
Recent progress in block copolymer soft-template-assisted synthesis of versatile mesoporous materials for energy storage systems
Keon-Woo
Kim
ab,
Bomi
Park
ab,
Jun
Kim
ab,
Changshin
Jo
*bc and
Jin Kon
Kim
 *ab
*ab
aNational Creative Research Initiative Center for Hybrid Nano Materials By High-Level Architectural Design of Block Copolymer, Republic of Korea
bDepartment of Chemical Engineering, Pohang University of Science and Technology (POSTECH), Pohang, Gyungbuk 790-784, Republic of Korea. E-mail: jochangsihn@postech.ac.kr; jkkim@postech.ac.kr
cGraduate Institute of Ferrous & Energy Materials Technology (GIFT), Pohang University of Science and Technology (POSTECH), Republic of Korea
First published on 6th March 2023
Abstract
Soft-templating methods, which utilize the block copolymer (BCP)-derived self-assembly with inorganic precursors, have been extensively applied to synthesize a wide range of mesoporous materials. Compared with other synthetic approaches including template-free and hard templating methods, the soft-templating method offers significant advantages for customizing various compositions, particle morphologies, and pore sizes/structures of mesoporous materials. During the last decade, various soft templating approaches have been developed to synthesize functional mesoporous materials for a variety of applications. In this review, we outline recent developments in synthetic approaches for mesoporous materials and their potential applications, particularly in energy storage systems (ESSs) such as batteries and supercapacitors. In addition, this review provides general information about soft-templating methods which can be applied to the tailored synthesis for the specific requirements of various applications.
 Keon-Woo Kim | Keon-Woo Kim received a B.S. degree in polymer science & engineering from Yeungnam University in Korea (2017). Currently he is a PhD candidate in chemical engineering at POSTECH, Republic of Korea, under the supervision of Prof. Jin Kon Kim since 2017. His research interests include synthesis of nanomaterials and their application to printing technologies and electrochemical devices. |
 Bomi Park | Bomi Park received a B.S. degree in chemistry from POSTECH (2022). Currently she is a M.S. candidate in chemical engineering at POSTECH, Republic of Korea, under the supervision of Prof. Jin Kon Kim since 2022. Her research interests include synthesis of mesoporous materials by using block copolymers and their application in energy storage systems. |
 Jun Kim | Jun Kim received a B.S. degree in chemical engineering from POSTECH (2017). Currently he is a PhD candidate in chemical engineering at POSTECH, Republic of Korea, under the supervision of Prof. Jin Kon Kim since 2017. His research interests include synthesis of mesoporous 3D graphene utilizing block copolymers and its application in energy storage systems. |
 Changshin Jo | Changshin Jo is an Assistant Professor at the Graduate Institute of Ferrous & Energy Materials Technology (GIFT) and the Department of Chemical Engineering at Pohang University of Science and Technology (POSTECH). He obtained his B.S. degree (2010) and PhD degree (2016) on synthesis of nanostructured electrode materials in the Department of Chemical Engineering at POSTECH. He then joined the University of Cambridge as a Research Associate and Marie Skłodowska-Curie Fellow (2017–2020) in the Department of Engineering. He also worked two years as an Assistant Professor in the Department of Chemical Engineering at Chung-Ang University (2020–2022). The main themes of his research in the NEDL group have been (i) synthesis of functional inorganic materials and (ii) applications of those materials in energy storage applications. |
 Jin Kon Kim | Jin Kon Kim is a full professor of Chemical Engineering at POSTECH. Also, he is the director of the Center for Smart Block Copolymer Self-Assembly funded by the National Creativity Research Initiative Program supported by the National Research Foundation in Korea. He received his B.S. in chemical engineering from Seoul National University (Korea) in 1980, M.S. in chemical engineering from Korea Advanced Institute of Science and Technology (Korea) in 1982, and PhD in chemical engineering from Polytechnic University in 1990. His research interests include phase behavior of block copolymers and development of new functional nanomaterials based on block copolymer self-assembly. |
1. Introduction
To provide a stable supply that meets the massive energy requirement of modern society, effective energy storage systems (ESSs) should be developed.1–13 Energy storage devices broadly consist of electrolytes, active materials, and current collectors.14,15 In general, energy storage is achieved through the redox reaction between the electrolyte and active materials.16–25 Therefore, increasing the interfacial contact area has been considered as an effective strategy to enhance device performance.3,26–36 This consideration has influenced many research groups to focus on controlling the physical architecture of active materials.27–30Porous materials have several advantages owing to the presence of pores and voids within their structure. First, the high surface area of porous materials provides numerous active sites that can interact with electrolytes in oxidation/reduction reactions. Second, the porous structure significantly reduces the diffusion distance for adsorption/desorption of ions, enabling stable charging/discharging under fast operating conditions. In addition, the characteristics of porous materials depend on the type of porous structure. For example, porous materials with bicontinuous structures allow efficient mass and ion transport to the interior of the materials, maximizing active sites. In contrast, porous materials with discrete pores are ineffective in mass and ion transport due to the low connectivity of pores.33,37–43
Porous materials are divided into three categories according to pore size, namely, macropores (>50 nm), mesopores (2–50 nm), and micropores (<2 nm),44–46 and show several (dis)advantages depending on the pore size. For instance, despite their extremely high surface areas, microporous materials are not suitable for efficient mass transport owing to their small pore size.15,47–49 Meanwhile, macroporous materials facilitate mass transport owing to their large pore sizes, but their surface area is not sufficient to achieve high energy storage performance.50–52 Mesoporous materials complement the features of the other two types. Because of their high surface area and large pore size, they have been widely used for catalysts,53,54 energy storage systems,55–59 and sensors.60,61 In particular, the high surface area allows the effective loading of host materials, which increases the energy storage capacity and allows effective charge transfer between the mesoporous framework and host materials. In addition, the interconnected large pores provide structural stability by relieving the volume expansion and strain applied to the material during repeated charge/discharge cycles.62–68
Several methods, such as hard and soft templating, have been developed to synthesize mesoporous materials. Hard templating typically uses mesoporous silica,48,62 carbon,48 and colloidal crystals69 as sacrificial templates. Benefiting from the thermal stability of the hard template, this method can produce various mesoporous materials with high crystallinities. However, this method requires the time-consuming process of preparation of a sacrificial template. In addition, the use of corrosive solvents for template removal restricts composition versatility and synthesis flexibility because they often dissolve several metal oxides and substrates.5,70–72 On the other hand, the soft-templating method can effectively synthesize mesoporous materials because it mainly uses organic materials such as small molecular surfactants or block copolymers (BCPs) as both structure directing agents (SDAs) and templates, which are easy to control their molecular weight and chemical composition and easy to remove. For example, precursors of a target material are filled in pre-assembled BCPs followed by induction of the reaction. After removing the BCPs through solvent extraction, mesoporous materials are obtained. Instead of the above sequential synthetic approach, direct co-assembly of the precursors with SDAs is mainly used to form a mesostructured composite, which does not require a pre-assembly process of BCPs. The obtained mesostructured composites are finally converted to mesoporous materials through post-treatments, such as calcination and solvent extraction.64,73–98
Because it enables tailoring of synthesis conditions, soft templating is a powerful synthetic approach capable of producing mesoporous materials with various compositions and morphologies. The properties of mesoporous materials play a vital role in determining the final performance; therefore, a number of synthetic approaches have been reported for tailoring various compositions (carbon,84,99,100 metal oxide,101,102 and metal-based materials103,104) and morphologies (hierarchical structures,82,105,106 particles,107,108 and patterned films109–111).
Herein, we focus on the recent developments in the mesoporous materials and their application to ESSs. In particular, the synthetic approaches and composition of carbon and metal-based materials (metal, metal oxide, and metal nitride) were covered, which have been intensively studied for ESSs. Following the introduction, the second section introduces the basic principles of synthetic approaches (hard and soft templating) for mesoporous materials. In the third section, various compositions of mesoporous materials prepared using the soft-templating method are reviewed. The fourth section discusses soft-templating methods for synthesizing various hierarchical architectures composed of mesoporous structures. The fifth section presents the potential applications of mesoporous materials in batteries and supercapacitors. Finally, the summary and perspective of the soft-templating method are described.
2. Synthetic approaches for mesoporous materials
Several synthetic approaches have been reported for preparing mesoporous materials with various compositions, such as metals, metal oxides, metal nitrides, and carbon.143–163 In general, the condensation and crystallization reactions of most precursors proceed in random directions to form a micrometer-scale bulk structure. Therefore, the morphological changes in most materials during the reaction are difficult to predict, thus limiting nano-scale structural control.112–115 To solve this issue, hard-template and soft-template methods that can maintain the mesoporous structure during the reaction of precursors have been proposed.116–119Hard-templating methods use thermally stable templates such as pre-synthesized mesoporous silica, carbon, and metal oxide-based nanocrystals (Fig. 1a). To obtain mesoporous materials, the reaction proceeds upon heat treatment after the hard template is filled with the precursors. Owing to the thermally stable hard template, the reaction can be terminated without structural collapse even at high temperatures,106,120–123 resulting in high-quality mesoporous materials having highly crystallinity and even single-crystallinity.70,71 However, this method has the disadvantages of being a time-consuming process and costly, because an additional process is required to prepare the sacrificial template. In the process of removing the sacrificial template, the use of a corrosive solvent is unavoidable, which is non-environment friendly and even harmful. In addition, it is difficult to control the pore size/shape because the resultant product is the inverted shape of the sacrificial template.
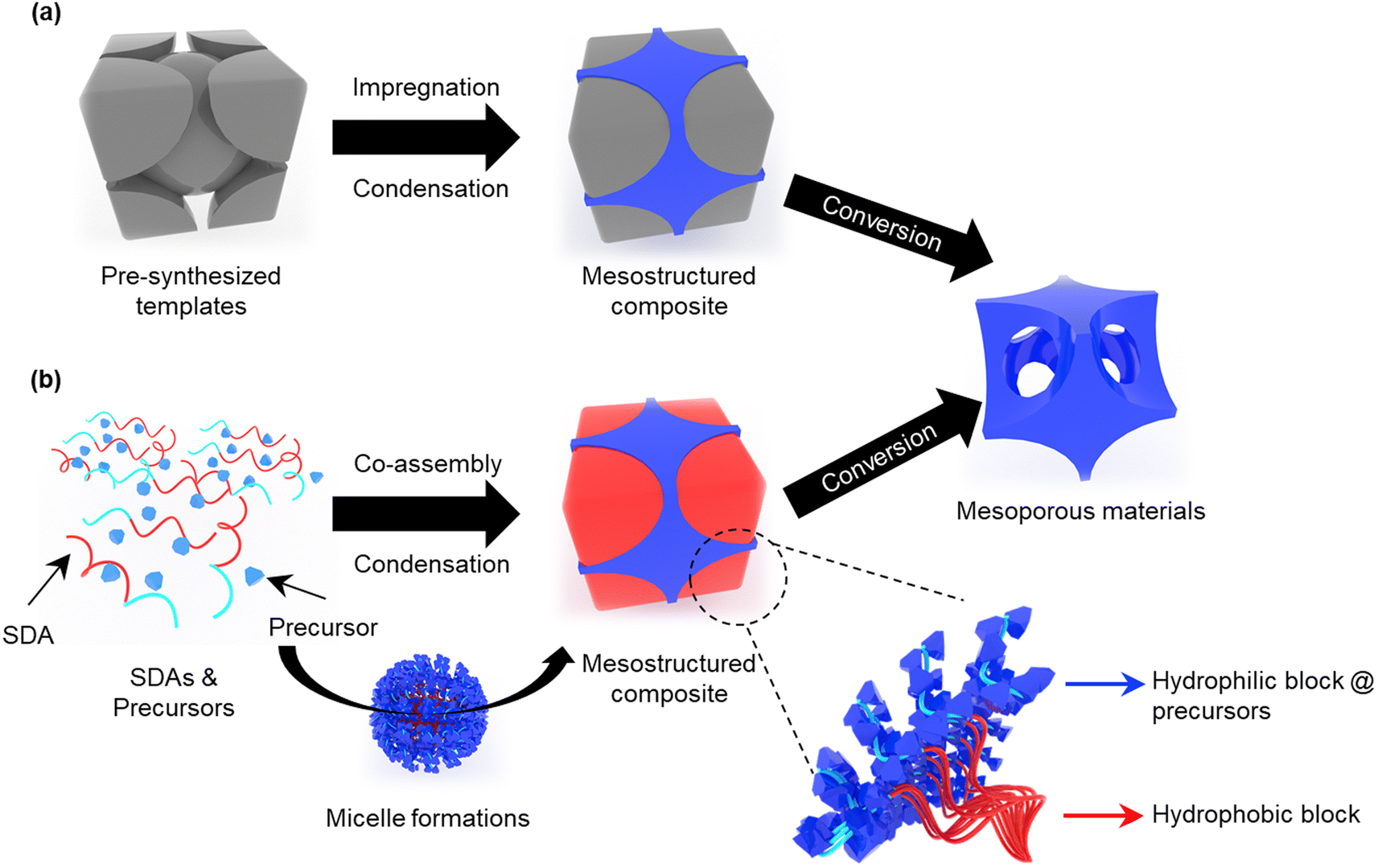 | ||
| Fig. 1 Schematic of (a) hard- and (b) soft-templating methods for fabricating mesoporous materials. | ||
Meanwhile, soft-templating has been regarded as a powerful synthetic approach for mesoporous materials because this method enables efficient control of mesoporous materials in a short time through the co-assembly of a SDA and precursors to form mesostructured composites (Fig. 1b). Here, SDAs act as both the structure-directing agent and the soft template. When a solution containing SDAs and precursors evaporates and reaches a concentration higher than the critical micelle concentration, the SDAs and precursors start self-assembling into spherical or cylindrical micelles. After the solvent completely evaporates, a mesostructured composite is formed. This process is called evaporation-induced self-assembly (EISA).124–128 When hydrophilic precursors are used, the obtained mesostructured composite consists of a stack of micelles in which the hydrophobic and hydrophilic parts containing the precursors are separated. After the template is removed (mainly by calcination), the hydrophilic and hydrophobic parts were converted into a skeleton and mesopores, respectively. The soft templating method can produce mesoporous materials with various compositions and morphologies by adjusting the precursors and molecular structures of the SDAs (Fig. 2). Despite their high flexibility and universal applicability, many parameters should be considered for soft-templating. Also, their structural stability and crystallinity are lower than those synthesized by hard-templating methods. For instance, co-assembly of SDAs and precursors requires sensitive synthesis conditions (e.g., precursor reactivity control, appropriate humidity, solvent composition, etc.), and collapse of the mesoporous structure often occurs during the template removal process. Therefore, many efforts have been made to address the above issues, which will be discussed in detail in the following section.
 | ||
| Fig. 2 Synthetic versatility of mesoporous materials based on the soft-templating method. | ||
3. Soft-template synthesis of mesoporous materials with various compositions
The soft-template synthetic process is divided into two steps: (i) formation of a mesostructured composite through micelle formation and (ii) conversion from a mesostructured composite to mesoporous materials by eliminating SDAs. During the formation of a mesostructured composite (step (i)), suitable SDAs should be chosen. Small-molecule surfactant SDAs are inexpensive and soluble in both water and organic solvents, enabling the easy synthesis of mesoporous materials. However, their low molecular weight limits the pore size control. Meanwhile, BCPs have easily controllable lengths of hydrophilic and hydrophobic blocks; thus, the pore size and wall thickness can be finely tuned. The most commonly used BCPs as SDAs comprise both hydrophilic and hydrophobic blocks. Poly(ethylene oxide) (PEO), poly(2-vinylpyridine) (P2VP), and poly(4-vinylpyridine) (P4VP) have been widely used as hydrophilic blocks, whereas poly(propylene oxide) (PPO), polystyrene (PS), poly(isoprene) (PI), and poly(butadiene) (PB) are used as hydrophobic blocks.129–133 During step (ii), mesoporous materials are obtained by removing the SDA (mainly using heat treatment) and improving the mechanical strength of the components. Because SDAs with small molecular weights have poor thermal stability, structural collapses often occur during high-temperature calcination. Meanwhile, BCPs can enhance thermal stability depending on the type of block, and an ordered mesoporous structure can be maintained without structural collapse even after calcination at high temperatures. Under these considerations, BCP-based SDAs can maintain structural stability even at high temperatures, enabling the synthesis of mesoporous carbon and metal oxides, as well as metal nitrides that require an additional high-temperature nitridation process. Compared to other compositions, mesoporous metals require a complementary soft-templating method because of their high surface energy and difficulty in co-assembling precursors and SDAs. In this section, we focus on synthetic approaches for mesoporous carbon, metal oxides, metals, and metal nitrides. The mesoporous materials covered in this review are summarized in Table 1.| Product | Template | Solvent | Precursor | Pore size (nm) | S BET (m2 g−1) | Ref. |
|---|---|---|---|---|---|---|
| Mesoporous carbon | PEO-b-PPO-b-PEO | EtOH | Resol | 6.8 | 590.0 | 146 |
| Mesoporous polymer–silica and carbon–silica nanocomposites | F127 | EtOH/HCl | Tetraethyl orthosilicate/resol | 6.7 | 2470.0 | 147 |
| Mesoporous carbon | PS-b-PEO | THF | Resol | 23.0 | 1510.0 | 117 |
| Mesoporous carbon | PAN-b-PMMA | — | PAN-b-PMMA | 5.96–17.42 | 427.6–213.1 | 28 |
| N and O dual-doped mesoporous carbon | PAN-b-PMMA | DMF | PAN-b-PMMA | 10.0 | 503.0 | 26 |
| N-Doped mesoporous carbon | PS-b-PAA | THF/H2O | Urea–formaldehyde | 9.5–17.2 | 458.0–476.0 | 29 |
| Mesoporous TiO2 | PI-b-PEO | THF | Titanium chloride | 23.6 | 75.0 | 137 |
| Mesoporous TiO2 | PS-b-PEO | THF | Titanium isopropoxide/titanium chloride | 20.7 | 72.0 | 162 |
| Mesoporous TiO2 | PS-b-PEO | THF/AcAc | Titanium isopropoxide | 16.0 | 112.0 | 68 |
| Mesoporous TiO2 | PS-b-P2VP-b-PEO | THF/HCl | Titanium isopropoxide | 35.0–45.0 | 90.0 | 89 |
| Mesoporous Nb2O5 | PI-b-PEO | Chloroform | Niobium chloride/niobium ethoxide | 24.0 | 69.0 | 137 |
| Mesoporous CoOx/C | PS-b-PEO | THF | Cobalt nitrate hexahydrate/resol | 13.4–16.0 | 394.0–483.0 | 165 |
| Mesoporous WO3 | PS-b-PEO | THF/AcAc | Tungsten chloride | 10.6–15.3 | 76.0–136.0 | 166 |
| Mesoporous TiNb2O7 | PS-b-PEO | THF/HCl | Titanium isopropoxide/niobium ethoxide/tetraethyl orthosilicate | 40.0 | 74.0 | 155 |
| Mesoporous CexZr1−xO2 | PS-b-PEO | THF/EtOH | Zirconium acethylacetonate/cerium chloride heptahydrate | 10.6–15.1 | 65.8–73.7 | 156 |
| Mesoporous Fe2O3 | PS-b-PAA-b-PEG | THF/NaOH | Iron nitrate nonahydrate | 39.0 | 86.9 | 177 |
| Mesoporous Pt | PI-b-PDMAEMA | Chloroform/Methanol | Stabilized Pt | 17.0 | 18.0 | 192 |
| Mesoporous Au | PS-b-PEO | — | Gold chloride trihydrate | 20.0–60.0 | — | 194 |
| Mesoporous Rh | PMMA-b-PEO | DMF/H2O | Sodium hexachlororhodate/ascorbic acid | 9.0 | 50.0 | 193 |
| Mesoporous TiN–C | F127 | EtOH/H2O | TiCl4/resol | 8.0 | 389.0 | 37 |
| Mesoporous TiN | PI-b-PS-b-PEO | THF/HCl | Pt | 18.8 | 60.0 | 206 |
| Mesoporous NbN | PI-b-PS-b-PEO | THF/HCl | Pt | 26.8 | 49.0 | 206 |
3.1. Carbon-based materials
Mesoporous carbons have been widely used as catalysts,134–136 fuel cells,53,137,138 and energy storage devices40,139–141 owing to their high surface area, light weight, and excellent thermal and chemical stability.142–145 To synthesize mesoporous carbon using the soft-template method, the following conditions must be satisfied. First, a mesostructured composite should be formed through the co-assembly of the carbon precursors and SDAs; for this purpose, low molecular weight precursors should be used. Second, during high-temperature heat treatment to convert the mesostructured composite to mesoporous carbon, the SDAs must be thermally decomposed through carbonization, while the precursors should retain their structure. Therefore, the selection of precursors and SDAs should be carefully considered.Meng et al. first demonstrated the synthesis of mesoporous carbon using the soft template method.146 They used PEO-b-PPO-b-PEO as an SDA and low-molecular-weight polymers made of phenol and formaldehyde as a precursor. As the solvent evaporated from the solution containing the SDAs and precursor, an ordered mesostructured composite was formed. Here, the low-molecular-weight polymer can interact selectively with the PEO block to successfully form a mesostructured composite. In the mesostructured composite, a thermally stable resol resin was formed via thermopolymerization at a relatively low temperature (100–140 °C). During the conversion process at high temperatures (350–500 °C), the resol resin stably maintained the carbon framework, while the SDA thermally decomposed, yielding mesoporous carbon. The obtained mesoporous carbon had pore diameters of 6.8 nm, as well as large surface area (590 m2 g−1) and pore volume (0.51 cm3 g−1). Although mesoporous carbon can be synthesized using this approach, the carbonization process induces structural shrinkage accompanied by a decrease in pore size, pore volume, and surface area. To solve this problem, Liu et al. proposed a triconstituent co-assembly approach.147 First, they prepared mesostructured composites using a silicate oligomer, PEO-PPO-PEO, and resol. Silicate served as a reinforcing material to relieve structural shrinkage during calcination. Framework shrinkage after calcination was minimized because of the presence of rigid silicates. For instance, large pore sizes reaching 8.5 and 6.7 nm for the mesostructured composite and for carbon–silica nanocomposite, respectively, were achieved. After removing the silica using an HF solution, ordered mesoporous carbon with a pore size of 6.7 nm, pore volume of 2.02 cm3 g−1, and large surface area of 2470 m2 g−1 was obtained. In addition to the original pores, small pores generated by the removal of silica contribute to the large surface area. Based on these approaches, many researchers have synthesized mesoporous carbons.131–138 However, the pore size is still limited to less than 10.0 nm because of the low molecular weight of Pluronic SDAs, which greatly limit their applications. Instead of using Pluronic SDAs, Deng et al. synthesized mesoporous carbon using PS-b-PEO as an SDA and low-molecular-weight resol as a carbon source.117 To increase the pore size, they synthesized PS-b-PEO with a larger molecular weight (PS230-b-PEO125, 29![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 700 g mol−1) than Pluronic F127 (12600 g mol−1). When the solvent evaporated from the solution containing resol and PS-b-PEO, co-assembly of resol and PEO occurred to form a mesostructured composite. The resultant mesoporous carbon had a relatively large pore size (∼23.0 nm) because of the high molecular weight of PS-b-PEO.
700 g mol−1) than Pluronic F127 (12600 g mol−1). When the solvent evaporated from the solution containing resol and PS-b-PEO, co-assembly of resol and PEO occurred to form a mesostructured composite. The resultant mesoporous carbon had a relatively large pore size (∼23.0 nm) because of the high molecular weight of PS-b-PEO.
Yan et al. reported a new synthetic approach for mesoporous carbon (Fig. 3a).28 Here, many parameters for co-assembly kinetics, such as the ratio of SDAs and carbon source and solvent evaporation rate, can be ignored because the soft template served as the carbon source. They utilized polyacrylonitrile-b-polymethylmethacrylate copolymer (PAN-b-PMMA) as both the carbon precursor and SDAs. PAN and PMMA served as carbon sources and sacrificial blocks, respectively. PAN-b-PMMA self-assembled through microphase separation to form a mesostructure at 250 °C and then converted to mesoporous carbon after subsequent carbonization at 800 °C. Then, PAN was converted to carbon, and PMMA was thermally decomposed to form mesopores. During thermal treatment at 250 °C, PAN remained thermally stable throughout the cross-linking reactions accompanied by partial dehydrogenation and cyclization of PAN (Fig. 3b). After carbonization, PAN-b-PMMA was converted to mesoporous carbon without structural collapse because the cross-linking of PAN offered sufficient thermal stability. Furthermore, the pore size was precisely controlled by adjusting the molecular weight or volume fraction of the sacrificial PMMA block.26,148–151 Recently, Zhou et al. demonstrated the synthesis of mesoporous carbon fibers by electrospinning PAN-b-PMMA with 64 vol% PAN (Fig. 3c).26 During electrospinning, the solvent rapidly evaporated, resulting in the formation of fibers. The oxidation process then stabilized the fiber structure through the crosslinking and cyclization of PAN. After pyrolysis, the fiber maintained its shape without agglomeration and exhibited a bicontinuous mesoporous structure (Fig. 3d and e).
 | ||
| Fig. 3 (a) Conversion process of PAN-b-PMMA into mesoporous carbon. (b) Mechanism of thermal chemistry of PAN carbonization: (left) thermal stabilization and (right) pyrolysis involving dehydrogenation and denitrogenation, which eventually lead to the partially graphitic structure.28 Copyright 2015, Royal Society of Chemistry (c) fabrication of the mesoporous carbon fiber. (d and e) SEM images of the mesoporous carbon fiber.26 Copyright 2019, The American Association for the Advancement of Science. | ||
Considerable effort has been focused on fabricating heteroatom-doped (such as N, B, P, O, and S) mesoporous carbons, particularly N-doping because N atoms can significantly enhance the wettability, surface polarity, electrical conductivity, and electron-donor affinity of carbon frameworks.134,152,153 Carbonization of organic self-assembled composites comprising SDAs and certain N-containing precursors (such as melamine, urea–phenol–formaldehyde resins and dicyandiamide) provides a versatile route for synthesizing N-doped mesoporous carbon.8,67 However, retaining the structural stability of the resulting mesoporous carbons requires a small amount of N-rich dopants, which greatly restricts the final N content in the framework. To overcome this limitation, Liu et al. reported a new synthetic route for rich N-doped mesoporous carbons based on a template-catalyzed in situ polymerization and co-assembly process.29 They used polystyrene-b-poly(acrylic acid) copolymer (PS-b-PAA) as the SDA, and catalyst and urea–formaldehyde (UF) precursors as both the carbon and nitrogen sources. The proposed mechanism is illustrated in Fig. 4. When PS-b-PAA, formaldehyde, and urea were dissolved in a THF/H2O solution, hydrogen ions were generated from the PAA block owing to ionization in water, thereby catalyzing the polymerization of UF to form UF precursors through hydroxymethylurea derivatives. As THF evaporated, the amount of H2O in the solution increased, resulting in spherical PS-b-PAA micelles, in which PS and PAA formed the core and shell, respectively. The UF precursors interact with the PAA block to form a UF precursor/PS-b-PAA composite. After all of the solvent evaporated, cross-linking of UF precursors occurred to form UF resin/PS-b-PAA, which was converted into N-rich mesoporous carbon via subsequent pyrolysis in nitrogen. Because the N-rich UF resin was used as the main framework in the composites, the obtained mesoporous carbon had a high N content (∼19 wt%).
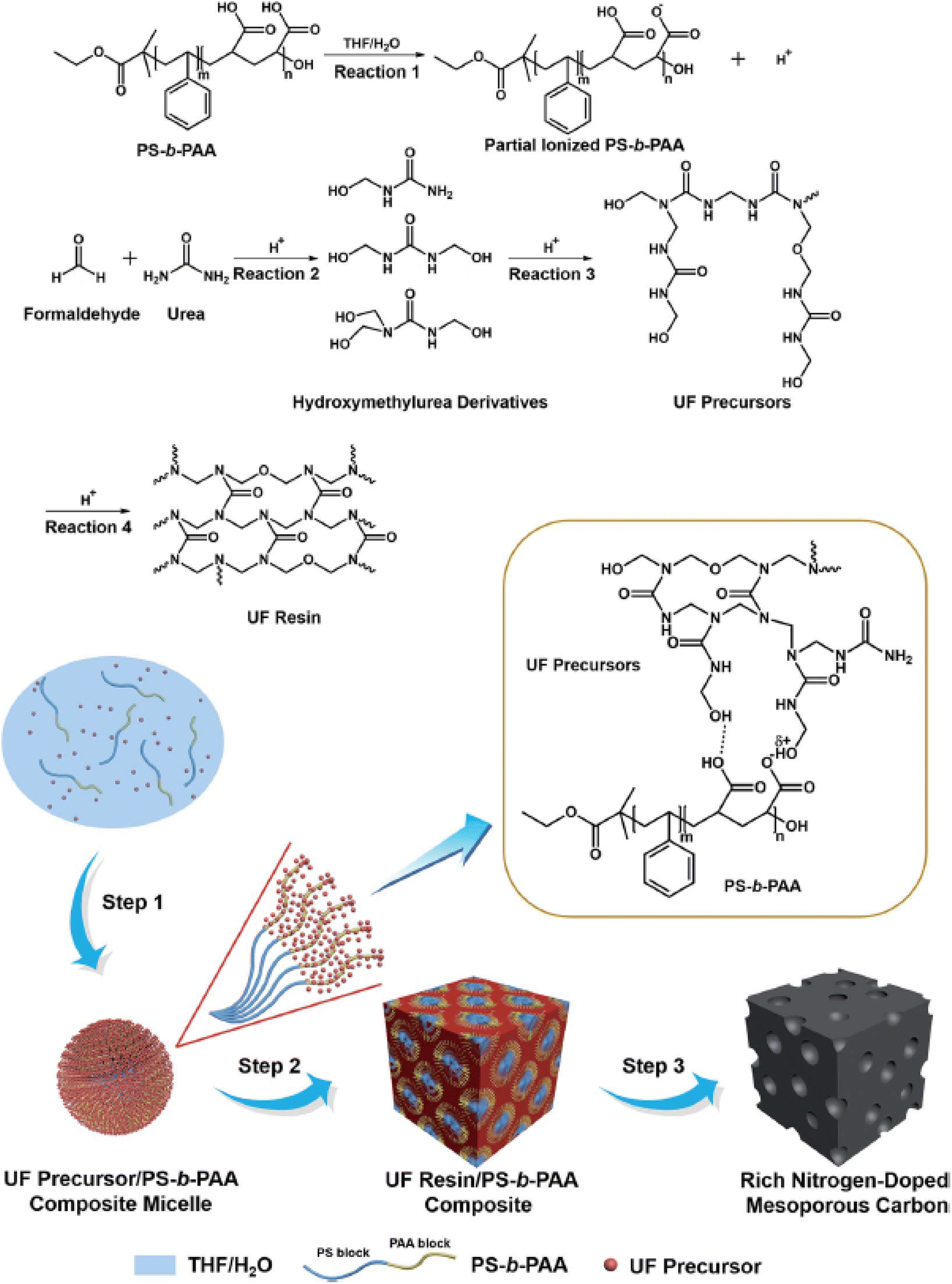 | ||
| Fig. 4 The formation of rich N-doped mesoporous carbon through a template-catalyzed in situ polymerization and co-assembly process.29 Copyright 2018, Royal Society of Chemistry. | ||
3.2. Mesoporous metal oxides
Mesoporous metal oxides, especially transition metal oxides, have received considerable interest because of their intrinsic physicochemical properties, high surface area, and wide range of applications in energy storage materials,154,155 sensors,92 and catalysts.156 In synthesizing mesoporous metal oxides, the thermal stability of the soft template, reactivity to hydrolysis and condensation of inorganic precursors, and crystallization of metal oxides should be considered.157,158Block copolymer-based SDAs have been widely employed as soft template materials for the synthesis of mesoporous metal oxides. The presence of water induces explosive hydrolysis and condensation of precursors, which cause difficulty in co-assembly with SDAs. Therefore, the co-assembly of the SDA and precursors in a nonaqueous solution is required. Yang et al. demonstrated a general procedure for synthesizing mesoporous metal oxides.159,160 They synthesized ordered mesoporous metal oxides, including TiO2, ZrO2, Nb2O5, Ta2O5, Al2O3, SnO2, WO3, HfO2, and their mixed oxides, using non-aqueous solutions containing metal chloride precursors and poly(alkylene oxide) containing BCPs (for instance, Pluronic). The pore walls of the obtained metal oxides were semi-crystalline and most metal oxides exhibited hexagonally packed mesophases. Although Pluronic has been widely used to synthesize mesoporous metal oxides, tunable pore sizes and highly crystalline phases are difficult to obtain owing to its low molecular weight and low thermal stability.159,160 Fattakhova-Rohlfing et al. reported the differences between TiO2 synthesized from Pluronic and non-Pluronic SDAs.80 They used P123, and poly(ethylene-co-buthylene)-b-poly(ethylene oxide) copolymer (KLE) as Pluonic and non-Pluronic SDAs, respectively. Mesoporous TiO2 obtained from KLE showed high crystallinity and maintained its mesoporous structure in the temperature range of 550–700 °C. By contrast, mesoporous TiO2 derived from P123 exhibited a collapsed mesoporous structure at 600 °C. This difference is attributed to the large molecular weight of KLE, which provides mesoporous TiO2 with thicker pore walls that can sufficiently support the mesostructures during calcination. Therefore, block copolymers with high molecular weights are suitable for producing crystalline mesoporous metal oxides because of high thermal stability owing to their large molecular weights. In addition, tailored pore sizes can be obtained by adjusting the volume ratio of the hydrophilic/hydrophobic blocks and molecular weight.
Although crystalline mesoporous metal oxides can be synthesized using non-Pluronic SDAs, certain metal oxides with high crystallization temperatures are still difficult to synthesize because crystallization and template decomposition simultaneously occur during calcination. Early template decomposition causes structural collapse; therefore, a thermally stable template at high temperature is required for the synthesis of highly crystalline mesoporous metal oxides.161 Lee et al. introduced the combined assembly by soft and hard (CASH) method to achieve highly crystalline mesoporous metal oxides with large-sized and uniform pores without structural collapse (Fig. 5a).137 They used PI-b-PEO as the SDA. A hydrophilic PEO block was easily decomposed upon heating and the hydrophobic PI block containing sp2 carbon was converted to a thermally stable amorphous carbon during thermal treatment in an inert (argon) environment. During this process, PEO was burned off, and the metal oxide crystals nucleated, grew, and were sintered into the wall material. Simultaneously, the PI block was converted to amorphous carbon, which can sufficiently support the mesoporous structure. The remaining amorphous carbon was removed by heating in air, and highly crystalline mesoporous metal oxides were thus obtained. Owing to the thermally stable amorphous carbon, the mesoporous metal oxides could be maintained without structural collapse, even at 1000 °C. Highly crystalline mesoporous TiO2 (Fig. 5b and c) and Nb2O5 (Fig. 5d and e) that crystallized at temperatures higher than 400 °C were synthesized. Notably, the CASH method can be extended to other polymers containing sp2 carbons; therefore, thermally stable mesoporous TiO2 and Nb2O5 were synthesized using PS-b-PEO.162
 | ||
| Fig. 5 (a) Schematic of the CASH method. HR-TEM images and SAED patterns of mesoporous TiO2 (b, c) and Nb2O5 (d and e).137 Copyright 2008, Springer Nature. (f) Process of forming ordered and highly crystallized large-pore mesoporous TiO2 from the ligand-assisted assembly method. FESEM (g, h) and TEM (i) images of mesoporous TiO2 and its corresponding SAED pattern (j).68 Copyright 2011, Wiley-VCH. | ||
The CASH method has been reported to be capable of stable conversion from composite to crystalline mesoporous metal oxides; however, the synthesis of certain mesoporous metal oxides with high reactivity to hydrolysis and condensation under non-hydrolytic sol–gel conditions remains difficult because solvent purity and relative humidity can affect the hydrolysis and condensation reactions of inorganic precursors, resulting in poor reproducibility of the co-assembly of inorganic precursors and SDAs.163 To solve this problem, Zhang et al. reported a novel ligand-assisted assembly method for preparing highly ordered crystalline mesoporous metal oxides (Fig. 5f).68 To control the hydrolysis and condensation of inorganic precursors, they employed acetylacetone (AcAc) as a coordination agent. AcAc interacted with the highly reactive titanium isopropoxide to retard the hydrolysis and condensation reactions, which increased the time for precursors to co-assemble with SDAs. Therefore, the co-assembly process was more controllable, leading to highly ordered and crystalline mesoporous TiO2 after calcination (Fig. 5g–j).
Based on the above two methods, various mesoporous metal oxides have been synthesized.88,89,164–166 Zhu et al. synthesized highly ordered mesoporous WO3 using ligand-assisted co-assembly to prepare a mesostructured composite and the CASH method to convert it to mesoporous oxides (Fig. 6a).166 They used PS-b-PEO, WCl6, and AcAc as the soft template, inorganic precursor, and chelating agent, respectively. AcAc strongly coordinates with tungsten ions and retards hydrolysis and thus can stabilize the co-assembly of the SDA and precursors. The co-assembled composite was then heated under a nitrogen atmosphere. Owing to the thermally stable amorphous carbon derived from the PS block, structural collapse was prevented during the high-temperature crystallization process. After calcination in air to remove residual carbon, ordered crystalline mesoporous WO3 was obtained. In addition, by using PEO-b-PS with different volume fractions of PEO, mesopores of various sizes were obtained (Fig. 6b–e). In addition, molecules capable of coordinating with metal ions have been used as chelating agents, such as polyols, amines, resol, and α-hydroxycarboxylic acids including ascorbic, lactic, citric, or glycolic acids.167–170 Another approach to controlling the reaction rate of the precursors is by using acidic conditions. Under highly acidic conditions, the hydrolysis of metal alkoxide precursors is accelerated, but condensation is hindered due to the protonation of M − OH moieties.155,171–173 In addition, the inverse of condensations is also promoted. These effects lead to the formation of small hydrophilic oligomers, thus providing a stable co-assembly with SDAs without chelating agents.173 Jo et al. synthesized mesoporous TiNb2O7 under highly acidic conditions.155 To control the condensation rate of precursors, they added an appropriate amount of concentrated HCl (35–37%) to a mixture of Ti and Nb-alkoxide (here, [H2O/M(OR)x] > 2). At a controlled condensation rate, the Ti and Nb sols were controlled to a small size that could sufficiently interact with the PEO block of PS-b-PEO. After mesostructured composite formation and subsequent calcination, ordered mesoporous TiNb2O7 was synthesized.155 The versatile soft-template processes enabled the synthesis of various mesoporous metal oxides in single and multiple compositions, as well as nanoparticle-decorated metal oxides.101,174–182 Recently, Yang et al. reported the synthesis of multi-composition and nanoparticle-decorated mesoporous metal oxides (Fig. 6f).156 They prepared precisely controlled mesoporous CexZr1−xO2 (0 < x < 1) by tuning the Ce/Zr molar ratio of the feeding precursors. In addition, Pt nanoparticles were decorated on the surface of mesoporous CexZr1−xO2 through the in situ reduction of an H2PtCl6·6H2O aqueous solution by NaBH4 (Fig. 6g–i).
 | ||
| Fig. 6 (a) Schematic of the formation of ordered mesoporous WO3via ligand-assisted co-assembly and the subsequent CASH method. (b–e) FESEM images of mesoporous WO3 with two different pore sizes. (b and d: top view and c and e: cross-sectional view)166 Copyright 2017, American Chemical Society. (f) The synthesis process of ordered mesoporous CexZr1−xO2 and Pt/CexZr1−xO2. (g) STEM and (h) HR-TEM images, and (i) energy-dispersive X-ray element mapping of O, Zr, Ce, and Pt in Pt/CexZr1−xO2.156 Copyright 2019, Wiley-VCH. | ||
Despite these significant advances in the synthesis of mesoporous metal oxides, some metal oxides such as CoO3, MoO3, and V2O5 are still difficult to synthesize into high-quality mesoporous structures because of their rapid thermal growth at high temperatures and requirement of multiple oxidation states. Lunkenbein et al. reported several strategies for template removal from the mesostructured composite to synthesize mesoporous MoO3 (Fig. 7).81 The composite comprised poly(butadiene)-b-poly(dimethylaminoethyl methacrylate) copolymer (PB-b-PDMAEMA) as a soft template and phosphomolybdic acid (H3PMo12O40) as a MoO3 precursor. They employed three methods to remove SDAs from the mesostructured composites: (1) direct calcination, (2) CASH method, and (3) sequential heat and H–O-plasma treatments. In the first method, direct calcination induced both the fast growth of MoO3 nanocrystals and template decomposition, resulting in agglomerated crystalline MoO3. In the second method, the mesostructured composite was subjected to heat treatment in an inert environment and subsequently calcined in air. The composite heat-treated in an inert environment maintained a mesoporous structure; however, the structure collapsed after the subsequent heat treatment to remove the template because of the change in the oxidation state of Mo. During heat treatment in an inert environment, the oxidation state of Mo was reduced because of its low oxygen fugacity. During heat treatment in air to remove carbon, the MoO3 crystal, which contained the highest oxidation state of Mo, rapidly grew, leading to the collapse of the mesoporous structure. In the third method, a mesoporous structure was obtained by subjecting the mesostructured composite to heat treatment in an inert environment and subsequent H–O-plasma treatment; however, the carbonaceous material still remained. Therefore, new methods (or strategies) for synthesizing mesoporous metal oxides in multiple oxidation states are needed.
 | ||
| Fig. 7 (a) Chemical structures of H3PMo and PB-b-PDMAEMA. (b) H3PMo clusters were selectively incorporated into the PDMAEMA domains. (c) Systematic calcination studies were conducted either directly in air (route 1) or by applying a two-step heat treatment (route 2): first in an argon atmosphere and second in an oxidative atmosphere. Alternatively, argon heat-treated samples were exposed to H–O plasma (route 3).81 Copyright 2013, Royal Society of Chemistry. | ||
3.3. Metals
Metallic materials exhibit distinctive physicochemical characteristics, such as inherent conductivity and electrochemical stability.183,184 Mesoporous metal structures show enhanced performance in catalysis, energy conversion and storage, and chemical and biological sensors by maximizing the rate of electron transfer inside a porous structure.185–187 Despite these advantages, the synthesis of mesoporous metallic materials remained a challenge for a long time owing to their high surface energies, which tend to minimize the surface area.188,189 To fabricate mesoporous metallic materials, various strategies based on soft templates have been developed.180–185To promote the self-assembly of metal nanoparticles (NP) and soft templates, the high surface energy of NPs should be reduced by capping them with ligand molecules.190,191 Warren et al. successfully synthesized mesoporous Pt via the co-assembly of ligand-stabilized Pt NPs and a block copolymer (Fig. 8).192 To synthesize mesoporous Pt, a specific block copolymer that meet the following requirements should be selected. First, NPs should be highly soluble in organic solvents to interact with the block copolymer. Second, ligand-stabilized NPs should possess a high metal volume fraction to prevent structural collapse during template removal. Third, the NPs should interact with only one block of the block copolymer. Fourth, the diameter of the NPs should be less than the microdomain size of the block to facilitate their incorporation into the target microdomain. They selected N,N-di-2-propoxyethyl-N-3-mercaptopropyl-N-methylammonium chloride as the ligand and PI-b-poly(dimethylaminoethyl methacrylate) copolymer (PI-b-PDMAEMA) as the SDA. The ligand-stabilized Pt NPs had a diameter of 1.4 nm and high solubility in organic solvents even after sufficient aging. Therefore, a macroscopically homogeneous solution was prepared by combining Pt NPs, PI-b-PDMAEMA, and a mixed solvent of chloroform and methanol. During solvent evaporation, the co-assembly of aged Pt NPs and PI-b-PDMAEMA occurred, leading to an ordered mesostructured composite which was finally converted to pure mesoporous Pt after Ar–O plasma treatment. Owing to the high metal content of the aged Pt NPs, ordered mesoporous Pt was successfully synthesized from the mesostructured composite without structural collapse. Li et al. successfully extended a single mesoporous metal to a binary mesoporous metal system via co-assembly of ligand-stabilized metal NPs and SDAs.193
 | ||
| Fig. 8 (a) Chemical structure of the ligand used to produce moderately hydrophilic Pt NPs with high solubility. (b) A true-scale model of a NP. (c) Chemical structure of PI-b-PDMAEMA. (d) Self-assembly of Pt NPs with the block copolymer. (e) Pyrolysis of the mesostructured composite under an inert atmosphere produces a mesoporous Pt–C composite. (f) Ar–O plasma treatment or acid etching of Pt–C produces ordered mesoporous Pt.192 Copyright 2008, The American Association for the Advancement of Science. | ||
Yamauchi’s group suggested facile synthetic approaches yielding mesoporous metal films and NPs by combining a soft-templating method with electrochemical deposition (Fig. 9a–d)194 or wet chemical synthesis (Fig. 9e–i).195 The combination of soft templating and electrochemical deposition enabled the synthesis of a mesoporous metal film on a conductive substrate (Fig. 9a). They first prepared a soft-template film composed of spherical micelles of PS-b-PEO on a conductive substrate. Then, electrochemical deposition was conducted by applying a potential to the substrate in an aqueous solution of HAuCl4. The hydrophilic PEO blocks in the PS-b-PEO films could interact with the Au precursor in an aqueous solution. As electrochemical deposition progressed, the mesostructured composite of PS-b-PEO and Au started from the bottom of the PS-b-PEO film. Mesoporous Au films were obtained after template removal using an oxygen plasma (Fig. 9b and c). Mesoporous Au films of various thicknesses and pore sizes were synthesized by adjusting the electrochemical deposition time and volume fraction (or length) of the PS block in PS-b-PEO. Moreover, this synthetic method was extended to various metals (Cu,196 Pd,197 Ni,198 Pt,199 and Rh195) and metal alloys (Pt–Pd,197,200 Pt–Cu,201 and AuCuNi) (Fig. 9d).43 They also reported a synthetic approach for mesoporous metal NPs using wet chemical synthesis (Fig. 9e).195 To synthesize mesoporous Rh, PEO-b-poly(methyl methacrylate) copolymer (PEO-b-PMMA), Na3RhCl6, and ascorbic acid (AA) were used as the SDA, metal precursor, and reducing agent, respectively. Mesostructured composites were successfully prepared after chemical reduction by assembling the above three compounds in DMF/H2O mixed solvents. After consecutive washing with an appropriate solvent, mesoporous Rh nanoparticles were obtained (Fig. 9f–i).
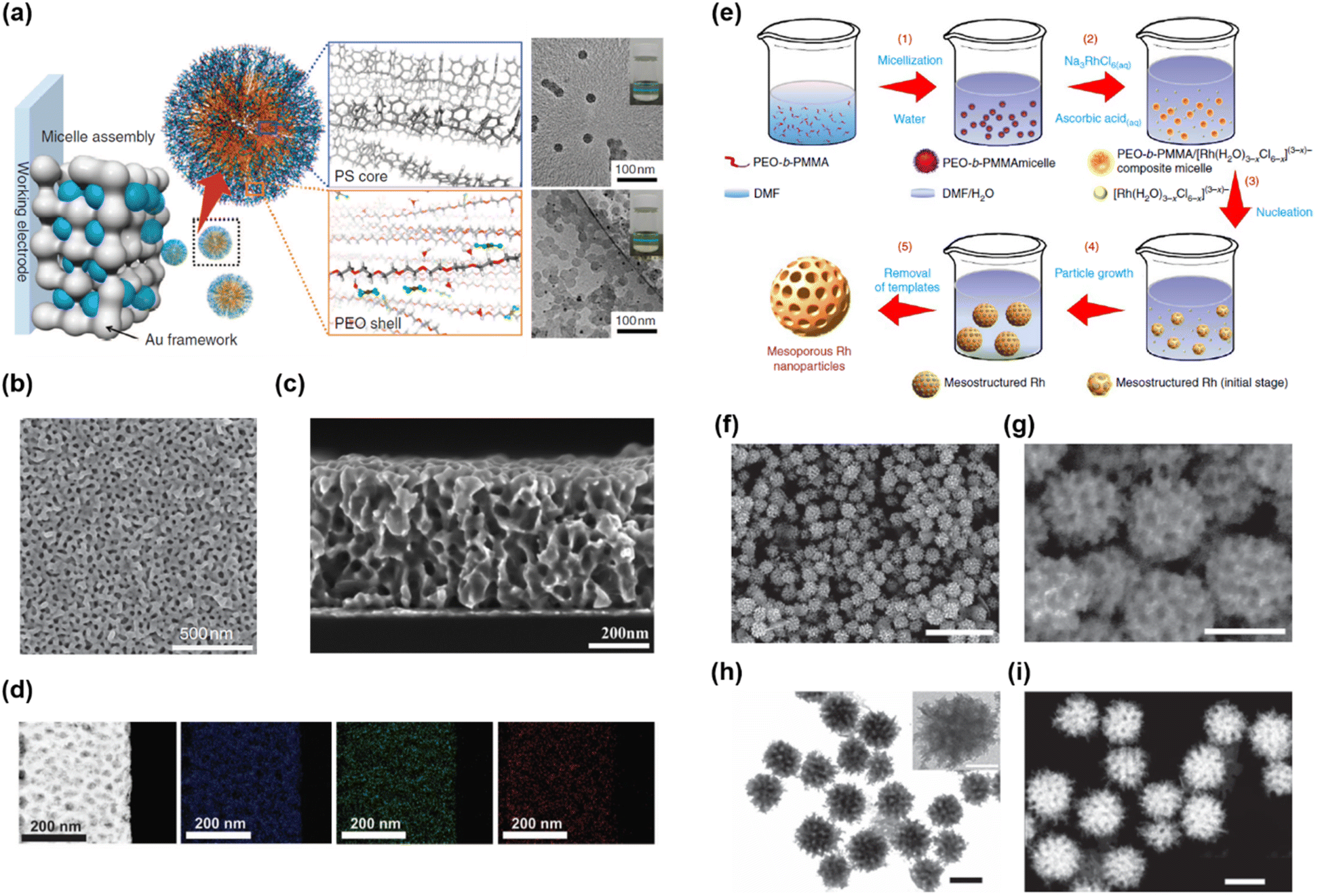 | ||
| Fig. 9 (a) Synthetic concept of mesoporous Au films. (b and c) Top and cross-sectional SEM images of mesoporous Au films. (d) STEM image and EDS elemental mapping of the mesoporous AuCuNi film.194 Copyright 2015, Nature Publishing Group. (e) Formation mechanism for mesoporous Rh nanostructures. (f and g) SEM images of mesoporous Rh nanoparticles. (h) TEM and (i) STEM images of mesoporous Rh nanoparticles.195 Copyright 2017, Nature Publishing Group. | ||
3.4. Metal nitrides
Metal nitrides have been extensively investigated because of their high electrical conductivity, outstanding thermal and electrochemical stability, and corrosion resistance.192 The synthesis of mesoporous metal nitrides can maximize the surface area and mass transfer, thereby improving their performance in various applications such as energy storage, energy conversion, catalysis, and sensors.202–205Because the synthesis of most metal nitrides involves a high-temperature nitridation process, the hard-template method has been used to maintain their mesoporous structure. Although hard template-based synthesis can produce high-quality mesoporous metal nitrides, this approach has disadvantages of long processing time and high cost. Thus, soft-template-based synthesis is preferred for synthesizing a wide range of mesoporous metal nitrides. However, nitridation without structural collapse remains a challenge because the template cannot be maintained during high-temperature nitridation.
To solve this problem, Ramasamy et al. introduced a carbon-supported synthetic approach using a block copolymer as a soft template.37 A mesostructured composite was prepared by co-assembling F127, TiCl4, and resol. After calcination at 700 °C under a nitrogen flow, a mesoporous TiO2–C composite was obtained. Mesoporous TiN–C was synthesized after nitridation at 700 °C, suggesting that TiO2 in TiO2–C was successfully converted to TiN without structural collapse. The amorphous carbon in TiO2–C prevented the aggregation and growth of TiN crystals during nitridation. Although successful nitridation was possible while maintaining the mesoporous structure, the pore size was relatively small (8 nm) because of the low molecular weight of F127. Meanwhile, when a high-molecular-weight BCP is used as the SDA, mesoporous nitride with a large pore size can be obtained.37 For example, Fritz et al. recently synthesized mesoporous TiN and NbN using a PI-b-PS-b-PEO copolymer (ISO) as the SDA (Fig. 10a).206 Calcination was performed to remove ISO at 450 °C after preparing a mesostructured composite through the co-assembly of ISO and metal oxide precursors. During calcination, ISO was decomposed, leading to the formation of mesoporous Nb2O5 and TiO2 and subsequent conversion to mesoporous NbN and TiN after nitridation at 700 °C under an NH3 flow (Fig. 10b and c).
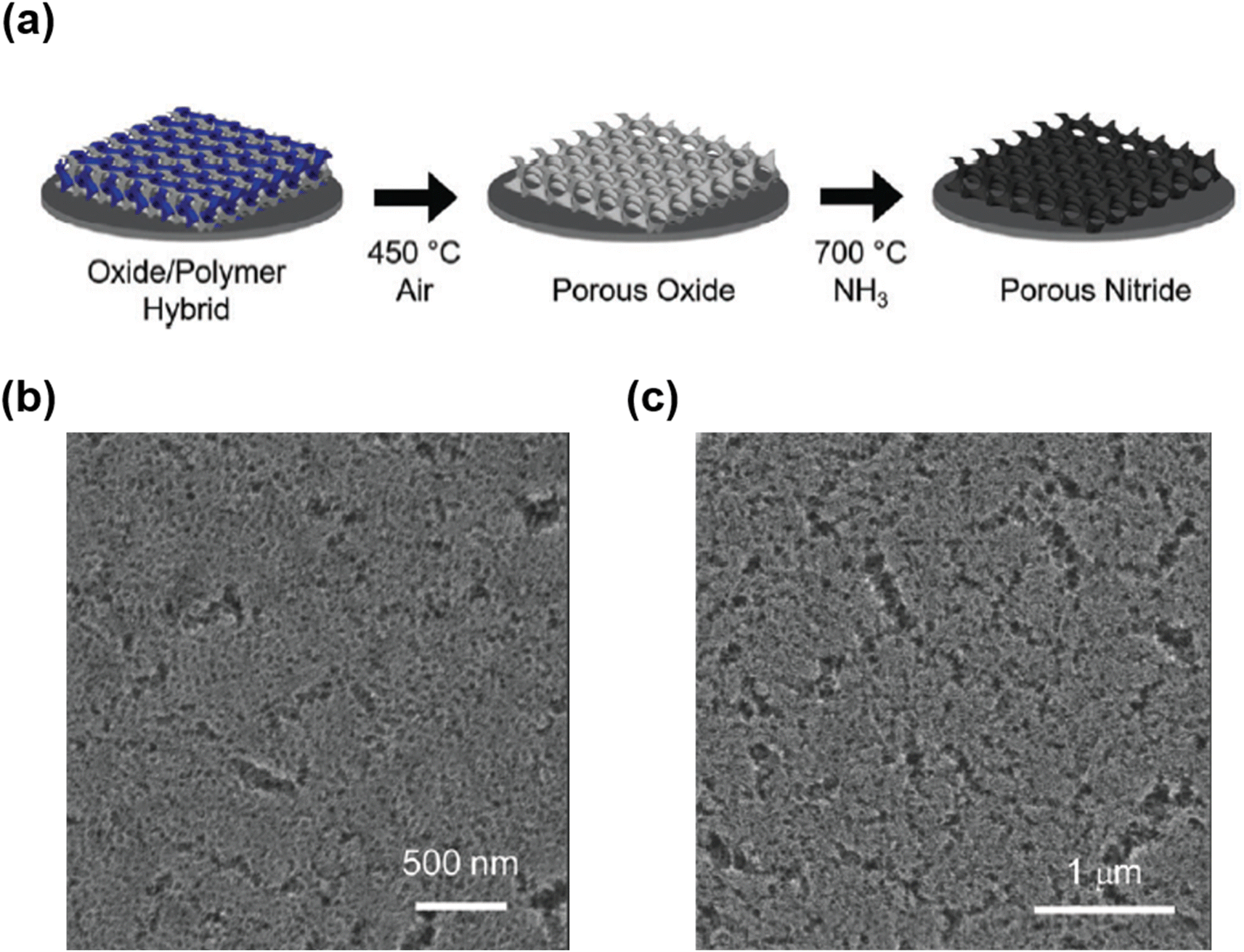 | ||
| Fig. 10 (a) Schematic of the two-step heat treatment used to convert a mesostructured composite to a mesoporous metal nitride thin film. In the first step, an air heat treatment is used to form a mesoporous metal oxide thin film, which is converted to a porous nitride film in the second step by heating under NH3. SEM images of (b) porous NbN and (c) TiN.206 Copyright 2017, Royal Society of Chemistry. | ||
4. Innovative soft-template synthesis for various hierarchical morphologies of mesoporous materials
4.1. Hierarchically structured mesoporous materials
Materials with hierarchical pores have synergetic benefits originating from different pore size regimes, such as highly improved mass transport through macropores and high specific surface area due to the presence of micropores and mesopores.207 Thus, hierarchical porous materials can be used for high-performance energy storage, energy conversion, catalysis, and sensors.208–211Hierarchical porous materials are synthesized by using a multiple templating method that combines hard and soft templates.212Fig. 11 shows a scheme of multiple-template synthetic methods. Although hierarchical porous materials with various macroporous structures are synthesized using hard (such as AAO, silica, colloidal crystals, and metal oxides)213,214 and soft templates (small molecule surfactants and BCPs), such synthetic routes require complicated time-consuming processes.
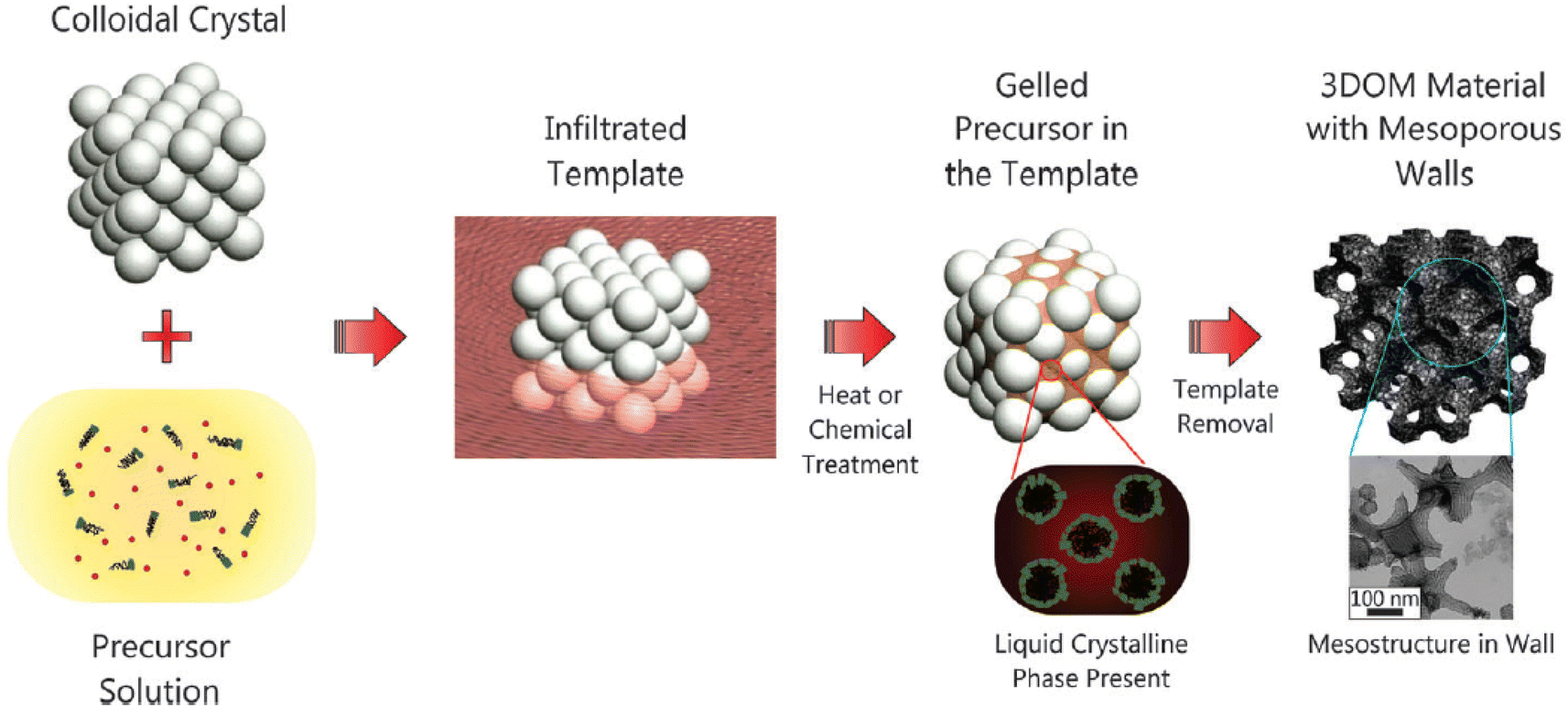 | ||
| Fig. 11 Schematic of a multiple templating approach.212 Copyright 2013, Royal Society of Chemistry. | ||
Sai et al. successfully fabricated a hierarchical porous scaffold consisting of mesopores and macropores by using only a soft-templating method.215 They dissolved an appropriate amount of PS-b-PEO and oligomer PEO in xylene and then induced solvent evaporation at 130 °C. During solvent evaporation, spinodal decomposition (SD) of PS-b-PEO and oligomer PEO and microphase separation of PS-b-PEO occurred. After the selective removal of the oligomer PEO by rinsing with water, they obtained a macroporous polymeric wall composed of cylindrical mesopores. Although hierarchically structured organic materials were fabricated using this approach, the hierarchical structure of inorganic materials was difficult to control because of the difficulty in simultaneously controlling both the sol–gel reaction of inorganic precursors and macro-/meso-phase separation behavior.
Lee’s group reported a versatile method for synthesizing hierarchically porous inorganic materials.216 They employed PS-b-PEO as a soft template, phenol–formaldehyde resin (resol) as an additive to induce macrophase separation, and tetraethyl orthosilicate (TEOS) as a silica source (Fig. 12a).216 During evaporation of the solvent from the solution, a resol-rich phase and a (resol/silicate/PS-b-PEO)-rich phase were formed through macrophase separation. In the (resol/silicate/PS-b-PEO)-rich phase, silicate and resol were incorporated into the PEO microdomains and thus formed an ordered mesostructure by microphase separation. Thermopolymerization (curing) of the resol in the resol-rich phase supports the overall framework. After calcination to remove the PS-b-PEO and cured resol, three-dimensionally interconnected meso/macroporous silica was obtained. The hierarchical porous structure exhibited mesopores with, a dominant pore size of 25 nm and a narrow size distribution (Fig. 12d), as well as macropores with diameters ranging from 50 nm to 400 nm (Fig. 12e). In addition, they synthesized hierarchically porous TiO2 and Nb2O5.217 Recently, they proposed another method to synthesize hierarchical porous structures using evaporation-induced SD (Fig. 12f).207 Multiple-phase separation was induced by controlling the evaporation conditions of THF and nitric acid in a mixed solution containing PS-b-PEO/precursors/THF/nitric acid. The selective evaporation of THF resulted in macrophase separation of the PS-b-PEO/precursors phase and nitric acid. Simultaneously, microphase separation of PS-b-PEO occurred, resulting in the formation of a hierarchical macro/mesostructured composite. After calcination, the composite was converted to hierarchical porous metal oxides. They also synthesized various hierarchical porous materials including SiO2, carbon, WO3, TiO2, and titanium–niobium oxides. In addition, they reported that macrophase separation could depend on the composition ratio of nitric acid to the PS-b-PEO/precursor phases (Fig. 12g), resulting in various macrostructures such as mesoporous structures containing isolated macropores, co-continuous macro/mesoporous structures, and spherical mesoporous structures (Fig. 12h–i).
 | ||
| Fig. 12 (a) Schematic of the hierarchical assembly of micro and macrophase separation. (b and c) SEM images of h-SiO2 on different length scales. (d) N2 physisorption isotherm and (inset) mesopore size distribution. (e) Macropore size distribution determined by mercury intrusion porosimetry.216 Copyright 2014, American Chemical Society. (f) Schematic of multiscale phase separation for the synthesis of hierarchically macro and mesoporous metal oxides. (g) Schematic of macrophase separation controlled by different ratios of acid phase to precursor/PS-b-PEO phase. (h and i) SEM images of various hierarchical porous structures obtained from multiple phase separation.207 Copyright 2018, Wiley-VCH. | ||
4.2. Mesoporous materials with confined morphologies
Confining the mesoporous structure to a macroscopic shaped cavity can enhance physiochemical properties that cannot be improved by conventional mesoporous materials.218 For example, synergistic effects can be obtained by combining high surface area, large pore volume, and enhanced mass/ion accessibility originating from the mesoporous structure with the advantage of macroscopic morphologies.219–221 To synthesize confined morphologies of mesoporous materials, Lee’s group reported various strategies based on SD and soft-templating methods.103,145,222 They designed prehydrolyzed aluminosilicate (AS)/PS-b-PEO/homo PS or PMMA blend systems (Fig. 13a and b).103 When the solvent evaporates from the blend solution, macrophase separation of homopolymers and AS/PS-b-PEO phases and the microphase separation of PS-b-PEO occur, creating a confined structure with a mesostructured sphere in the homopolymer matrix. Here, the pore orientation and particle shape were adjusted by changing the homopolymer type. For the PS homopolymer, owing to the preferential interaction between the PS block of PS-b-PEO and the PS matrix, the AS/PS-b-PEO phase showed a parallel orientation of the cylindrical microdomain to the interface (Fig. 13a). Meanwhile, for the PMMA homopolymer, because PMMA homopolymers are enthalpically neutral favorable for both PEO/precursors and PS blocks, the AS/PS-b-PEO phase exhibited a perpendicular orientation of the cylindrical microdomain to the interface of the PMMA matrix. Furthermore, they implemented a higher level of multiscale structure by mixing homo PS and homo PMMA binary blends and AS/PS-b-PEO. Here, PS-b-PEO acted as both a compatibilizer and SDA. In PS-b-PEO, the PS block is compatible with homo-PS, whereas the PEO block is more compatible with homo-PMMA. Consequently, the mesostructured AS/PS-b-PEO could be located at the interface between PMMA and PS to minimize the PS/PMMA interfacial contact area. | ||
| Fig. 13 Effect of the homopolymer matrix on the pore orientation and shape of inorganic particles prepared in (a) PS and (b) PMMA homopolymers.103 Copyright 2018, Wiley-VCH. (c) Schematic of the preparation of bowl-like inorganic particles and 2D mesoporous nanosheets.222 Copyright 2020, The American Association for the Advancement of Science. (d) Patterning of 2D surfaces with mesoporous conducting polymer nanosheets. (e) Patterning of various functional free-standing surfaces, including MoS2, titania, exfoliated graphene, and CNTs.223 Copyright 2015, Nature Publishing Group. | ||
Using ternary-polymer blend systems, they also synthesized mesostructured materials confined in bowls and 2D nanosheets (Fig. 13c).145,222 To fabricate confined mesoporous materials, they used PS-b-PEO as the SDA, excess homo PMMA as the organic matrix, homo PS as one of the minor components, and AS as an inorganic precursor. During solvent evaporation from the solution containing the above components, macrophase separation of the homo PS, homo PMMA, and PS-b-PEO/AS phases occurred, resulting in dispersed homo PS and PS-b-PEO/AS phases in the homo PMMA matrix. PS-b-PEO is immiscible with both high-molecular-weight homo PS and homo PMMA, while homo PS and homo PMMA are immiscible with each other. Therefore, macrophase separation occurred because of the immiscibility of the three phases. The PS-b-PEO/AS and homo PS phases were in contact with each other to reduce the interfacial tension and existed in dispersed phases in the homo PMMA matrix. Owing to the compatibility of the PS block of PS-b-PEO with homo PS, the surface of the PS-b-PEO/AS phase was partially covered with homo PS. In addition, because the homo PMMA matrix is neutral to both PS and PEO/AS of the PS-b-PEO/AS phase, the microdomains of PS-b-PEO/AS were oriented perpendicular to the interface of the PMMA matrix. Therefore, the confined mesoporous AS particles obtained after calcination consisted of highly accessible, open cylindrical pores. Furthermore, by changing the relative molecular weight of homo PS with respect to that of PS-b-PEO (rM = MhPS/MBCP), various confined shapes were obtained. For example, in the range of 0.2 ≤ rM ≤ 2, the size of particles and concavities increased as rM increased, maintaining mesopore sizes. At a high rM value, the PS-b-PEO/AS rich phase became extremely thin with monolayered micelles, resulting in mesoporous inorganic nanosheets. Various compositions comprising carbon, TiO2, and Nb2O5/C have also been synthesized.139,145,217
Liu et al. demonstrated a universal strategy for synthesizing confined mesoporous conducting polymers with 2D morphology by inducing the orientation of SDAs and monomers on the surface of 2D nanosheets (Fig. 13d).223 First, PS-b-PEO (SDA) forms spherical micelles in a mixture of THF and H2O, which are organized on the 2D surface of graphene oxide through hydrogen bonding. The formed micelles serve as guides for the polymerization of the conducting polymer monomer (pyrrole or aniline). After the removal of SDAs, mesoporous conducting polymers (polypyrrole (PPy) and polyaniline (PANi)) confined to the 2D surfaces of graphene oxide were obtained. This approach can be applied to various functional surfaces, including 2D exfoliated graphene (EG), MoS2, titania nanosheets, and 1D carbon nanotubes (CNTs), yielding confined mesoporous materials with various functional surfaces.
4.3. User-customized mesoporous materials
To date, soft-templating methods have been mainly investigated for the synthesis of mesoporous materials in bulk. However, to expand the application of mesoporous materials to microelectronics or mobile devices, the production of materials with various customized shapes, including thin films, patterned shapes, and complex 3D architectures are preferable.Tan et al. demonstrated a simple pattern of mesoporous thin films by combining soft templating and laser-induced transient heating (Fig. 14a).33 They used PI-b-PS-b-PEO or Pluronic F127 as the SDA and oligomeric resols as precursors. PI-b-PS-b-PEO and resols were first dissolved in a solvent and spin-coated on a boron-doped silicon substrate to form a mesostructured hybrid thin film, which was subsequently irradiated with a continuous-wave CO2 laser. During laser irradiation, the boron-doped Si substrate absorbed most of the laser energy and dissipated heat, leading to simultaneous thermopolymerization of the resol and decomposition of BCP. Therefore, the mesoporous carbon thin film remained in the laser-passed region. After the nonirradiated region was selectively rinsed, a patterned mesoporous carbon thin film was obtained (Fig. 14b). Additionally, various pore sizes were implemented by tailoring the mass ratio of resol and the SDA, as well as the molecular structure of the SDA. Yu et al. fabricated patterned mesoporous thin films by combining photolithography and soft templating (Fig. 14c).110 They prepared a mesostructured composite comprising PI-b-PS-b-PEO and niobium oxide precursor thin films by spin-coating. The as-made composite films were then readily patterned through a conventional photolithography process. After etching and photoresist removal, a mesoporous thin film with a well-defined pattern was obtained (Fig. 14d and e). However, this method requires multiple processes, including selective rinsing and conventional photolithography. In contrast, printing technology that directly deposits a solution on a desired region of a substrate can easily provide a patterned thin film without multiple processes. Soft-templating methods are based on solution-phase synthesis and are relatively easy to combine with printing technology. However, to prevent nozzle clogging during printing, the evaporation rate of the solvent and reactivity of the precursors should be carefully controlled. Devabharathi et al. recently fabricated mesoporous metal oxides in a selective area using inkjet printing.224 They prepared a homogeneous ink by mixing Pluronic F-127 as the SDA and metal chloride precursors with an appropriate solvent. During printing, the solvent evaporated and co-assembly of F-127 and the precursors occurred, resulting in a mesostructured composite film. Subsequently, a mesoporous metal-oxide film was obtained after calcination. Moreover, they synthesized various mesoporous metal oxides, including In2O3, ITO, CuO, and SnO2. Inkjet printing is based on drop-on-demand; therefore, this approach is not suitable for large-scale and continuous line production. By contrast, extrusion-based printing is more suitable for covering a large area or a large amount of material because of continuous ink ejection.
 | ||
| Fig. 14 (a) Schematic of the transient laser heating process for synthesizing patterned mesoporous carbon. (b) Optical image and cross sectional and plane SEM images (inset) of macroscopic trenches after CO2 laser irradiations.33 Copyright 2015, The American Association for the Advancement of Science. (c) Schematic of the combination of photolithography and soft templating. (d and e) SEM images of a patterned niobium carbonitride thin film.110 Copyright 2021, American Chemical Society. (f) Schematic illustration of printing-assisted evaporation-induced self-assembly. (g) OM (left) and SEM (right) images of a patterned mesoporous WO3 film.13 Copyright 2020, Nature Publishing Group. | ||
Kim et al. demonstrated that mesoporous WO3 was successfully patterned over a large area by combining the EISA protocol with electrostatic-force-assisted dispensing printing (Fig. 14f).13 They mixed PS-b-PEO as a SDA and WCl6 as a precursor in a mixture of tetrahydrofuran and ethanol. When the obtained solution was printed on fluorine-doped tin oxide (FTO)-coated glass, co-assembly of PS-b-PEO and WCl6 occurred as the solvent evaporated, resulting in a mesostructured composite. Patterned mesoporous WO3 was then formed after template removal through calcination and oxygen plasma treatment (Fig. 14g). As the sample stage was controlled by using a computer, user-customized patterning was easily achieved over a large area (10 cm2). In addition to the synthesis of patterned mesoporous thin films, achieving the desired shape of mesoporous materials in three dimensions can extend their applications. In particular, the synthesis of 3D geometry architecture consisting of interconnected mesopores is important for biomedical applications such as bone regeneration226 and drug delivery.227,228 Several groups have attempted to synthesize 3D structured mesoporous materials using 3D printing but failed to achieve high surface area and complex geometry.229–231 Recently, Shukrun Farrell et al. introduced a method for synthesizing mesoporous silica with complex 3D geometry.225 They integrated a soft-templating method with digital light processing, which is a stereolithography 3D printing technology that applies patterned UV light to cure sequential 2D layers of UV-polymerizable ink to create 3D objects. First, TEOS was added to a solution containing Pluronic F-127. At this time, hydrolysis and condensation of TEOS occurred near F-127 micelles, and mesoporous silica skeletons were thus formed. Next, UV-curable ink containing an epoxy aliphatic acrylate as a monofunctional monomer, aliphatic urethane diacrylate as a cross-linker, and a photoinitiator was added to the above solution to enable photopolymerization. During UV irradiation, photopolymerization occurred to form a user-customized shape, resulting in an elastomer containing mesostructured silica. Finally, after high-temperature calcination, the elastomer and organic template decomposed, leaving pure silica. The synthesis of mesoporous materials in user-customized shapes (selective area or desired architecture) is expected to not only save production costs but also expand the existing standardized shapes of ESSs in the future.
5. Application of mesoporous materials to energy storage systems
Mesoporous materials prepared by the soft-templating method confer considerable advantages to ESSs owing to their high surface area, large pore volume, tunable pore size, and shape.232 The high surface area provides abundant active sites by maximizing the interface between the electrolyte and active materials.233 In addition, the large pore volume provides the accommodation of volume expansion and strain relaxation of materials during the repeated charge/discharge of ESSs.142 Device performance can be maximized by tailoring the pore size and shape of mesoporous materials to meet the needs of various ESSs. Although many research groups have successfully enhanced the performance of ESSs by synthesizing various mesoporous materials, we only focused on batteries and supercapacitors based on mesoporous materials.105,234,2355.1. Batteries
Increasing energy requirements necessitate the development of ESSs for sustainable energy supply. For such a purpose, rechargeable batteries are believed to be the most feasible option among various ESSs because of their long cycle life, high energy efficiency, and simple maintenance. Diverse rechargeable batteries with different charge carriers such as Li+, Na+, K+, Ca2+, Mg2+, Zn2+ and Al3+ have been demonstrated.236–242 Numerous research studies on various mesoporous cathode142,243–251 and anode55,145,155,252–260 materials have been reported to improve the energy storage performance of batteries.Graphite is the most popular anode material for lithium-ion batteries (LIBs) owing to its low cost, excellent electrical conductivity, and high chemical stability. However, the development of LIBs with high energy density is hampered by the low capacity of graphite (372 mA h g−1). Thus, ordered mesoporous carbons (OMCs) have attracted considerable attention as alternatives to graphite because of their high specific surface area and large pore volume, providing numerous active sites for Li+ adsorption and storage.234,235 Velez et al. synthesized mesoporous carbon materials and compared them with graphite when applied as anodes for LIBs.34 The synthesized mesoporous carbon consisted of mesopores (∼7 nm) and exhibited large surface area (769 m2 g−1) and pore volume (1.4 cm3 g−1). These characteristics provided abundant mesopores for storing large amounts of Li ions, resulting in notable electrochemical properties as an anode material for LIBs with a high capacity (670 mA h g−1) exceeding that of graphite (358 mA h g−1). In addition, the pore size and wall thickness of the mesoporous carbon play a major role in determining energy storage performance. Mesoporous metal oxides have been developed as anode materials in alkaline-ion battery systems because of their high theoretical capacities.261 For example, Ti-based oxides are particularly interesting because they prevent the formation of solid electrolyte interface (SEI) layers and guarantee structural stability. Furthermore, Ti is an abundant element, thus making it a cost-effective material.154 For the first time, Kang et al. synthesized a mesoporous Li4Ti5O12 (LTO)–carbon nanocomposite (Meso-LTO–C) and reported its excellent performance as an anode material in LIB systems.154 The synthesized mesoporous LTO consisted of mesopores (∼20 nm) and exhibited a large pore volume (0.148 cm3 g−1) and surface area (68.7 m2 g−1). When Meso-LTO–C and bulk-LTO were applied as anode materials for LIBs, Meso-LTO–C exhibited a superior electrochemical performance with a capacity of 115 mA h g−1 at a 10C rate compared to a bulk-LTO LIB because of its large surface area and easy ion accessibility. Despite the high stability and rate capability of Ti-based oxides, the theoretically low lithium charge capacity (<200 mA h g−1) and loss of energy density from the relatively high Li+ insertion voltage (>1.5 vs. Li/Li+) remain limitations to the use of anodes with a Ti4+/Ti3+ redox couple. Titanium–niobium oxides have been introduced as alternative anode materials because of their high lithium charge capacity and broad lithium charge potential (1.7–1.0 V).155 Guo et al. synthesized mesoporous TiNb2O7 and demonstrated its successful application as a high-capacity anode in LIBs.262 The large mesopores (∼22 nm) and high surface area (48 m2 g−1) of the mesoporous TNO (m-TNO) electrode yielded a high reversible capacity (281 mA h g−1) that was almost twice that of LTO (∼160 mA h g−1). In addition, the performance of the m-TNO electrode was markedly superior to those of reported Ti- and Nb-based electrodes.262
The simultaneous integration of multiscale porous structures can produce synergistic properties. For example, the combination of macropores and mesopores can benefit from the effective mass transport from macropores and abundant reaction sites from mesopores. Recently, Jo et al. demonstrated a high-performance LIB by applying hierarchically structured anode materials.207 They synthesized co-continuous macro/mesoporous titanium–niobium oxides (ccm-TNO) and then compared the LIB electrode performance of the ccm-TNO with m-TNO without macropores. Although the two electrode materials had similar structural properties (surface area, pore volume, pore size, and crystal size), their electrochemical performances showed significant differences. For example, at a rate of 0.2C, ccm-TNO exhibited a higher capacity (257 mA h g−1) than m-TNO (215 mA h g−1). Additionally, the cmm-TNO electrode retained 44% of its maximal capacity (112 mA h g−1) at 60C rate, whereas the m-TNO electrode only retained 33% of its capacity. These results indicate that the introduction of macropores into the mesoporous structure could improve electrochemical performance by providing enhanced ionic transport compared with the mesoporous structure alone.
Mesoporous structures provide high surface area and fast ion transport, but stable loading of host materials is restricted due to their small pore size. In contrast, macroporous structures have a relatively small surface area, but their large pore volume allows a large amount of host material loading. Therefore, these two advantages can be synergistically combined by introducing macropores into the mesoporous structure. Lim et al. applied these advantages to a lithium–sulfur battery (LSB) system (Fig. 15a and b).105 LSBs have been recognized as an alternative to LIBs because of their high theoretical energy density (2600 W h kg−1), but their practical use is limited by their low conductivity and the sluggish redox kinetics of sulfur. To solve these problems, stable loading of sulfur into the porous material is crucial, requiring careful control of the porous architecture to improve the performance of LSBs. Co-continuous hierarchical macro/mesoporous titanium nitride (h-TiN) was synthesized as a sulfur host. The obtained h-TiN consisted of mesopores (32 nm) and had a surface area and pore volume of 47 m2 g−1 and 0.3 cm3 g−1, respectively. To evaluate the effect of macropores on LSB performance, they also prepared mesoporous TiN (m-TiN) with virtually identical structural properties (pore size, pore volume, and surface area). When the same amount of sulfur (∼72 wt%) was impregnated into both TiN samples, h-TiN was stably filled without the growth of sulfur particles; by contrast, m-TiN showed sulfur particles aggregated on the surface of the mesopores (Fig. 15a). This aggregation was due to the large space of macropores, which provided sufficient space for sulfur loading. The initial discharge and charge capacities of h-TiN were 1040 and 1037 mA h g−1, respectively, which were higher than those of m-TiN (739 and 741 mA h g−1, respectively). In addition, h-TiN showed superior electrochemical performance at a high rate of 5C compared to m-TiN (Fig. 15b) because sulfur was stably loaded in the interconnected macropores.
 | ||
| Fig. 15 (a) SEM images of h-TiN and m-TiN before and after sulfur impregnation. (b) The galvanostatic test of h-TiN and m-TiN at a high current density of 5C rate.105 Copyright 2019, Wiley-VCH. (c) Schematic of the synthetic pathway. SEM images of (d) GDMC, (e) PPO-coated GDMC, and (f) gyroidal nanohybrid. (g) Potential during external lithiation of the gyroidal sulfur–PEDOT phase vs. lithium metal in liquid electrolyte. (h) Open-circuit voltage of the solid-state gyroidal hybrid over the first 4 h after charge.142 Copyright 2018, Royal Society of Chemistry. | ||
Miniaturization of ESSs enables efficient usage of space and maximizes volumetric or areal energy storage performance, providing many opportunities for integration with other electronic devices. Conventional planar 2D stacked configurations of ESSs entail a waste of space on a macroscopic scale. For example, the electrolyte/separator layer between the anode and cathode occupies a micrometer-scale space. The fabrication of a bicontinuous anode–electrolyte–cathode layer at the nanometer scale can solve this issue. Although this concept has already been established,142 its implementation has been difficult because of strict material requirements and the complexity of their nanofabrication. Recently, Werner et al. successfully fabricated a cathode–electrolyte–anode system with a 3D bicontinuous network with a size less than 50 nm and demonstrated its battery operation (Fig. 16c–h).142 First, they synthesized gyroidal mesoporous carbon (GDMC) monoliths that satisfied certain prerequisites (lithium redox, electrical conductivity, and structural integrity) to act as anodes and provided sufficient space for nanoconfinement of the electrolyte and cathode layers. Subsequently, a thin film of poly(phenylene oxide) (PPO) (thickness of less than 10 nm) was uniformly coated on the surface of the GDMC monoliths by electropolymerization. This PPO thin film allowed ionic transport while ensuring electronic insulation between the anode and cathode layers. Finally, bifunctional sulfur–poly(3,4-ethylenedioxythiophene) (PEDOT) was filled into PPO-coated GDMC through an in situ infiltration–polymerization method. Here, sulfur and PEDOT were selected as the redox-active material and current collector, respectively. To introduce lithium, the sulfur–PEDOT phase was electrochemically reduced using lithium metal as the counter electrode (Fig. 15g). After charging to 3.5 V, the gyroidal nanohybrid exhibited a stable open-circuit voltage of 2.8 V for several hours (Fig. 15h). In addition, a well-defined discharge plateau was maintained for 20 cycles. Nanoscale 3D bi-continuous network ESSs are in an early state of research but they would provide help to fabricate a new type of battery in the future.
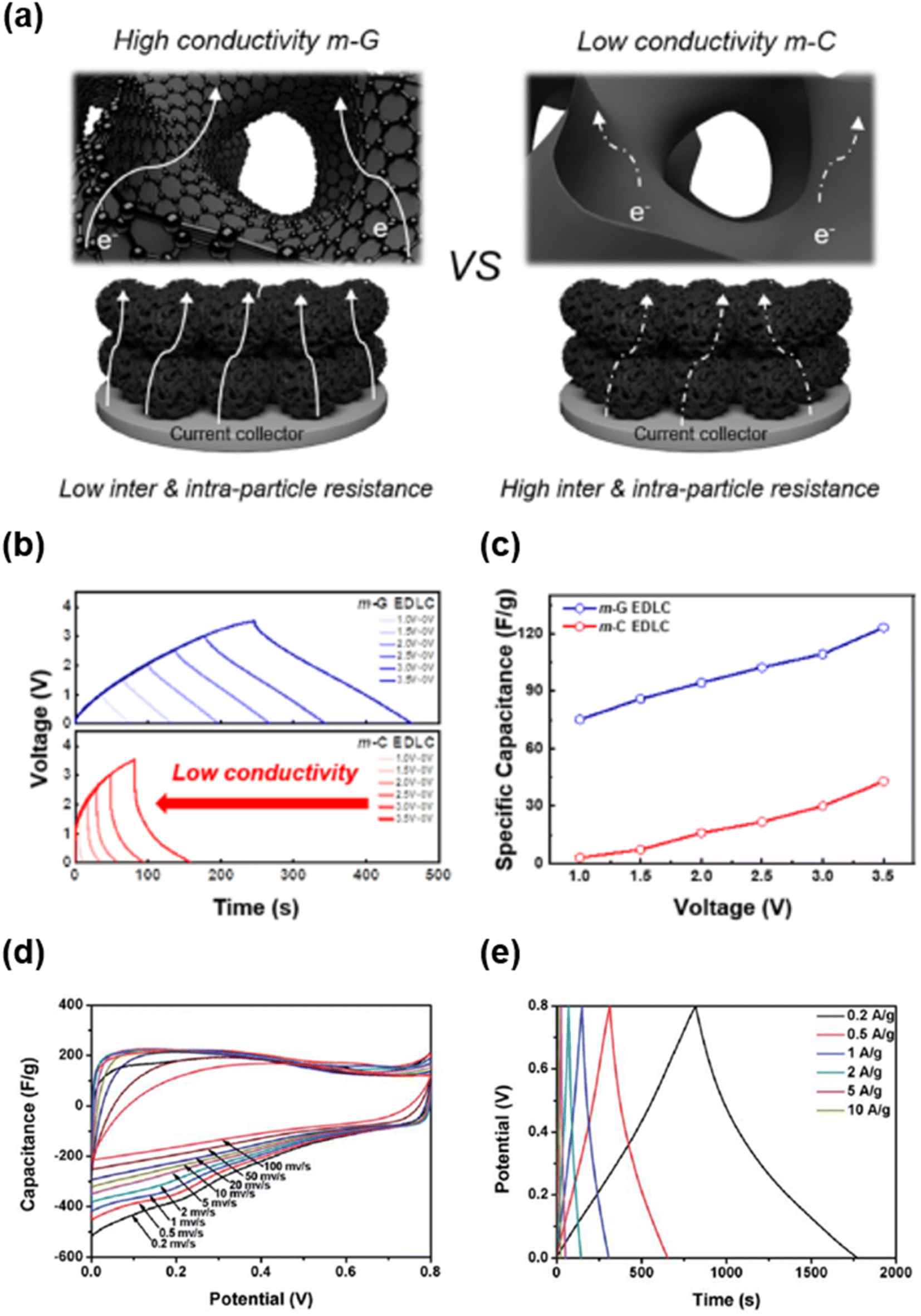 | ||
| Fig. 16 (a) Schematic of electron transport in m-G (left) and m-C (right) EDLCs. (b) Galvanostatic charge/discharge curves of EDLCs at various operation voltages up to 3.5 V. (c) Specific capacitance of EDLCs at various operation voltages.233 Copyright 2022, American Chemical Society. (d) CV curves and (e) galvanostatic charge/discharge curves of the N-doped mesoporous carbon.28 Copyright 2018, Royal Society of Chemistry. | ||
5.2. Supercapacitors
Compared to batteries, supercapacitors (SCs) exhibit higher power densities, superior long-term stability, and higher rate capabilities.263–266 Therefore, they are mainly used for applications that require high power density and long lifetimes that cannot be achieved by batteries. The energy storage mechanisms of SC materials can be divided into two categories: non-faradaic and faradaic reactions. In non-faradaic materials, energy is stored by forming an electric double-layer through physical adsorption and desorption of ions at the interface between electrolytes and active materials;267–269 thus, these materials are mainly called electric double layer capacitors (EDLCs). Energy storage in EDLCs is fast and stable for a long time because it usually relies on the physical adsorption and desorption of ions.270–272 Meanwhile, the energy storage mechanism of faradaic materials involves a redox reaction.263,273 These materials exhibit inferior stability to non-faradaic materials but have high specific capacitance.In an EDLC system, mesoporous carbon exhibits high capacitance and excellent rate capability. Graphitization of mesoporous carbon is another effective approach for improving the electrochemical performance of EDLCs by enhancing the electrical conductivity of active materials.65,135 Kim et al. synthesized mesoporous graphene (m-G) and mesoporous carbon (m-C) with almost identical structures (pore size and surface area) to evaluate the effect of electrical conductivity (Fig. 16a–c).233 Because interconnected graphene channels were distributed throughout its mesoporous structure, m-G exhibited lower inter/intra-particle resistance, thereby facilitating fast electron transport (Fig. 16a). With increasing operation voltage (0–3.5 V), the galvanic charging/discharging (GCD) curve of m-G maintained a symmetrical shape, whereas that of m-C was significantly distorted (Fig. 16b). In addition, at the highest operation voltage (3.5 V), the specific capacitance (123.3 F g−1) of m-G was considerably higher than that (42.8 F g−1) of m-C (Fig. 16c). These results imply that graphitization of a mesoporous carbon framework is an effective approach to significantly improve the electrochemical performance.
Heteroatom doping on the carbon surface is an effective strategy for enhancing the electrochemical performance by the addition of a pseudo-capacitive reaction. Many studies on doping mesoporous carbon with hetero atoms such as B, N, O, P, and S have been reported.274–278 Liu et al. synthesized N-doped mesoporous carbon and prepared high-performance EDLCs (Fig. 16d and e).29 The synthesized N-doped mesoporous carbon exhibited a high nitrogen content of 19 wt%, mesopores with pore sizes of 9.5–17.2 nm and large surface area (458–476 m2 g−1). The cyclic voltammetry (CV) curve of the N-doped mesoporous carbon showed a rectangular shape over the entire voltage range owing to the EDLC and reversible humps due to the faradaic reaction between 0.2 and 0.6 V (Fig. 16d). In addition, the symmetrical GCD curve indicated stable device operation (Fig. 16e). The specific capacitance was 225 F g−1 at 0.5 A g−1, which was much higher than that of neat mesoporous carbon. These results are attributed to both the pseudocapacitive reaction of the nitrogen content and high surface area of the mesoporous structure. Many transition metal oxides exhibit pseudocapacitive behavior and have been widely utilized as pseudo-capacitor electrodes.279–284 Kim et al. showed that a mesoporous tungsten trioxide (WO3) film exhibited mesopores (∼30 nm) and high specific surface area (32.14 m2 g−1).13 At a current density of 0.02 mA cm−2, the areal capacitances of a mesoporous WO3 film were similar to that of a compact WO3 film without mesopores. However, the mesoporous WO3 film exhibited a much higher rate capability. The capacitance retentions of mesoporous and compact WO3 were 68% and 34%, respectively, when the current density was changed from 0.02 mA cm−2 to 1.0 mA cm−2. This difference is attributed to the effective and fast ion transport owing to the mesoporous structure.
6. Summary and perspective
This paper summarizes the recent progress and achievements in the soft-templating synthesis of mesoporous materials. First, we described the basic synthetic approaches, such as hard and soft templating methods, for fabricating mesoporous materials, as well as their (dis)advantages. Second, we discussed various compositions of mesoporous materials prepared by using the soft-templating method. Through the design of controllable synthesis conditions, such as self-assembly of SDAs and precursors, types of SDAs or precursors, and post-treatment, the soft-templating method has achieved tremendous progress in various applications with a wide range of mesoporous structures. Third, we summarized the various architectures of mesoporous materials synthesized by new soft-templating methods. Finally, we discussed the recent progress of mesoporous materials for energy storage systems such as batteries and supercapacitors.Despite notable achievements in soft-templating synthetic approaches, certain challenges and opportunities remain as follows. (i) In the synthesis of mesoporous materials, especially metal oxides and nitrides, the conversion from mesostructured composites to mesoporous materials relies on high-temperature thermal treatment, which restrict their direct integration on plastic substrates for flexible electrode applications. Therefore, innovative synthetic approaches capable of template removal while inducing the hydrolysis and condensation reactions of inorganic materials at low temperatures are highly required. (ii) Although great advances have been achieved in the synthesis of mesoporous materials with hierarchical structures, precise tailoring of the macrostructures of most hierarchical structures remains difficult. For example, hierarchical macro/mesoporous materials can be obtained using an SD of a polymer blend, but the resultant synthesized macroporous shape is not diverse and unpredictable. To expand the functionality of mesoporous materials, it is necessary to realize various macrostructures (complex architecture, sheets, spheres, and janus) by precisely controlling the formation of mesopores and macrostructure during self-assembly. (iii) For electrode applications for ESSs, mesoporous materials should have specific structures for satisfying high mass loading and effective loading of active materials in mesoporous materials. To achieve high mass loading on a current collector, spherical mesoporous particles with uniform size should be realized. In addition, for efficient interaction between the active materials and the electrolyte, active materials such as high-capacity active materials, alkali metals and sulfur, should be uniformly loaded into the conductive and physically/chemically stable mesopores without the restriction of electrolyte penetration. Therefore, efforts for the precisely controlled structures would attract great attention for high performance ESSs. In this regard, soft-templating methods have been effectively applied in various research fields because of the diversity of methods and structures.
Author contribution
K.-W. Kim designed and wrote the draft of the manuscript. B. Park and J. Kim helped to design the manuscript and to arrange figures and references. J. K. Kim and C. J. supervised and wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Creative Research Initiative Program supported by the National Research Foundation of Korea (NRF) (grant no. 2022R1A3A3002149) funded by the Korean Government. This work was supported by the Korea Institute of Energy Technology Evaluation and Planning (KETEP) grant funded by the Korea Government (MOTIE) (20228510070100, development of long-life sodium ion batteries using a low-cost Prussian blue analogue).References
- C. Li, Q. Li, Y. V. Kaneti, D. Hou, Y. Yamauchi and Y. Mai, Chem. Soc. Rev., 2020, 49, 4681–4736 RSC.
- E. Lim, C. Jo and J. Lee, Nanoscale, 2016, 8, 7827–7833 RSC.
- Y. Deng, J. Wei, Z. Sun and D. Zhao, Chem. Soc. Rev., 2013, 42, 4054–4070 RSC.
- T. Kim, W. Song, D.-Y. Son, L. K. Ono and Y. Qi, J. Mater. Chem. A, 2019, 7, 2942–2964 RSC.
- M. R. Benzigar, S. N. Talapaneni, S. Joseph, K. Ramadass, G. Singh, J. Scaranto, U. Ravon, K. Al-Bahily and A. Vinu, Chem. Soc. Rev., 2018, 47, 2680–2721 RSC.
- M. Peer, M. Lusardi and K. F. Jensen, Chem. Mater., 2017, 29, 1496–1506 CrossRef CAS.
- S. Yin, T. Tian, K. S. Wienhold, C. L. Weindl, R. Guo, M. Schwartzkopf, S. V. Roth and P. Müller-Buschbaum, Adv. Mater. Interfaces, 2020, 7, 2001002 CrossRef CAS.
- K. Liang, W. Wang, Y. Yu, L. Liu, H. Lv, Y. Zhang and A. Chen, New J. Chem., 2019, 43, 2776–2782 RSC.
- J. Cherusseri and K. K. Kar, J. Mater. Chem. A, 2016, 4, 9910–9922 RSC.
- N. K and C. S. Rout, J. Mater. Chem. A, 2021, 9, 8248–8278 RSC.
- J. Balach, J. Linnemann, T. Jaumann and L. Giebeler, J. Mater. Chem. A, 2018, 6, 23127–23168 RSC.
- H. Ao, Y. Zhao, J. Zhou, W. Cai, X. Zhang, Y. Zhu and Y. Qian, J. Mater. Chem. A, 2019, 7, 18708–18734 RSC.
- K.-W. Kim, T. Y. Yun, S.-H. You, X. Tang, J. Lee, Y. Seo, Y.-T. Kim, S. H. Kim, H. C. Moon and J. K. Kim, NPG Asia Mater., 2020, 12, 84 CrossRef CAS.
- L. Peng, Z. Fang, J. Li, L. Wang, A. M. Bruck, Y. Zhu, Y. Zhang, K. J. Takeuchi, A. C. Marschilok, E. A. Stach, E. S. Takeuchi and G. Yu, ACS Nano, 2018, 12, 820–828 CrossRef CAS PubMed.
- J. Jiang, Y. Li, J. Liu, X. Huang, C. Yuan and X. W. Lou, Adv. Mater., 2012, 24, 5166–5180 CrossRef CAS PubMed.
- Y. Zou, X. Zhou, J. Ma, X. Yang and Y. Deng, Chem. Soc. Rev., 2020, 49, 1173–1208 RSC.
- B. K. Wheatle, J. R. Hampton, G. G. Rodríguez-Calero, J. G. Werner, Y. Gu, U. Wiesner and H. D. Abruña, J. Electroanal. Chem., 2020, 871, 114284 CrossRef CAS.
- Q. Lu and Y. Zhou, J. Power Sources, 2011, 196, 4088–4094 CrossRef CAS.
- M. R. Lukatskaya, S. Kota, Z. Lin, M.-Q. Zhao, N. Shpigel, M. D. Levi, J. Halim, P.-L. Taberna, M. W. Barsoum, P. Simon and Y. Gogotsi, Nat. Energy, 2017, 2, 17105 CrossRef CAS.
- G. Feng, S. Li, V. Presser and P. T. Cummings, J. Phys. Chem. Lett., 2013, 4, 3367–3376 CrossRef CAS.
- Y. Wang, Y. Song and Y. Xia, Chem. Soc. Rev., 2016, 45, 5925–5950 RSC.
- R. Ma, Z. Chen, D. Zhao, X. Zhang, J. Zhuo, Y. Yin, X. Wang, G. Yang and F. Yi, J. Mater. Chem. A, 2021, 9, 11501–11529 RSC.
- R. Wang, X. Yan, J. Lang, Z. Zheng and P. Zhang, J. Mater. Chem. A, 2014, 2, 12724–12732 RSC.
- M. Zhang, H. Mei, P. Chang and L. Cheng, J. Mater. Chem. A, 2020, 8, 10670–10694 RSC.
- Y. Yang, W. Wang, G. Meng and J. Zhang, J. Mater. Chem. A, 2022, 10, 14137–14170 RSC.
- Z. Zhou, T. Liu, A. U. Khan and G. Liu, Sci. Adv., 2019, 5, eaau6852 CrossRef PubMed.
- Z. Li, K. Guo and X. Chen, RSC Adv., 2017, 7, 30521–30532 RSC.
- K. Yan, L.-B. Kong, Y.-H. Dai, M. Shi, K.-W. Shen, B. Hu, Y.-C. Luo and L. Kang, J. Mater. Chem. A, 2015, 3, 22781–22793 RSC.
- Y. Liu, Z. Wang, W. Teng, H. Zhu, J. Wang, A. A. Elzatahry, D. Al-Dahyan, W. Li, Y. Deng and D. Zhao, J. Mater. Chem. A, 2018, 6, 3162–3170 RSC.
- C. Liang, Z. Li and S. Dai, Angew. Chem., Int. Ed., 2008, 47, 3696–3717 CrossRef CAS PubMed.
- J. Fan, X. Ran, Y. Ren, C. Wang, J. Yang, W. Teng, L. Zou, Y. Sun, B. Lu, Y. Deng and D. Zhao, Langmuir, 2016, 32, 9922–9929 CrossRef CAS PubMed.
- H. Xiong, T. Gao, K. Li, Y. Liu, Y. Ma, J. Liu, Z.-A. Qiao, S. Song and S. Dai, Adv. Sci., 2019, 6, 1801543 CrossRef PubMed.
- K. W. Tan, B. Jung, J. G. Werner, E. R. Rhoades, M. O. Thompson and U. Wiesner, Science, 2015, 349, 54–58 CrossRef CAS PubMed.
- V. Velez, G. Ramos-Sánchez, B. Lopez, L. Lartundo-Rojas, I. González and L. Sierra, Carbon, 2019, 147, 214–226 CrossRef CAS.
- Z. Su, C. Yang, C. Xu, H. Wu, Z. Zhang, T. Liu, C. Zhang, Q. Yang, B. Li and F. Kang, J. Mater. Chem. A, 2013, 1, 12432–12440 RSC.
- A. Singh and V. Kalra, J. Mater. Chem. A, 2019, 7, 11613–11650 RSC.
- E. Ramasamy, C. Jo, A. Anthonysamy, I. Jeong, J. K. Kim and J. Lee, Chem. Mater., 2012, 24, 1575–1582 CrossRef CAS.
- E. Ramasamy, J. Chun and J. Lee, Carbon, 2010, 48, 4563–4565 CrossRef CAS.
- H. Qutaish, S. Tanaka, Y. V. Kaneti, J. Lin, Y. Bando, A. A. Alshehri, S.-I. Yusa, Y. Yamauchi, M. S. A. Hossain and J. Kim, Microporous Mesoporous Mater., 2018, 271, 16–22 CrossRef CAS.
- C. Jo, Y. Park, J. Jeong, K. T. Lee and J. Lee, ACS Appl. Mater. Interfaces, 2015, 7, 11748–11754 CrossRef CAS PubMed.
- S. Shubhashish, A. S. Amin, Y. Dang, S. J. Karasik, H. S. Khanna and S. L. Suib, ACS Appl. Nano Mater., 2022, 5, 7078–7091 CrossRef CAS.
- E. Kamali-Heidari and A. Kamyabi-Gol, Phys. Rev. B Condens. Matter, 2019, 570, 176–181 CrossRef CAS.
- A. S. Nugraha, V. Malgras, J. Kim, J. Bo, C. Li, M. Iqbal, Y. Yamauchi and T. Asahi, Small Methods, 2018, 2, 1800283 CrossRef.
- T. Soboleva, X. Zhao, K. Malek, Z. Xie, T. Navessin and S. Holdcroft, ACS Appl. Mater. Interfaces, 2010, 2, 375–384 CrossRef CAS PubMed.
- Y. Wang, Y. Zhu, S. Chen and W. Li, Energy Fuels, 2014, 28, 945–955 CrossRef CAS.
- Y. Gogotsi, A. Nikitin, H. Ye, W. Zhou, J. E. Fischer, B. Yi, H. C. Foley and M. W. Barsoum, Nat. Mater., 2003, 2, 591–594 CrossRef CAS PubMed.
- T. Kotaka, Y. Tabuchi and P. P. Mukherjee, J. Power Sources, 2015, 280, 231–239 CrossRef CAS.
- V. Malgras, J. Tang, J. Wang, J. Kim, N. L. Torad, S. Dutta, K. Ariga, M. S. A. Hossain, Y. Yamauchi and K. C. W. Wu, J. Nanosci. Nanotechnol., 2019, 19, 3673–3685 CrossRef CAS PubMed.
- X. Ren, H. Li, J. Chen, L. Wei, A. Modak, H. Yang and Q. Yang, Carbon, 2017, 114, 473–481 CrossRef CAS.
- L. Wu, Y. Li, Z. Fu and B.-L. Su, Natl. Sci. Rev., 2020, 7, 1667–1701 CrossRef CAS PubMed.
- K. J. De France, F. Xu and T. Hoare, Adv. Healthc. Mater., 2018, 7, 1700927 CrossRef PubMed.
- Y. Ding, C. Wang, R. Zheng, S. Maitra, G. Zhang, T. Barakat, S. Roy, B.-L. Su and L.-H. Chen, J. Energy Chem., 2022, 4, 100081 CAS.
- S. Lee, M. Choun, Y. Ye, J. Lee, Y. Mun, E. Kang, J. Hwang, Y.-H. Lee, C.-H. Shin, S.-H. Moon, S.-K. Kim, E. Lee and J. Lee, Angew. Chem., Int. Ed., 2015, 54, 9230–9234 CrossRef CAS PubMed.
- Y. Mun, M. J. Kim, S.-A. Park, E. Lee, Y. Ye, S. Lee, Y.-T. Kim, S. Kim, O.-H. Kim, Y.-H. Cho, Y.-E. Sung and J. Lee, Appl. Catal., B, 2018, 222, 191–199 CrossRef CAS.
- M. G. Fischer, X. Hua, B. D. Wilts, I. Gunkel, T. M. Bennett and U. Steiner, ACS Appl. Mater. Interfaces, 2017, 9, 22388–22397 CrossRef CAS PubMed.
- J. Hwang, C. Jo, M. G. Kim, J. Chun, E. Lim, S. Kim, S. Jeong, Y. Kim and J. Lee, ACS Nano, 2015, 9, 5299–5309 CrossRef CAS PubMed.
- C. Xu, D. Niu, N. Zheng, H. Yu, J. He and Y. Li, ACS Sustain. Chem. Eng., 2018, 6, 5999–6007 CrossRef CAS.
- G. Lee, C. Lee, C.-M. Yoon, M. Kim and J. Jang, ACS Appl. Mater. Interfaces, 2017, 9, 5222–5230 CrossRef CAS PubMed.
- Y. Qiu, M. Hou, J. Gao, H. Zhai, H. Liu, M. Jin, X. Liu and L. Lai, Small, 2019, 15, 1903836 CrossRef CAS PubMed.
- T. Wagner, S. Haffer, C. Weinberger, D. Klaus and M. Tiemann, Chem. Soc. Rev., 2013, 42, 4036–4053 RSC.
- Y. Shimizu, A. Jono, T. Hyodo and M. Egashira, Sens. Actuators, B, 2005, 108, 56–61 CrossRef CAS.
- S. Zeng, S. Liu, Y. Qi, L. Cui, Q. Dai and C. Bai, RSC Adv., 2018, 8, 11462–11468 RSC.
- W. Zhang, R.-r. Cheng, H.-h. Bi, Y.-h. Lu, L.-b. Ma and X.-j. He, New Carbon Mater., 2021, 36, 69–81 CrossRef CAS.
- T. Ghoshal, A. Thorat, N. Prochukhan and M. A. Morris, Nanomaterials, 2022, 12, 2223 CrossRef CAS PubMed.
- J. Lee, R. Nankya, A. Kim and H. Jung, Electrochim. Acta, 2018, 290, 496–505 CrossRef CAS.
- J. Lee, M. Kang, I.-K. Shim, D. H. Lee, A. Kim and H. Jung, J. Nanosci. Nanotechnol., 2018, 18, 6995–7003 CrossRef CAS PubMed.
- J. Wei, D. Zhou, Z. Sun, Y. Deng, Y. Xia and D. Zhao, Adv. Funct. Mater., 2013, 23, 2322–2328 CrossRef CAS.
- J. Zhang, Y. Deng, D. Gu, S. Wang, L. She, R. Che, Z.-S. Wang, B. Tu, S. Xie and D. Zhao, Adv. Energy Mater., 2011, 1, 241–248 CrossRef CAS.
- T. Matsuno, Y. Kuroda, M. Kitahara, A. Shimojima, H. Wada and K. Kuroda, Angew. Chem., Int. Ed., 2016, 55, 6008–6012 CrossRef CAS PubMed.
- Q. Wu, C. Liu, J. Peng and F. Liu, RSC Adv., 2017, 7, 19557–19564 RSC.
- Z. Wu, Q. Li, D. Feng, P. A. Webley and D. Zhao, J. Am. Chem. Soc., 2010, 132, 12042–12050 CrossRef CAS PubMed.
- P. Strubel, S. Thieme, T. Biemelt, A. Helmer, M. Oschatz, J. Brückner, H. Althues and S. Kaskel, Adv. Funct. Mater., 2015, 25, 287–297 CrossRef CAS.
- M. R. J. Scherer, L. Li, P. M. S. Cunha, O. A. Scherman and U. Steiner, Adv. Mater., 2012, 24, 1217–1221 CrossRef CAS PubMed.
- L. Xiang, S. Yuan, F. Wang, Z. Xu, X. Li, F. Tian, L. Wu, W. Yu and Y. Mai, J. Am. Chem. Soc., 2022, 144, 15497–15508 CrossRef CAS PubMed.
- Y. La, J. Song, M. G. Jeong, A. Cho, S.-M. Jin, E. Lee and K. T. Kim, Nat. Commun., 2018, 9, 5327 CrossRef CAS PubMed.
- S. Tanaka, N. Nishiyama, Y. Egashira and K. Ueyama, Chem. Commun., 2005, 16, 2125–2127 RSC.
- M. Trivedi, F. Peng, X. Xia, P. I. Sepulveda-Medina and B. D. Vogt, Langmuir, 2019, 35, 14049–14059 CrossRef CAS PubMed.
- H.-F. Fei, Y. Long, H.-J. Yu, B. M. Yavitt, W. Fan, A. Ribbe and J. J. Watkins, ACS Appl. Mater. Interfaces, 2020, 12, 57322–57329 CrossRef CAS PubMed.
- J. Choma, J. Jagiello and M. Jaroniec, Carbon, 2021, 183, 150–157 CrossRef CAS.
- D. Fattakhova-Rohlfing, M. Wark, T. Brezesinski, B. M. Smarsly and J. Rathouský, Adv. Funct. Mater., 2007, 17, 123–132 CrossRef CAS.
- T. Lunkenbein, M. Kamperman, M. Schieder, S. With, Z. Li, H. Sai, S. Förster, U. Wiesner and J. Breu, J. Mater. Chem. A, 2013, 1, 6238–6248 RSC.
- T. M. Bennett, G. He, R. R. Larder, M. G. Fischer, G. A. Rance, M. W. Fay, A. K. Pearce, C. D. J. Parmenter, U. Steiner and S. M. Howdle, Nano Lett., 2018, 18, 7560–7569 CrossRef CAS PubMed.
- F. Meng, Z. Fang, Z. Li, W. Xu, M. Wang, Y. Liu, J. Zhang, W. Wang, D. Zhao and X. Guo, J. Mater. Chem. A, 2013, 1, 7235–7241 RSC.
- W.-C. Chu, B. P. Bastakoti, Y. V. Kaneti, J.-G. Li, H. R. Alamri, Z. A. Alothman, Y. Yamauchi and S.-W. Kuo, Eur. J. Chem., 2017, 23, 13734–13741 CrossRef CAS PubMed.
- Y. Oka, Y. Kuroda, T. Matsuno, K. Kamata, H. Wada, A. Shimojima and K. Kuroda, Eur. J. Chem., 2017, 23, 9362–9368 CrossRef CAS PubMed.
- J. Wei, Z. Sun, W. Luo, Y. Li, A. A. Elzatahry, A. M. Al-Enizi, Y. Deng and D. Zhao, J. Am. Chem. Soc., 2017, 139, 1706–1713 CrossRef CAS PubMed.
- Y. Zou, X. Zhou, Y. Zhu, X. Cheng, D. Zhao and Y. Deng, Acc. Chem. Res., 2019, 52, 714–725 CrossRef CAS PubMed.
- T. Nagaura, H.-P. Phan, V. Malgras, T.-A. Pham, H. Lim, A. Ashok, J. Kim, J. You, N.-T. Nguyen, J. Na and Y. Yamauchi, Angew. Chem., Int. Ed., 2021, 60, 9660–9665 CrossRef CAS PubMed.
- O. Olatidoye, D. Thomas and B. P. Bastakoti, New J. Chem., 2021, 45, 15761–15766 RSC.
- P. Li, Y. Feng, D. Cheng and J. Wei, J. Colloid Interface Sci., 2022, 625, 435–445 CrossRef CAS PubMed.
- B. P. Bastakoti, S. Ishihara, S.-Y. Leo, K. Ariga, K. C. W. Wu and Y. Yamauchi, Langmuir, 2014, 30, 651–659 CrossRef CAS PubMed.
- Y. Li, W. Luo, N. Qin, J. Dong, J. Wei, W. Li, S. Feng, J. Chen, J. Xu, A. A. Elzatahry, M. H. Es-Saheb, Y. Deng and D. Zhao, Angew. Chem., Int. Ed., 2014, 53, 9035–9040 CrossRef CAS PubMed.
- W. S. Chi, C. S. Lee, H. Long, M. H. Oh, A. Zettl, C. Carraro, J. H. Kim and R. Maboudian, ACS Appl. Mater. Interfaces, 2017, 9, 37246–37253 CrossRef CAS PubMed.
- S. Tanaka, B. P. Bastakoti, Y. L. S.-i. Yusa, D. Ishii, K. Kani, A. Fatehmulla, W. A. Farooq, M. J. A. Shiddiky, Y. Bando, Y. V. Kaneti, Y. Yamauchi and M. S. A. Hossain, Eur. J. Inorg. Chem., 2017, 2017, 1328–1332 CrossRef CAS.
- X. Xiao, L. Liu, J. Ma, Y. Ren, X. Cheng, Y. Zhu, D. Zhao, A. A. Elzatahry, A. Alghamdi and Y. Deng, ACS Appl. Mater. Interfaces, 2018, 10, 1871–1880 CrossRef CAS PubMed.
- V. Malgras, Y. Shirai, T. Takei and Y. Yamauchi, J. Am. Chem. Soc., 2020, 142, 15815–15822 CrossRef CAS PubMed.
- A. Balint, M. Papendick, M. Clauss, C. Müller, F. Giesselmann and S. Naumann, Chem. Commun., 2018, 54, 2220–2223 RSC.
- F. Markus, J. R. Bruckner and S. Naumann, Macromol. Chem. Phys., 2020, 221, 1900437 CrossRef CAS.
- J. Shim, J. Lee, Y. Ye, J. Hwang, S.-K. Kim, T.-H. Lim, U. Wiesner and J. Lee, ACS Nano, 2012, 6, 6870–6881 CrossRef CAS PubMed.
- T. Liu, J. Serrano, J. Elliott, X. Yang, W. Cathcart, Z. Wang, Z. He and G. Liu, Sci. Adv., 2020, 6, eaaz0906 CrossRef CAS PubMed.
- A. A. S. Gonçalves and M. Jaroniec, J. Colloid Interface Sci., 2019, 537, 725–735 CrossRef PubMed.
- Y. Ren, X. Yang, X. Zhou, W. Luo, Y. Zhang, X. Cheng and Y. Deng, Chin. Chem. Lett., 2019, 30, 2003–2008 CrossRef CAS.
- J. Hwang, S. Kim, U. Wiesner and J. Lee, Adv. Mater., 2018, 30, 1801127 CrossRef PubMed.
- A. Fischer, M. Antonietti and A. Thomas, Adv. Mater., 2007, 19, 264–267 CrossRef CAS.
- W.-G. Lim, C. Jo, A. Cho, J. Hwang, S. Kim, J. W. Han and J. Lee, Adv. Mater., 2019, 31, 1806547 CrossRef PubMed.
- J. Shi, H. Cui, J. Xu, N. Yan and Y. Liu, Chem. Eng. J., 2020, 389, 124459 CrossRef CAS.
- Y. Mun, J. Shim, K. Kim, J. W. Han, S.-K. Kim, Y. Ye, J. Hwang, S. Lee, J. Jang, Y.-T. Kim and J. Lee, RSC Adv., 2016, 6, 88255–88264 RSC.
- H.-S. Hsueh, C.-T. Yang, J. I. Zink and M. H. Huang, J. Phys. Chem. B, 2005, 109, 4404–4409 CrossRef CAS PubMed.
- K. Brezesinski, J. Haetge, J. Wang, S. Mascotto, C. Reitz, A. Rein, S. H. Tolbert, J. Perlich, B. Dunn and T. Brezesinski, Small, 2011, 7, 407–414 CrossRef CAS PubMed.
- F. Yu, R. P. Thedford, K. R. Hedderick, G. Freychet, M. Zhernenkov, L. A. Estroff, K. C. Nowack, S. M. Gruner and U. B. Wiesner, ACS Appl. Mater. Interfaces, 2021, 13, 34732–34741 CrossRef CAS PubMed.
- B. T. Sone, S. S. Nkosi, M. M. Nkosi, E. Coetsee-Hugo, H. C. Swart and M. Maaza, AIP Conf. Proc., 2018, 1962, 040003 CrossRef.
- T. W. Hansen, A. T. DeLaRiva, S. R. Challa and A. K. Datye, Acc. Chem. Res., 2013, 46, 1720–1730 CrossRef CAS PubMed.
- M. S. Song, S. Nahm, W. I. Cho and C. Lee, Phys. Chem. Chem. Phys., 2015, 17, 23496–23502 RSC.
- L. Almar, T. Andreu, A. Morata, M. Torrell, L. Yedra, S. Estradé, F. Peiró and A. Tarancón, J. Mater. Chem. A, 2014, 2, 3134–3141 RSC.
- Y. Ito, Y. Tanabe, H. J. Qiu, K. Sugawara, S. Heguri, N. H. Tu, K. K. Huynh, T. Fujita, T. Takahashi, K. Tanigaki and M. Chen, Angew. Chem., 2014, 126, 4922–4926 CrossRef.
- F. Markus, C. Vogler, J. R. Bruckner and S. Naumann, ACS Appl. Nano Mater., 2021, 4, 3486–3492 CrossRef CAS.
- Y. Deng, T. Yu, Y. Wan, Y. Shi, Y. Meng, D. Gu, L. Zhang, Y. Huang, C. Liu, X. Wu and D. Zhao, J. Am. Chem. Soc., 2007, 129, 1690–1697 CrossRef CAS PubMed.
- W. Zhong, T. Jiang, T. Jafari, A. S. Poyraz, W. Wu, D. A. Kriz, S. Du, S. Biswas, M. Thompson Pettes and S. L. Suib, Microporous Mesoporous Mater., 2017, 239, 328–335 CrossRef CAS.
- S.-z. Niu, S.-d. Wu, W. Lu, Q.-h. Yang and F.-y. Kang, New Carbon Mater., 2017, 32, 289–296 CrossRef CAS.
- X. Deng, K. Chen and H. Tüysüz, Chem. Mater., 2017, 29, 40–52 CrossRef CAS.
- W. Luc and F. Jiao, Acc. Chem. Res., 2016, 49, 1351–1358 CrossRef CAS PubMed.
- Z. Liu, C. Li, M. Kuang, B. Liu and B. Yang, Environ. Sci. Pollut. Res., 2021, 28, 31630–31639 CrossRef CAS PubMed.
- Z. Zhang, Y. Zhu, H. Asakura, B. Zhang, J. Zhang, M. Zhou, Y. Han, T. Tanaka, A. Wang, T. Zhang and N. Yan, Nat. Commun., 2017, 8, 16100 CrossRef CAS PubMed.
- C. Wang, Y. Zhao, L. Zhou, Y. Liu, W. Zhang, Z. Zhao, W. N. Hozzein, H. M. S. Alharbi, W. Li and D. Zhao, J. Mater. Chem. A, 2018, 6, 21550–21557 RSC.
- Y. Dong, X. Zhan, X. Niu, J. Li, F. Yuan, Y. Zhu and H. Fu, Microporous Mesoporous Mater., 2014, 185, 97–106 CrossRef CAS.
- B. E. Wilson, S. G. Rudisill and A. Stein, Microporous Mesoporous Mater., 2014, 197, 174–179 CrossRef CAS.
- K. Nanaji, A. Jyothirmayi, U. Varadaraju, T. N. Rao and S. Anandan, J. Alloys Compd., 2017, 723, 488–497 CrossRef CAS.
- J. H. Pan, X. S. Zhao and W. I. Lee, Chem. Eng. J., 2011, 170, 363–380 CrossRef CAS.
- C. T. Kresge, M. E. Leonowicz, W. J. Roth, J. C. Vartuli and J. S. Beck, Nature, 1992, 359, 710–712 CrossRef CAS.
- J. S. Beck, J. C. Vartuli, W. J. Roth, M. E. Leonowicz, C. T. Kresge, K. D. Schmitt, C. T. W. Chu, D. H. Olson, E. W. Sheppard, S. B. McCullen, J. B. Higgins and J. L. Schlenker, J. Am. Chem. Soc., 1992, 114, 10834–10843 CrossRef CAS.
- Y. Wan, Y. Shi and D. Zhao, Chem. Commun., 2007, 9, 897–926 RSC.
- Y. Wan and D. Zhao, Chem. Rev., 2007, 107, 2821–2860 CrossRef CAS PubMed.
- D. Zhao, J. Feng, Q. Huo, N. Melosh, G. H. Fredrickson, B. F. Chmelka and G. D. Stucky, Science, 1998, 279, 548–552 CrossRef CAS PubMed.
- J. Shi, N. Lin, H.-b. Lin, J. Yang and W.-l. Zhang, New Carbon Mater., 2020, 35, 401–409 CrossRef CAS.
- S. Zhu, H. Tian, N. Wang, B. Chen, Y. Mai and X. Feng, Small, 2018, 14, 1702755 CrossRef PubMed.
- D. Hou, J. Zhang, H. Tian, Q. Li, C. Li and Y. Mai, Adv. Mater. Interfaces, 2019, 6, 1901476 CrossRef CAS.
- J. Lee, M. Christopher Orilall, S. C. Warren, M. Kamperman, F. J. DiSalvo and U. Wiesner, Nat. Mater., 2008, 7, 222–228 CrossRef CAS PubMed.
- H. Wakayama, H. Yonekura and Y. Kawai, Int. J. Nanotechnol., 2018, 15, 798–803 CrossRef CAS.
- C. Jo, W.-G. Lim, A. H. Dao, S. Kim, S. Kim, S. Yoon and J. Lee, J. Mater. Chem. A, 2017, 5, 24782–24789 RSC.
- S. Choudhury, M. Agrawal, P. Formanek, D. Jehnichen, D. Fischer, B. Krause, V. Albrecht, M. Stamm and L. Ionov, ACS Nano, 2015, 9, 6147–6157 CrossRef CAS PubMed.
- B. Xu, F. Wu, D. Mu, L. Dai, G. Cao, H. Zhang, S. Chen and Y. Yang, Int. J. Hydrogen Energy, 2010, 35, 632–637 CrossRef CAS.
- J. G. Werner, G. G. Rodríguez-Calero, H. D. Abruña and U. Wiesner, Energy Environ. Sci., 2018, 11, 1261–1270 RSC.
- Y. Korenblit, M. Rose, E. Kockrick, L. Borchardt, A. Kvit, S. Kaskel and G. Yushin, ACS Nano, 2010, 4, 1337–1344 CrossRef CAS PubMed.
- T. N. Phan, M. K. Gong, R. Thangavel, Y. S. Lee and C. H. Ko, J. Alloys Compd., 2019, 780, 90–97 CrossRef CAS.
- S. Kim, M. Ju, J. Lee, J. Hwang and J. Lee, J. Am. Chem. Soc., 2020, 142, 9250–9257 CrossRef CAS PubMed.
- Y. Meng, D. Gu, F. Zhang, Y. Shi, H. Yang, Z. Li, C. Yu, B. Tu and D. Zhao, Angew. Chem., Int. Ed., 2005, 44, 7053–7059 CrossRef CAS PubMed.
- R. Liu, Y. Shi, Y. Wan, Y. Meng, F. Zhang, D. Gu, Z. Chen, B. Tu and D. Zhao, J. Am. Chem. Soc., 2006, 128, 11652–11662 CrossRef CAS PubMed.
- A. Zhang, A. Li, Y. Wang, M. Liu, H. Ma, Z. Song and J. Liu, RSC Adv., 2016, 6, 103843–103850 RSC.
- J. M. Serrano, T. Liu, A. U. Khan, B. Botset, B. J. Stovall, Z. Xu, D. Guo, K. Cao, X. Hao, S. Cheng and G. Liu, Chem. Mater., 2019, 31, 8898–8907 CrossRef CAS.
- T. Liu, Z. Zhou, Y. Guo, D. Guo and G. Liu, Nat. Commun., 2019, 10, 675 CrossRef CAS PubMed.
- Z. Zhou, T. Liu, A. U. Khan and G. Liu, Mol. Syst. Des. Eng., 2020, 5, 153–165 RSC.
- A. F. M. El-Mahdy, T.-E. Liu and S.-W. Kuo, J. Hazard. Mater., 2020, 391, 122163 CrossRef CAS PubMed.
- X. Yang, H. Ma and G. Zhang, Langmuir, 2017, 33, 3975–3981 CrossRef CAS PubMed.
- E. Kang, Y. S. Jung, G.-H. Kim, J. Chun, U. Wiesner, A. C. Dillon, J. K. Kim and J. Lee, Adv. Funct. Mater., 2011, 21, 4349–4357 CrossRef CAS.
- C. Jo, Y. Kim, J. Hwang, J. Shim, J. Chun and J. Lee, Chem. Mater., 2014, 26, 3508–3514 CrossRef CAS.
- X. Yang, X. Cheng, J. Ma, Y. Zou, W. Luo and Y. Deng, Small, 2019, 15, 1903058 CrossRef PubMed.
- K. Yu, A. J. Hurd, A. Eisenberg and C. J. Brinker, Langmuir, 2001, 17, 7961–7965 CrossRef CAS.
- K. Yu, B. Smarsly and C. J. Brinker, Adv. Funct. Mater., 2003, 13, 47–52 CrossRef CAS.
- P. Yang, D. Zhao, D. I. Margolese, B. F. Chmelka and G. D. Stucky, Nature, 1998, 396, 152–155 CrossRef CAS.
- P. Yang, D. Zhao, D. I. Margolese, B. F. Chmelka and G. D. Stucky, Chem. Mater., 1999, 11, 2813–2826 CrossRef CAS.
- H. Xiong, H. Zhou, C. Qi, Z. Liu, L. Zhang, L. Zhang and Z.-A. Qiao, Chem. Eng. J., 2020, 398, 125527 CrossRef CAS.
- J. Hwang, J. Kim, E. Ramasamy, W. Choi and J. Lee, Microporous Mesoporous Mater., 2011, 143, 149–156 CrossRef CAS.
- D. Xu, K. Ge, S. Qi, Y. Chen, J. Qiu, S. Wang, Y. Tian, S. Fang, C. Liu and Q. Liu, J. Mater. Sci., 2020, 55, 7645–7651 CrossRef CAS.
- J. Lee, J. Kim and T. Hyeon, Adv. Mater., 2006, 18, 2073–2094 CrossRef CAS.
- Z. Wang, Y. Zhu, W. Luo, Y. Ren, X. Cheng, P. Xu, X. Li, Y. Deng and D. Zhao, Chem. Mater., 2016, 28, 7773–7780 CrossRef.
- Y. Zhu, Y. Zhao, J. Ma, X. Cheng, J. Xie, P. Xu, H. Liu, H. Liu, H. Zhang, M. Wu, A. A. Elzatahry, A. Alghamdi, Y. Deng and D. Zhao, J. Am. Chem. Soc., 2017, 139, 10365–10373 CrossRef CAS PubMed.
- D. Feng, T.-N. Gao, M. Fan, A. Li, K. Li, T. Wang, Q. Huo and Z.-A. Qiao, NPG Asia Mater., 2018, 10, 800–809 CrossRef CAS.
- B. Eckhardt, E. Ortel, D. Bernsmeier, J. Polte, P. Strasser, U. Vainio, F. Emmerling and R. Kraehnert, Chem. Mater., 2013, 25, 2749–2758 CrossRef CAS.
- W. Luo, Y. Li, J. Dong, J. Wei, J. Xu, Y. Deng and D. Zhao, Angew. Chem., Int. Ed., 2013, 52, 10505–10510 CrossRef CAS PubMed.
- S. Kim, C. Choi, J. Hwang, J. Park, J. Jeong, H. Jun, S. Lee, S.-K. Kim, J. H. Jang, Y. Jung and J. Lee, ACS Nano, 2020, 14, 4988–4999 CrossRef CAS PubMed.
- B. L. Cushing, V. L. Kolesnichenko and C. J. O'Connor, Chem. Rev., 2004, 104, 3893–3946 CrossRef CAS PubMed.
- A. E. Danks, S. R. Hall and Z. Schnepp, Mater. Horiz., 2016, 3, 91–112 RSC.
- G. J. d. A. A. Soler-Illia, C. Sanchez, B. Lebeau and J. Patarin, Chem. Rev., 2002, 102, 4093–4138 CrossRef PubMed.
- X. Zhang, M. Wang, G. Zhu, D. Li, D. Yan, T. Lu and L. Pan, Ceram. Int., 2017, 43, 2398–2402 CrossRef CAS.
- J. N. Kondo, Y. Hiyoshi, R. Osuga, A. Ishikawa, Y.-H. Wang and T. Yokoi, Microporous Mesoporous Mater., 2018, 262, 191–198 CrossRef CAS.
- U. Mgwetyana, M. Elizabeth Makhatha, M. Mamo and P. Ndungu, Mater. Today: Proc., 2018, 5, 10585–10591 CAS.
- S. Tanaka, Y. V. Kaneti, R. Bhattacharjee, M. N. Islam, R. Nakahata, N. Abdullah, S.-i. Yusa, N.-T. Nguyen, M. J. A. Shiddiky, Y. Yamauchi and M. S. A. Hossain, ACS Appl. Mater. Interfaces, 2018, 10, 1039–1049 CrossRef CAS PubMed.
- R. Azoulay, N. Shomrat, I. Weisbord, G. Atiya and T. Segal-Peretz, Small, 2019, 15, 1904657 CrossRef CAS PubMed.
- B. K. Barick, A. Simon, I. Weisbord, N. Shomrat and T. Segal-Peretz, J. Colloid Interface Sci., 2019, 557, 537–545 CrossRef CAS PubMed.
- D. H. Yi, C.-Y. Nam, G. Doerk, C. T. Black and R. B. Grubbs, ACS Appl. Bio Mater., 2019, 1, 672–683 CAS.
- Y. Wang, N. Li, C. Hou, B. He, J. Li, F. Dang, J. Wang and Y. Fan, Ceram. Int., 2020, 46, 9119–9128 CrossRef CAS.
- G. He, P. Wang, K. Feng, H. Dong, H. Zhao, F. Sun, H. Yin, W. Li and G. Li, Macromolecules, 2021, 54, 906–918 CrossRef CAS.
- L. Tang, B. Han, K. Persson, C. Friesen, T. He, K. Sieradzki and G. Ceder, J. Am. Chem. Soc., 2010, 132, 596–600 CrossRef CAS PubMed.
- L. Tang, X. Li, R. C. Cammarata, C. Friesen and K. Sieradzki, J. Am. Chem. Soc., 2010, 132, 11722–11726 CrossRef CAS PubMed.
- X. Zhang, N. Peng, T. Liu, R. Zheng, M. Xia, H. Yu, S. Chen, M. Shui and J. Shu, Nano Energy, 2019, 65, 104049 CrossRef CAS.
- Y.-P. Zhu, T.-Z. Ren and Z.-Y. Yuan, Catal. Sci. Technol., 2015, 5, 4258–4279 RSC.
- V. Malgras, H. Ataee-Esfahani, H. Wang, B. Jiang, C. Li, K. C. W. Wu, J. H. Kim and Y. Yamauchi, Adv. Mater., 2016, 28, 993–1010 CrossRef CAS PubMed.
- Y. Yamauchi, M. Komatsu, A. Takai, R. Sebata, M. Sawada, T. Momma, M. Fuziwara, T. Osaka and K. Kuroda, Electrochim. Acta, 2007, 53, 604–609 CrossRef CAS.
- Y. Yamauchi and K. Kuroda, Chem.–Asian J., 2008, 3, 664–676 CrossRef CAS PubMed.
- L. Palanikumar, E. S. Choi, J. Y. Cheon, S. H. Joo and J.-H. Ryu, Adv. Funct. Mater., 2015, 25, 957–965 CrossRef CAS.
- E. N. Kusumawati and T. Sasaki, Chem. Rec., 2019, 19, 2058–2068 CrossRef CAS PubMed.
- S. C. Warren, L. C. Messina, L. S. Slaughter, M. Kamperman, Q. Zhou, S. M. Gruner, F. J. DiSalvo and U. Wiesner, Science, 2008, 320, 1748–1752 CrossRef CAS PubMed.
- Z. Li, K. Hur, H. Sai, T. Higuchi, A. Takahara, H. Jinnai, S. M. Gruner and U. Wiesner, Nat. Commun., 2014, 5, 3247 CrossRef PubMed.
- C. Li, Ö. Dag, T. D. Dao, T. Nagao, Y. Sakamoto, T. Kimura, O. Terasaki and Y. Yamauchi, Nat. Commun., 2015, 6, 6608 CrossRef CAS PubMed.
- B. Jiang, C. Li, Ö. Dag, H. Abe, T. Takei, T. Imai, M. S. A. Hossain, M. T. Islam, K. Wood, J. Henzie and Y. Yamauchi, Nat. Commun., 2017, 8, 15581 CrossRef CAS PubMed.
- J. Mondal, P. Borah, A. Modak, Y. Zhao and A. Bhaumik, Org. Process Res. Dev., 2014, 18, 257–265 CrossRef CAS.
- L. Wang, Y. Nemoto and Y. Yamauchi, J. Am. Chem. Soc., 2011, 133, 9674–9677 CrossRef CAS PubMed.
- P. Karthika, H. Ataee-Esfahani, Y.-H. Deng, K. C. W. Wu, N. Rajalakshmi, K. S. Dhathathreyan, D. Arivuoli, K. Ariga and Y. Yamauchi, Chem. Lett., 2013, 42, 447–449 CrossRef CAS.
- Y. Kuroda, Y. Yamauchi and K. Kuroda, Chem. Commun., 2010, 46, 1827–1829 RSC.
- S. Guo, S. Dong and E. Wang, ACS Nano, 2010, 4, 547–555 CrossRef CAS PubMed.
- Z. Zhang, Y. Yang, F. Nosheen, P. Wang, J. Zhang, J. Zhuang and X. Wang, Small, 2013, 9, 3063–3069 CrossRef CAS PubMed.
- Y. Shi, Y. Wan, R. Zhang and D. Zhao, Adv. Funct. Mater., 2008, 18, 2436–2443 CrossRef CAS.
- S. W. Robbins, H. Sai, F. J. DiSalvo, S. M. Gruner and U. Wiesner, ACS Nano, 2014, 8, 8217–8223 CrossRef CAS PubMed.
- P. A. Beaucage, E. M. Susca, S. M. Gruner and U. B. Wiesner, Chem. Mater., 2017, 29, 8973–8977 CrossRef CAS.
- B. G. Kim, C. Jo, J. Shin, Y. Mun, J. Lee and J. W. Choi, ACS Nano, 2017, 11, 1736–1746 CrossRef CAS PubMed.
- K. E. Fritz, P. A. Beaucage, F. Matsuoka, U. Wiesner and J. Suntivich, Chem. Commun., 2017, 53, 7250–7253 RSC.
- C. Jo, J. Hwang, W.-G. Lim, J. Lim, K. Hur and J. Lee, Adv. Mater., 2018, 30, 1703829 CrossRef PubMed.
- J. Wang, J. Tang, B. Ding, V. Malgras, Z. Chang, X. Hao, Y. Wang, H. Dou, X. Zhang and Y. Yamauchi, Nat. Commun., 2017, 8, 15717 CrossRef CAS PubMed.
- S. A. Hesse, K. E. Fritz, P. A. Beaucage, R. P. Thedford, F. Yu, F. J. DiSalvo, J. Suntivich and U. Wiesner, ACS Nano, 2020, 14, 16897–16906 CrossRef CAS PubMed.
- C. M. A. Parlett, M. A. Isaacs, S. K. Beaumont, L. M. Bingham, N. S. Hondow, K. Wilson and A. F. Lee, Nat. Mater., 2016, 15, 178–182 CrossRef CAS PubMed.
- Y. Li, X. Zhou, W. Luo, X. Cheng, Y. Zhu, A. M. El-Toni, A. Khan, Y. Deng and D. Zhao, Adv. Mater. Interfaces, 2019, 6, 1801269 CrossRef.
- N. D. Petkovich and A. Stein, Chem. Soc. Rev., 2013, 42, 3721–3739 RSC.
- Y. Hu, X. Wang, M. Zhang, S. Wang, S. Li and G. Chen, Nano Lett., 2021, 21, 250–257 CrossRef CAS PubMed.
- D. Lee, J.-Y. Jung, M.-J. Jung and Y.-S. Lee, Chem. Eng. J., 2015, 263, 62–70 CrossRef CAS.
- H. Sai, K. W. Tan, K. Hur, E. Asenath-Smith, R. Hovden, Y. Jiang, M. Riccio, D. A. Muller, V. Elser, L. A. Estroff, S. M. Gruner and U. Wiesner, Science, 2013, 341, 530–534 CrossRef CAS PubMed.
- J. Hwang, C. Jo, K. Hur, J. Lim, S. Kim and J. Lee, J. Am. Chem. Soc., 2014, 136, 16066–16072 CrossRef CAS PubMed.
- S. Kim, I. Jeong, J. Hwang, M. J. Ko and J. Lee, Chem. Commun., 2017, 53, 4100–4103 RSC.
- W. Zhu, S. Tao, C.-a. Tao, W. Li, C. Lin, M. Li, Y. Wen and G. Li, Langmuir, 2011, 27, 8451–8457 CrossRef CAS PubMed.
- F. Li, Z. Wang, N. S. Ergang, C. A. Fyfe and A. Stein, Langmuir, 2007, 23, 3996–4004 CrossRef CAS PubMed.
- J. Du, X. Lai, N. Yang, J. Zhai, D. Kisailus, F. Su, D. Wang and L. Jiang, ACS Nano, 2011, 5, 590–596 CrossRef CAS PubMed.
- H.-J. Liu, X.-M. Wang, W.-J. Cui, Y.-Q. Dou, D.-Y. Zhao and Y.-Y. Xia, J. Mater. Chem., 2010, 20, 4223–4230 RSC.
- S. Kim, J. Hwang, J. Lee and J. Lee, Sci. Adv., 2020, 6, eabb3814 CrossRef CAS PubMed.
- S. Liu, P. Gordiichuk, Z.-S. Wu, Z. Liu, W. Wei, M. Wagner, N. Mohamed-Noriega, D. Wu, Y. Mai, A. Herrmann, K. Müllen and X. Feng, Nat. Commun., 2015, 6, 8817 CrossRef CAS PubMed.
- N. Devabharathi, S. K. Mondal and S. Dasgupta, Nanoscale, 2019, 11, 13731–13740 RSC.
- E. Shukrun Farrell, Y. Schilt, M. Y. Moshkovitz, Y. Levi-Kalisman, U. Raviv and S. Magdassi, Nano Lett., 2020, 20, 6598–6605 CrossRef CAS PubMed.
- C. Li, C. Jiang, Y. Deng, T. Li, N. Li, M. Peng and J. Wang, Sci. Rep., 2017, 7, 41331 CrossRef CAS PubMed.
- Z. Wang, Y. Wang, J. Yan, K. Zhang, F. Lin, L. Xiang, L. Deng, Z. Guan, W. Cui and H. Zhang, Adv. Drug Deliv. Rev, 2021, 174, 504–534 CrossRef CAS PubMed.
- P. Pei, Z. Tian and Y. Zhu, Microporous Mesoporous Mater., 2018, 272, 24–30 CrossRef CAS.
- F. Putz, S. Scherer, M. Ober, R. Morak, O. Paris and N. Hüsing, Adv. Mater. Technol., 2018, 3, 1800060 CrossRef.
- D.-S. Moon and J.-K. Lee, Langmuir, 2012, 28, 12341–12347 CrossRef CAS PubMed.
- X. Zuo, Y. Xia, Q. Ji, X. Gao, S. Yin, M. Wang, X. Wang, B. Qiu, A. Wei, Z. Sun, Z. Liu, J. Zhu and Y.-J. Cheng, ACS Nano, 2017, 11, 889–899 CrossRef CAS PubMed.
- Y. Yan, G. Chen, P. She, G. Zhong, W. Yan, B. Y. Guan and Y. Yamauchi, Adv. Mater., 2020, 32, 2004654 CrossRef CAS PubMed.
- K.-W. Kim, J. Kim, C. Choi, H. K. Yoon, M. C. Go, J. Lee, J. K. Kim, H. Seok, T. Kim, K. Wu, S. H. Kim, Y. M. Kim, J. H. Kwon and H. C. Moon, ACS Appl. Mater. Interfaces, 2022, 14, 46994–47002 CrossRef CAS PubMed.
- J. Xu, Y. Dou, Z. Wei, J. Ma, Y. Deng, Y. Li, H. Liu and S. Dou, Adv. Sci., 2017, 4, 1700146 CrossRef PubMed.
- G. He, B. Mandlmeier, J. Schuster, L. F. Nazar and T. Bein, Chem. Mater., 2014, 26, 3879–3886 CrossRef CAS.
- N. Nitta, F. Wu, J. T. Lee and G. Yushin, Mater. Today, 2015, 18, 252–264 CrossRef CAS.
- L. Wang, Y. Lu, J. Liu, M. Xu, J. Cheng, D. Zhang and J. B. Goodenough, Angew. Chem., 2013, 125, 2018–2021 CrossRef.
- T. Hosaka, K. Kubota, A. S. Hameed and S. Komaba, Chem. Rev., 2020, 120, 6358–6466 CrossRef CAS PubMed.
- S. Gheytani, Y. Liang, F. Wu, Y. Jing, H. Dong, K. K. Rao, X. Chi, F. Fang and Y. Yao, Adv. Sci., 2017, 4, 1700465 CrossRef PubMed.
- J. Niu, Z. Zhang and D. Aurbach, Adv. Energy Mater., 2020, 10, 2000697 CrossRef CAS.
- M. Song, H. Tan, D. Chao and H. J. Fan, Adv. Funct. Mater., 2018, 28, 1802564 CrossRef.
- G. A. Elia, K. V. Kravchyk, M. V. Kovalenko, J. Chacón, A. Holland and R. G. A. Wills, J. Power Sources, 2021, 481, 228870 CrossRef CAS.
- S. Kim, W.-G. Lim, H. Im, M. Ban, J. W. Han, J. Lee, J. Hwang and J. Lee, J. Am. Chem. Soc., 2021, 143, 15644–15652 CrossRef CAS PubMed.
- Y. Li, C. Wu, Y. Bai, L. Liu, H. Wang, F. Wu, N. Zhang and Y. Zou, ACS Appl. Mater. Interfaces, 2016, 8, 18832–18840 CrossRef CAS PubMed.
- C. Liu, Z. G. Neale and G. Cao, Mater. Today, 2016, 19, 109–123 CrossRef CAS.
- G. Wang, H. Liu, J. Liu, S. Qiao, G. M. Lu, P. Munroe and H. Ahn, Adv. Mater., 2010, 22, 4944–4948 CrossRef CAS PubMed.
- F. Jiao, J. Bao, A. H. Hill and P. G. Bruce, Angew. Chem., Int. Ed., 2008, 47, 9711–9716 CrossRef CAS PubMed.
- C. Chen, H. Xu, B. Zhang, Q. Jiang, Y. Zhang, L. Li and Z. Lin, Chem. Commun., 2020, 56, 786–789 RSC.
- H. Tian, H. Tian, S. Wang, S. Chen, F. Zhang, L. Song, H. Liu, J. Liu and G. Wang, Nat. Commun., 2020, 11, 5025 CrossRef CAS PubMed.
- K.-H. Kwak, H. J. Suh, A. Kim, S. Park, J. Song, S. Li, Y. Kim, G. Jeong, H. Kim and Y.-J. Kim, Nano Energy, 2019, 66, 104138 CrossRef CAS.
- J. Zhang, X. Zhou, Y. Wang, J. Qian, F. Zhong, X. Feng, W. Chen, X. Ai, H. Yang and Y. Cao, Small, 2019, 15, 1903723 CrossRef CAS PubMed.
- Y. Xu, Y. Zhu and C. Wang, J. Mater. Chem. A, 2014, 2, 9751–9757 RSC.
- J. Jeong, J. Chun, W.-G. Lim, W. B. Kim, C. Jo and J. Lee, Nanoscale, 2020, 12, 11818–11824 RSC.
- R. Mo, F. Li, X. Tan, P. Xu, R. Tao, G. Shen, X. Lu, F. Liu, L. Shen, B. Xu, Q. Xiao, X. Wang, C. Wang, J. Li, G. Wang and Y. Lu, Nat. Commun., 2019, 10, 1474 CrossRef PubMed.
- K. Lan, Y. Liu, W. Zhang, Y. Liu, A. Elzatahry, R. Wang, Y. Xia, D. Al-Dhayan, N. Zheng and D. Zhao, J. Am. Chem. Soc., 2018, 140, 4135–4143 CrossRef CAS PubMed.
- H. Liu, Z. Bi, X.-G. Sun, R. R. Unocic, M. P. Paranthaman, S. Dai and G. M. Brown, Adv. Mater., 2011, 23, 3450–3454 CrossRef CAS PubMed.
- W. Li, Z. Wu, J. Wang, A. A. Elzatahry and D. Zhao, Chem. Mater., 2014, 26, 287–298 CrossRef CAS.
- W. Li, F. Wang, Y. Liu, J. Wang, J. Yang, L. Zhang, A. A. Elzatahry, D. Al-Dahyan, Y. Xia and D. Zhao, Nano Lett., 2015, 15, 2186–2193 CrossRef CAS PubMed.
- J. Lee, S. Kim, J.-H. Park, C. Jo, J. Chun, Y.-E. Sung, E. Lim and J. Lee, J. Mater. Chem. A, 2020, 8, 3119–3127 RSC.
- J.-H. Kwon, K. N. Chaudhari, E. Coy, J. H. Seo, S. J. Ahn, Y.-H. Lee, S. Lee, Y. C. Cho, O. Choi, K. S. Lee, D. I. Son and Y. Kim, ACS Sustain. Chem. Eng., 2021, 9, 16627–16636 CrossRef CAS.
- Y. Zhao, X. Li, B. Yan, D. Xiong, D. Li, S. Lawes and X. Sun, Adv. Energy Mater., 2016, 6, 1502175 CrossRef.
- B. Guo, X. Yu, X.-G. Sun, M. Chi, Z.-A. Qiao, J. Liu, Y.-S. Hu, X.-Q. Yang, J. B. Goodenough and S. Dai, Energy Environ. Sci., 2014, 7, 2220–2226 RSC.
- J. B. Cook, H.-S. Kim, Y. Yan, J. S. Ko, S. Robbennolt, B. Dunn and S. H. Tolbert, Adv. Energy Mater., 2016, 6, 1501937 CrossRef.
- R. R. Salunkhe, Y. V. Kaneti, J. Kim, J. H. Kim and Y. Yamauchi, Acc. Chem. Res., 2016, 49, 2796–2806 CrossRef CAS PubMed.
- M. S. Kim, E. Lim, S. Kim, C. Jo, J. Chun and J. Lee, Adv. Funct. Mater., 2017, 27, 1603921 CrossRef.
- L. Kuang, F. Ji, X. Pan, D. Wang, X. Chen, D. Jiang, Y. Zhang and B. Ding, Chem. Eng. J., 2017, 315, 491–499 CrossRef CAS.
- A. G. Pandolfo and A. F. Hollenkamp, J. Power Sources, 2006, 157, 11–27 CrossRef CAS.
- K. Jost, G. Dion and Y. Gogotsi, J. Mater. Chem. A, 2014, 2, 10776–10787 RSC.
- R. R. Salunkhe, Y. V. Kaneti and Y. Yamauchi, ACS Nano, 2017, 11, 5293–5308 CrossRef CAS PubMed.
- J. Du, Y. Zhang, H. Wu, S. Hou and A. Chen, Carbon, 2020, 156, 523–528 CrossRef CAS.
- M. Xie, H. Meng, J. Chen, Y. Zhang, C. Du, L. Wan and Y. Chen, ACS Appl. Energy Mater., 2021, 4, 1840–1850 CrossRef CAS.
- X. Feng, Y. Bai, M. Liu, Y. Li, H. Yang, X. Wang and C. Wu, Energy Environ. Sci., 2021, 14, 2036–2089 RSC.
- K. Jayaramulu, S. Mukherjee, D. M. Morales, D. P. Dubal, A. K. Nanjundan, A. Schneemann, J. Masa, S. Kment, W. Schuhmann, M. Otyepka, R. Zbořil and R. A. Fischer, Chem. Rev., 2022, 122, 17241–17338 CrossRef CAS PubMed.
- L.-N. Han, X. Wei, Q.-C. Zhu, S.-M. Xu, K.-X. Wang and J.-S. Chen, J. Mater. Chem. A, 2016, 4, 16698–16705 RSC.
- H. Peng, B. Yao, X. Wei, T. Liu, T. Kou, P. Xiao, Y. Zhang and Y. Li, Adv. Energy Mater., 2019, 9, 1803665 CrossRef.
- J. Zhou, J. Lian, L. Hou, J. Zhang, H. Gou, M. Xia, Y. Zhao, T. A. Strobel, L. Tao and F. Gao, Nat. Commun., 2015, 6, 8503 CrossRef CAS PubMed.
- X.-H. Li and M. Antonietti, Angew. Chem., Int. Ed., 2013, 52, 4572–4576 CrossRef CAS PubMed.
- T.-N. Ye, L.-B. Lv, X.-H. Li, M. Xu and J.-S. Chen, Angew. Chem., Int. Ed., 2014, 53, 6905–6909 CrossRef CAS PubMed.
- X. Xie, C. Hou, D. Wu, X. Sun, X. Yang, Y. Zhang, R. Yu, S. Zhang, H. Kimura and W. Du, J. Alloys Compd., 2022, 891, 161967 CrossRef CAS.
- J. Liu, J. Jiang, M. Bosman and H. J. Fan, J. Mater. Chem., 2012, 22, 2419–2426 RSC.
- H. J. Lee, J. H. Lee, S.-Y. Chung and J. W. Choi, Angew. Chem., Int. Ed., 2016, 55, 3958–3962 CrossRef CAS PubMed.
- M. Khot and A. Kiani, Int. J. Energy Res., 2022, 46, 21757–21796 CrossRef CAS.
- Y. Wang, J. Guo, T. Wang, J. Shao, D. Wang and Y.-W. Yang, Nanomaterials, 2015, 5, 1667–1689 CrossRef CAS PubMed.
- E. Lim, J. Chun, C. Jo and J. Hwang, Korean J. Chem. Eng., 2021, 38, 227–247 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2023 |