
Bulk depolymerization of graft polymers based on trans-cyclobutane-fused cyclooctene†
Zeyu
Wang
 and
Junpeng
Wang
*
and
Junpeng
Wang
*
School of Polymer Science and Polymer Engineering, The University of Akron, Akron, Ohio 44325, USA. E-mail: jwang6@uakron.edu
First published on 16th October 2023
Abstract
Polymers capable of undergoing facile and selective depolymerization are highly desired for achieving a fully circular polymer economy. To advance the design of next-generation chemically recyclable polymers, it is essential to understand how the structure and architecture of polymers influence the thermodynamics of depolymerization. In this study, we investigate the structure-depolymerizability relationship of a series of graft polymers with varied sidechain lengths and grafting densities. The precision graft polymers are prepared through ring-opening metathesis polymerization of highly reactive trans-cyclooctene macromonomers and can be depolymerized into the more stable cis-cyclooctene form. Increasing side-chain length or grafting density is found to promote depolymerization. Ball-milling-assisted depolymerization is also pursued but results in significant chain scissions and low monomer yield. The findings here can aid the development of new depolymerizable polymers with useful properties.
Introduction
The commercial success of the world's first synthetic plastic, Bakelite, marked the introduction of the Polymer Age and inspired rapid development of other synthetic polymers. Owing to their high specific strength (strength divided by density), polymers are employed in nearly every facet of modern living, including clothing, packaging, and automobiles. A significant proportion of polymer products are meant for single use and are subsequently discarded. The improper collection and low recycling rates of post-consumer polymer waste have created significant environmental challenges. As of 2015, an estimated 79% of this waste was found to accumulate in landfills or the natural environment.1 Contamination and thermomechanical degradation during mechanical recycling result in inferior recycled products with compromised color, reduced food safety, and degraded mechanical properties.2Chemical recycling, by means of depolymerization, serves as an ideal complement to traditional mechanical recycling, particularly for tackling mixed plastics, crosslinked polymers, and polymer composites.3,4 However, the unselective thermodynamic processes during the depolymerization of conventional vinyl polymers often result in a mixture of low-value products, along with a small fraction of the desired monomers.5–8 A promising solution is to design polymers capable of selectively depolymerizing into the monomeric constituents, which can then be used for constructing new polymers with equivalent properties. The thermodynamics of depolymerization is expressed by the Gibbs free energy (ΔGdp = ΔHdp – TΔSdp, where ΔHdp and ΔSdp are the enthalpy change and entropy change during depolymerization, respectively). In a typical depolymerization reaction, both ΔHdp and ΔSdp are positive, and it is the relative magnitude of enthalpic and entropic factors that determine how favorable the reaction is.
There has been a growing number of examples that utilize ring-opening polymerization of cyclic monomers to develop depolymerizable polymers.9–15 The size, geometry, stereochemistry, and substituents of these cyclic monomers offer vast opportunities to tune the thermodynamics of (de)polymerization. While the relationship between monomer structure, ring strain, and (de)polymerization thermodynamics is becoming better understood, there is a lack of attention regarding the effect of polymer architecture on (de)polymerization thermodynamics. Additionally, most of the depolymerization studies were conducted in dilute solutions to drive the formation of monomers, but the extensive use of solvents limits practicality.
A variety of cyclic monomers have been utilized to access depolymerizable polymers, including lactones,12,16 cyclic carbonates,15,17–19 cyclic acetals,14,20 and cycloalkenes.9,11,13,21,22 Amongst all these types, cycloalkenes form polymers without heteroatoms along the backbone, providing greater resistance to hydrolytic degradation. The polymerization of cycloalkenes is often achieved through ring-opening metathesis polymerization (ROMP) using Grubbs catalysts.23–26 The high reactivity and functional group tolerance make such systems convenient for constructing complex and sophisticated polymers.27–29
We recently reported the synthesis of depolymerizable graft polymers using controlled grafting-through polymerization.30 The seemingly contradictory demands between grafting-through (which requires highly exergonic polymerization) and depolymerization was reconciled in a system featuring trans-cyclobutane-fused cyclooctene (tCBCO, Fig. 1a): The trans-cyclobutane fused ring reduces the ring strain energy of the cis-cyclooctene monomer, i.e., tCBcCO, allowing the corresponding polymer to be depolymerized.21cis-to-trans isomerization of alkene increases ring strain energy by >10 kcal mol−1, and the corresponding trans-cyclooctene monomer, i.e., tCBtCO, can undergo controlled polymerization, at a concentration as low as 5 mM.31 Regardless of whether the polymer is made from tCBcCO or tCBtCO, it depolymerizes into tCBcCO, which is the therodynamically more stable form. The high reactivity of tCBtCO enables grafting-through polymerization of tCBtCO-based macromonomer (MM), allowing the precise synthesis of depolymerizable graft polymers.
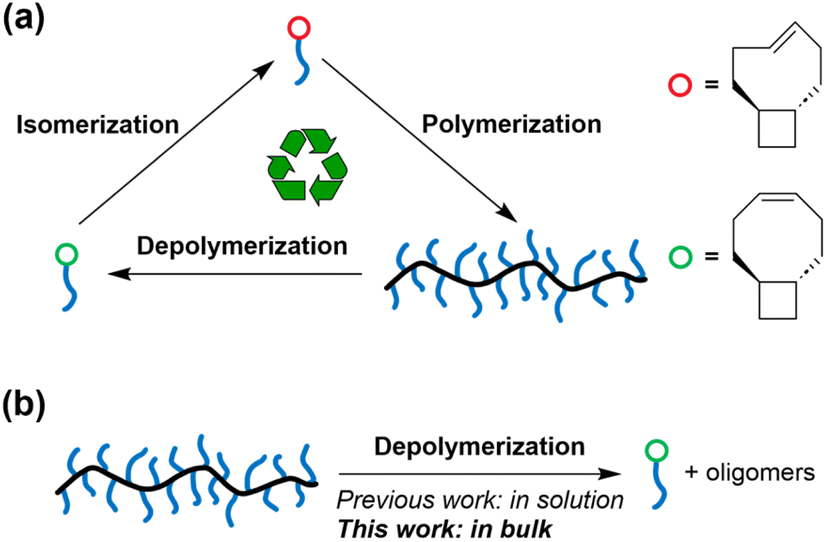 | ||
| Fig. 1 (a) Isomerization of tCBcCO-MM into tCBtCO-MM increases the driving force of polymerization, and the resulting graft polymer can be depolymerized back to tCBcCO-MM. (b) Depolymerization of graft polymers in bulk is enabled by the dilution by side chains. | ||
We envisioned that the side chains would serve as a diluent for the backbone, which favors depolymerization. In addition, steric repulsion between the sidechains can also drive the depolymerization. Motivated by these hypotheses, we tested the feasibility of solvent-free depolymerization of tCBCO-based graft polymers (Fig. 1b) and investigated the impact of side chain length and grafting density on the extent of depolymerization. In addition, ball milling-assisted depolymerization was also explored.
Results and discussion
Three tCBtCO-based MMs with different side-chain numbers and lengths were synthesized from esterification between either a tCBtCO alcohol or a tCBtCO-diol and a carboxyl end-functionalized poly(ethylene glycol) (PEG-CO2H) of 2 kDa or 5 kDa molecular weight to afford MM-2k, MM-2-2k, and MM-5k (Fig. 2; see ESI† for synthetic details). MM-2k and MM-5k were synthesized according to our previously report procedure.30 Because of the steric demands in attaching two 2 kDa PEG-CO2H to one tCBtCO-diol to form MM-2-2k, excess amount of PEG-CO2H (3.33 equiv. to tCBtCO diol, or 1.67 equiv. to –OH) was used to ensure full consumption of the alcohol functionalities on tCBtCO. Two peaks were observed in the SEC trace, the Mn of which measured by MALS was 4.6 and 2.5 kDa, respectively (Fig. 2 and Fig. S11†). By integrating the area of each peak, the ratio of the 4.6 kDa peak to the 2.5 kDa peak was 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2, which is consistent with quantitative conversion of hydroxyl groups to PEG esters, leaving 40 mol% of the starting PEG-CO2H still unreacted or as the N-acylurea derivative from the EDC reagent.
:2, which is consistent with quantitative conversion of hydroxyl groups to PEG esters, leaving 40 mol% of the starting PEG-CO2H still unreacted or as the N-acylurea derivative from the EDC reagent.
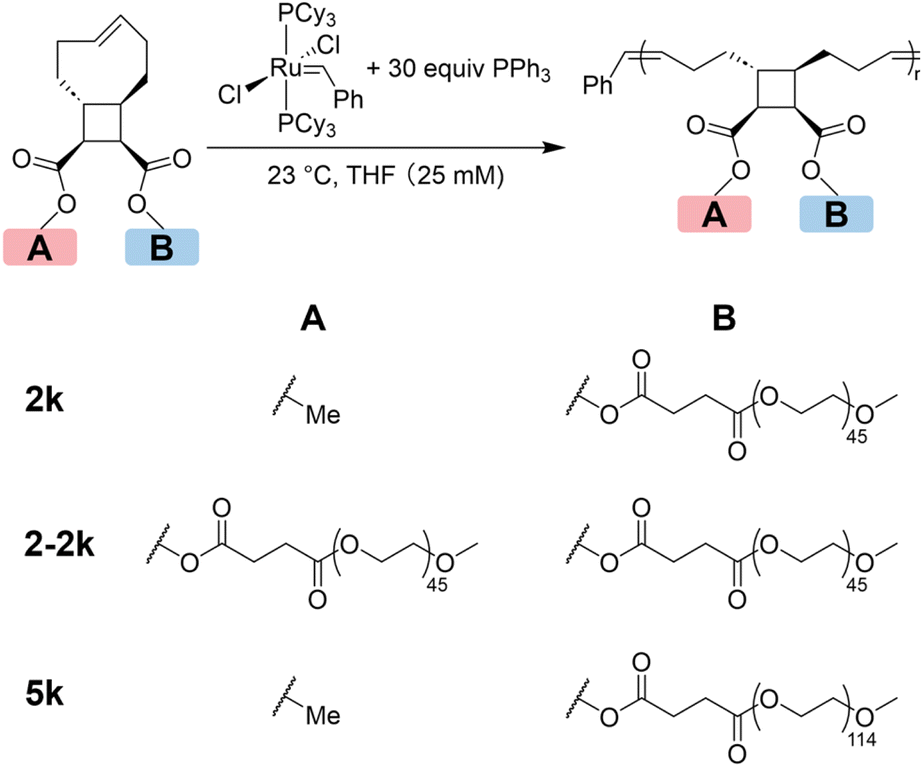 | ||
| Fig. 2 Synthesis of graft polymers with different grafting densities via ring-opening metathesis polymerization. | ||
Due to the difficulty in separating MM-2-2k and the excess PEG-CO2H, the mixture was directly used for polymerization. ROMP of MM-2-2k generated a new peak at shorter retention time, corresponding to the graft polymer g-2-2k, which should contain two 2k PEG side chains in each repeating unit. The relative area of the 4.6 kDa peak declined from 60% to 7%, while that of the 2.5 kDa species remained unchanged after polymerization, which is consistent with the presumption that the 2.5 kDa species was not functionalized with tCO. Polymers g-2k and g-5k were prepared by ROMP of MM-2k and MM-5k, respectively. Any remaining linear PEG was removed by dialysis. The SEC traces of 2k, 2-2k and 5k MMs before and after ROMP are shown in Fig. 3. The information of the three graft polymers is summarized in Table 1; narrow molecular weight distributions and good agreement with the theoretical molecular weights were obtained for all graft polymers.
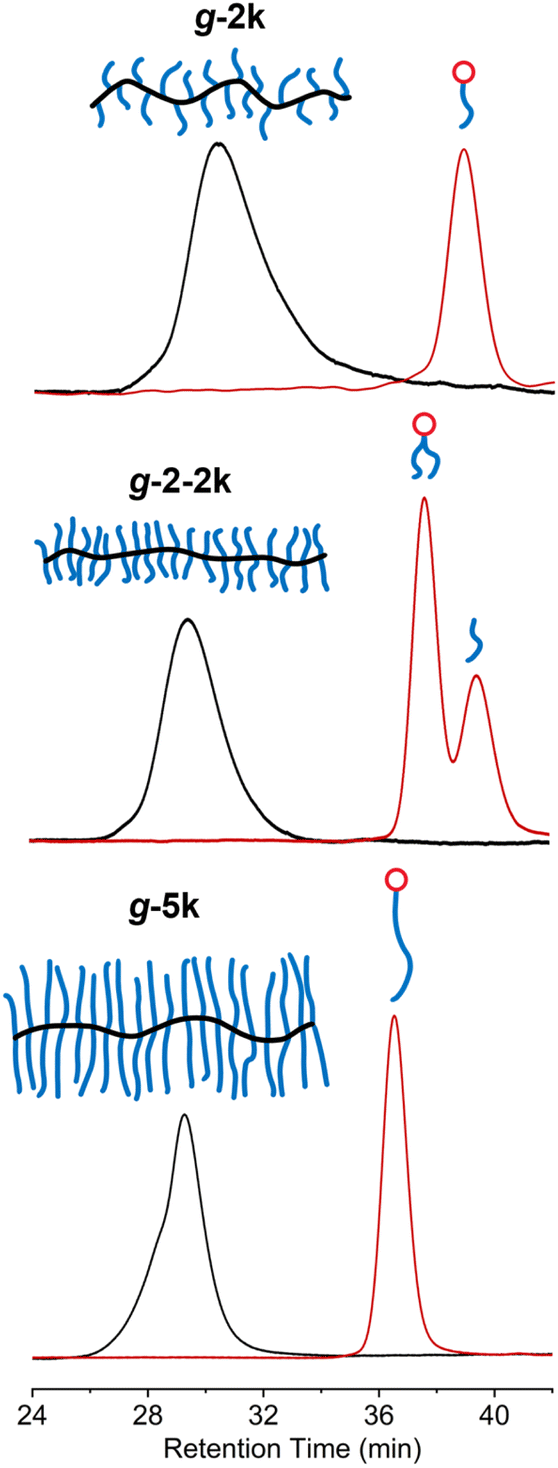 | ||
| Fig. 3 SEC analysis of graft polymers with varying grafting densities synthesized from different macromonomers. | ||
| Sample | Conversiona (%) |
M
n,theob (kDa) |
M
nc (kDa) |
Đc |
|---|---|---|---|---|
| a Obtained from SEC analysis of the relative peak areas of MM and graft polymer. b Calculated from the product of SEC conversion and feed ratio [MM]0/[G2]0. c Measured by SEC-RI/MALS. | ||||
| g-2k | 97 | 229 | 206 | 1.16 |
| g-2-2k | 88 | 393 | 417 | 1.11 |
| g-5k | 91 | 487 | 510 | 1.16 |
We first monitored the depolymerization of g-2-2k in the melt state to determine the time needed for the depolymerization to reach a thermodynamic equilibrium. A solution of Grubbs second-generation catalyst (G2) in DCM (10 μL, 5 mol% to the backbone olefin) was added to the bottom of a 1 mL vial and was let dry under nitrogen flow. The solid g-2-2k was transferred onto the dried G2 and was melted at 100 °C to form a homogeneous reddish-orange liquid (Fig. S1†). The amount of g-2-2k decreased by 73% after 1 day of depolymerization, while the fractions of tCBcCO-MM and oligomers increased to 57% and 16%, respectively (Fig. 4a and b). Thereafter, the depolymerization slowed down and eventually reached an equilibrium (59% MM, 32% oligomers, and 9% graft polymer) after 1 week. The conversion to monomer in bulk was not as high as that in solution, albeit the bulk depolymerization was done at a higher temperature, because of the much higher olefin concentration in bulk ([olefin] = ∼0.2–0.5 M in bulk, and 0.01 M in solution). Due to the slow diffusion of the polymer and the catalyst in the highly viscous melt state, the depolymerization rate in bulk was much slower than that in the solution state, where the depolymerization was complete within 2 h at 50 °C in toluene.30 During the course of depolymerization, the formation of monomer plateaued quickly while oligomers were continuously generated until a thermodynamic equilibrium was reached. The profile of molecular weight change over the extent of depolymerization (Fig. 4c) resembles that of the solution study in that no abrupt drop in Mn at the beginning stage, which indicates a end-to-end unzipping mechanism, owing to the difficulty for a G2 molecule to access the congested internal olefins on the backbone of the graft polymer.28,30
 | ||
| Fig. 4 Monitoring bulk depolymerization of g-2-2k for 1 week in the presence of G2 ([G2]0/[olefin]0 = 5/100) at 100 °C. (a) Evolution of SEC traces over the course of 1-week depolymerization in bulk. The number of repeating units is labeled on the corresponding peak. (b) The change in fractions of graft polymer, oligomers and macromonomer during the depolymerization. (c) The profile of normalized Mn (Mn/Mn,0) and dispersity (Đ) over the extent of depolymerization. | ||
To investigate the effect of grafting density and side-chain length on the depolymerization of graft polymers, g-2k, g-2-2k and g-5k were subjected to the same depolymerization conditions (5 mol% G2, 100 °C, and for 7 days) and the resulting depolymerization products were analyzed by SEC (Fig. 5). To demonstrate the stability of G2 under this condition, we made a mixture of G2 and PEG 2k (2 wt%) and heated it at 100 °C under nitrogen for 7 days. Note that here an unfunctionalized PEG 2k was used to simplify the NMR characterization. Comparison between the 1H NMR spectra before and after the heat treatment showed that one third of G2 remained intact (Fig. S2†). To confirm that equilibrium had been reached after heating the polymer in the presence of 5 mol% G2 at 100 °C for 7 days, after the mixture was treated under this condition, another 5 mol% G2 was added to the system, and the mixture was heated at 100 °C for another 7 days. The resulting mixture was subjected to 1H NMR and SEC characterization, which was nearly identical to the characterization of the mixture right after the first 7 days of heating (Fig. S3†), indicating that equilibrium had been established before adding the second portion of G2 and heating. As discussed previously,30 the MM/oligomer should be cyclic, as a result of ring-closing metathesis that produced eight-membered rings or larger macrocycles with n PEG tail(s), where n is the number of repeating units of the MM/oligomer, which was determined by SEC-RI/MALS. The depolymerization of g-2k yielded 46.2% of tCBcCO-MM (n = 1), and the rest 53.8% should be oligomeric and polymeric species (n ≥ 2). When the grafting density was doubled by having two 2k PEG chains per repeating units, the graft polymer g-2-2k depolymerized into a higher amount of MM, 68.4%, 21.5% oligomers (mostly as dimer as seen in the SEC trace), and about a 10.1% graft polymer fraction that was 58.6% of the initial Mn. When the side-chain length was increased from 2k to 5k, there was 68.0% monomer generated after the depolymerization of g-5k, with the rest being oligomers. In comparison to other graft polymers, no residual graft polymer was observed after depolymerization of g-5k. Although g-5k contains a higher mass in each repeat unit than g-2-2k does, the conversion to tCBcCO-MM was about the same in the two cases. This could be attributed to a stronger repulsion effect from two 2k PEG chains tethered to one backbone repeating unit than that from one 5k PEG chain. Compared to our previous work where depolymerization was conducted in solutions and almost quantitative yield of MM was obtained,30 the lower conversion (<70%) of monomer in the bulk depolymerization can be attributed to less entropic gain as a result of increased concentration. It should be noted that the addition of 5 mol% G2 (∼2 wt% for g-2k and ∼1 wt% for g-2-2k and g-5k) could add cost and introduce toxicity to the depolymerized materials. The toxicity concern could be alleviated by purification through precipitation into diethyl ether, manifested by a disappearance of the distinct brown colour.
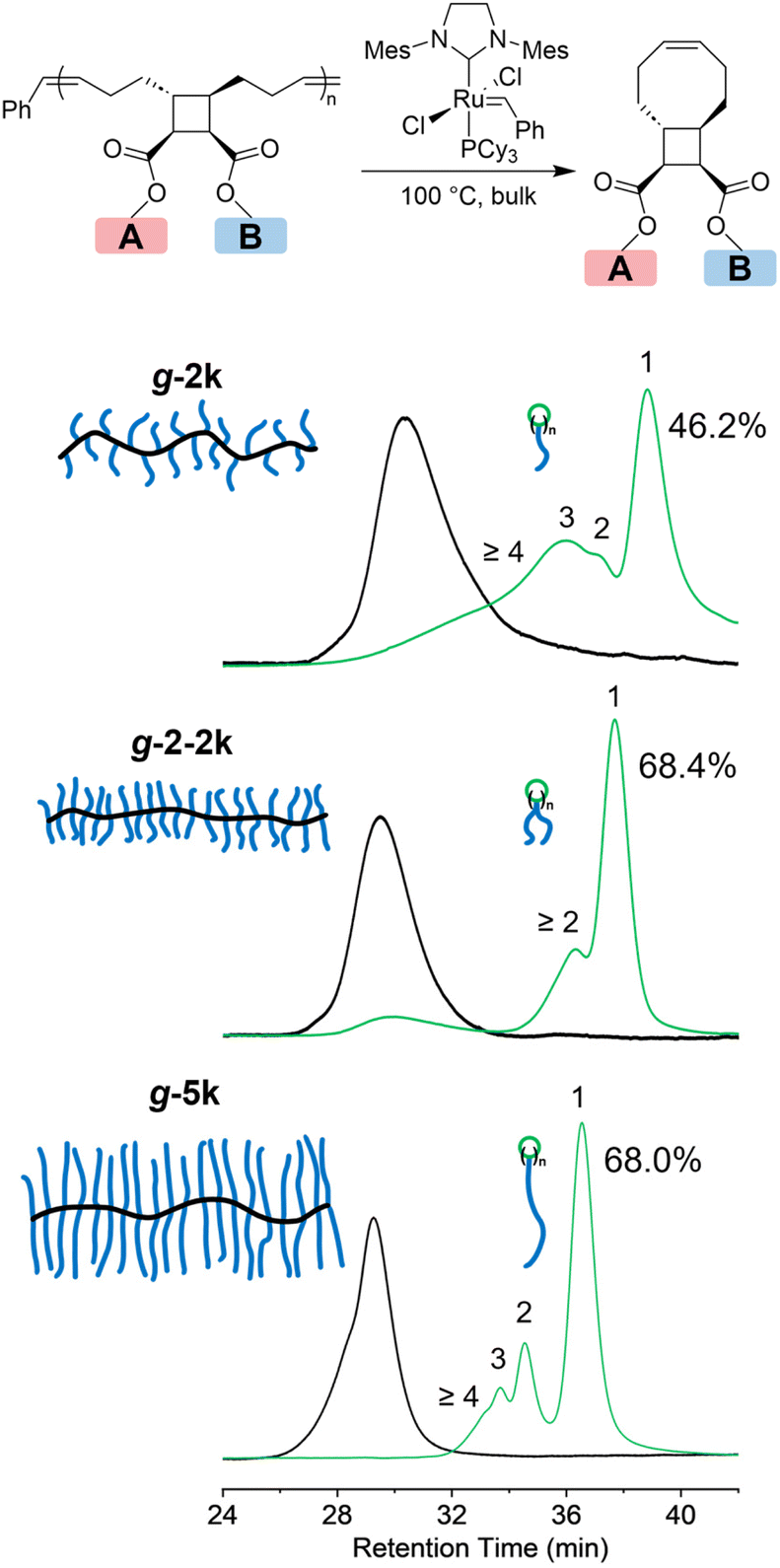 | ||
| Fig. 5 Bulk depolymerization of graft polymers with different grafting densities. The number of repeating units is labeled on the corresponding peak. The percentages denote the conversion of depolymerization product cis-cyclooctene macromonomer. | ||
Ball milling has been demonstrated for its efficacy in depolymerizing a variety of polymers, including poly(α-methyl styrene),32 polystyrene,33 and poly(ethylene terephthalate).34,35 Motivated by these successes, we investigated the depolymerization of tCBCO-based graft polymer under ball milling. The g-2-2k was charged with 5 mol% G2 and subjected to a ball milling at room temperature. After a milling cycle of on–off–on–off–on with 10 min intervals (50 min in total: 30 min milling and 20 min resting), the resulting mixture was quenched with EVE and analyzed by SEC to obtain the molecular weight information. The peak corresponding to the graft polymer shifted to a longer retention time, corresponding to a reduction in Mn from 393 kDa to 50 kDa (Fig. 6). However, the yield of depolymerization in this case was low, with only 1.5% monomer and 4.4% dimer generated. The large Mn decrease, combined with low depolymerization yield, can be attributed to a combination of extensive mechanochemical backbone scissions on the extended brush backbone36 and the limited accessibility of G2 to backbone or terminal olefins. These results are preliminary in nature, and we anticipate that optimization of the conditions will lead to higher conversions.
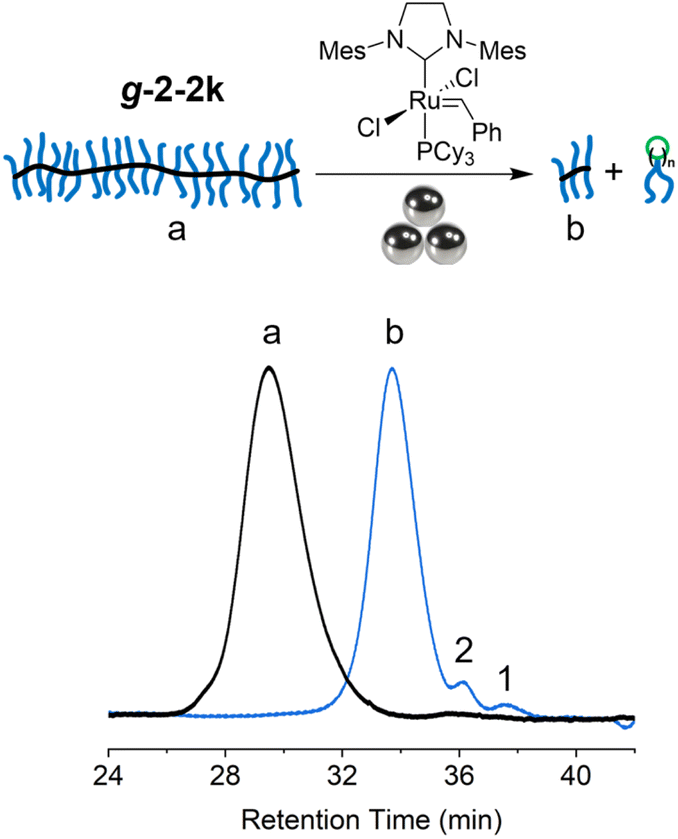 | ||
| Fig. 6 Depolymerization of g-2-2k in the presence of G2 via ball milling. | ||
Conclusions
We investigated the bulk depolymerization of tCBCO-based graft polymers, leveraging the dilution effect of side chains. The extent of depolymerization increased with longer side chain or higher grafting density, as a result of increased entropic gain. Although ball milling has been shown as an effective tool for depolymerizing poly(α-methyl)styrene, it was found to significantly reduce the Mn of our graft polymer while yielding little monomer. These results here offer valuable insights into designing next-generation highly depolymerizable polymers from a polymer conformational perspective.Author contributions
Z. W. and J. W. conceived and designed the experiments. Z. W. conducted the synthesis, polymerization, and depolymerization studies. Z. W. and J. W. wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Science Foundation under Grant No. DMR-2042494 and the American Chemical Society Petroleum Research Fund under Grant No. 65048-DNI7. We thank Dr James M. Eagan for glovebox access. We thank The Ohio Board of Regents and The National Science Foundation (CHE-0341701 and DMR-0414599) for funds used to purchase the NMR instrument used in this work.References
- R. Geyer, J. R. Jambeck and K. L. Law, Sci. Adv., 2017, 3, e1700782 CrossRef PubMed.
- Z. O. Schyns and M. P. Shaver, Macromol. Rapid Commun., 2021, 42, 2000415 CrossRef CAS PubMed.
- M. Zeller, N. Netsch, F. Richter, H. Leibold and D. Stapf, Chem. Ing. Tech., 2021, 93, 1763–1770 CrossRef CAS.
- R. P. Lee, M. Tschoepe and R. Voss, Waste Manage., 2021, 125, 280–292 CrossRef CAS PubMed.
- L. S. Diaz-Silvarrey, K. Zhang and A. N. Phan, Green Chem., 2018, 20, 1813–1823 RSC.
- R. R. Guddeti, R. Knight and E. D. Grossmann, Ind. Eng. Chem. Res., 2000, 39, 1171–1176 CrossRef CAS.
- R. Miranda, J. Yang, C. Roy and C. Vasile, Polym. Degrad. Stab., 2001, 72, 469–491 CrossRef CAS.
- J. Yu, L. Sun, C. Ma, Y. Qiao and H. Yao, Waste Manage., 2016, 48, 300–314 CrossRef CAS PubMed.
- J. Zhou, D. Sathe and J. Wang, J. Am. Chem. Soc., 2022, 144, 928–934 CrossRef CAS PubMed.
- D. Sathe, H. Chen and J. Wang, Macromol. Rapid Commun., 2023, 44, 2200304 CrossRef CAS PubMed.
- W. J. Neary and J. G. Kennemur, Macromolecules, 2017, 50, 4935–4941 CrossRef CAS.
- L. Zhou, Z. Zhang, C. Shi, M. Scoti, D. K. Barange, R. R. Gowda and E. Y.-X. Chen, Science, 2023, 380, 64–69 CrossRef CAS.
- C. Shi, R. W. Clarke, M. L. McGraw and E. Y.-X. Chen, J. Am. Chem. Soc., 2022, 144, 2264–2275 CrossRef CAS PubMed.
- B. A. Abel, R. L. Snyder and G. W. Coates, Science, 2021, 373, 783–789 CrossRef CAS PubMed.
- W. C. Ellis, Y. Jung, M. Mulzer, R. Di Girolamo, E. B. Lobkovsky and G. W. Coates, Chem. Sci., 2014, 5, 4004–4011 RSC.
- L. Cederholm, J. Wohlert, P. Olsen, M. Hakkarainen and K. Odelius, Angew. Chem., Int. Ed., 2022, 61, e2022045 Search PubMed.
- Y. Liu, H. Zhou, J. Z. Guo, W. M. Ren and X. B. Lu, Angew. Chem., Int. Ed., 2017, 56, 4862–4866 CrossRef CAS.
- D. J. Saxon, E. A. Gormong, V. M. Shah and T. M. Reineke, ACS Macro Lett., 2020, 10, 98–103 CrossRef.
- W. Zhang, J. Dai, Y.-C. Wu, J.-X. Chen, S.-Y. Shan, Z. Cai and J.-B. Zhu, ACS Macro Lett., 2022, 11, 173–178 CrossRef CAS PubMed.
- H. G. Hester, B. A. Abel and G. W. Coates, J. Am. Chem. Soc., 2023, 145, 8800–8804 CrossRef CAS PubMed.
- D. Sathe, J. Zhou, H. Chen, H.-W. Su, W. Xie, T.-G. Hsu, B. R. Schrage, T. Smith, C. J. Ziegler and J. Wang, Nat. Chem., 2021, 13, 743–750 CrossRef CAS PubMed.
- J. D. Feist and Y. Xia, J. Am. Chem. Soc., 2019, 142, 1186–1189 CrossRef PubMed.
- R. Walker, R. M. Conrad and R. H. Grubbs, Macromolecules, 2009, 42, 599–605 CrossRef CAS PubMed.
- M. Yasir, P. Liu, I. K. Tennie and A. F. Kilbinger, Nat. Chem., 2019, 11, 488–494 CrossRef CAS PubMed.
- Y. Xia, B. D. Olsen, J. A. Kornfield and R. H. Grubbs, J. Am. Chem. Soc., 2009, 131, 18525–18532 CrossRef CAS PubMed.
- T. L. Choi and R. H. Grubbs, Angew. Chem., Int. Ed., 2003, 42, 1743–1746 CrossRef CAS PubMed.
- G. I. Peterson, K.-T. Bang and T.-L. Choi, J. Am. Chem. Soc., 2018, 140, 8599–8608 CrossRef CAS PubMed.
- W. J. Neary, T. A. Isais and J. G. Kennemur, J. Am. Chem. Soc., 2019, 141, 14220–14229 CrossRef CAS PubMed.
- J. L. Self, C. S. Sample, A. E. Levi, K. Li, R. Xie, J. R. De Alaniz and C. M. Bates, J. Am. Chem. Soc., 2020, 142, 7567–7573 CrossRef CAS.
- Z. Wang, S. Yoon and J. Wang, Macromolecules, 2022, 55, 9249–9256 CrossRef CAS.
- H. L. Chen, Z. Shi, T. G. Hsu and J. P. Wang, Angew. Chem., Int. Ed., 2021, 60, 25493–25498 CrossRef CAS PubMed.
- E. Jung, D. Yim, H. Kim, G. I. Peterson and T. L. Choi, J. Polym. Sci., 2023, 61, 553–560 CrossRef CAS.
- V. P. Balema, I. Z. Hlova, S. L. Carnahan, M. Seyedi, O. Dolotko, A. J. Rossini and I. Luzinov, New J. Chem., 2021, 45, 2935–2938 RSC.
- V. Štrukil, ChemSusChem, 2021, 14, 330–338 CrossRef PubMed.
- A. W. Tricker, A. A. Osibo, Y. C. Chang, J. X. Kang, A. Ganesan, E. Anglou, F. Boukouvala, S. Nair, C. W. Jones and C. Sievers, ACS Sustainable Chem. Eng., 2022, 10, 11338–11347 CrossRef CAS.
- J. Noh, G. I. Peterson and T. L. Choi, Angew. Chem., Int. Ed., 2021, 60, 18651–18659 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3py00812f |
| This journal is © The Royal Society of Chemistry 2023 |