
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePolymorph screening at surfaces of a benzothienobenzothiophene derivative: discovering new solvate forms†
Ann Maria
James
 a,
Nemo
McIntosh
b,
Félix
Devaux
c,
Patrick
Brocorens
b,
Jérôme
Cornil
b,
Alessandro
Greco
d,
Lucia
Maini
e,
Priya
Pandey
e,
Lorenzo
Pandolfi
f,
Birgit
Kunert
a,
Elisabetta
Venuti
f,
Yves Henri
Geerts
cg and
Roland
Resel
*a
a,
Nemo
McIntosh
b,
Félix
Devaux
c,
Patrick
Brocorens
b,
Jérôme
Cornil
b,
Alessandro
Greco
d,
Lucia
Maini
e,
Priya
Pandey
e,
Lorenzo
Pandolfi
f,
Birgit
Kunert
a,
Elisabetta
Venuti
f,
Yves Henri
Geerts
cg and
Roland
Resel
*a
aInstitute of Solid State Physics, Graz University of Technology, Petersgasse 16, 8010 Graz, Austria. E-mail: roland.resel@tugraz.at
bLaboratory for Chemistry of Novel Materials, University of Mons, 7000 Mons, Belgium
cLaboratoire de Chimie des Polymères, Université Libre de Bruxelles (ULB), 1050 Bruxelles, Belgium
dMax Planck Institute for Polymer Research, 55128 Mainz, Germany
eDipartimento di Chimica “G. Ciamician”, University Bologna, 40126 Bologna, Italy
fDipartimento di Chimica Industriale “Toso Montanari”, Università di Bologna viale del Risorgimento, 4, 40136, Bologna, Italy
gInternational Solvay Institutes of Physics and Chemistry, Université Libre de Bruxelles, 1050 Bruxelles, Belgium
First published on 12th July 2023
Abstract
The discovery of new polymorphs opens up unique applications for molecular materials since their physical properties are predominantly influenced by the crystal structure type. The deposition of molecules at surfaces offers great potential in the variation of the crystallization conditions, thereby allowing access to unknown polymorphs. With our surface crystallization approach, four new phases are found for an oligoethylene glycol-benzothienobenzothiophene molecule, and none of these phases could be identified via classical polymorph screening. The corresponding crystal lattices of three of the new phases were obtained via X-ray diffraction (XRD). Based on the volumetric considerations together with X-ray fluorescence and Raman spectroscopy data, the phases are identified as solvates containing one, two or three solvent molecules per molecule. The strong interaction of dichloromethane with the oligoethylene glycol side chains of the molecules may be responsible for the formation of the solvates. Temperature-dependent XRD reveals the low thermal stability of the new phases, contrary to the thermodynamically stable bulk form. Nevertheless, the four solvates are stable under ambient conditions for at least two years. This work illustrates that defined crystallization at surfaces enables access to multiple solvates of a given material through precise and controlled variations in the crystallization kinetics.
New conceptsPolymorph screening, as an established technique to discover new crystal structures of molecular materials, is routinely applied to pharmaceutical compounds and organic semiconductors. However, the method can be improved substantially by including substrate surfaces as templates for crystallization. Especially in the case of thin-film applications for a specific molecular material, crystallization at surfaces must be taken into account to identify the relevant phases. This work demonstrates that combining classical polymorph screening with surface crystallization reveals the possibility of finding new phases of molecular crystal materials. |
Introduction
Polymorphism is the ability of a compound to possess distinct crystal structures with the same molecular composition, which occurs as a consequence of the complex interplay between thermodynamics and kinetics during the crystallization process.1–3 For a given compound, different polymorphic forms emerge due to the alterations that occur either in the packing arrangement of a defined molecular conformation or through a change in the molecular conformation by itself.4,5 A wide range of different technologies is highly impacted by polymorphism, as physical properties are strongly dependent on the crystal structure.6–9 The development of novel molecular materials for specific applications (e.g., pharmaceuticals) starts with the design and synthesis of new molecules, and is often accompanied by a polymorph screening process.10,11 The underlying idea behind the screening process is to gain access to a maximum number of phases and to choose the best crystal forms in terms of the processability, performance and stability for the desired properties.12 The strategy adopted is often one of crystallization under a broad range of conditions, mainly via solution processing. Starting from a specific solvent library, single/mixed solvents are used to prepare solutions, and via supersaturation by cooling, evaporation or anti-solvent use, nucleation is initiated and crystal growth occurs.13,14During the crystallization processes, the systems have the tendency to move towards thermodynamic equilibrium.15 However, the route towards equilibrium passes through local energy minima, and hence the possibilities of stable molecular packing are highly diverse.16,17 During the solidification process, if kinetic processes dominate, metastable polymorphic forms with a larger free energy may eventually emerge.18,19 A less stabilising free energy is associated with metastable forms. The minima could be reached by varying the crystal growth kinetics, which are highly dependent on the external conditions such as the temperature, the type of solvent, molecular additives and impurities.20,21 However, the meticulous role of kinetic factors is far from being understood.22
Crystallization can also be carried out under spatial confinement, such as in nano-pores23,24 or at substrate surfaces.25–27 Utilizing solid surfaces as nucleation templates is undoubtedly a promising strategy for discovering new forms.28–30 In some systems, the adaptation of the molecular packing to a substrate surface leads to an entropy reduction and thereby results in the formation of new phases.31–33 Thus molecular crystals nucleating in the proximity of a surface may organise differently, and these new phases are not accessible otherwise.27 Therefore, it is vital to integrate this feature into the polymorph screening process.
One additional aspect of crystallization at surfaces is that the growth kinetics can be varied over a range that is considerably compared with other techniques. There exist a number of diverse thin-film preparation techniques which result in crystalline phases away from the thermodynamic equilibrium. Solution-based methods such as spin-coating, drop-casting, dip-coating and blade-coating are good examples of such thin-film techniques.34–36 One key parameter in the solution-based deposition technique is the evaporation rate of the solvent during the crystallization process.37 Solvent evaporation can also be controlled relatively well by monitoring the experimental conditions such as the substrate temperature during the deposition process or by maintaining a solvent atmosphere around the crystallization site.38–40
The present work illustrates that the classical polymorph screening technique carried out using a defined set of solvents can be combined with thin-film crystallization. The method is demonstrated here using an organic semiconducting small molecule, OEG-BTBT, whose chemical structure is depicted in Fig. 1. Recently, a classical polymorph screening investigation on this molecule was published.41 Two enantiotropically related phases were found (Form I, and Form II) together with a solvate with dichloromethane (Form III). Our surface crystallization approach identifies four additional solvates of OEG-BTBT. Please note that the formation of a solvate is quite rare for organic electronic molecules.
 | ||
| Fig. 1 Chemical structure of 2,7-bis(2-(2-methoxyethoxy)ethoxy)benzo[b]-benzo[4,5]thieno[2,3-d]thiophene (OEG-BTBT). | ||
Experimental methods
Crystallization of the 2,7-bis(2-(2-methoxyethoxy)ethoxy)benzo[b]benzo[4,5]thieno[2,3-d]thiophene (OEG-BTBT) molecule was investigated on silicon oxide surfaces via solution processing. Ten different solvents with boiling points in the range of 312.7 K (dichloromethane) to 532 K (chloronaphthalene) were selected based on the recommendation used in the established polymorph screening methodology.11 The as-synthesized material42 was dissolved up to the solubility limit and was either spin-coated or drop-cast onto 2 × 2 cm silicon wafers with a 150 nm thick thermally grown oxide coating. Prior to the deposition, the substrates were chemically cleaned via ultrasonication in acetone for 15 min, followed by rinsing with isopropanol and drying with CO2 gas. The surface energy of the cleaned substrates was determined via contact angle measurements using four different solvents.43 A total surface energy of 49 mN m−1 was found with a polar part of 24 mN m−1 and a dispersive part of 25 mN m−1.New phases were found only in the case of dichloromethane (DCM) and 1,2-dichlorobenzene (DCB) solvents. The new phases were compared with the well-characterized stable form of OEG-BTBT prepared from chloroform (CF) solutions. The solubility limit is 0.8 g l−1, 0.5 g l−1 and 0.9 g l−1 for DCM, DCB and CF, respectively.
For the spin-coating process, the VTC-100 PA spin coater (Shenyang Kejing Auto-instrument Co. Ltd) was used and 200 μL of the solution was drop cast onto the substrates. The spin coater was programmed to do a sequential 2-step spinning process with a coating speed of 1000 rpm for 9 s followed by spinning at 1500 rpm for 30 s. For drop casting, 250 μL of the as-prepared solutions were cast onto the substrate surface, and the evaporation of the solvent was carried out in both open and closed environments. In order to drop cast at elevated temperatures, a heating chamber was used for temperature equilibration of the substrate.
The surface-deposited OEG-BTBT crystals were characterized using two different X-ray based methods: specular X-ray diffraction (XRD) and grazing incidence X-ray diffraction (GIXD). The XRD measurements were carried out using a PANalytical Empyrean diffractometer. The primary side is equipped with a sealed copper tube and a multilayer mirror for monochromatization (λ = 1.5418 Å) and to produce a parallel beam. The secondary side has an anti-scatter slit, a 0.02 rad Soller slit, and a PANalytical PIXcel 3D detector operating as a point detector. The measurements were converted to the reciprocal space using the expression qz = 4π/λ![[thin space (1/6-em)]](https://www.rsc.org/images/entities/i_char_2009.gif) sin
sin![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) θ, where qz is the scattering vector perpendicular to the substrate and 2θ is the scattering angle. The observation of Bragg's peaks was analysed in terms of the peak positions, from which the corresponding interplanar distance (d) of a net plane series can be calculated via d = 2π/qz. The crystal size L in the vertical (z) direction is related to the peak width Δqz by L = 2π/Δqz. In situ crystallization studies were performed using an AntonPaar DHS900 heating stage under inert (nitrogen) conditions.44 A defined heating rate of 4° min−1 was used during the heating and cooling experiments. Once the required annealing temperature had been reached, the system was maintained under the same conditions for 30 minutes so that the system could be in equilibrium.45
θ, where qz is the scattering vector perpendicular to the substrate and 2θ is the scattering angle. The observation of Bragg's peaks was analysed in terms of the peak positions, from which the corresponding interplanar distance (d) of a net plane series can be calculated via d = 2π/qz. The crystal size L in the vertical (z) direction is related to the peak width Δqz by L = 2π/Δqz. In situ crystallization studies were performed using an AntonPaar DHS900 heating stage under inert (nitrogen) conditions.44 A defined heating rate of 4° min−1 was used during the heating and cooling experiments. Once the required annealing temperature had been reached, the system was maintained under the same conditions for 30 minutes so that the system could be in equilibrium.45
GIXD investigations were performed under ambient conditions at the XRD1 beamline, Elettra synchrotron, Trieste, Italy. The primary beam had an incidence angle of 1.5° relative to the substrate surface with a beam dimension of 50 × 50 μm and a wavelength of 1.400 Å. A stationary Dectris Pilatus 2M detector was used for collecting the diffracted intensity. Calibration of the experimental set up was performed on polycrystalline LaB6. In order to improve the statistics, the samples were rotated during the measurement, and the diffracted intensities were integrated over an exposure time of 10 s for a sample rotation of 10°. Real space detector images were converted into reciprocal space as a function of qz (the component of the scattering vector perpendicular to the substrate surface) and of qxy (the component of the scattering vector parallel to the substrate surface). The data were assessed using the in-house GIDVis software package,46 which creates 2-D contour plots with a linearly scaled colour code ranging from purple (representing low intensity) to yellow (representing high intensity). The resultant reciprocal space maps are corrected based on geometrical correction factors, such as Lorentz and polarization factor. GIDVis provides tools to determine the peak positions and peak intensities. The information on the peak positions was used for indexing of the observed Bragg peaks using GIDInd software.47
Results
Thin films of known phases
As a first step, thin films were prepared via spin coating and drop casting from dissolutions prepared with ten different solvents, and in eight cases clear Bragg peaks were observed. As an example, the results obtained with chloroform (CF) solutions are discussed hereafter. The XRD pattern of the samples deposited via spin coating versus drop casting are depicted in Fig. 2a. The diffraction peaks observed at qz = 0.342 Å−1 for both samples can be assigned to the 100 peak of the stable bulk phase of the OEG-BTBT molecule at room temperature (Form I). The contrast in peak width for these two films corresponds to the different crystal sizes: values of 9 nm and 56 nm are estimated for spin coating and drop casting, respectively. No clear Bragg peak could be observed for thin films prepared from dichloromethane (DCM) via spin coating, and only thickness interference features (Kiessig fringes arising from a thickness of 5 nm) were observed, as shown in Fig. 2b. In a subsequent step, the thin film was annealed at a temperature of 318 K for 20 minutes in order to induce crystallization. Fig. 2b clearly shows the amorphous-to-crystalline transformation from post-deposition heat treatment due to the development of Bragg peaks.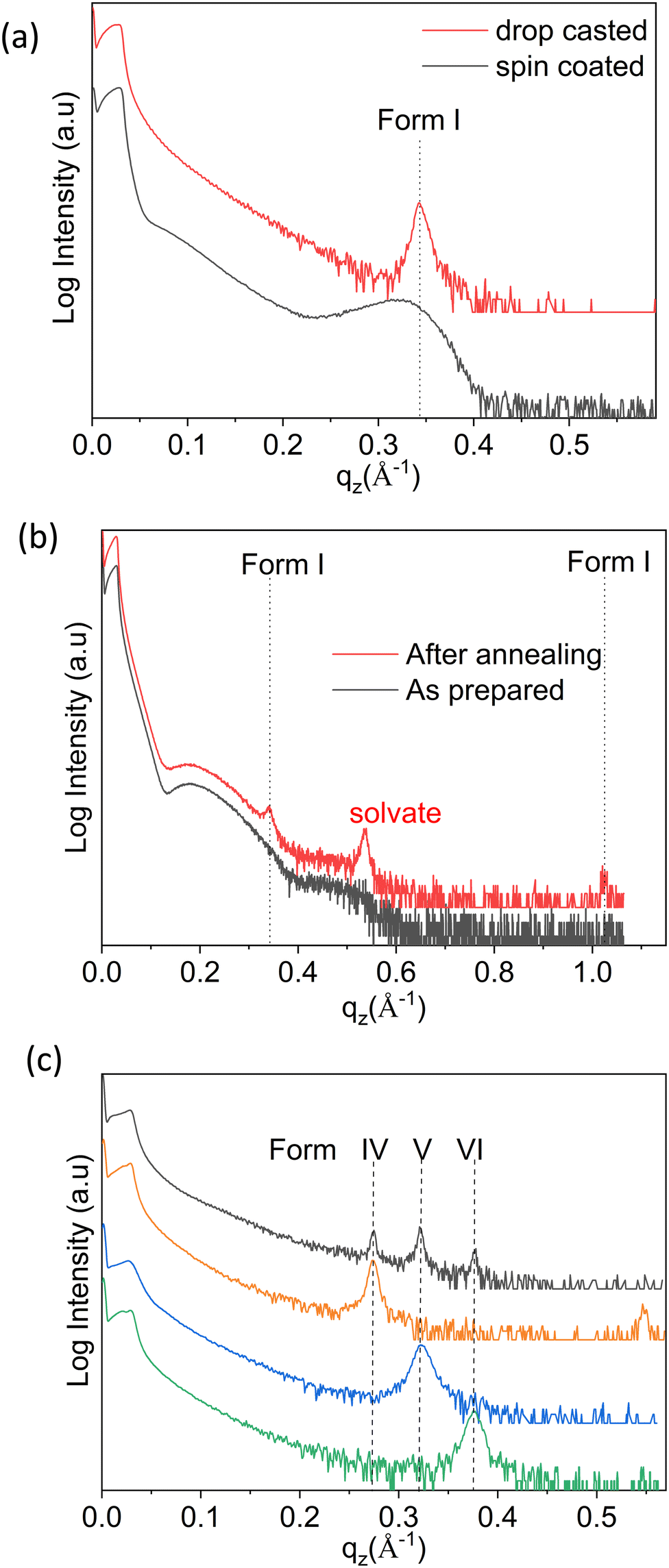 | ||
| Fig. 2 Specular X-ray diffraction curves of (a) a spin-coated and a drop-cast film from chloroform solution, and (b) a spin-coated film from dichloromethane solution in the as-prepared state and after annealing (318 K for 20 min). (c) XRD curves of films drop cast from dichloromethane revealing three unknown phases (Forms IV, V, and VI). Details of the different preparation conditions are given in the text. Throughout, the respective characteristic peak positions are given by the dotted vertical lines. | ||
Besides the thickness the interference fringes, the annealed film shows a number of Bragg peaks that can be unambiguously assigned to already known phases of OEG-BTBT. The dominant features are a Bragg peak of a DCM solvate41 and the 100/200 peaks of Form I. This film shows limited stability over time, i.e., within days, the diffraction peak of the solvate disappears and the peaks of Form I increase in intensity. This is in full agreement with previous observations that a metastable solvate phase converts into Form I within a day.41 The prerequisites for the appearance of the already known phases of OEG-BTBT in the thin films are collected in Table 1.
| Phase | Thin-film preparation conditions |
|---|---|
| Form I | From chloroform. |
| From dichloromethane (DCM): – spin coating + annealing at 318 K + aging | |
| Form II | From Form I, stable 403–438 K |
| Form III | From DCM (1 g L−1): – solvothermal41 |
| Form IV | From DCM (1 g L−1): – drop casting at room temperature |
| Form V | From DCM (1 g L−1): – drop casting at 333 K; – drop casting at a reduced evaporation rate |
| Form VI | From DCM (4 g L−1): – drop casting at room temperature |
| Form VII | From 1,2-dichlorobenzene (DCB; 1 g L−1): – drop casting at a reduced evaporation rate |
Thin films of new phases
The next sample series were prepared via the drop casting of DCM solutions onto the substrates under different deposition conditions, facilitating a broader variation of the crystallization kinetics. Different evaporation rates were achieved during the solidification process: (i) at different temperatures (from 303 to 353 K in steps of 10 K); (ii) by covering the sample with a Petri dish; and (iii) placing the sample in a closed chamber. The XRD pattern of the film deposited at a temperature of 303 K is shown in Fig. 2c (black curve), and the peak patterns corresponding to depositions at other temperatures are given in Fig. S1 (ESI†). Three independent X-ray diffraction peaks are assigned to three new phases of OEG-BTBT, denoted here as Form IV, Form V and Form VI. Reduction of the solvent evaporation rate, and variation of the deposition temperature and the concentration of the solution provides access to the phase pure forms. The defined preparation conditions are listed in Table 1 and the corresponding XRD patterns are presented in Fig. 2c.These new phases appear to be stable under ambient conditions over long periods, as tested repeatedly over the course of two years. The diffraction patterns at large qz values (large scattering angles 2θ) are depicted in Fig. S2 (ESI†). Peaks are found at equidistant positions, which implies that they can be assigned to higher order diffraction caused by a single interplanar distance. This means that a strong preferred orientation of the crystals is present with a defined crystallographic plane parallel to the substrate surface. In addition to the phase pure forms, films with different combinations of coexisting (concomitant) phases could be prepared. Their respective XRD patterns are shown in Fig. S3 (ESI†), and the corresponding processing conditions required to obtain these specific phase mixtures are listed in Table S1 (ESI†).
The three new phases were investigated via GIXD to obtain a larger data set for crystal structure determination. The collected GIXD patterns are shown in Fig. 3; for comparison, the GIXD pattern of a Form I sample is shown in Fig. S4a (ESI†). In all cases, a pronounced preferred orientation (or texture) of the crystals is observed, which is shown by spot-like or arc-like diffraction features. Indexation of the individual diffraction pattern was performed using 12 peaks for Form IV, 11 peaks for Form V and 8 peaks for Form VI. The associated crystallographic lattices of the three new phases could be identified; the lattice constants are listed in Table S2 (ESI†) together with those of the known phases for comparison. The differences between the three new phases and the stable phase of OEG-BTBT were also investigated using high-frequency Raman spectroscopy, probing intramolecular vibrations. Explicit differences are observed between all four different phases (Fig. S5, ESI†). Unfortunately, a defined molecular packing cannot be provided for the three presented crystal structures. Therefore, only qualitative considerations can be made based on the detected crystalline lattices.
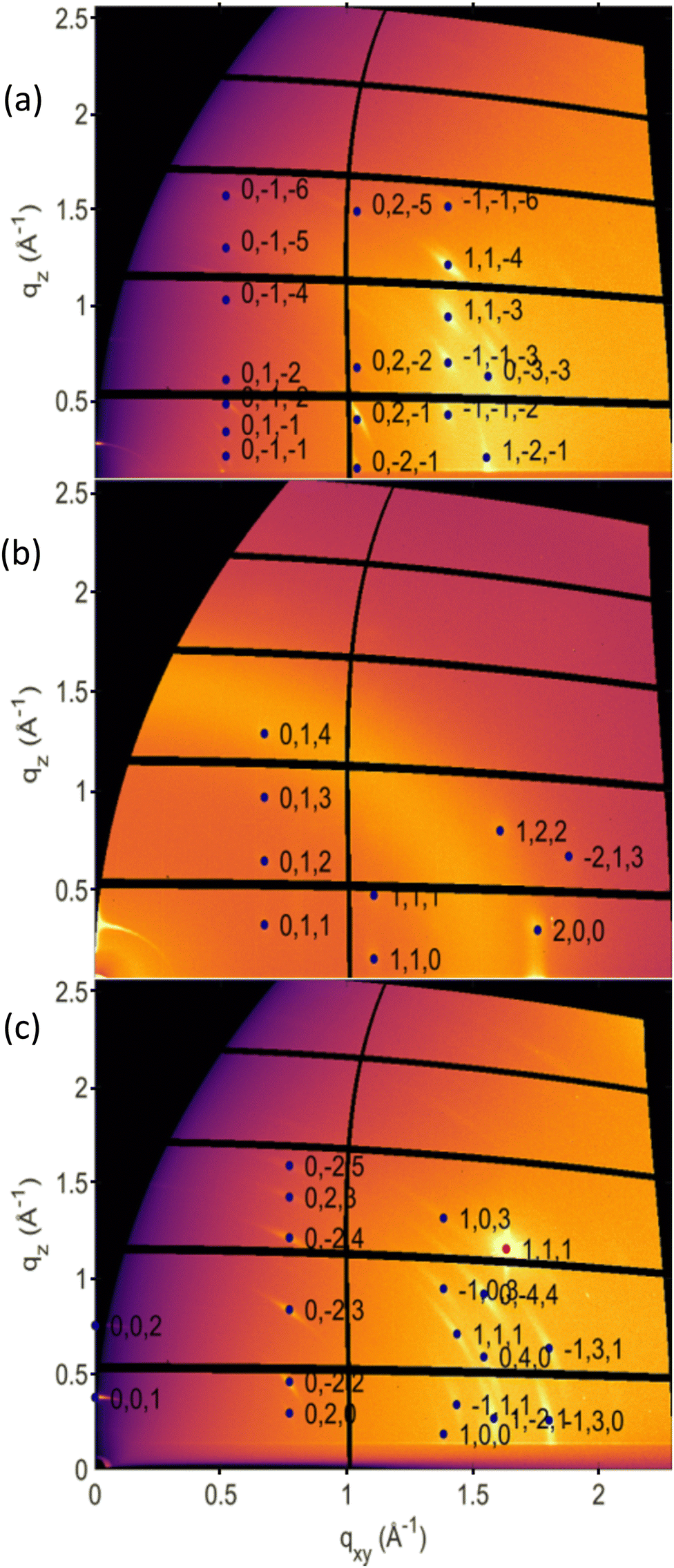 | ||
| Fig. 3 Grazing incidence X-ray diffraction images recorded on three thin-film phases of OEG- BTBT: (a) Form IV, (b) Form V, and (c) Form VI. Indexing of the Bragg peaks (blue points) reveals the corresponding crystallographic lattices; a single diffraction peak of the silicon substrate is observed (red point). | ||
The analysis of the crystal structure was performed on the basis of the volumes of the crystallographic unit cells, whose values are given in Table 2. Despite significant differences in the lattice constants of the new phases, the volumes of the crystallographic unit cells are comparable to Form I. Therefore, we assign two molecules per unit cell (Z = 2) to the three new phases. In a subsequent step, the observed unit cell volumes were considered in more detail by assuming the required space of one OEG-BTBT molecule in the crystalline environment to be 584.5 Å3, i.e., the value obtained from Form I. With the knowledge that the composition of the DCM solvate (Form III) is one DCM molecule per OEG-BTBT molecule, we estimated the required volume of a single DCM molecule to be 64.8 Å3. This value fits reasonably well with the estimated van der Waals volumes, depending on the van der Waals radii, and values between 58 and 83 Å3 were obtained.48 Assuming three, two and one DCM molecule per OEG-BTBT unit incorporated into Forms IV, V and VI, respectively, comparable DCM volumes are yielded within our three new phases (compare Table 2). Therefore, we are led to the conclusion that Forms IV, V and VI are solvates that include different fractions of solvent molecules within their crystalline structures. The presence of DCM is further supported by observing the characteristic dichloromethane peak at 1423 cm−1 in the Raman spectra (Fig. S5, ESI†) for Forms IV and VI,49 as well as by the detection of chlorine atoms in the films via X-ray fluorescence, as shown in Fig. S6a (ESI†). However, no clear identification of the DCM solvent molecule could be obtained via infrared spectroscopy in the characteristic spectral range between 700 and 1500 cm−1 (Fig. S6b, ESI†).
| Form I | Form II | Form III | Form IV | Form V | Form VI | |
|---|---|---|---|---|---|---|
| Temperature [K] | 298 | 404 | 100 | 298 | 298 | 298 |
| Volume [Å3] | 1169.1 | 3619.7 | 2597.3 | 1399.0 | 1312.7 | 1236.6 |
| Z | 2 | 6 | 4 | 2 | 2 | 2 |
| Solvent molecules per unit cell | 0 | 0 | 4 | 6 | 4 | 2 |
| Volume for one OEG-BTBT molecule [Å3] | 584.5 | 603.3 | ||||
| Estimated volume per solvent molecule [Å3] | 0 | 0 | 64.8 | 76.6 | 71.8 | 67.5 |
| Transition temperature [K] | 403.0 | 429.0 | Unstable | 347.2 | 347.9 | 366.0 |
Thermal stabilities
The thermal stability of the solvates was studied using temperature-dependent XRD, and the results are shown in Fig. 4. Upon heating the phase pure film of Form I up to a temperature of 443 K, a phase transition to Form II is observed at 403 K, and the final melting is witnessed at 429 K (Fig. 4a). These transition temperatures are in complete agreement with previous work carried out on Form I.56 Recrystallization from the melt (starting at 443 K) regenerates Form II at 423 K and finally Form I at 398 K (Fig. S4b, ESI†). The observed reversibility confirms that Form I and Form II are enantiotropically related and can be considered thermodynamically stable in their specific temperature range.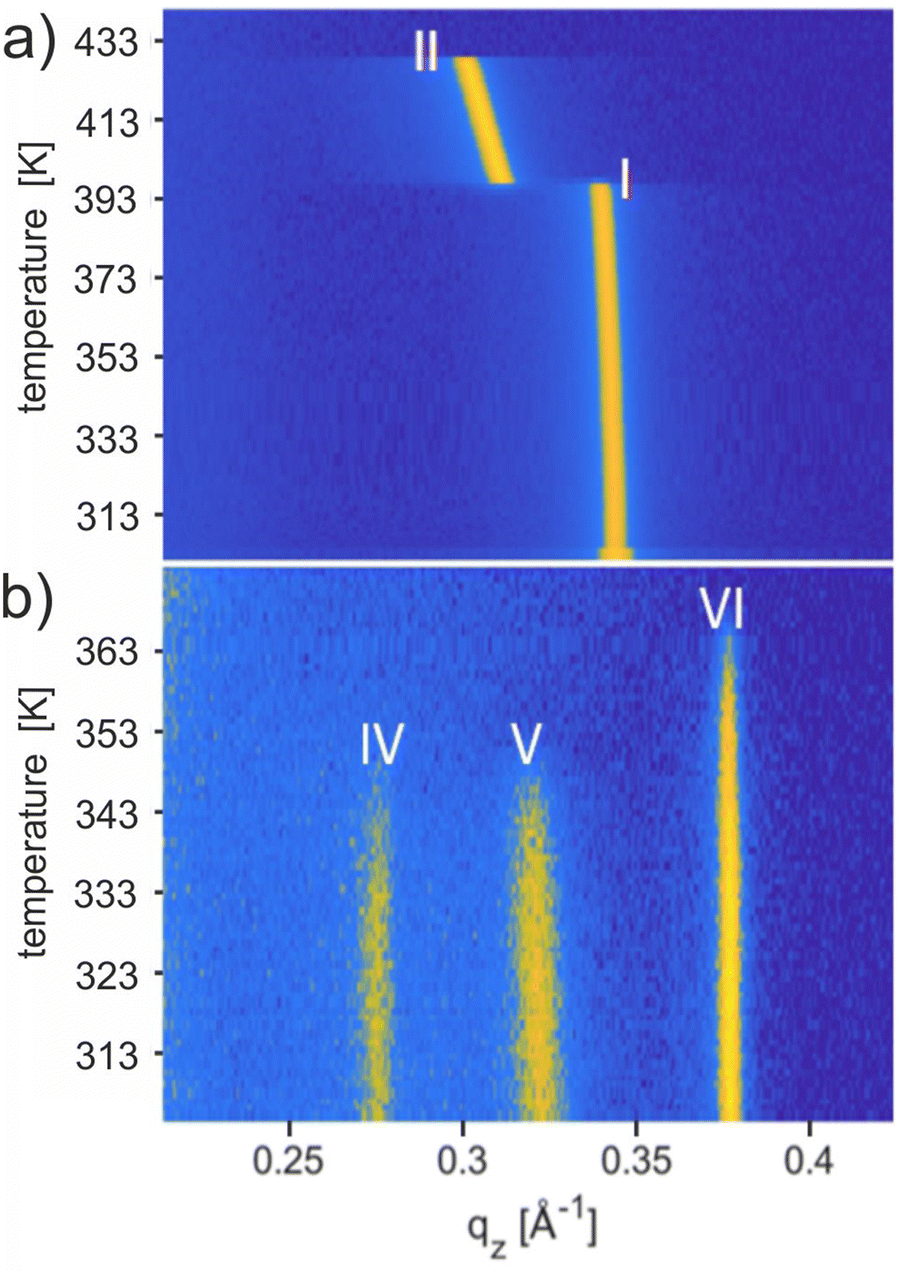 | ||
| Fig. 4 Temperature-dependent in situ XRD measurements for OEG-BTBT films, prepared via drop casting, in a waterfall plot. (a) Thin film of Form I prepared from chloroform solution, and (b) thin film containing all three new solvates simultaneously. | ||
Results from temperature-dependent XRD investigations of the new solvates are shown in Fig. 4b performed on a sample with concomitant solvates. Form IV and Form V show the disappearance of the Bragg peaks at 347.2 K and 347.9 K, respectively, whereas, for Form VI, the peak vanishes at 366 K. The absence of any diffraction peaks above these temperatures indicates that phase transitions are reached. The observed thermal stability for all three phases is far below the melting temperature of Form II (429 K) and clearly below the phase transition of Form I (403 K). The transition temperatures are given in Table 2. Note that a change from Form IV to Form I is observed by annealing a phase pure thin film of Form IV at a temperature of 403 K and 413 K for 30 minutes and subsequent cooling to room temperature (Fig. S7, ESI†).50
Using the 1,2-dichlorobenzene solvent for thin-film fabrication gave one additional unknown phase of OEG-BTBT, which is denoted as Form VII in Table 1. Drop-casting experiments with a reduced solvent evaporation rate during the crystallization process resulted in thin films with two coexisting phases: Form VII and Form I. However, attempts to prepare phase pure films of Form VII were unsuccessful. The major challenge lies in further reducing the evaporation rate of the high-boiling-point solvent 1,2 dichlorobenzene. Fig. S8 (ESI†) shows the corresponding experimental results: XRD curves, GIXD, Raman spectroscopy, and temperature-dependent XRD measurements. The temperature-dependent investigations carried out on Form VII reveal its stability up to 308 K.
Discussion
Recently, the OEG-BTBT molecule was subjected to a systematic search process via polymorph screening.41 As a result, three different types of bulk crystal structure were identified: the thermodynamically stable Form I at room temperature, the high-temperature polymorph Form II, and the metastable solvate with dichloromethane Form III. Moreover, the preparation of thin films of OEG-BTBT via solution processing unveiled four additional phases: three new phases from dichloromethane and one new phase from 1,2-dichlorobenzene. In the first three cases the phases could be unambiguously identified as a solvate. In all four cases, the strong preferred orientation of the crystallites in the GIXD image indicates that the presence of the surface impacts the crystallization kinetics.The observed thermal stability of the new phases is observed to range from 347 K (Form IV) to 366 K (Form VI), values that are far below the phase transition temperature of the thermodynamically stable phase at room temperature (403 K) or the high-temperature phase (429 K). Normally, phase pure crystalline polymorphs of molecular compounds show a weak spread of melting temperatures: for example, a difference of 13 K is observed in the case of the molecule 5-methyl-2-[(2-nitrophenyl)amino]-3-thiophenecarbonitrile (known as ROY; 7 polymorphs)2,51 or 14 K in the case of 5H-dibenz[b,f]azepine-carboxamine (carbamazepine, C15H12N2O; 5 polymorphs).52 In specific cases, differences in the melting temperature of up to 30 K can be observed.53,54 The literature suggests that a difference in the melting point above 50 K is scarce among the various crystalline polymorphs of a given compound.55 However, in the case of solvates, the thermal stability is drastically reduced in comparison with the not-solvated phase, such that reduced phase transition temperatures of 80 K can be observed.56 The observed substantial reduction of the thermal stability agrees with our suggestion that these three phases from DCM solutions are solvates. Furthermore, a simple packing analysis based on the required volume of a single OEG-BTBT molecule within Form I and of DCM molecules within Form III reveals that the three new solvates differ by the number of DCM molecules per OEG-BTBT molecule (Table 2). It is also interesting that we see this large difference in the transition temperature for polymorph (Form VII) prepared from yet another solvent, 1,2-dichlororbenzene, which has a much higher boiling point (453 K).
In principle, crystal structure solutions based on GIXD experiments could be performed using a combined theoretical/experimental approach. However, molecules comparable in size to OEG-BTBT with flexible moieties are on the edge for obtaining a crystal structure solution.57,58 In the current case, only speculation can be offered about the crystalline packing of DCM and OEG-BTBT molecules within the new phases. It is observed that the new phases are more stable than the known solvate (Form III). Therefore, considerably different packings are expected for Form III versus Forms IV, V and VI. One possibility would be the embedding of DCM molecules into the OEG side chains. It is well known that OEG and its polymer have a polar character and are thus prone to interact with polar solvents. Moreover, an exceptionally high solubility is found for polyethylene glycol in the solvent DCM.59 The high flexibility in the chain conformation, together with the specific polar centres, is thus beneficial for the incorporation of polar molecules into the crystalline environment.60,61 Generally, the preparation of solvates could be a first step towards the preparation of unknown polymorphs obtained through a careful desolvation process.62,63
Conclusions
This work unambiguously demonstrates the potentiality of incorporating surfaces into the state-of-the-art polymorph screening technique to discover new polymorphs. The introduction of a substrate surface into the crystallization process gives the opportunity to vary the crystallization kinetics over a wide range through the simultaneous presence of a surface for templating crystallization. The molecule OEG-BTBT, which is known for its polymorphism in the bulk phase, was chosen for the discovery of new phases at the proximity of a surface by varying the deposition conditions. The factors that were varied in the screening process are the deposition technique (spin coating, drop casting), the type of solvent (10 solvents), the solution concentration (1–4 g L−1), the deposition temperature (room temperature to 353 K) and the evaporation rate of the solvent (e.g., achieved by covering the sample). In addition to the three polymorphs found via classical polymorph screening, we found four additional solvates, of which three are obtained from dichloromethane and one is obtained from 1,2-dichlorobenzene, which are used as the processing solvents. In all four cases, the substrate has a decisive influence on the crystallization process as a strong preferred orientation of the crystals relative to the substrate surface is observed. The crystal lattices of the solvates from dichloromethane were determined through indexing the GIXD diffraction pattern, and quantitative packing analysis reveals that the inclusion of solvent molecules into the crystalline lattices occurs. This is confirmed by the unusual low thermal stabilities of the solvates with respect to the known polymorphs of the molecule. This work shows that combining polymorph screening with crystallization at defined surfaces unlocks the possibility of finding new unknown phases of a particular molecular material.Author contributions
Y. G. proposed the project and OEG-BTBT molecule was synthesized in his group by F. D. R. R. supervised the project. R. R. and L. M. conceived the idea. A. M. J., L. P. and A. G. performed the experiments. A. M. J., N. M. and P. P. analysed the data. J. C. and P. B. gave important input for interpretation. A. M. J. and R. R. organized and wrote the manuscript. All authors contributed to the discussion and commented on the manuscript.Conflicts of interest
The authors declare no competing interests.Acknowledgements
This work was funded by the European Union's Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie grant agreement no. 811284 (UHMob). The support by the Excellence of Science programme (EOS) of FNRS and FWO, through the 2D to 3D (no. 30489208), and CHISUB (no. 40007495) projects, is acknowledged. The authors acknowledge Elettra Sincrotrone Trieste for providing access to its synchrotron radiation facilities and we thank Luisa Barba for assistance in using beamline XRD1. J. C. is a research director of the Belgian National Fund for Scientific Research (FNRS).References
- W. C. McCrone, Phys. Chem. Org. Solid State, 1965, 2, 725–767 Search PubMed.
- L. Yu, Acc. Chem. Res., 2010, 43, 1257–1266 CrossRef CAS PubMed.
- S. Riera-Galindo, A. Tamayo and M. Mas-Torrent, ACS Omega, 2018, 3, 2329–2339 CrossRef CAS PubMed.
- L. Yu, S. M. Reutzel-Edens and C. A. Mitchell, Org. Process Res. Dev., 2000, 4, 396–402 CrossRef CAS.
- D. E. Braun, T. Gelbrich, V. Kahlenberg, G. Laus, J. Wieser and U. J. Griesser, New J. Chem., 2008, 32, 1677–1685 RSC.
- H. G. Brittain, Drugs Pharm. Sci., 1999, 95, 183–226 Search PubMed.
- H. Chung and Y. Diao, J. Mater. Chem. C, 2016, 4, 3915–3933 RSC.
- D. Gentili, M. Gazzano, M. Melucci, D. Jones and M. Cavallini, Chem. Soc. Rev., 2019, 48, 2502–2517 RSC.
- G. R. Desiraju, Crystal engineering: the design of organic solids, Materials Science Monographs, Elsevier, 1989, vol. 54 Search PubMed.
- S. Morissette, Adv. Drug Delivery Rev., 2004, 56, 275–300 CrossRef CAS PubMed.
- C.-H. Gu, H. Li, R. B. Gandhi and K. Raghavan, Int. J. Pharm., 2004, 283, 117–125 CrossRef CAS PubMed.
- E. H. Lee, Asian J. Pharm. Sci., 2014, 9, 163–175 CrossRef.
- M. Allesø, F. van den Berg, C. Cornett, F. S. Jørgensen, B. Halling-Sørensen, H. L. de Diego, L. Hovgaard, J. Aaltonen and J. Rantanen, J. Pharm. Sci., 2008, 97, 2145–2159 CrossRef PubMed.
- S.-L. Lee, J. Adisoejoso, Y. Fang, K. Tahara, Y. Tobe, K. S. Mali and S. De Feyter, Nanoscale, 2015, 7, 5344–5349 RSC.
- I. V. Markov, Crystal growth for beginners: fundamentals of nucleation, crystal growth and epitaxy, World scientific, 2016 Search PubMed.
- J. Chen, B. Sarma, J. M. B. Evans and A. S. Myerson, Cryst. Growth Des., 2011, 11, 887–895 CrossRef CAS.
- S. L. Price, D. E. Braun and S. M. Reutzel-Edens, Chem. Commun., 2016, 52, 7065–7077 RSC.
- D. P. Otto and M. M. De Villiers, Curr. Drug Discovery Technol., 2017, 14, 72–105 CrossRef CAS PubMed.
- L. Nicoud, F. Licordari and A. S. Myerson, Cryst. Growth Des., 2018, 18, 7228–7237 CrossRef CAS.
- I. D. Tevis, L. C. Palmer, D. J. Herman, I. P. Murray, D. A. Stone and S. I. Stupp, J. Am. Chem. Soc., 2011, 133, 16486–16494 CrossRef CAS PubMed.
- L. Padrela, J. Zeglinski and K. M. Ryan, Cryst. Growth Des., 2017, 17, 4544–4553 CrossRef CAS.
- M. Kitamura, CrystEngComm, 2009, 11, 949 RSC.
- J.-M. Ha, J. H. Wolf, M. A. Hillmyer and M. D. Ward, J. Am. Chem. Soc., 2004, 126, 3382–3383 CrossRef CAS PubMed.
- M. Beiner, G. Rengarajan, S. Pankaj, D. Enke and M. Steinhart, Nano Lett., 2007, 7, 1381–1385 CrossRef CAS PubMed.
- R. Resel, J. Phys.: Condens. Matter, 2008, 20, 184009 CrossRef.
- T. Salzillo and A. Brillante, Adv. Mater. Interfaces, 2022, 9, 2200815 CrossRef CAS.
- M. Kaltenegger, S. Hofer, R. Resel, O. Werzer, H. Riegler, J. Simbrunner, C. Winkler, Y. Geerts and J. Liu, CrystEngComm, 2022, 27, 4921–4931 RSC.
- S. J. Bonafede and M. D. Ward, J. Am. Chem. Soc., 1995, 117, 7853–7861 CrossRef CAS.
- X. Yang, B. Sarma and A. S. Myerson, Cryst. Growth Des., 2012, 12, 5521–5528 CrossRef CAS.
- D. H. Case, V. K. Srirambhatla, R. Guo, R. E. Watson, L. S. Price, H. Polyzois, J. K. Cockcroft, A. J. Florence, D. A. Tocher and S. L. Price, Cryst. Growth Des., 2018, 18, 5322–5331 CrossRef CAS.
- J. J. De Yoreo and P. G. Vekilov, Rev. Mineral. Geochem., 2003, 54, 57–93 CrossRef CAS.
- J. V. Parambil, S. K. Poornachary, J. Y. Y. Heng and R. B. H. Tan, CrystEngComm, 2019, 21, 4122–4135 RSC.
- J. V. Parambil, S. K. Poornachary, S. J. Hinder, R. B. H. Tan and J. Y. Y. Heng, CrystEngComm, 2015, 17, 6384–6392 RSC.
- A. I. Koptyaev, V. V. Travkin, Y. I. Sachkov, Y. V. Romanenko and G. L. Pakhomov, J. Mater. Sci.: Mater. Electron., 2021, 32, 17791–17799 CrossRef CAS.
- Y.-M. Xie, Q. Sun, T. Zhu, L.-S. Cui, F. Liang, S.-W. Tsang, M.-K. Fung and L.-S. Liao, Org. Electron., 2018, 55, 1–5 CrossRef CAS.
- Y. Xiao, C. Zuo, J. Zhong, W. Wu, L. Shen and L. Ding, Adv. Energy Mater., 2021, 11, 2100378 CrossRef CAS.
- B. Wedl, R. Resel, G. Leising, B. Kunert, I. Salzmann, M. Oehzelt, N. Koch, A. Vollmer, S. Duhm and O. Werzer, RSC Adv., 2012, 2, 4404–4414 RSC.
- H. Yang, S. W. LeFevre, C. Y. Ryu and Z. Bao, Appl. Phys. Lett., 2007, 90, 172116 CrossRef.
- G. De Luca, A. Liscio, F. Nolde, L. M. Scolaro, V. Palermo, K. Müllen and P. Samorì, Soft Matter, 2008, 4, 2064–2070 RSC.
- C. S. Kim, S. Lee, E. D. Gomez, J. E. Anthony and Y.-L. Loo, Appl. Phys. Lett., 2008, 93, 327 Search PubMed.
- P. Pandey, N. Demitri, L. Gigli, A. M. James, F. Devaux, Y. H. Geerts, E. Modena and L. Maini, Cryst. Growth Des., 2022, 22, 1680–1690 CrossRef CAS.
- N. Turetta, M.-A. Stoeckel, R. Furlan de Oliveira, F. Devaux, A. Greco, C. Cendra, S. Gullace, M. Gicevičius, B. Chattopadhyay and J. Liu, J. Am. Chem. Soc., 2022, 144, 2546–2555 CrossRef CAS PubMed.
- D. K. Owens and R. C. Wendt, J. Appl. Polym. Sci., 1969, 13, 1741–1747 CrossRef CAS.
- R. Resel, E. Tamas, B. Sonderegger, P. Hofbauer and J. Keckes, J. Appl. Crystallogr., 2003, 36, 80–85 CrossRef CAS.
- M. Dohr, O. Werzer, Q. Shen, I. Salzmann, C. Teichert, C. Ruzié, G. Schweicher, Y. H. Geerts, M. Sferrazza and R. Resel, ChemPhysChem, 2013, 14, 2554–2559 CrossRef CAS PubMed.
- B. Schrode, S. Pachmajer, M. Dohr, C. Röthel, J. Domke, T. Fritz, R. Resel and O. Werzer, J. Appl. Crystallogr., 2019, 52, 683–689 CrossRef CAS PubMed.
- M. P. Kainz, L. Legenstein, V. Holzer, S. Hofer, M. Kaltenegger, R. Resel and J. Simbrunner, J. Appl. Crystallogr., 2021, 54, 1256–1267 CrossRef CAS PubMed.
- W. L. DeLano, CCP4 Newsl. Protein Crystallogr, 2002, 40, 82–92.
- X. Xiang, X. He, W. Xia, J. Yin, X. Yuan and X. Zhou, Anal. Methods, 2020, 12, 988–995 RSC.
- A. M. James, A. Greco, N. Mclntosh, F. Devaux, P. Brocorens, J. Cornil, P. Pandey, L. Maini, Y. H. Geerts and R. Resel, submitted.
- A. Levesque, T. Maris and J. D. Wuest, J. Am. Chem. Soc., 2020, 142, 11873–11883 CrossRef CAS PubMed.
- J.-B. Arlin, L. S. Price, S. L. Price and A. J. Florence, Chem. Commun., 2011, 47, 7074–7076 RSC.
- P. Tipduangta, K. Takieddin, L. Fabian, P. Belton and S. Qi, Cryst. Growth Des., 2015, 15, 5011–5020 CrossRef CAS.
- J. Nyman and G. M. Day, Phys. Chem. Chem. Phys., 2016, 18, 31132–31143 RSC.
- I. B. Rietveld, M. Barrio, P. Lloveras, R. Céolin and J.-L. Tamarit, Int. J. Pharm., 2018, 552, 193–205 CrossRef CAS PubMed.
- G. Perlovich, L. Hansen and A. Bauer-Brandl, J. Therm. Anal. Calorim., 2003, 73, 715–725 CrossRef CAS.
- C. Lercher, C. Röthel, O. M. Roscioni, Y. H. Geerts, Q. Shen, C. Teichert, R. Fischer, G. Leising, M. Sferrazza and G. Gbabode, Chem. Phys. Lett., 2015, 630, 12–17 CrossRef CAS PubMed.
- S. Hofer, A. Hofer, J. Simbrunner, M. Ramsey, M. Sterrer, A. Sanzone, L. Beverina, Y. Geerts and R. Resel, J. Phys. Chem. C, 2021, 125, 28039–28047 CrossRef CAS PubMed.
- M. Kawakami, M. Egashira and S. Kagawa, Bull. Chem. Soc. Jpn., 1976, 49, 3449–3453 CrossRef CAS.
- M. J. Blandamer, M. F. Fox, E. Powell and J. W. Stafford, Makromol. Chem.: Macromol. Chem. Phys., 1969, 124, 222–231 CrossRef CAS.
- B. Ensing, A. Tiwari, M. Tros, J. Hunger, S. R. Domingos, C. Pérez, G. Smits, M. Bonn, D. Bonn and S. Woutersen, Nat. Commun., 2019, 10, 1–8 CrossRef CAS PubMed.
- B. Fours, Y. Cartigny, S. Petit and G. Coquerel, Faraday Discuss., 2015, 179, 475–488 RSC.
- S. Bhattacharya and B. K. Saha, Cryst. Growth Des., 2013, 13, 606–613 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3mh00764b |
| This journal is © The Royal Society of Chemistry 2023 |