
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTriazaphosphaadamantane-functionalized terpyridine metal complexes: cyclohexane oxidation in homogeneous and carbon-supported catalysis†
Ivy L.
Librando
 a,
Anup
Paul
*a,
Abdallah G.
Mahmoud
ab,
Atash V.
Gurbanov
ac,
Sónia A. C.
Carabineiro
*ad,
M. Fátima C.
Guedes da Silva
ae,
Carlos F. G. C.
Geraldes
fg and
Armando J. L.
Pombeiro
ah
a,
Anup
Paul
*a,
Abdallah G.
Mahmoud
ab,
Atash V.
Gurbanov
ac,
Sónia A. C.
Carabineiro
*ad,
M. Fátima C.
Guedes da Silva
ae,
Carlos F. G. C.
Geraldes
fg and
Armando J. L.
Pombeiro
ah
aCentro de Química Estrutural, Institute of Molecular Sciences, Instituto Superior Técnico, Universidade de Lisboa, 1049-001 Lisboa, Portugal. E-mail: anuppaul@tecnico.ulisboa.pt; sonia.carabineiro@fct.unl.pt
bDepartment of Chemistry, Faculty of Science, Helwan University, Ain Helwan, Cairo 11795, Egypt
cExcellence Center, Baku State University, Z. Xalilov Str. 23, Az 1148 Baku, 117198, Azerbaijan
dLAQV-REQUIMTE, Department of Chemistry, NOVA School of Science and Technology, Universidade NOVA de Lisboa, 2829-516 Caparica, Portugal
eDepartamento de Engenharia Química, Instituto Superior Técnico, Universidade de Lisboa, 1049-001 Lisboa, Portugal
fDepartment of Life Sciences, Faculty of Science and Technology, 3000-393 Coimbra, Portugal. E-mail: geraldes@uc.pt
gCoimbra Chemistry Center, University of Coimbra, 3004-535 Coimbra, Portugal
hPeoples' Friendship University of Russia (RUDN University), Research Institute of Chemistry, Moscow, 117198, Russian Federation
First published on 2nd December 2022
Abstract
In accordance with UN's Sustainable Development Goal (UN's SDG) 12 which encompasses the sustainable use of chemical products and a sound circular economy, this work is focused on the synthesis of Co(II), Ni(II) and Mn(II) complexes bearing combined 1,3,5-triaza-7-phosphaadamantane and benzyl terpyridine core moieties (PTA–Bztpy) as ligand, followed by their evaluation as catalysts for the microwave-assisted cyclohexane oxidation using tert-butyl hydroperoxide (TBHP) as oxidant. The most active catalyst, with a manganese metal center, was heterogenized on six different carbon materials. The results disclosed the influence of several reaction parameters, such as catalyst loading, temperature, reaction time and solvent, on the catalytic activity and selectivity of the homogeneous and carbon-supported catalysts. Recyclability of the carbon-supported catalyst allowed facile separations, recovery and reuse for five consecutive cycles.
Sustainability spotlightThe oxidation product KA oil is an irreplaceable chemical precursor in the Nylon manufacturing industries. However, the industrial thermal catalytic process has raised some ecological concerns due to the harsh reaction conditions and high energy utilization. These drawbacks have inspired our research group to develop effective methods to catalyze cyclohexane oxidation under mild conditions. In this paper, the catalytic activity of unsupported and carbon-supported organometallic complexes based on the water-soluble 1,3,5-triaza-7-phosphaadamantane (PTA) incorporating a terpyridine substructure was described. In pursuit of a cost-effective and sustainable reaction, this work was able to design a recyclable catalyst (inexpensive and easy to handle), use water as a reaction promoter (elimination of toxic promoters), employ microwave irradiation (for efficient energy use) and obtain moderate KA oil yields under mild reaction conditions. Furthermore, this work is aligned with the UN's Sustainable Development Goal 12 regarding the sustainable consumption and production of chemical products. |
Introduction
The oxidative functionalization of hydrocarbons is an important route for the transformation of petroleum-based feedstocks to products of commercial significance from both practical and fundamental perspectives.1–5 Cyclohexane oxidation remains a highly significant research field, as evidenced by the constant output of new strategies to improve selectivity and efficiency, due to its important role in the industrial-scale production of cyclohexanone and cyclohexanol, also known as KA oil, as chemical precursors in nylon manufacturing via oxidation to adipic acid.6,7 In many industrial processes, the reaction utilizes cobalt-based homogenous catalysts and is known to proceed by a free radical autoxidation mechanism8,9 with a general conversion of 5% and low selectivity for KA oil at 150–170 °C and 1–2 MPa.10,11 Such harsh conditions, along with low efficiency and ecological considerations, have limited its further development.12,13 This prompted several research groups, including ours, to explore alternative pathways to synthesize better catalysts for the selective oxidation of cyclohexane.14–16Research towards the improved activity and selectivity of a catalytic system is at the core of sustainable practices for the reduction of costs, time, and waste.17,18 However, the progress in transition metal catalytic applications depends to a large extent on the appropriate selection of ligands, which provide electronically and sterically well-defined scaffolds for catalysts.19–21 Both metal and the ligand can cooperate synergistically and their interplay facilitates a chemical process.22 Within this context, a number of homo- and hetero-donor ligands has been developed, mainly P- and N-containing moieties with different and electronically diverging donor sites. These ligands and their derivatives have made some of the most fundamental contributions to the development of catalysis.23,24 Among these, terpyridine (tpy)25,26 and triazaphosphaadamantane (PTA)27,28 scaffolds have been widely used in the field of homogeneous catalysis and continue to receive widespread attention as their resulting complexes have been utilized in a range of cutting-edge applications. Moreover, various first-row transition metal complexes with P- and N-containing ligands were exploited in the context of oxidation reactions. In particular, the copper(II) complexes of tpy and PTA derivatives for the oxidation of benzyl alcohol and alkanes were performed by our research group.29–31 Beyond the aforementioned copper(II) catalysts, other oxidation reactions involving such ligands have also been reported.25,26,32,33
Many transition metal-based heterogeneous catalysts have been investigated as catalysts for oxidation reactions.34–36 These reactions have flourished and are proposed as viable methods due to the stability, recoverability and reusability of the catalyst, which are still a challenge in sustainable chemical process development.37–39 Solid systems such as zeolites, metal–organic frameworks and carbon materials have been reported to be active and have increasing prominence in the field of metal-supported heterogeneous catalysts for chemical synthesis and/or industrial-relevant transformations.40–42
Inspired by the potential of tpy-based complexes and the success of PTA-derived ligands in catalysis, the combination of the two scaffolds in a single polydentate ligand and exploring its catalytic potential are of our utmost interest. Taking into consideration the UN's SDGs regarding responsible production of chemical products for sustainability, herein we report the first example of incorporating a PTA moiety into a benzyl terpyridine derivative (Fig. 1) and the synthesis of its Co(II), Ni(II) and Mn(II) complexes. The catalytic activity of the compounds bearing such a novel ligand, as homogeneous and carbon-supported catalysts, was evaluated towards cyclohexane oxidation for KA oil production under microwave irradiation.
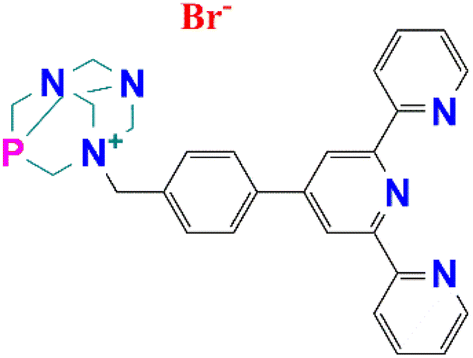 | ||
| Fig. 1 Schematic representation of the PTA–terpyridine ligand PTA–Bztpy. | ||
Results and discussion
Synthesis and characterization of ligand and complexes
The synthesis and characterization of the ligand precursor, 4′-[4-(bromomethyl)phenyl]-2,2′:6′,2′′-terpyridine (BrBztpy), was adapted according to a reported work30 while the N-alkylated PTA derivative (PTA–Bztpy) was obtained following the modified procedures described in the literature.43,44 BrBztpy was reacted with PTA in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mole ratio and refluxed in acetone (Scheme 1) to afford the monoalkylated PTA ligand (PTA–Bztpy) as a beige solid in moderate yield (60%). The obtained product was characterized by FTIR, elemental analysis (EA), 1H, 13C{1H}, 31P{1H}, DEPT NMR spectroscopy, and ESI-MS. In the 1H NMR spectrum of PTA–Bztpy (Fig. S1†), the terpyridine-based moiety exhibits signals in the 7.5–8.7 ppm range assigned to the aromatic protons. The protons of the methylene group bridging the PTA cage and the tpy moiety appear at 4.24 ppm as a singlet.
:1 mole ratio and refluxed in acetone (Scheme 1) to afford the monoalkylated PTA ligand (PTA–Bztpy) as a beige solid in moderate yield (60%). The obtained product was characterized by FTIR, elemental analysis (EA), 1H, 13C{1H}, 31P{1H}, DEPT NMR spectroscopy, and ESI-MS. In the 1H NMR spectrum of PTA–Bztpy (Fig. S1†), the terpyridine-based moiety exhibits signals in the 7.5–8.7 ppm range assigned to the aromatic protons. The protons of the methylene group bridging the PTA cage and the tpy moiety appear at 4.24 ppm as a singlet.
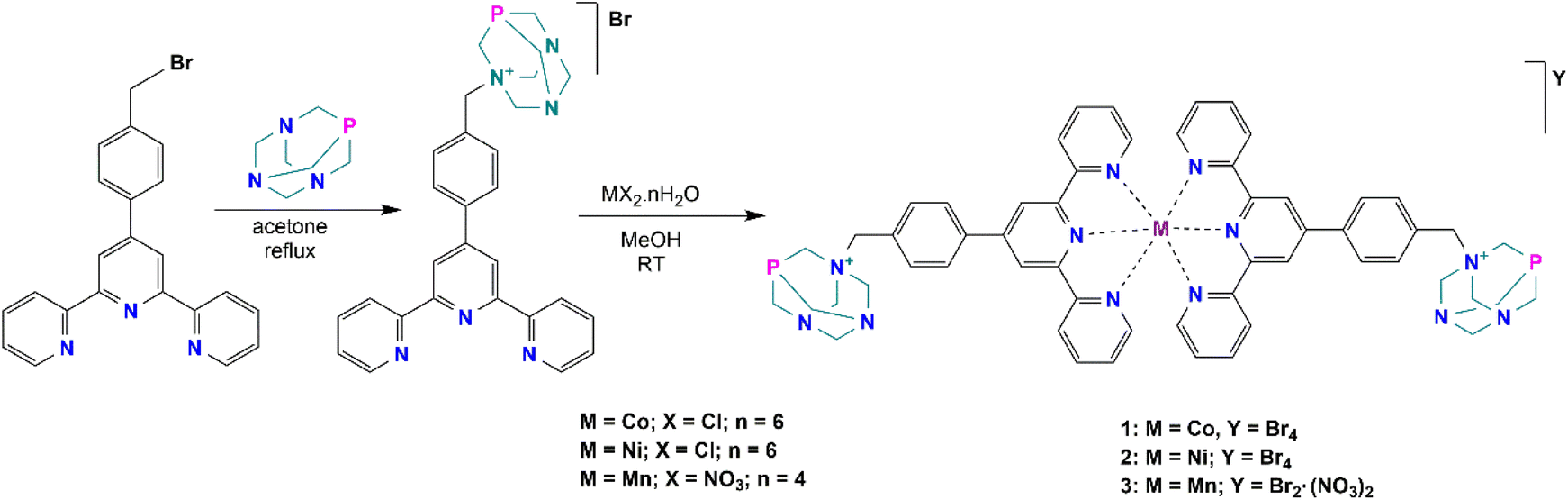 | ||
| Scheme 1 Synthesis of complexes 1–3 with the PTA–Bztpy ligand. | ||
The quaternization of PTA was confirmed by the splitting pattern of the –CH2 proton signals in the cage system that appears at δ 3.8–5.1.
In the 13C{1H} NMR spectrum of PTA–Bztpy (Fig. S2†), the aromatic carbons were observed in the 118–156 ppm range while the methylene carbon of the benzyl group was observed at δ 64.72. The spectrum also reveals four signals confirming the N-alkylation of the PTA moiety, at δ 79.27 for N–CH2–N+, δ 69.82 for N–CH2–N, δ 52.4 for N+–CH2–P and doublets at δ 45.74 (J = 46 Hz) for N–CH2–P. The DEPT NMR spectrum of PTA–Bztpy is also shown in Fig. S3.† The 2D NMR (HSQC) spectrum correlates the assignment of proton peaks to the corresponding carbon atoms, as shown in Fig. S4.†
The 31P{1H} NMR spectroscopy of PTA–Bztpy (Fig. S5†) confirms the presence of a single phosphorus species with a singlet at δ −83.44. The ESI(+)MS spectrum (Fig. S6†) displayed a peak at ca. 479.12 (calcd 479.5) assigned to [p-tpy-C6H4-CH2–PTA]+.
Complexes 1–3 were synthesised by reacting PTA–Bztpy with either CoCl2·6H2O, NiCl2·6H2O or Mn(NO3)2·4H2O in MeOH, in this order (Scheme 1). Characterization of the complexes was carried out by elemental analysis (EA), FTIR spectroscopy and ESI-Mass spectrometry. The complexes are soluble in polar solvents, including water. The terpyridine moiety binds the metal in a κ-N,N,N mode. Analytical data are consistent with the proposed structures in Scheme 1. Elemental analysis reveals an empirical formula of M(PTA–Bztpy)2Br2 (M = Co, Ni, Mn), suggesting a 1:2 metal–ligand complex.
The structure of 2 was confirmed by single crystal X-ray diffraction (see ESI, Fig. S7 and Table S1†). The results were obtained at low temperature (150 K) from very low-quality crystals. Despite many attempts, we were unable to improve the quality of the crystals and, therefore, the discussion of the structure will be limited to only a few generic characteristics. The compound crystallized in the triclinic space group, the asymmetric unit containing a Ni(II) cation coordinated by two PTA–Bztpy through the N-tpy moieties, and four bromide anions.
The metal centre defines a distorted octahedral geometry. In one of the ligands the benzyl group is coplanar with the tpy moiety while in the other the PTA-benzyl assembly is twisted by ca. 40° relative to the attached tpy. The solid-state structure of the compound appears to be highly stabilized by several H⋯Br interactions.
In the FTIR analysis, the ν(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N) stretching vibrations of 1–3 are observed at higher wavenumbers (1613, 1603, 1602 cm−1, respectively) than that of the free PTA–Bztpy (1585 cm−1), what indicates metal chelation through the pyridinic nitrogen atoms. The dominant peak envelope in the ESI-MS (Fig. S8–S10†) of each complex (described in detail in the Experimental section) arose from the [p-tpy–C6H4–CH2–PTA]+ ion and the observed isotope mass matched that of the calculated one (calcd = 479.5, found 479.12). These results agree with the stoichiometry given by microanalysis and proved that the desired structure has been achieved.
N) stretching vibrations of 1–3 are observed at higher wavenumbers (1613, 1603, 1602 cm−1, respectively) than that of the free PTA–Bztpy (1585 cm−1), what indicates metal chelation through the pyridinic nitrogen atoms. The dominant peak envelope in the ESI-MS (Fig. S8–S10†) of each complex (described in detail in the Experimental section) arose from the [p-tpy–C6H4–CH2–PTA]+ ion and the observed isotope mass matched that of the calculated one (calcd = 479.5, found 479.12). These results agree with the stoichiometry given by microanalysis and proved that the desired structure has been achieved.
Characterization of carbon supports and heterogenized materials
The carbon materials used in this study were prepared and characterized in accordance with our previous work.44 The commercially purchased activated carbon (AC) and multi-walled carbon nanotubes (MWCNTs) underwent chemical surface modification by oxidation with HNO3 (-ox materials) followed by NaOH to give the corresponding sodium carboxylates (-ox-Na materials).The morphology of the six carbon supports was analysed by scanning electron microscopy (SEM). The SEM images of AC-based materials are shown in Fig. 2a–c, while the CNT-based ones are depicted in Fig. 2d–f. From the SEM micrographs, the microporosity of AC-based materials can be clearly seen and apparently the morphology of AC-ox (Fig. 2b) does not differ significantly from the pristine activated carbon (Fig. 2a), but AC-ox-Na (Fig. 2c) has smaller pores than that of the commercial AC and AC-ox materials.
 | ||
| Fig. 2 SEM images of carbon supports (a) AC, (b) AC-ox, (c) AC-ox-Na, (d) CNT, (e) CNT-ox, and (f) CNT-ox-Na. | ||
CNT materials (Fig. 2d–f), having filamentous appearance, display pores resulting from the 3D stacking of the fibers. The surface modified CNTs (Fig. 2e and f) have more tangled appearance that clump together as compared to the more elongated threadlike morphology of the untreated CNT (Fig. 2d).
The carbon-supported manganese complex 3 was subjected to transmission electron microscopy (TEM), as shown in Fig. 3a–f. The tangled appearance might be due to the MWCNT shortening and agglomeration after oxidation–reduction treatments. Upon surface modification, CNTs possess a high density of oxygen bearing functional groups that affect its adsorption and catalytic activity.45,46 Likewise, these surface defects are predominant anchoring sites for metals.
 | ||
| Fig. 3 TEM micrographs of complex 3 immobilized on (a) AC, (b) AC-ox, (c) AC-ox-Na, (d) CNT, (e) CNT-ox, and (f) CNT-ox-Na. | ||
Fig. S11† shows the N2 adsorption–desorption isotherm of 3-CNT-ox-Na. The isotherm exhibits the characteristics of the type IV classification which indicates a mesoporous material. The hysteresis loops are of H1 type which is often associated with a narrow distribution of cylindrical pores,47,48 resulting from the free space in the bundles.49 The uptrend of hysteresis was observed at P/Po > 0.6. This implies that the hysteresis, as well as the capillary condensation at P/Po > 0.6, are due to the existence of intertubular spaces on CNTs.50 The Brunauer–Emmett–Teller (BET) surface area (SBET) and pore volume of 3-CNT-ox-Na are 214 m2 g−1 and 1.26 cm3 g−1, respectively, while those of carbon supports are shown in Table S2.†
The heterogenization efficiency of the supports to anchor complex 3 was determined by ICP-AES, as shown in Table S3.† The most efficient carbon materials are those where the surface was modified, preferably the carboxylated (-ox-Na) ones. The % Mn loaded onto the support ranges from 0.1 to 3.8%.
In a catalytic reaction, particularly those in a liquid-phase where less mass-transport limitations are required, CNTs are ideal alternatives.51 The highly disordered and principally “empty” porous structures are usually preferred in order to avoid transport restrictions.52,53 The porous structure of CNTs, with pores resulting from the free space in the bundles, improves diffusion and the accessibility of the reactants, thus influencing the activity and selectivity of the reaction.51
Cyclohexane oxidation catalysis under microwave irradiation
The catalytic oxidation of cyclohexane to the corresponding alcohol and ketone products is a critical organic transformation that has required a great attention in the search for the most effective combination of metal sources and organic ligands. Our group has been interested in exploring the influence of both nitrogen- and phosphorus-containing metal complexes on the catalytic reactivity of these systems in aerobic oxidation reactions.30,32 Despite the significant progress in this field, heterogeneous catalysts involving metal–tpy–PTA substructures have been scarcely investigated. In this work, we explore the catalytic ability of PTA–Bztpy–M (M = Co, Ni, Mn) hybrid materials together with the homogeneous counterparts for the cyclohexane oxidation by tBuOOH (TBHP, 70% aqueous solution) under microwave irradiation conditions (Scheme 2). It has been established that the reaction rates are accelerated and product selectivity is changed when microwave radiation is used rather than conventional heating.54,55 However, PTA and terpyridine combined nitrogen rich species have not been tried before for this kind of catalytic reactions. | ||
| Scheme 2 Cyclohexane oxidation to ketone and alcohol. | ||
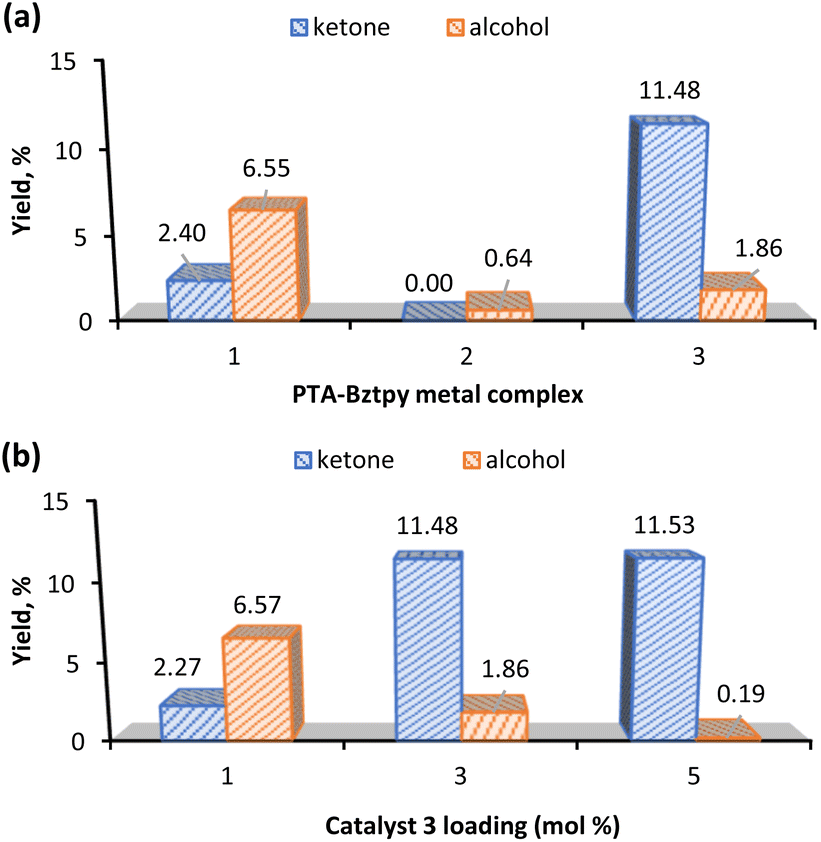 | ||
| Fig. 4 (a) Preliminary screening among PTA–Bztpy complexes (3 mol% relative to the substrate) for the homogeneous cyclohexane oxidation. (b) Complex 3 at different catalyst loadings. Reaction conditions: cyclohexane (5.0 mmol), 70% aqueous TBHP (10 mmol), 2 h, MW (30 W, 100 °C), 2 mL MeCN. | ||
| Entry | Catalyst | Solvent (2 mL) | Yieldb (%) | K/Ac | ||
|---|---|---|---|---|---|---|
| K | A | Total | ||||
| a Reaction conditions: cyclohexane (5.0 mmol), 70% aqueous TBHP (10 mmol), CH3NO2 (100 μL), 3 mol% catalyst, 2 h, MW (30 W), 100 °C, 2 mL solvent. b Molar yields based on substrate determined by GC analysis (after PPh3 treatment), i.e., moles of products (K + A) per 100 mol of cyclohexane; K = cyclohexanone, A = cyclohexanol. c Ratio between the molar concentrations of K and A. d 1 mol% catalyst. e 5 mol% catalyst. f 75 °C. g 125 °C. h 135 °C. i 1 h. j 1.5 h. k 2.5 h. l 3 h. | ||||||
| 1 | 1 (Co) | MeCN | 2.40 | 6.55 | 8.94 | 0.4 |
| 2 | 2 (Ni) | MeCN | 0.00 | 0.64 | 0.64 | 0.0 |
| 3 | 3 (Mn) | MeCN | 11.5 | 1.86 | 13.3 | 6.2 |
| 4d | 3 (Mn) | MeCN | 2.27 | 6.57 | 8.84 | 0.3 |
| 5 | 3 (Mn) | MeCN | 11.5 | 1.86 | 13.3 | 6.2 |
| 6e | 3 (Mn) | MeCN | 11.5 | 0.19 | 11.7 | 60.5 |
| 7f | 3 (Mn) | MeCN | 2.62 | 3.82 | 6.44 | 0.7 |
| 8 | 3 (Mn) | MeCN | 11.5 | 1.86 | 13.3 | 6.2 |
| 9g | 3 (Mn) | MeCN | 7.74 | 4.76 | 12.5 | 1.6 |
| 10h | 3 (Mn) | MeCN | 5.54 | 4.78 | 10.3 | 1.2 |
| 11i | 3 (Mn) | MeCN | 4.66 | 0.48 | 5.14 | 9.7 |
| 12j | 3 (Mn) | MeCN | 6.05 | 0.51 | 6.56 | 11.9 |
| 13 | 3 (Mn) | MeCN | 11.5 | 1.86 | 13.3 | 6.2 |
| 14k | 3 (Mn) | MeCN | 10.3 | 2.66 | 12.9 | 3.9 |
| 15l | 3 (Mn) | MeCN | 9.54 | 2.71 | 12.2 | 3.5 |
| 16 | 3 (Mn) | MeCN | 11.5 | 1.86 | 13.3 | 6.2 |
| 17 | 3 (Mn) | MeCN:acetone (1:1 v/v) |
5.00 | 3.23 | 8.23 | 1.5 |
| 18 | 3 (Mn) | MeCN:H2O (1:1 v/v) |
11.8 | 9.32 | 21.1 | 1.3 |
Fig. 4a shows that complex 3 gave the highest yield of KA oil and favours the ketone product. This could be due to the propensity of Mn to participate in radical chemistry and to the versatile redox behaviour of this metal.56,57 Interestingly, when the effect of the catalyst loading was evaluated (Fig. 4b), complex 3 with a loading of 1 mol% preferred the alcohol to the ketone, while an increase to 5 mol% gave an insignificant amount of cyclohexanol (0.19%) (Table 1, entry 6). Moreover, the catalyst preferably promotes the cyclohexanone production (see K/A ratio, Table 1). This could be an important improvement in the production process of polyamide 6 by avoiding additional non-sustainable oxidation step of KA oil to pure cyclohexanone.58
Since the highest total KA oil yield is obtained with a catalyst 3 amount of 3 mol% (Fig. 4b), this catalyst loading was used in further studies.
The effect of temperature in the KA oil yield is shown in Fig. S12.† The model reaction was set at 100 °C and temperature variations were carried out ranging from 75 °C to 135 °C. The homogeneous catalyst 3 gave an optimum yield at 100 °C which decreases when the temperature was raised to 125 °C. Further temperature increase to 135 °C leads to the total yield decrease to ca. 10% (Table 1, entry 10). Degradation of the catalyst at higher temperatures could be a possible reason for this decline in the yields. Considering the product selectivity, the temperature increase from 100 °C to 135 °C raises the cyclohexanol yields from 1.86% to 4.78%, respectively (Table 1, entries 8–10). At 75 °C the catalyst is selective to the alcohol component of KA oil (Table 1, entry 7).
In Fig. S13† various times were tested from 1 to 3 h (Table 1, entries 11–15) and the catalysis was optimal at 2 h. The homogeneous Mn complex 3 was selective to ketone regardless of the tested reaction time; however, it is also observed that as the time increases from 1 to 3 h, the yield of the alcohol product also increases while the ketone yield reaches a maximum after 2 h. These results suggest that the increase in reaction time has allowed the substrate to be in good contact with the active sites leading to increased conversion and improved selectivity toward the ketone with more alcohol being oxidized. However, the prolonged reactions have a negative effect on the KA oil yield. This could be attributed to the partial decomposition of the catalyst or the possible accumulation of intermediates that hinder adsorption on the catalytic sites.
The effect of the reaction medium in the KA oil yield is presented in Fig. 5. Acetonitrile (MeCN) was chosen as the solvent in our model reaction due to its high resistance to oxidizing agents and in view of the good solubility of the substrate. The use of MeCN in oxidation reaction has been often reported.14,16 Different co-solvents such as acetone and water were tested.
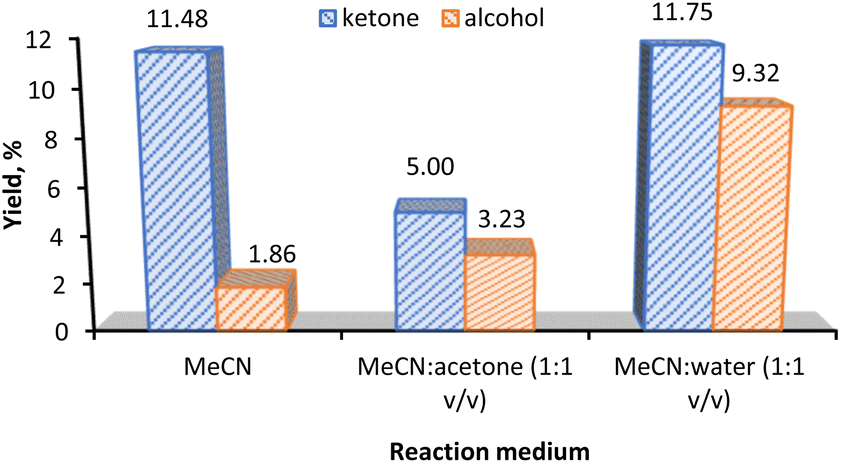 | ||
| Fig. 5 Effect of solvent in the KA oil yield using 3 as (3 mol% relative to the substrate) catalyst. Reaction conditions: cyclohexane (5.0 mmol), 70% aqueous TBHP (10 mmol), 2 h, MW (30 W, 100 °C), 2 mL solvent. | ||
The KA oil yield increases from ca. 13% to ca. 21% upon a solvent change from MeCN to 1:1 mixtures (v/v) of MeCN:water. It is noticeable that the large increase in the KA oil total yield is due to the increase in the cyclohexanol product from 1.86% (MeCN) to 9.32% (MeCN/water, 1:1 v/v). On the other hand, the mixture of MeCN/acetone only gave an 8.2% KA oil yield.
| Entry | Catalyst | Load (mol%) | Solvent | Yieldb (%) | K/Ac | ||
|---|---|---|---|---|---|---|---|
| K | A | Total | |||||
| a Reaction conditions: cyclohexane (5.0 mmol), 70% aqueous TBHP (10 mmol), CH3NO2 (100 μL), 2 h, MW (30 W), 100 °C, 2 mL solvent. b Molar yields based on substrate determined by GC analysis (after PPh3 treatment), i.e., moles of products (K + A) per 100 mol of cyclohexane; K = cyclohexanone, A = cyclohexanol. c Ratio between the molar concentrations of K and A. d 75 °C. e 125 °C. f 1 h. g 2.5 h. | |||||||
| 1 | 3-AC | 0.1 | MeCN | 0.09 | 0.99 | 1.08 | 0.1 |
| 2 | 3-AC-ox | 0.1 | MeCN | 0.88 | 4.36 | 5.24 | 0.2 |
| 3 | 3-AC-ox-Na | 0.1 | MeCN | 1.06 | 5.31 | 6.37 | 0.2 |
| 4 | 3-CNT | 0.1 | MeCN | 0.0 | 0.67 | 0.67 | 0.0 |
| 5 | 3-CNT-ox | 0.1 | MeCN | 1.11 | 4.90 | 6.01 | 0.2 |
| 6 | 3-CNT-ox-Na | 0.1 | MeCN | 4.30 | 4.31 | 8.61 | 1.0 |
| 7 | CNT-ox-Na | — | MeCN | 0.47 | 0.57 | 1.04 | 0.8 |
| 8 | Blank | — | MeCN | 0.00 | 0.06 | 0.06 | 0.0 |
| 9 | 3-CNT-ox-Na | 0.1 | MeCN | 4.30 | 4.31 | 8.61 | 1.0 |
| 10 | 3-CNT-ox-Na | 0.5 | MeCN | 5.39 | 4.62 | 10.0 | 1.2 |
| 11 | 3-CNT-ox-Na | 1.0 | MeCN | 4.64 | 4.33 | 8.96 | 1.1 |
| 12d | 3-CNT-ox-Na | 0.5 | MeCN | 1.77 | 3.73 | 5.50 | 0.5 |
| 13 | 3-CNT-ox-Na | 0.5 | MeCN | 5.39 | 4.62 | 10.0 | 1.2 |
| 14e | 3-CNT-ox-Na | 0.5 | MeCN | 5.8 | 4.60 | 10.4 | 1.3 |
| 15f | 3-CNT-ox-Na | 0.5 | MeCN | 3.24 | 5.53 | 8.77 | 0.6 |
| 16 | 3-CNT-ox-Na | 0.5 | MeCN | 5.39 | 4.62 | 10.0 | 1.2 |
| 17g | 3-CNT-ox-Na | 0.5 | MeCN | 5.03 | 4.84 | 9.87 | 1.0 |
| 18 | 3-CNT-ox-Na | 0.5 | MeCN | 5.39 | 4.62 | 10.0 | 1.2 |
| 19 | 3-CNT-ox-Na | 0.5 | MeCN:acetone (1:1 v/v) |
5.56 | 4.69 | 10.3 | 1.2 |
| 20 | 3-CNT-ox-Na | 0.5 | MeCN:H2O (1:1 v/v) |
16.1 | 13.6 | 29.6 | 1.2 |
| 21 | 3-CNT-ox-Na | 0.5 | MeCN:H2O (3:1 v/v) |
13.8 | 11.4 | 25.2 | 1.2 |
Fig. 6a shows the yield comparison of complex 3 supported on the six different carbon materials (Table 2, entries 1–6). Control experiments in the absence of catalyst and support (Table 2, entry 8) and the use of bare CNT-ox-Na support (Table 2, entry 7) led to the formation of insignificant amounts of products. Despite the heterogenization of 3 leads to a KA oil yield of 10%, a value lower than that obtained under homogeneous conditions (13%), the heterogenization method has its own advantages which include the facile separation and reuse of the catalyst. The results depicted in Fig. 6a reveal that the alcohol is the preferred product under heterogeneous conditions, but the ketone is favoured when using complex 3 alone as catalyst. These results can suggest that in the heterogenized systems there are not enough active sites for the ready oxidation of alcohol because the catalyst loading is very low and the immobilized complex was at 2–3 wt% Mn only, apart from steric effects of the support, thus leading to the decline of selectivity towards ketone.
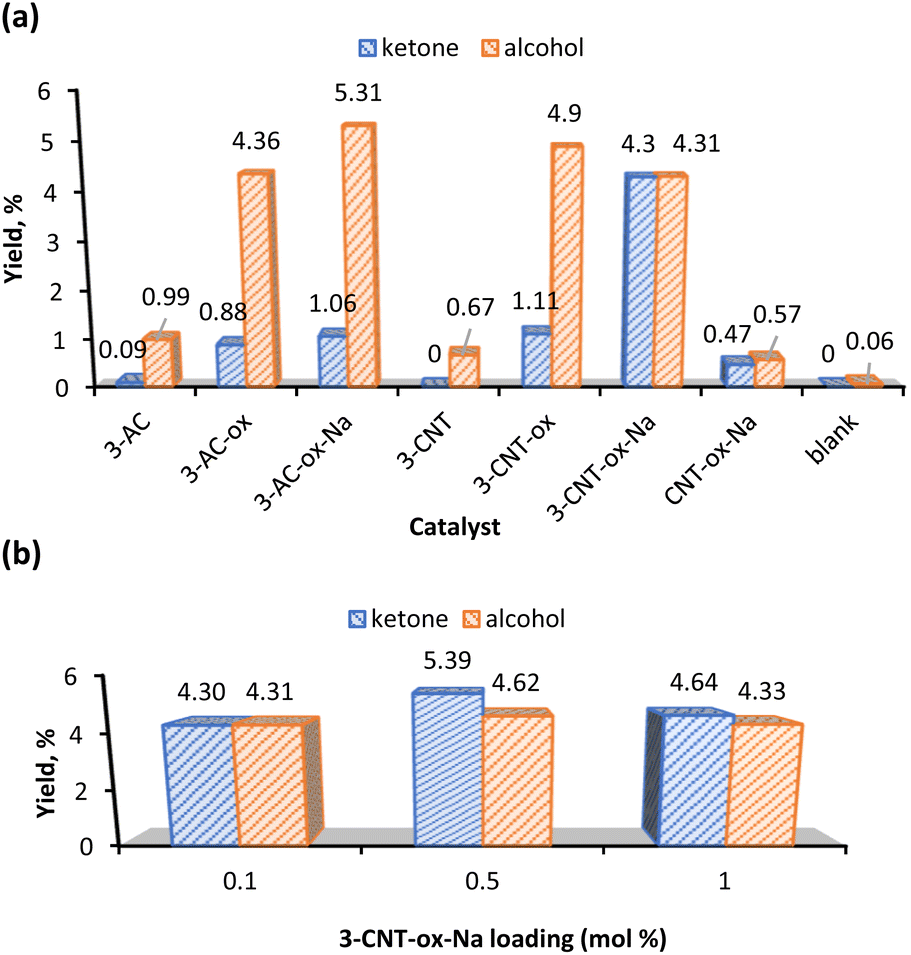 | ||
| Fig. 6 (a) Effect of catalyst (0.1 mol% relative to the substrate) on the yield product of heterogeneous cyclohexane oxidation (the blank test included for comparison). (b) Effect of 3-CNT-ox-Na catalyst loading. Reaction conditions: cyclohexane (5.0 mmol), 70% aqueous TBHP (10 mmol), 2 h, MW (30 W, 100 °C), 2 mL MeCN. | ||
As reported, the results also indicate that high KA oil yields were obtained in surface-modified materials, proving the importance of surface oxygen groups as anchorage sites.16,36
Fig. 6b shows that the optimal catalyst load of 3-CNT-ox-Na that gave the highest KA oil yield was 0.5 mol% and the favourability towards the ketone compound is enhanced as compared to the loading of 0.1 mol%. Increasing the catalyst loading to 1 mol% decreases the corresponding yield of product conceivably because of mixing problems of larger amounts of catalyst and possible agglomeration of its particles (Table 2, entry 11).44,59,60
As shown in Fig. S15,† in the heterogenized system the alcohol is the major product after 1 h reaction. Although the ketone yield slightly increases after 2 h, the total yield of products was almost the same when the reaction time was extended to 2.5 h. Aside from the lower number of catalytic sites in heterogeneous systems due to low catalyst loading and hampering steric effects, the short reaction time would not be enough to oxidize the alcohol to ketone, thus the observed results. As the reaction progresses to 2.5 h, the KA oil total yield decreases with the possibility of degradation as observed also with increasing reaction temperatures (Table 2, entry 17).
Similarly to what was obtained for the homogeneous system, the use of 3-CNT-ox-Na as catalyst also gives the highest KA oil yield when the reaction proceeds in MeCN:water solvent mixtures instead of MeCN alone (Table 2; compare entries 20 and 18, values of 30% and 10% in this order).
Moreover, the heterogenized system showed no product selectivity when MeCN was employed as solvent (Fig. 7). With a 3:1 v/v MeCN/water solvent ratio only 25% of the KA oil yield was obtained (Table 2, entry 21). The addition of acetone as co-solvent does not lead to a significant improvement of KA oil yield (10.3%, Table 2, entry 19). The rate acceleration by addition of water could be possibly attributed to the efficient solvation of radical species and propagation through the solution by a chain reaction.61 Solvent proticity and acidity of protons could be the contributing factors for these experimental findings. Since the electron cloud in a radical is enhanced, it needs a solvent with an acidic or more positive protons for efficient solvation. The hydrogens in CH3CN, although classified as aprotic, possess a slight acidity (Abraham's hydrogen bond acidity, αMeCN = 0.07)62 and the addition of water (Abraham's hydrogen bond acidity, αwater = 0.82)63 supplements the solvent mixture with more acidic protons than CH3CN alone.1,64,65 The possible promoting effect of water in alkane oxidation by assisting proton transfer steps has been rationalized by DFT calculations.66,67
 | ||
| Fig. 7 Effect of reaction medium on the KA oil yield using 3-CNT-ox-Na (0.5 mol% relative to the substrate) as catalyst. Reaction conditions: cyclohexane (5.0 mmol), 70% aqueous TBHP (10 mmol), 2 h, MW (30 W, 100 °C), 2 mL solvent. | ||
The oxidation of cyclohexane catalyzed by various metal complexes is expected to follow a radical mechanism (Scheme 3). In fact, detailed models, based on these mechanisms, that include a number of radical species and chain reaction steps, have been proposed.68–73 The first steps (initiation) concern the reactions of TBHP with Mn active sites of the heterogenized catalyst to produce the active radicals tBuO˙ and tBuOO˙ in accordance with the Haber–Weiss route (eqn (1) and (2)).74 The radical tBuOO˙ can undergo dismutation to tBuO˙ and O2. The tert-butoxyl radical abstracts H atom from cyclohexane (CyH) to form cyclohexyl radical (Cy˙) which then reacts with molecular oxygen from air (eqn (3)) to give cyclohexyl peroxyl radical (CyOO˙) (eqn (4)). This reacts with TBHP forming alkyl hydroperoxide (CyOOH) and tBuOO˙ (eqn (5)).The metal-assisted decomposition of CyOOH to CyO˙ leads to the formation of cyclohexanol (CyOH) and Cy˙ by H abstraction from cyclohexane (eqn (6) and (7)). In the final steps, CyOO˙ would either decompose to alcohol (CyOH) and ketone (Cy′O) (eqn (8)) or regenerate CyOOH and Cy˙ (eqn (5)), as proposed for some metal-catalyzed alkane oxidations by O2.75–77 The CyOO˙ radical can also undergo the mixed bimolecular Russell termination to form ketone and tBuOH (eqn (9)) increasing the ketone selectivity.74
 | ||
| Scheme 3 Proposed mechanism for cyclohexane oxidation catalyzed by an immobilized Mn species. | ||
In comparison to other carbon-supported transition metal complexes used as catalysts for cyclohexane oxidation (Table 3), the 3-CNT-ox-Na catalyst was able to produce KA oil with moderate/good yields using a low catalyst loading (0.5 mol%), usually a shorter reaction time and without further activation by co-catalysts. With the C-scorpionate iron(II) complex [FeCl2(Tpm)] [Tpm = κ3-HC(C3H3N2)3] (Fe) supported on oxidized single-walled carbon nanohorns (Fe@SWCNH-oxi),16 although the heterogenized system showed an interesting activity under mild conditions, the reaction needed the addition of pyrazine carboxylic acid (Hpca) as a promoter, whereas the current catalytic system under study used water as such.
| Catalyst | Conditions | Yield (%) | Ref. | ||
|---|---|---|---|---|---|
| K | A | Total | |||
| 3 | Homogeneous Mn(II)–tpy–PTA complex (3 mol%), MW (30 W), 100 °C, 2 h, 2 mL MeCN/H2O (1:1 v/v) |
12 | 9 | 21 | This work |
| 3-CNT-ox-Na | Carbon-supported Mn(II)–tpy–PTA complex (0.5 mol%), MW (30 W), 100 °C, 2 h, 2 mL MeCN/H2O (1:1 v/v) |
16 | 14 | 30 | This work |
| Au/CNT-COL | Carbon-supported Au nanoparticles, Hpca (co-catalyst, n Hpca/n catalyst = 50), 6 h at RT, 3 mL MeCN | 1.6 | 2 | 3.6 | 3 |
![[V with combining low line]](https://www.rsc.org/images/entities/char_0056_0332.gif) @CNT-ox @CNT-ox |
Carbon-supported V(V) complex (5.0 μmol), MW (20 W), 80 °C, 2 h, 3 mL MeCN | 5 | 7 | 12 | 14 |
F![[e with combining low line]](https://www.rsc.org/images/entities/char_0065_0332.gif) @SWCNH-oxi @SWCNH-oxi |
Carbon-supported C-scorpionate Fe(II) complex (0.1 mol%), Hpca (co-catalyst, n Hpca/n catalyst = 40), MW (20 W), 50 °C, 1.5 h, 3 mL MeCN | 12 | 17 | 29 | 16 |
| Ag6-GO | Graphene oxide-supported silver clusters, 110 °C, 17 h, solvent-free, TBHP/O2 | 62 | 0.6 | 63 | 80 |
| Cu2O-BGC | Bougainvillea glabra (B. glabra) derived carbon supported copper composite (Cu2O/BGC), 70 °C, 18 h reflux | 32 | 14 | 46 | 68 |
The dioxidovanadium(V) () complex bearing an aroylhydrazone Schiff base ligand and immobilized on oxidized carbon nanotubes (@CNT-ox) was able to produce 12% KA oil, with preference for the alcohol product,14 under microwave irradiation (20 W) at 80 °C for 2 h with no addition of acid and/or co-catalysts. Metal nanoparticles (NPs), in particular Au NPs, immobilized on carbon materials were tested as catalysts for the cyclohexane oxidation.78,79 The most active catalyst was prepared by supporting Au NPs on CNT by the colloidal method (Au/CNT-COL), and achieved an overall yield of products of 3.6% after 6 h at ambient temperature and with the promoting effects of Hpca.3 Glutathione (GSH)-protected Ag6 clusters supported on graphene oxide (Ag6-GO) using TBHP as oxidant in molecular oxygen atmosphere was highly effective and selective to the cyclohexanone product for 17 h of stirring at 110 °C.80 Likewise, the in situ synthesized Cu NPs supported on Bougainvillea glabra (B. glabra)-derived carbon (Cu2O-BGC) exhibited a high catalytic activity for cyclohexane oxidation with TBHP as oxidant under solvent-free condition. This system showed some selectivity towards cyclohexanone at 70 °C for 18 h reflux.68
Recyclability and leaching experiments
For the recyclability test of the 3-CNT-ox-Na catalyst, after completion of the reaction (1st cycle) the catalyst was separated from the reaction mixture by centrifugation, washed several times with acetonitrile and distilled water, dried at 100 °C overnight and reused; this procedure was repeated until a total of five successive runs with the same catalyst. As shown in Fig. 8, there occurs a significant decrease in activity on the third cycle and subsequent drops in the following ones. This decline can be attributed to the possible desorption of the catalyst from the surface (leaching) with subsequent deactivation or decomposition on the support as implied by the decrease in the % Mn from 2.91% (before the 1st cycle) to 0.087% (after the 5th cycle) based on ICP results. This may also be the reason for the change in selectivity towards the alcohol product after the first recycling and agrees with what was observed above, that alcohol is preferred at lower catalyst amount in both homo- and heterogeneous systems (Fig. 4b and 6b).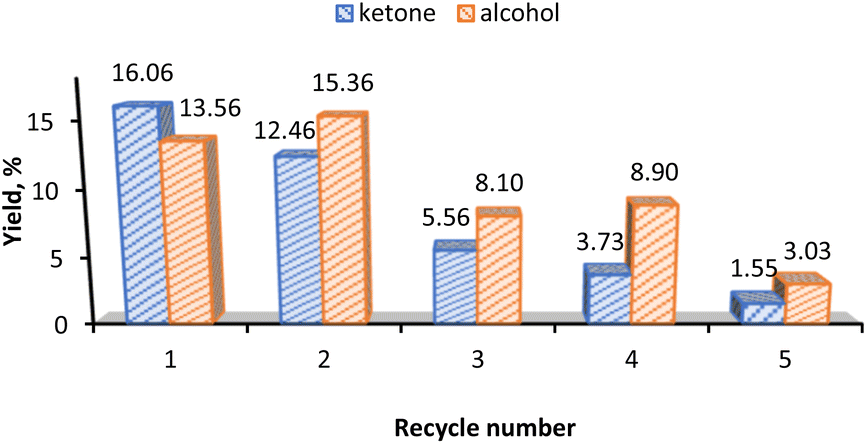 | ||
| Fig. 8 Recycle studies for 3-CNT-ox-Na. Reaction conditions: cyclohexane (5.0 mmol), 70% aqueous TBHP (10 mmol), 2 h, MW (30 W, 100 °C), 2 mL solvent (MeCN/H2O, 1:1 v/v), 0.5 mol% catalyst (relative to the substrate). | ||
Conclusions
A series of mononuclear metal complexes containing a PTA-functionalized terpyridine derivative was prepared and characterized. NMR, FTIR and ESI-MS data confirmed the quaternization of PTA ligand to the benzylated terpyridine. The complexation of the novel ligand to the metals (Co(II), Ni(II), Mn(II)) occurred in a κ3-N,N′,N′′ coordination mode of the terpyridine moiety, what was also confirmed by the single-crystal X-ray diffraction analysis for 2.The catalytic activity of the compounds was investigated for the cyclohexane oxidation under microwave irradiation. Complex 3 bearing the manganese metal gave the highest yield of KA oil and is more selective towards the ketone product. The heterogenization of 3 on carboxylated CNTs afforded 3-CNT-ox-Na which was also used as catalyst for the same oxidation reaction. In both the homogeneous and heterogeneous system, cyclohexanone is the favoured product at the higher catalyst loading and higher temperature, while a longer reaction time and the use of the solvent mixture acetonitrile/water (1:1 v/v) favours the cyclohexanol product. The optimum total KA oil yield of 30% was obtained using 0.5 mol% of the heterogeneous catalyst 3-CNT-ox-Na, under microwave irradiation (30 W) at 100 °C for 2 h and using the aforementioned solvent mixture. Recyclability of the heterogenized catalyst led to reuse for five successive cycles. It is also noteworthy to mention the accelerating effect of water which thus can be used as a reaction promoter thereby eliminating the addition of hazardous chemicals.
The findings in this work provide a new method to achieve robust catalysts based on PTA–terpyridine complexes for the oxidation of cyclohexane in a facile, one-pot procedure under microwave irradiation. This work highlights the sustainability development goals for the safe use of chemicals, substantial recyclability and more economical use of water and energy in accordance with the agreed international frameworks. Moreover, the strategy to combine PTA and tpy ligand moieties is also expected to be applied successfully towards the preparation of supramolecular catalysts, and further investigation is underway, as well as towards an improved catalyst stability so that prospects for eventual industrial application can be considered.
Experimental
Materials and instrumentation
All reagents were purchased commercially and used as received. Elemental (C, H, N) and ICP analyses were carried out by Laboratório de Análises of Instituto Superior Técnico. The 1H-, 13C-, DEPT-, and 31P NMR spectra were obtained using the Bruker Avance (Bruker, Billerica, MA, USA) 300 MHz spectrometer at ambient temperature. The chemical shifts were reported in ppm using tetramethylsilane as internal reference. Mass spectra (ESI-MS) were obtained on a Varian 500-MS LC Ion Trap Mass Spectrometer (Agilent Technologies, Santa Clara, CA, USA) equipped with electrospray ion source. Infrared spectra (4000–400 cm−1) were recorded on a Bruker Vertex 70 instrument (Bruker Corporation, Ettlingen, Germany) in KBr pellets. Reactions under microwave irradiation were performed using an Anton Paar Monowave 300 reactor (Anton Paar GmbH, Graz, Austria). Gas chromatography (GC) analyses were done using Clarus 500 gas chromatograph (PerkinElmer, Waltham, MA, USA) with a BP-20 capillary column (30 m × 0.22 mm × 25 μm; SGE, Australia), a flame ionization detector (FID) and run by the Total Chrom software.Preparation of PTA–Bztpy ligand and complexes
:C 46.52, H 4.74, N 11.63; found: C 46.57, H 4.40, N 11.43. FTIR (KBr, cm−1): 3402 (br), 1613 (s), 1472 (s), 1307 (s), 1183 (s), 793 (s). ESI-MS(+) in MeOH (m/z): 157 (calcd 157.1) [PTA + H]+, 479.12 (calcd 479.5) [p-tpy–C6H4–CH2–PTA]+, 698 (calcd 698.3) [Co(κ3-N,N′,N′′-tpy-p–C6H4–CH2–PTA)Br2]+.
:C 46.53, H 4.74, N 11.63; found: C 46.50, H 4.32, N 11.08. FTIR (KBr, cm−1): 3384 (br), 1603 (s), 1473 (s), 1406 (s), 1309 (s), 1248 (s), 1015 (s) 793 (s). ESI-MS(+) in MeOH (m/z): 172 (calcd 172.2) [PTA–CH3]+, 479.12 (calcd 479.5) [p-tpy–C6H4–CH2–PTA]+, 697 (calcd 698) [Ni(κ3-N,N′,N′′-tpy-p–C6H4–CH2–PTA)Br2]+.
X-ray diffraction analysis
A low quality crystal of 2 was immersed in cryo-oil, mounted in a Nylon loop, and measured at 150 K. A Bruker APEX-II PHOTON 100 diffractometer (Bruker AXS GmbH, Karlsruhe, Germany) was used (graphite monochromated Mo-Kα radiation of 0.71073) and a full sphere of data was collected using phi and omega scans of 0.5° per frame. Cell parameters were retrieved using Bruker SMART and refined on all the observed reflections using Bruker SAINT.81 Absorption corrections were applied using SADABS.82 Structures were solved by direct methods using SHELXS-2014/4 and refined with SHELXL-2018/3.83 Calculations were performed using the WinGX-Version 2020.1.84 All atoms (except hydrogen) were refined anisotropically. The aromatic and the methylene hydrogen atoms were included in the model at geometrically calculated positions and refined using a riding model. Resulting from instability during refinement, some distances and thermal parameters were restrained by using the DFIX and SIMU instructions. There were disordered molecules in the structure that could not be modelled. Running Platon Squeeze routine85 revealed 270 electrons per unit cell and a total potential solvent accessible void volume of 655 Å3 which fit well for 15 water molecules. These were removed from the model and not included in the final refinement of the structure.Preparation of carbon materials
The carbon materials used in this study as catalyst supports were the activated carbon (AC) and multi-walled carbon nanotube (CNT). These materials were used as received or subjected to surface treatments with nitric acid (HNO3) and sodium hydroxide (NaOH). The AC-ox and CNT-ox supports were obtained by refluxing 1 g of carbon material in 75 mL of 5 M HNO3 for 3 h. After the indicated time, the oxidized materials were separated by filtration and washed with distilled water until neutral pH.2,4,15,86 To prepare the AC-ox-Na and CNT-ox-Na supports, the oxygenated materials (AC-ox and CNT-ox) were further derivatized by adding 75 mL of 20 mM NaOH and refluxed for 1 h, followed by filtration and distilled water washing until neutral pH was obtained.2,4,15,86 A total of six different catalyst supports were produced and utilized in this work.Textural characterization of carbon supports
The six carbon-based supports were degassed at 150 °C for 48 h and subsequently characterized by N2 adsorption/desorption at 77 K using the Micrometrics ASAP 2060 gas sorption instrument (Hidden Isochema, Warrington, UK). The specific surface area (SBET), the total pore volume (P/Po = 0.99), micropore volume (t-method) and the average pore size were determined. The surface morphology was performed by scanning electron microscopy (SEM) in a Hitachi S-2400 (Tokyo, Japan) instrument and transmission electron microscopy (TEM) in a Hitachi 8100 (Tokyo, Japan) equipment at IST MicroLab.Immobilization procedure
The heterogenization of complex 3 (0.050 g) onto the six carbon materials (0.15 g per support) was done by dissolving complex 3 with 30 mL methanol and stirring at RT for 72 h. The resulting solid was then separated by filtration, washed with MeOH and dried overnight at 120 °C. The Mn loading was determined by ICP at the Laboratório de Análises of Instituto Superior Técnico.Microwave-assisted oxidation of cyclohexane
The oxidation reactions were carried out according to the following procedure: a 10 mL borosilicate glass vial equipped with a magnetic stirring bar containing the catalyst, homogeneous or carbon-supported, was added with 2 mL MeCN, 70% aq. TBHP (10 mmol), cyclohexane (5 mmol) and nitromethane (100 μL) as an internal standard. The vial was tightly capped and placed in the MW reactor where the mixture was irradiated (30 W) and stirred (600 rpm) for 2 h at 100 °C. After the reaction, the mixture was cooled to RT and centrifuged to separate the catalyst. The supernatant was then subjected to GC analysis.In a mixture containing water as a co-solvent, after the reaction was cooled at room temperature, 4 mL of n-hexane was added to the mixture and stirred vigorously to extract the substrate and the organic products from the reaction. The organic phase was then separated and analyzed for GC.
For the analysis of products, an internal standard method was used. An aliquot of the supernatant or organic phase was added with excess triphenylphosphine to reduce the formed cyclohexyl hydroperoxide to the corresponding alcohol, following a method developed by Shul'pin.75,87,88 After PPh3 addition, a 0.5 μL of the sample was taken and injected to the GC instrument. The injection temperature was 240 °C and helium was used as the carrier gas (mobile phase). After the injection, a temperature-programmed method was employed, wherein the reaction temperature was maintained at 100 °C for 1 min followed by a gradient increase of 10 °C min−1 until 160 °C and held at this temperature for 1 min. The products were identified by comparison of the retention time to that of the known reference compounds. Blank experiments, either no catalyst or with carbon support only (no heterogenized complex), were also performed and no significant amounts of products were detected. GC-MS analyses were done using a Clarus 600 C instrument (PerkinElmer, Waltham, MA, USA). The ionization voltage was 70 eV and helium was used as the carrier gas. GC was conducted in a temperature-programmed analysis by using a ZB-5 capillary column (30 m × 0.25 mm × 25 μm). Reaction products were identified by comparison of the mass spectra to fragmentation patterns obtained from the NIST spectral library stored in the computer software of the mass spectrometer.
Recyclability of the high-yielding catalyst using suitable reaction conditions was performed up to five consecutive cycles. After completion of each run, the products were separated and analyzed while the catalyst was recovered, washed with acetonitrile and water, dried overnight and reused.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful for the financial support from Fundação para a Ciência e a Tecnologia (FCT), Portugal, through project UIDB/00100/2020 of Centro de Quίmica Estrutural. The work was also funded by national funds through FCT, under the Scientific Employment Stimulus-Institutional Call (CEEC-INST/00102/2018). We also acknowledge the Associate Laboratory for Green Chemistry – LAQV financed by national funds from FCT/MCTES (UIDB/50006/2020 and UIDP/5006/2020) and Base-UIDB/50020/2020 and Programmatic-UIDP/50020/2020 funding of the Associate Laboratory LSRE-LCM. I. L. L. acknowledges the CATSUS Ph.D. Program from FCT for her grant PD/BD/135555/2018. A. P. is grateful to FCT and Instituto Superior Técnico (IST), Portugal through DL/57/2017 (Contract no. IST-ID/197/2019). AGM is grateful to Associação do Instituto Superior Técnico para Investigação e Desenvolvimento for his post-doctoral fellowship through grant no. BL133/2021-IST-ID. This publication is also supported by the RUDN University Strategic Academic Leadership Program (recipient AJLP, preparation). A. V. G. thanks FCT, Instituto Superior Técnico (DL 57/2016, L 57/2017 and CEEC Institutional 2018 Programs, Contract no: IST-ID/110/2018) and Baku State University for financial support. The authors also acknowledge the Portuguese NMR Network (IST-UL Centre) for access to the NMR facility. CFGCG thanks the FCT for funding the Coimbra Chemistry Centre through the programmes UIDB/00313/2020 and UIDP/00313/2020, also co-founded by FEDER/COMPETE 2020-EU. The authors are also thankful to Benjoe Rey B. Visayas (University of Massachusetts Dartmouth) for the help with the graphics.References
- F. Liang, W. Zhong, L. Xiang, L. Mao, Q. Xu, S. R. Kirk and D. Yin, J. Catal., 2019, 378, 256–269 CrossRef CAS.
- M. P. de Almeida, L. M. D. R. S. Martins, S. A. C. Carabineiro, T. Lauterbach, F. Rominger, A. S. K. Hashmi, A. J. L. Pombeiro and J. L. Figueiredo, Catal. Sci. Technol., 2013, 3, 3056 RSC.
- S. A. C. Carabineiro, L. M. D. R. S. Martins, M. Avalos-Borja, J. G. Buijnsters, A. J. L. Pombeiro and J. L. Figueiredo, Appl. Catal., A, 2013, 467, 279–290 CrossRef CAS.
- L. M. D. R. S. Martins, M. P. de Almeida, S. A. C. Carabineiro, J. L. Figueiredo and A. J. L. Pombeiro, ChemCatChem, 2013, 5, 3847–3856 CrossRef CAS.
- Alkane Functionalization, ed., A. J. L. Pombeiro and M. F. C. Guedes da Silva, John Wiley & Sons, Inc., Hoboken, New Jersey, 2019 Search PubMed.
- X. Wang, Z. Feng, J. Liu, Z. Huang, J. Zhang, J. Mai and Y. Fang, J. Colloid Interface Sci., 2020, 580, 377–388 CrossRef CAS PubMed.
- M. A. Andrade and L. M. D. R. S. Martins, Catalysts, 2019, 10, 2 CrossRef.
- M. Conte, X. Liu, D. M. Murphy, K. Whiston and G. J. Hutchings, Phys. Chem. Chem. Phys., 2012, 14, 16279–16285 RSC.
- M. L. Kuznetsov and A. J. L. Pombeiro, J. Catal., 2021, 399, 52–66 CrossRef CAS.
- J. Liu, R. Liu, H. Li, W. Kong, H. Huang, Y. Liu and Z. Kang, Dalton Trans., 2014, 43, 12982–12988 RSC.
- S. Rana and S. B. Jonnalagadda, ChemistrySelect, 2017, 2, 2277–2281 CrossRef CAS.
- U. Schuchardt, W. A. Carvalho and E. V. Spinach, Synlett, 1993, 1993, 713–718 CrossRef.
- A. K. Suresh, M. M. Sharma and T. Sridhar, Ind. Eng. Chem. Res., 2000, 39, 3958–3997 CrossRef CAS.
- M. Sutradhar, M. A. Andrade, S. A. C. Carabineiro, L. M. D. R. S. Martins, M. de F. C. G. da Silva and A. J. L. Pombeiro, Nanomaterials, 2021, 11, 1456 CrossRef CAS PubMed.
- S. A. C. Carabineiro, L. M. D. R. S. Martins, A. J. L. Pombeiro and J. L. Figueiredo, ChemCatChem, 2018, 10, 1804–1813 CrossRef CAS.
- A. P. C. Ribeiro, L. M. D. R. S. Martins, S. A. C. Carabineiro, J. G. Buijnsters, J. L. Figueiredo and A. J. L. Pombeiro, ChemCatChem, 2018, 10, 1821–1828 CrossRef CAS.
- G. Centi and S. Perathoner, Catal. Today, 2003, 77, 287–297 CrossRef CAS.
- P. T. Anastas, L. B. Bartlett, M. M. Kirchhoff and T. C. Williamson, Catal. Today, 2000, 55, 11–22 CrossRef CAS.
- P. C. J. Kamer, P. W. N. M. Van Leeuwen and J. N. H. Reek, Acc. Chem. Res., 2001, 34, 895–904 CrossRef CAS PubMed.
- W. Wang, G. B. Hammond and B. Xu, J. Am. Chem. Soc., 2012, 134, 5697–5705 CrossRef CAS PubMed.
- O. R. Luca and R. H. Crabtree, Chem. Soc. Rev., 2013, 42, 1440–1459 RSC.
- J. R. Khusnutdinova and D. Milstein, Angew. Chem., Int. Ed., 2015, 54, 12236–12273 CrossRef CAS PubMed.
- P. J. Guiry and C. P. Saunders, Adv. Synth. Catal., 2004, 346, 497–537 CrossRef CAS.
- M. P. Carroll and P. J. Guiry, Chem. Soc. Rev., 2014, 43, 819–833 RSC.
- A. Winter and U. S. Schubert, ChemCatChem, 2020, 12, 2890–2941 CrossRef CAS.
- C. Wei, Y. He, X. Shi and Z. Song, Coord. Chem. Rev., 2019, 385, 1–19 CrossRef CAS PubMed.
- A. Guerriero, M. Peruzzini and L. Gonsalvi, Coord. Chem. Rev., 2018, 355, 328–361 CrossRef CAS.
- A. G. Mahmoud, M. F. C. Guedes da Silva and A. J. L. Pombeiro, Coord. Chem. Rev., 2021, 429, 213614 CrossRef CAS.
- Z. Ma, L. Wei, E. C. B. A. Alegria, L. M. D. R. S. Martins, M. F. C. Guedes da Silva and A. J. L. Pombeiro, Dalton Trans., 2014, 43, 4048–4058 RSC.
- A. Paul, A. P. C. Ribeiro, A. Karmakar, M. F. C. Guedes da Silva and A. J. L. Pombeiro, Dalton Trans., 2016, 45, 12779–12789 RSC.
- A. G. Mahmoud, M. F. C. Guedes da Silva, E. I. Śliwa, P. Smoleński, M. L. Kuznetsov and A. J. L. Pombeiro, Chem.−Asian J., 2018, 13, 2868–2880 CrossRef CAS PubMed.
- L. M. D. R. S. Martins, E. C. B. A. Alegria, P. Smoleński, M. L. Kuznetsov and A. J. L. Pombeiro, Inorg. Chem., 2013, 52, 4534–4546 CrossRef CAS PubMed.
- E. I. Śliwa, D. S. Nesterov, J. Kłak, P. Jakimowicz, A. M. Kirillov and P. Smoleński, Cryst. Growth Des., 2018, 18, 2814–2823 CrossRef.
- S. A. C. Carabineiro, B. F. Machado, R. R. Bacsa, P. Serp, G. Draić, J. L. Faria and J. L. Figueiredo, J. Catal., 2010, 273, 191–198 CrossRef CAS.
- S. A. C. Carabineiro, P. B. Tavares and J. L. Figueiredo, Appl. Nanosci., 2012, 2, 35–46 CrossRef CAS.
- S. A. C. Carabineiro, A. P. C. Ribeiro, J. G. Buijnsters, M. Avalos-Borja, A. J. L. Pombeiro, J. L. Figueiredo and L. M. D. R. S. Martins, Catal. Today, 2020, 357, 22–31 CrossRef CAS.
- H. B. Khosravi, R. Rahimi, M. Rabbani, A. Maleki and A. Mollahosseini, Silicon, 2021, 13, 451–465 CrossRef CAS.
- C. Solórzano, F. J. Méndez, J. L. Brito, P. Silva, J. R. Anacona and E. Bastardo-González, J. Organomet. Chem., 2020, 908, 121073 CrossRef.
- E. Pakrieva, A. P. C. Ribeiro, L. M. D. R. S. Martins, I. A. S. Matias, S. A. C. Carabineiro, E. Kolobova, A. J. L. Pombeiro, J. L. Figueiredo and A. Pestryakov, Catal. Today, 2020, 357, 39–45 CrossRef CAS.
- L. M. D. R. S. Martins, A. Martins, E. C. B. A. Alegria, A. P. Carvalho and A. J. L. Pombeiro, Appl. Catal., A, 2013, 464–465, 43–50 CrossRef CAS.
- O. Kholdeeva and N. Maksimchuk, Catalysts, 2021, 11, 1–23 CrossRef.
- Y. Xiao, J. Liu, K. Xie, W. Wang and Y. Fang, Mol. Catal., 2017, 431, 1–8 CrossRef CAS.
- F. P. Pruchnik and P. Smoleński, Appl. Organomet. Chem., 1999, 13, 829–836 CrossRef CAS.
- I. L. Librando, A. G. Mahmoud, S. A. C. Carabineiro, M. F. C. Guedes da Silva, C. F. G. C. Geraldes and A. J. L. Pombeiro, Nanomaterials, 2021, 11, 1–29 CrossRef PubMed.
- J. L. Figueiredo and M. F. R. Pereira, J. Energy Chem., 2013, 22, 195–201 CrossRef CAS.
- A. Schaetz, M. Zeltner and W. J. Stark, ACS Catal., 2012, 2, 1267–1284 CrossRef CAS.
- Y. H. Hu and E. Ruckenstein, Ind. Eng. Chem. Res., 2004, 43, 708–711 CrossRef CAS.
- M. Thommes and C. Schlumberger, Annu. Rev. Chem. Biomol. Eng., 2021, 12, 137–162 CrossRef CAS PubMed.
- S. A. C. Carabineiro, T. Thavorn-Amornsri, M. F. R. Pereira and J. L. Figueiredo, Water Res., 2011, 45, 4583–4591 CrossRef CAS PubMed.
- J. J. Niu, J. N. Wang, Y. Jiang, L. F. Su and J. Ma, Microporous Mesoporous Mater., 2007, 100, 1–5 CrossRef CAS.
- J. Zhu, A. Holmen and D. Chen, ChemCatChem, 2013, 5, 378–401 CrossRef CAS.
- D. R. Rolison, Science, 2003, 299, 1698–1701 CrossRef CAS PubMed.
- G. Prieto, A. Martínez, R. Murciano and M. A. Arribas, Appl. Catal., A, 2009, 367, 146–156 CrossRef CAS.
- J. Robinson, S. Kingman, D. Irvine, P. Licence, A. Smith, G. Dimitrakis, D. Obermayer and C. O. Kappe, Phys. Chem. Chem. Phys., 2010, 12, 4750–4758 RSC.
- A. Kokel, C. Schäfer and B. Török, Green Chem., 2017, 19, 3729–3751 RSC.
- R. J. Trovitch, Acc. Chem. Res., 2017, 50, 2842–2852 CrossRef CAS PubMed.
- P. Chandra, T. Ghosh, N. Choudhary, A. Mohammad and S. M. Mobin, Coord. Chem. Rev., 2020, 411, 213241 CrossRef CAS.
- D. Ottaviani, V. Van-Dúnem, A. P. Carvalho, A. Martins and L. M. D. R. S. Martins, Catal. Today, 2020, 348, 37–44 CrossRef CAS.
- I. L. Librando, A. G. Mahmoud, S. A. C. Carabineiro, M. F. C. Guedes da Silva, C. F. G. C. Geraldes and A. J. L. Pombeiro, Catalysts, 2021, 11, 1–15 CrossRef.
- I. L. Librando, A. G. Mahmoud, S. A. C. Carabineiro, M. F. C. G. da Silva, F. J. Maldonado-Hódar, C. F. G. C. Geraldes and A. J. L. Pombeiro, Catalysts, 2022, 12, 45 CrossRef CAS.
- B. Ensing, F. Buda, P. Blöchl and E. J. Baerends, Angew. Chem., Int. Ed., 2001, 40, 2893–2895 CrossRef CAS PubMed.
- P. Winget, D. M. Dolney, D. J. Giesen, C. J. Cramer and D. G. Truhlar, Minnesota Solvent Descriptor Database, 1999, https://comp.chem.umn.edu/solvation/mnsddb.pdf Search PubMed.
- M. H. Abraham, J. Andonian-Haftvan, G. S. Whiting, A. Leo and R. S. Taft, J. Chem. Soc., Perkin Trans. 2, 1994, 1777–1791 RSC.
- S. Mitroka, S. Zimmeck, D. Troya and J. M. Tanko, J. Am. Chem. Soc., 2010, 132, 2907–2913 CrossRef CAS PubMed.
- B. R. B. Visayas, S. K. Pahari, T. C. Gokoglan, J. A. Golen, E. Agar, P. J. Cappillino and M. L. Mayes, Chem. Sci., 2021, 12, 15892–15907 RSC.
- M. L. Kuznetsov and A. J. L. Pombeiro, Inorg. Chem., 2009, 48, 307–318 CrossRef CAS PubMed.
- M. V. Kirillova, M. L. Kuznetsov, V. B. Romakh, L. S. Shul'pina, J. J. R. Fraústo da Silva, A. J. L. Pombeiro and G. B. Shul’pin, J. Catal., 2009, 267, 140–157 CrossRef CAS.
- C. Xie, Q. Xiong, L. Jiang, Y. Wang, Q. Tang, J. He and J. Wang, Appl. Surf. Sci., 2022, 576, 151833 CrossRef CAS.
- T. R. Barman, M. Sutradhar, E. C. B. A. Alegria, M. F. C. Guedes da Silva and A. J. L. Pombeiro, Catalysts, 2020, 10, 1–16 Search PubMed.
- A. R. Kim, S. Ahn, T. U. Yoon, J. M. Notestein, O. K. Farha and Y. S. Bae, ChemCatChem, 2019, 11, 5650–5656 CrossRef CAS.
- M. Sutradhar, T. Roy Barman, E. C. B. A. Alegria, H. M. Lapa, M. F. C. Guedes da Silva and A. J. L. Pombeiro, New J. Chem., 2020, 44, 9163–9171 RSC.
- T. C. O. Mac Leod, M. V. Kirillova, A. J. L. Pombeiro, M. A. Schiavon and M. D. Assis, Appl. Catal., A, 2010, 372, 191–198 CrossRef CAS.
- R. Kumar, S. Sithambaram and S. L. Suib, J. Catal., 2009, 262, 304–313 CrossRef CAS.
- M. Nowotny, L. N. Pedersen, U. Hanefeld and T. Maschmeyer, Chem.–Eur. J., 2002, 8, 3724–3731 CrossRef CAS PubMed.
- G. B. Shul’pin, C. R. Chim., 2003, 6, 163–178 CrossRef.
- G. Shul’pin, Mini-Rev. Org. Chem., 2009, 6, 95–104 CrossRef.
- M. V. Kirillova, A. M. Kirillov, P. M. Reis, J. A. L. Silva, J. J. R. Fraústo da Silva and A. J. L. Pombeiro, J. Catal., 2007, 248, 130–136 CrossRef CAS.
- A. S. K. Hashmi and G. J. Hutchings, Angew. Chem., Int. Ed., 2006, 45, 7896–7936 CrossRef PubMed.
- A. S. K. Hashmi, Chem. Rev., 2007, 107, 3180–3211 CrossRef CAS PubMed.
- R. Anumula, C. Cui, M. Yang, J. Li and Z. Luo, J. Phys. Chem. C, 2019, 123, 21504–21512 CrossRef CAS.
- Bruker APEX2, Bruker AXS Inc., Madison, Wisconsin, USA, 2012 Search PubMed.
- G. M. Sheldrick, SADABS. Program for Empirical Absorption Correction, University of Gottingen, Germany, 2000 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- L. J. Farrugia, J. Appl. Crystallogr., 2012, 45, 849–854 CrossRef CAS.
- A. L. Spek, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 9–18 CrossRef CAS PubMed.
- L. M. D. R. S. Martins, A. P. C. Ribeiro, S. A. C. Carabineiro, J. L. Figueiredo and A. J. L. Pombeiro, Dalton Trans., 2016, 45, 6816–6819 RSC.
- G. B. Shul'Pin, Catalysts, 2016, 6, 50 CrossRef.
- G. B. Shul’pin, J. Mol. Catal. A: Chem., 2002, 189, 39–66 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2183127. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2su00017b |
| This journal is © The Royal Society of Chemistry 2023 |