
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Quinone-functionalised carbons as new materials for electrochemical carbon dioxide capture†
Niamh A.
Hartley
 a,
Suzi M.
Pugh
a,
Zhen
Xu
a,
Daniel C. Y.
Leong
a,
Adam
Jaffe
b and
Alexander C.
Forse
*a
a,
Suzi M.
Pugh
a,
Zhen
Xu
a,
Daniel C. Y.
Leong
a,
Adam
Jaffe
b and
Alexander C.
Forse
*a
aYusuf Hamied Department of Chemistry, University of Cambridge, Cambridge, Cambridgeshire, UK. E-mail: acf50@cam.ac.uk
bDepartment of Chemistry and Biochemistry, University of Notre Dame, Notre Dame, IN, USA
First published on 30th June 2023
Abstract
The need for cost-effective carbon dioxide capture technology is rapidly increasing. To limit the global temperature increase to 1.5 °C within the next century, the level of CO2 mitigation needs to increase drastically. Current capture technology, i.e., amine scrubbing, provides several challenges that limit widespread deployment: high regeneration energy, high operational costs and degradation issues. An emerging energy-efficient technology that can address some of the limitations of amines is electrochemically driven carbon dioxide capture. For example, redox-active quinone molecules are capable of capturing carbon dioxide following electrochemical reduction, and can then be regenerated upon electrochemical oxidation. Despite great advances in the chemistry of quinones for electrochemical CO2 capture, however, the integration of quinones in carbon capture devices remains an ongoing challenge. Here we present a new class of quinone-functionalized electrodes for electrochemical CO2 capture, using the diazonium radical reaction to graft quinone molecules to a porous carbon surface. By grafting redox-active molecules to this conductive surface, not only is carbon dioxide capture significantly enhanced when the bound quinone species are electrochemically reduced, but the functionalization process also improves the energy storage of the carbon material. Through constant current experiments in the presence of CO2, reversible carbon capture was observed with initial uptake capacities at 0.4 mmol g−1 which stabilizes to 0.2 mmol g−1 over 100 cycles with an energy consumption of 254 kJ mol−1 per cycle. Our facile low-cost synthesis of quinone-functionalised carbons is highly tunable since both the carbon and redox-active molecule can be modified, and our work therefore paves the way for the design and discovery of improved electrode materials for electrochemical CO2 capture.
 Alexander C. Forse | Dr Alexander Forse is Assistant Professor in Materials Chemistry at the University of Cambridge. The Forse Group researches new materials that help with climate change mitigation, with a focus on energy storage and carbon dioxide capture. Dr Forse holds a UKRI Future Leaders Fellowship and an ERC starting grant, and he recently received the Anatole Abragram Prize for pioneering applications of NMR spectroscopy to the characterization of new materials for electrochemical energy storage and carbon dioxide capture. Before joining the faculty at Cambridge, Dr Forse was a Philomathia fellow at U.C. Berkeley. |
Introduction
Due to the rapid increase in anthropogenic CO2 emissions over the past century, a number of technologies have been developed to reduce CO2 emissions from point sources.1–3 The most mature technology is amine scrubbing, which employs amine molecules to capture CO2 at a high concentration.4 Limitations of this technology include low CO2 capacities, parasitic side-reactions and large regeneration energies.5,6 Amine scrubbing utilizes a temperature-swing process and requires high temperatures to release CO2 and regenerate the amines. Electrochemically driven carbon dioxide capture is emerging as an energy-efficient alternative to thermally driven processes.7 Electrochemical systems can be operated under isothermal conditions, limiting energy loss via heat. A range of electrochemical carbon dioxide capture systems have been reported, including those based on electrochemically driven pH swings,8 capacitive systems,9,10 and systems that employ redox-active molecules such as quinones, both in protic and aprotic electrolytes.11–15 Using redox-active quinone molecules is one of the largest areas of research for new technologies due to their highly reversible redox-activity and stoichiometric capture of CO2 upon reduction.Electrochemically driven carboxylation of reduced quinones has been well documented.16,17 While neutral anthraquinone does not show carbon dioxide binding, electrochemical reduction to form a radical anion (1 electron reduction) or dianion (2 electron reduction) activates this molecule to bind CO2 in either an EECC or ECEC (E = electron transfer, C = chemical reaction) mechanism (Scheme 1).18 While most early studies used analytical electrochemistry techniques to study the carbon dioxide capture of quinones and other redox-active molecules,19,20 more recent work has begun to develop practical devices that can capture carbon dioxide in an electrochemically driven process.21
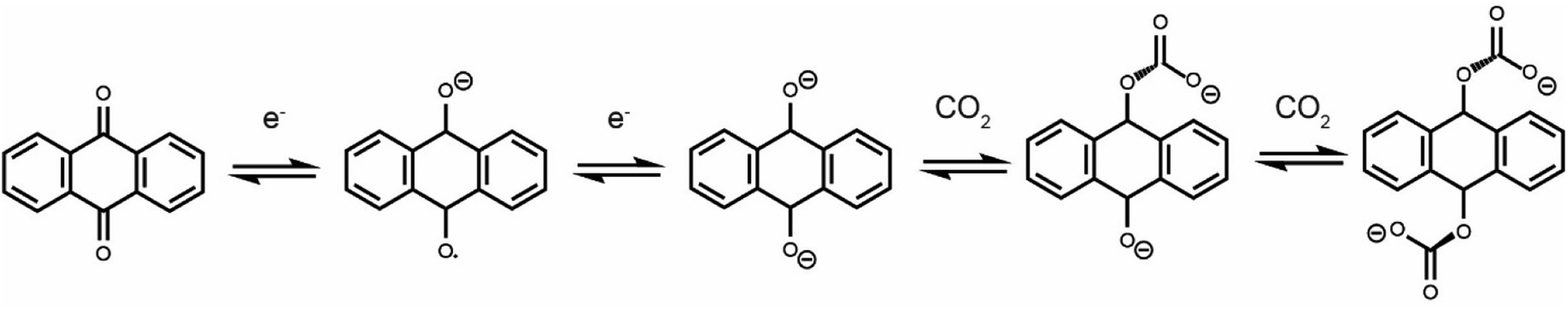 | ||
| Scheme 1 Anthraquinone redox activity in the presence of CO2. This scheme shows an EECC mechanism, though an ECEC mechanism is also thought to operate. | ||
One approach for making devices is to use quinones dissolved in solution in a flow battery-like system,22,23 though these systems suffer from low quinone solubility as well as the crossover of redox-active molecules through the separator membrane, causing capacity fade. An alternative approach that can address these issues is to immobilize quinones in a solid-state electrode in a battery-like architecture. Initial approaches in this direction used solid-state quinone electrodes to electrochemically capture carbon dioxide, yet they suffered from significant instability due to quinone dissolution into the organic electrolytes used,24,25 leading to a capacity loss when cycling. Research by Voskian et al.26 addressed this challenge by developing electrodes comprised of poly(anthraquinone) and carbon nanotubes and demonstrated electrochemical CO2 capture at a variety of CO2 concentrations. Notably, the poly(anthraquinone) electrodes could be stacked, allowing higher CO2 adsorption capacities to be achieved. However, a limitation of this approach is the complex synthesis of the polymer electrode, which requires expensive coupling agents. This motivates the need for the development of new classes of inexpensive, easily synthesized quinone-functionalized electrodes that are broadly tunable for electrochemical CO2 capture.
In the search for new quinone-based electrodes for electrochemical carbon dioxide capture, we were inspired by previous work on the covalent functionalisation of carbon electrodes for energy storage applications.27 Quinone functionalisation provides additional energy storage capacity through faradaic processes, adding to the initial capacity of the carbon from non-faradaic electrochemical double-layer capacitance.28 The use of a covalent attachment between quinone molecules and the carbon backbone prevents the dissolution of the redox-active molecule into the electrolyte, increasing the cycle life of the electrode. Furthermore, this attachment ensures facile electron transfer to and from grafted molecules. A popular approach for covalently attaching quinones to activated carbon is to use the spontaneous diazonium radical reaction.29,30 This well-understood reaction allows a range of materials to be investigated, as the reactions are tolerant to a diverse set of quinones and conductive carbon materials,31,32 and only require an inexpensive radical initiator to be stirred with the activated carbon material and quinone molecule present in solution. Quinone-functionalised activated carbons have previously been reported as improved energy storage materials, however, they have not yet been implemented in carbon capture technology.33
In this work we develop quinone-functionalised porous carbon materials for electrochemically driven carbon dioxide capture. We demonstrate that the incorporation of quinones increases the energy storage capacity of the carbon electrodes, while at the same time introducing electrochemically mediated carbon dioxide capture. Our work paves the way for the development of new materials that can help mitigate climate change.
Experimental
Material synthesis and purification
CMK-3 mesoporous carbon (type B, >99%) was purchased from ACS materials LLC. Microporous YP-80F was purchased from Kuraray. Polytetrafluoroethylene preparation (PTFE) (60 wt% dispersion in H2O), tert-butyl nitrite (90%) and 9,10-anthraquinone (97%) were purchased from Sigma-Aldrich.2-Aminoanthraquinone (>80%) was purchased from Tokyo Chemical Industry and further purified by sulphuric acid recrystallization.34 Analytical grade sulphuric acid (18 M, >95%) and methanol (99.9%) were purchased from Fischer Scientific. Acetonitrile, extra dry over molecular sieves (99.9%), was purchased from Acros Organics. N,N-Dimethylformamide (99%) was purchased from Alfa Aesar. Acetone and ethanol (>99%) were purchased from VWR Chemicals. Nylon membrane disks (diam. 47 mm, nylon 0.22 μm) were purchased from GVS North America.
1-Butyl-3-methylimidazolium bis(trifluoromethyl sulfonyl)imide (99%) was purchased from Iolitec. Carbon dioxide vapour withdrawal cylinder was purchased from BOC. The 3-electrode cell set-up was purchased from Swagelok (PFA-820-3) and stainless steel current collectors were custom-made.
Synthesis of functionalized CMK-3 (f-CMK-3)
Pognon et al. have previously shown that anthraquinone-grafted carbons can be synthesized by adding a diazonium radical initiator and 2-aminoanthraquinone (2-AAQ) to a porous carbon material.29 This study found that the anthraquinone loading on the surface could be changed using a variety of concentrations and mixing times. Our synthesis adapted the conditions that showed the highest anthraquinone loading from the literature protocol by mixing 2-AAQ and CMK-3 together for 24 hours prior to the addition of a radical initiator, allowing time for the 2-AAQ to diffuse into the inner pores in the carbon network, promoting an even distribution and reducing side reactions such as polymerization.35–38 For a microporous carbon, the diffusion of anthraquinone into the carbon network can block some pores, leading to a loss of surface area and a lower quinone loading.39 Pore blockages also affect the electrochemical behaviour, as it limits ionic transport of the electrolyte to the charged surface, thus leading to a loss in electrochemical capacity.40 This can be overcome by using mesoporous carbons with pore sizes between 2 and 10 nm to facilitate electrolyte accessibility. As such, CMK-3, a mesoporous carbon with a pore diameter >4 nm, was ultimately chosen for electrochemical quinone grafting.Mesoporous CMK-3 (50 mg, 1 equiv.) and 2-AAQ (0.28 g, 3 equiv.) were stirred in degassed acetonitrile for 16 hours under N2. Tert-butyl nitrite (0.13 g, 1 equiv. compared to 2-AAQ) was added and the mixture was stirred for 30 min, and then another 1 equiv. of tert-butyl nitrite was added to the organic suspension. This process was repeated until a total of 3 equiv. of tert-butyl nitrite were added. The suspension was kept at ambient temperature while stirring under N2. After 4 h, the reaction mixture was vacuum filtered on a nylon filtration membrane and the f-CMK-3 powder was washed with successive aliquots of acetonitrile (500 mL), DMF (500 mL), acetone (100 mL) and methanol (100 mL) and dried in a vacuum oven at 100 °C for 12 h. The final weight percentage increase after functionalisation was 22 ± 7 wt%.
For a control experiment, the procedure was repeated without including the tert-butyl nitrite reagent (which is required to form the diazonium species, and for covalent functionalisation to take place).
The same procedure was used for all freestanding composite films and all films had a thickness of ca. 250 μm. A similar procedure was used to make YP-80F films, except the carbon material used was as-purchased microporous YP-80F.
For a control experiment, CMK-3 (50 mg) was prepared in the presence of dissolved anthraquinone (5 mg, 10 wt%) in ethanol (5 mL). The suspension was stirred for 2 hours then kneaded to create an electrode film. Similarly, another control experiment followed the diazonium radical reaction without the addition of the radical initiator. CMK-3 (50 mg, 1 equiv.) and 2-AAQ (0.28 g, 0.3 equiv.) were stirred in degassed acetonitrile for 16 hours under N2 and then washed following the synthetic procedure explained above.
Two-electrode cells for electrochemical analysis were prepared as coin cells in Cambridge Energy Solutions CR2032 SS316 coin cell cases. Film electrodes were cut from freestanding composite f-CMK-3 and YP-80F films with areal mass loadings of f-CMK-3 ranging between 5–11 mg cm−2. The minimum weight ratio between the negative (f-CMK-3) and positive (YP80F) electrode was 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :>2.5, respectively. This ensured excess charging capacity on the YP-80F electrode (relative to the f-CMK-3 electrode). Therefore, the potential on the working f-CMK-3 electrode is expected to limit the stable working voltage range for the cell. Whatman glass microfiber filter (GF/A) was used as a separator and dried in vacuo at 100 °C for 24 h prior to use. 1-Butyl-3-methylimidazolium bis(trifluoromethyl sulfonyl)imide ionic liquid (200 μm) was used as an electrolyte in all experiments. All electrochemical cells were assembled inside a glovebox under an inert nitrogen (N2) atmosphere. Each coin cell contained two SS316 spacer disks and one SS316 spring to ensure sufficient and consistent pressure in the cells. The coin cells were sealed in the glovebox using a Compact Hydraulic Coin Cell Crimper (Cambridge Energy Solutions) before being removed for testing.
:>2.5, respectively. This ensured excess charging capacity on the YP-80F electrode (relative to the f-CMK-3 electrode). Therefore, the potential on the working f-CMK-3 electrode is expected to limit the stable working voltage range for the cell. Whatman glass microfiber filter (GF/A) was used as a separator and dried in vacuo at 100 °C for 24 h prior to use. 1-Butyl-3-methylimidazolium bis(trifluoromethyl sulfonyl)imide ionic liquid (200 μm) was used as an electrolyte in all experiments. All electrochemical cells were assembled inside a glovebox under an inert nitrogen (N2) atmosphere. Each coin cell contained two SS316 spacer disks and one SS316 spring to ensure sufficient and consistent pressure in the cells. The coin cells were sealed in the glovebox using a Compact Hydraulic Coin Cell Crimper (Cambridge Energy Solutions) before being removed for testing.
Electrochemical cell characterisation
Cyclic voltammetry (CV) and constant current experiments were performed on Biologic potentiostats (VSP-3e and VMP-3e) and the electrochemical set-up was controlled with Biologic EC-Lab V11.43 software. The CV scans were performed at a scan rate of 5 mV s−1 for all cells unless otherwise stated.Electrochemical gas cell experiments
Electrochemical gas adsorption experiments were carried out with a custom-made device reported previously (Fig. S1.1†).44 Electrochemical cells for gas measurements were assembled in meshed coin cells from MTI Corporation (CR2032-CASE-MESH, 1 mm holes) and placed in gas manifold devices with f-CMK3-K electrodes exposed to the gas reservoir. Devices were then evacuated for 10 min and filled with pure CO2 gas (>1.01 bar). This was repeated three times to fully exchange the N2 gas (from the glovebox environment in which the cells were prepared) with CO2. The electrochemical cells were placed in an incubator oven (SciQuip Incu-80S) at 35 °C. This temperature was consistent with similar literature systems, enabling accurate comparison.26 The constant current was applied across the cell and the increase and decrease in pressure in the device, upon release and capture, respectively, was monitored using a pressure transducer (Omega, PX309-030A5V) which was connected to the potentiostat as an external device, enabling simultaneous electrochemistry and cell gas pressure measurements.Constant-current experiments with constant voltage holding steps were carried out while simultaneously monitoring how the CO2 pressure in the system varied. A decrease in pressure is then characteristic of CO2 capture by the functionalized electrode and an increase in pressure is CO2 release. The pressure difference between capture and release was then used to calculate the number of CO2 molecules captured using the ideal gas law.
Results and discussion
Electrode fabrication and characterization
Targeting a quinone-functionalised carbon material for electrochemical CO2 capture, we adapted a previously developed synthetic route that employs a diazonium radical reaction (Scheme 2). We selected CMK-3 mesoporous carbon and 2-aminoanthraquinone to yield the anthraquinone functionalised carbon, f-CMK-3. We hypothesized that the use of a mesoporous carbon for the functionalisation reaction would avoid pore blockage, leaving sufficient porosity following functionalization for electrolyte and CO2 access during electrochemical CO2 capture.45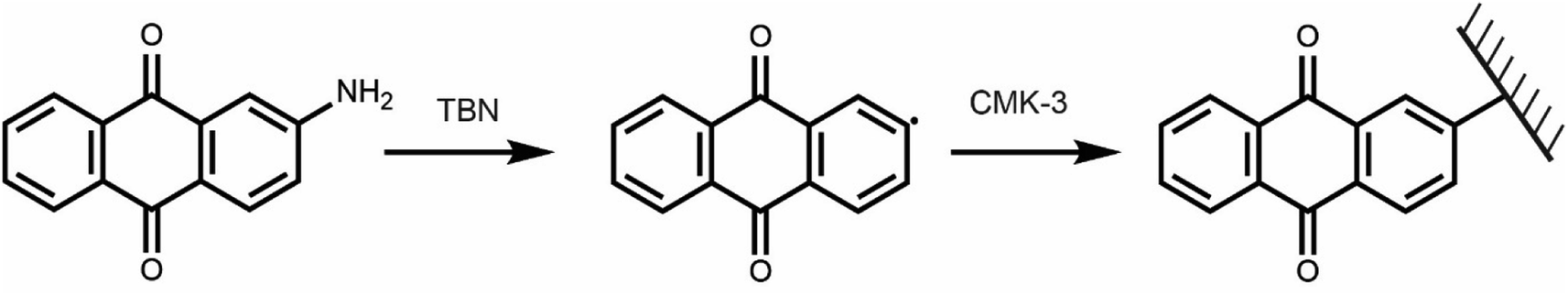 | ||
| Scheme 2 Functionalisation of CMK-3 using 2-AAQ and tert-butyl nitrite (TBN) used as the radical initiator. | ||
N2 gas sorption isotherms at 77 K showed typical type IV adsorption–desorption hysteresis loops for u-CMK-3 and f-CMK-3, consistent with the presence of mesopores,46 as well as a steep uptake at low relative pressures (0.0 to 0.1 P/P0), consistent with the presence of micropores (Fig. 1A & Section S2†). Upon quinone grafting, f-CMK-3 shows a decrease in adsorbed volume at relative pressures greater than 0.5 P/P0, suggesting that anthraquinone molecules have been grafted in the carbon mesopores, rather than in the micropores. Our findings are consistent with previous studies, that also attributed a decrease in surface area after quinone grafting to the presence of quinones on the carbon structure.32 However, gas sorption alone cannot be used to infer whether chemical grafting of anthraquinone has taken place, as a similar sorption profile may be seen regardless of whether anthraquinone was physisorbed or chemisorbed to the carbon surface. Furthermore, as molecular nitrogen has a significantly smaller radius than ions used in the ionic liquid electrolyte, 1-butyl-3-methylimidazolium bis(trifluoromethyl sulfonyl)imide ([bmim][TFSI]), gas sorption cannot provide information about the electrochemical accessibility of pores.47 In summary, the gas sorption measurements support the presence of anthraquinones within the mesoporous carbon and show that significant porosity is retained, which should enable CO2 species to enter the pores in electrochemical carbon capture measurements.
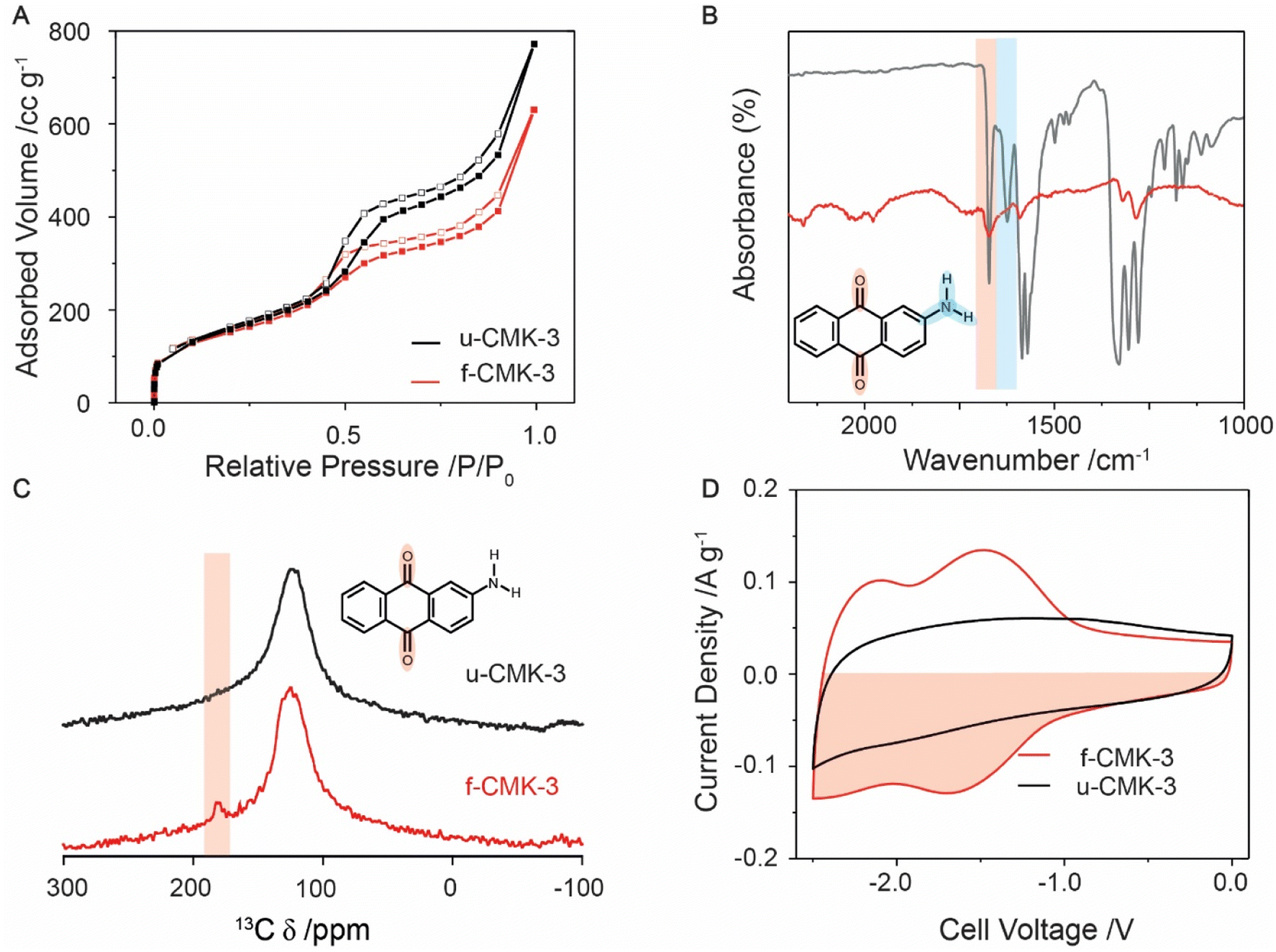 | ||
| Fig. 1 Characterization of u-CMK-3 (black line) and f-CMK-3 (red line). (A) N2 gas sorption adsorption (filled squares) and desorption (hollow squares) isotherms at 110 °C. (B) FT-IR comparison of the absorbance difference spectrum between f-CMK-3 and u-CMK-3 (red line) and 2-aminoanthraquinone (grey line). (C) Solid state 13C NMR of u-CMK-3 (black) and f-CMK-3 (red). Both samples were mixed with Cr(acac)3 to enhance longitudinal (T1) relaxation, see experimental section.59 Red shaded area shows a peak in the carbonyl region for f-CMK-3. (D) CVs of CMK-3, in [bmim][TFSI], at 1 mV s−1. The counter electrode was an oversized YP80F:PTFE (95:5 wt%) electrode, as described in the Experimental section. The capacity was calculated from the red shaded area. | ||
To ascertain whether anthraquinone was covalently attached to the CMK-3 support, FT-IR was used to monitor the loss of amine groups from 2-AAQ following functionalization (Fig. 1B & S3.1†) as well as the presence of the quinone carbonyls.48 Activated carbon is highly absorbing of IR light, resulting in low signal intensity for both CMK-3 samples. However, weak anthraquinone peaks can still be identified in f-CMK-3. The peaks for N–H and C–N bonds, shown at 3500 (Fig. S3.2†), 1650–1580 (blue shaded) and 1342–1266 cm−1, respectively,49 were observed in the 2-AAQ spectrum but were absent in u-CMK-3 and f-CMK-3. Though the signal intensities are weak for these samples, C–N modes are predicted to have strong transmittance due to aryl resonance and their absence, as well as the lack of N–H peaks in the f-CMK-3 spectrum, support the loss of amine groups during anthraquinone grafting. This was also supported by energy-dispersive X-ray spectroscopy (EDX) measurements where nitrogen was absent in the chemical composition of f-CMK-3 (Fig. S4.1 and S4.2†). X-ray photoelectron spectroscopy (XPS) did show a trace amount of nitrogen however, the origin was unknown (Fig. S5.1†). Meanwhile, the stretch of the carbonyl group at 1650 cm−1 (red shaded) was seen in both the 2-AAQ and f-CMK-3 spectra but not the u-CMK-3 spectrum, confirming the presence of quinone in the functionalised carbon. Peaks seen in f-CMK-3 at 1290 cm−1 are also present in anthraquinone and therefore are not assigned to C–N bond in 2-AAQ. The absence of N–H peaks in the f-CMK-3 spectrum supports the loss of the amine group during the radical synthesis, resulting in anthraquinone being covalently grafted to the CMK-3 structure. Solid state 13C NMR was used to further confirm the presence of anthraquinone in the f-CMK-3 structure (Fig. 1C). An intense broad peak was observed at approximately 120 ppm for both samples, and was assigned to graphitic-like sp2-hybridised carbon. The highlighted peak at 180 ppm for f-CMK-3 structure supports the additional presence of carbonyl bonds in anthraquinone units after functionalisation. Consistent with this, X-ray photoelectron spectroscopy (XPS) showed an increase in the carbonyl signal following functionalisation (Fig. S5.1†).
Electrochemical measurements further support the success of the functionalisation reaction and allow the amount of electrochemically accessible anthraquinone to be quantified. Cyclic voltammetry (CV) curves on two-electrode cells show clear differences in the charge storage behaviours of f-CMK-3 and u-CMK-3 (Fig. 1D). In these measurements, a microporous carbon, YP80F, with excess charge storage capacity was used as the positively charged electrode. The CV curve of the u-CMK-3 cell (black line) displays typical electrochemical double-layer capacitive behaviour, identified by a square-like CV with the absence of obvious redox peaks. This so-called “double-layer capacity” seen in activated carbon materials arises from capacitor-like energy storage at the interface between the electronically charged carbon surface and oppositely charged ions in the electrolyte. In contrast, the CV curve of f-CMK-3 exhibits two charge storage mechanisms: (a) double-layer capacitance, evidenced by a rectangular contribution, and (b) faradaic charge storage, evidenced by two redox couples centered at cell voltages of −1.5 V and −2.2 V which correspond to the two 1 e− reduction/oxidation processes of anthraquinone.50 A slight loss of double-layer capacity was seen at low cell voltages (e.g., −0.5 V) upon functionalization, likely owing to the reduction in surface area and pore blockage after anthraquinone functionalization on CMK-3, as seen by gas sorption. However, the overall charge-storage capacity increased by 25% from 39 to 52 mA h g−1 owing to the addition of the faradaic contributions.
Assuming any difference in charge storage for the two carbons could be attributed to anthraquinone, the estimated quinone loading was calculated to be 9 ± 1 wt% (Section S6†), which was within the range of what is expected from the literature synthesis.29 In this case, the small loss of double-layer capacity upon functionalization was ignored for simplification. The cell voltage window was limited by the stability of the electrolyte therefore two full reduction peaks could not be obtained. This suggests the quinone loading reported above is an underestimation of the amount of AQ present. Indeed, a simple estimation of the anthraquinone loading based on the sample mass increase following functionalisation gave a higher value of 22 ± 7 wt% (see experimental). This means that we cannot rule out the presence of some electrochemically inactive anthraquinone in f-CMK-3. We also explored further techniques to explore the incorporation of anthraquinones in f-CMK-3, including EDX (Table S4.1†), XPS (Table S5.2†) and TGA, (Fig. S7.1†). However, accurate quantification of anthraquinone loading from these techniques was not possible.51 Ultimately, we believe the electrochemical techniques are the best method to determine quinone loading as they indicate the quantity of quinone that is electrochemically accessible and therefore would partake in an electrochemical carbon capture process.
To further confirm anthraquinone was chemisorbed to CMK-3, control experiments were carried out. When the reaction was carried out without the addition of the TBN radical initiator, the resulting electrode material showed a cyclic voltammogram very similar to u-CMK-3, with no obvious faradaic contributions from anthraquinone observed, suggesting that physisorbed anthraquinone and 2-AAQ is removed during the washing steps of the synthesis (Fig. S8.1†). Similarly, when u-CMK-3 was mixed with dissolved anthraquinone during the electrode film-making process, clear redox peaks were again not observed (Fig. S8.1†). In summary, the grafting of anthraquinone onto the carbon surface was confirmed using a range of characterization techniques and control experiments, with the amount of electrochemically accessible anthraquinone on the surface estimated with a lower limit of 9 wt%.29
Electrochemical carbon dioxide capture measurements
Having successfully prepared anthraquinone-functionalised CMK-3, i.e., f-CMK-3, electrochemistry measurements were carried out in the presence of CO2 to assess whether this material could capture CO2 in an electrochemically driven process. Preliminary evidence for electrochemical CO2 capture was obtained from CV experiments, where differences in the two-electrode CV profiles were observed under N2 and CO2 (Fig. 2A). Under N2, the CV curve of the cell shows two redox events, while under CO2, the redox peaks are shifted, coalescing into a single broad peak at −1.9 V. A smaller peak at −0.75 V is also observed and has been previously linked to a stabilized quinone intermediate.13 Peak shifts have previously been reported in other quinone-based systems under the presence of CO2,11,52 with the second reduction peak generally reported to shift to more positive potentials while the potential of the first reduction peak remains largely unchanged. A shift in the second reduction peak to more positive potentials is caused by the reaction of the reduced quinone species with carbon dioxide. This is thought to make the second reduction more thermodynamically favorable through the stabilization of the products, leading to a shift of the second reduction peak to more positive potentials. The movement of both redox peaks that we observe for f-CMK-3 has not been previously reported in quinone-based CO2 capture chemistry. The cause for the coalescence of the two redox events is currently unknown, however, a shifting of peaks in the CV curves gave preliminary evidence for carbon dioxide capture by f-CMK-3.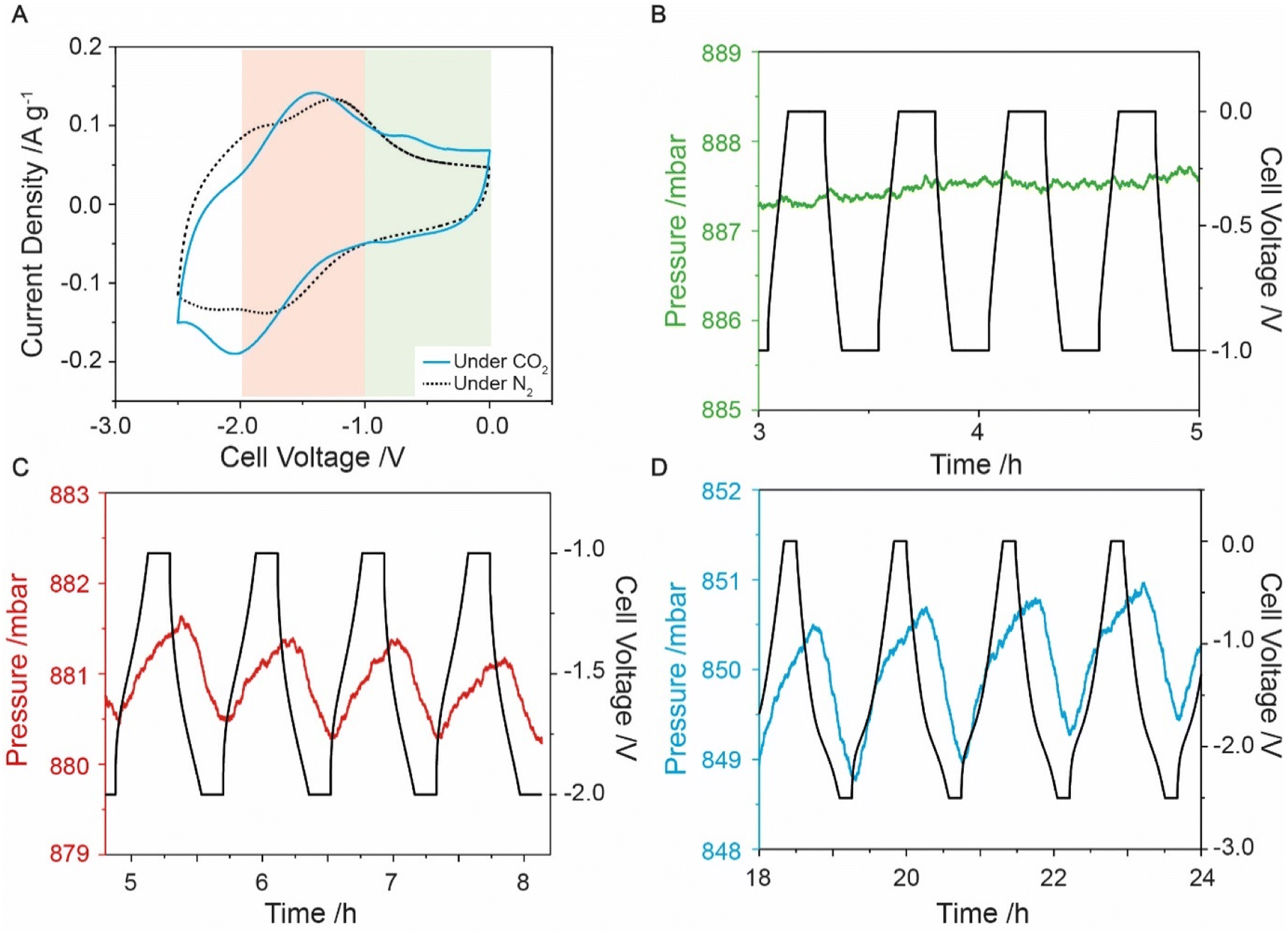 | ||
| Fig. 2 Data from f-CMK-3 cell in gas manifold under CO2 atmosphere. (A) CV under N2 (black dashed line) and CO2 (blue line) at 1 mV s−1. (B)–(D) show pressure and voltage data of f-CMK-3 under CO2 with different cell voltage windows. Experiments ran was a constant current at 0.1 A g−1 then voltage was held for 10 min. (B) 0 to −1 V, pressure – green line. (C) −1 to −2 V, pressure – red line. (D) 0 to −2.5 V, pressure – blue line. | ||
More conclusive evidence for electrochemically driven CO2 capture was provided by charging experiments during which the CO2 pressure was monitored in a custom electrochemical cell. To isolate the faradaic and double-layer capacity contributions to CO2 capture, three different voltage windows were explored. A voltage window of 0 to −1 V was first used to avoid anthraquinone redox-activity on f-CMK-3, which meant any pressure variation seen would not be due to anthraquinone redox (Fig. 2B). The measurements in this voltage range show negligible pressure variations, similar to what is seen for u-CMK-3 in the same voltage range (Fig. S9.2†). Experiments under an N2 environment also showed minimal pressure variation (Fig. S10.2 & S11.2†).
A voltage range of −1 to −2 V was then used to isolate anthraquinone redox activity and monitor electrochemically driven adsorption (Fig. 2C). When the f-CMK-3 cell was charged to −2 V, a decrease in pressure (red line) is observed, indicating the capture of CO2. When the f-CMK-3 electrode is then discharged to 0 V, the pressure in the system increases owing to the release of CO2. The pressure oscillations are synchronous with the electrochemical charging and discharging, demonstrating that the reduction and oxidation of anthraquinone drive the capture and release of carbon dioxide. This cell voltage range shows that targeting the redox activity of anthraquinone leads to carbon capture. Finally, experiments where the voltage was cycled between 0 and −2.5 V again revealed reversible capture and release of carbon dioxide, and showed larger magnitude pressure variations, corresponding to a larger CO2 capacity (Fig. 2D).
For optimization experiments, the voltage was limited to −2 V to minimize any parasitic side reactions at large cell voltages. Degradation was seen to increase when a larger voltage range was used (Fig. S12.1 and S12.2†) therefore a cell voltage window between −1 to −2 V was used. To optimize the CO2 capture capability of the electrode, the current density was varied and a value of 0.1 A g−1 was found to be slow enough for carbon dioxide to be captured by reduced anthraquinones but fast enough to limit any additional degradation of the cell (Fig. S13.1†). Adsorption capacity values of around 0.4 mmol g−1 were seen with an energy consumption of 256 kJ mol−1 (Section S14†). This energy consumption is higher than for the polyanthraquinone system developed by Voskian et al., however, the corresponding adsorption capacities were not reported for that system.26 These capacities were comparable to the theoretical value for 1 equivalent of CO2 per anthraquinone molecule, calculated as 0.46 mmol g−1 from our estimated quinone loading (Sections S15 & S16†). Previous anthraquinone capture systems were able to obtain a 1.5 CO2:quinone ratio,26 suggesting there is potential to optimize f-CMK-3 to obtain higher CO2 capacities. Other studies obtained an adsorption capacity of 5.9 mmolCO2 gAQ−1 which is comparable to our experimental value of 4.44 mmolCO2 gAQ−1 assuming a quinone loading of 9 wt%. However, these literature values were only seen in the initial 50 cycles before the cell degraded.24
The electronic charge stored was calculated from the electrochemistry measurements, providing information on the electrochemical behaviour of f-CMK-3 under CO2 (Section S17, Fig. S17.3†). Electronic charge values were approximately 1.0 mmol g−1 (i.e., 1.0 mmol of electrons per gram of f-CMK-3)—much higher than the CO2 adsorption values. The faradaic efficiency, the percentage of total charge extracted during cell discharge relative to during cell charging, was seen to increase above 90% during cycling (Fig. S17.4†). However, the charge-capture efficiency of the cell—i.e., the proportion of electronic charges that were used for CO2 capture—was ca. 20% after cycling (Fig. S17.4†). In other words, approximately 80% of the electronic charge stored is not utilized for CO2 capture and is used for other processes, e.g., capacitive and faradaic charging. A downward pressure slope was present in this limited voltage range (Fig. S17.2†), indicating some degradation may be taking place. Open circuit voltage (OCV) experiments were used to confirm no gas leakage in the system (Fig. S9.1, S10.1, S11.1 & 17.1†). In the future, more stable quinone molecules and carbon supports could reduce the amount of degradation seen within the system.53
Further experiments were run between 0 to −2.5 V with a 10 minutes voltage hold after each cycle to allow maximum adsorption (Fig. 2D). From the initial cycles, f-CMK-3 had an adsorption capacity of around 1 mmol g−1, which rapidly decreased upon cycling, presumably due to degradation processes (see below).54,55 After the system began to stabilise, the adsorption capacity leveled to around 0.4 mmol g−1, similar to what is seen for a limited cell voltage range of −1 to −2 V, but this larger voltage window proved more energy intensive, exhibiting a higher energy consumption of 830 kJ mol−1. Additionally, a steeper negative pressure slope for this larger voltage window suggests increased material degradation (Fig. S17.5†). Observe electronic charge values in the larger voltage window were around 2.5 mmol g−1 and are larger than the adsorption values, indicating poorer faradaic efficiency (Fig. S17.6 & S17.7†). A charge-capture efficiency of below 20% indicates a lower proportion of charge was used for CO2 capture processes compared to the more limited voltage window of −1 to −2 V. In summary, carbon capture has been shown to be facilitated by the presence of anthraquinone on CMK-3 however degradation occurs even in limited potential windows.
Interestingly, control experiments on the unfunctionalized carbon, u-CMK-3, also showed a small amount of reversible CO2 capture with an initial adsorption capacity of 0.15 mmol g−1 when cycled within a −1 to −2 V window (Fig. S9.3†), which is higher than what is seen under N2 atmosphere (Fig. S11.2†). It can be assumed that some redox-active functionalities are present on the u-CMK-3 surface, shown by a high oxygen content in elemental analysis and XPS (Tables S6.1 and S18.1†).56 These functionalities are likely to only give broad features in the CV experiments, indistinguishable from the double-layer capacitance, but may contribute to electrochemical CO2 capture when anthraquinone is not present.57 This unexpected electrochemical CO2 adsorption may also be due to the carbon electrode polarization causing enhanced CO2 physisorption. This would not be seen under N2 due to the lack of polarizability. The difference in adsorption capacity between f-CMK-3 and u-CMK-3 was assigned to grafted anthraquinone—around 0.2 mmol g−1—confirming grafting of anthraquinone on CMK-3 improves carbon adsorption.
Finally, long cycling studies were carried out between −1 to −2 V to monitor the cell degradation over 200 cycles (Fig. 3A). Following initial losses, the CO2 adsorption capacity began to stabilize around 0.2 mmol g−1. u-CMK-3 showed much smaller adsorption capacities, with no overlap seen between u-CMK-3 and f-CMK-3 within error (Fig. 3A). We hypothesize that some anthraquinone was lost from f-CMK-3 during cycling under CO2. The loss of anthraquinone after CO2 capture was further investigated using electrochemical techniques. The CV curve of f-CMK-3 showed a large decrease in faradaic contribution with minimal loss to double-layer capacity after 200 cycles (Fig. 3B). Anthraquinone loading was recalculated to be 3.4%; this accounted for the difference in electronic charge of u-CMK-3 and f-CMK-3 after 200 cycles. The lack of stability under CO2 was not seen under an N2 atmosphere. When cycling up to 3000 cycles, electronic charge capacity retention was 84% under N2 whereas under CO2 this was limited to 54% (Fig. S20.1†). The observed lack of long-term stability could be due to an irreversible reaction between f-CMK-3 and CO2. Fig. 3C shows that the electronic charge values were maintained at around 1 mmol g−1 with faradaic efficiencies above 95% after 200 cycles. The charge-capture efficiency was slightly lower than the value of 20% observed in the above experiment (Fig. 2C), indicating degradation is still present in f-CMK-3 materials.
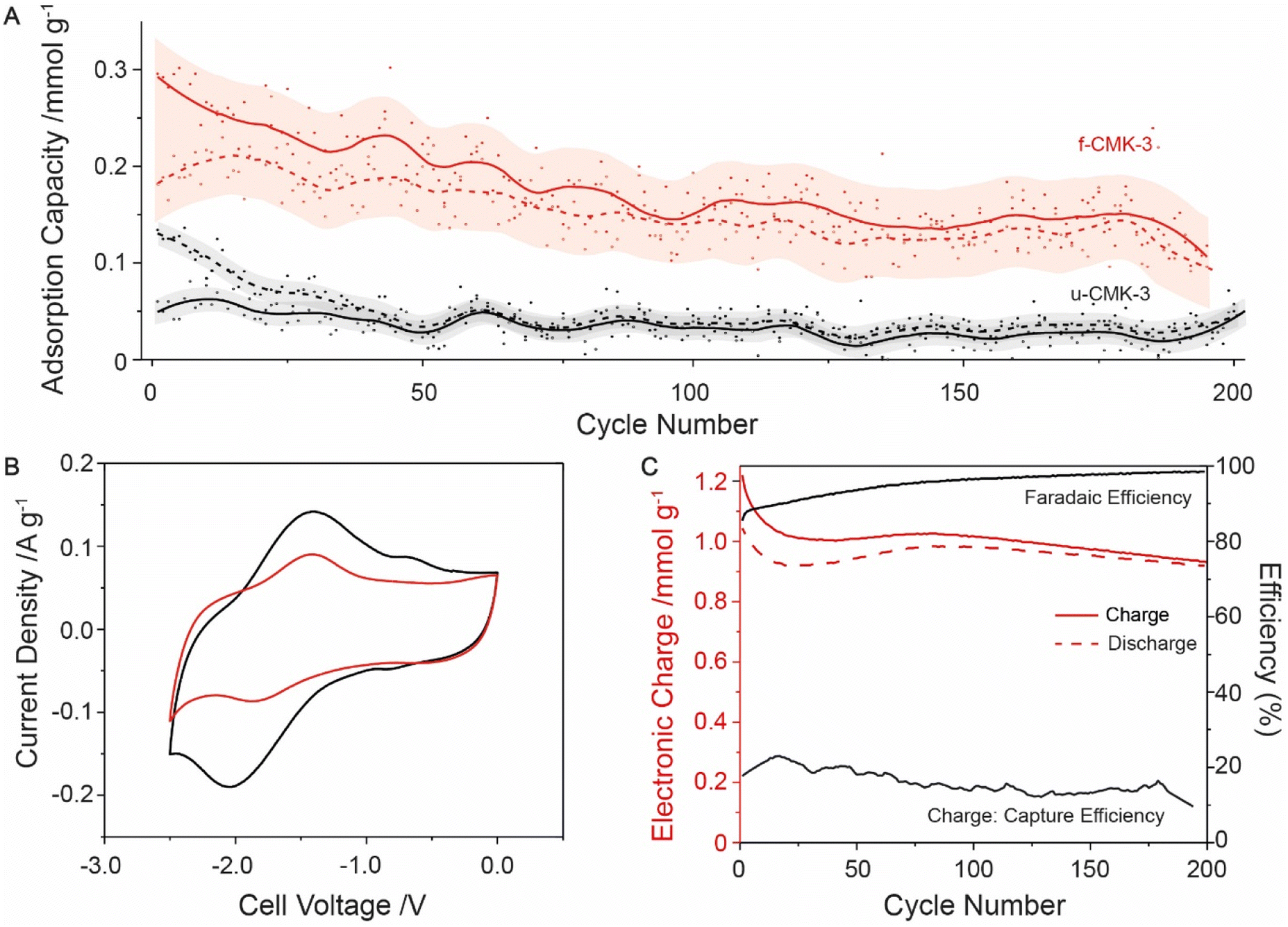 | ||
| Fig. 3 Long cycling experiment of f-CMK-3 and u-CMK-3 under CO2, cycled between −1 to −2 V at 0.1 A g−1 with voltage hold for 10 minutes. (A) Adsorption capacity of f-CMK-3 (red) and u-CMK-3 (black). The moving average for capture (solid line) and release (dashed) were calculated from raw data (filled and hollow circles respectively). Errors of moving average (transparent) were also calculated (Section S19†). (B) CV cycles of f-CMK-3 before (black) and after (red) cycling under CO2, showing a capacity decrease of 28%. (C) Charge and efficiency data given for f-CMK-3 experiment. Charge (solid red line) and discharge (dashed red line) plotted. Faradaic efficiency (solid black line) and charge-capture efficiency (solid black line) also plotted. | ||
The loss of electronic charge from anthraquinone degradation under CO2 was investigated using 1H NMR (Fig. S21.1†). After cycling, the ionic liquid electrolyte was collected and NMR spectra showed the presence of the [bmim][TFSI] electrolyte (as expected) as well as some unknown aromatic peaks. These were compared to anthraquinone (Fig. S21.1†), but could not be accurately assigned. This could suggest a small amount of anthraquinone has degraded and was released into the electrolyte. However, it is unknown if this degradation can account for an electronic capacity loss of 28%, thus still allowing for the possibility that some anthraquinone becomes electrochemically inaccessible after a reaction with CO2. In the future, a full investigation of the factors determining stability in these systems will be carried out, so that more stable sorbents can be obtained. Finally, we note that our materials are readily tunable since both the porous carbon and the redox-active support can be readily varied. Indeed, we functionalised a second porous carbon which also showed evidence for electrochemical CO2 capture (Fig. S22.1†).
Conclusion and outlook
In this work we have developed a new class of quinone-functionalised carbon materials for electrochemically driven CO2 capture. Structural characterization and electrochemistry measurements showed that anthraquinones were successfully grafted to CMK-3 mesoporous carbon using the diazonium radical reaction to yield f-CMK-3. Following quinone functionalisation, clear increases in electrochemical charge storage capacity were obtained due to anthraquinone redox reactions. By incorporating f-CMK-3 in an electrochemical gas cell and applying a cell voltage in the range of −1 to −2 V in the presence of CO2, CO2 adsorption capacities ca. 0.4 mmol g−1 were demonstrated that utilize the faradaic redox reactions of anthraquinone on the carbon surface. The addition of anthraquinone therefore improves both the carbon capture and energy storage abilities of the solid-state electrode. Upon cycling, however, a loss of carbon dioxide adsorption is seen which could be due to an irreversible reaction between anthraquinone and CO2. Our facile synthesis method lends itself to the preparation of a large family of sorbents for electrochemical CO2 capture. Indeed, the porous carbon support and the redox-active molecules can readily be varied to prepare a large family of materials. Research beyond the scope of this study into the carbon-capture mechanism within these systems is ongoing. Furthermore, more work to investigate the efficiency of electrochemical capture systems in more realistic conditions, in lower concentrations of CO2, and in the presence of oxygen and moisture needs to be carried out. These conditions have yet to be investigated for this material but have been carried out for other systems.15,58 In the future, we envisage that optimization of these materials will lead to higher electrochemical CO2 capture capacities, as well as improved stability under operating conditions. Our work opens the door to studies of an entirely new class of materials for electrochemical CO2 capture.Data availability
All raw experimental data files are available in the Cambridge Research Repository, Apollo, with the identifier https://doi.org/10.17863/CAM.95424.Author contributions
N. A. H, A. J. and A. C. F designed the research. N. A. H performed the material synthesis and electrode film fabrication. N. A. H. performed and analysed the FT-IR, TGA, SEM-EDX and gas experiments. N. A. H and D. C. Y. L performed and analysed the electrochemistry experiments. S. M. P performed the solid state 13C NMR experiments. Z. X and N. A. H. interpreted the XPS data. All authors interpreted the results and contributed to the writing of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by an EPSRC New Horizons award (EP/V048090/1), a UKRI Future Leaders Fellowship to A. C. F. (MR/T043024/1), and an EPSRC doctoral training award to N. A. H. (ref EP/T517847/1). D. C. Y. L. acknowledges the RSC for the award of an Undergraduate Research Bursary. We thank Israel Temprano for their support with equipment for electrochemical gas sorption measurements. We thank Jamie W. Gittins and Emma J. Latchem for their collaboration and technical expertise. We thank Mengnan Wang at the Department of Chemical Engineering, Imperial College London for running the XPS samples. For the purpose of open access, the author has applied a Creative Commons Attribution (CC BY) licence to any Author Accepted Manuscript version arising.References
- J. Tollefson and K. R. Weiss, Nations Adopt Historic Global Climate Accord, Nature, 2015, 528, 315–316 CrossRef CAS PubMed.
- V. Masson-Delmotte, P. Zhai, A. Pirani, S. L. Connors, C. Pean, S. Berger, N. Caud, Y. Chen, L. Goldfarb, M. I. Gomis, M. Huang, K. Leitzell, E. Lonnoy, J. B. R. Matthews, T. K. Maycock, T. Waterfield, O. Yelekci, R. Yu and B. Zhou, IPCC, 2021: Summary for Policymakers, in Climate Change 2021: the Physical Science Basis. Contribution of Working Group I to the Sixth Assessment Report of the Intergovernmental Panel on Climate Change, Cambridge, 2021, DOI:10.1017/9781009157896.001.
- W. Gao, S. Liang, R. Wang, Q. Jiang, Y. Zhang, Q. Zheng, B. Xie, C. Y. Toe, X. Zhu, J. Wang, L. Huang, Y. Gao, Z. Wang, C. Jo, Q. Wang, L. Wang, Y. Liu, B. Louis, J. Scott, A.-C. Roger, R. Amal, H. He and S.-E. Park, Industrial Carbon Dioxide Capture and Utilization: State of the Art and Future Challenges, Chem. Soc. Rev., 2020, 49, 8584–8686, 10.1039/d0cs00025f.
- G. T. Rochelle, Amine Scrubbing for CO2 Capture, Carbon Capture and Greenhouse Gases, 2010, 325, 1652–1654, DOI:10.1007/978-3-642-28036-8_100191.
- S. Vasudevan, S. Farooq, I. A. Karimi, M. Saeys, M. C. G. Quah and R. Agrawal, Energy Penalty Estimates for CO2 Capture: Comparison between Fuel Types and Capture-Combustion Modes, Energy, 2016, 103, 709–714, DOI:10.1016/j.energy.2016.02.154.
- A. C. Forse and P. J. Milner, New Chemistry for Enhanced Carbon Capture: Beyond Ammonium Carbamates, Chem. Sci., 2021, 14, 508–516, 10.1039/d0sc06059c.
- L. E. Clarke, M. E. Leonard, T. A. Hatton and F. R. Brushett, Thermodynamic Modeling of CO2 Separation Systems with Soluble, Redox-Active Capture Species, Ind. Eng. Chem. Res., 2022, 61(29), 10531–10546, DOI:10.1021/acs.iecr.1c04185.
- S. Jin, M. Wu, R. G. Gordon, M. J. Aziz and D. G. Kwabi, PH Swing Cycle for CO2 capture Electrochemically Driven through Proton-Coupled Electron Transfer, Energy Environ. Sci., 2020, 13(10), 3706–3722, 10.1039/d0ee01834a.
- C. Liu and K. Landskron, Design, Construction, and Testing of a Supercapacitive Swing Adsorption Module for CO2 Separation, Chem. Commun., 2017, 53(26), 3661–3664, 10.1039/c7cc01055a.
- B. Kokoszka, N. K. Jarrah, C. Liu, D. T. Moore and K. Landskron, Supercapacitive Swing Adsorption of Carbon Dioxide, Angew. Chem., Int. Ed., 2014, 53(14), 3698–3701, DOI:10.1002/anie.201310308.
- J. M. Barlow and J. Y. Yang, Oxygen-Stable Electrochemical CO2 Capture and Concentration with Quinones Using Alcohol Additives, J. Am. Chem. Soc., 2022, 144(31), 14161–14169, DOI:10.1021/jacs.2c04044.
- B. Gurkan, F. Simeon and T. A. Hatton, Quinone Reduction in Ionic Liquids for Electrochemical CO2 Separation, ACS Sustainable Chem. Eng., 2015, 3(7), 1394–1405, DOI:10.1021/acssuschemeng.5b00116.
- F. Simeon, M. C. Stern, K. M. Diederichsen, Y. Liu, H. J. Herzog and T. A. Hatton, Electrochemical and Molecular Assessment of Quinones as CO2-Binding Redox Molecules for Carbon Capture, J. Phys. Chem. C, 2022, 126(3), 1389–1399, DOI:10.1021/acs.jpcc.1c09415.
- J. S. Kang, S. Kim and T. A. Hatton, Redox-Responsive Sorbents and Mediators for Electrochemically Based CO2 Capture, Curr. Opin. Green Sustainable Chem., 2021, 100504, DOI:10.1016/j.cogsc.2021.100504.
- Y. Liu, H. Z. Ye, K. M. Diederichsen, T. Van Voorhis and T. A. Hatton, Electrochemically Mediated Carbon Dioxide Separation with Quinone Chemistry in Salt-Concentrated Aqueous Media, Nat. Commun., 2020, 11(1), 1–11, DOI:10.1038/s41467-020-16150-7.
- T. Journal, M. B. Mizen and M. S. Wrighton, Reductive Addition of CO2 to 9,10-Phenanthrenequinone, J. Electrochem. Soc., 1989, 136(4), 941–946 CrossRef.
- D. H. Apaydin, E. D. Głowacki, E. Portenkirchner and N. S. Sariciftci, Direct Electrochemical Capture and Release of Carbon Dioxide Using an Industrial Organic Pigment: Quinacridone, Angew. Chem., Int. Ed., 2014, 53(26), 6819–6822, DOI:10.1002/anie.201403618.
- D. L. Dubois, A. Miedaner, W. Bell and J. C. Smart, Chapter 4 - ELECTROCHEMICAL CONCENTRATION OF CARBON DIOXIDE, in Electrochemical and Electrocatalytic Reactions of Carbon Dioxide, 1993, pp. 94–117, DOI:10.1016/B978-0-444-88316-2.50008-5.
- T. Comeau Simpson and R. R. Durand, Reactivity of Carbon Dioxide with Quinones, Electrochim. Acta, 1990, 35(9), 1399–1403, DOI:10.1016/0013-4686(90)85012-C.
- T. Nagaoka, N. Nishii, K. Fujii and K. Ogura, Mechanisms of Reductive Addition of CO2, to Quinones in Acetonitrile, J. Electroanal. Chem., 1992, 322, 383–389 CrossRef CAS.
- A. Hemmatifar, J. S. Kang, N. Ozbek, K. J. Tan and T. A. Hatton, Electrochemically Mediated Direct CO2 Capture by a Stackable Bipolar Cell, ChemSusChem, 2022, 15(6) DOI:10.1002/cssc.202102533.
- F. Pan and Q. Wang, Redox Species of Redox Flow Batteries: A Review, Molecules, 2015, 20(11), 20499–20517, DOI:10.3390/molecules201119711.
- K. M. Diederichsen, Y. Liu, N. Ozbek, H. Seo and T. A. Hatton, Toward Solvent-Free Continuous-Flow Electrochemically Mediated Carbon Capture with High-Concentration Liquid Quinone Chemistry, Joule, 2022, 6(1), 221–239, DOI:10.1016/j.joule.2021.12.001.
- D. Wielend, D. H. Apaydin and N. S. Sariciftci, Anthraquinone Thin-Film Electrodes for Reversible CO2 Capture and Release, J. Mater. Chem. A, 2018, 6(31), 15095–15101, 10.1039/c8ta04817g.
- Y. Liang, Z. Tao and J. Chen, Organic Electrode Materials for Rechargeable Lithium Batteries, Adv. Energy Mater., 742–769, DOI:10.1002/aenm.201100795.
- S. Voskian and T. A. Hatton, Faradaic Electro-Swing Reactive Adsorption for CO2 Capture, Energy Environ. Sci., 2019, 12(12), 3530–3547, 10.1039/c9ee02412c.
- D. Wielend, Y. Salinas, F. Mayr, M. Bechmann, C. Yumusak, H. Neugebauer, O. Brüggemann and N. S. Sariciftci, Immobilized Poly(Anthraquinones) for Electrochemical Energy Storage Applications: Structure-Property Relations, ChemElectroChem, 2021, 8(22), 4360–4370, DOI:10.1002/celc.202101315.
- K. Kalinathan, D. P. DesRoches, X. Liu and P. G. Pickup, Anthraquinone Modified Carbon Fabric Supercapacitors with Improved Energy and Power Densities, J. Power Sources, 2008, 181(1), 182–185, DOI:10.1016/j.jpowsour.2008.03.032.
- G. Pognon, T. Brousse, L. Demarconnay and D. Bélanger, Performance and Stability of Electrochemical Capacitor Based on Anthraquinone Modified Activated Carbon, J. Power Sources, 2011, 196(8), 4117–4122, DOI:10.1016/j.jpowsour.2010.09.097.
- A. Jaffe, A. Saldivar Valdes and H. I. Karunadasa, Quinone-Functionalized Carbon Black Cathodes for Lithium Batteries with High Power Densities, Chem. Mater., 2015, 27(10), 3568–3571, DOI:10.1021/acs.chemmater.5b00990.
- G. Schmidt, S. Gallon, S. Esnouf, J.-P. Bourgoin and P. Chenevier, Mechanism of the Coupling of Diazonium to Single-Walled Carbon Nanotubes and Its Consequences, Chem. - Eur. J., 2009, 15, 2101–2110 CrossRef CAS PubMed.
- D. Malka, O. Hanna, T. Hauser, S. Hayne, S. Luski, Y. Elias, R. Attias, T. Brousse and D. Aurbach, Improving the Capacity of Electrochemical Capacitor Electrode by Grafting 2-Aminoanthraquinone over Kynol Carbon Cloth Using Diazonium Chemistry, J. Electrochem. Soc., 2018, 165(14), A3342–A3349, DOI:10.1149/2.0531814jes.
- Y. R. Hu, X. L. Dong, H. K. Zhuang, D. Yan, L. Hou and W. C. Li, Introducing Electrochemically Active Oxygen Species to Boost the Pseudocapacitance of Carbon-Based Supercapacitor, ChemElectroChem, 2021, 8(16), 3073–3079, DOI:10.1002/celc.202100641.
- P. H. Groggins and H. P. Newton, 2-Aminoanthraquinone from Chlorobenzene and Phthalic Anhydride Further Studies in Its Preparation, Ind. Eng. Chem., 1929, 21(4), 369–375, DOI:10.1021/ie50232a032.
- J. K. Kariuki and M. T. McDermott, Formation of Multilayers on Glassy Carbon Electrodes via the Reduction of Diazonium Salts, Langmuir, 2001, 17(19), 5947–5951, DOI:10.1021/la010415d.
- J. K. Kariuki and M. T. McDermott, Nucleation and Growth of Functionalized Aryl Films on Graphite Electrodes, Langmuir, 1999, 15(19), 6534–6540, DOI:10.1021/la990295y.
- F. Anariba, S. H. DuVall and R. L. McCreery, Mono-and Multilayer Formation by Diazonium Reduction on Carbon Surfaces Monitored with Atomic Force Microscopy “Scratching", Anal. Chem., 2003, 75(15), 3837–3844, DOI:10.1021/ac034026v.
- M. Delamar, G. Desarmot, O. Fagebaume, R. Hitmi, J. Pinson and J. M. Saveant, Modification of carbon fiber surfaces by electrochemical reduction of aryl diazonium salts: Application to carbon epoxy composites, Carbon, 1997, 35, 801–807 CrossRef CAS.
- T. Brousse, C. Cougnon and D. Bélanger, Grafting of Quinones on Carbons as Active Electrode Materials in Electrochemical Capacitors, J. Braz. Chem. Soc., 2018, 989–997, DOI:10.21577/0103-5053.20180015.
- E. Lebègue, C. Benoit, T. Brousse, J. Gaubicher and C. Cougnon, Effect of the Porous Texture of Activated Carbons on the Electrochemical Properties of Molecule-Grafted Carbon Products in Organic Media, J. Electrochem. Soc., 2015, 162(12), A2289–A2295, DOI:10.1149/2.0481512jes.
- S. Brunauer, P. H. Emmett and E. Teller, Adsorption of Gases in Multimolecular Layers, J. Am. Chem. Soc., 1938, 60, 309–319 CrossRef CAS.
- M. Thommes, K. Kaneko, A. v. Neimark, J. P. Olivier, F. Rodriguez-Reinoso, J. Rouquerol and K. S. W. Sing, Physisorption of Gases, with Special Reference to the Evaluation of Surface Area and Pore Size Distribution (IUPAC Technical Report), Pure Appl. Chem., 2015, 87(9–10), 1051–1069, DOI:10.1515/pac-2014-1117.
- J. Chmiola, G. Yushin, Y. Gogotsi, C. Portet, P. Simon and P. L. Taberna, Anomalous Increase in Carbon Capacitance at Pore Sizes Less Than 1 Nanometer, Science, 2006, 313(5794), 1760–1763, DOI:10.1126/science.113219.
- T. B. Binford, G. Mapstone, I. Temprano and A. C. Forse, Enhancing the Capacity of Supercapacitive Swing Adsorption CO2 Capture by Tuning Charging Protocols, Nanoscale, 2022, 14(22), 7980–7984, 10.1039/d2nr00748g.
- A. C. Forse, J. M. Griffin, C. Merlet, J. Carretero-Gonzalez, A. R. O. Raji, N. M. Trease and C. P. Grey, Direct Observation of Ion Dynamics in Supercapacitor Electrodes Using in situ Diffusion NMR Spectroscopy, Nat. Energy, 2017, 2(3), 16216, DOI:10.1038/nenergy.2016.216.
- M. Kruk and M. Jaroniec, Gas Adsorption Characterization of Ordered Organic-Inorganic Nanocomposite Materials, Chem. Mater., 2001, 3169–3183, DOI:10.1021/cm0101069.
- J. C. Groen, L. A. A. Peffer and J. Perez-Ramirez, Pore Size Determination in Modified Micro- and Mesoporous Materials. Pitfalls and Limitations in Gas Adsorption Data Analysis, Microporous Mesoporous Mater., 2003, 60, 1–17 CrossRef CAS.
- C. M. Hussain, C. Saridara and S. Mitra, Modifying the Sorption Properties of Multi-Walled Carbon Nanotubes via Covalent Functionalization, Analyst, 2009, 134(9), 1928–1933, 10.1039/b823316k.
- Sigma Aldrich, IR Spectrum Table & Chart, https://www.sigmaaldrich.com/GB/en/technical-documents/technical-article/analytical-chemistry/photometry-and-reflectometry/ir-spectrum-table, accessed 2022-12-05.
- C. Han, H. Li, R. Shi, T. Zhang, J. Tong, J. Li and B. Li, Organic Quinones towards Advanced Electrochemical Energy Storage: Recent Advances and Challenges, J. Mater. Chem. A, 2019, 7(41), 23378–23415, 10.1039/c9ta05252f.
- M. Toupin and D. Bélanger, Spontaneous Functionalization of Carbon Black by Reaction with 4-Nitrophenyldiazonium Cations, Langmuir, 2008, 24(5), 1910–1917, DOI:10.1021/la702556n.
- A. T. Bui, N. A. Hartley, A. J. W. Thom and A. C. Forse, Trade-Off between Redox Potential and the Strength of Electrochemical CO2 Capture in Quinones, J. Phys. Chem. C, 2022, 126(33), 14163–14172, DOI:10.1021/acs.jpcc.2c03752.
- D. P. Tabor, R. Gómez-Bombarelli, L. Tong, R. G. Gordon, M. J. Aziz and A. Aspuru-Guzik, Mapping the Frontiers of Quinone Stability in Aqueous Media: Implications for Organic Aqueous Redox Flow Batteries, J. Mater. Chem. A, 2019, 7(20), 12833–12841, 10.1039/c9ta03219c.
- R. G. Compton and C. E. Banks, Equilibrium Electrochemistry and the Nernst Equation, in Understanding Voltammetry, 2010, pp. 1–34 Search PubMed.
- A. Gennaro, A. A. Isse and E. Vianeuo, Solubility and Electrochemical Determination of CO2 in Some Dipolar Aprotic Solvents, J. Electroanal. Chem. Interfacial Electrochem., 1990, 289, 203–215 CrossRef CAS.
- İ. Demiral, C. Samdan and H. Demiral, Enrichment of the Surface Functional Groups of Activated Carbon by Modification Method, Surf. Interfaces, 2021, 22, 100873, DOI:10.1016/j.surfin.2020.100873.
- C. M. Ghimbeu, R. Gadiou, J. Dentzer, D. Schwartz and C. Vix-Guterl, Influence of Surface Chemistry on the Adsorption of Oxygenated Hydrocarbons on Activated Carbons, Langmuir, 2010, 26(24), 18824–18833, DOI:10.1021/la103405j.
- J. M. Kolle, M. Fayaz and A. Sayari, Understanding the Effect of Water on CO2 Adsorption, Chem. Rev., 2021, 121(13), 7280–7345, DOI:10.1021/acs.chemrev.0c00762.
- G. C. Levy and J. D. Cargioli, Spin-Lattice Relaxation in Solutions Containing Cr(III) Paramagnetic Relaxation Agents, J. Magn. Reson., 1973, 10, 231–234 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ta02213g |
| This journal is © The Royal Society of Chemistry 2023 |