
The N, P co-doped carbon-loading Ni3P@Ni heterojunction nanocomposites derived from polybenzimidazoles grafted with oxygen-phosphorus group as high-efficiency electrocatalyst for oxygen evolution reaction†
Gang
Wang
 *a,
Wenshuai
Tang
a,
Shuai
Yang
a,
Mingxia
Lu
a,
Hongliang
Wei
a,
Lifeng
Cui
*b and
Xiaodong
Chen
*c
*a,
Wenshuai
Tang
a,
Shuai
Yang
a,
Mingxia
Lu
a,
Hongliang
Wei
a,
Lifeng
Cui
*b and
Xiaodong
Chen
*c
aCollege of Chemistry and Chemical Engineering, Henan University of Technology, Zhengzhou 450001, PR China. E-mail: gwang198@gmail.com
bCollege of Smart Energy, Shanghai Jiao Tong University, Shanghai 200240, PR China. E-mail: lcui@usst.edu.cn
cDepartment of Mechanical Engineering, City University of Hong Kong, Tat Chee Avenue, Kowloon, Hong Kong SAR, China. E-mail: chenxd19891115@163.com
First published on 27th November 2023
Abstract
Interfacial engineering-induced structural optimization is considered an expectant strategy to elevate the electrochemical activities of earth-abundant electrocatalysts. Herein, the pioneering architecture of Ni3P@Ni heterojunctions embedded onto N, P co-doped C substrates was effectuated by a combination of the wet-impregnation process of Ni/polybenzimidazoles containing an oxygen-phosphorus group (PBI-OP) precursor and phosphating/carbonization treatment to obtain the Ni3P@Ni/CNP electrocatalyst. It provides substantial Ni3P@Ni heterojunction interfacial sites, and the strong interaction between Ni3P@Ni nanosheets and CNP microspheres promotes the rearrangement of the electronic configuration to modify the electrochemically active sites while enhancing the structural stability and promoting interfacial electron transfer, thus improving the electrocatalytic activity of OER. In addition, the large number of pyridine N species on the electrocatalyst accelerated the diffusion of O2, further reducing the overpotential of OER. DFT calculation unveiled that Ni3P@Ni/CNP has a lower activation energy than IrO2, indicating its extremely positive electrocatalytic effect on the OER kinetics. Ni3P@Ni/CNP-M exhibits a low overpotential of 239 mV, a low Tafel slope of 52 mV dec−1, and good stability at a current density of 10 mA cm−2 in 1 M KOH electrolyte. Therefore, interfacial engineering through multi-component structures can provide new inspiration for designing advanced OER electrocatalysts.
1. Introduction
The increasing energy crisis and environmental pollution brought on by the overconsumption of traditional fossil fuels have made it imperative to find eco-friendly and cost-effective renewable energy sources. Hydrogen is gaining attention globally as a clean and renewable energy source. The electrocatalytic decomposition of water to produce hydrogen is considered as a method to alleviate the current energy and environmental difficulties.1–4 The anodic oxygen evolution reaction (OER), rather than the hydrogen evolution reaction (HER), is a complicated quadrielectron transfer reaction, and its sluggish reaction kinetics is a bottleneck in water electrolysis technology.5 Despite numerous measures taken to improve OER kinetics, the results are still unsatisfactory, and there is an urgent need to push forward the exploration of high-efficiency OER electrocatalysts. At present, iridium, ruthenium, and the corresponding oxides (Ir, Ru, IrO2, and RuO2) are well-known for the benchmarked OER electrocatalysts, but their large-scale commercialization is limited due to their scarce reserves.6,7 Accordingly, the development of low-cost, earth-abundant, and high-performance OER electrocatalysts is still the key to realizing large-scale hydrogen production by electrochemical water electrolysis.Currently, the investigation for efficacious and cost-effective nanomaterials as OER electrocatalysts has motivated a great deal of investment in transition metal phosphides,8–11 nitrides,12–14 carbides,15,16 and other earth-abundant alternatives. Among these, nickel phosphide (Ni3P) with suitable d-electron configurations and metallic properties has been extensively considered as a promising non-precious-metal OER electrocatalyst on account of its advantages of desirable intrinsic activity, chemical stability, and especially low polarization.17–20 Nevertheless, the practical application of the Ni3P electrocatalyst has been severely restricted by several obstacles, such as the poor apparent activity, terrible conductivity, and insufficient exposure of electrocatalytically active sites triggered by the uncontrollable aggregation and metastable ability during the long-term OER proceedings.21 To respond to these challenges, multiple strategies have been executed to accelerate the electrocatalytic OER performance of Ni3P nanomaterials, including the architecture of heterostructure interfaces,22 heteroatom doping,23 nanostructure tailoring,24 and hybridization.25 Among them, refining ultrafine heterojunctions has been widely confirmed as one of the most approvable and feasible methods for optimizing local electronic structures, enhancing electrical conductivity, boosting charge transformation, and strengthening structural stability, which comprehensively facilitates majorizations in the adsorption energy of OER intermediates (O*, OH*, and OOH*) and thus giving rise to the increased intrinsic activity.26 In particular, the controlled integration of Ni3P with metals at the atomic/nano dimension to generate Janus-type metal/semiconductor heterojunctions enabled not only a favorable inheritance of the electrochemical properties of both contiguous components but also exerted significant charge redistribution and interface modification between them, which initiated the amelioration of electrocatalytically active sites and the compositional synergistic synergy, thus contributing to the enhancement of OER kinetics.22 Therefore, the integrated electrochemical performance of hetero-structured electrocatalysts was generally better than that of single-component counterparts or physical mixtures.27 Aside from electrocatalytic activity promotion via heterostructure engineering, the compositing active components with the carbonaceous nanomaterial would upgrade the conductivity while also further anchoring, decentralizing, and assembling the electrocatalytically active sites, leading to the improvement of electron transfer ability, reinforcement of structural ability, and increment in the quantity of accessible active sites.28 Accordingly, the controlled fabrication of composites of well-defined Janus-structured metal/Ni3P hetero-nanostructures with carbonaceous materials as high-efficiency OER electrocatalysts is feasible, even though it is challenging.
Enlightened by the aforementioned statement, we legitimately synthesized the well-defined and neatly distributed Janus-architectured Ni/Ni3P heterojunction nanosheets immobilized over the N, P co-doped carbon substrate (Ni3P@Ni/CNP) through a wet-impregnation treatment of Ni/polybenzimidazoles containing an oxygen-phosphorus group (PBI-OP) precursor, followed by thermal annealing. The well-refined heterojunction formed by the robust bonding of Ni3P and Ni could inherit exclusive chemical properties of both phases well to strengthen the synergistic effect and structural stability and simultaneously could effectively induce electron rearrangement to modify the electrocatalytically active sites, thus contributing to the enhancement of overall OER performance. In addition, the introduction of N, P co-doped C nanomaterial by the self-consumption of PBI-OP as a substrate for the Ni3P@Ni heterojunction encouraged the dispersion and exposure of electrocatalytically active sites and also increased the electrical conductivity. It was also demonstrated that a significant amount of pyridinic N species survive in the Ni3P@Ni/CNP electrocatalysts, which could favor the release of O2 molecules to further bring down the OER overpotential.29 After the electrochemical test, our synthesized Ni3P@Ni/CNP electrocatalyst displayed better performance than similar products and even commercial IrO2, particularly Ni3P@Ni/CNP-M, which only needed a low overpotential of 239 mV to reach the current density of 10 mA cm−2 in 1 M KOH electrolyte solution, and the Tafel slope was only 52 mV dec−1. Moreover, the current density of Ni3P@Ni/CNP-M remained almost unchanged after 20 h of continuous operation at 10 mA cm−2, demonstrating its excellent stability.
2. Experiments
2.1. Materials
3,3′-Diaminobenzidine (DAB) and bis(4-carboxyphenyl) phenyl phosphine oxide (BCPPO) were purchased from ACROS ORGANICS and SCIENTIFIC COMPASS, respectively. Nickel(II) chloride hexahydrate (NiCl2·6H2O, 98%) was purchased from Alab (Shanghai, China). Polyphosphoric acid (PPA, 80%) and other chemicals were purchased from Nanjing Chemical Reagent Co. The chemicals used in the study were used without any further purification.2.2. Preparation of PBI-OP
P2O5 and PPA were added to a three-neck flask with a mechanical stirrer and N2 inlet and outlet. Under the N2 atmosphere, the contents were warmed to 175 °C, stirred to dissolve P2O5 completely, and then cooled down to room temperature. DAB was added and stirred at 80 °C and 120 °C for 1 h each and then cooled to room temperature. BCPPO was added and stirred at 120 °C, 150 °C, 170 °C, and 190 °C for 12 h. The resulting mixture was cooled slightly and then poured into deionized water to obtain a brown product. The product was washed several times with deionized water to neutral and then soaked in 5 wt% Na2CO3 solution for 24 h. After washing with deionized water, the product was dried under vacuum at 100 °C for 36 h. PBI-OP was characterized by 1H NMR, and as shown in Fig. 1, all H ions in the PBI-OP structure could be matched to the corresponding signal peaks, which proved the successful synthesis of PBI-OP in our preparation conditions (yield: 97%. 1H NMR (DMSO-d6, ppm): 13.31, 8.40, 7.88, 7.87, 7.76, 7.74, 7.67, 7.61, 7.59).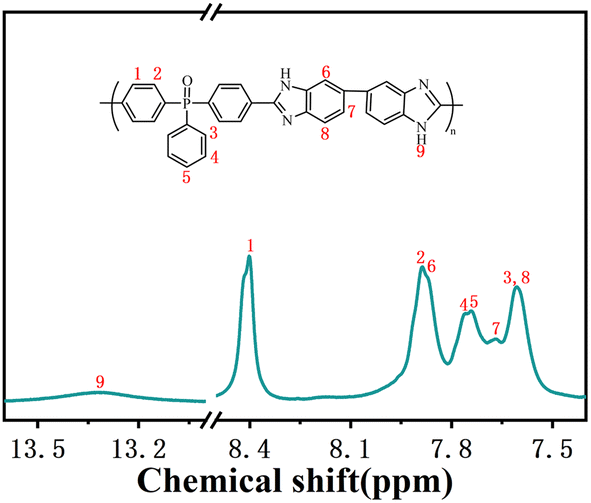 | ||
| Fig. 1 1H NMR spectrum and the corresponding chemical structure of PBI-OP. | ||
2.3. Preparation of Ni3P@Ni/CNP samples
Nickel(II) chloride hexahydrate (NiCl2·6H2O) was dissolved in deionized water and sonicated for 15 min to form a homogeneous solution. Then, the PBI-OP powder was added to the NiCl2 solution, stirred at 75 °C until the water was completely evaporated, and dried at 100 °C for 24 h using a vacuum drying oven to obtain the Ni/PBI-OP precursor. Next, the precursor was calcined in a tube furnace and roasted under N2 atmosphere at a set annealing temperature of 700 °C, 800 °C, and 900 °C for 3 h with a heating rate of 5 °C min−1, respectively, and the obtained samples were denoted as Ni3P@Ni/CNP-L, Ni3P@Ni/CNP-M, and Ni3P@Ni/CNP-H.2.4. Material characterization
The FTIR spectra of samples were obtained by Thermo Scientific Nicolet iS20 in the range of 400–4000 cm−1. The Raman spectra of samples were recorded with HORIBA Scientific LabRAM HR Evolution using a 532 nm laser diode as the excitation source. XRD patterns were obtained at 2θ values between 10° and 90° using a Japanese Rigaku Ultima IV with Cu Kα radiation (λ = 0.15418 nm) at a current of 40 mA and voltage of 40 kV. The morphology of samples was observed by scanning electron microscopy (SEM, Zeiss Gemini 300) and transmission electron microscopy (TEM, JEM 2100F). XPS analysis was performed on a Thermo ESCALAB 250XI instrument. Curve fitting and background subtraction were performed using Advantage software. The specific surface area of the powder samples was calculated using the BET model on a Micromeritics ASAP 2460 3.01 instrument. Based on the nitrogen adsorption data, the pore size distribution was calculated using the non-local density functional theory (NLDFT) equilibrium model. The thermal decomposition of ∼6 mg powder samples was carried out with a thermogravimetric analysis (STA 449F3) instrument under a flowing nitrogen atmosphere (20 cm3 min−1) at a heating rate of 10.0 °C min−1 up to 1000 °C. The first-principles method was utilized to perform density functional theory (DFT) calculations within the generalized gradient approximation (GGA) using the Perdew–Burke–Ernzerhof (PBE) formulation, which could quantitatively calculate the reaction energy barrier of the elementary step of the OER process, thus further exploring the OER mechanism of Ni3P@Ni/CNP electrocatalysts.2.5. Electrocatalytic measurements
All the samples were tested for electrocatalytic activity using a Corrtest CS350M Electrochemical workstation with a standard three-electrode system. The electrocatalyst ink was obtained by dispersing 5 mg of the sample into 650 μL of ethanol and 95 μL of Nafion solution (5% mass fraction) through ultrasonicating treatment for 30 min. Hg/HgO electrodes, graphite rods, and polished glassy carbon electrodes were selected as reference, counter, and working electrodes, respectively, for the electrochemical tests. The glassy carbon electrodes need to be cleaned before they can be used for electrochemical testing. Firstly, the glassy carbon electrodes were polished with 0.3 μm polishing powder, then put into an ultrasonic water bath for 2–3 min each time and repeated three times, and finally cleaned in anhydrous ethanol and deionized water. After cleaning, the glassy carbon electrode was activated using cyclic voltammetry in 0.5 M H2SO4 solution with a scanning range of 1.0 to −1.0 V until a stable pattern was obtained before use. 5 μL of the catalyst ink was deposited on a polished glass carbon electrode with a diameter of 3 mm with a catalyst loading of 0.475 mg cm−2, dried at room temperature and tested.Linear sweep voltammetry (LSV) was used to record polarization curves at a scan rate of 10 mV s−1. The electrochemical impedance spectroscopy (EIS) measurements were measured at 0.005 V vs. RHE over a frequency range of 0.1–106 Hz. The double layer capacitance (Cdl) of the samples was measured by cyclic voltammetry (CV) at different scan rates in the non-Faraday region. Electrochemical surface area (ECSA) was calculated by recording j (j = ja − jc) curves at different scan rates at 1.26 V (vs. RHE) using a linear slope (equivalent to 2Cdl). The stability of PBI-OP and Ni3P@Ni/CNP catalysts was tested by the chronopotentiometry method, in which the voltage values of PBI-OP, Ni3P@Ni/CNP-L, Ni3P@Ni/CNP-M, and Ni3P@Ni/CNP-H were set as 1.794 V, 1.589 V, 1.469 V, and 1.566 V, respectively. Their current densities can reach 10 mA cm−2 at the corresponding voltage values, and then they were run continuously for 48 h to observe the changes in the period current density.
3. Results and discussion
The categories of Ni3P@Ni/CNP samples could be successfully fabricated by a combination of the wetness-impregnation method and thermal annealing treatment. Briefly stated, the trailblazing PBI-OP was first synthesized by the copolymerization of DAB and BCPPO in the mixed liquids of P2O5 and PPA, and then the Ni2+ of NiCl2·6H2O was immobilized on its surface through Ni–N coordination bonds to form the Ni/PBI-OP precursor.30 Eventually, going through the phosphating/carbonization processes, the Ni/PBI-OP precursor could be transformed into the Ni3P@Ni/CNP samples as expected (Fig. 2a). The XRD patterns of the synthesized PBI-OP, Ni/PBI-OP precursor, and Ni3P@Ni/CNP samples are depicted in Fig. 2b. More concretely, the PBI-OP presented a non-crystallinity peak at 21.8°, which indicated the periodicity and parallelism of the PBI-OP backbone and also proved its amorphous structure.30,31 In contrast, the Ni/PBI-OP precursor also exhibited a similar but weak diffraction peak, suggesting that the polymer chain of PBI-OP did not suffer any damage when PBI-OP was successfully anchored with Ni2+ ions during the wet-impregnation process. No other diffraction peaks were observed for the Ni/PBI-OP precursor, suggesting that the Ni2+ ions were highly decentralized without forming crystal species over the PBI-OP substate. Whereas for Ni3P@Ni/CNP samples, there were no peaks mentioned above, but several novel distinctive diffraction peaks appeared in the XRD spectrum, disclosing that the structure of PBI-OP has been decomposed and carbonized during the annealing process. The three prominent peaks located at 44.50°, 51.85°, and 76.38° corresponded to the (111), (200), and (220) crystal planes of metallic Ni (JCPDS No. 87-0712), respectively, while the cognizable peaks at 41.76°, 42.82°, 43.63°, 46.32°, 47.14°, 50.44°, 52.52°, and 75.24° respectively, were indexed to the (231), (330), (112), (202), (141), (222), (132), and (233) crystal planes of Ni3P (JCPDS No. 34-0501). The diffraction peak detected at about 25° for the Ni3P@Ni/CNP samples indicated the formation of a carbon substrate.31 Realistically, these results manifested that the annealing temperatures set at 700 °C, 800 °C, and 900 °C were reasonable and capable for the generation of Ni, Ni3P, and carbon phase, which were further confirmed by the TG/DTA analysis (Fig. S2†). This consequence could be perceived as a prerequisite for the fabrication of the targeted Ni3P@Ni/CNP heterojunction samples.22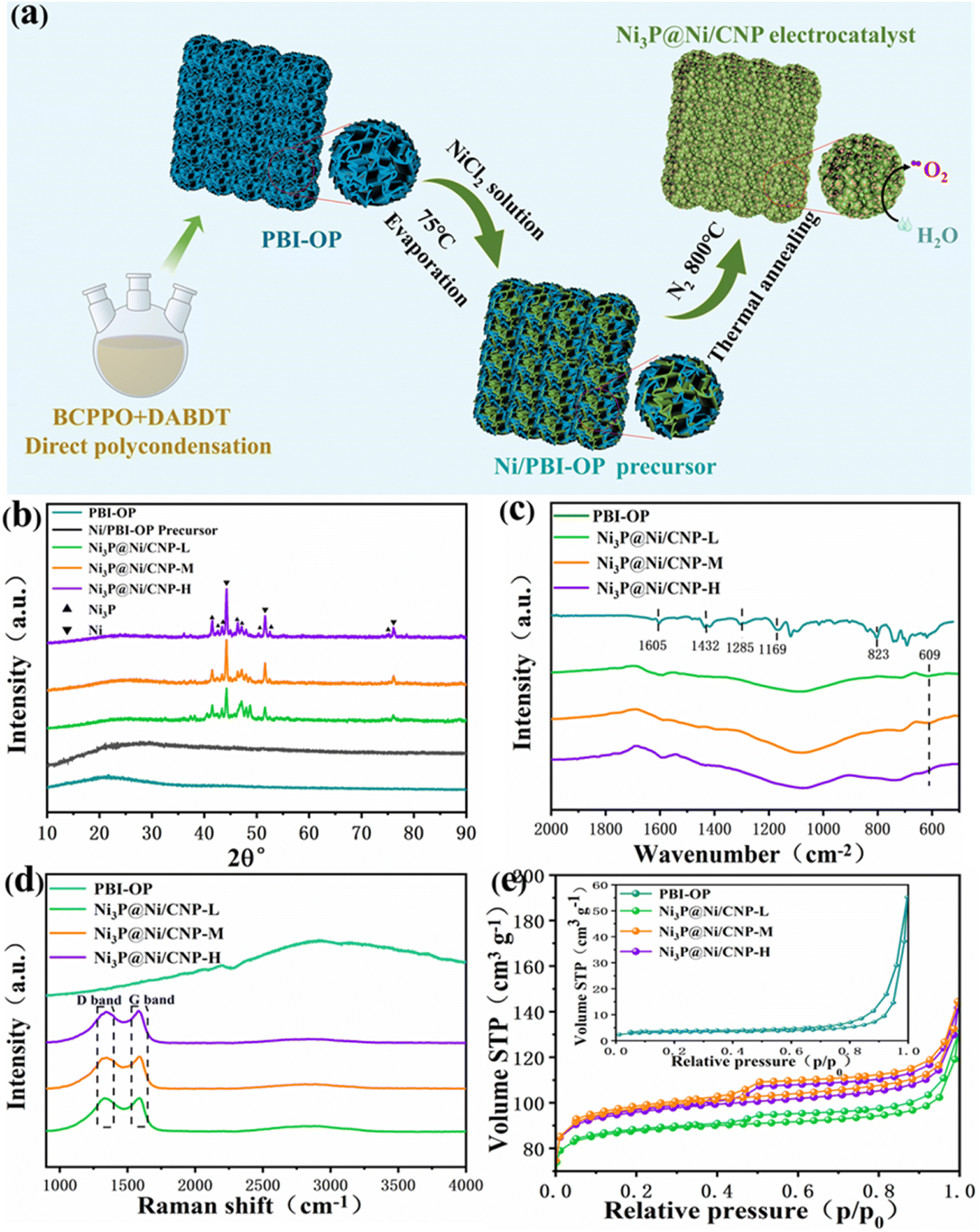 | ||
| Fig. 2 (a) Schematic illustration of the preparation process of Ni3P@Ni/CNP samples. (b) XRD patterns of PBI-OP, Ni/PBI-OP precursor, Ni3P@Ni/CNP-L, Ni3P@Ni/CNP-M, and Ni3P@Ni/CNP-H. (c) FT-IR spectrum, (d) Raman spectrum, and (e) N2 adsorption–desorption isotherms of PBI-OP, Ni3P@Ni/CNP-L, Ni3P@Ni/CNP-M, and Ni3P@Ni/CNP-H. | ||
The chemical structures of PBI-OP and Ni3P@Ni/CNP samples were further characterized by FTIR and Raman spectroscopies. As displayed in Fig. 2c, several prominent FTIR characteristic peaks of PBI-OP were detected at 1605 cm−1, 1432 cm−1, 1285 cm−1, 1169 cm−1, and 823 cm−1 respectively, which were ascribed to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C/CN stretching vibration of the benzimidazole ring, CC stretching vibration of the benzene ring, respiratory vibration of the imidazole ring, oxygen phosphorus group, and C–H out of plane bending vibration in the aromatic ring, thus confirming the successful synthesis of PBI-OP in our preparation conditions.32–34 Nevertheless, the FTIR characteristic peak belonging to PBI-OP was not observed in the Ni3P@Ni/CNP samples, which was mainly attributed to the fact that the structure of PBI-OP was subjected to complete decomposition during the annealing proceedings. Noticeably, a new emergent characteristic peak was recognized at 609 cm−1 and assigned to the stretching vibration of the Ni–P bond, thus confirming the successful synthesis of the Ni3P phase, which was consistent with XRD results.35–37 The Raman spectra of the synthesized PBI-OP and Ni3P@Ni/CNP samples are shown in Fig. 2d. It is illustrated that there are no obvious characteristic peaks in the Raman spectra of PBI-OP. Whereas for Ni3P@Ni/C samples, two dominant peaks at 1342 cm−1 (D band) and 1588 cm−1 (G-band) were well recognized, corresponding to the graphite structural defects and tangential vibrations of the graphite phase.38,39 These confirmed the formation of a microcrystalline graphite structure, which could impart the Ni3P@Ni/CNP samples with high electrical conductivity. It was worth noting that the ID/IG value of Ni3P@Ni/CNP-M was calculated to be as high as about 1.17, much larger than those of Ni3P@Ni/CNP-L (1.04) and Ni3P@Ni/CNP-H (1.08). This disclosed the generation of more crystal defects over Ni3P@Ni/CNP-M, which could effectively excite the conductivity band to elevate the electronic conductivity. This is of great importance as it would be dedicated to the enhancement of OER electrocatalytic performance.40
C/CN stretching vibration of the benzimidazole ring, CC stretching vibration of the benzene ring, respiratory vibration of the imidazole ring, oxygen phosphorus group, and C–H out of plane bending vibration in the aromatic ring, thus confirming the successful synthesis of PBI-OP in our preparation conditions.32–34 Nevertheless, the FTIR characteristic peak belonging to PBI-OP was not observed in the Ni3P@Ni/CNP samples, which was mainly attributed to the fact that the structure of PBI-OP was subjected to complete decomposition during the annealing proceedings. Noticeably, a new emergent characteristic peak was recognized at 609 cm−1 and assigned to the stretching vibration of the Ni–P bond, thus confirming the successful synthesis of the Ni3P phase, which was consistent with XRD results.35–37 The Raman spectra of the synthesized PBI-OP and Ni3P@Ni/CNP samples are shown in Fig. 2d. It is illustrated that there are no obvious characteristic peaks in the Raman spectra of PBI-OP. Whereas for Ni3P@Ni/C samples, two dominant peaks at 1342 cm−1 (D band) and 1588 cm−1 (G-band) were well recognized, corresponding to the graphite structural defects and tangential vibrations of the graphite phase.38,39 These confirmed the formation of a microcrystalline graphite structure, which could impart the Ni3P@Ni/CNP samples with high electrical conductivity. It was worth noting that the ID/IG value of Ni3P@Ni/CNP-M was calculated to be as high as about 1.17, much larger than those of Ni3P@Ni/CNP-L (1.04) and Ni3P@Ni/CNP-H (1.08). This disclosed the generation of more crystal defects over Ni3P@Ni/CNP-M, which could effectively excite the conductivity band to elevate the electronic conductivity. This is of great importance as it would be dedicated to the enhancement of OER electrocatalytic performance.40
The N2 adsorption–desorption isotherms were employed to determine the specific surface area, total pore volume, and pore size distribution for PBI-OP and Ni3P@Ni/CNP samples. As shown in Fig. 2e, all samples presented type IV isotherms with a significant H3 hysteresis loop, thus revealing the abundant existence of mesopores and macropores in these samples.22 The pore size profiles of all the samples (Fig. S3†) indicated that their pore size distributions were mainly concentrated in the 2–10 nm range. Correspondingly, the BET specific surface area of PBI-OP, Ni3P@Ni/CNP-L, Ni3P@Ni/CNP-M, and Ni3P@Ni/CNP-H was calculated to be 12.03 m2 g−1, 345.60 m2 g−1, 374.01 m2 g−1, and 370.29 m2 g−1, respectively (Table S1†). To be noted, the BET specific surface area of PBI-OP was much lower than those of the Ni3P@Ni/CNP samples, suggesting that during the phase transformation process from PBI-OP to Ni3P@Ni/CNP, the samples underwent giant morphological changes, and large amounts of mesopores and micropores were generated after the annealing treatment. Ni3P@Ni/CNP-M possessed the largest BET specific surface area, which originated from its abundant generation of mesoporous and microporous nanostructures at the appropriate annealing temperature of 800 °C.22,41 On these grounds, it was more sensible to infer that there were more intersecting micropores surviving in Ni3P@Ni/CNP-M, which could furnish more available channels for the diffusion and permeation of the electrolyte, formation of exposed active sites, and release of O2 bubbles during the OER process, hence guaranteeing highly efficient OER electrochemical behavior.42
The microscopic morphologies and structural features of PBI-OP and Ni3P@Ni/CNP samples were determined by SEM and TEM characterizations. As shown in Fig. 3a and b, the microscopic morphology of PBI-OP was made up of a large number of wrinkled and clustered nanosheets. When PBI-OP was loaded with Ni2+ ions, there was no conspicuous variation in its morphologies, and no distinguishable species resulting from Ni2+ aggregation was detected over the Ni/PBI-OP precursor (Fig. S4–S6†). Nevertheless, from the SEM images of Ni3P@Ni/CNP samples, as depicted in Fig. 3c and S7,† it could be discerned that when compared to PBI-OP and Ni/PBI-OP precursors, their nanostructures have changed significantly, exhibiting aggregated microspheres. In addition, the corresponding TEM, selected electron diffraction (SAED), high-resolution TEM (HRTEM), and EDS element mapping images of Ni3P@Ni/C are shown in Fig. 3d–i, respectively. From the HRTEM, SAED, and AFM images of Ni3P@Ni/CNP-M (Fig. 3d–g), it could be clearly observed that the Ni3P@Ni nanosheets are regularly immobilized in the CNP matrix, and their sizes and thicknesses are systematically measured to be about 20–30 nm and 1.5–2 nm (Fig. S8†) respectively, considerably smaller than those of counterparts, allowing more exposed active sites to be easily generated and permeated in the electrolyte.43 Besides, combined with the SAED results, the discernible lattice fringes with spacings of 1.97 Å and 2.03 Å, also recognized in the HRTEM image of Ni3P@Ni/CNP-M, belonged to the (202) plane of Ni3P phase and the (111) plane of Ni phase, respectively, which proved the coexistence of Ni and Ni3P phases,40 as also confirmed by XRD results. Furthermore, it was also indisputable proof that the Ni and Ni3P components were contiguous to each other, affirming the generation of Janus-type structures of the ultrafine heterointerfaces. Moreover, the corresponding EDS element mapping (Fig. 3h and i) demonstrated that the Ni element was completely distributed on the Ni3P@Ni nanosheets while the P element is mainly distributed on a portion of Ni3P@Ni nanosheets, further identifying the existence of Ni3P@Ni hetero interface structures. Besides, there were similar Ni3P@Ni heterostructures that could also be observed over Ni3P@Ni/CNP-L (Fig. S9†) and Ni3P@Ni/CNP-H (Fig. S10†), suggesting the effectiveness of the phosphating strategy with a polymeric compound with low phosphorus content for generating the Ni3P@Ni heterojunction in our preparation conditions, which is more advantageous to inheriting the electrochemical properties of both constituent components, while also triggering charge redistribution and interface modification between them, thus integrally bringing on the compositional synergy effect for the promotion of OER kinetics.44 Noticeably, the EDS mapping results also manifested that the C, N, and P elements have the same distribution profiles, indicating the successful formation of a CNP substate as expected due to the effective doping of N, P heteroatoms into the C matrix, which could offer the functionalities to modulate the electronic structure, improve the electrical conductivity, and optimize the adsorption energy of reactants and intermediates, thus accelerating the electrochemical reactions kinetics. Moreover, as can be seen from the results of the elemental and ICP-OES analyses (Table S2†), Ni3P@Ni/CNP-L, Ni3P@Ni/CNP-M and Ni3P@Ni/CNP-H have basically the same content of each element, and the most abundant one is Ni, which is 47.82%, 48.28%, and 48.03%, respectively.
 | ||
| Fig. 3 SEM images of (a) PBI-OP, (b) Ni/PBI-OP precursor, and (c) Ni3P@Ni/CNP-M. (d) TEM image of Ni3P@Ni/CNP-M, (e) SAED pattern of Ni3P@Ni/CNP-M, (f) AFM image of Ni3P@Ni/CNP-M, (g) HRTEM images of Ni3P@Ni/CNP-M, (h) and (i) EDS elemental mapping images of C, N, Ni, and P elements of Ni3P@Ni/CNP-M. | ||
XPS was employed to investigate further the composition and chemical states of PBI-OP and Ni3P@Ni/CNP samples. As depicted in Fig. 4a, S11a, and S12a,† the obtained XPS survey spectra clearly demonstrated the signals of C, N, O, and P for PBI-OP, while the signals of C, N, O, P, and Ni were found for Ni3P@Ni/CNP samples. The high-resolution Ni2p XPS spectrum of Ni3P@Ni/CNP-M (Fig. 4b, S11b, and S12b†) could be well deconvoluted into six prominent peaks attributed to Niδ+ (855.1 eV, 872.6 eV) in the Ni3P phase, Ni0 (852.2 eV, 868.9 eV) in the Ni phase, and satellite peaks (860.1 eV and 878.3 eV).23,36,45,46 The high-resolution C1s XPS spectrum of PBI-OP (Fig. S13a†) contained four main peaks centered at 284.1 eV, 284.9 eV, 285.9 eV, and 290.7 eV, representing C–C/CC, C–N, C–O–C and satellite peaks, respectively.47 In striking contrast, the high-resolution C1s XPS spectra of Ni3P@Ni/CNP-L, Ni3P@Ni/CNP-M, and Ni3P@Ni/CNP-H (Fig. 4c, S11c, and S12c†) were also divided into four individual component peaks at 284.8 eV, 285.9 eV, 286.8 eV and 289.1 eV, which corresponded to C–C/CC, C–N, C–P, and satellite peaks,23,24 further testifying the successful introduction of both N and P heteroatoms into the C matrix, which corroborated with the TEM results.48–50 In the high-resolution N1s XPS spectrum of PBI-OP (Fig. S13b†), two peaks at 397.3 eV and 398.1 eV were attributed to –N and –NH–, while the other two peaks at 399.4 eV and 400.5 eV were assigned to positively charged imines. The high-resolution N1s XPS spectrum of Ni3P@Ni/CNP samples could be fitted into four distinctive peaks (Fig. 4d, S11d, and S12d†). Apart from the three peaks at 397.8 eV for pyridinic-N species, 400.1 eV for pyrrolic-N species, and 401.1 eV for graphite-N species, respectively,40 a crucial peak at 399.3 eV could be observed indexed to the Ni–N species, disclosing that covalent bonds were generated at the interface between the Ni3P@Ni heterojunction nanosheets and CNP microspheres, which significantly strengthened the interaction of these two components.51 More impressively, the content of Ni–N species of Ni3P@Ni/CNP-M was measured to be about 28.31%, much higher than those of Ni3P@Ni/CNP-H and Ni3P@Ni/CNP-L (Table S3†), manifesting the existence of the strongest chemical interactions among them, which was expected to further reinforcing the structural durability and enhancing electrochemical performances.46 In addition, the content of pyridinic-N species in Ni3P@Ni/CNP-M was calculated to be as high as 43.1%, higher than those in Ni3P@Ni/CNP-L (40.15%) and Ni3P@Ni/CNP-H (32.81%), which is of great significance to accelerate the OER electrocatalytic activity because the pyridinic-N species are believed to encourage the release of O2 molecules for the reduction of OER overpotential.29 The high-resolution O1s XPS spectra of Ni3P@Ni/CNP samples (Fig. 4e, S11e, and S12e†) could be fitted to four distinctive peaks at 528.7 eV, 530.3 eV, 531.4 eV, and 532.6 eV, respectively, which are associated with the M–O bond, OH− species, O2 and H2O adsorbed on the surface.47 In the high-resolution P2p XPS spectrum of PBI-OP (Fig. S13d†), only one peak appeared at 132.9 eV, representing the P–O bond. In striking contrast, in the high-resolution P2p XPS spectra of Ni3P@Ni/CNP samples (Fig. 4f, S11f, and S12f†), besides the existing peak of the P–O bond, there were two additional peaks of P2p1/2 and P2p3/2 recognized at 129.8 eV and 128.9 eV, which were brought on by the presence of the P–Ni bond in Ni3P, also further validating the existence of the Ni3P phase.37,52
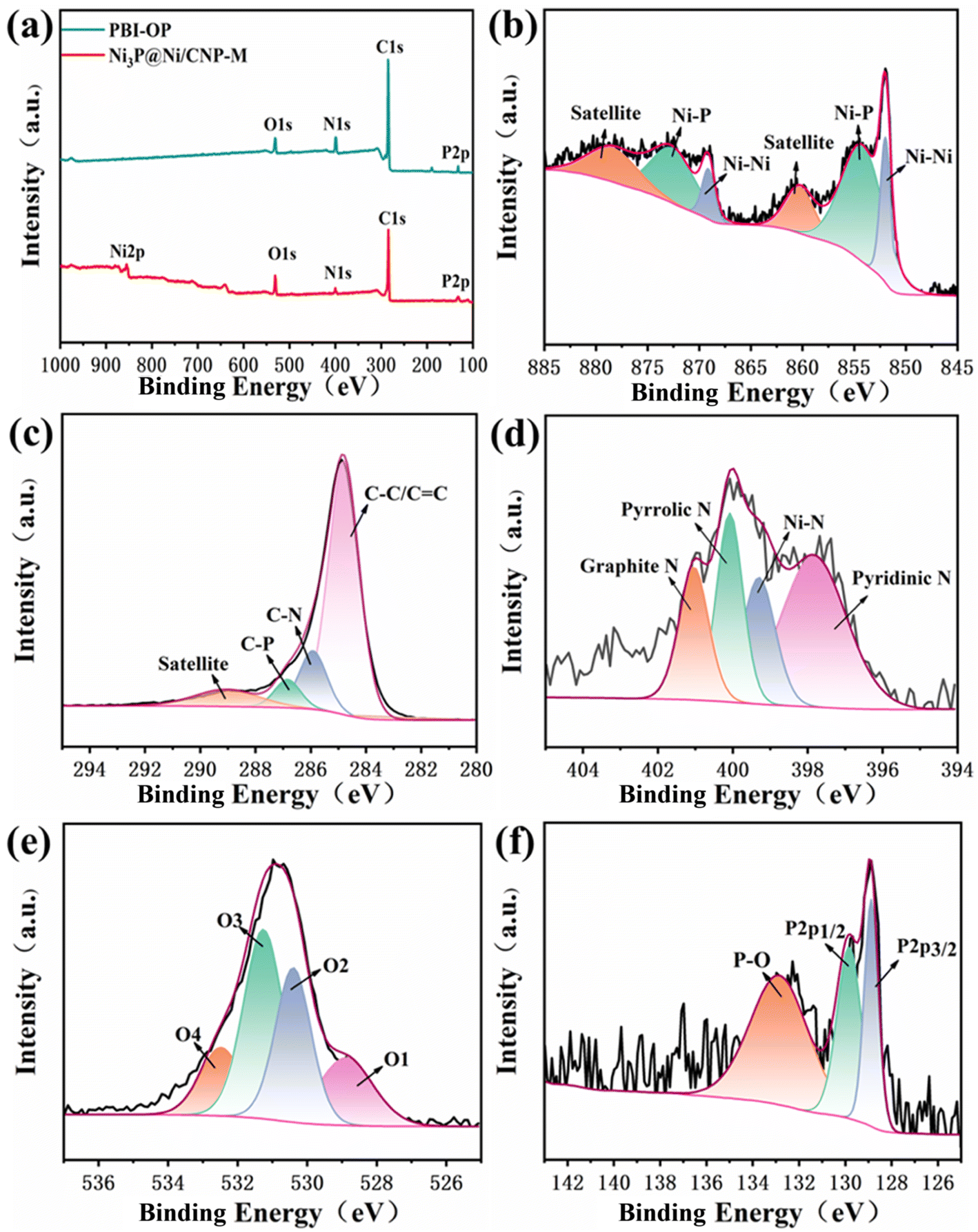 | ||
| Fig. 4 (a) XPS survey, (b) Ni2p, (c) C1s, (d) N1s, (e) O1s, and (f) P2p spectra of Ni3P@Ni/CNP-M. | ||
Linear scanning voltammetry (LSV) tests were performed using a 3 mm diameter glassy carbon electrode as the working electrode at a scanning rate of 10 mV s−1 and corrected using ohmic drop compensation. During testing, magnets were added to the electrolytic cell and slowly stirred to remove the O2 bubbles generated on the glassy carbon electrode. All potentials in this work were converted to reversible hydrogen electrode (RHE) through the equation of ERHE = EHg/HgO + 0.059 pH + 0.098. As illustrated in Fig. 5a and S14,† the polarization curves of Ni3P@Ni/CNP-M gave a much lower onset potential of 1.20 V vs. RHE than those of PBI-OP (1.56 eV), Ni3P@Ni/CNP-L (1.25 eV), Ni3P@Ni/CNP-H (1.30 eV), and even IrO2 (1.46 eV). Besides, Ni3P@Ni/CNP-M only needed an overpotential of 239 mV to achieve a current density of 10 mA cm−2, significantly lower than those of PBI-OP (564 mV), Ni3P@Ni/CNP-L (359 mV), Ni3P@Ni/CNP-H (336 mV), and IrO2 (347 mV), thus declaring a prominent enhancement in OER electrocatalytic performance. This consequence suggested that the formation and optimization of well-defined Ni3P@Ni heterojunction structures by adjusting the phosphating temperature could indeed accelerate electron transfer and significantly speed up the OER dynamics.26,53,54 It was pleasing to note that the electrocatalytic performance of Ni3P@Ni/CNP electrocatalysts was comparable to or even superior than those of the representative NiPx containing OER electrocatalysts reported recently (Fig. 5b and Table S4†), such as Ni/Ni2P@N–CNF (285 mV),22 N–Ni3P–Ni/N–CNTs/NF (270 mV),23 Ni3P/MnOOH (300 mV),25 Ni2P/Co–NF (316 mV),27 NiFeP (277 mV),55 Ni/Ni3C (350 mV),56 Ni2P/NiCo2O4 (250 mV),57 Ni2P/NPCM NCs (255 mV),58 Ni5P4 NSs (290 mV),59 CNT@NiCoP/C (297 mV),60 Co2P/Ni2P@NF (310 mV),61 Fe2P/Ni2P HS/NF (185 mV),62 Fe–CoNiP (280 mV),63 NiFeP@CNT (254 mV),64 NiFeP@GNS (211 mV),65 NFP@NG (295 mV),66 NiP2@FeP@CNT (261 mV),67 FeP/Ni2P@CNT (240 mV),68 and NiMn–P (190 mV).69 To explore the OER kinetics, the Tafel slope of IrO2, PBI-OP, and Ni3P@Ni/CNP electrocatalysts was calculated, as shown in Fig. 5c. Not surprisingly, compared with IrO2 (91 mV dec−1), PBI-OP (112 mV dec−1), Ni3P@Ni/CNP-L (68 mV dec−1), and Ni3P@Ni/CNP-H (64 mV dec−1), Ni3P@Ni/CNP-M has a smaller Tafel slope of only 52 mV dec−1, indicating that Ni3P@Ni/CNP-M could greatly accelerate the rate-determining step to acquire the more desirable OER kinetics. Moreover, the electrochemical double-layer capacitance (Cdl), representing the electrochemical surface area (ECSA), could be obtained by using CVs with different scan rates in the non-Faraday region and recording the j (j = ja − jc) curves at different scan rates at 1.26 V (vs. RHE) (Fig. S15†). From Fig. 5d, it was noteworthy that the linear slope (equivalent to twice the Cdl) of Ni3P@Ni/CNP-M was calculated to be as high as 8.9 mF cm−2, much greater than those of PBI-OP (0.08 mF cm−2), Ni3P@Ni/CNP-L (2.2 mF cm−2), and Ni3P@Ni/CNP-H (4.8 mF cm−2), verifying that the effective Ni/Ni3P heterojunction construction could greatly enlarge the exposure of electrochemically active sites, thus promoting the OER electrocatalytic activity.70,71 Taking into consideration the electrocatalytic durability of the above-mentioned samples, the Ni3P@Ni/CNP-M delivered the most stable performance with almost no attenuation in the current density during the consecutive 48 h operation (Fig. 5e), further identifying a favorable electrocatalytic stability owing to the strong interaction of the Ni3P@Ni heterojunction nanosheets and the CNP substrate of Ni3P@Ni/CNP-M, which has been authenticated by XPS analysis. In addition, as can be seen from the inset of Fig. 5e, the trends of the LSV curves of Ni3P@Ni/CNP-M after the 48 h stability test are basically the same, and the overpotential change required for the same current density is negligible, which further proves that Ni3P@Ni/CNP-M has excellent operational stability for OER during long-term electrochemical processes. More impressively, we characterized the Ni3P@Ni/CNP-M samples after stability testing by ICP-OES, XRD, TEM, and XPS. Table S5† indicates the content of each element of Ni3P@Ni/CNP-M before and after the stability test, and it can be seen that the composition of Ni3P@Ni/CNP-M did not change significantly after the test, showing good stability. From the XRD spectra shown in Fig. S16,† it can be seen that the Ni3P@Ni/CNP-M electrocatalysts showed no obvious crystal structure changes before and after the test. In addition, from the TEM images shown in Fig. S17,† it can be seen that the micro-morphology of Ni3P@Ni/CNP samples after the stability test was still highly consistent with that before the test. Moreover, from the analysis of the XPS spectra of Ni3P@Ni/CNP-M after the stability test (Fig. S18†), it was found that there was no significant change in the C, N, O, Ni, and P elements, and the above results verified the excellent stability of the Ni3P@Ni/CNP-M material.
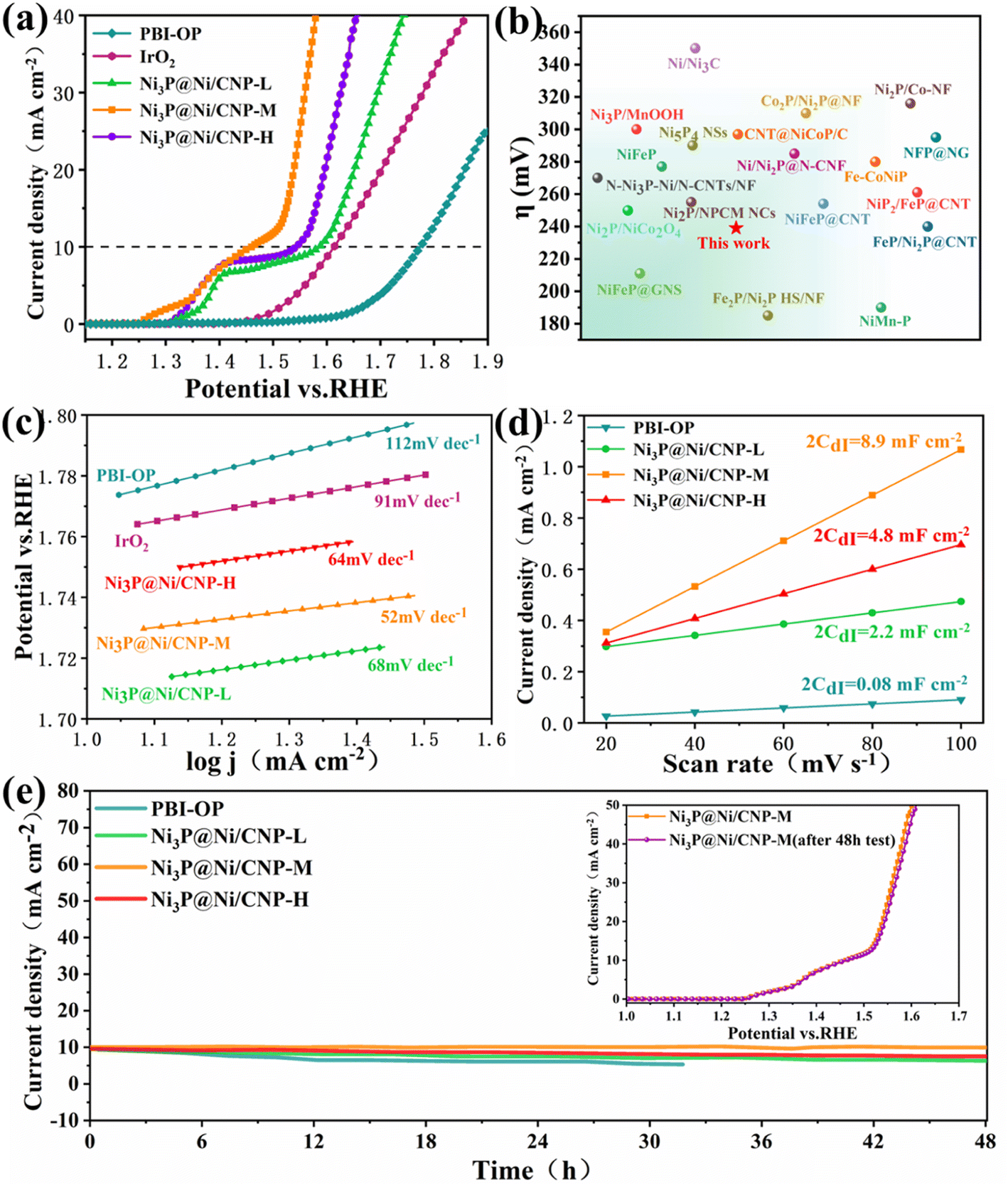 | ||
| Fig. 5 (a) Polarization curves of PBI-OP, IrO2, Ni3P@Ni/CNP-L, Ni3P@Ni/CNP-M, and Ni3P@Ni/CNP-H, (b) comparing the overpotential of Ni3P@Ni/CNP-M with the literature values. (c) The corresponding Tafel plots of PBI-OP, IrO2, Ni3P@Ni/CNP-L, Ni3P@Ni/CNP-M, and Ni3P@Ni/CNP-H. (d) The current density as a function of the scan rate. (e) Time-dependent current density curves of PBI-OP and Ni3P@Ni/CNP electrocatalyst; LSV curves of Ni3P@Ni/CNP-M before and after a 48 h test. | ||
To gain a thorough insight into the OER electrocatalytic kinetics, electrochemical impedance spectroscopy (EIS) was employed for the above-mentioned electrocatalysts in 1 M KOH electrolyte. As shown in Fig. 6a, the EIS Nyquist curves of all the samples displayed similar curves with a semi-circular curve in the high-frequency region, whose diameter represented the charge-transfer resistance and the equivalent resistance of the intermediate at the electrode/electrolyte interface. It could be detected that when compared with PBI-OP, Ni3P@Ni/CNP-L, and Ni3P@Ni/CNP-H, Ni3P@Ni/CNP-M delivered the least relaxation time of semicircle, indicating its lowest resistance to electron transfer, which could be attributed to the synergistic effect of Ni and Ni3P brought about by their formation of heterojunction interfaces, thus devoting to OER kinetics.42,54,55,72 Moreover, these impedance spectra were well-fitted and analyzed using ZView software and the EIS Spectrum Analyzer program and found to fit well with the Rs (RctCPE) equivalent circuit (Fig. 6b). Thereinto, Rs represented the solution resistance, CPE represented the constant phase element, and Rct represented the reactive charge transfer resistance at the electrode/electrolyte interface. The results elaborated that the data obtained by the simulation were basically consistent with the experimental data, and the corresponding EIS fitting parameters are exhibited in Table S6.† In the fitted data, Rct was the largest in the overall impedance, indicating that the charge transfer resistance was the main factor determining the OER kinetics of Ni3P@Ni/CNP electrocatalysts. More importantly, the Rct value of Ni3P@Ni/CNP-M was measured to be 5.367 Ω cm2, much lower than those of Ni3P@Ni/CNP-L (10.920 Ω cm2) and Ni3P@Ni/CNP-H (9.544 Ω cm2), manifesting its favorable charge transfer capability during the OER process. Noticeably, numerous research studies have demonstrated that the OER performances of electrocatalysts are mainly determined by the adsorption strength of *OH intermediates over the electrocatalysts.55,56 In response to this, the corresponding CV curves of OER were extensively examined in KOH electrolyte before and after adding 1 M CH3OH, and the results are depicted in Fig. 6c and S19.† After the addition of CH3OH, the current density of PBI-OP showed a small increase, while the current density of the Ni3P@Ni/CNP electrocatalyst showed a significant increase, with that of Ni3P@Ni/CNP-M being the most obvious. This was attributed to the weak adsorption strength of PBI-OP on *OH intermediates and the poor coverage of *OH intermediates on the surface, which limited the occurrence of the oxidation reaction (MOR) between CH3OH and *OH intermediates,40 whereas Ni3P@Ni/CNP-M provided a large number of sites for the coverage of *OH intermediates, which allowed the MOR to occur rapidly. Besides, the results also demonstrated the high surface coverage of *OH intermediates and strong adsorption strength over the Ni3P@Ni/CNP-M heterojunction interface, which was crucial to accelerate the oxidation deprotonation reaction process from *OH to *O intermediates (Fig. 6d), thus speeding up the overall OER kinetics.73
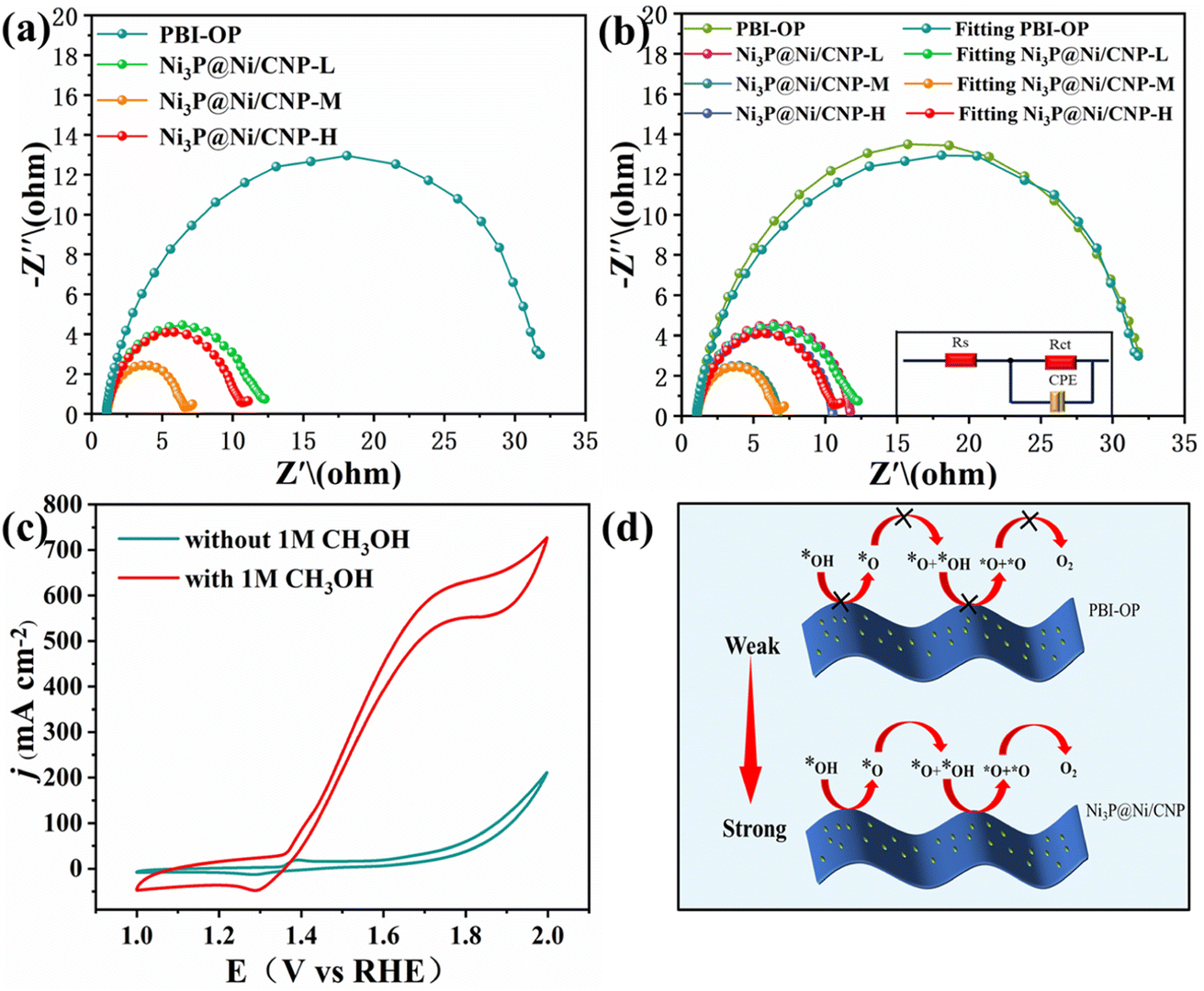 | ||
| Fig. 6 (a) EIS Nyquist plots. (b) Simulated equivalent circuit diagram and fitted Nyquist plots of PBI-OP, Ni3P@Ni/CNP-L, Ni3P@Ni/CNP-M, and Ni3P@Ni/CNP-H. (c) CV curves of Ni3P@Ni/CNP-M in 1 M KOH before and after the addition of 1 M CH3OH. (d) Schematic display of the accelerated OER kinetics of Ni3P@Ni/CNP samples by turning the adsorption strength of *OH intermediates. | ||
To further unveil the feasibility of the heterojunction construction and electrocatalysis process of Ni3P@Ni/CNP and IrO2, their relevant nano-structural properties and electrocatalytic OER kinetics were extendedly investigated by DFT calculations. More concretely, the Ni3P@Ni structure supported by the CNP substrate was constructed as a heterojunction with Ni3P (202) and Ni (111). The stable structure of Ni3P@Ni heterojunction after geometry optimizations by DFT calculation is exhibited in Fig. S20.† Whereas for the IrO2, the triple-layer (110) crystal plane of IrO2 served to acquire the optimized IrO2 structure, as illustrated in Fig. S21.† For the electrocatalytic process, a four-step OER mechanism was proposed for IrO2, including *OH, *O, and *OOH intermediates. However, what is completely different is that a five-step OER mechanism74,75 occurred over the Ni3P@Ni/CNP, including *OH, *O, *O + *OH, *O + *O intermediates (Fig. 7a). This was mainly because the formation of a heterojunction over Ni3P@Ni/CNP could effectively strengthen the synergistic effect of electrocatalytic active sites and ameliorate the adsorption strength of intermediate species, enabling a change in the OER intermediate steps as compared with IrO2. Furthermore, the charge density distribution of the Ni3P@Ni heterojunction is described in Fig. 7b. It is noticeable that superfluous electrons were gathered at the Ni3P@Ni heterojunction interface, which could make the rearrangement of electron configuration at the interface and adjust the adsorption/desorption energy of intermediates, thus reducing the overall energy barrier of OER.76 From the calculated free energy diagram of the OER process (Fig. 7c), it was noteworthy that for Ni3P@Ni, the coupling process step from *O intermediates to the O2 product (*O + *O → O2) required the largest energy barrier to overcome and could be perceived as the rate-determining step with an energy potential barrier of 1.5217 eV. Whereas for IrO2, the rate-determining step was the protonation step of *OOH to O2 (*OOH → O2) with an energy barrier of 1.5479 eV. The calculations disclosed that Ni3P@Ni/CNP had a lower OER activation energy than IrO2, thereby identifying the above experimental phenomenon that the OER activity of Ni3P@Ni/CNP is better than IrO2 as expected.
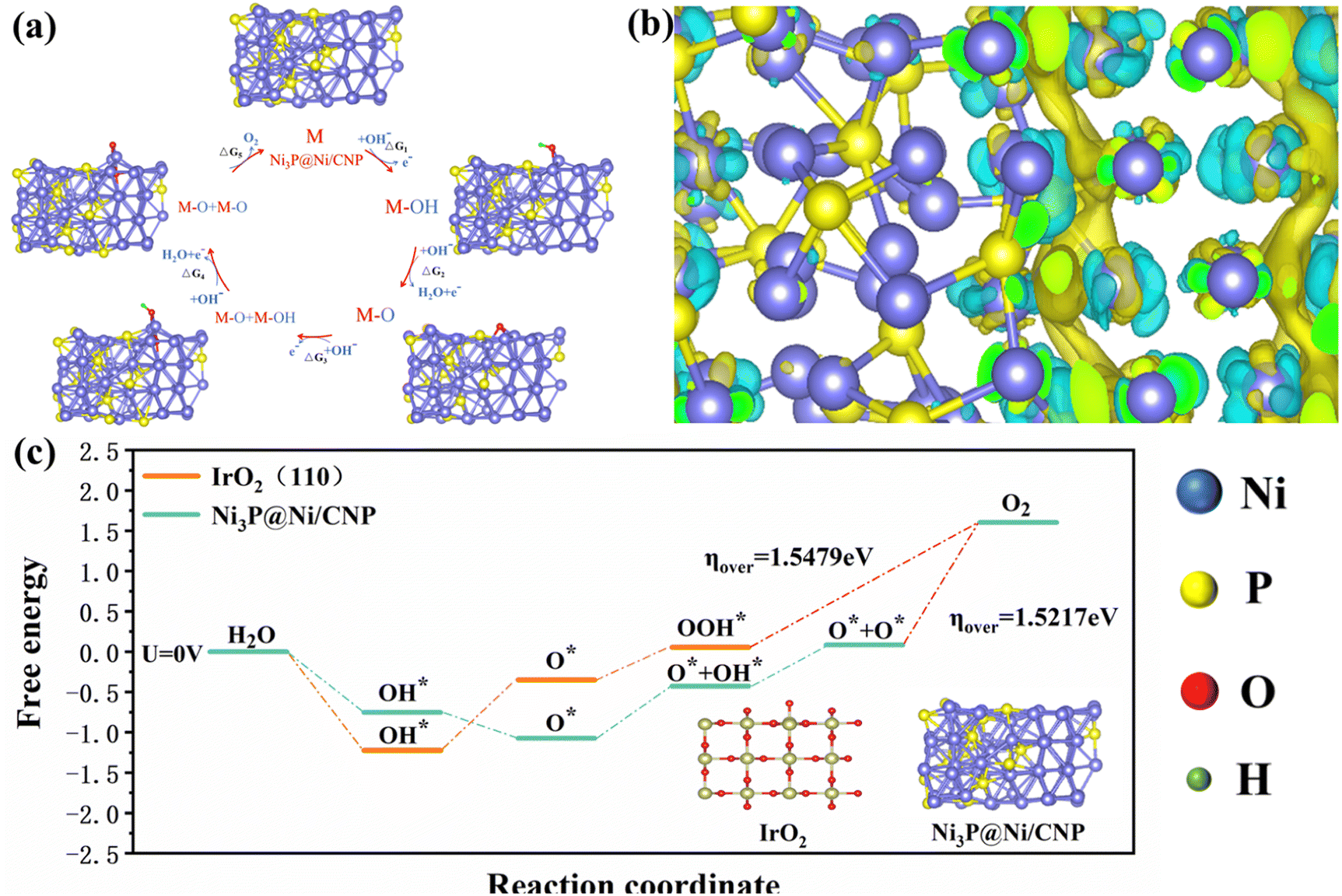 | ||
| Fig. 7 (a) OER mechanism of Ni3P@Ni/CNP in alkaline medium. (b) Schematic diagram of the interface charge. (c) Gibbs free energy plots of the OER steps on Ni3P@ Ni/CNP and IrO2 at U = 0 V. | ||
4. Conclusions
In conclusion, Ni3P@Ni/CNP heterojunction electrocatalysts were successfully synthesized by a wet-impregnation process of the Ni/PBI-OP precursor and subsequent phosphating/carbonization treatment. To this end, this exceptional microstructure could provide rich Ni3P@Ni heterojunction interfacial sites and strong interaction with CNP microspheres, which not only optimized and rearranged the electron configuration to elevate the intrinsic activity of electrochemically active sites but also boosted the structural stability and facilitated the interfacial electron transfer. Besides, there were affluent crystal defects that could be detected over the Ni3P@Ni/CNP samples, which could initiate the conductivity band to upgrade the electronic conductivity for the enhancement of OER kinetics. Furthermore, XPS illustrated that a large amount of pyridinic N species survived over the Ni3P@Ni/CNP samples, which could accelerate the O2 release and reduce the OER overpotential. Noticeably, DFT calculation also demonstrated that Ni3P@Ni/CNP presented a lower OER activation energy than IrO2, thereby further approving the OER kinetics. Among these samples, Ni3P@Ni/CNP-M perfectly inherited the advantages of a larger specific surface area, more exposed electrocatalytic active sites, higher conductivity, and a satisfactory heterojunction structure, thus accelerating the OER performance. Thus, Ni3P@Ni/CNP-M only needs a low overpotential of 239 mV in 1 M KOH electrolyte to reach the current density of 10 mA cm−2, and the low Tafel slope of 52 mV dec−1 is much lower than that of commercial supplies such as IrO2, showing excellent OER catalytic performance. Eventually, Ni3P@Ni/CNP-M presented excellent stability when tested continuously for 48 h at a current density of 10 mA cm−2.Conflicts of interest
The authors declared that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
This research was financially supported by the National Natural Science Foundation of China (No. U1904171), the Science and Technology Foundation of Henan Province (No. 222102240032), the Young Backbone Teachers Training Program Foundation of Henan University of Technology, and the Innovative Funds Plan of Henan University of Technology (No. 2021ZKCJ08).References
- A. Buttler and H. Spliethoff, Renewable Sustainable Energy Rev., 2018, 82, 2440–2454 CrossRef CAS
.
- J. Chi and H. Yu, Chin. J. Catal., 2018, 39, 390–394 CrossRef CAS
- H. Sun, Z. Yan, F. Liu, W. Xu, F. Cheng and J. Chen, Adv. Mater., 2020, 32, 1806326 CrossRef CAS PubMed
- Y. Xu and B. Zhang, ChemElectroChem, 2019, 6, 3214–3226 CrossRef CAS
- V. Charles, A. O. Anumah, K. A. Adegoke, M. O. Adesina, I. P. Ebuka, N. A. Gaya, S. Ogwuche and M. O. Yakubu, Sustainable Mater. Technol., 2021, 28, e00252 CrossRef CAS
- S. Chen, H. Huang, P. Jiang, K. Yang, J. Diao, S. Gong, S. Liu, M. Huang, H. Wang and Q. Chen, ACS Catal., 2020, 10, 1152–1160 CrossRef CAS
- A. Zagalskaya and V. Alexandrov, ACS Catal., 2020, 10, 3650–3657 CrossRef CAS
- A. Dutta and N. Pradhan, J. Phys. Chem. Lett., 2017, 8, 144–152 CrossRef CAS PubMed
- H. Li, P. Wen, D. S. Itanze, M. W. Kim, S. Adhikari, C. Lu, L. Jiang, Y. Qiu and S. M. Geyer, Adv. Mater., 2019, 31, 1900813 CrossRef PubMed
- R. Wang, X.-Y. Dong, J. Du, J.-Y. Zhao and S.-Q. Zang, Adv. Mater., 2018, 30, 1703711 CrossRef PubMed
- J. Yang, F. Zhang, X. Wang, D. He, G. Wu, Q. Yang, X. Hong, Y. Wu and Y. Li, Angew. Chem., Int. Ed., 2016, 55, 12854–12858 CrossRef CAS PubMed
- D. Gao, J. Zhang, T. Wang, W. Xiao, K. Tao, D. Xue and J. Ding, J. Mater. Chem. A, 2016, 4, 17363–17369 RSC
- X. Shen, W. Tan, Z. Wei, X. Wang, H. Zhou, J. Lv, M. Zhao, M. Zhang, J. Yang, G. He and L. Yang, J. Mater. Sci.: Mater. Electron., 2021, 32, 22974–22983 CrossRef CAS
- F. Zhang, S. Xi, G. Lin, X. Hu, X. W. Lou and K. Xie, Adv. Mater., 2019, 31, 1806552 CrossRef PubMed
- Q. Gao, W. Zhang, Z. Shi, L. Yang and Y. Tang, Adv. Mater., 2019, 31, 1802880 CrossRef PubMed
- Q. Hu, X. Liu, B. Zhu, L. Fan, X. Chai, Q. Zhang, J. Liu, C. He and Z. Lin, Nano Energy, 2018, 50, 212–219 CrossRef CAS
- S.-J. Guan, P. Zhang, S.-J. Ji, Y. Cao and N.-T. Suen, Inorg. Chem., 2022, 61, 12772–12780 CrossRef CAS PubMed
- L. Jin, H. Xia, Z. Huang, C. Lv, J. Wang, M. G. Humphrey and C. Zhang, J. Mater. Chem. A, 2016, 4, 10925–10932 RSC
- S. P. Keerthana, B. J. Rani, G. Ravi, R. Yuvakkumar, S. I. Hong, D. Velauthapillai, B. Saravanakumar, M. Thambidurai and C. Dang, Int. J. Hydrogen Energy, 2020, 45, 18859–18866 CrossRef CAS
- M. B. Stevens, C. D. M. Trang, L. J. Enman, J. Deng and S. W. Boettcher, J. Am. Chem. Soc., 2017, 139, 11361–11364 CrossRef CAS PubMed
- A. Ray, S. Sultana, L. Paramanik and K. M. Parida, J. Mater. Chem. A, 2020, 8, 19196–19245 RSC
- X. Li, J. Zhou, C. Liu, L. Xu, C. Lu, J. Yang, H. Pang and W. Hou, Appl. Catal., B, 2021, 298, 120578 CrossRef CAS
- B. Lu, Y. Wang, W. Li, S. Zhou, H. Gao, Q. Zou, J. Li and J. Zang, ACS Appl. Nano Mater., 2021, 4, 7443–7453 CrossRef CAS
- F. Wang, J. Chen, X. Qi, H. Yang, H. Jiang, Y. Deng and T. Liang, Appl. Surf. Sci., 2019, 481, 1403–1411 CrossRef CAS
- Y. Tan, Q. Li, Q. Che, X. Chen, X. Xu and Y. Chen, Electrochim. Acta, 2019, 324, 134897 CrossRef CAS
- J. Liu, Y. Wang, Y. Liao, C. Wu, Y. Yan, H. Xie and Y. Chen, ACS Appl. Mater. Interfaces, 2021, 13, 26948–26959 CrossRef CAS PubMed
- S. Li, Y. Zhang, Y. Yuan, F. Chang, K. Zhu, G. Li, Z. Bai and L. Yang, Int. J. Hydrogen Energy, 2023, 48, 3355–3363 CrossRef CAS
- Z. Li, M. Hu, P. Wang, J. Liu, J. Yao and C. Li, Coord. Chem. Rev., 2021, 439, 213953 CrossRef CAS
- X. Chen, Y. Chen, Z. Shen, C. Song, P. Ji, N. Wang, D. Su, Y. Wang, G. Wang and L. Cui, Appl. Surf. Sci., 2020, 529, 147173 CrossRef CAS
- Y. Zhuang, J. G. Seong, W. H. Lee, Y. S. Do, M. J. Lee, G. Wang, M. D. Guiver and Y. M. Lee, Macromolecules, 2015, 48, 5286–5299 CrossRef CAS
- Q. Wu, G. Xiao and D. Yan, e-Polym., 2008, 073 Search PubMed
- G. Wang, G. Xiao and D. Yan, J. Mater. Sci., 2011, 369, 388–396 CAS
- G. Wang, Y. Yao, G. Xiao and D. Yan, J. Membr. Sci., 2013, 425–426, 200–207 CrossRef CAS
- G. Wang, S. Yang, Y. Ding, M. Lu, B. Hua, J. Kang, W. Tang, H. Wei, L. Zhu and X. Cao, Chem. Commun., 2022, 58, 12333–12336 RSC
- T. Chen, B. Yang, S. Li, K. Wang, X. Jiang, Y. Zhang and G. He, Ind. Eng. Chem. Res., 2011, 50, 11043–11048 CrossRef CAS
- C. Du, M. Shang, J. Mao and W. Song, J. Mater. Chem. A, 2017, 5, 15940–15949 RSC
- J. Wojewoda-Budka, A. Wierzbicka-Miernik, L. Litynska-Dobrzynska, M. J. Szczerba, G. Mordarski, M. Mosialek, Z. Huber and P. Zieba, Electrochim. Acta, 2016, 209, 183–191 CrossRef CAS
- D. A. L. Almeida, A. B. Couto and N. G. Ferreira, J. Alloys Compd., 2019, 788, 453–460 CrossRef CAS
- M. Zhang, Q. Dai, H. Zheng, M. Chen and L. Dai, Adv. Mater., 2018, 30, 1703711 CrossRef PubMed
- G. Wang, S. Yang, M. Lu, B. Hua, Z. Zhang, J. Kang, W. Tang, H. Wei, L. Cui and X. Chen, Electrochim. Acta, 2023, 443, 141939 CrossRef CAS
- J. Yu, G. Cheng and W. Luo, J. Mater. Chem. A, 2017, 5, 11229–11235 RSC
- M. Yan, Z. Zhao, P. Cui, K. Mao, C. Chen, X. Wang, Q. Wu, H. Yang, L. Yang and Z. Hu, Nano Res., 2021, 14, 4220–4226 CrossRef CAS
- G. Shi, C. Yu, Z. Fan, J. Li and M. Yuan, ACS Appl. Mater. Interfaces, 2019, 11, 2662–2669 CrossRef CAS PubMed
- D. Li, Z.-F. Zhang, Z.-Y. Yang, W.-Y. Wu, M.-H. Zhang, T.-R. Yang, Q.-S. Zhang and J.-Y. Xie, J. Alloys Compd., 2022, 921, 166204 CrossRef CAS
- Q. Dong, M. Li, M. Sun, F. Si, Q. Gao, X. Cai, Y. Xu, T. Yuan, S. Zhang, F. Peng, Y. Fang and S. Yang, Small Methods, 2021, 5, 2100878 CrossRef CAS PubMed
- G. Li, J. Wang, J. Yu, H. Liu, Q. Cao, J. Du, L. Zhao, J. Jia, H. Liu and W. Zhou, Appl. Catal., B, 2020, 261, 118147 CrossRef CAS
- X. Chen, Y. Chen, X. Luo, H. Guo, N. Wang, D. Su, C. Zhang, T. Liu, G. Wang and L. Cui, Appl. Surf. Sci., 2020, 526, 146626 CrossRef CAS
- Y. Du, G. Pan, L. Wang and Y. Song, Appl. Surf. Sci., 2019, 469, 61–67 CrossRef CAS
- M. Li, L. Zhang, Q. Xu, J. Niu and Z. Xia, J. Catal., 2014, 314, 66–72 CrossRef CAS
- X. Li, L. Ni, J. Zhou, L. Xu, C. Lu, G. Yang, W. Ding and W. Hou, Nanoscale, 2020, 12, 13987–13995 RSC
- P. Ding, H. Song, J. Chang and S. Lu, Nano Res., 2022, 15, 7063–7070 CrossRef CAS
- J. Y. Xiang, J. P. Tu, X. L. Wang, X. H. Huang, Y. F. Yuan, X. H. Xia and Z. Y. Zeng, J. Power Sources, 2008, 185, 519–525 CrossRef CAS
- X.-D. Wang, Y. Cao, Y. Teng, H.-Y. Chen, Y.-F. Xu and D.-B. Kuang, ACS Appl. Mater. Interfaces, 2017, 9, 32812–32819 CrossRef CAS PubMed
- B. You, N. Jiang, M. Sheng, M. W. Bhushan and Y. Sun, ACS Catal., 2016, 6, 714–721 CrossRef CAS
- C. G. Read, J. F. Callejas, C. F. Holder and R. E. Schaak, ACS Appl. Mater. Interfaces, 2016, 8, 12798–12803 CrossRef CAS PubMed
- Q. Qin, J. Hao and W. Zheng, ACS Appl. Mater. Interfaces, 2018, 10, 17827–17834 CrossRef CAS PubMed
- L. Wang, C. Gu, X. Ge, J. Zhang, H. Zhu and J. Tu, Adv. Mater. Interfaces, 2017, 4, 1700481 CrossRef
- L. Yan, H. Jiang, Y. Wang, L. Li, X. Gu, P. Dai, D. Liu, S.-F. Tang, G. Zhao, X. Zhao and K. M. Thomas, Electrochim. Acta, 2019, 297, 755–766 CrossRef CAS
- M. Ledendecker, S. K. Calderon, C. Papp, H.-P. Steinrueck, M. Antonietti and M. Shalom, Angew. Chem., Int. Ed., 2015, 54, 12361–12365 CrossRef CAS PubMed
- Y. Zhao, G. Fan, L. Yang, Y. Lin and F. Li, Nanoscale, 2018, 10, 13555–13564 RSC
- H. Zhao, J. Liang and Y. Zhao, J. Alloys Compd., 2022, 907, 164479 CrossRef CAS
- X. Wang, B. Wang, Y. Chen, M. Wang, Q. Wu, K. Srinivas, B. Yu, X. Zhang, F. Ma and W. Zhang, J. Mater. Sci. Technol., 2022, 105, 266–273 CrossRef CAS
- M. Ramadoss, Y. Chen, X. Chen, Z. Su, M. Karpuraranjith, D. Yang, M. A. Pandit and K. Muralidharan, J. Phys. Chem. C, 2021, 125, 20972–20979 CrossRef CAS
- Y. Liu, B. Wang, K. Srinivas, M. Wang, Z. Chen, Z. Su, D. Liu, Y. Li, S. Wang and Y. Chen, Int. J. Hydrogen Energy, 2022, 47, 12903–12913 CrossRef CAS
- D. Yang, Z. Su, Y. Chen, Y. Lu, B. Yu, K. Srinivas, B. Wang and W. Zhang, J. Mater. Chem. A, 2020, 8, 22222–22229 RSC
- H. Wang, X. Wang, B. Zheng, D. Yang, W. Zhang and Y. Chen, Electrochim. Acta, 2019, 318, 449–459 CrossRef CAS
- Y. Liu, B. Wang, Y. Lu, Z. Su, Y. Li, Q. Wu, D. Yang, Y. Chen and S. Wang, J. Mater. Sci., 2021, 56, 16000–16009 CrossRef CAS
- Z. Su, Y. Lu, K. Srinivas, B. Wang, W. Zhang, Y. Chen and D. Yang, J. Alloys Compd., 2021, 883, 160926 CrossRef CAS
- D. Yang, Z. Su, Y. Chen, K. Srinivas, J. Gao, W. Zhang, Z. Wang and H. Lin, Small, 2020, 17, 2006881 CrossRef PubMed
- L. S. Bezerra and G. Maia, J. Mater. Chem. A, 2020, 8, 17691–17705 RSC
- Q. Mou, Z. Xu, G. Wang, E. Li, J. Liu, P. Zhao, X. Liu, H. Li and G. Cheng, Inorg. Chem. Front., 2021, 8, 2889–2899 RSC
- Y. Wang, S. Tao, H. Lin, G. Wang, K. Zhao, R. Cai, K. Tao, C. Zhang, M. Sun, J. Hu, B. Huang and S. Yang, Nano Energy, 2021, 81, 105606 CrossRef CAS
- Q. Hu, Z. Wang, X. Huang, Y. Qin, H. Yang, X. Ren, Q. Zhang, J. Liu, M. Shao and C. He, Appl. Catal., B, 2021, 286, 119920 CrossRef CAS
- C. Hu, L. Zhang and J. Gong, Energy Environ. Sci., 2019, 12, 2620–2645 RSC
- M. Najibah, E. Tsoy, H. Khalid, Y. Chen, Q. Li, C. Bae, J. Hnát, M. Plevová, K. Bouzek and J. H. Jang, J. Membr. Sci., 2021, 640, 119832 CrossRef CAS
- H. Liu, J. Gao, X. Xu, Q. Jia, L. Yang, S. Wang and D. Cao, Chem. Eng. J., 2022, 448, 137706 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ta05422e |
| This journal is © The Royal Society of Chemistry 2023 |