopants, namely: inorganic, organic, and polymeric.19Most of the inorganic compounds used as dopants for CPs are gaseous molecules (e.g., halogens, oxygen), metals (e.g., Li, Na, Mg), Lewis acids (e.g. FeCl3, SbCl5, VOCl3), and inorganic acids (e.g. HCl, H2SO4). Although the oxidizing nature of a dopant determines doping efficiency, an important consideration regarding inorganic dopants is that they are the smallest among different types of doping compounds. This results in a higher mobility of their counter ions within a polymeric network and consequently higher conductivity.19 In solution processing, a challenge for inorganic dopants has been their processability with common solvents. CPs are hydrophobic in nature and inorganic dopants are hydrophilic, making it difficult to find a common solvent.
To overcome the solubility limitation of inorganic dopants, one approach was using organic dopants which are hydrophobic as well. It could also help with better environmental stability of CPs oxidation in an ambient environment. However, they have comparably bigger counter ions which restrict their mobility in CPs and lower the conductivity. Some examples of organic dopants include sulfonate derivatives of anthraquinone or naphthalene, acetic, citric, oxalic, and tartaric acid, and copper phthalocyanine. Obviously, polymeric dopants offer the biggest size of compounds in the above classification, though the most stable one. Examples are polyaniline doped with polyacrylic acid and PEDOT doped with PSS.19
Another perspective is how the interaction between the dopant and CP impacts its microstructure. Ma et al. reported that by using an organic molecule (N-DMBI) for doping of n-type CP (FDBPPV), better carrier mobility was achieved by enhancing molecular packing.215 Another example is PEDOT with preferential edge-on orientation when doped with PSS, Tos, or OTf,216–220 while favoring face-on orientation when doped with smaller counter ions (like chloride Cl−).52,140
Although there is no limitation in exploiting different doping agents in the oCVD method, it generally was not a center of attention and only a fraction of the dopants are used for synthesis in practice. It should be noted that in oCVD, the same oxidant that is used for synthesis would remain in the polymer structure and serve as a dopant. Possibly a post-processing step could be added to improve the properties with the aid of a second oxidizing agent, as reported in the case of PEDOT.140
On the other hand, it is safe to conclude that the solvent incompatibility for inorganic dopants with CPs is resolved by oCVD. Along with the superior performance of inorganic dopants, this might be the reason that FeCl365 has been widely used as a dopant in oCVD of CPs. Other dopants used are as follows: Br2,176,221 CuCl2,102,222 VOCl3,125,223 SbCl5,125,174,223,224 MOCl5225 and H2SO4.150
5. Polymer–substrate interface
An important aspect of surface coating is how the deposited coating interacts with the underlying substrate. Such an interaction not only contributes to the functionality of the top layer but is also important for the long-term stability of the coating. Below, we review the most relevant aspects of coating–substrate interfacial properties from the viewpoint of CPs and oCVD.a. Uniformity
Uniformity is a critical aspect of coating CPs on a surface. The processing history of the coating along with the polymer–substrate interaction determines the thin film uniformity of a specific polymer–substrate pair. CVD methods in general are well-recognized to result in outstanding uniformity compared to solution-processed thin film coatings.67 The reasons could be understood in a number of limitations that solution coating techniques suffer when compared to solvent-free techniques, which of course may be different for each specific wet method. For example, drop casting, while being an easy method, is negatively influenced by the “coffee ring effect”. When the solvent evaporation rate is higher at the edge of the droplets compared with its center, it results in a non-uniform film.226,227 Spin coating on the other hand has been able to give highly uniform films compared to drop-casting or other wet-processing techniques and as a result has often been the method of choice.228,229 However, during the drying stage, the solution gradually becomes more viscous and the coating enters a so-called shrinkage stage. This sometimes results in the formation of meniscus and film thinning at the side walls. On the other hand, spin coating fails to result in high-ordered conjugated thin films as the solvent usually evaporates too fast and does not allow enough relaxation time to the polymeric chain ordering. To partially compensate for this problem, high boiling point solvents could be used.205 However, this could end in residual solvents trapping in the coating microstructure, up to 20%.230 It has been reported that this could be mostly resolved by exposing the film to high temperatures (annealing).231 Yet even with annealing, the residual solvent negatively affects the film properties and this may be exacerbated by using high boiling point chlorinated solvents, which are not a favorable choice for the environment.199b. Conformality
Conformality refers to how a thin film homogeneously covers or “shrink wraps” on a surface. A surface could be qualitatively categorized into smooth and rough. Rough in here stands for nano/micro-structured surfaces which have a texture and/or 3D structure. Two modes of coverage are conformal and planar.232Conformal coverage features a film with uniform thickness resembling the geometry of the underneath substrate. This type of coverage is challenging for solution processing primarily because of surface tension effects.233,234 This is particularly a limitation for textured surfaces due to dewetting.235 Solution processing may result in surface bridges inhibiting the liquid to penetrate the porous three-dimensional features of the geometry.232 For example, spin coating as a highly exploited method, is controlled by two driving forces when coating a surface: centrifugal and capillary.236 In case a geometry has 3D features, centrifugal force tries to “push” the solution into the 3D network and make the film conformal; while the capillary forces want to make the flow level. In the beginning, the centrifugal forces are the main driving force; however, as the solvent evaporates and the solution becomes more viscous, the capillary forces dominate.236
CVD on the other hand is widely known to result in conformal coverage on different surfaces including porous 3D surfaces.125,232 This is the result of a number of factors. First, from a mass transport point of view, vapors have several orders of magnitude higher diffusion coefficients than liquids. This means that in the case of a 3D geometry, vapors could better diffuse into the surface sites which are not easily accessible for liquids. Additionally, the surface adsorption of gaseous molecules is much lower than the solution in a single “collision” with the substrate. This results in a slower morphology development and as a result higher relaxation time of the polymer chains that favors conformal coverage. Finally, in CVD practice in general, the tendency of any surface spot to be coated would gradually decrease upon film formation. This feature is called “self-limiting surface reaction” and contributes to conformality, which could be imagined as the opposite of capillary levelling in solution processing.233Fig. 8(a) shows a porous textile that is coated with PEDOT:PSS solution using spin coating, resulting in poor uniformity and conformality, as well as coated substrate using oCVD resulted in highly uniform and conformal PEDOT films.103 In the same manner, Fig. 8(b) and (c) shows uncoated and PPy-coated microporous carbon felt.237
 | ||
| Fig. 8 (a) Corduroy swatch with 2 in2 surface coated with PEDOT: PSS solution (left) and with 4 in2 surface deposited with PEDOT via oCVD; reproduced from ref. 103 with permission from The Royal Society of Chemistry. (b) and (c) Microporous carbon felt uncoated and coated with PPy via oCVD, respectively.237 | ||
c. Interfacial adhesion
As a practical aspect, it is necessary for a CP film to firmly adhere to the underlying substrate during the lifespan of the relevant application. Here, adhesive strength is the work required to separate a CP film from its substrate.238 Therefore, the bonding strength between the film and the substrate determines the force of adhesion, the failure of which is a result of extrinsic and/or intrinsic stresses.239 Extrinsic stresses, by definition, arise from external sources such as UV irradiation, chemical oxidation, mechanical stress etc. On the other hand, intrinsic stresses are a result of the interfacial features of any substrate-thin film pairs which are determined by the nature of thin film/substrate on the one hand and the history of thin film formation on the other hand, which are all affected by surface wetting behavior, thermodynamic work of adhesion, and interfacial tension.238,239 These intrinsic aspects could be further affected by extrinsic stresses as well. Relevant discussion in this review is how oCVD differs from solution processing at the film–substrate interfacial adhesion from the intrinsic point of view. To discuss this, we briefly discuss about physical or chemical bonding.Next comes the interfacial tension between a CP film and a substrate. The adhesive force is maximum when the difference between the surface free energy of the film and the substrate (called interfacial tension) is minimum.238 This surface tension is formed during the coating process, the control of which is crucial for adhesion. Therefore, a CP film on polymeric substrate results in a lower surface tension than a CP/metal pair. This aspect is the same for both oCVD and solution coating. However, for any two layers, the process by which they come into contact will determine the level of internal stress. This will be the source of interfacial failure upon exposure to external stresses over time. For example, if there is mechanical compressive stress, the film is susceptible to delamination or cracking in case of tensile stress.239,242,243
During solution processing, the solvent evaporates, and the polymeric film has to shrink. However, the polymer at the solid–liquid interface is already physically adhered to the substrate. As a result, it could not “flow” in parallel with the volume change.
Therefore, polymer chains are locked in configurations that are thermodynamically unfavorable,244–246 which causes internal stress in the plane of the coating.247 Additionally, the physical mismatch between a polymer and a potential metallic substrate prompts interfacial failure during time as a result of different responses to external stresses.248
In polymer CVD practice, there is no report addressing the phenomenon of internal stress. However, from the inorganic CVD process, it is well known that internal stresses would form as a result of non-equilibrium deposition conditions.249 The mechanism is such that the first adsorbed molecules on a substrate occupy the spots that do not possess the lowest energy, which is then trapped by the new incoming molecules resulting in internal stress. Such a process could be partially controlled by the deposition rate.239 At the time of writing this review, a systematic study comparing the degree of internal stress during polymer oCVD and solution processing is missing. Therefore, further comparison is not possible.
In solution processing, different surface-initiated polycondensation methods have been exploited to prepare well-defined conjugated polymer brushes. This is a bottom-up approach that results in CP grafting from a substrate.252 The other method is when an already developed polymer chain is grafted to a surface. In an example, the cyclopentadiene–maleimide Diels–Alder ligation was used to graft poly(3-hexylthiophene) to a pre-treated silicon substrate.253
In oCVD, inspired by the work of Kovacic,254 it was observed that when a substrate has aromatic groups in its structure, EDOT makes a chemical bond with the substrate, following the proposed mechanism (Scheme 3).255 In another report, a simple and straightforward procedure, (3-aminopropyl) trimethoxy silane129 was attached to a silicon wafer forming Si–O–Si, and the molecule was used as a linker to PPy. Using the same principles, linker molecules such as 4-aminothiophenol, methyl 4-mercaptobenzoate, or 4-mercaptobenzoic acid were used as a linker molecule to graft poly(EDOT-co-3-TE) and poly(EDOT-co-TAA) grown by oCVD.145 These methods could be compared with a grafting-to approach in solution processing. Unlike solution processing, the grafting-from approach has not been replicated in oCVD yet.
 | ||
| Scheme 3 The proposed mechanism of PEDOT grafting to a substrate having aromatic groups in its structure.255 | ||
6. Substrates
The wide variety of substrates that could be used in CVD processing is probably one of the most outstanding features it offers. Discussion on substrates may be conducted in three directions, namely (i) the substrate surface geometry, (ii) the substrate material and (iii) processing concerns.As a solvent-free procedure, oCVD provides the opportunity for thin film deposition of CPs on a diverse range of substrate materials and surface geometries. Examples are the reports by Wang et al.145, describing a methanol sensor development using printed circuit boards as the substrate and coated with poly(3,4-ethylenedioxythiopheneco-thiophene-3-aceticacid) [poly(EDOT-co-TAA)] and/or poly((3,4-ehylenedioxythiophene-co-thiophene-3-ethanol) ethanol) [poly(EDOT-co-3TE)]. Smolin et al. used micro/mesoporous carbide-derived carbon (CDC) and porous Mo2C CDC as substrates and coated them with PANI for energy storage applications.174,224 In another example, Nejati et al. deposited PANI onto nanostructured AAO, titanium oxide, and activated carbon and used the composite for energy storage applications with enhanced properties in performance. Their result showed that the ultrathin coating of PANI could adhere to the underlying nanostructure surface, which ensures a high surface area that is crucial for the specific application.125 Furthermore, the possibility of coating an ultrathin layer in the order of 4 nm could significantly add to the long-term stability of the modified electrode.125,258 Exploiting the possibility to coat a textured substrate with nano/meso/micro features with a CP and preserve its geometry has shown to be a versatile approach for improved performance across a wide range of applications. For example, Kim et al. deposited 20 nm of PEDOT on mesoporous ITO and validated it as a Pt-free cathode for application in dye-sensitized solar cells (DSSCs) with a performance similar to a Pt-based cathode.259 In another example Howden et al. grew PEDOT by oCVD on a range of macro/nano-scale textured surfaces and demonstrated that this procedure increased light absorption in photovoltaics by different light trapping mechanisms.260 Conformal PEDOT layer of 8 to 17 nm on vertically aligned carbon nanotube (VA-CNT) showed a 10-fold increased sensitivity for volatile organic compound (VOC) sensing application.134 Depositing of 10 nm of PEDOT on VA-CNT showed also showed a 10-fold increase in pseudocapacitance when the hybrid material was used as a supercapacitive electrode.133
The oCVD method is generally categorized as a substrate-independent method due to the possibility of using a diverse range of materials and surface features, as mentioned above.66,141–143 This substrate-independence of oCVD is especially beneficial in a wide range of CPs applications, where multiple layers are successively deposited. In these cases, the interaction of solvent with all the layers underneath becomes relevant. In this regard, one important concern is that the step of depositing a subsequent layer should not damage the pre-existing layer. This is achieved by choosing orthogonal solvents in device fabrication using wet processing.261
On merit of oCVD, PEDOT was deposited on both sides of a porous paper and the composite material was tested for its supercapacitive performance and flexible energy storage application.138 The same strategy was used to fabricate flexible paper-based organic photovoltaics.131 Also, a convenient wearable electronic device could be fabrics/textiles due to their widespread use in daily life. Yet such materials are susceptible to deformation and damage when in contact with solvents.262 On the other hand, oCVD proved to be a feasible method for depositing conjugated polymers for different applications for sensitive substrates.263 Andrew et al. reported employing different types of textiles as substrates for deposition of conjugated polymers across different applications such as supercapacitors, optoelectronic, transistors, and sensors.157,264–267 The same research group also reported depositing PProDOT on microtextured living plants for health monitoring applications, which takes research in this area to the next level.268 A list of substrates reported in the literature for oCVD is provided in Table 2 along with their proposed applications.
| Substrate | Applications | Polymer | Ref. |
|---|---|---|---|
| Cotton towel/corduroy fabric | Wearable electronics | P(DMT) | 103,149 |
| Printed circuit boards | Methanol sensor | Poly(EDOT-co-TAA) | 145 |
| Electro-spun mat | Chemiresistive biosensor | Poly(EDOT-co-3TE) | 146 |
| Porous CDC/Mo2C CDC | Energy storage | PANI | 174,224 |
| Unmodified paper | Organic photovoltaic | PEDOT | 131 |
| Graphene | Organic solar cell | PEDOT | 132 |
| Aligned carbon nanotube | Energy storage, sensor | PEDOT | 133,134 |
| n-doped ZnO layer | Organic optoelectronic | PEDOT, PPy | 135,269 |
| Cellulose filter paper/porous nylon membrane | Supercapacitor | PEDOT | 138 |
| Sponge-type carbon felt | Electrocatalytic system | Polydopamine | 150 |
| Porous AAO/TiO2/activated carbon | Energy storage | PT | 125 |
| Microporous polyurethane fibers | Piezoresistive sensor | PPy | 44 |
Despite the benefits oCVD provides in substrate choice, it is recognized that in one case oCVD is not substrate-independent and that is when the oxidant reacts with the substrate. In some of these cases, surface cationic species are formed which could provide the opportunity for covalent linking and hence improve the interfacial adhesion of the coating.65 Yet, given that strong oxidants are used in synthesis of conjugated polymers, the possibility for surface degradation of vulnerable substrates such as polymers could not be excluded. In fact, the interaction of metal halides including FeCl3 (as the most common oxidant so far) with a number of polymers such as nylon or PVC is well documented decades ago.270,271 It is necessary for future research on oCVD to take into account any possible substrate deterioration during synthesis especially for more fragile materials including polymers, a topic that is mostly overlooked until now.
7. Industrialization and scale-up
As a milestone in oCVD development, an initial scale-up attempt was reported in 2015.272 PEDOT was successfully deposited on larger than usual areas of ∼20 × 30 cm2 textile/PET foil for the first time. The oCVD process was adopted in the roll-to-roll (R2R) processing procedure considering its industrial relevance (Fig. 9(a)). Oxidant and monomer are vaporized and directed onto the surface in a vertical manner with an angle of 0°. The rolling helps with uniformity of thin film deposition on the substrate. Uniformity of PEDOT characteristics was monitored by the change in film conductivity on different areas of ∼ an A4-sized PET foil. The result showed a standard deviation of up to 10% in conductivity values (Fig. 9(b)).272 The above result provides an initial indication of the oCVD success for scaling up.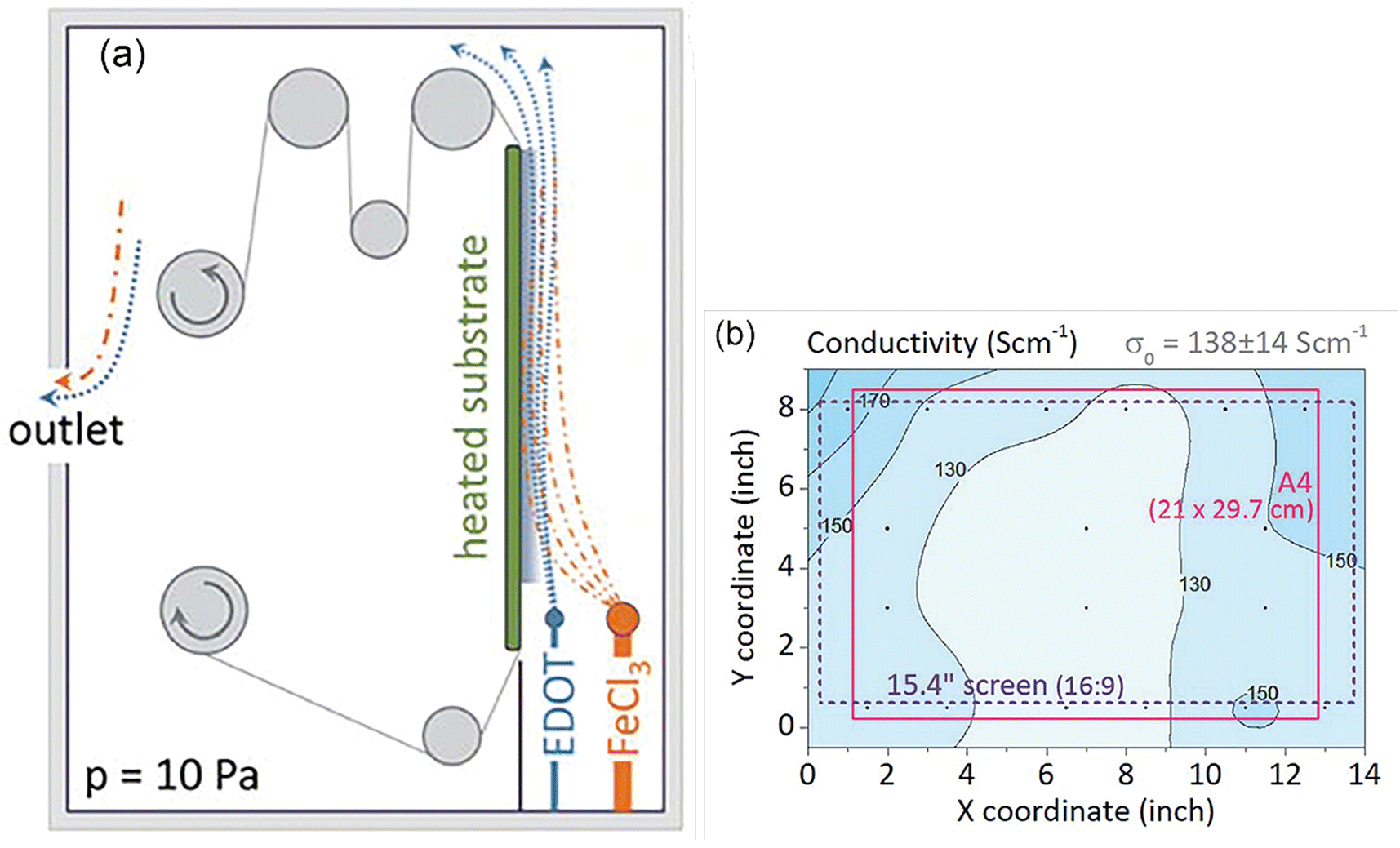 | ||
| Fig. 9 (a) An illustration of the R2R oCVD reactor constructed in MIT; (b) PEDOT conductivity distribution deposited in an area of ∼20 cm × 30 cm at a deposition temperature of 100 °C. Reproduced from ref. 272 with permission from The Royal Society of Chemistry. | ||
However, there are certain aspects that are not yet understood. One primary aspect is batch-to-batch variation between the synthesized polymers using oCVD, which is a critical aspect for the commercialization of any chemical process in terms of process reliability.273,274 This aspect becomes particularly pressing considering that as a step-growth polymerization technique, oxidative polymerization is inherently not a controlled procedure, giving rise to batch-to-batch variance.275 To address this in the field of CPs, controlled chain-growth polymerization has been proposed which could successfully control the polymerization kinetics. These reactions again get the advantage of catalysts in their synthesis.276,277 In this regard, it looks absolutely necessary to work toward catalytic chemical vapor deposition reactions of CPs, possibly by getting inspiration from the field of inorganic semiconductors. An immediate idea could be the deposition of a thin catalyst layer on a substrate to enhance the chemical environment and further exploit the layer as a substrate.278
Another aspect worth noting is recording and ultimately modelling the deposition rate of CPs grown by oCVD. Within the chemical vapor deposition community, it is well known and documented that relative concentration of precursors, deposition temperature, and vacuum pressure could also be used to fine-tune film growth temperature.279,280 Also, the fluid dynamics of the reaction environment which is associated with reactor size and configuration as well as flow channels all play a role. For industrial adaptation, it is crucial to record, quantify and model these aspects. Such an endeavor has been followed in the inorganic CVD field (which has long been adopted in industry) and to a good extent for initiated chemical vapor deposition of polymers.281,282 Yet, it is apparent that for oCVD, this has yet to be developed. For this purpose, a good start could be a systematic inclusion of deposition rate data along with reactor configurational features. Reviewing the literature, it could be concluded that such data exists for some reports, yet is not included in many reports.
8. Future perspective and concluding remarks
The oCVD process is reviewed and compared with solution-based techniques for synthesis and film formation of conjugated polymers. As a synthesis method, oCVD offers the possibility of direct polymerization of the most common conjugated monomers without any additional step to modify the monomer. Yet, compared to solution-based techniques, it is limited in terms of monomer versatility as it does not have the freedom of using a catalyst to facilitate the polymerization of certain conjugated monomers as well as the challenges arising from the inherent vapor pressure properties of certain monomers. Solvent-based techniques on the other hand accommodate more diverse synthesis routes, which makes it possible to control the structure–property relationship of CPs to a greater extent. Solvent-based techniques also offer bulk synthesis of CPs as well as the diversity in their chemical structure, which might be at the cost of cumbersome synthesis routes.In terms of processing and thin film formation, oCVD generally outperforms solvent-based coating techniques as it offers the possibility of producing highly uniform, conformal, and robust CP films with good control over thickness and over a diverse range of substrates (planar or with a 3D texture). On the other hand, solvent-based coating methods offer versatility, economic feasibility, and technological maturity, which give rise to their ease of adaptation in industrial settings. Yet solvent-based techniques face challenges when precision coating on non-planar and textured surfaces is required, combined with the inherent insolubility issue related to conjugated polymer chemistry.
Future research should be aimed at widening the “monomer library” of the oCVD method, which might become possible by the adaptation of new polymerization mechanisms with the aid of catalysts or photo-induced processes, details of which were introduced in the current review. Also, attempts should be made to overcome synthesis challenges arising from low vapor pressure of certain conjugated monomers. This could be possibly by use of a new generation of high-temperature oCVD reactors, derived from inorganic CVD practice. Additionally, investigation of thin film properties in terms of microstructure could significantly contribute to the film performance of the CPs produced by oCVD and pave the way for their adaptation for device fabrication.
Conflicts of interest
The authors have no conflicts to declare.Notes and references
- J. Mort, Science, 1979, 1980(208), 819–825 Search PubMed.
- F. Lincoln Vogel, Synth. Met., 1979, 1, 1 CrossRef.
- https://www.oxfordreference.com/ .
- E. P. T. Kerja, Conducting Polymers: A New Era in Electrochemistry, 1967, vol. 13 Search PubMed.
- M. Kraft, S. Adamczyk, A. Polywka, K. Zilberberg, C. Weijtens, J. Meyer, P. Görrn, T. Riedl and U. Scherf, ACS Appl. Mater. Interfaces, 2014, 6, 11758–11765 CrossRef CAS PubMed.
- N. Casado and D. Mecerreyes, RSC Polym. Chem. Ser., 2021, 1–26 CAS.
- R. Schroot, M. Jäger and U. S. Schubert, Chem. Soc. Rev., 2017, 46, 2754–2798 RSC.
- F. Szillat, B. V. K. J. Schmidt, A. Hubert, C. Barner-Kowollik and H. Ritter, Macromol. Rapid Commun., 2014, 35, 1293–1300 CrossRef CAS PubMed.
- G. Inzelt, Conducting Polymers A New Era in Electrochemistry, Springer-Verlag, Berlin Heidelberg, 2008 Search PubMed.
- J. Kim, J. H. Kim and K. Ariga, Joule, 2017, 1, 739–768 CrossRef CAS.
- D. A. Buttry and F. C. Anson, J. Electroanal. Chem., 1981, 130, 333–338 CrossRef CAS.
- O. Inganläs, Chem. Soc. Rev., 2010, 39, 2633–2642 RSC.
- Y. Nagasaki, Polym. J., 2018, 50, 821–836 CrossRef CAS.
- A. Dianatdar, O. Akin, I. Mongatti, J. Momand, G. Ruggeri, F. Picchioni and R. K. Bose, RSC Adv., 2021, 11, 35187–35196 RSC.
- A. McNaught and A. D. Wilkinson, The IUPAC Compendium of Chemical Terminology, International Union of Pure and Applied Chemistry (IUPAC), Research Triangle Park, NC, 2019 Search PubMed.
- H. Shirakawa, E. J. Louis, A. G. MacDiarmid, C. K. Chiang and A. J. Heeger, J. Chem. Soc., Chem. Commun., 1977, 578–580 RSC.
- Editorial, Nat. Mater., 2020, 19, 921 CrossRef PubMed.
- W. Deits, P. Cukor, M. Rubner and H. Jopson, Synth. Met., 1982, 4(3), 199–210 CrossRef CAS.
- P. Kar, Doping in Conjugated Polymers, John Wiley & Sons, Inc., Scrivener Publishing LLC, Hoboken, New Jersey, Salem, Massachusetts, 2013 Search PubMed.
- A. J. Heeger, Chem. Soc. Rev., 2010, 39, 2354–2371 RSC.
- A. Dianatdar, M. Miola, O. De Luca, P. Rudolf, F. Picchioni and R. Bose, J. Mater. Chem. C, 2022, 10, 557–570 RSC.
- J. Heinze, Electronically conducting polymers, in Electrochemistry IV. Topics in Current Chemistry, ed. E. Steckhan, 1990, vol. 152, pp.1–47 Search PubMed.
- J. R. Skotheim, T. A. Elsenbaumer and R. L. Reynolds, Handbook of Conducting Polymers, Marcel Dekker, Inc, New York, NY, USA, 1998 Search PubMed.
- T. Onggar, I. Kruppke and C. Cherif, Polymers, 2020, 12, 1–46 CrossRef PubMed.
- R. S. Potember, R. C. Hoffman, H. S. Hu, J. E. Cocchiaro, C. A. Viands, R. A. Murphy and T. O. Poehler, Polymer, 1987, 28, 574–580 CrossRef CAS.
- T. Yokozawa and Y. Ohta, in Semiconducting Polymers: Controlled Synthesis and Microstructure, ed. C. Luscombe, The Royal Society of Chemistry, 2016, pp.1–37 Search PubMed.
- J. Pecher and S. Mecking, Chem. Rev., 2010, 110, 6260–6279 CrossRef CAS PubMed.
- E. F. Woods, A. J. Berl and J. A. Kalow, ChemPhotoChem, 2021, 5, 4–11 CrossRef CAS.
- R. K. Sadhir and K. F. Schoch, Thin Solid Films, 1993, 223, 154–160 CrossRef CAS.
- D. Bilger, S. Z. Homayounfar and T. L. Andrew, J. Mater. Chem. C, 2019, 7, 7159–7174 RSC.
- M. Wan, Conducting Polymers with Micro or Nanometer Structure, Tsinghua University Press, Berlin, Beijing, 2008 Search PubMed.
- J. M. D’Arcy, H. D. Tran, V. C. Tung, A. K. Tucker-Schwartz, R. P. Wong, Y. Yang and R. B. Kaner, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 19673–19678 CrossRef PubMed.
- T. Yamamoto, T. Yokozawa, I. Osaka, R. D. McCullough, J. Liu, J. W. Y. Lam, B. Z. Tang, Y. Morisaki, Y. Chujo, M. Takase, M. Iyoda, A. Nagai, P. W. Siu, D. P. Gates, K. Naka, A. Wild, A. Winter, M. D. Hager, U. S. Schubert and K. Akagi, Conjugated Polymer Synthesis: Methods and Reactions, WILEY-VCH Verlag & Co. KGaA, Weinheim, Germany, 2010 Search PubMed.
- A. Mohammadi, M. Hasan, B. Liedberg and I. Lundstrom, Synth. Met., 1986, 14, 189–197 CrossRef CAS.
- J. P. Lock, S. G. Im and K. K. Gleason, Macromolecules, 2006, 39, 5326–5329 CrossRef CAS.
- L. Dou, Y. Liu, Z. Hong, G. Li and Y. Yang, Chem. Rev., 2015, 115, 12633–12665 CrossRef CAS PubMed.
- O. Ostroverkhova, Chem. Rev., 2016, 116, 13279–13412 CrossRef CAS PubMed.
- J. F. Mike and J. L. Lutkenhaus, J. Polym. Sci., Part B: Polym. Phys., 2013, 51, 468–480 CrossRef CAS.
- A. C. Grimsdale, K. L. Chan, R. E. Martin, P. G. Jokisz and A. B. Holmes, Chem. Rev., 2009, 109, 897–1091 CrossRef CAS PubMed.
- S. Ghosh and R. N. Basu, Conjugated Polym. Nanostruct. Energy Convers. Storage Appl., 2021, 207–232 CAS.
- T. Nezakati, A. Seifalian, A. Tan and A. M. Seifalian, Chem. Rev., 2018, 118, 6766–6843 CrossRef CAS PubMed.
- C. Zhu, L. Liu, Q. Yang, F. Lv and S. Wang, Chem. Rev., 2012, 112, 4687–4735 CrossRef CAS PubMed.
- E. Smela, Adv. Mater., 2003, 15, 481–494 CrossRef CAS.
- A. Mukherjee, A. Dianatdar, M. Z. Gładysz, H. Hemmatpour, M. Hendriksen, P. Rudolf, M. K. Włodarczyk-Biegun, M. Kamperman, A. G. Prakash Kottapalli and R. K. Bose, ACS Appl. Mater. Interfaces, 2023, 15, 31899–31916 CrossRef CAS PubMed.
- M. F. De Riccardis, Fundam. Conjugated Polym. Blends, Copolym. Compos., 2015, 519–579 CAS.
- P. P. Deshpande, N. G. Jadhav, V. J. Gelling and D. Sazou, J. Coat. Technol. Res., 2014, 11, 473–494 CrossRef CAS.
- M. Ates, J. Adhes. Sci. Technol., 2016, 30, 1510–1536 CrossRef CAS.
- L. Allison, S. Hoxie and T. L. Andrew, Chem. Commun., 2017, 53, 7182–7193 RSC.
- M. Sairam, S. K. Nataraj, T. M. Aminabhavi, S. Roy and C. D. Madhusoodana, Sep. Purif. Rev., 2006, 35, 249–283 CrossRef CAS.
- P. Lindemann, M. Tsotsalas, S. Shishatskiy, V. Abetz, P. Krolla-Sidenstein, C. Azucena, L. Monnereau, A. Beyer, A. Gölzhäuser, V. Mugnaini, H. Gliemann, S. Bräse and C. Wöll, Chem. Mater., 2014, 26, 7189–7193 CrossRef CAS.
- G. Zhang, Z. A. Lan and X. Wang, Angew. Chem., Int. Ed., 2016, 55, 15712–15727 CrossRef PubMed.
- M. Heydari Gharahcheshmeh and K. K. Gleason, Mater. Today Adv., 2020, 8, 100086 CrossRef.
- M. Armand, Adv. Mater., 1990, 2, 278–286 CrossRef CAS.
- J. A. Shetzline and S. E. Creager, J. Electrochem. Soc., 2014, 161, H917–H923 CrossRef CAS.
- Z. Qiu, B. A. G. Hammer and K. Müllen, Prog. Polym. Sci., 2020, 100, 101179 CrossRef CAS.
- M. H. Harun, E. Saion, A. Kassim, N. Yahya and E. Mahmud, Sensors, 2007, 2, 63–68 Search PubMed.
- T. H. Le, Y. Kim and H. Yoon, Polymers, 2017, 9, 150 CrossRef PubMed.
- M. G. Sumdani, M. R. Islam, A. N. A. Yahaya and S. I. Safie, Polym. Eng. Sci., 2022, 62, 269–303 CrossRef CAS.
- L. Zhai, S. I. Khondaker, J. Thomas, C. Shen and M. McInnis, Nano Today, 2014, 9, 705–721 CrossRef CAS.
- M. Chang, G. T. Lim, B. Park and E. Reichmanis, Polymers, 2017, 9, 23–31 CrossRef.
- C. I. Awuzie, Mater. Today: Proc., 2017, 4, 5721–5726 CrossRef.
- K. Namsheer and C. S. Rout, RSC Adv., 2021, 11, 5659–5697 RSC.
- R. J. Waltman and J. Bargon, Can. J. Chem., 1986, 64, 76–95 CrossRef CAS.
- J. Heinze, B. A. Frontana-Uribe and S. Ludwigs, Chem. Rev., 2010, 110, 4724–4771 CrossRef CAS PubMed.
- M. Heydari Gharahcheshmeh and K. K. Gleason, Adv. Mater. Interfaces, 2019, 6, 1–27 CAS.
- A. M. Coclite, R. M. Howden, D. C. Borrelli, C. D. Petruczok, R. Yang, J. L. Yagüe, A. Ugur, N. Chen, S. Lee, W. J. Jo, A. Liu, X. Wang and K. K. Gleason, Adv. Mater., 2013, 25, 5392–5423 CrossRef CAS PubMed.
- K. K. Gleason, Nat. Rev. Phys., 2020, 2, 347–364 CrossRef CAS.
- D. Bilger, S. Z. Homayounfar and T. L. Andrew, J. Mater. Chem. C, 2019, 7, 7159–7174 RSC.
- T. K. Das and S. Prusty, Polym.-Plast. Technol. Eng., 2012, 51, 1487–1500 CrossRef CAS.
- Y. Tan and K. Ghandi, Synth. Met., 2013, 175, 183–191 CrossRef CAS.
- E. M. Genies, G. Bidan and A. F. Diaz, J. Electroanal. Chem., 1983, 149, 101–113 CrossRef CAS.
- T. Yamamoto, J. Organomet. Chem., 2002, 653, 195–199 CrossRef CAS.
- T. Yamamoto, Macromol. Rapid Commun., 2002, 23, 583–6006 CrossRef CAS.
- S. Sadki, P. Schottland, N. Brodie and G. Sabouraud, Chem. Soc. Rev., 2000, 29, 283–293 RSC.
- T. L. Kelly and M. O. Wolf, Chem. Soc. Rev., 2010, 39, 1526–1535 RSC.
- T. Iyoda, M. Kitano and T. Shimidzu, J. Chem. Soc., Chem. Commun., 1991, 1618–1619 RSC.
- Y. Yagci, F. Yilmaz, S. Kiralp and L. Toppare, Macromol. Chem. Phys., 2005, 206, 1178–1182 CrossRef CAS.
- Y. Yagci, S. Jockusch and N. J. Turro, Macromolecules, 2007, 40, 4481–4485 CrossRef CAS.
- E. M. Genies, G. Bidan and A. F. Diaz, J. Electroanal. Chem., 1983, 149, 101–113 CrossRef CAS.
- X. Liu, V. Sharapov, Z. Zhang, F. Wiser, M. A. Awais and L. Yu, J. Mater. Chem. C, 2020, 8, 7026–7033 RSC.
- E. F. Woods, A. J. Berl and J. A. Kalow, Angew. Chem., Int. Ed., 2020, 59, 6062–6067 CrossRef CAS PubMed.
- D. Bhattacharyya, R. M. Howden, D. C. Borrelli and K. K. Gleason, J. Polym. Sci., Part B: Polym. Phys., 2012, 50, 1329–1351 CrossRef CAS.
- L. M. H. Groenewoud, A. E. Weinbeck, G. H. M. Engbers and J. Feijen, Synth. Met., 2002, 126, 143–149 CrossRef CAS.
- L. Martin, J. Esteve and S. Borrós, Thin Solid Films, 2004, 451–452, 74–80 CrossRef CAS.
- C. Nastase, F. Nastase, A. Dumitru, M. Ionescu and I. Stamatin, Composites, Part A, 2005, 36, 481–485 CrossRef.
- X. Y. Zhao, M. Z. Wang and J. Xiao, Eur. Polym. J., 2006, 42, 2161–2167 CrossRef CAS.
- X. Y. Zhao, M. Zhu Wang, Z. Wang and B. Zhu Zhang, Thin Solid Films, 2008, 516, 8272–8277 CrossRef CAS.
- P. Yang, J. Zhang and Y. Guo, Appl. Surf. Sci., 2009, 255, 6924–6929 CrossRef CAS.
- T. Barman and A. R. Pal, Appl. Surf. Sci., 2012, 259, 691–697 CrossRef CAS.
- M. Rajabi, A. R. Ghassami, M. Abbasi Firouzjah, S. I. Hosseini and B. Shokri, Plasma Chem. Plasma Process., 2013, 33, 817–826 CrossRef CAS.
- C. S. Park, D. H. Kim, B. J. Shin, D. Y. Kim, H. K. Lee and H. S. Tae, Materials, 2016, 9, 1–11 Search PubMed.
- C. S. Park, E. Y. Jung, D. H. Kim, D. Y. Kim, H. K. Lee, B. J. Shin, D. H. Lee and H. S. Tae, Materials, 2017, 10, 1272 CrossRef PubMed.
- S. Kim, J. S. Oh, T. Hwang, H. W. Seo, D. C. Jeong, J. H. Lee, L. Wen, C. Song, J. G. Han and J. Do Nam, Macromol. Res., 2019, 243–249 CrossRef CAS.
- M. N. Subramaniam, P. S. Goh, W. J. Lau, A. F. Ismail, M. Gürsoy and M. Karaman, Appl. Surf. Sci., 2019, 484, 740–750 CrossRef CAS.
- P. D. Dapkus, Annu. Rev. Mater. Sci., 1982, 12, 243–269 CrossRef CAS.
- M. Meyyappan, J. Phys. D: Appl. Phys., 2009, 42, 213001 CrossRef.
- S. M. George, Chem. Rev., 2010, 110, 111–131 CrossRef CAS PubMed.
- S. J. Yu, K. Pak, M. J. Kwak, M. Joo, B. J. Kim, M. S. Oh, J. Baek, H. Park, G. Choi, D. H. Kim, J. Choi, Y. Choi, J. Shin, H. Moon, E. Lee and S. G. Im, Adv. Eng. Mater., 2018, 20(3), 1700622 CrossRef.
- S. G. Im and K. K. Gleason, Macromolecules, 2007, 40, 6552–6556 CrossRef CAS.
- S. Nejati and K. K. S. Lau, Langmuir, 2011, 27, 15223–15229 CrossRef CAS PubMed.
- Y. Y. Smolin, M. Soroush and K. K. S. Lau, Beilstein J. Nanotechnol., 2017, 8, 1266–1276 CrossRef CAS PubMed.
- K. Baba, G. Bengasi, D. El Assad, P. Grysan, E. Lentzen, K. Heinze, G. Frache and N. D. Boscher, Eur. J. Org. Chem., 2019, 2368–2375 CrossRef CAS.
- N. Cheng, L. Zhang, J. Joon Kim and T. L. Andrew, J. Mater. Chem. C, 2017, 5, 5787–5796 RSC.
- G. Kaur, R. Adhikari, P. Cass, M. Bown and P. Gunatillake, RSC Adv., 2015, 5, 37553–37567 RSC.
- D. Kumar and R. C. Sharma, Eur. Polym. J., 1998, 34, 1053–1060 CrossRef CAS.
- G. Bengasi, K. Baba, G. Frache, J. Desport, P. Gratia, K. Heinze and N. D. Boscher, Angew. Chem., Int. Ed., 2019, 58, 2103–2108 CrossRef CAS PubMed.
- H. Goktas, X. Wang, N. D. Boscher, S. Torosian and K. K. Gleason, J. Mater. Chem. C, 2016, 4, 3403–3414 RSC.
- O. Schäfer, A. Greiner, J. Pommerehne, W. Guss, H. Vestweber, H. Y. Tak, H. Bässler, C. Schmidt, G. Lüssem, B. Schartel, V. Stümpflen, J. H. Wendorff, S. Spiegel, C. Möller and H. W. Spiess, Synth. Met., 1996, 82, 1–9 CrossRef.
- K. M. Vaeth and K. F. Jensen, Macromolecules, 1998, 31, 6789–6793 CrossRef CAS.
- K. M. Vaeth and K. F. Jensen, Appl. Phys. Lett., 1997, 71, 2091–2093 CrossRef CAS.
- K. Kim, M. Y. Jung, G. L. Zhong, J. Il Jin, T. Y. Kim and D. J. Ahn, Synth. Met., 2004, 144, 7–11 CrossRef CAS.
- T. Yamamoto and T. Aki Koizumi, Polymer, 2007, 48, 5449–5472 CrossRef CAS.
- T. Yamamoto, Bull. Chem. Soc. Jpn., 2010, 83, 431–455 CrossRef CAS.
- D. Cardenas-Morcoso, E. Vey, M. Heiderscheid, G. Frache and N. D. Boscher, J. Mater. Chem. C, 2022, 10, 2194–2204 RSC.
- J. Xu and K. K. Gleason, ACS Appl. Mater. Interfaces, 2011, 3, 2410–2416 CrossRef CAS PubMed.
- R. W. Johnson, A. Hultqvist and S. F. Bent, Mater. Today, 2014, 17, 236–246 CrossRef CAS.
- T. Q. Nguyen, R. Y. Yee and B. J. Schwartz, J. Photochem. Photobiol., A, 2001, 144, 21–30 CrossRef CAS.
- Mezrlow, An Automated Irrigation System Using Arduino Microcontroller, 2011, 1908, 2–6.
- R. Borjas and D. A. Buttry, Chem. Mater., 1991, 3, 872–878 CrossRef CAS.
- W. J. M. J. S. R. Jayasundara and G. Schreckenbach, J. Phys. Chem. C, 2020, 124, 17528–17537 CrossRef CAS.
- Y. Sui, Y. Deng, T. Du, Y. Shi and Y. Geng, Mater. Chem. Front., 2019, 3, 1932–1951 RSC.
- M. Goel, C. D. Heinrich, G. Krauss and M. Thelakkat, Macromol. Rapid Commun., 2019, 40, 1–31 CrossRef PubMed.
- Z. Li, C. C. Chueh and A. K. Y. Jen, Prog. Polym. Sci., 2019, 99, 101175 CrossRef CAS.
- H. Jia and T. Lei, J. Mater. Chem. C, 2019, 7, 12809–12821 RSC.
- S. Nejati, T. E. Minford, Y. Y. Smolin and K. K. S. Lau, ACS Nano, 2014, 8, 5413–5422 CrossRef CAS PubMed.
- D. C. Borrelli, S. Lee and K. K. Gleason, J. Mater. Chem. C, 2014, 2, 7223–7231 RSC.
- S. Lee, D. C. Borrelli and K. K. Gleason, Org. Electron., 2016, 33, 253–262 CrossRef CAS.
- S. Lee, D. C. Borrelli, W. J. Jo, A. S. Reed and K. K. Gleason, Adv. Mater. Interfaces, 2018, 5, 1–8 Search PubMed.
- S. Vaddiraju, K. Seneca and K. K. Gleason, Adv. Funct. Mater., 2008, 18, 1929–1938 CrossRef CAS.
- A. Castro-Carranza, J. C. Nolasco, S. Bley, M. Rückmann, F. Meierhofer, L. Mädler, T. Voss and J. Gutowski, J. Polym. Sci., Part B: Polym. Phys., 2016, 54, 1537–1544 CrossRef CAS.
- M. C. Barr, J. A. Rowehl, R. R. Lunt, J. Xu, A. Wang, C. M. Boyce, S. G. Im, V. Bulović and K. K. Gleason, Adv. Mater., 2011, 23, 3500–3505 CrossRef CAS PubMed.
- H. Park, R. M. Howden, M. C. Barr, V. Bulović, K. Gleason and J. Kong, ACS Nano, 2012, 6, 6370–6377 CrossRef CAS PubMed.
- N. Lachman, H. Xu, Y. Zhou, M. Ghaffari, M. Lin, D. Bhattacharyya, A. Ugur, K. K. Gleason, Q. M. Zhang and B. L. Wardle, Adv. Mater. Interfaces, 2014, 1, 1–6 Search PubMed.
- X. Wang, A. Ugur, H. Goktas, N. Chen, M. Wang, N. Lachman, E. Kalfon-Cohen, W. Fang, B. L. Wardle and K. K. Gleason, ACS Sens., 2016, 1, 374–383 CrossRef CAS.
- S. Bley, M. Rückmann, A. Castro-Carranza, F. Meierhofer, L. Mädler, T. Voss and J. Gutowski, Phys. Status Solidi C, 2016, 13, 614–617 CrossRef CAS.
- R. M. Howden, E. D. McVay and K. K. Gleason, J. Mater. Chem. A, 2013, 1, 1334–1340 RSC.
- J. P. Lock, J. L. Lutkenhaus, N. S. Zacharia, S. G. Im, P. T. Hammond and K. K. Gleason, Synth. Met., 2007, 157, 894–898 CrossRef CAS.
- A. Liu, P. Kovacik, N. Peard, W. Tian, H. Goktas, J. Lau, B. Dunn and K. K. Gleason, Adv. Mater., 2017, 29, 1606091 CrossRef PubMed.
- E. F. G. Meysam Heydari Gharahcheshmeh, M. T. Robinson and K. K. Gleason, Adv. Funct. Mater., 2021, 31, 2008712 CrossRef.
- X. Wang, X. Zhang, L. Sun, D. Lee, S. Lee, M. Wang, J. Zhao, Y. Shao-Horn, M. Dinca, T. Palacios and K. K. Gleason, Sci. Adv., 2018, 4, 5780 CrossRef PubMed.
- D. Bhattacharyya and K. K. Gleason, J. Mater. Chem., 2012, 22, 405–410 RSC.
- W. J. Jo, D. C. Borrelli, V. Bulović and K. K. Gleason, Org. Electron., 2015, 26, 55–60 CrossRef CAS.
- W. J. Jo, J. T. Nelson, S. Chang, V. Bulović, S. Gradečak, M. S. Strano and K. K. Gleason, Adv. Mater., 2016, 28, 6399–6404 CrossRef CAS PubMed.
- S. Vaddiraju and K. K. Gleason, Nanotechnology, 2010, 21, 125503 CrossRef PubMed.
- X. Wang, S. Hou, H. Goktas, P. Kovacik, F. Yaul, A. Paidimarri, N. Ickes, A. Chandrakasan and K. Gleason, ACS Appl. Mater. Interfaces, 2015, 7, 16213–16222 CrossRef CAS PubMed.
- D. Bhattacharyya, K. Senecal, P. Marek, A. Senecal and K. K. Gleason, Adv. Funct. Mater., 2011, 21, 4328–4337 CrossRef CAS.
- D. C. Borrelli and K. K. Gleason, Macromolecules, 2013, 46, 6169–6176 CrossRef CAS.
- S. Lee and K. K. Gleason, Adv. Funct. Mater., 2015, 25, 85–93 CrossRef CAS.
- T. L. Andrew, L. Zhang, N. Cheng, M. Baima, J. J. Kim, L. Allison and S. Hoxie, Acc. Chem. Res., 2018, 51, 850–859 CrossRef CAS PubMed.
- H. Coskun, A. Aljabour, P. De Luna, D. Farka, T. Greunz, D. Stifter, M. Kus, X. Zheng, M. Liu, A. W. Hassel, W. Schöfberger, E. H. Sargent, N. S. Sariciftci and P. Stadler, Sci. Adv., 2017, 3, 1–9 Search PubMed.
- H. Coskun, A. Aljabour, L. Uiberlacker, M. Strobel, S. Hild, C. Cobet, D. Farka, P. Stadler and N. S. Sariciftci, Thin Solid Films, 2018, 645, 320–325 CrossRef CAS.
- Y. Xu, X. Wang, J. Zhou, B. Song, Z. Jiang, E. M. Y. Lee, S. Huberman, K. K. Gleason and G. Chen, Sci. Adv., 2018, 4, 3031 CrossRef PubMed.
- M. Nojima, Y. Ohta and T. Yokozawa, Kobunshi, 2013, 62, 236–238 CAS.
- M. K. Jeong, E. H. Suh, K. Lee, J. Jang and I. H. Jung, Org. Electron., 2020, 86, 105921 CrossRef CAS.
- D. Qu, T. Qi and H. Huang, J. Energy Chem., 2021, 59, 364–387 CrossRef CAS.
- E. F. Woods, A. J. Berl and J. A. Kalow, Angew. Chem., 2020, 132, 6118–6123 CrossRef.
- D. Bilger, K. W. Park and T. L. Andrew, Synth. Met., 2019, 250, 1–6 CrossRef CAS.
- H. Kuzmany and J. Kürti, Synth. Met., 1987, 21, 95–102 CrossRef.
- J. L. Brédas, R. Silbey, D. S. Boudreaux and R. R. Chance, J. Am. Chem. Soc., 1983, 105, 6555–6559 CrossRef.
- J. Mei and Z. Bao, Chem. Mater., 2014, 26, 604–615 CrossRef CAS.
- K. Chung, A. McAllister, D. Bilby, B. G. Kim, M. S. Kwon, E. Kioupakis and J. Kim, Chem. Sci., 2015, 6, 6980–6985 RSC.
- Z. Fan, D. Du, H. Yao and J. Ouyang, ACS Appl. Mater. Interfaces, 2017, 9, 11732–11738 CrossRef CAS PubMed.
- S. Hayashi, S. I. Yamamoto and T. Koizumi, Sci. Rep., 2017, 7, 1–8 CrossRef PubMed.
- J. S. Wu, Y. J. Cheng, M. Dubosc, C. H. Hsieh, C. Y. Chang and C. S. Hsu, Chem. Commun., 2010, 46, 3259–3261 RSC.
- A. T. Lawal and G. G. Wallace, Talanta, 2014, 119, 133–143 CrossRef CAS PubMed.
- R. Brooke, P. Cottis, P. Talemi, M. Fabretto, P. Murphy and D. Evans, Prog. Mater. Sci., 2017, 86, 127–146 CrossRef CAS.
- K. G. Neoh, E. T. Kang and K. L. Tan, Polymer, 1993, 34, 3921–3928 CrossRef CAS.
- C. Kranz, M. Ludwig, H. E. Gaub and W. Schuhmann, Adv. Mater., 1995, 7, 38–40 CrossRef CAS.
- P. P. Sengupta, P. Kar and B. Adhikari, Thin Solid Films, 2009, 517, 3770–3775 CrossRef CAS.
- F. Ghamouss, A. Brugère, A. C. Anbalagan, B. Schmaltz, E. Luais and F. Tran-Van, Synth. Met., 2013, 168, 9–15 CrossRef CAS.
- N. Y. Abu-Thabit, J. Chem. Educ., 2016, 93, 1606–1611 CrossRef CAS.
- D. L. Meyer, N. Schmidt-Meinzer, C. Matt, S. Rein, F. Lombeck, M. Sommer and T. Biskup, J. Phys. Chem. C, 2019, 123, 20071–20083 CrossRef CAS.
- L. Zhu, C. Jiang, G. Chen, Z. Zhou and Q. Li, Org. Electron., 2017, 49, 278–285 CrossRef CAS.
- Y. Y. Smolin, K. L. Van Aken, M. Boota, M. Soroush, Y. Gogotsi and K. K. S. Lau, Adv. Mater. Interfaces, 2017, 4, 1601201 CrossRef.
- J. Stejskal and M. Trchová, Polym. Int., 2012, 61, 240–251 CrossRef CAS.
- H. Chelawat, S. Vaddiraju and K. Gleason, Chem. Mater., 2010, 22, 2864–2868 CrossRef CAS.
- R. D. McCullough, R. D. Lowe, M. Jayaraman and D. L. Anderson, J. Org. Chem., 1993, 58, 904–912 CrossRef CAS.
- T. A. Chen, X. Wu and R. D. Rieke, J. Am. Chem. Soc., 1995, 117, 233–244 CrossRef CAS.
- R. D. Mccullough and R. D. Lowe, J. Chem. Soc., Chem. Commun., 1992, 70–72 RSC.
- R. D. McCullough, P. C. Ewbank and R. S. Loewe, J. Am. Chem. Soc., 1997, 119, 633–634 CrossRef CAS.
- R. D. McCullough, S. P. Williams, R. D. Lowe, M. Jayaraman and S. Tristram-Nagle, J. Am. Chem. Soc., 1993, 115, 4910–4911 CrossRef CAS.
- T. A. Chen and R. D. Rieke, J. Am. Chem. Soc., 1992, 114, 10087–10088 CrossRef CAS.
- M. R. Andersson, D. Seise, M. Berggren, H. Járvinen, I. T. Hjertberg, O. Inganás, O. Wennerstrom and J. Ósterholm, Macromolecules, 1994, 27, 6503–6506 CrossRef CAS.
- X. Shen, W. Hu and T. P. Russell, Macromolecules, 2016, 49, 4501–4509 CrossRef CAS.
- D. Alberga, A. Perrier, I. Ciofini, G. F. Mangiatordi, G. Lattanzi and C. Adamo, Phys. Chem. Chem. Phys., 2015, 17, 18742–18750 RSC.
- X. Wang, X. Zhang, L. Sun, D. Lee, S. Lee, M. Wang, J. Zhao, Y. Shao-Horn, M. Dinca, T. Palacios and K. K. Gleason, Sci. Adv., 2018, 4, 1–10 Search PubMed.
- R. Noriega, J. Rivnay, K. Vandewal, F. P. V. Koch, N. Stingelin, P. Smith, M. F. Toney and A. Salleo, Nat. Mater., 2013, 12, 1038–1044 CrossRef CAS PubMed.
- D. T. Scholes, P. Y. Yee, J. R. Lindemuth, H. Kang, J. Onorato, R. Ghosh, C. K. Luscombe, F. C. Spano, S. H. Tolbert and B. J. Schwartz, Adv. Funct. Mater., 2017, 27, 1702654 CrossRef.
- K. Ueji, M. Ohno, J. Takeya and S. Watanabe, Appl. Phys. Express, 2018, 12, 011004 CrossRef.
- D. T. Scholes, P. Y. Yee, G. R. McKeown, S. Li, H. Kang, J. R. Lindemuth, X. Xia, S. C. King, D. S. Seferos, S. H. Tolbert and B. J. Schwartz, Chem. Mater., 2019, 31, 73–82 CrossRef CAS.
- H. Y. Park, H. Yang, S. K. Choi and S. Y. Jang, ACS Appl. Mater. Interfaces, 2012, 4, 214–221 CrossRef CAS PubMed.
- K. Lee, M. K. Jeong, E. H. Suh, W. J. Jeong, J. G. Oh, J. Jang and I. H. Jung, Adv. Electron. Mater., 2022, 8, 1–11 Search PubMed.
- F. Zhang, E. Mohammadi, X. Luo, J. Strzalka, J. Mei and Y. Diao, Langmuir, 2018, 34, 1109–1122 CrossRef CAS PubMed.
- A. Zen, M. Saphiannikova, D. Neher, J. Grenzer, S. Grigorian, U. Pietsch, U. Asawapirom, S. Janietz, U. Scherf, I. Lieberwirth and G. Wegner, Macromolecules, 2006, 39, 2162–2171 CrossRef CAS.
- M. A. Ruderer, S. Guo, R. Meier, H. Y. Chiang, V. Körstgens, J. Wiedersich, J. Perlich, S. V. Roth and P. Müller-Buschbaum, Adv. Funct. Mater., 2011, 21, 3382–3391 CrossRef CAS.
- J. Liu, Y. Sun, X. Gao, R. Xing, L. Zheng, S. Wu, Y. Geng and Y. Han, Langmuir, 2011, 27, 4212–4219 CrossRef CAS PubMed.
- M. H. Rezvani, F. Farajollahi, A. Nikfarjam, P. Bakhtiarpour and E. Saydanzad, Materials, 2013, 6, 1994–2006 CrossRef CAS PubMed.
- D. Alberga, G. F. Mangiatordi, L. Torsi and G. Lattanzi, J. Phys. Chem. C, 2014, 118, 8641–8655 CrossRef CAS.
- S. Sanda, R. Nakamichi, T. Nagase, T. Kobayashi, K. Takimiya, Y. Sadamitsu and H. Naito, Org. Electron., 2019, 69, 181–189 CrossRef CAS.
- T. J. M. Choongik Kim and A. Facchetti, Science, 2007, 318, 76–80 CrossRef PubMed.
- E. Mohammadi, G. Qu, P. Kafle, S. H. Jung, J. K. Lee and Y. Diao, Mol. Syst. Des. Eng., 2020, 5, 125–138 RSC.
- G. Jo, J. Jung and M. Chang, Polymers, 2019, 11, 332 CrossRef PubMed.
- J. S. Kim, Y. Park, D. Y. Lee, J. H. Lee, J. H. Park, J. K. Kim and K. Cho, Adv. Funct. Mater., 2010, 20, 540–545 CrossRef CAS.
- P. L. Danielsen, J. Phys. C: Solid State Phys., 1986, 19, L741 CrossRef CAS.
- Y. Yao, H. Dong and W. Hu, Polym. Chem., 2013, 4, 5197–5205 RSC.
- K. S. Ahn, H. Jo, J. B. Kim, I. Seo, H. H. Lee and D. R. Lee, ACS Appl. Mater. Interfaces, 2020, 12, 1142–1150 CrossRef CAS PubMed.
- X. Wang, W. Deng, Y. Chen, X. Wang, P. Ye, X. Wu, C. Yan, X. Zhan, F. Liu and H. Huang, J. Mater. Chem. A, 2017, 5, 5585–5593 RSC.
- M. S. Chen, J. R. Niskala, D. A. Unruh, C. K. Chu, O. P. Lee and J. M. J. Fréchet, Chem. Mater., 2013, 25, 4088–4096 CrossRef CAS.
- M. S. Chen, J. R. Niskala, D. A. Unruh, C. K. Chu, O. P. Lee and J. M. J. Fréchet, Chem. Mater., 2013, 25, 4088–4096 CrossRef CAS.
- R. Rieger, D. Beckmann, A. Mavrinskiy, M. Kastler and K. Müllen, Chem. Mater., 2010, 22, 5314–5318 CrossRef CAS.
- M. Pandey, N. Kumari, S. Nagamatsu and S. S. Pandey, J. Mater. Chem. C, 2019, 7, 13323–13351 RSC.
- G. Giri, D. M. Delongchamp, J. Reinspach, D. A. Fischer, L. J. Richter, J. Xu, S. Benight, A. Ayzner, M. He, L. Fang, G. Xue, M. F. Toney and Z. Bao, Chem. Mater., 2015, 27, 2350–2359 CrossRef CAS.
- M. Brinkmann, L. Hartmann, L. Biniek, K. Tremel and N. Kayunkid, Macromol. Rapid Commun., 2014, 35, 9–26 CrossRef CAS PubMed.
- M. H. Gharahcheshmeh, M. M. Tavakoli, E. F. Gleason, M. T. Robinson, J. Kong and K. K. Gleason, Sci. Adv., 2019, 5, 1–13 Search PubMed.
- W. Ma, K. Shi, Y. Wu, Z. Y. Lu, H. Y. Liu, J. Y. Wang and J. Pei, ACS Appl. Mater. Interfaces, 2016, 8, 24737–24743 CrossRef CAS PubMed.
- C. M. Palumbiny, F. Liu, T. P. Russell, A. Hexemer, C. Wang and P. Müller-Buschbaum, Adv. Mater., 2015, 27, 3391–3397 CrossRef CAS PubMed.
- I. Petsagkourakis, E. Pavlopoulou, G. Portale, B. A. Kuropatwa, S. Dilhaire, G. Fleury and G. Hadziioannou, Sci. Rep., 2016, 6, 1–8 CrossRef PubMed.
- S. Kee, N. Kim, B. S. Kim, S. Park, Y. H. Jang, S. H. Lee, J. Kim, J. Kim, S. Kwon and K. Lee, Adv. Mater., 2016, 28, 8625–8631 CrossRef CAS PubMed.
- T. Lee, W. Kwon and M. Park, Org. Electron., 2019, 67, 26–33 CrossRef CAS.
- M. N. Gueye, A. Carella, N. Massonnet, E. Yvenou, S. Brenet, J. Faure-Vincent, S. Pouget, F. Rieutord, H. Okuno, A. Benayad, R. Demadrille and J. P. Simonato, Chem. Mater., 2016, 28, 3462–3468 CrossRef CAS.
- D. Bhattacharyya and K. K. Gleason, Chem. Mater., 2011, 23, 2600–2605 CrossRef CAS.
- S. G. Im, D. Kusters, W. Choi, S. H. Baxamusa, M. C. M. van de Sanden and K. K. Gleason, ACS Nano, 2008, 2, 1959–1967 CrossRef CAS PubMed.
- S. Kaviani, M. Mohammadi Ghaleni, E. Tavakoli and S. Nejati, ACS Appl. Polym. Mater., 2019, 1, 552–560 CrossRef CAS.
- Y. Y. Smolin, M. Soroush and K. K. S. Lau, Ind. Eng. Chem. Res., 2017, 56, 6221–6228 CrossRef CAS.
- S. E. Atanasov, M. D. Losego, B. Gong, E. Sachet, J. P. Maria, P. S. Williams and G. N. Parsons, Chem. Mater., 2014, 26, 3471–3478 CrossRef CAS.
- R. G. Larson, Nature, 2017, 550, 466–467 CrossRef CAS PubMed.
- J. Chang, Z. Lin, J. Li, S. L. Lim, F. Wang, G. Li, J. Zhang and J. Wu, Adv. Electron. Mater., 2015, 1, 1–9 Search PubMed.
- A. Teichler, J. Perelaer and U. S. Schubert, J. Mater. Chem. C, 2013, 1, 1910–1925 RSC.
- C. S. Lee, W. Yin, A. P. Holt, J. R. Sangoro, A. P. Sokolov and M. D. Dadmun, Adv. Funct. Mater., 2015, 25, 5848–5857 CrossRef CAS.
- K. B. Burke, W. J. Belcher, L. Thomsen, B. Watts, C. R. McNeill, H. Ade and P. C. Dastoor, Macromolecules, 2009, 42, 3098–3103 CrossRef CAS.
- R. Meier, M. Schindler, P. Müller-Buschbaum and B. Watts, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 84, 1–6 Search PubMed.
- P. Moni, A. Al-Obeidi and K. K. Gleason, Beilstein J. Nanotechnol., 2017, 8, 723–735 CrossRef CAS PubMed.
- K. K. Gleason, CVD Polymers: Fabrication of Organic Surfaces and Devices, 2015, 1, pp.1–11 Search PubMed.
- D. E. Weidner, L. W. Schwartz and R. R. Eley, J. Colloid Interface Sci., 1996, 179, 66–75 CrossRef CAS.
- F. Zhang, G. Baralia, A. Boborodea, C. Bailly, B. Nysten and A. M. Jonas, Langmuir, 2005, 21, 7427–7432 CrossRef CAS PubMed.
- L. E. Stillwagon and R. G. Larson, Phys. Fluids A, 1992, 4, 895–903 CrossRef CAS.
- A. Dianatdar, PhD thesis, University of Groningen, 2022.
- K. L. Mittal, Polym. Eng. Sci., 1977, 17, 467–473 CrossRef CAS.
- J. O. Carneiro, V. Teixeira and S. Azevedo, Encyclopedia of Thermal Stresses, 2014, pp.4222–4231 Search PubMed.
- X. Wang, K. Chen, L. S. de Vasconcelos, J. He, Y. C. Shin, J. Mei and K. Zhao, Nat. Commun., 2020, 11, 1–10 CrossRef CAS PubMed.
- M. Lane, Annu. Rev. Mater. Res., 2003, 33, 29–54 CrossRef CAS.
- G. Yan and J. R. White, Polym. Eng. Sci., 1999, 39, 1856–1865 CrossRef CAS.
- R. Katsumata, S. Ata, K. Kuboyama and T. Ougizawa, J. Appl. Polym. Sci., 2013, 128, 60–65 CrossRef CAS.
- J. Y. Chung, T. Q. Chastek, M. J. Fasolka, H. W. Ro and C. M. Stafford, ACS Nano, 2009, 3, 844–852 CrossRef CAS PubMed.
- H. Zhang and L. Zhang, Appl. Phys. Lett., 2016, 106, 033102 CrossRef.
- P. T. Chen, Y. W. Yang, G. Reiter and A. C. M. Yang, Polymer, 2020, 204, 122753 CrossRef CAS.
- S. G. Croll, J. Appl. Polym. Sci., 1979, 23, 847–858 CrossRef CAS.
- M. Ree, C. W. Chu and M. J. Goldberg, J. Appl. Phys., 1994, 75, 1410–1419 CrossRef CAS.
- G. Abadias, E. Chason, J. Keckes, M. Sebastiani, G. B. Thompson, E. Barthel, G. L. Doll, C. E. Murray, C. H. Stoessel and L. Martinu, J. Vac. Sci. Technol., A, 2018, 36, 020801 CrossRef.
- L. Ouyang, B. Wei, C. Chen Kuo, S. Pathak, B. Farrell and D. C. Martin, Sci. Adv., 2017, 3, 1–12 Search PubMed.
- B. Wei, J. Liu, L. Ouyang, C. C. Kuo and D. C. Martin, ACS Appl. Mater. Interfaces, 2015, 7, 15388–15394 CrossRef CAS PubMed.
- S. K. Sontag, N. Marshall and J. Locklin, Chem. Commun., 2009, 3354–3356 RSC.
- B. Yameen, C. Rodriguez-Emmenegger, C. M. Preuss, O. Pop-Georgievski, E. Verveniotis, V. Trouillet, B. Rezek and C. Barner-Kowollik, Chem. Commun., 2013, 49, 8623–8625 RSC.
- P. Kovacic and M. B. Jones, Chem. Rev., 1987, 87, 357–379 CrossRef CAS.
- S. G. Im, P. J. Yoo, P. T. Hammond and K. K. Gleason, Adv. Mater., 2007, 19, 2863–2867 CrossRef CAS.
- A. J. Clancy, M. K. Bayazit, S. A. Hodge, N. T. Skipper, C. A. Howard and M. S. P. Shaffer, Chem. Rev., 2018, 118, 7363–7408 CrossRef CAS PubMed.
- S. Byun, J. Choi, Y. Roh, D. Song, M. H. Ryou and Y. M. Lee, Electrochim. Acta, 2020, 332, 135471 CrossRef CAS.
- M. A. del Valle, M. A. Gacitúa, F. Hernández, M. Luengo and L. A. Hernández, Polymers, 2023, 15, 1450 CrossRef CAS PubMed.
- D. H. Kim, S. E. Atanasov, P. Lemaire, K. Lee and G. N. Parsons, ACS Appl. Mater. Interfaces, 2015, 7, 3866–3870 CrossRef CAS PubMed.
- R. M. Howden, E. J. Flores, V. Bulović and K. K. Gleason, Org. Electron., 2013, 14, 2257–2268 CrossRef CAS.
- A. M. Gaikwad, Y. Khan, A. E. Ostfeld, S. Pandya, S. Abraham and A. C. Arias, Org. Electron., 2016, 30, 18–29 CrossRef CAS.
- E. Singh, M. Meyyappan and H. S. Nalwa, ACS Appl. Mater. Interfaces, 2017, 9, 34544–34586 CrossRef CAS PubMed.
- R. Fan and T. L. Andrew, J. Electrochem. Soc., 2020, 167, 037542 CrossRef CAS.
- L. Zhang and T. L. Andrew, ACS Appl. Mater. Interfaces, 2018, 10, 38574–38580 CrossRef CAS PubMed.
- L. Zhang, W. Viola and T. L. Andrew, ACS Appl. Mater. Interfaces, 2018, 10, 36834–36840 CrossRef CAS PubMed.
- W. Viola, C. Jin and T. L. Andrew, Synth. Met., 2020, 268, 116483 CrossRef CAS.
- L. K. Allison, S. Rostaminia, A. Kiaghadi, D. Ganesan and T. L. Andrew, ACS Omega, 2021, 6, 31869–31875 CrossRef CAS PubMed.
- J. J. Kim, L. K. Allison and T. L. Andrew, Sci. Adv., 2019, 5, 0463 Search PubMed.
- A. Castro-Carranza, J. C. Nolasco, S. Bley, M. Rückmann, F. Meierhofer, L. Mädler, T. Voss and J. Gutowski, J. Polym. Sci., Part B: Polym. Phys., 2016, 54, 1537–1544 CrossRef CAS.
- L.-C. Chao and E.-P. Chang, J. Appl. Polym. Sci., 1981, 26, 603–610 CrossRef CAS.
- D. C. Dragunski, A. R. Freitas, A. F. Rubira and E. C. Muniz, Polym. Degrad. Stab., 2000, 67, 239–247 CrossRef CAS.
- P. Kovacik, G. del Hierro, W. Livernois and K. K. Gleason, Mater. Horiz., 2015, 2, 221–227 RSC.
- H. K. H. Lee, Z. Li, I. Constantinou, F. So, S. W. Tsang and S. K. So, Adv. Energy Mater., 2014, 4, 1400768 CrossRef.
- L. Mockus, J. J. Peterson, J. M. Lainez and G. V. Reklaitis, Org. Process Res. Dev., 2015, 19, 908–914 CrossRef CAS.
- W. Shin, W. Ko, S. H. Jin, T. Earmme and Y. J. Hwang, Chem. Eng. J., 2021, 412, 128572 CrossRef CAS.
- M. P. Aplan and E. D. Gomez, Ind. Eng. Chem. Res., 2017, 56, 7888–7901 CrossRef CAS.
- H. Seyler, D. J. Jones, A. B. Holmes and W. W. H. Wong, Chem. Commun., 2012, 48, 1598–1600 RSC.
- P. Serp, P. Kalck and R. Feurer, Chem. Rev., 2002, 102, 3085–3128 CrossRef CAS PubMed.
- M. Mirabedin, H. Vergnes, N. Caussé, C. Vahlas and B. Caussat, Appl. Surf. Sci., 2021, 554, 149501 CrossRef CAS.
- G. Drewelow, H. Wook Song, Z. T. Jiang and S. Lee, Appl. Surf. Sci., 2020, 501, 144105 CrossRef CAS.
- S. H. Baxamusa, S. G. Im and K. K. Gleason, Phys. Chem. Chem. Phys., 2009, 11, 5227–5240 RSC.
- K.-H. Dahmen, inEncyclopedia of Physical Science and Technology, ed. R. A. Meyers, Academic Press, San Diego, 3rd edn, 2002, vol. 2, pp.787–808 Search PubMed.
| This journal is © The Royal Society of Chemistry 2023 |