
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of a light-responsive platinum curcumin complex, chemical and biological investigations and delivery to tumor cells by means of polymeric nanoparticles
Viviana
Vergaro
 *ab,
Maria Michela
Dell'Anna
c,
Hamid R.
Shahsavari
cd,
Francesca
Baldassarre
ab,
Danilo
Migoni
a,
Piero
Mastrorilli
c,
Francesco Paolo
Fanizzi
a and
Giuseppe
Ciccarella
*ab
*ab,
Maria Michela
Dell'Anna
c,
Hamid R.
Shahsavari
cd,
Francesca
Baldassarre
ab,
Danilo
Migoni
a,
Piero
Mastrorilli
c,
Francesco Paolo
Fanizzi
a and
Giuseppe
Ciccarella
*ab
aBiological and Environmental Sciences Department, UdR INSTM of Lecce University of Salento, Via Monteroni, 73100 Lecce, Italy. E-mail: viviana.vergaro@unisalento.it; giuseppe.ciccarella@unisalento.it
bInstitute of Nanotechnology, CNR NANOTEC, Consiglio Nazionale delle Ricerche, Via Monteroni, 73100 Lecce, Italy
cDICATECh, Politecnico di Bari, via Orabona, 4, 70125 Bari, Italy
dDepartment of Chemistry, Institute for Advanced Studies in Basic Sciences (IASBS), Zanjan, 45137-66731, Iran
First published on 30th August 2023
Abstract
Platinum-based anticancer drugs are common in chemotherapy, but problems such as systemic toxicity and acquired resistance of some tumors hamper their clinical applications and therapeutic efficacy. It is necessary to synthesize Pt-based drugs and explore strategies to reduce side effects and improve pharmacokinetic profiles. Photo-responsive chemotherapeutics have emerged as an alternative strategy against several cancers, as photoactivation offers spatial selectivity and fewer side effects. Here, we combine chemical synthesis and nanotechnology to create a multifunctional platinum drug delivery system based on the novel metal complex [Pt(ppy)(curc)] (ppy = deprotonated 2-phenylpyridine, curc = deprotonated curcumin)] embodying the naturally occurring bioactive molecule, curcumin. The ultrasonication method coupled with the layer-by-layer technology was employed to produce nanocolloids, which demonstrated a good biocompatibility, higher solubility in aqueous solution, stability, large drug loading, and good biological activity in comparison with the free drug. In vitro release experiments revealed that the polymeric nanoformulation is relatively stable under physiological conditions (pH = 7.4 and 37 °C) but sensitive to acidic environments (pH = 5.6 and 37 °C) which would trigger the release of the loaded drug. Our approach modifies the bioavailability of this Pt-based drug increasing its therapeutic action in terms of both cytotoxic and anti-metastasis effects.
Introduction
Cisplatin, cis-[PtCl2(NH3)2], is a famous anticancer chemotherapeutic agent found by Barnett Rosenberg and his colleagues in 1969.1 Since it was first approved by the Food and Drug Administration, FDA, in 1978, cisplatin has been widely used in clinics to treat a wide range of cancers, including testicular, bladder, ovarian, head and neck cancers.2–5 The major limitations of chemotherapeutic agents are often difficulties with solubility, formulation, biodistribution and ability to cross cell membranes. These problems have prompted the exploration of various scaffolds to act as vectors for targeted delivery of platinum-based complexes. Thus, it is of great significance to develop new platinum-based drugs by investigating nanoformulations able to improve the low hydrophilicity of these molecules, cellular uptake, and antitumor efficacy.6,7The interaction of metal ions with coordinating ligands is useful to enhance biological properties, resulting in improved chemical stability, increased pharmacological activity, and reduction of side effects.8–11 Metal complexes based on naturally occurring bioactive molecules are now considered promising drugs and many studies are dedicated to the investigation of their biological activities.
Within bioactive metal chelating ligands, particular interest has been devoted to curcumin, [1,7-bis(4-hydroxy-3-methoxyphenyl)-1,6-heptane-3,5-dione], a naturally occurring pigmented component of the spice turmeric, which is well-known for its therapeutic properties.12,13
Curcumin is a lipophilic polyphenol and poorly water-soluble and in vitro studies demonstrated its very low bioavailability and photodegradation, features that limit its use in the biomedical field, despite its promising biological preclinical and clinical effects.14–17 The latter effects concern its (i) antiproliferative and antimetastatic properties via mitochondrial pathways involving caspase and the Bcl-2 family of proteins; (ii) the inhibition of angiogenesis by interfering with the activity of NF-kB; and (iii) the inhibition of carcinogenesis and tumor growth through free-radical scavenging properties.18
Curcumin exists in keto–enol forms, and the equilibrium depends on the nature of the solvent,19 but in order to improve the pharmacokinetic profile, it can be stabilized through its encapsulation in nanocarriers20–25 or through complexation with a transition metal ion.26–30
In previous studies, curcumin-based metal complexes have been widely probed for their anticancer properties, showing improved stability, better bioavailability, and greater cytotoxicity in their effectiveness as anticancer agents with respect to both pure curcumin and the combined treatment of cisplatin and curcumin.31–37 An interesting aspect is that metal bonded curcumin confers photochemical properties to the complex. This makes curcumin suitable as a ligand-photosensitizer in photoactivated chemotherapeutic studies. Phototherapy represents a new frontier for cancer treatment to overcome the limitations of conventional chemotherapy and, certainly, a curcumin metal-based drug shows potent cytotoxicity and photocytotoxicity in visible light with low toxicity under dark conditions.38–43 Selective photo-release of the O,O-donor therapeutically active curcumin as a β-diketonate ligand along with the active DNA crosslinking platinum(II) species makes this conjugate dually active, showing an improvement in antitumor efficacy based on the synergistic effects and enhancement of anti-metastasis activity. Thus, in the present study, aiming at enhancing the potential phototoxicity of the system and formulating a cisplatin-like platinum complex containing the anion of curcumin as the leaving group, we exploited the use of the photosensitizer chelating C^N ligand 2-phenylpyridinate (ppy), as the carrier ligand. The obtained platinum compound belongs to the category of cycloplatinated(II) complexes, that have attracted much attention from the scientific community for their interesting biological activities attributed to C^N ligands.44,45
The advantages of using the Pt(ppy) backbone for the novel complex lie in its lipophilic character useful for passing the lipidic cellular membrane, in the possibility of the ppy ligand having hydrophobic interactions with DNA bases,46 and in its photoluminescence properties.47,48
In addition, loading light-stimuli small molecule drugs into delivery systems, such as polymeric nanoparticles or micelles, liposomes or mesoporous silica, allows drug accumulation and therapeutic effects to be increased.49,50 Natural biopolymers such as pectin, alginate and chitosan have recently found wide applications in food and pharmaceutical fields.51 The presence of functional groups in biopolymer structures improves the encapsulation of bioactive compounds and drugs as well as the formation of a cross-linked dense network after encapsulation.52 Polymeric nanoparticles prepared with biodegradable and biocompatible materials have been used as tools for metal-based drug delivery to improve drug stability and efficiency, decreasing the frequency of administration of the drug while maintaining a steady and effective concentration at the target site.
Recently, pectin-based delivery systems have been registered for colon-specific delivery purposes.53 Pectin is a natural, anionic polysaccharide obtained from the cell walls of fruits such as apples, oranges and pears.54 Although it is resistant to the enzymes acting in the upper gastro-intestinal tract, it undergoes complete degradation by colonic bacterial enzymes.55
Chitosan is another linear polysaccharide with cationic properties which is obtained from chitin, a major component of the shells of shrimp, lobsters, and crabs. Pectin and chitosan are known to combine to form a polyelectrolyte complex which can be used to encapsulate bioactive molecules and drugs.56
Following our work in the field of bioactive Pt(II) complexes,31,32,49,50 we prepared and studied a Pt(II) complex of formula [Pt(ppy)(curc)] (ppy = deprotonated 2-phenylpyridine and curc = deprotonated curcumin) with the aim to extend studies on the potential application of transition metal complexes as a pharmacological tool. The ultrasonication method coupled with the layer-by-layer technology was used to prepare chitosan and pectin polymeric nanocolloids to encapsulate [Pt(ppy)(curc)] affording the material denoted as Pt(ppy)(curc)-NCs.
Nanocolloids are stable in biological fluids; however, upon reaching the intracellular compartment, degradation of biopolymers could occur, leading to changes in the physical structure of nanoparticles and then to the rapid release of encapsulated drugs. The light activation of [Pt(ppy)(curc)], a molecule embodying two light-sensitive ligands (ppy and curc), was evaluated in vitro and resulted in improved toxicity and enhanced anti-metastasis activity as demonstrated by transwell migration assay. All biological activities were evaluated against the breast carcinoma cell lines (MCF-7), which have been extensively used to assess the effectiveness of various anticancer drugs and are commonly used for in vitro studies in medicine due to their peculiar features reviewed in ref. 57.
Results and discussion
Synthesis of a Pt(II) complex
Recently, cyclometalated platinum(II) complexes have been studied for their anticancer properties,58,59 which are often enhanced by the presence of different auxiliary ligands, such as phosphane groups60 and N-heterocyclic aromatics.58 In this framework, we designed the synthesis of a cyclometalated platinum(II) complex with structure (C^N)Pt(O^O), in which C^N is the monoanionic cyclometalating ligand 2-phenylpyrinidate (ppy) and (O^O) is curcuminate (curc) in its β-diketonate form, aiming at merging the biological activities of the cyclometalated platinum(II) complexes with those of curcumin. The synthetic strategy stems from the consideration that in protic solvents curcumin is predominantly in the enol form through an intramolecular hydrogen transfer. Therefore, the acidic enol proton of curcumin can be simply taken off by α-deprotonation under alkaline conditions, promoting the O,O′-bidentate coordination to a metal center. Thus, we easily obtained [Pt(ppy)(curc)] by the reaction of [PtCl(ppy)(dmso)] with an equimolar amount of curcumin in the presence of a slight excess of KOH (Scheme 1) reaching a yield of the desired product (82%) much higher than those reported for other β-diketonate complexes with similar structures, but prepared from cyclometalated chloride-bridged platinum dimers and acetylacetonate functionalities.61 The HR ESI-MS spectrogram in positive mode of a diluted methanol solution of [Pt(ppy)(curc)] showed an intense peak at m/z 739.1384, whose isotope pattern is superimposable to that calculated for the cation [Pt(ppy)(curc) + Na]+.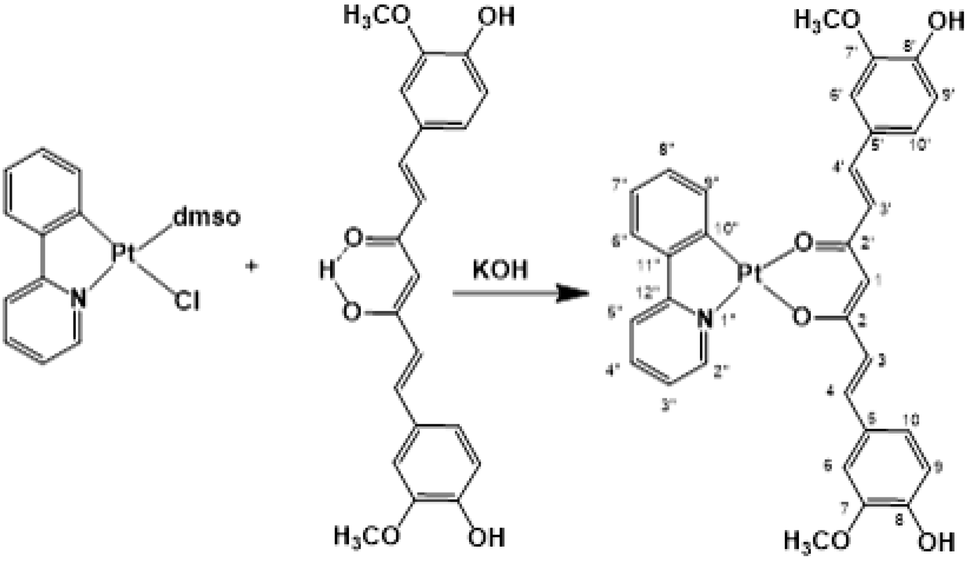 | ||
| Scheme 1 Synthesis of [Pt(ppy)(curc)]. | ||
The 1H–195Pt HMQC NMR spectrum (DMSO-d6) of [Pt(ppy)(curc)] revealed that both proton doublets at 9.18 ppm (3JHH = 5.2 Hz) and 7.67 ppm (3JHH = 7.6 Hz) were coupled to platinum (3JHPt ≈ 20 Hz); thus, they were attributed to H2′′ and H9′′ (see Scheme 1 for atom numbering) of the ppy ligand, respectively, confirming the chelation of ppy to the metal. A substantial high-field shift of the signal belonging to H9′′ upon coordination was observed as in other complexes,62 being 8.34 ppm for the H9′′ resonance in the 1H NMR spectrum of the starting complex [Pt(ppy)(dmso)Cl]. The other proton signals of the pyridine moiety of ppy appeared as a pseudotriplet at 8.07 ppm (3JHH = 7.6 Hz) for H4′′ and a doublet at 7.98 ppm (3JHH = 7.6 Hz) for H5′′, the H3′′ signal being at 7.09 ppm. The other proton signals of the phenyl moiety of ppy appeared as a doublet at 6.97 ppm (3JHH = 8.0 Hz) for H6′′ and as a pseudotriplet at 7.24 ppm (3JHH = 7.4 Hz) for H8′′, while H7′′ and H3′′ signals overlapped at 7.09 ppm. Regarding the curcuminate ligand, the γ-proton signal of the coordinated diketonate moiety was observed at 5.66 ppm, high-field shifted with respect to the analogous resonance of free curcumin in DMSO-d6 (6.15 ppm), as already observed for other curcumin-based metal chelate complexes.32,63,64 As expected, the intramolecular hydrogen bonded enolic proton signal, observed at 10.05 ppm for free curcumin, was not detected.65 Furthermore, a split in chemical shift between the two halves of the coordinated curcuminate was revealed for all proton signals (for example, H3 and H3′ doublets fell at δ 7.57 and δ 7.50 ppm, respectively, and H4 and H4′ doublets fell at δ 6.39 and δ 6.35 ppm, respectively), due to loss of chemical equivalency upon metal coordination. As for 13C{1H} NMR signals, the most important occurrence that confirmed the proposed structure for [Pt(ppy)(curc)] was the up-field shift of the β-diketonate 13C carbonyl signals (δ 176.4 and 174.4 ppm) with respect to the carbonyl peak of free curcumin (δ 183.6 ppm). The other NMR 13C and 195Pt signals are reported in the Experimental section.
The IR spectrum of [Pt(ppy)(curc)] showed two characteristic bands at 1620 cm−1 and 1607 cm−1 (ref. 32) due to νC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O coupled with νCC and to νCC coupled with νCO, respectively, typical for metal chelate β-diketonate systems.32,66
O coupled with νCC and to νCC coupled with νCO, respectively, typical for metal chelate β-diketonate systems.32,66
Physical chemical characterization of nanocolloids
Ultrasonication assisted layer by layer technology was employed for the preparation of nanocolloids.49,50 Dynamic light scattering (DLS) measurements revealed that the size of nanocolloids was uniform, and the average diameter was 190 ± 10.32 nm (Fig. 1a). The colloidal stability was investigated by measuring the size and the zeta potential for 25 days after the synthesis procedure. The good stability of our nanocolloids was demonstrated by the negligible fluctuations of the measured values (Fig. 1b).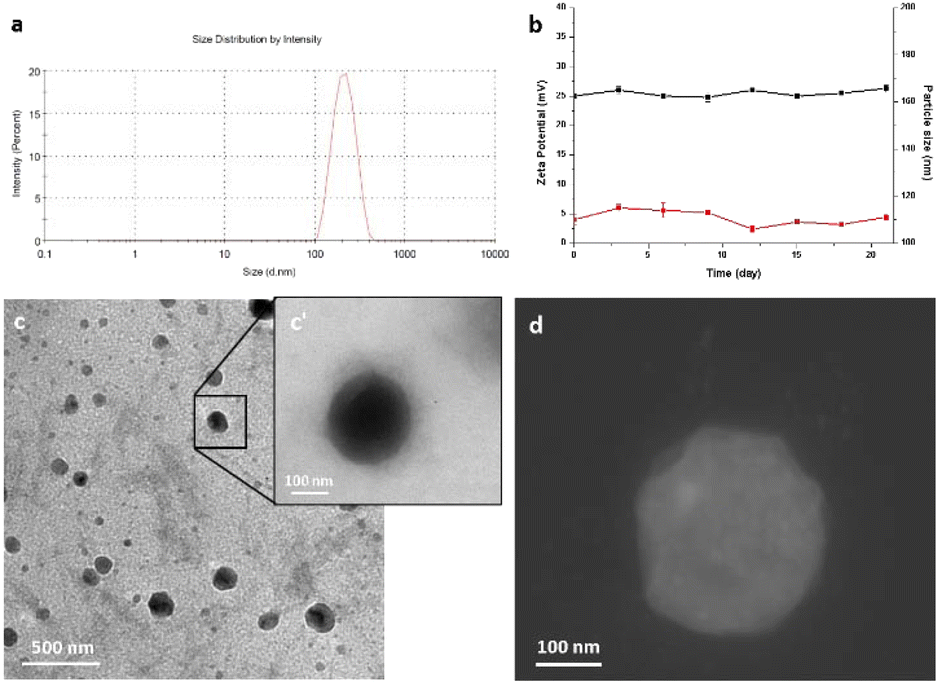 | ||
| Fig. 1 Physical chemical characterization of Pt(ppy)(curc)-NCs obtained after 20 min of ultrasonication in chitosan solution. (a) Size distribution; (b) colloidal stability by measuring the size (red line) and the zeta potential (black line) during 25 days of zeta potential measurements; (c) TEM image of Pt(ppy)(curc)-NCs, and the inset (c′) shows a magnification of highlighted nanocolloids; (d) STEM image. | ||
The morphology and surface of nanocolloids were observed and imaged using a transmission electron microscope (TEM) and STEM (Fig. 1c, c′ and d). TEM analysis revealed good sample preparation and dispersion of nanoparticles with round-shaped nanostructures. The particle size observed by TEM was slightly smaller than that measured by the DLS method. Furthermore, a halo around nanocolloids was observed, indicating the presence of coating layers. The zeta potential and polydispersity index were +25.6 ± 2.6 and 0.015 ± 0.012 mV, respectively, indicating good colloidal dispersion. Drug encapsulation efficiency measured by ICP was 68.3 ± 2.3%.
In vitro release kinetics
The pH-dependent release profiles were studied by the dynamic dialysis method at pH 7.4 or 5.5 in the presence or absence of light irradiation. A pH of 7.4 was used to simulate the value of blood, while pH 5.5 corresponded to the physiological environment in the endosomes of cancer cells, respectively. As expected, Pt(ppy)(curc)-NCs exhibited a pH-sensitive release (Fig. 2). In PBS solution at pH 7.4, without light irradiation, about 20% encapsulated drug was released from the nanocolloids after 24 h and less than 35% was released within 72 h. However, under dark conditions an initial burst release took place at pH 5.5, and more than 50% of the incorporated complex was released within 8 h. In the following hours of incubation, the cumulative release percentages reached about 80%.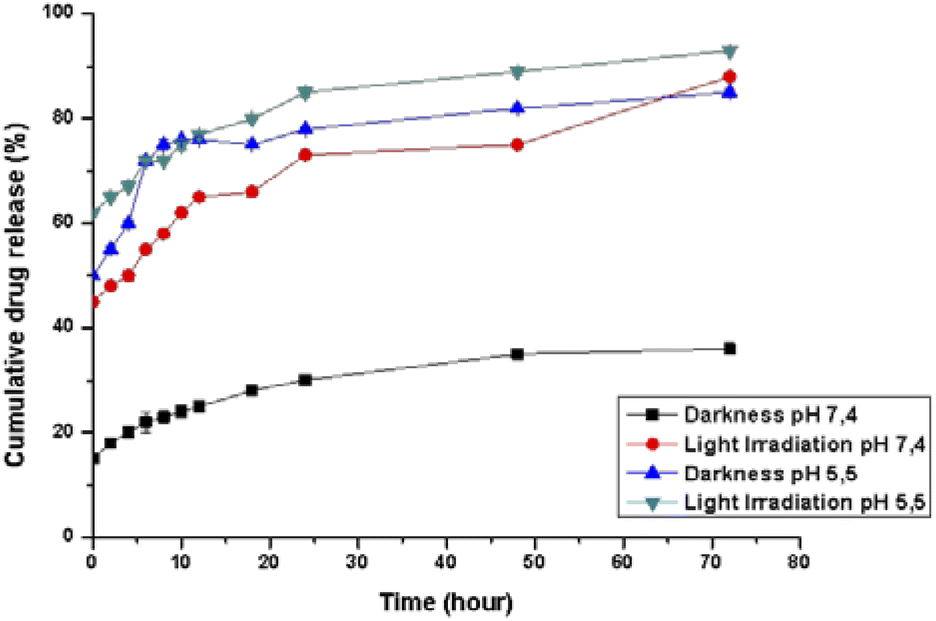 | ||
| Fig. 2 Cumulative platinum release profile (%) from Pt(ppy)(curc)-NCs. The kinetics was studied both in the dark and under visible light irradiation conditions, at 37 °C in phosphate buffer solution at pH 5.5 and 7.4. Black and blue lines represent the kinetics under dark conditions at pH 7.4 and pH 5.5, respectively. Red and green lines represent the kinetics after visible light irradiation at pH 7.4 and pH 5.5, respectively. Values represent mean ± SD, and were obtained from five independent experiments. | ||
Moreover, at pH 5.5, the release rate increased both in the dark and under light irradiation, because the acid pH affects the polysaccharidic shell composition. In particular, in the dark a release of about 45% occurred already in the first 30 minutes, reaching 75% in the following 10 hours of incubation. In an acidic environment and under light irradiation, the complex was released more quickly, and about 60% of the entrapped drug was released after 2 h, with more than 90% being released after 72 h.
Biological activity
For all these reasons, tests on cellular uptake were carried out on tumour cells only, but deeper studies regarding normal cells are also warranted.
Fluorescence microscopy and fluorescence activated cell sorting (FACS) analyses were performed to investigate the cellular uptake of free [Pt(ppy)(curc)] and Pt(ppy)(curc)-NCs in MCF-7 cells based on the intrinsic green fluorescence of drugs, due to the presence of curcumin. As shown in Fig. 3, [Pt(ppy)(curc)] could enter the cells after 3 h of incubation in vitro but the fluorescence intensity was very weak. As expected, a more intense green fluorescence was observed with Pt(ppy)(curc)-NCs. The cells maintained the fluorescence signal over 48 h. Similar results were reported by Chen and co-workers,69 who used polymeric nanoparticles to deliver a platinum–curcumin complex obtained by the nanoprecipitation method. They found a stronger green fluorescence associated with curcumin in the synthesized nanoparticles in comparison to the free complex. In our previous work,49,50 we have already demonstrated that also the fluorescence of curcumin alone decays after 1 h of incubation.
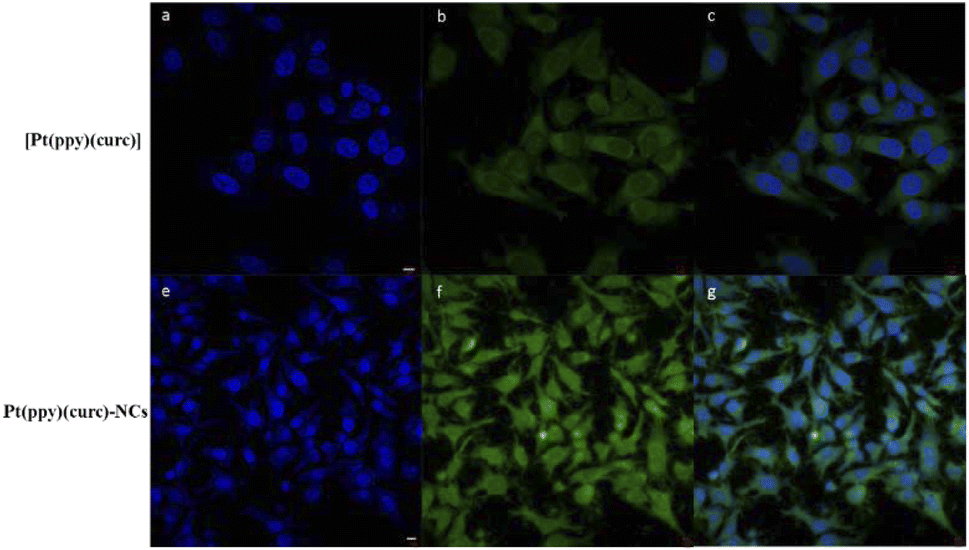 | ||
| Fig. 3 Confocal images of MCF-7 cells treated with 50 μM of [Pt(ppy)(curc)] (a–c) and 50 μM of Pt(ppy)(curc)-NCs (e–f) in the dark for 2 h. The green fluorescence signal is associated with curcumin, and blue represents nuclei stained with DAPI. Scale bar = 5 μm. | ||
The polymeric shell acts as a physical barrier preventing the passage of light inside the complex and provides prolonged protection for hydrophobic bioactive compounds.70,71 The above experiments proved that metal complexation and consequent drug encapsulation could improve the photostability of curcumin.
In order to further quantify the drug uptake, the intracellular drug concentration was measured using a flow cytometer at different times (0.5, 2 and 6 h, Fig. 4). Free curcumin was used as the reference. It was reported that the fluorescence associated with free curcumin is dampened when it is complexed with platinum.72 In fact, in accordance with the literature, FACS analysis revealed that intracellular green fluorescence intensity of free curcumin was the highest in comparison to that of the free complex and Pt(ppy)(curc)-NCs, but it strongly decayed during time of incubation, while the fluorescence associated with [Pt(ppy)(curc)] was nearly steady, while that of Pt(ppy)(curc)-NCs increased significantly with exposure time. However, the intracellular uptake of Pt(ppy)(curc)-NCs was much higher than that of cells treated with [Pt(ppy)(curc)], which is consistent with the result of fluorescence microscopy, previously shown.
 | ||
| Fig. 4 Intracellular drug concentration. The intensity of green fluorescence associated with curcumin was calculated by FACS analysis. MCF-7 cells were treated with curcumin (black histogram), [Pt(ppy)(curc)] (red histogram) and Pt(ppy)(curc)-NCs (blue histogram) and the cytometer test was performed at different times of 0.5, 2 and 6 h. Values represent mean ± SD and were obtained from five independent experiments. | ||
Cell cytotoxicity and apoptosis study
To evaluate the in vitro cytotoxicity of different drug formulations, MTT assay was employed on MCF-7 cells. As illustrated in Fig. 5, all drugs show inhibitory effects on the tumor cells. The IC50 of free cisplatin and free curcumin are 60 μM and 80 μM, respectively. When combining the two free drugs, the IC50 reduced to 45 μM. Free [Pt(ppy)(curc)] showed a slight cytotoxicity with an IC50 of 90 μM, but a significant reduction in IC50 was observed after loading [Pt(ppy)(curc)] into the nanocolloids. Pt(ppy)(curc)-NCs exhibited four times stronger cell inhibition than free [Pt(ppy)(curc)] (IC50 20 μM).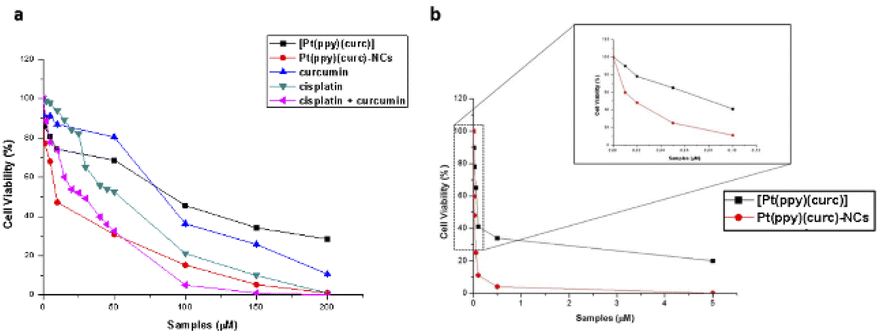 | ||
| Fig. 5 Cell cytotoxicity study. Panel (a): effect of curcumin (blue line), cisplatin (green line), cisplatin + curcumin (pink line), [Pt(ppy)(curc)] (black line), and Pt(ppy)(curc)-NCs (red line) on the viability of MCF-7 cells treated for 24 h. The concentrations tested are in the range from 0 to 200 μM for each condition. Values represent mean ± SD and were obtained from five independent experiments. Panel (b): effect of [Pt(ppy)(curc)] (black line) and Pt(ppy)(curc)-NCs (red line) on viability of MCF-7 cells treated for 24 h and after visible light irradiation. The concentrations tested are in the range from 0 up to 5 μM for each condition. Values represent mean ± SD and were obtained from five independent experiments. | ||
We further examined the phototoxicity of [Pt(ppy)(curc)] measuring the cytotoxicity after 1 h of light irradiation of MCF-7 cells treated with [Pt(ppy)(curc)] and Pt(ppy)(curc)-NCs. Fig. 5b shows a significant increase in cytotoxicity reaching an IC50 for [Pt(ppy)(curc)] and Pt(ppy)(curc)-NCs of 0.10 μM and 0.02 μM, respectively.
To detect quantitative apoptosis in MCF-7 cells, annexin V/PI double staining was conducted. As shown in Fig. 6, the experiment was carried out in the dark and under visible light conditions using the IC50 of free [Pt(ppy)(curc)] and Pt(ppy)(curc)-NCs. All formulations showed increased cytotoxicity under visible light irradiation compared to those with formulations under dark conditions. The highest percentage of late apoptotic cells was detected for the Pt(ppy)(curc)-NC treatment group, which is consistent with the result of MTT. All these data indicate that Pt(ppy)(curc)-NCs could exert a higher inhibition of tumor cell growth in vitro with respect to free [Pt(ppy)(curc)].
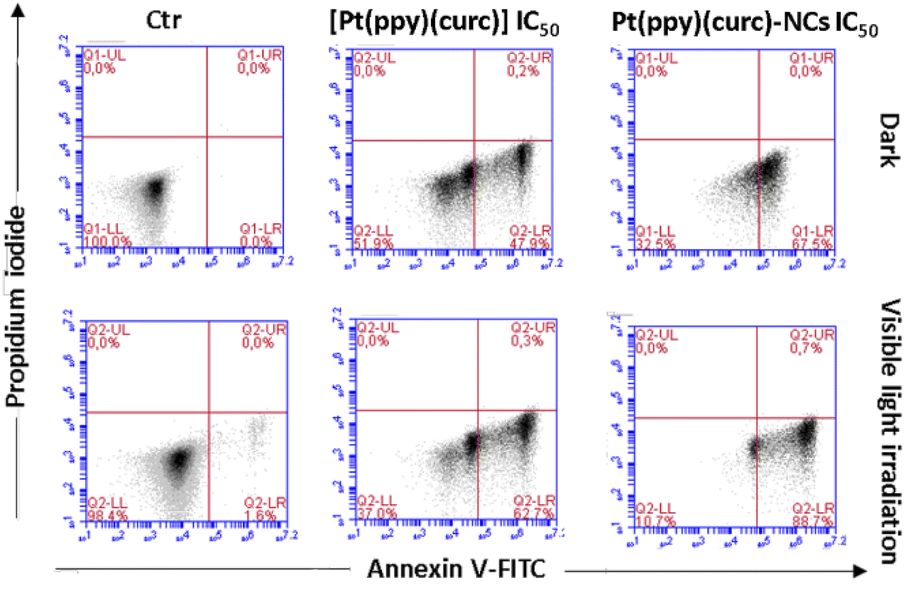 | ||
| Fig. 6 Apoptosis study of MCF-7 cells mediated by [Pt(ppy)(curc)] and Pt(ppy)(curc)-NCs at IC50 concentrations calculated by the MTT test both in the dark and under visible light irradiation. Apoptosis was measured by flow cytometry after PI/annexin V-FITC staining. Q1-UL, PI+ (cells undergoing necrosis); Q1-UR, annexin V-FITC+ PI+ (cells in the late period of apoptosis and undergoing secondary necrosis); Q1-LR, annexin V-FITC+ PI− (cells in the early period of apoptosis); Q1-LL, annexin V-FITC− PI− (living cells). | ||
Similar results have been reported by Tabatabaei and co-workers,73 who used a Pt(II) complex immobilized on polymer-modified magnetic carbon nanotubes. In their study they employed a magnetic field instead of visible light irradiation, indicating that the guiding of the carrier by the magnetic field is very effective in increasing the amount of cellular penetration, where, due to the acidity of the tumor cell microenvironment, platinum drugs can be released quickly and penetrate the nucleus to kill proliferating tumor cells by causing DNA damage.
An enhancement, in terms of cytotoxicity, of nanodrug delivery systems was also demonstrated using specific ligands such as folate,74,75 epidermal growth factor76–78 and transferrin receptors.79 On the other side, it has been reported that both platinum complexes binding ppy ligands have shown lower toxicity with respect to cisplatin against normal cells (MCF-10A), and the platinum complex containing diketonate, as the leaving group, is more cytotoxic in cancer than in normal breast cells.80,81
Determination of reactive oxygen species (ROS) production
To gain insights into the role of irradiation, intracellular production of reactive oxygen species (ROS) determination experiments, using 2′,7′-dichlorodihydrofluorescein diacetate (H2DCFDA) as the probe, were carried out in the dark and after light exposure. The relevant results (Fig. 7) revealed that ROS production in MCF-7 cells treated with curcumin (with and without cisplatin), [Pt(ppy)(curc)] and [Pt(ppy)(curc)]-NCs increased after light irradiation. In contrast, light irradiation had negligible effects on ROS production in MCF-7 cells treated with only cisplatin. | ||
| Fig. 7 H2DCFDA assay showing ROS levels in MCF-7 cells incubated with curcumin, cisplatin, cisplatin and curcumin, [Pt(ppy)(curc)] and Pt(ppy)(curc)-NCs. The concentration tested was the IC50 for all conditions. The incubation period was 24 h for dark conditions (black bars) and 4 h in the dark and then exposed for 1 h to light (400–700 nm, 2.5 J cm−2) for photoexposure experiments (gray bars). Values represent mean + SD and were obtained from three experiments. Very statistically significant p < 0.001 (***) and extremely statistically significant p < 0.0001 (****). | ||
Pt–DNA adduct evaluation
Pt(II)-complexes, as intercalating ligands, are promising drugs in the treatment of human cancers with acquired or intrinsic resistance to drugs currently applied in clinics. This class of compounds can act as intercalant molecules and interact with the DNA double helix by aromatic π-stacking between base pairs, forcing the sequential base pairs apart, increasing the helix length and stiffness, and through this, preventing DNA transcription and replication.2,82So, cisplatin and all other Pt drugs are believed to exert their cytotoxic effects by forming bifunctional covalent Pt adducts with DNA.83–85 Pt concentrations in DNA isolated from cisplatin, cisplatin + curcumin, [Pt(ppy)(curc)] and Pt(ppy)(curc)-NC treated MCF-7 cells were determined using ICP-OES.
The cells were previously incubated for 24 h at IC50 concentrations previously calculated. The experiments were carried out both in the dark and under light irradiation, and the Pt content was evaluated in the nuclear fraction.
Pt–DNA adduct estimation (Fig. 8) confirmed that the light irradiation conditions led to an enhancement of the quantity of platinum reaching the nucleus, under all conditions tested. In the case of nanoformulation this effect is more evident.
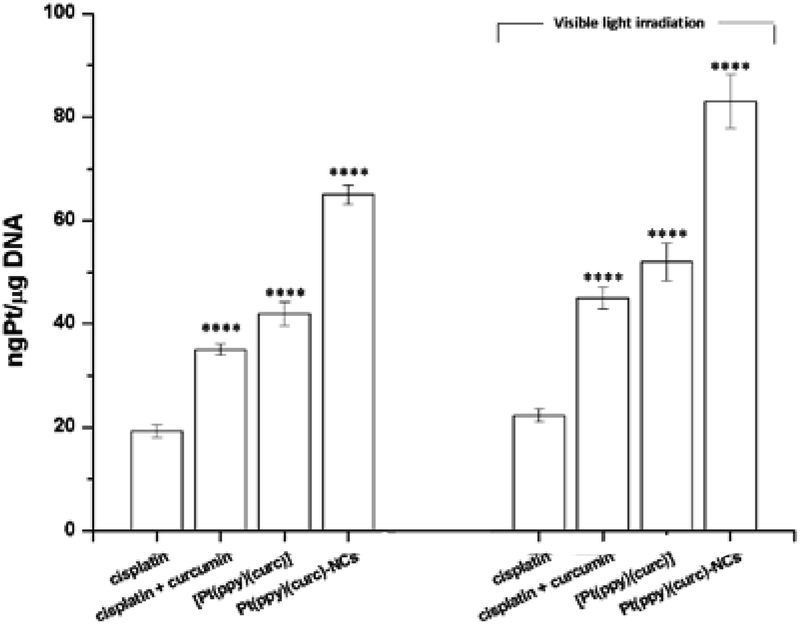 | ||
| Fig. 8 DNA platination of MCF-7 cells in the dark and under visible light irradiation. The cells were incubated with cisplatin, cisplatin + curcumin, [Pt(ppy)(curc)] and Pt(ppy)(curc)-NCs. The total incubation period was 24 h either in the dark or exposed to light (400–700 nm, 2.5 J cm−2) for 1 h after 4 h in the dark. The tested concentration was the IC50 for all conditions. Values represent mean ± SD and were obtained from three independent experiments. Statistical analysis: *p < 0.05 and ****p < 0.0001 vs. the cisplatin group. | ||
The addition of curcumin to the system increased the nuclear uptake of the metal (either in the dark or under visible light irradiation) due to the lipophilic nature of curcumin, that facilitated the crossing of the lipidic cellular membrane by the metal drug. In fact, the intermolecular interactions between the β-diketone ring and/or the electron rich π-systems of curcumin and the metal center of cisplatin could drag the platinum complex into the cell (Fig. 8). The nuclear uptake of Pt increased for Pt(curc)(ppy) in the presence of two coordinated lipophilic ligands that enhanced the metal complex capability to pass the lipidic biological barrier (Fig. 8). Finally, the Pt nuclear uptake reached its maximum value by using NCs, which are known to increase the cellular drug uptake by passive or active delivery processes.86
Anti-metastasis effect
Migration assay was used to investigate the synergistic anti-metastasis effect of formulations. As shown in Fig. 9, free cisplatin and free curcumin alone did not retard migration of MCF-7 cells, but the combined treatment of curcumin with cisplatin inhibited this phenomenon. Free [Pt(ppy)(curc)] significantly suppressed cell migration and a greater inhibitory effect was measured with Pt(ppy)(curc)-NCs, especially under visible light irradiation.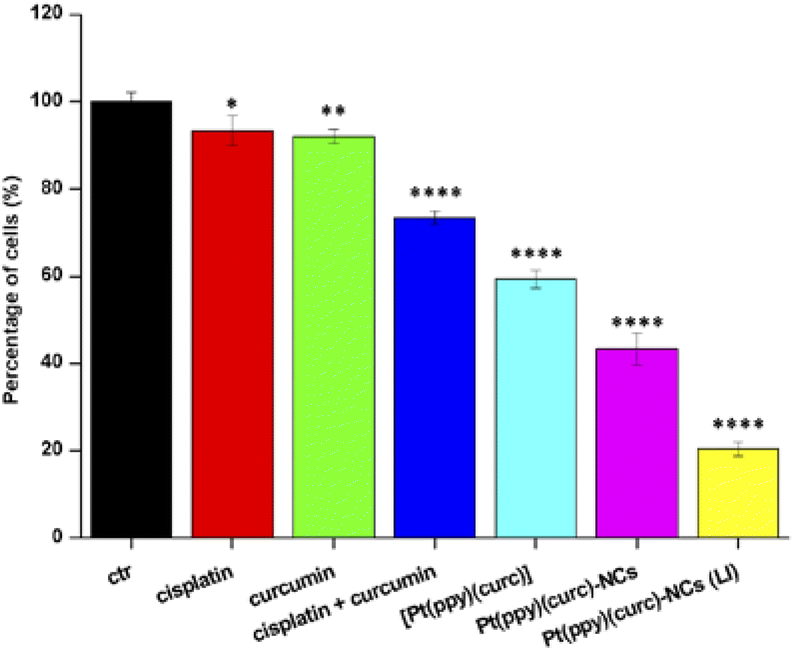 | ||
| Fig. 9 Cell migration rate. MCF-7 cells were treated with at IC50 of cisplatin (red histogram), curcumin (green histogram), cisplatin + curcumin (blue histogram), [Pt(ppy)(curc)] (light blue histogram), Pt(ppy)(curc)-NCs (pink histogram) and Pt(ppy)(curc)-NCs after visible light irradiation (yellow histogram). Error bars represent three independent experiments, each performed three times. Statistical analysis: *p < 0.05, **p < 0.01, and ****p < 0.0001, vs. the control group. | ||
These results indicate that coordinated curcumin, nanoformulation and light irradiation concur in sensitizing the cellular microenvironment toward the Pt-chemotherapeutic drug to retard cell migration. An improvement in chemotherapeutic efficacy, based on the enhanced anti-metastasis activity of a platinum complex of curcumin delivered by dual-responsive polymeric nanoparticles has been reported by Chen and co-workers.87
Plausible mechanisms acting on the cytotoxic effect
All reported experiments suggested that simultaneous mechanisms might act in cancer cells treated with Pt(ppy)(curc)-NCs exposed to light irradiation, the latter being responsible for the significant decrease in the IC50 value (a thousand times lower after light exposure with respect to the dark). First, the NC formulation and the presence of two lipophilic metal ligands facilitate the crossing of the biological barriers, and thus the migration of the drug to the cellular nucleus. The toxicity of platinum complexes bearing the curcumin ligand is hypothesized starting with the formation of an aquo-Pt complex after partial or total detachment of curcumin from the metal in physiological solution.88 This detachment is favored by both the light and the high trans influence of the C atom of the C^N cycloplatinated(II) complex,89 that enhances the capability of the trans unit (O atom of curc) to act as a desirable leaving group. In this way, the main mode of toxicity of the Pt–aquo complex would be the dissociation of the aquo ligand and the formation of a coordination bond between Pt and DNA as it occurs for cisplatin.90 Moreover, since the 2-phenylpyridinate group is covalently linked to the metal, the Pt(ppy) side of the platinum complex should remain unaltered after curcumin detachment. Theoretical studies have shown that ligands similar or equal to ppy have hydrophobic interactions with the base pairs of DNA through noncovalent π–π stacking,81 thus contributing to increased cytotoxicity. The light irradiation increased the ROS production in cancer cells treated with Pt(ppy)(curc)-NCs due to the presence of two photosensitizer groups: curcumin and the Pt(ppy) moiety, the latter endowed with luminescence properties attributed to π–π* and metal-to-ligand charge transfer (MLCT) transitions.91 A third mechanism could also enhance the cytotoxicity of Pt(ppy)(curc)-NCs after light irradiation: ROS formed upon light exposure could damage the polymeric NC structure, thus accelerating the delivery of the drug.68Experimental
Materials and instruments
IR spectra were recorded on a Bruker-Vector 22 spectrometer. NMR spectra were recorded on a Bruker Advance 400 spectrometer or on a Bruker Avance III 700 MHz instrument equipped with a cryoprobe; chemical shifts are in ppm and coupling constants in Hz; the frequencies are referred to Me4Si for 1H and for 13C and to H2PtCl6 for 195Pt NMR. The signal attributions and coupling constant assessment were made based on multinuclear NMR analyses including 1H–195Pt HMQC, 1H–13C HMQC, and 1H–13C HSQC experiments. Elemental analyses were obtained on a EuroVector CHNS EA3000 elemental analyser using acetanilide as the analytical standard material. Triplicate runs have been performed to ensure reproducibility. High-resolution mass spectrometry (HR-MS) analyses were performed using a time-of-flight mass spectrometer equipped with an electrospray ion source (Bruker micrOTOF). The sample solutions were introduced by continuous infusion with the aid of a syringe pump at a flow rate of 180 μL min−1. The instrument was operated at end plate offset −500 V and capillary voltage −4500 V. Nebulizer pressure was 1.5 bar (N2) and the drying gas (N2) flow was 10 L min−1. The capillary exit and skimmer 1 were 120 and 40 V, respectively. The drying gas temperature was set at 220 °C. The calculated (exact mass) and the experimental (accurate) m/z values were compared considering the isotope pattern of the main ion (which gives the most intense peak), by using the software Bruker Daltonics Data Analysis (version 3.3). All solvents and reagents were obtained from commercial sources. N-(2-Hydroxyethyl)piperazine-N′-(2-ethanesulfonic acid) (HEPES) and 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-tetrazolium bromide (MTT) were purchased from Sigma-Aldrich. All reagents were of molecular biology grade and used without further purification. The MCF-7 cell line was purchased from the American Type Culture Collection (ATCC). TEM images were collected with a JEOL JEM 1400 with a LaB6 source at 80 kV. The zeta potential and the hydrodynamic diameter of the Pt(ppy)(curc)-NCs were measured with a Malvern Zetasizer Nano ZS (Dynamic Light Scattering analysis). The UV-Vis absorption and emission spectra were recorded with a Varian-Cary 500 spectrophotometer and a Varian Cary Eclipse spectrofluorometer, respectively. Absorption and fluorescence spectra were measured in deionized water, with a final concentration of 50 μM. The quartz cuvettes used were of 1.0 cm path length. ICP experiments were performed with an ICP-OES Thermo Scientific instrument. Biological imaging tests were carried out with a Zeiss LSM700 confocal microscope (Zeiss, Germany) equipped with a Zeiss Axio Observer Z1 inverted microscope using a 63× objective with a 1.46 numerical aperture oil immersion lens for imaging.Synthesis and characterization of [Pt(ppy)(curc)] (ppy = deprotonated 2-phenylpyridine, curc = deprotonated curcumin)
Under inert atmospheric conditions, complex [PtCl(ppy)(dmso)]62 (100 mg, 0.216 mmol) was added to an ethanolic solution (10 mL) of potassium curcuminate [K(curc)], prepared by dissolving KOH (15 mg, 0.260 mmol) in 10 mL of absolute ethanol and subsequent treatment with curcumin (80 mg, 0.216 mmol). The resulting dark red mixture was allowed to react under stirring for 12 h at room temperature. Afterwards, the solvent was removed under reduced pressure and the residue was extracted with CH2Cl2 (20 mL). The obtained orange solution was filtered through Celite to remove solid impurities and the filtrate was concentrated to a small volume (1.0 mL). Finally, n-pentane (5 mL) was added. The resulting dark red precipitate was filtered, washed with n-pentane (2 × 3 mL) and dried under high vacuum. Yield = 127 mg, 82%. The obtained complex was air stable and was soluble in DMSO and slightly soluble in CHCl3 and CH2Cl2.Anal. calcd for [Pt(ppy)(curc)], C32H27NO6Pt: C, 53.63; H, 3.80; N, 1.95. Found: C, 53.23; H, 3.51; N, 1.61.
1H NMR (400 MHz DMSO-d6, 298 K) δ = 9.18 (d, 3JHPt ≈ 20 Hz, 3JHH = 5.5 Hz, 1 H, H2′′), 8.07 (pseudo t, 3JHH = 7.6 Hz, 1 H, H4′′), 7.98 (d, 3JHH = 7.6 Hz, 1 H, H5′′), 7.67 (d, 3JHH ≈ 7.6 Hz, 3 H, H9′′, 3JHPt ≈ 20 Hz, overlapped to H9 and H9′), 7.57 (d, 3JHH = 14.9 Hz, 1 H, H3), 7.50 (d, 3JHH = 14.0 Hz, 1 H, H3′), 7.48 (d, 3JHH ≈ 7.0 Hz, 1 H, H10), 7.47 (d, 3JHH ≈ 7.0 Hz, 1 H, H10′), 7.24 (pseudo t, 3JHH = 7.4 Hz, 1 H, H8′′), 7.09 (pseudo t, 3JHH ≈ 7.3 Hz, 1 H, H3′′), 7.09 (pseudo t, 3JHH ≈ 7.3 Hz, 1 H, H7′′), 7.09 (s, 1 H, H6), 7.05 (s, 1 H, H6′), 6.97 (d, 8.0 Hz, 1 H, H6′′), 6.39 (d, 3JHH = 14.9 Hz, 1 H, H4), 6.35 (d, 3JHH = 14.0 Hz, 1 H, H4′), 5.66 (s, 1 H, H1), 3.75 (s, 3 H, OCH3), 3.74 (s, 3 H, OCH3′).
13C{1H} NMR (176 MHz DMSO-d6, 298 K) δ = 176.4 (C2), 174.4 (C2′), 167.2 (C12′′), 147.5 (C2′′), 145.1 (C8 and C8′)141.0 (C11′′), 140.9 (C7), 140.7 (C7′) 140.5 (C10′′), 139.4 (C4′′), 130.6 (C3), 130.5 (C3′), 129.3 (C8′′), 125.0 (C6′′), 124.7 (C6′), 123.6 (C9′′) 123.55 (C7′′), 123.5 (C6), 122.3 (C5 and C5′), 119.4 (C5′′), 117.3 (C4), 117.2 (C4′), 105.0 (C1), 55.4 (OCH3).
195Pt{1H} NMR (86 MHz, DMSO-d6, 298 K) δ = −2825 ppm (s).
HRMS(+) (ESI, methanol) m/z: calcd for C32H27NNaO6Pt [M + Na]+ 739.1381, found 739.1384.
IR (KBr, cm−1): 3449 (broad, m, νO–H), 1620 (m, νCO + νCC), 1607 (m, νCC + νCO), 1513 (s, νCC), 1287 (m, δC–O–C), 1273 (m, νC–O), and 1162 (m, νO–H).
Preparation of nanocolloids
Nanocolloids were built by the sonication assisted layer-by-layer technique. Biodegradable chitosan (CHI, a polycation) and pectin (PEC, polyanion) were chosen for a biocompatible and biodegradable coating on drug nanoparticles.CHI solution was prepared at a 0.10% concentration in acid acetic 0.5 M; PEC aqueous solutions (0.5 mg mL−1) were prepared in ultra-pure water. The pH of the polymer solutions was adjusted to 5.0. Nanocolloids were prepared in a two-step procedure. In the first step, 2.0 mg of [Pt(ppy)(curc)] was added to 20 mL of a chitosan solution, stirred for 5 min and then ultrasonicated for 20 min at 20% power (150 watt) in a water/ice bath, to make [Pt(ppy)(curc)]/chitosan nanocores. The excess polycation was removed in three centrifugation/washing steps with deionized water. The following layer-by-layer self-assembly of pectin and chitosan was performed using the LbL method. Thereafter, 5.0 mL of solution containing the polyanion was added and the dispersion was continuously shaken for 20 min, followed again by three centrifugation/washing steps. This procedure was repeated 2.5 times for the couple of polymers resulting in the deposition of five polyelectrolyte layers on the Pt(ppy)(curc)-NCs.
Platinum-loading of nanocolloids
Drug loadings were quantified by inductively coupled plasma optical emission spectrometry (ICP-OES). The experimental setup provides for the construction of a calibration line, using four points. The loading percentage is defined as the Pt concentration (in ppt) in solution after loading divided by the Pt concentration (in ppt) in solution before loading.Release kinetics
The release kinetics was evaluated in phosphate buffer at physiological pH (pH 7.4) and acidic pH (pH 5.5). Pt(ppy)(curc)-NCs were constantly stirred to increase the release rate and to establish equilibrium conditions. Separate tubes were used each time. At each time point, 500 μL of release media was replaced with the same volume of fresh release media. At selected time intervals, Pt(ppy)(curc)-NCs were separated by centrifugation. The supernatant was collected and the platinum content in the supernatant was determined by ICP-OES analysis. The same experimental setup was used to study the release kinetics after light irradiation.Visible light irradiation
The cells were placed in a flat-bottom 6-well microplate above a xenon lamp (Spectral Products mod.1118263) for 1 h (at a distance of 3.5 cm). During irradiation, the temperature was kept at 23 ± 2 °C. Native samples to be irradiated were obtained from the same stock. After irradiation, the samples were kept in a CO2 incubator for 20 h at 37 °C before further analysis.Staining and confocal imaging of living cells
The MCF-7 cancer cell line was maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with fetal bovine serum (FBS) (10%), penicillin (100 U mL−1 culture medium), streptomycin (100 mg mL−1 culture medium), and glutamine (5%). Cells were grown in a humidified incubator at 37 °C, 5% CO2, and 95% relative humidity. The cell line was serum-starved for 24 h before any test. For staining experiments MCF-7 (5 × 104) cells were seeded onto a 35 mm glass bottom Petri dish and incubated in complete media for 24 h. Then, the cells were incubated with the [Pt(ppy)(curc)] complex and Pt(ppy)(curc)-NCs at a concentration of 50 μM for 30 min or 2 h in the dark in a humidified incubator at 37 °C, 5% CO2, and 95% relative humidity. After rinsing with phosphate buffered saline (PBS) twice, cells were stained with Hoechst and imaged by confocal microscopy immediately after adding complete media without red phenol. Laser beams were used for the excitation of Hoechst (λ = 405 nm) and [Pt(ppy)(curc)] (λ = 488 nm) or Pt(ppy)(curc)-NCs (λ = 488 nm).Cytotoxicity determination by MTT assay
The MCF-7 cancer cell line was used in the general cytotoxicity test. The MTT method was used to measure the activity of living cells via mitochondrial dehydrogenase activity. The key component was 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide or MTT. Mitochondrial dehydrogenases in viable cells cleave the tetrazolium ring, yielding purple MTT formazan crystals, which are insoluble in aqueous solutions. The MTT method is most effective when cultures are prepared in multi-well plates. Cells (3,5 × 104 cells per mL) were added to 24-well culture plates at 0.5 mL per well, serum-starved for 24 h, and incubated at 37 °C, 5% CO2, and 95% relative humidity. In order to establish the IC50, MCF-7 cells were treated for 24 h with curcumin, cisplatin or the [Pt(ppy)(curc)] complex and Pt(ppy)(curc)-NCs at different concentrations from 1.0 up to 200 μM. The control was a complete culture medium. After 24 h of incubation, cultures were removed from the incubator and a MTT solution (10% of the culture volume) was aseptically added, reaching a final concentration of 0.50 mg mL−1. Cultures were returned to the incubator and incubated for another 3 h. After the incubation period, cultures were removed from the incubator and the resulting MTT formazan crystals were dissolved in DMSO (using the same volume of the culture). The plates were ready within 15 min after adding DMSO. After the incubation time, pipetting up and down was required to completely dissolve the MTT formazan crystals. Absorbance at a wavelength of 570 nm was measured using a spectrophotometer. The results were expressed as mean ± SD of three separate trials.To evaluate the cytotoxicity under light irradiation, MCF-7 cells were treated for 4 h with [Pt(ppy)(curc)] and Pt(ppy)(curc)-NCs at different concentrations from 0.10 up to 5.0 μM. Then the samples were subjected to light irradiation for 1 h. After this, the samples were kept in the CO2 incubator for 20 h at 37 °C. The subsequent experimental procedures were those detailed above.
Flow cytometric analysis of apoptosis
An annexin V-fluorescein isothiocyanate (FITC) kit (Thermo Fisher Scientific, Inc.) was used to determine cellular apoptosis, both under dark and light irradiation conditions. MCF-7 cells were treated with [Pt(ppy)(curc)] and Pt(ppy)(curc)-NCs at IC50 concentrations established in the MTT assay for both conditions. Then, MCF-7 cells were collected, washed twice with PBS and subjected to centrifugation at 1200 rpm for 5 min at room temperature. Subsequently, the cell pellet was resuspended and treated with annexin V-FITC and propidium iodide (PI) solutions. After incubating for 15 min at room temperature in the dark, additional annexin V binding buffer (10 mM Hepes, 140 mM NaCl, 2.5 mM CaCl2 in deionized water (dH2O)) was added to each tube and the cells were analyzed. Flow cytometry was performed with a flow cytometer (BD Biosciences, San Jose, CA, USA) equipped with FlowJo software.H2DCFDA assay
2′,7′-Dichlorodihydrofluorescein diacetate (H2DCFDA) assay was used to detect generation of cellular reactive oxygen species (ROS). H2DCFDA, under oxidation by cellular ROS, generates fluorescent 2,7-dichlorofluorescein (DCF) having an emission spectral maximum at 525 nm. The percentage of the cell population generating ROS was determined by fluorescence-activated cell sorting (FACS). About 3.0 × 105 MCF-7 cells were plated in two 6-well plates and treated with compounds: cisplatin, curcumin, cisplatin and curcumin, [Pt(ppy)(curc)] complex and Pt(ppy)(curc)-NCs, and then incubated for 4 h in the dark. One plate was irradiated with visible light of 400–700 nm for 1 h in PBS, while the other was kept in the dark. The cells were subsequently trypsinized and a clear suspension of ∼105 cells per mL was made. The suspensions were then treated with 1.0 μM H2DCFDA solution and kept in the dark for ∼10 min at room temperature. The samples along with untreated controls were analyzed by FACS. The experiment was performed in triplicate.Cellular uptake from Pt estimation
To measure the cellular platinum uptake, about 106 MCF-7 cells were seeded in 75 mm tissue culture dishes and treated with cisplatin, cisplatin and curcumin, [Pt(ppy)(curc)] complex and Pt(ppy)(curc)-NCs at IC50 concentrations for 24 h both in the dark and under visible light irradiation conditions. After incubation, we proceeded with DNA extraction using a PROMEGA kit (Wizard® Genomic DNA Purification Kit). The cells were harvested in the medium and then washed twice with PBS and suspended in nuclei lysis solution by pipetting to lyse the cells. Then few μL of RNase solution were added to the nuclear lysate. After incubation at 37 °C, the protein precipitation solution was added at room temperature. After centrifugation, the precipitated protein formed a tight white pellet. The supernatant (containing the DNA) was carefully removed and transferred to a clean microcentrifuge tube containing 600 μL of room temperature iso-propanol. After centrifugation, 70% ethanol was added. The DNA pellet was collected after centrifugation and it was incubated with DNA rehydration solution, at 65 °C for 1 h. DNA was then quantified using a NanoDrop spectrophotometer. The amount of platinum in the DNA was determined by ICP-OES. The Pt concentration was expressed as the percentage of platinum content with respect to the platinum estimated in the fed solution. All experiments were performed in duplicate along with untreated controls.In vitro cellular uptake
MCF-7 cells were seeded in six-well plates at a density of 2 × 105 cells per well and incubated overnight at 37 °C. Free curcumin, cisplatin + curcumin, [Pt(ppy)(curc)] and Pt(ppy)(curc)-NCs were then added to the media and incubated at 37 °C at 0.5, 2 and 6 h. For measurement of uptake, cells were washed with cold PBS three times and then trypsinized, redispersed and enumerated using a BD Biosciences cytometer (San Jose, CA, USA) equipped with FlowJo software.Transwell migration assay
Transwell insert chambers with 8 mm pore size and 6.5 mm diameter (Corning, San Diego, CA, USA) were used for migration assay. Briefly, 5 × 104 MCF-7 cells were placed in the inner chambers and were exposed to a serum-free medium containing PBS or different drug formulations (curcumin, cisplatin, cisplatin + curcumin or [Pt(ppy)(curc)] and Pt(ppy)(curc)-NCs). A medium containing 10% serum was added to the lower chambers to stimulate cell migration. Following incubation for 24 h, non-migrated cells on the upper surface of the polycarbonate filter were gently removed using a cotton swab. Migratory cells in the lower compartment were stained with 1% crystal violet solution. After 20 min of incubation, the unbound crystal violet was removed by washing the wells four times with tap water. The bound crystal violet was eluted by adding 400 μL of 33% acetic acid solution (33% v/v in ddH2O) into each insert and shaking for 10 min. The eluent from the lower chamber was transferred into a 96-well clear microplate (Corning 3599), and the absorbance at 590 nm was measured using a plate reader. Absorbance at 590 nm was subtracted from the data of blank Transwell® without cells. The absorbance values of MCF-7 cells seeded at various cell densities were obtained by measuring the OD at 590 nm. The standard curves of cell numbers versus absorbance were plotted, and an equation for each cell line was generated. The R2 values of these standard curves were greater than 0.99. The total number of cells passing through the Transwell® membrane was determined by converting absorbance values to cell numbers using the equation from the standard curve and multiplying by the dilution factor. The percentages of migration were determined by dividing the number of migrated cells by the number of plated cells.Statistical analysis
Statistical differences between controls and drug-treated cells were determined by one-way ANOVA (Sidak). P-values < 0.05 were considered statistically significant. Data were analyzed using the Stata 8.2/SE package (StataCorp LP).Conclusions
The rationale of this study was the design of nano-drug delivery systems for a platinum complex, with a therapeutic index better than those of cisplatin and its derivatives. We have successfully designed and synthesized nanoscale polymeric colloids to deliver the Pt–curcumin complex [Pt(ppy)(curc)]. The resulting nanocolloids, Pt(ppy)(curc)-NCs, were stable under physiological conditions and showed a slow and sustainable release of platinum over several hours. The cytotoxicity studies revealed the enhanced antitumor activity of the nanocolloids with respect to the free complex, or cisplatin, or cisplatin + curcumin against breast cancer cell lines, MCF-7. Enhanced efficacy was demonstrated by the nanocolloids with respect to the free metal-drug, with an increase in Pt uptake. The amount of Pt incorporated in the nuclear DNA of the cells treated with the curcumin-based drug was found to be higher under visible light conditions with respect to darkness. In the case of cells treated with Pt(ppy)(curc)-NCs, a satisfactory 85 ngPt μg−1 DNA nuclear concentration was reached. These results indicate that nanostructuration had a direct effect on cellular uptake, and the complexation with curcumin implied in some way a more facile conversion of the pro-drug to active cisplatin and curcumin molecules, leading to an enhancement of cytotoxicity. The presented results indicate how the nanostructuration of a therapeutic agent or a prodrug can modify the bioavailability of bioactive molecules and increase therapeutic action. Furthermore, the nanoformulation demonstrated a good influence on cell migration, especially after visible-light activation. This parameter is very important, because metastasis to other secondary organs is the main cause of treatment failure. Chemoresistance and metastasis are reported to be closely correlated in human cancer patients.92This combinatorial strategy, which involves the use of photosensitive natural molecules complexed with platinum and nanoformulations, may pave the way for a more effectual cure for treatment-resistant human carcinomas.
Author contributions
Viviana Vergaro performed conceptualization, methodology, and data curation and wrote the original draft. Maria Michela Dell'Anna and Hamid Reza Shahsavari performed the methodology and investigation and wrote the original draft. Francesca Baldassarre and Danilo Migoni performed the investigation. Piero Mastrorilli, Francesco Paolo Fanizzi and Giuseppe Ciccarella revised the manuscript and funded the research activity.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was supported by “Tecnopolo di Nanotecnologia e Fotonica per la Medicina di Precisione” (TECNOMED)-FISR/MIUR-CNR: delibera CIPE no. 3449 del 7/08/2017, CUP: B83B17000010001; “Tecnopolo per la Medicina di precisione” (TecnoMed Puglia) – Regione Puglia: DGR no. 2117 del 21/11/2018, CUP: B84I18000540002.Notes and references
- I. Kostova, Recent Pat. Anti-Cancer Drug Discovery, 2006, 1, 1–22 CrossRef CAS PubMed.
- T. C. Johnstone, K. Suntharalingam and S. J. Lippard, Chem. Rev., 2016, 116, 3436–3486 CrossRef CAS PubMed.
- Y. W. Kong, E. C. Dreaden, S. Morandell, W. Zhou, S. S. Dhara, G. Sriram, F. C. Lam, J. C. Patterson, M. Quadir, A. Dinh, K. E. Shopsowitz, S. Varmeh, Ö. H. Yilmaz, S. J. Lippard, H. C. Reinhardt, M. T. Hemann, P. T. Hammond and M. B. Yaffe, Nat. Commun., 2020, 11, 1–12 CrossRef PubMed.
- J. Kim, S. Pramanick, D. Lee, H. Park and W. J. Kim, Biomater. Sci., 2015, 3, 1002–1017 RSC.
- P. Ma, H. Xiao, C. Li, Y. Dai, Z. Cheng, Z. Hou and J. Lin, Mater. Today, 2015, 18, 554–564 CrossRef CAS.
- S. J. Tabatabaei Rezaei, V. Amani, M. R. Nabid, N. Safari and H. Niknejad, Polym. Chem., 2015, 6, 2844–2853 RSC.
- X. Wang and Z. Guo, Chem. Soc. Rev., 2013, 42, 202–224 RSC.
- A. K. Renfrew, N. S. Bryce and T. Hambley, Chem.–Eur. J., 2015, 21, 15224–15234 CrossRef CAS PubMed.
- K. Karami, Z. Mehri Lighvan, H. Farrokhpour, M. Dehdashti Jahromi and A. A. Momtazi-borojeni, J. Biomol. Struct. Dyn., 2018, 36, 3324–3340 CrossRef CAS PubMed.
- P. Štarha and Z. Trávníček, Coord. Chem. Rev., 2019, 395, 130–145 CrossRef.
- M. G. A. El-Wahed, M. S. Refat and S. M. El-Megharbel, J. Mol. Struct., 2008, 892, 402–413 CrossRef CAS.
- M. Heger, R. F. van Golen, M. Broekgaarden and M. C. Michel, Pharmacol. Rev., 2014, 66, 222–307 CrossRef PubMed.
- R. Wilken, M. S. Veena, M. B. Wang and E. S. Srivatsan, Mol. Cancer, 2011, 10, 12 CrossRef CAS PubMed.
- S. C. Gupta, B. Sung, J. H. Kim, S. Prasad, S. Li and B. B. Aggarwal, Mol. Nutr. Food Res., 2013, 57, 1510–1528 CrossRef CAS PubMed.
- A. B. Kunnumakkara, D. Bordoloi, G. Padmavathi, J. Monisha, N. K. Roy, S. Prasad and B. B. Aggarwal, Br. J. Pharmacol., 2017, 174, 1325–1348 CrossRef CAS PubMed.
- S. Tai, J. Wang, F. Sun, S. Xutian, T. Wang and M. King, BMC Complementary Altern. Med., 2006, 6, 4 CrossRef PubMed.
- P. Anand, A. B. Kunnumakkara, R. A. Newman and B. B. Aggarwal, Mol. Pharm., 2007, 4, 807–818 CrossRef CAS PubMed.
- S. Prasad, S. C. Gupta, A. K. Tyagi and B. B. Aggarwal, Biotechnol. Adv., 2014, 32, 1053–1064 CrossRef CAS PubMed.
- X. Mei, D. Xu, S. Xu, Y. Zheng and S. Xu, Chem.-Biol. Interact., 2012, 197, 31–39 CrossRef CAS PubMed.
- Z. Hussain, H. E. Thu, S.-F. Ng, S. Khan and H. Katas, Colloids Surf., B, 2017, 150, 223–241 CrossRef CAS PubMed.
- A. C. da Silva, P. D. d. F. Santos, J. T. d. P. Silva, F. V. Leimann, L. Bracht and O. H. Gonçalves, Trends Food Sci. Technol., 2018, 72, 74–82 CrossRef.
- M. F. S. Jadid, B. Shademan, R. Chavoshi, N. Seyyedsani, E. Aghaei, E. Taheri, P. Goleij, S. Hajazimian, V. Karamad, J. Behroozi, M. N. Sabet, A. Isazadeh and B. Baradaran, Environ. Toxicol., 2021, 36, 1043–1051 CrossRef CAS PubMed.
- A. Zarrabi, A. Zarepour, A. Khosravi, Z. Alimohammadi and V. K. Thakur, Fibers, 2021, 9(3), 19 CrossRef CAS.
- S. Sun, X. Du, M. Fu, A. R. Khan, J. Ji, W. Liu and G. Zhai, Int. J. Pharm., 2021, 595, 120227 CrossRef CAS PubMed.
- M. Bagheri, M. H. Fens, T. G. Kleijn, R. B. Capomaccio, D. Mehn, P. M. Krawczyk, E. M. Scutigliani, A. Gurinov, M. Baldus, N. C. H. van Kronenburg, R. J. Kok, M. Heger, C. F. van Nostrum and W. E. Hennink, Mol. Pharm., 2021, 18, 1247–1263 CrossRef CAS PubMed.
- A. Shakeri, Y. Panahi, T. P. Johnston and A. Sahebkar, BioFactors, 2019, 45, 304–317 CrossRef CAS PubMed.
- S. Wanninger, V. Lorenz, A. Subhan and F. T. Edelmann, Chem. Soc. Rev., 2015, 44, 4986–5002 RSC.
- Y. Lyu, M. Yu, Q. Liu, Q. Zhang, Z. Liu, Y. Tian, D. Li and M. Changdao, Carbohydr. Polym., 2020, 230, 115573 CrossRef CAS PubMed.
- D. K. Singh, R. Jagannathan, P. Khandelwal, P. M. Abraham and P. Poddar, Nanoscale, 2013, 5, 1882–1893 RSC.
- S. A. Ahmed, Md. N. Hasan, D. Bagchi, H. M. Altass, M. Morad, R. S. Jassas, A. M. Hameed, J. Patwari, H. Alessa, A. Alharbi and S. K. Pal, ACS Omega, 2020, 5, 15666–15672 CrossRef CAS PubMed.
- M. M. Dell'Anna, V. Censi, B. Carrozzini, R. Caliandro, N. Denora, M. Franco, D. Veclani, A. Melchior, M. Tolazzi and P. Mastrorilli, J. Inorg. Biochem., 2016, 163, 346–361 CrossRef PubMed.
- V. Censi, A. B. Caballero, M. Pérez-Hernández, V. Soto-Cerrato, L. Korrodi-Gregório, R. Pérez-Tomás, M. M. Dell'Anna, P. Mastrorilli and P. Gamez, J. Inorg. Biochem., 2019, 198, 110749 CrossRef CAS PubMed.
- T. Q. Hieu and D. T. T. Thao, J. Chem., 2019, 2019, 8082195 Search PubMed.
- E. Halevas, A. Pekou, R. Papi, B. Mavroidi, A. G. Hatzidimitriou, G. Zahariou, G. Litsardakis, M. Sagnou, M. Pelecanou and A. A. Pantazaki, J. Inorg. Biochem., 2020, 208, 111083 CrossRef CAS PubMed.
- G. Mazzone, S. Scoditti, R. Caligiuri, L. Ricciardi, E. Sicilia, M. G. Lupo, I. Rimoldi, N. Godbert, M. La Deda, A. Ionescu, M. Ghedini, I. Aiello and G. Facchetti, Inorg. Chem., 2022, 61, 7188–7200 CrossRef CAS PubMed.
- A. Ionescu, R. Caligiuri, N. Godbert, L. Ricciardi, M. La Deda, M. Ghedini, N. Ferri, M. G. Lupo, G. Facchetti, I. Rimoldi and I. Aiello, Appl. Organomet. Chem., 2020, 34, e5455 CrossRef CAS.
- A. Ionescu, N. Godbert, L. Ricciardi, M. La Deda, I. Aiello, M. Ghedini, I. Rimoldi, E. Cesarotti, G. Facchetti, G. Mazzeo, G. Longhi, S. Abbate and M. Fusè, Inorg. Chim. Acta, 2017, 461, 267–274 CrossRef CAS.
- A. Singh and P. K. Dutta, Int. J. Biol. Macromol., 2020, 156, 514–521 CrossRef CAS PubMed.
- P. Jahangoshaei, L. Hassani, F. Mohammadi, A. Hamidi and K. Mohammadi, JBIC, J. Biol. Inorg. Chem., 2015, 20, 1135–1146 CrossRef CAS PubMed.
- A. Kunwar, H. Narang, K. I. Priyadarsini, M. Krishna, R. Pandey and K. B. Sainis, J. Cell. Biochem., 2007, 102, 1214–1224 CrossRef CAS PubMed.
- G. Seeta Rama Raju, E. Pavitra, G. Purnachandra Nagaraju, K. Ramesh, B. F. El-Rayes and J. S. Yu, Dalton Trans., 2014, 43, 3330–3338 RSC.
- A. L. Capodilupo, V. Vergaro, G. Accorsi, E. Fabiano, F. Baldassarre, G. A. Corrente, G. Gigli and G. Ciccarella, Tetrahedron, 2016, 72, 2920–2928 CrossRef CAS.
- A. L. Capodilupo, V. Vergaro, F. Baldassarre, A. Cardone, G. A. Corrente, C. Carlucci, S. Leporatti, P. Papadia, G. Gigli and G. Ciccarella, Biochim. Biophys. Acta, Gen. Subj., 2015, 1850, 385–392 CrossRef CAS PubMed.
- G. L. Edwards, D. S. C. Black, G. B. Deacon and L. P. G. Wakelin, Can. J. Chem., 2005, 83, 980–989 CrossRef CAS.
- M. V. Babak, M. Pfaffeneder-Kmen, S. M. Meier-Menches, M. S. Legina, S. Theiner, C. Licona, C. Orvain, M. Hejl, M. Hanif, M. A. Jakupec, B. K. Keppler, C. Gaiddon and C. G. Hartinger, Inorg. Chem., 2018, 57, 2851–2864 CrossRef CAS PubMed.
- H.-K. Liu and P. J. Sadler, Acc. Chem. Res., 2011, 44, 349–359 CrossRef CAS PubMed.
- T. Zou, J. Liu, C. T. Lum, C. Ma, R. C. T. Chan, C. N. Lok, W. M. Kwok and C. M. Che, Angew. Chem., Int. Ed., 2014, 53, 10119–10123 CrossRef CAS PubMed.
- M. Fereidoonnezhad, B. Kaboudin, T. Mirzaee, R. Babadi Aghakhanpour, M. Golbon Haghighi, Z. Faghih, Z. Faghih, Z. Ahmadipour, B. Notash and H. R. Shahsavari, Organometallics, 2017, 36, 1707–1717 CrossRef CAS.
- F. De Castro, V. Vergaro, M. Benedetti, F. Baldassarre, L. Del Coco, M. M. Dell'Anna, P. Mastrorilli, F. P. Fanizzi and G. Ciccarella, ACS Appl. Bio Mater., 2020, 3, 6836–6851 CrossRef CAS PubMed.
- V. Vergaro, F. Baldassarre, F. De Castro, D. Migoni, M. M. Dell'Anna, P. Mastrorilli, F. P. Fanizzi and G. Ciccarella, Bioinorg. Chem. Appl., 2022, 2022, 9571217 Search PubMed.
- G. A. Martău, M. Mihai and D. C. Vodnar, Polymers, 2019, 11(11), 1837 CrossRef PubMed.
- V. B. V. Maciel, C. M. P. Yoshida and T. T. Franco, Carbohydr. Polym., 2015, 132, 537–545 CrossRef CAS PubMed.
- V. Vergaro, P. Papadia, P. Petrini, F. P. Fanizzi, S. A. De Pascali, F. Baldassarre, L. Pastorino and G. Ciccarella, Int. J. Biol. Macromol., 2017, 99, 187–195 CrossRef CAS PubMed.
- S. T. Minzanova, V. F. Mironov, D. M. Arkhipova, A. V. Khabibullina, L. G. Mironova, Y. M. Zakirova and V. A. Milyukov, Polymers, 2018, 10(12), 1407 CrossRef PubMed.
- E. A. Günter, O. V. Popeyko, P. A. Markov, E. A. Martinson, S. G. Litvinets, E. A. Durnev, S. V. Popov and Y. S. Ovodov, Carbohydr. Polym., 2014, 103, 550–557 CrossRef PubMed.
- V. B. V. Maciel, C. M. P. Yoshida, S. M. S. S. Pereira, F. M. Goycoolea and T. T. Franco, Molecules, 2017, 22(10), 1707 CrossRef PubMed.
- S. Comsa, A. M. Cimpean and M. Raica, Anticancer Res., 2015, 35, 3147–3154 CAS.
- J. R. Berenguer, J. G. Pichel, N. Giménez, E. Lalinde, M. T. Moreno and S. Piñeiro-Hermida, Dalton Trans., 2015, 44, 18839–18855 RSC.
- M. Fereidoonnezhad, M. Niazi, Z. Ahmadipour, T. Mirzaee, Z. Faghih, Z. Faghih and H. R. Shahsavari, Eur. J. Inorg. Chem., 2017, 2017, 2247–2254 CrossRef CAS.
- M. Frezza, Q. P. Dou, Y. Xiao, H. Samouei, M. Rashidi, F. Samari and B. Hemmateenejad, J. Med. Chem., 2011, 54, 6166–6176 CrossRef CAS PubMed.
- J. Brooks, Y. Babayan, S. Lamansky, P. I. Djurovich, I. Tsyba, R. Bau and M. E. Thompson, Inorg. Chem., 2002, 41, 3055–3066 CrossRef CAS PubMed.
- A. Esmaeilbeig, H. Samouei, S. Abedanzadeh and Z. Amirghofran, J. Organomet. Chem., 2011, 696, 3135–3142 CrossRef CAS.
- M. S. Refat, Spectrochim. Acta, Part A, 2013, 105, 326–337 CrossRef CAS PubMed.
- F. Kühlwein, K. Polborn and W. Beck, Z. Anorg. Allg. Chem., 1997, 623, 1211–1219 CrossRef.
- R. Benassi, E. Ferrari, S. Lazzari, F. Spagnolo and M. Saladini, J. Mol. Struct., 2008, 892, 168–176 CrossRef CAS.
- K. Nakamoto, in Handbook of Vibrational Spectroscopy, 2001 Search PubMed.
- S. Kwiatkowski, B. Knap, D. Przystupski, J. Saczko, E. Kędzierska, K. Knap-Czop, J. Kotlińska, O. Michel, K. Kotowski and J. Kulbacka, Biomed. Pharmacother., 2018, 106, 1098–1107 CrossRef PubMed.
- J. Seung Lee, J. Kim, Y.-s. Ye and T.-i. Kim, Adv. Drug Delivery Rev., 2022, 186, 114339 CrossRef CAS PubMed.
- Y. Chen, C. Chen, X. Zhang, C. He, P. Zhao, M. Li, T. Fan, R. Yan, Y. Lu, R. J. Lee, M. W. Khan, M. Sarfraz, X. Ma, T. Yang and G. Xiang, Acta Pharm. Sin. B, 2020, 10, 1106–1121 CrossRef CAS PubMed.
- L. Chen, G. Bai, S. Yang, R. Yang, G. Zhao, C. Xu and W. Leung, Food Res. Int., 2014, 62, 1147–1153 CrossRef CAS.
- J. A. Ramos-Hernández, J. A. Ragazzo-Sánchez, M. Calderón-Santoyo, R. I. Ortiz-Basurto, C. Prieto and J. M. Lagaron, Nanomaterials, 2018, 8(11), 868 CrossRef PubMed.
- K. Mitra, S. Gautam, P. Kondaiah and A. R. Chakravarty, Angew. Chem., Int. Ed., 2015, 54, 13989–13993 CrossRef CAS PubMed.
- S. J. Tabatabaei Rezaei, A. Hesami, H. Khorramabadi, V. Amani, A. M. Malekzadeh, A. Ramazani and H. Niknejad, Appl. Organomet. Chem., 2018, 32, e4401 CrossRef.
- H. Chen, S. Pazicni, N. L. Krett, R. W. Ahn, J. E. Penner-Hahn, S. T. Rosen and T. V. O'Halloran, Angew. Chem., Int. Ed., 2009, 48, 9295–9299 CrossRef CAS PubMed.
- N. V. Nukolova, H. S. Oberoi, S. M. Cohen, A. V. Kabanov and T. K. Bronich, Biomaterials, 2011, 32, 5417–5426 CrossRef CAS PubMed.
- A. A. Bhirde, V. Patel, J. Gavard, G. Zhang, A. A. Sousa, A. Masedunskas, R. D. Leapman, R. Weigert, J. S. Gutkind and J. F. Rusling, ACS Nano, 2009, 3, 307–316 CrossRef CAS PubMed.
- C.-L. Tseng, W.-Y. Su, K.-C. Yen, K.-C. Yang and F.-H. Lin, Biomaterials, 2009, 30, 3476–3485 CrossRef CAS PubMed.
- X.-H. Peng, Y. Wang, D. Huang, Y. Wang, H. J. Shin, Z. Chen, M. B. Spewak, H. Mao, X. Wang, Y. Wang, Z. Chen, S. Nie and D. M. Shin, ACS Nano, 2011, 5, 9480–9493 CrossRef CAS PubMed.
- C. Xu, B. Wang and S. Sun, J. Am. Chem. Soc., 2009, 131, 4216–4217 CrossRef CAS PubMed.
- A. Muscella, C. Vetrugno, F. P. Fanizzi, C. Manca, S. A. De Pascali and S. Marsigliante, Cell Death Dis., 2014, 4, 796 CrossRef PubMed.
- H. R. Shahsavari, J. Hu, S. Chamyani, Y. Sakamaki, R. B. Aghakhanpour, C. Salmon, M. Fereidoonnezhad, A. Mojaddami, P. Peyvasteh and H. Beyzavi, Organometallics, 2021, 40, 72–82 CrossRef CAS PubMed.
- S. Kemp, N. J. Wheate, S. Wang, J. G. Collins, S. F. Ralph, A. I. Day, V. J. Higgins and J. R. Aldrich-Wright, JBIC, J. Biol. Inorg. Chem., 2007, 12, 969–979 CrossRef CAS PubMed.
- G. Weber, J. Messerschmidt, A. C. Pieck, A. M. Junker, A. Wehmeier and U. Jaehde, Anal. Bioanal. Chem., 2004, 380, 54–58 CrossRef CAS PubMed.
- A. Ghezzi, M. Aceto, C. Cassino, E. Gabano and D. Osella, J. Inorg. Biochem., 2004, 98, 73–78 CrossRef CAS PubMed.
- S. Arnould, I. Hennebelle, P. Canal, R. Bugat and S. Guichard, Eur. J. Cancer, 2003, 39, 112–119 CrossRef CAS PubMed.
- A. Shah, S. Aftab, J. Nisar, M. N. Ashiq and F. J. Iftikhar, J. Drug Delivery Sci. Technol., 2021, 62, 102426 CrossRef CAS.
- Y. Chen, C. Chen, X. Zhang, C. He, P. Zhao, M. Li, T. Fan, R. Yan, Y. Lu, R. J. Lee, M. W. Khan, M. Sarfraz, X. Ma, T. Yang and G. Xiang, Acta Pharm. Sin. B, 2020, 10, 1106–1121 CrossRef CAS PubMed.
- K. Mitra, S. Gautam, P. Kondaiah and A. R. Chakravarty, Angew. Chem., 2015, 127, 14195–14199 CrossRef.
- E. Lalinde, R. Lara, I. P. López, M. T. Moreno, E. Alfaro-Arnedo, J. G. Pichel and S. Piñeiro-Hermida, Chem.–Eur. J., 2018, 24, 2440–2456 CrossRef CAS PubMed.
- K. Suntharalingam, O. Mendoza, A. A. Duarte, D. J. Mann and R. Vilar, Metallomics, 2013, 5, 514–523 CrossRef CAS PubMed.
- S. Paziresh, V. Sicilia, I. Ara, A. Martín and S. Fuertes, Organometallics, 2019, 38, 3804–3815 CrossRef CAS.
- S.-Q. Liang, T. M. Marti, P. Dorn, L. Froment, S. R. R. Hall, S. Berezowska, G. Kocher, R. A. Schmid and R.-W. Peng, Cell Death Dis., 2015, 6, e1824 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2023 |