DOI:
10.1039/D2QM01199A
(Research Article)
Mater. Chem. Front., 2023,
7, 1591-1598
Exceptionally flexible quinodimethanes with multiple conformations: polymorph-dependent colour tone and emission of crystals†
Received
21st November 2022
, Accepted 4th February 2023
First published on 8th February 2023
Abstract
Tetraarylated quinodimethanes, which are one of the overcrowded ethylenes, can adopt multiple conformations, such as folded, twisted, and other forms. However, it is difficult to obtain all of these conformations due to energy differences among them. To tackle this issue, we designed flexible polyaryl-substituted tetraazaanthraquinodimethanes (N4AQDs), in which a steric hindrance in the overcrowded fjord region was moderately reduced. As a result, X-ray analyses revealed that N4AQDs could adopt not only folded and twisted forms but also intermediate structures, e.g., planar and twisted-folded forms, in pseudopolymorphs. Since there is a large difference in the electronic configuration among conformers, the colour tone of crystals varied from yellow to red depending on the conformation of overcrowded N4AQDs. Notably, red to near-infrared emission was observed for the crystal containing twisted conformers. This is the first example of exceptionally flexible quinodimethanes with tunable photophysical properties and multi-chromic behaviour based on multiple conformations.
Introduction
Stereoisomers (e.g., enantiomers and diastereomers) with topologically equivalent structures but different conformations and configurations often exhibit inherent physical properties. Since some diastereomers can be switched among two or more stable states by macroscopic external stimulation such as by light and/or heat, the microscopic properties based on the conformation/configuration can be controlled.1–3 These have been widely studied for potential applications in molecular switches, memory devices, and sensor materials.4–9
With regard to such response systems, the conformations of overcrowded ethylenes (OCEs), in which a C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond is surrounded by bulky substituents, can be switched between two or more conformers [e.g., folded (F) and twisted (T) forms] by external stimuli,10–13 especially for use in molecular motors.14–18 Generally, the T-form has a higher HOMO and lower LUMO than the F-form, resulting in a significant difference in absorption and emission wavelengths between conformers. Recently, there has been steady progress toward obtaining the T-form in addition to the F-form for their potential application as stimuli-responsive molecules;19–26 however, investigations on the T-form remain insufficient due to its potential instability or shorter lifetime.
C double bond is surrounded by bulky substituents, can be switched between two or more conformers [e.g., folded (F) and twisted (T) forms] by external stimuli,10–13 especially for use in molecular motors.14–18 Generally, the T-form has a higher HOMO and lower LUMO than the F-form, resulting in a significant difference in absorption and emission wavelengths between conformers. Recently, there has been steady progress toward obtaining the T-form in addition to the F-form for their potential application as stimuli-responsive molecules;19–26 however, investigations on the T-form remain insufficient due to its potential instability or shorter lifetime.
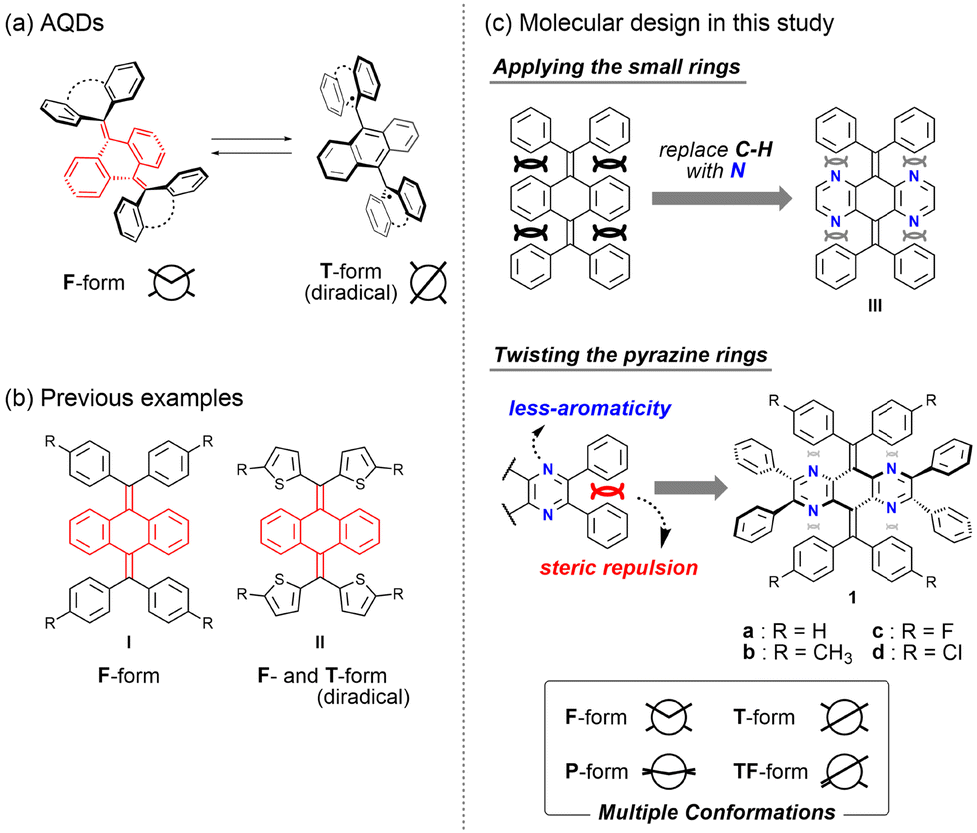
Among various OCEs, anthraquinodimethanes (AQDs) have thus far been used as a common way to obtain the T-form in addition to the F-form by introducing tricyclic aromatic units (Fig. 1a). Notably, since the T-form as seen in AQDs is the open-shell diradical species, various properties such as colour and magnetic properties can be controlled by external stimuli based on a change in the conformation between the closed-shell F-form and the open-shell T-form.27,28 We also reported several tetraaryl-AQD derivatives such as I29 and II12 (Fig. 1b). In particular, for II, we have realised the control of redox properties based on the thermal equilibrium between the closed-shell F-form and the open-shell T-form. As shown in these AQDs, while open-shell species are interesting from the viewpoint of their potential applications, their high reactivity often causes a problem in handling. Therefore, we envisaged that further functional molecules that exhibit drastic changes in absorption and emission based on the conformers with different electronic configurations should be obtained when quinodimethane derivatives could exhibit the “closed-shell” T-form in addition to the F-form.
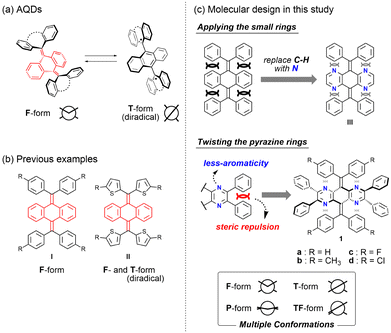 |
| | Fig. 1 (a) Structural change of AQDs. (b) Previous examples of AQDs. (c) Molecular design in this study to make multiple conformations stable by decreasing the steric hindrance between aromatic rings (smaller aromaticity). | |
Since it is difficult to isolate more than two structures for the same molecule in OCEs, there are very few examples where the existence of both F- and T-forms in (pseudo)polymorphs was confirmed by X-ray analyses.30–33 As exemplified by these molecules, dramatic chromic behaviour based on the difference in the electronic configuration between F- and T-forms is expected; however, the molecular design strategy is still limited to such molecules with rigid and planar aromatic groups. Since these molecules are very attractive, we envisaged that we could obtain unexplored conformations in addition to these two conformers by an appropriate molecular design. Therefore, we aimed to construct a unique molecule that could adopt not only F- and T-forms but also other conformations [e.g., planar (P) and twisted-folded (TF) forms], so that unprecedented switching behaviour could be achieved with the involvement of multiple states.
Herein, we report tetraazaanthraquinodimethane (N4AQD) derivatives 1a–d, which can adopt both the closed-shell T- and F-forms in crystals by reducing the steric hindrance in the overcrowded fjord region. Furthermore, we have also succeeded in creating not only these two structures, but also several intermediate structures in polymorphs. In other words, we modulated the photophysical properties based on the conformation observed in the crystal. Generally, since dynamically averaged physical properties are observed in solution, the present results represent the first example where colour tone and emission colour are controlled based on multiple conformations in the crystal that can be generated by recrystallization from appropriate solvents.
Results and discussion
Design strategy
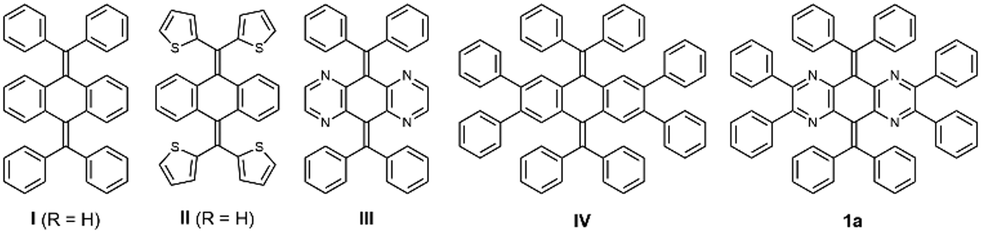
On the basis of the difference in structures found in quinodimethane derivatives I and II, we expected that the conformations could be significantly affected by a change in steric hindrance around the central CC double bond. Therefore, we considered that the decrease in steric hindrance in the fjord region would be key to creating the T-form for AQDs. At the same time, to control the conformations of AQDs, we propose a new strategy: reducing the steric hindrance in the overcrowded fjord region by replacing the C–H moiety of the central AQD skeleton with nitrogen atoms (derivative III, Fig. 1c). Instead of an approach that uses thienyl groups, which contribute to the stabilization of open-shell species, the introduction of pyrazine rings into the central AQD skeleton is designed to reduce the aromaticity of the tricyclic core and allow the skeleton itself to twist.
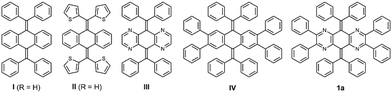
Before starting the synthesis, we performed density functional theory (DFT) calculations at the (U)B3LYP/6-31G* level for N4AQDs III (as well as I and II for comparison). The results showed that the closed-shell T-form in III is still calculated as a transition state with one imaginary frequency as well as I (Table 1). Thus, to obtain the T-form as a stable structure, we decided to use an additional approach to perturb the molecular structure by the introduction of two vic-diphenyl groups into both sides of the central N4AQD skeleton (compound 1, Fig. 1c). This strategy can induce a further twisting of the less-aromatic pyrazine ring due to steric repulsion between adjacent phenyl groups, resulting in a decrease in steric hindrance between the N4AQD core and the aryl groups on the exocyclic bonds to make the T-form more stable. DFT calculations were then performed on the N4AQD derivative 1, which suggested that the T-form would be obtained as the most stable structure and the F-form would also exist as a metastable structure (Table 1 and Table S9, ESI†). The results of the DFT study for non-aza analogue IV confirmed the validity of our molecular design. Thus, we focused on N4AQDs with peripheral phenyl groups, with the expectation that both the F- and T-forms could appear in crystals and designed four derivatives 1a–d to investigate the effect of the steric and electronic properties of the substituent at the 4-position on each of the aryl groups (Table S6, ESI†).
Table 1 Relative energies of I, II, III, IV, and 1a based on the optimised structures obtained by DFT calculations [(U)B3LYP/6-31G*]
|
|
| ΔErel [kcal mol−1] |
|
|

|
|
F-form |
T-form |
T-form (diradical) |
| Singlet |
Triplet |
|
Optimised as a transition-state structure.
|
|
I
|
0 |
14.0a |
13.6 |
13.9 |
|
II
|
0 |
9.40 |
2.31 |
2.35 |
|
III
|
0 |
3.35a |
14.3 |
16.1 |
|
IV
|
0 |
11.3 |
12.8 |
13.4 |
|
1a
|
3.15 |
0 |
10.9 |
13.9 |
Preparation
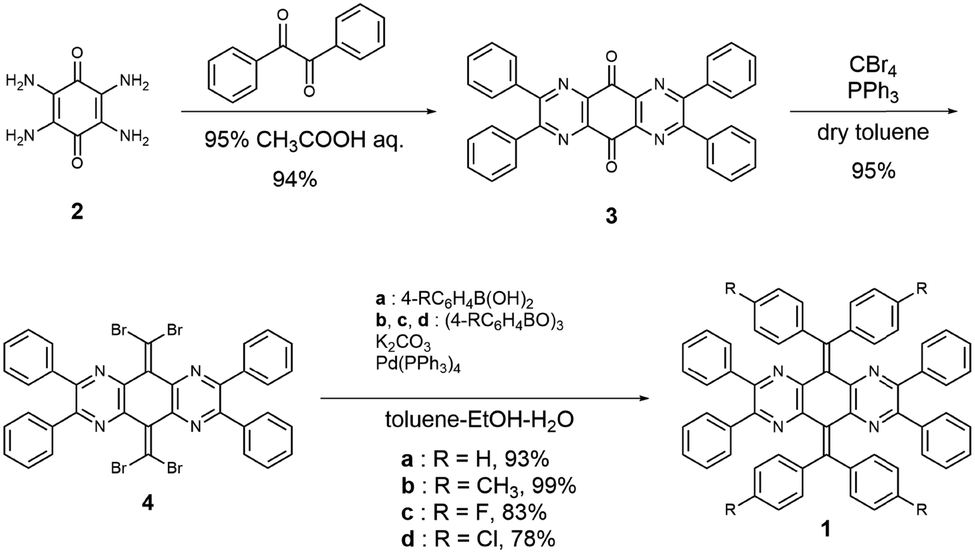
The N4AQD derivatives 1a–d were synthesised as follows (Scheme 1). First, the dehydrative condensation of 2,3,5,6-tetraamino-1,4-benzoquinone 234 with benzil to give 3,35 and subsequent double dibromo-olefination led to the precursor 4.36,37 Next, Suzuki–Miyaura cross-coupling reactions of 4 with the corresponding boronic acid or boroxin afforded the desired N4AQD derivatives 1a–d in 78–99% yields. Reference compounds I,38III,39 and IV were synthesised in a similar manner (Scheme S1, ESI†). These seven AQD derivatives were characterised by 1H/13C NMR and IR spectroscopies, and field desorption (FD)-mass spectrometry (see the ESI†).
 |
| | Scheme 1 Preparation of 1. | |
Structure in solution
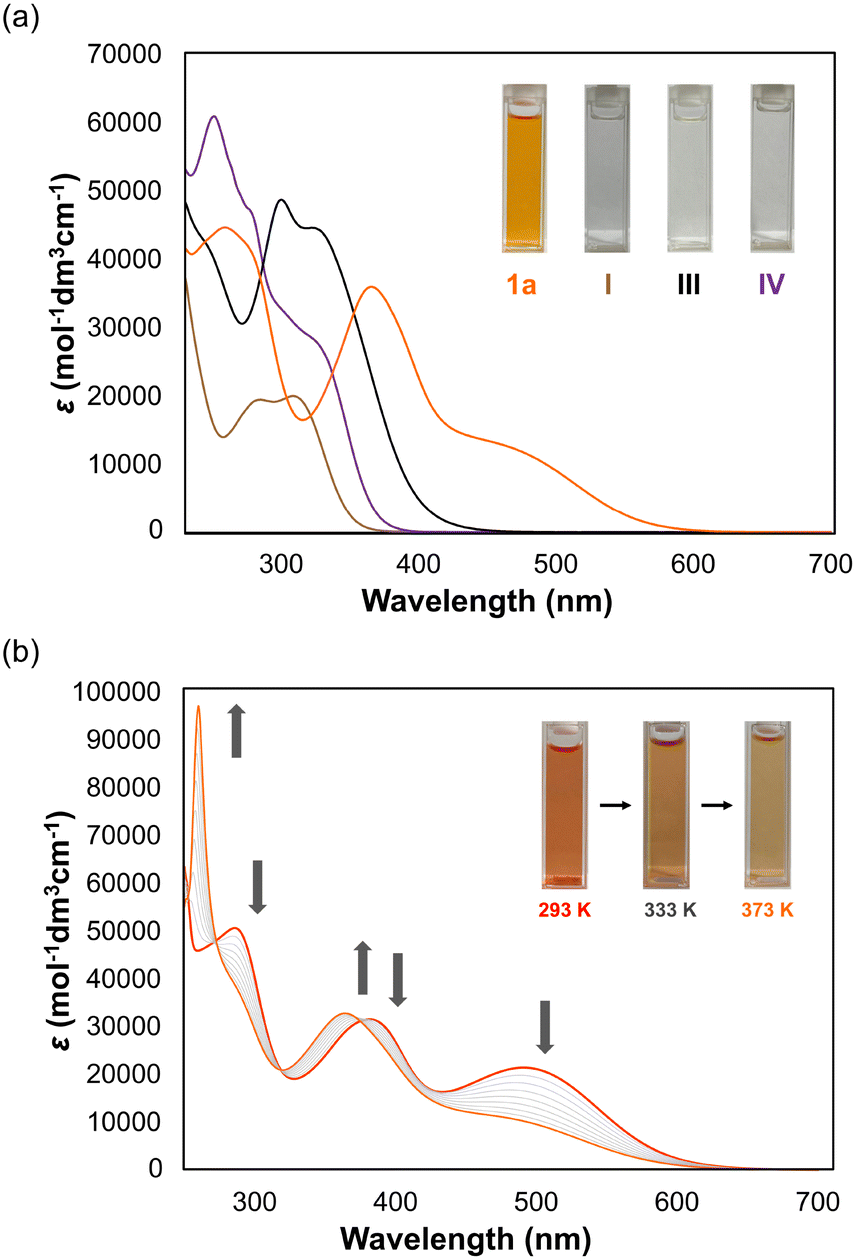
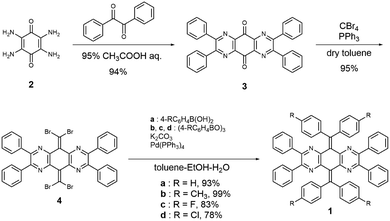
To gain insight into the solution-phase structure of the N4AQD derivatives, the UV/Vis spectrum of the parent phenyl derivative 1a (R = H) was measured in CH2Cl2. Compared to the spectra of the reference compounds I, III, and IV, a red shift of the absorption maxima was clearly observed (λmax = 470 nm for 1a and λmax ≈ 320 nm for I, III, and IV), and the solution of 1a is reddish-orange whereas those of I, III and IV are colourless to pale yellow (Fig. 2a and Table 2). The HOMO–LUMO gaps of each derivative predicted by DFT calculations were estimated to be 4.11 eV, 3.67 eV, and 3.87 eV for the F-forms of I, III, and IV, respectively, and 2.38 eV for the T-form of 1a, which is in good agreement with the trend of the absorption end of each derivative. These results suggest that only 1a can have a contribution from a twisted structure in solution. In addition, the UV/Vis spectra of 1b (R = CH3), 1c (R = F), and 1d (R = Cl) in CH2Cl2 were almost the same as that of 1a (Fig. S15 and Table S7, ESI†), indicating that all N4AQD derivatives 1 have contributions from the T-form. According to variable-temperature (VT) 1H NMR analysis of derivative 1b, no broadening of signals was observed upon heating from 303 K to 393 K in DMSO-d6 (Fig. S16, ESI†). Thus, there is no contribution of open-shell species. On the other hand, some proton signals shifted upon heating in DMSO-d6 and cooling in CD2Cl2 (Fig. S17, ESI†). To clarify the reason why some proton signals shifted upon elevating the temperature, UV/Vis spectra of 1b were measured in the range from 293 K to 373 K in DMSO (Fig. 2b). Upon elevating the temperature, a decrease in the absorption band around 500 nm was observed, accompanied by a change in the colour of the solution from reddish-orange to nearly yellow. These observations clearly indicate that the F- and T-forms are in equilibrium in solution. Thus, a thermochromic behaviour was observed for 1b, since the contribution of T-form is more dominant at lower temperature while that of the metastable F-form increases at higher temperatures according to the Boltzmann distribution. Due to the rapid change in structures, all N4AQD derivatives 1 are non-fluorescent in solution. Such fluorescence quenching can often be observed in tetraarylethylenes,40,41 which are the representative molecules with aggregation-induced emission properties.42
 |
| | Fig. 2 (a) UV/Vis spectra of 1a, I, III, and IV in CH2Cl2. (b) VT UV/Vis spectra of 1b in DMSO from 293 K to 373 K (every 10 K). | |
Table 2 Experimental and theoretical absorption properties (in CH2Cl2, at 293 K) of 1a, I, III, and IV. Shoulder peaks are marked with “sh”. λend is the absorption end of each spectrum. The energy gap ΔEDFTLUMO–HOMO was estimated by DFT calculations (B3LYP/6-31G*)
|
|
λ
max [nm] (log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ε) ε) |
λ
end [nm] |
ΔEDFTLUMO–HOMO [eV]/[nm] |
|
1a (Ph8N4AQD) |
sh 470 (4.09) |
657 |
|
| 366 (4.56) |
3.50/354 (F-form) |
| 259 (4.65) |
2.38/520 (T-form) |
|
I (Ph4AQD) |
308 (4.30) |
453 |
4.11/302 |
| 285 (4.29) |
|
III (Ph4N4AQD) |
322 (4.65) |
498 |
3.67/338 |
| 300 (4.69) |
|
IV (Ph8AQD) |
sh 320 (4.46) |
409 |
3.88/319 |
| sh 275 (4.68) |
| 251 (4.78) |
X-Ray analyses
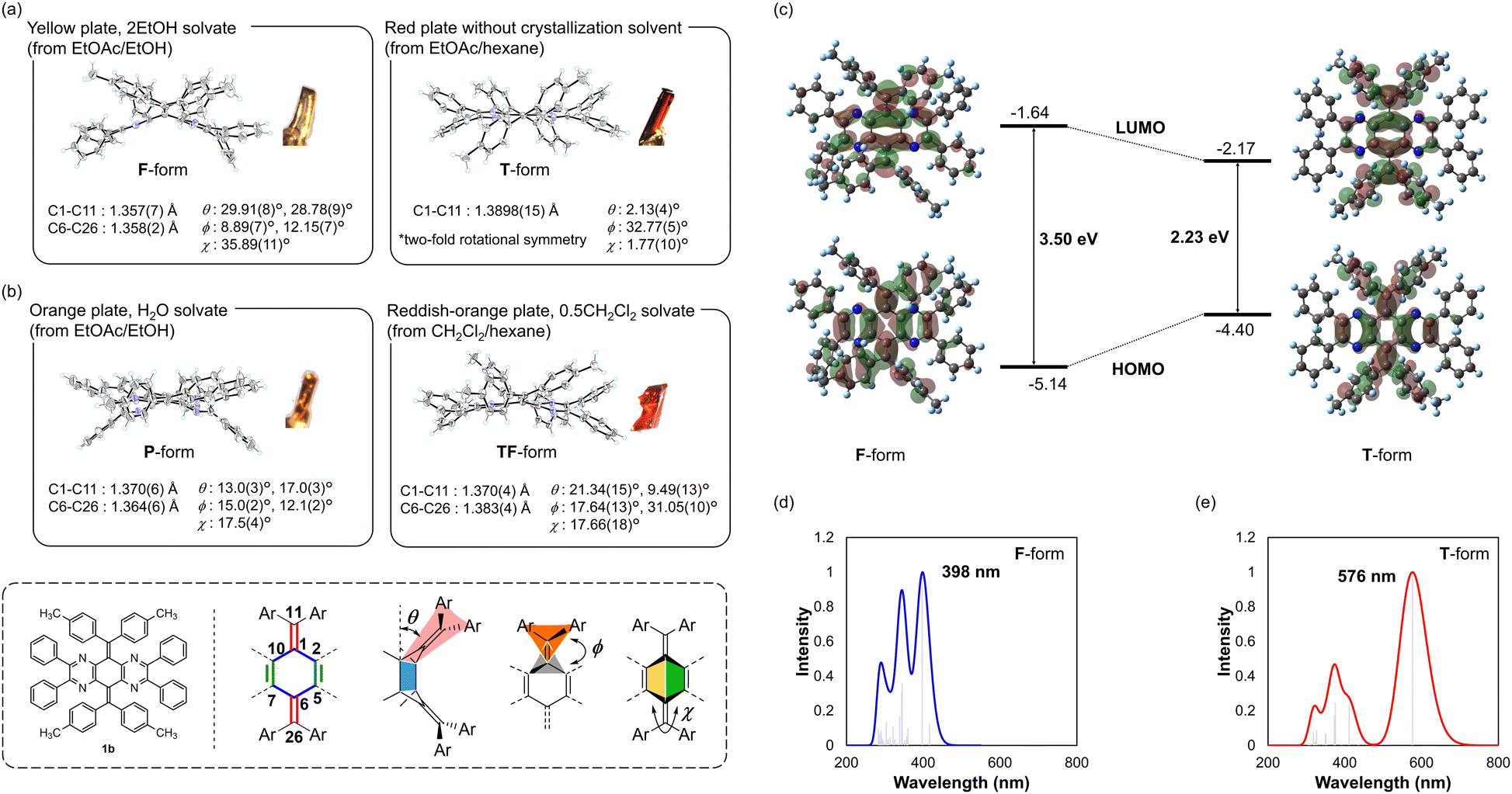
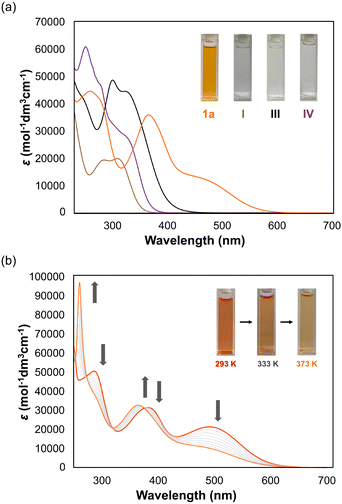
Since the presence of the T-form was indicated for N4AQD derivatives (1) in solution, we turned our attention to their structures and photophysical properties in the solid states. X-ray structures were investigated using single crystals of N4AQD derivatives 1a–d, which were obtained by recrystallization from various solvents. For the tolyl derivative 1b, recrystallization from EtOAc/EtOH produced yellow crystals as EtOH solvate, while that from EtOAc/hexane provided red crystals without containing any solvent. X-Ray analyses of both crystals revealed that 1b adopts the F- and T-forms in yellow crystals containing EtOH and red crystals, respectively (Fig. 3a). To investigate the origin of their colour, we performed DFT calculations (B3LYP/6-31G*) using the obtained crystallographic coordinates, and the results indicated that the T-form has a higher HOMO and lower LUMO than the F-form, i.e., the electronic structures of these conformers are significantly different (Fig. 3c and Table S10, ESI†). Actually, time-dependent (TD)-DFT calculations (B3LYP/6-31G*) predicted a ca. 180 nm difference in the simulated absorption maxima between these conformers (398 nm for F-form and 576 nm for T-form), which well reproduces the colour of each crystal (Fig. 3d, e and Fig. S21, ESI†). On the other hand, the only X-ray structures obtained for the reference compounds III and IV were folded (Fig. S10, ESI†). These results emphasise that both the nitrogen atoms and the vic-diphenyl groups introduced into the central AQD skeleton contribute to stabilization of the T-form.
 |
| | Fig. 3 ORTEP drawings of 1b [(a) F-form in 2EtOH solvate (recrystallised from EtOAc/EtOH) and T-form without any solvent (recrystallised from EtOAc/hexane), and (b) P-form in H2O solvate (recrystallised from EtOAc/EtOH) and TF-form in 0.5CH2Cl2 solvate (recrystallised from CH2Cl2/hexane)]. The solvent molecules are omitted for clarity. (c) HOMO and LUMO levels calculated using the DFT method (B3LYP/6-31G*). Simulated UV/Vis spectra of (d) the F-form and (e) the T-form obtained by TD-DFT calculations (B3LYP/6-31G*) based on the crystallographic coordinates. | |
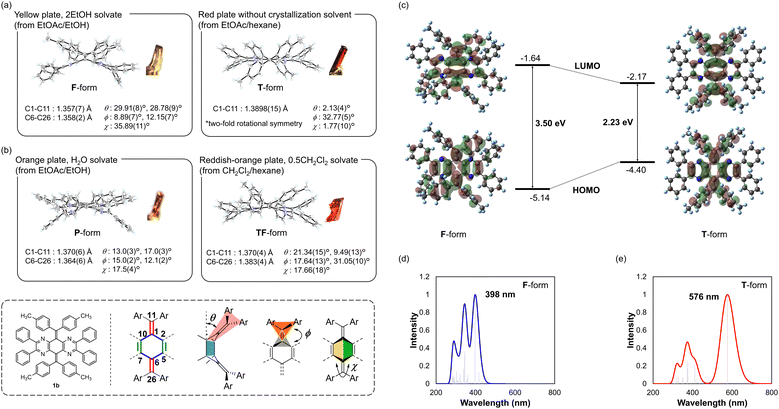
The most important finding is that the obtained crystal structures were not only the F- and T-forms but also several other conformations, which could not be classified as either of them. For orange-coloured H2O-solvated crystal and reddish-orange-coloured CH2Cl2-solvated crystal of 1b, the P-form with an almost normal planar geometry of the CC double bond and the TF-form with an intermediary structure between F- and T-forms were observed, respectively (Fig. 3b and Table S5, ESI†). Furthermore, for other derivatives (e.g., phenyl derivative 1a, fluorophenyl derivative 1c, and chlorophenyl derivative 1d), multiple conformations were observed in their crystals, regardless of the difference in the steric and electronic properties of the substituents on the aryl groups (Table 3 and Fig. S6–S9, ESI†). Since these intermediate structures are considered to be unstable in solution, their abundance ratio is generally zero. In fact, optimization from the obtained crystallographic coordinates as the initial structure yielded the T-form, which is the most stable structure. Even though there are some intermolecular contacts between molecules in each crystal, no significant interactions were observed to stabilise the specific conformation (Fig. S11–S14, ESI†). Based on these results, the intermediate structures observed in the pseudopolymorphs are stabilised by the crystal lattice, which is modified by the solvent molecules filling a void while forming a crystal. Accordingly, we clarified that multiple intermediate structures as well as the F- and T-forms can arise in the same molecule for AQD-type OCEs. Although a few examples have been described previously, where multiple conformations with a slight difference in dihedral angles were observed in (pseudo)polymorphs,43,44 this study is unique because a drastic change in colour has been achieved for multiple conformers with a large difference in molecular geometry such as the F-, T-, P-, and TF-forms. Thus, specific electronic properties such as HOMO–LUMO gaps based on the structures of the pseudopolymorphs can be determined, whereas dynamically averaged physical properties are generally observed in solution (Table 4 and Fig. S20, ESI†).
Table 3 List of the single crystals of 1a, 1b, 1c, and 1d analysed by X-ray in this study
|
|
Conformation |
Colourb |
Shape |
Solvate |
Recrystallization solvent |
|
In a crystal, there are two crystallographically independent molecules.
Colours are determined visually.
|
|
1a (R = H) |
F-form |
Yellow |
Plate |
CH2Cl2 |
CH2Cl2/hexane |
|
F-form |
Yellow |
Plate |
CHCl3 |
CHCl3/EtOH |
|
T-form |
Red |
Block |
None |
CH2Cl2/EtOH |
|
1b (R = CH3) |
F-form |
Yellow |
Plate |
2EtOH |
EtOAc/EtOH |
|
P-form |
Orange |
Plate |
H2O |
EtOAc/EtOH |
|
P-form |
Orange |
Plate |
CH2Cl2 |
CH2Cl2/hexane |
|
P-form |
Orange |
Plate |
0.5CHCl3 |
CHCl3/EtOH |
|
TF-form |
Reddish-orange |
Plate |
0.5CH2Cl2 |
CH2Cl2/hexane |
|
TF-form |
Reddish-orange |
Block |
0.5hexane |
EtOAc/hexane |
|
TF-form |
Reddish-orange |
Plate |
0.5EtOAc |
EtOAc/EtOH |
|
TF-form |
Reddish-orange |
Plate |
CH2Cl2 |
CH2Cl2/EtOH |
|
T-form |
Red |
Plate |
None |
EtOAc/hexane |
|
1c (R = F) |
F-form |
Yellow |
Block |
EtOAc |
EtOAc |
|
F-form |
Yellow |
Plate |
0.25CH2Cl2 |
CH2Cl2/hexane |
|
F-form |
Yellow |
Plate |
1.25CH2Cl2 |
CH2Cl2/EtOH |
|
F-form |
Yellow |
Plate |
2CHCl3 |
CHCl3/EtOH |
|
F-form + semi-P-forma |
Orange |
Plate |
H2O |
CHCl3/hexane |
|
TP-form |
Red |
Needle |
CHCl3 |
CHCl3/hexane |
|
TP-forma |
Red |
Needle |
CH2Cl2 |
CH2Cl2/hexane |
|
TP-forma |
Red |
Needle |
EtOH |
EtOAc/EtOH |
|
TF-form |
Reddish-orange |
Plate |
CHCl3 |
CHCl3/hexane |
|
1d (R = Cl) |
P-forma |
Orange |
Plate |
CHCl3 |
CHCl3/EtOH |
|
P-form |
Orange |
Plate |
CH2Cl2 |
CH2Cl2/hexane |
|
TF-form |
Reddish-orange |
Plate |
CHCl3 |
CHCl3/hexane |
|
T-form |
Red |
Needle |
None |
CHCl3/hexane |
Table 4 Estimated electronic properties of 1b (F-, P-, TF-, and T-forms). All data were calculated using the (TD-)DFT method (B3LYP/6-31G*)
|
|
E
DFTHOMO [eV] |
E
DFTLUMO [eV] |
E
DFTLUMO–HOMO [eV] |
λ
TD-DFTmax [nm] |
|
F-form |
−5.14 |
−1.64 |
3.50 |
375 |
|
T-form |
−4.40 |
−2.17 |
2.23 |
576 |
|
P-form |
−4.79 |
−1.91 |
2.88 |
454 |
|
TF-form |
−4.68 |
−2.04 |
2.64 |
489 |
Emission properties
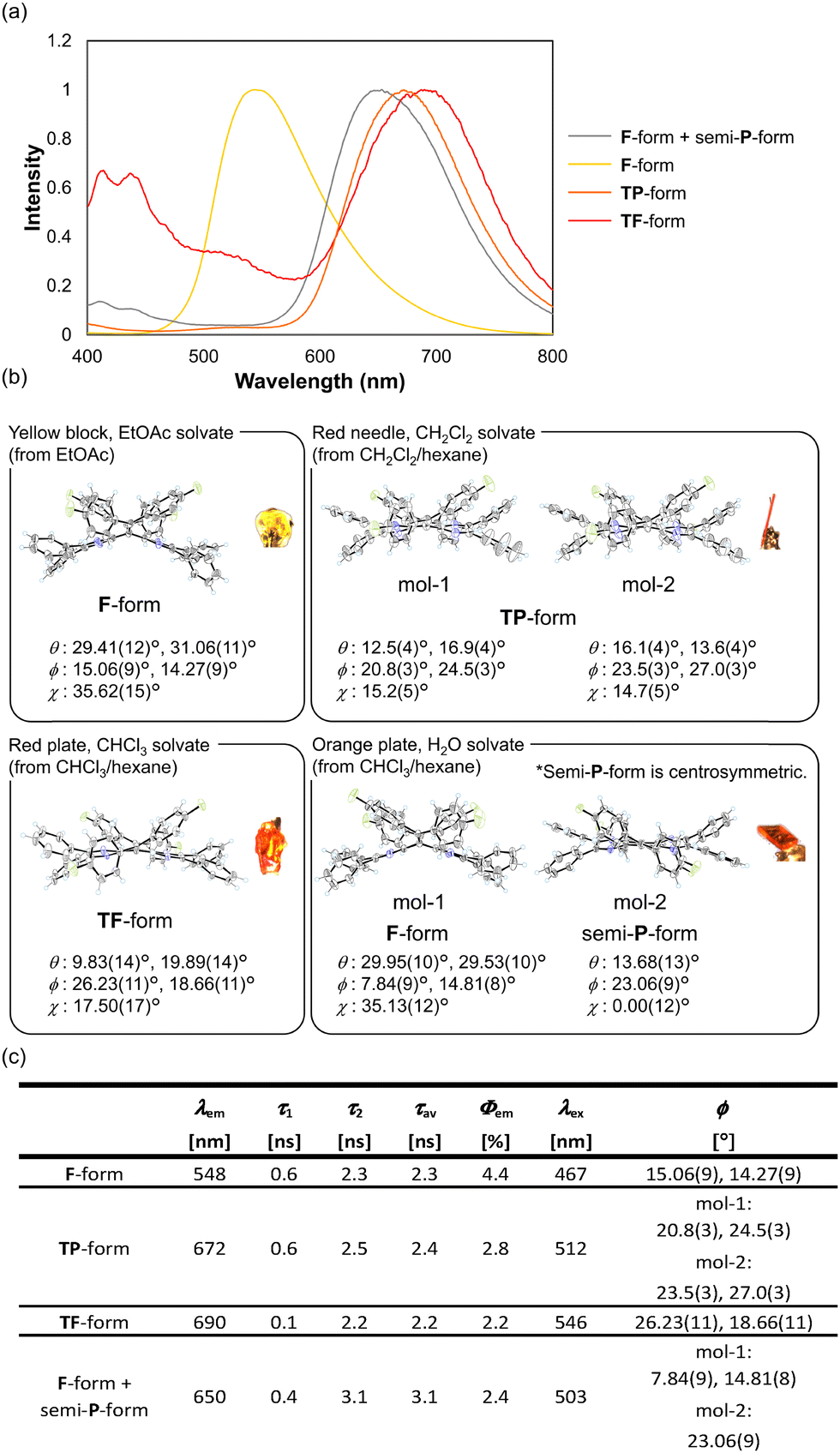
N4AQD derivatives 1 are non-fluorescent in solution yet become fluorescent in crystals. Notably, the luminescence of the crystals is strongly affected by the difference in the conformations of N4AQD derivatives 1. Fig. 4a shows the emission spectra of crystals for the fluorophenyl derivative 1c under excitation at 380 nm. The emission wavelength changed depending on the conformations of the crystals (λem = 548 nm and Φem = 4.4% for F-form, λem = 672 nm and Φem = 2.8% for the twisted-planar (TP) form, λem = 690 nm and Φem = 2.2% for the TF-form, λem = 650 nm and Φem = 2.4% for the F-form + semi-P-form), and a red shift of the emission maximum was observed as the dihedral angle ϕ increased [ϕ = 15.06(9)°, 14.27(9)° for the F-form, mol-1: ϕ = 20.8(3)°, 24.5(3)°, mol-2: ϕ = 23.5(3)°, 27.0(3)° for the TP-form, ϕ = 26.23(11)°, 18.66(11)° for the TF-form, mol-1: ϕ = 7.84(9)°, 14.81(8)°, mol-2: ϕ = 23.06(9)° for the F-form + semi-P-form] (Fig. 4b). Interestingly, there is about a 140 nm difference in the emission maximum between the F-form and the TF-form, even though they are the same molecule, whereas there is an 80-nm difference in the excitation spectra between them (Fig. 4c and Fig. S18, ESI†). In addition, the emission of the TF-form extends to the red to near-infrared (NIR) region although N4AQDs are the neutral species without any charges. Long-wavelength emission of 1c is unlikely to be in a polarised form45,46 but is solely due to the twisted conformation of the molecule. In the case of 1a, 1b, and 1d, the emission spectra also depend on the conformation in the crystal. Additionally, no significant intermolecular interactions which can be enough to influence their photophysical properties were observed in the pseudopolymorphs of N4AQD derivatives 1 (Fig. S11–S14, ESI†), though the change in the colour of luminescence in ordinary polymorphs is mostly due to changes in intermolecular interactions.47–49 These results indicate that the changes in the colour of luminescence in N4AQDs 1 depend only on the conformations of the molecules in the crystals, and a shift of the emission maximum up to 140 nm was accomplished by dynamic structural changes based on the flexibility of molecules.
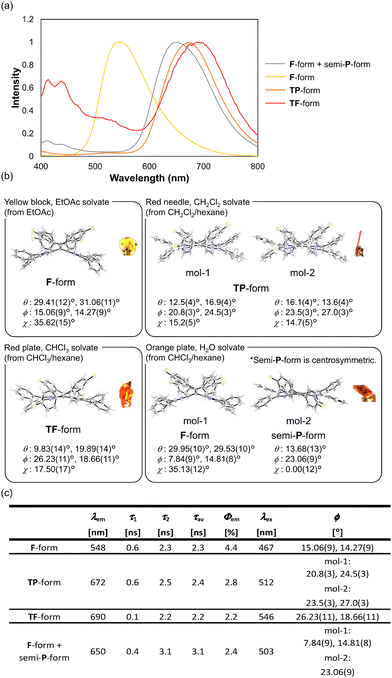 |
| | Fig. 4 (a) Emission spectra of each conformation of 1c in the solid state. (b) ORTEP drawings of 1c [F-form in EtOAc solvate (recrystallised from EtOAc), TP-form in CH2Cl2 solvate (recrystallised from CH2Cl2/hexane), TF-form in CHCl3 solvate (recrystallised from CHCl3/hexane), and those in which the F- and semi-P-forms exist as two crystallographically independent molecules in H2O solvate (recrystallised from CHCl3/hexane)]. Each of them was classified by the difference in dihedral angle θ, ϕ, and χ) for which emission spectra were measured. The solvent molecules are omitted for clarity. (c) Luminescence properties and dihedral angle ϕ of each conformation of 1c were recorded in the solid state. All crystals were excited at 380 nm. Emission lifetimes were detected at the longer emission maximum. λex is the value of the peak in the excitation spectra of 1c. | |
Mechanochromic behaviour
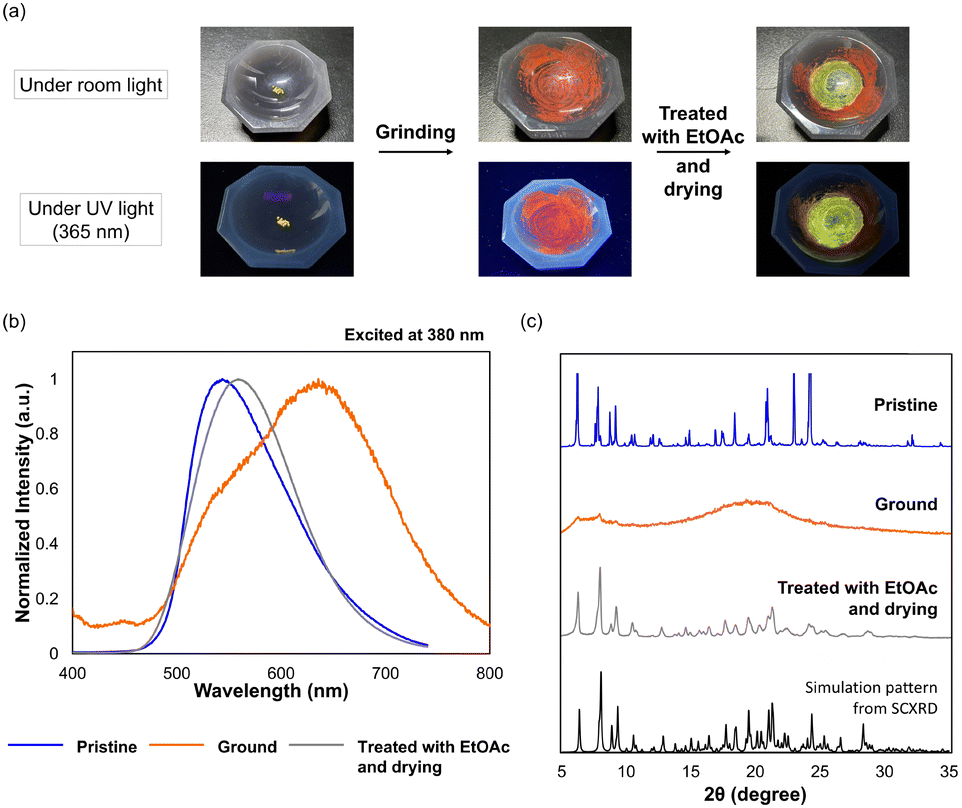
We then investigated a stimuli-responsive behaviour of crystals for 1c. Actually, colour changes were observed under visible and UV light of fluorophenyl derivative 1c when the yellow EtOAc-solvated crystals (F-form) were ground in an agate mortar and pestle, as shown in Fig. 5a. After yellow crystals with a folded conformation were ground, the colour of the samples became reddish-orange (Fig. S19, ESI†) and the maximum emission wavelength shifted from λem = 544 nm to λem = 636 nm (Fig. 5b and Table S8, ESI†), suggesting that a twisted conformation is produced. Furthermore, the original crystal with yellow emission (λem = 559 nm) can be recovered by dissolving the ground powder with EtOAc followed by drying, which indicates that the HOMO–LUMO gap can be modulated based on the conformations by being exposed to external stimuli. To investigate the origin of the change in the photophysical properties upon mechanical stimulation, powder X-ray diffraction (PXRD) analyses were conducted for each solid-state sample of 1c. As shown in Fig. 5c, since the PXRD pattern of the ground sample (orange line) has no sharp peaks, a phase transition from crystal to amorphous occurred upon grinding the pristine crystal. As a result, the metastable F-form could no longer be maintained in an amorphous state, and thus the more stable twisted conformation appeared. Next, the sharp diffraction pattern was regenerated by treating the sample with EtOAc followed by drying (Fig. 5c, from the orange line to the grey line), which demonstrates that 1c exhibits mechanochromism in the ground and excited states based on the crystal-to-amorphous phase transition. This phenomenon was realised due to the “flexibility” granted to the AQD-type OCE, i.e., not only the reduction of steric hindrance by the introduction of nitrogen atoms into the central framework but also the introduction of vic-diphenyl groups on the fused pyrazine rings are important for creating multiple conformations with tunable photophysical properties.
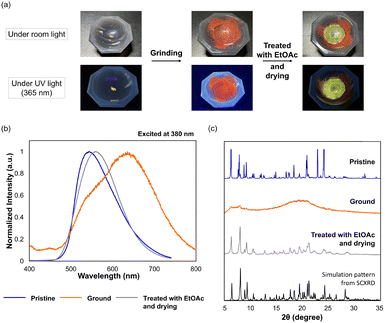 |
| | Fig. 5 (a) Photographs of 1c under room light (upper) and UV light (lower) in various states. (b) Changes in the emission spectrum for 1c. (c) Changes in the PXRD pattern of 1c. The black line is the simulation pattern from single-crystal X-ray diffraction (SCXRD). | |
Conclusions
In summary, we have designed and synthesised four kinds of tetraaza AQD derivative 1 with moderately reduced steric hindrance in the overcrowded fjord region with the introduction of vic-diphenyl groups. We succeeded in observing not only the F- and T-forms but also many other conformers, e.g., the P-form, TF-form, and TP-form, in pseudopolymorphs of 1, and this is the first demonstration of such multiple conformations for AQD-type OCEs. In addition, we demonstrated that the HOMO/LUMO levels can be modulated by the conformations in crystals. Thus, the changes in colour tone and emission of the crystals were accounted for by the difference in the electronic configurations of the conformers, and these quinodimethanes should be candidates for the development of advanced materials exhibiting conformation-dependent multi-chromism. It is particularly significant that the formation of twisted conformations with long-wavelength absorption/NIR-emission was achieved without the involvement of charge-separation states. This study has realised the control of absorption and luminescence properties derived from conformational changes, which is a very rare phenomenon in organic molecules, and revealed the potential value of novel sensor materials composed of simple but flexible organic molecules exhibiting an unprecedented gradual change in colour.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by JSPS KAKENHI and JSPS Grant-in-Aid for Research Fellow (Grant Numbers JP20H02719, JP20K21184, JP21H01912, JP21H05468, and JP20J20972). T. O and Y. I. acknowledge Toyota Riken Scholar. Y. I. is grateful for the 2020 DIC Award in Synthetic Organic Chemistry, Japan.
Notes and references
- H. M. D. Bandara and S. C. Burdette, Chem. Soc. Rev., 2012, 41, 1809–1825 RSC.
- R. Klajn, Chem. Soc. Rev., 2014, 43, 148–184 RSC.
- M. Irie, T. Fukaminato, K. Matsuda and S. Kobatake, Chem. Rev., 2014, 114, 12174–12277 CrossRef CAS PubMed.
- Y. Sakata, S. Fukushima, S. Akine and J. Setsune, Chem. Commun., 2016, 52, 1278–1281 RSC.
- P. Lentes, E. Stadler, F. Röhricht, A. Brahms, J. Gröbner, F. D. Sönnichsen, G. Gescheidt and R. Herges, J. Am. Chem. Soc., 2019, 141, 13592–13600 CrossRef CAS PubMed.
- L. Bai, P. Bose, Q. Gao, Y. Li, R. Ganguly and Y. Zhao, J. Am. Chem. Soc., 2017, 139, 436–441 CrossRef CAS PubMed.
- Y. Tang, Y. Zeng and R. Xiong, J. Am. Chem. Soc., 2022, 144, 8633–8640 CrossRef CAS PubMed.
- M. Raisch, D. Genovese, N. Zaccheroni, S. B. Schmidt, M. L. Focarete, M. Sommer and C. Gualandi, Adv. Mater., 2018, 30, 1802813 CrossRef PubMed.
- W. Li, Q. Huang, Z. Mao, J. Zhao, H. Wu, J. Chen, Z. Yang, Y. Li, Z. Yang, Y. Zhang, M. P. Aldred and Z. Chi, Angew. Chem., Int. Ed., 2020, 59, 3739–3745 CrossRef CAS PubMed.
- J. Luo, K. Song, F. Long Gu and Q. Miao, Chem. Sci., 2011, 2, 2029–2034 RSC.
- Y. Ishigaki, Y. Hayashi and T. Suzuki, J. Am. Chem. Soc., 2019, 141, 18293–18300 CrossRef CAS PubMed.
- Y. Ishigaki, T. Hashimoto, K. Sugawara, S. Suzuki and T. Suzuki, Angew. Chem., Int. Ed., 2020, 59, 6581–6584 CrossRef CAS PubMed.
- Y. Adachi, T. Nomura, S. Tazuhara, H. Naito and J. Ohshita, Chem. Commun., 2021, 57, 1316–1319 RSC.
- N. Koumura, R. W. Zijlstra, R. A. van Delden, N. Harada and B. L. Feringa, Nature, 1999, 401, 152–155 CrossRef CAS PubMed.
- B. L. Feringa, Acc. Chem. Res., 2001, 34, 504–513 CrossRef CAS PubMed.
- T. Kudernac, N. Ruangsupapichat, M. Parschau, B. Maciá, N. Katsonis, S. R. Harutyunyan, K.-H. Ernst and B. L. Feringa, Nature, 2011, 479, 208–211 CrossRef CAS PubMed.
- S. Kassem, T. van Leeuwen, A. S. Lubbe, M. R. Wilson, B. L. Feringa and D. A. Leigh, Chem. Soc. Rev., 2017, 46, 2592–2621 RSC.
- M. Baroncini, S. Silvi and A. Credi, Chem. Rev., 2020, 120, 200–268 CrossRef CAS PubMed.
- H. Meyer, Monatsh. Chem., 1909, 30, 165–177 CrossRef CAS.
- P. U. Biedermann, J. J. Stezowski and I. Agranat, Eur. J. Org. Chem., 2001, 15–34 CrossRef CAS.
- W. R. Browne, M. M. Pollard, B. de Lange, A. Meetsma and B. L. Feringa, J. Am. Chem. Soc., 2006, 128, 12412–12413 CrossRef CAS PubMed.
- T. Suzuki, H. Okada, T. Nakagawa, K. Komatsu, C. Fujimoto, H. Kagi and Y. Matsuo, Chem. Sci., 2018, 9, 475–482 RSC.
- Y. Hirao, Y. Hamamoto, N. Nagamachi and T. Kubo, Phys. Chem. Chem. Phys., 2019, 21, 12209–12216 RSC.
- X. Yin, J. Z. Low, K. J. Fallon, D. W. Paley and L. M. Campos, Chem. Sci., 2019, 10, 10733–10739 RSC.
- T. Nishiuchi, S. Aibara, H. Sato and T. Kubo, J. Am. Chem. Soc., 2022, 144, 7479–7488 CrossRef CAS PubMed.
- Y. Hirao, Y. Hamamoto and T. Kubo, Chem. – Asian J., 2022, 17, e202200121 CAS.
- T. Nishiuchi, R. Ito, E. Stratmann and T. Kubo, J. Org. Chem., 2020, 85, 179–186 CrossRef CAS PubMed.
- M. B. S. Wonink, B. P. Corbet, A. A. Kulago, G. B. Boursalian, B. de Bruin, E. Otten, W. R. Browne and B. L. Feringa, J. Am. Chem. Soc., 2021, 143, 18020–18028 CrossRef CAS PubMed.
- Y. Ishigaki, K. Sugawara, M. Yoshida, M. Kato and T. Suzuki, Bull. Chem. Soc. Jpn., 2019, 92, 1211–1217 CrossRef CAS.
- T. Suzuki, T. Fukushima, T. Miyashi and T. Tsuji, Angew. Chem., Int. Ed. Engl., 1997, 36, 2495–2497 CrossRef CAS.
- P. U. Biedermann, J. J. Stezowski and I. Agranat, Chem. – Eur. J., 2006, 12, 3345–3354 CrossRef CAS PubMed.
- H. Takezawa, T. Murase and M. Fujita, J. Am. Chem. Soc., 2012, 134, 17420–17423 CrossRef CAS PubMed.
- Y. Wang, Y. Ma, K. Ogumi, B. Wang, T. Nakagawa, Y. Fu and Y. Matsuo, Commun. Chem., 2020, 3, 93 CrossRef CAS PubMed.
- K. Wallenfels and W. Draber, Justus Liebigs Ann. Chem., 1963, 667, 55–71 CrossRef CAS.
- Y. Yamashita, T. Suzuki, G. Saito and T. Mukai, Chem. Lett., 1986, 715–718 CrossRef CAS.
- T. Suzuki, Y. Umezawa, Y. Sakano, H. Tamaoki, R. Katoono and K. Fujiwara, Chem. Lett., 2015, 44, 905–907 CrossRef CAS.
- T. Suzuki, Y. Ishigaki, K. Sugawara, Y. Umezawa, R. Katoono, A. Shimoyama, Y. Manabe, K. Fukase and T. Fukushima, Tetrahedron, 2018, 74, 2239–2244 CrossRef CAS.
- S. Pola, C.-H. Kuo, W.-T. Peng, M. M. Islam, I. Chao and Y.-T. Tao, Chem. Mater., 2012, 24, 2566–2571 CrossRef CAS.
- K. Sun, K. Sugawara, A. Lyalin, Y. Ishigaki, K. Uosaki, T. Taketsugu, T. Suzuki and S. Kawai, Angew. Chem., Int. Ed., 2021, 60, 9427–9432 CrossRef CAS PubMed.
- Q. Qi, J. Zhang, B. Xu, B. Li, S. X.-A. Zhang and W. Tian, J. Phys. Chem. C, 2013, 117, 24997–25003 CrossRef CAS.
- T. Machida, R. Taniguchi, T. Oura, K. Sada and K. Kokado, Chem. Commun., 2017, 53, 2378–2381 RSC.
- Y. Hong, J. W. Y. Lam and B. Z. Tang, Chem. Soc. Rev., 2011, 40, 5361–5388 RSC.
- X. Gu, J. Yao, G. Zhang, Y. Yan, C. Zhang, Q. Peng, Q. Liao, Y. Wu, Z. Xu, Y. Zhao, H. Fu and D. Zhang, Adv. Funct. Mater., 2012, 22, 4862–4872 CrossRef CAS.
- T. Nishiuchi, S. Aibara, T. Yamakado, R. Kimura, S. Saito, H. Sato and T. Kubo, Chem. – Eur. J., 2022, 28, e202200286 CAS.
- Y. Chandrasekaran, G. K. Dutta, R. B. Kanth and S. Patil, Dyes Pigm., 2009, 83, 162–167 CrossRef CAS.
- Q. Li, C. Yan, J. Zhang, Z. Guo and W.-H. Zhu, Dyes Pigm., 2019, 162, 802–807 CrossRef CAS.
- T. Mutai, H. Shono, Y. Shigemitsu and K. Araki, CrystEngComm, 2014, 16, 3890–3895 RSC.
- D. Kitagawa, T. Nakahama, K. Mutoh, Y. Kobayashi, J. Abe, H. Sotome, S. Ito, H. Miyasaka and S. Kobatake, Dyes Pigm., 2017, 139, 233–238 CrossRef CAS.
- K. Zheng, F. Ni, Z. Chen, C. Zhong and C. Yang, Angew. Chem., Int. Ed., 2020, 59, 9972–9976 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2176740–2176766. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2qm01199a |
|
| This journal is © the Partner Organisations 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *b,
Yoshio
Yano
b,
Takanori
Suzuki
*b,
Yoshio
Yano
b,
Takanori
Suzuki