
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Investigation of storage environments on aminopolymer stabilization within UiO-67(Zr) for CO2 capture†
Rachel A.
Yang
 a,
Darius R.
Ganza
a,
Michael R.
Smith
ab,
Stanley
Cho
a,
Jacqueline A.
Vandermel
a,
Elizabeth
Jiang
a and
Michele L.
Sarazen
*a
a,
Darius R.
Ganza
a,
Michael R.
Smith
ab,
Stanley
Cho
a,
Jacqueline A.
Vandermel
a,
Elizabeth
Jiang
a and
Michele L.
Sarazen
*a
aDepartment of Chemical and Biological Engineering, Princeton University, 41 Olden Street, Princeton, New Jersey 08544, USA. E-mail: msarazen@princetone.edu
bDepartment of Chemistry, Princeton University, Frick Chemistry Laboratory, Princeton, New Jersey 08544, USA
First published on 10th January 2023
Abstract
Aminopolymers, poly(ethylene imine) (PEI) and poly(propylene imine) (PPI), are supported within nanoporous UiO-67(Zr) and evaluated for CO2 capture from simulated flue gas (10% CO2) and air (400 ppm CO2). N2 physisorption and Fourier-transform infrared spectroscopy indicate that PEI and PPI are physisorbed in UiO-67(Zr) voids but not tethered to metal nodes or carboxylate backbones. Mass-normalized CO2 capacity increases with repeated CO2 uptake-regeneration cycles for 30–50 wt% PPI, suggesting CO2 diffusion limitations at these higher loadings. 20 wt% PPI/UiO-67(Zr) and 20 wt% PEI/UiO-67(Zr) exposed to ambient air for 7 days demonstrate higher oxidative degradation in PEI than PPI, though PEI oxidation is reduced when stabilized within UiO-67(Zr) relative to when unconfined. Exposure of composites to liquid storage environments of varying polarity, hydrogen-bonding capability, and size indicate that 20 wt% PPI/UiO-67(Zr) amine efficiencies are minimally affected by solvent-induced aminopolymer conformational changes. Conversely, 20 wt% PEI/UiO-67(Zr) is more sensitive to surrounding solvent environments, exhibiting the greatest amine efficiency in methanol and the lowest in acetone due to solvent-induced aminopolymer swelling and reaction, respectively. Overall, this work provides insight into CO2 capture efficacies and chemical stabilities of composite PEI and PPI materials under various storage environments to inform future adsorbent and system design.
Introduction
Anthropogenic CO2 emissions are a primary cause of global warming effects and are on an increasing trend.1,2 CO2 from fossil fuel combustion is the largest source of United States greenhouse gas emissions with point sources, including fossil-fuel based power generation and industrial processes, comprising more than 40%.3 As such, most CO2 capture, storage, and utilization technologies are centered on post-combustion CO2 from these large point sources. Conventionally, CO2 absorption is performed in large-scale scrubbing units utilizing liquid amines, including aqueous monoethanolamine (MEA) and diethanolamine (DEA) solutions that suffer from high energy costs associated with amine regeneration.4,5 Solid amine adsorbents such as aminopolymers supported within mesoporous silicas,6–10 aluminas,11,12 and metal–organic frameworks1,13–15 have shown efficacy for CO2 capture from flue gas (5–15% CO2) and directly from air (DAC; ∼400 ppm CO2).Branched poly(ethylenimine) (PEI) is one of the most widely studied aminopolymers,8,11 and has been predominantly incorporated into mesoporous oxides,7,8,11,12,16 but also into a variety of other nanoporous solids, including metal–organic frameworks (MOFs) for CO2 adsorption.1,13–15 Additionally, PEI is less volatile compared to non-tethered lighter amines like ethylenediamine,17 triethylenediamine,18 and tetraethylenepentamine,19 which results in lower evaporative losses of active amine sites from the support.1 However, reaction events between PEI amine functionalities and CO2 after initial chemisorption irreversibly form covalent urea-like linkages at elevated temperatures.12,20 Further, secondary amines are particularly susceptible to degradation during temperature swing-adsorption regeneration methods under oxidative environments (∼373 K) due to the formation of moieties (imines, imides, etc.) with decreased basicity that result in lower CO2 capacities and amine efficiencies.12,21,22 In contrast, poly(propylene imine) (PPI) is a similar branched, aliphatic aminopolymer compared to PEI, but the propyl backbone demonstrates enhanced resistance to degradation under oxidative environments.7,8,23,24 PPI supported in inorganic oxides have demonstrated notable amine efficiencies,7,8 but is otherwise underexplored within other more complex nanoporous solids, including MOFs, which represent some of the highest-performing CO2 adsorbents (>2 mmolCO2 gadsorbent−1) even in the absence of amine functionalization.25 MOFs are promising due to high pore volumes for aminopolymer incorporation to create adsorbents with high amine densities per unit volume without severe mass transfer limitations and due to high densities of potentially open metal and/or acidic sites for amine tethering to make more stable hybrid adsorbents. Additionally, some families like MIL-101 exhibit stability against poisoning by sulfur- or nitrogen-containing species commonly found in flue gas.26 Finally, the synthetic physicochemical tunability of MOF confining voids and relative homogeneity of active sites compared to more conventional supports like metal oxides provides the opportunity to probe aminopolymer–MOF interactions to ultimately optimize parameters that influence polymer conformation and stability for both PPI and PEI. In this work, we leverage the tunability of MOF structures to elucidate interactions between PEI and PPI within nanoporous MOFs to understand the ramifications of molecular-scale features on CO2 uptake, amine efficiency, and material stability, especially in the context of storage conditions, which are not well-understood for aminopolymer/MOF systems.
Specifically, we focus on UiO-67(Zr) (Fig. 1), which contains Zr6O4(OH)4 nodes coordinated to twelve biphenyl-4,4′-dicarboxylic acid linkers to form mesoporous octahedral (d = 2.3 nm) and microporous tetrahedral (d = 1.15 nm) cages.27 For CO2 capture, UiO-67(Zr)'s open mesoporous and microporous framework allows facile CO2 diffusion to adsorption sites and incorporation of guest moieties, including aminopolymers that can be potentially stabilized through interaction with Brønsted acid moieties (μ3-OH) on UiO-67(Zr) nodes. UiO-67(Zr) is also characterized by thermal stability in air at high temperatures (up to 723 K), making this material amenable for flue gas streams (>393 K), and by chemical stability in various solvents,28 including chloroform, pyridine, acetone, and methanol.29,30 However, it is unclear if these observations extend to longer time scales (>24 h), other solvents, and what the ramifications are for CO2 uptake; these insights are imperative for the design of future adsorbents and also for commercial usage, where synthesized materials must retain their structures and functions even after storage for extended times.
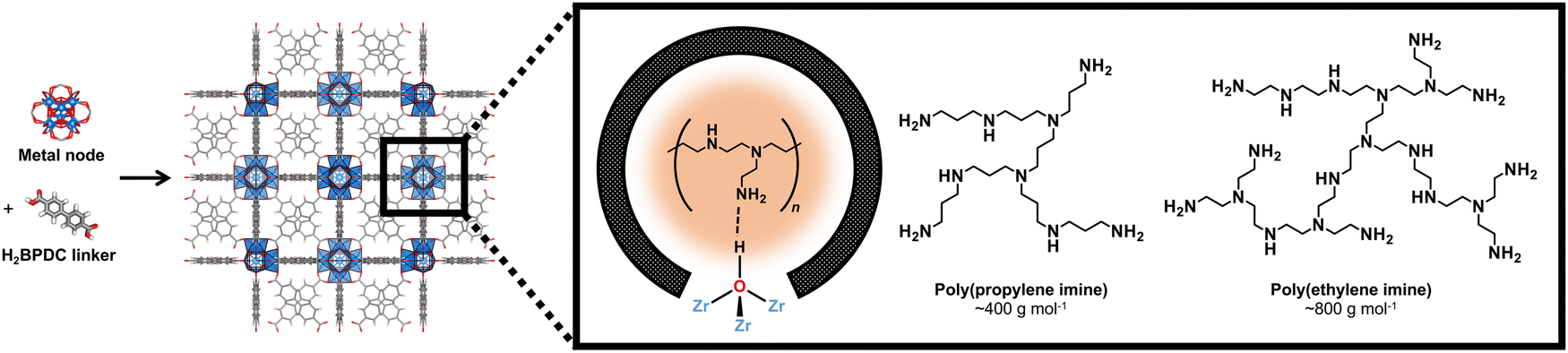 | ||
| Fig. 1 Zr6O4(OH)4 nodes and biphenyl-4,4′-dicarboxylic acid linker species that form the UiO-67(Zr) nanoporous framework structure containing larger octahedral and smaller tetrahedral cages that can host aminopolymer (poly(propylene imine) and poly(ethylene imine)) guests, which serve as CO2 adsorption sites. | ||
UiO-67(Zr) functionalization has increased mass-normalized CO2 capacities,31 but to our knowledge, there are no studies that investigate PEI and PPI supported within UiO-67(Zr) or probe the longer term stability of these composite adsorbent systems. In this work, PEI and PPI are supported in UiO-67(Zr) at varied weight loadings to probe the nature of aminopolymer packing within pores and the resulting ramifications for CO2 diffusion to amine adsorption sites during repeated uptake-regeneration cycles for simulated flue gas (10% CO2) and air (400 ppm CO2) streams. Exposure of PEI and PPI composite materials to varying gaseous and liquid environments at ambient temperature and pressure demonstrate the benefits of confinement within UiO-67(Zr) pores for aminopolymer stabilization against oxidative degradation; aminopolymer–solvent reactions and solvent-induced aminopolymer conformational changes are also suggested and impact observed amine efficiencies, with a more pronounced effect on PEI. Material characterization via N2 physisorption, Fourier-transform infrared spectroscopy, scanning electron microscopy, and thermogravimetric analysis further elucidate the degree of aminopolymer interaction with and mobility within UiO-67(Zr) pores. Overall, PEI and PPI within UiO-67(Zr) are shown to be promising solid adsorbents for CO2 capture, with PPI demonstrating enhanced chemical tenability. The insights into aminopolymer–MOF interactions and composite material stability in diverse storage conditions gleaned here will inform future adsorbent system design considerations for both laboratory and commercial scales.
Experimental
Materials
Zirconium(IV) chloride (ZrCl4; Thermo Scientific, 99.5 + %), biphenyl-4,4′-dicarboxylic acid (H2BPDC; Acrōs Organics, 98%), hydrochloric acid (HCl; EMD Millipore Corporation, 37% in water), dimethylformamide (DMF; Fisher Chemical, 99.8 + %), and ethanol (EtOH; Koptec, 200 proof) were utilized for UiO-67(Zr) synthesis. Azetidine (Thermo Scientific, 98%), hydrobromic acid (HBr; Alfa Aesar, 48% in water), methanol (MeOH; Fisher Chemical, 99.9%), and Ambersep r900(OH) ion exchange resin (Ambersep; AlfaAesar) were used for poly(propylene imine) (PPI) synthesis. Branched poly(ethylene imine) (PEI; Sigma-Aldrich) with an average molecular weight of 800 g mol−1 and ethylenediamine (EDA; Sigma-Aldrich, ≥99%) were acquired commercially. Acetone (Fisher Chemical, 99.5 + %), dichloromethane (DCM; Fisher Chemical, 99.9 + %), hexanes (EMD Millipore Corporation, ≥98.5%), acetonitrile (MeCN; J.T. Baker, ≥99.9%), methanol (Fisher Chemical, 99.9%), and toluene (LabChem, 100%) were utilized for solvent storage experiments. CO2 uptake experiments were performed using argon (Ar; Airgas, UHP), 10% CO2 in He (Airgas), and 400 ppm CO2 in He (Airgas). All starting materials were utilized as received without additional purification.Synthesis of UiO-67(Zr)
UiO-67(Zr) was synthesized in accordance with established procedure,27 with some modifications. In a typical synthesis, 27 mmol ZrCl4, 75 mL DMF, and 5 mL 37% HCl(aq) were first added to a 250 mL Erlenmeyer flask. The mixture was then sonicated for 20 minutes until clear and colorless. To this mixture, 38 mmol H2BPDC and 75 mL DMF were added and sonicated for 20 minutes, resulting in an opaque white mixture. The flask was then sealed with a glass stopcock and put into a convection oven at 353 K for 16 h without stirring to allow for crystallization. After cooling naturally to ambient temperature, the white solid was isolated via centrifugation (8000 × g, 10 min) and then washed with DMF (2×, 150 mL each), followed by EtOH (2×, 150 mL each). Finally, the white solid was dried at 363 K overnight under mild vacuum (125 torr).Polymerization of azetidine to yield branched poly(propylene imine)
Branched poly(propylene imine) (PPI) with an average molecular weight of ∼400 g mol−1 was synthesized following literature precedent.8 Briefly, 1.9 mmol azetidine and 54 μL 48% HBr(aq) were added sequentially to 2 mL MeOH in a 25 mL glass pressure tube. The tube was then capped and placed into a heated oil bath at 353 K for 72 h. After, the tube was removed from heat and cooled to ambient temperature. MeOH was then removed via rotary evaporation (60 rpm, 10−3 torr, 323 K) utilizing a 50 mL round bottom flask to concentrate the aminopolymer product. Ambersep resin and 10 mL deionized water were added to the viscous, very pale-yellow product and the mixture was stirred magnetically for 24 h. Vacuum filtration was utilized to remove the Ambersep resin and the clear filtrate was concentrated by rotary evaporation (60 rpm, 10−3 torr, 343 K) to yield viscous, pale-yellow PPI. 1H NMR in D2O was utilized to determine primary, secondary, and tertiary amine distributions (Fig. S2, ESI†).Synthesis of composite aminopolymer/UiO-67(Zr) adsorbents
Nominal weight percentages of PEI and PPI were first massed and put into clean 20 mL glass scintillation vials. MeOH was then added in a ratio of 50 μL/1 mg aminopolymer and stirred magnetically until PEI/PPI was fully dissolved, as evidenced by a clear mixture. After, UiO-67(Zr) was added, the vial was capped to prevent solvent loss, and the mixture was allowed to stir for 24 h. The resulting white composite aminopolymer/UiO-67(Zr) materials were rotovapped (60 rpm, 10−3 torr, 323 K) to remove MeOH and aid aminopolymer migration into UiO-67(Zr) pores through capillary action. Samples referred to as “fresh” were then utilized immediately for CO2 uptake tests or for storage experiments.Synthesis of EDA-UiO-67(Zr)
In a typical synthesis, 60 mg UiO-67(Zr) was dried at 423 K under flowing N2 for 1 h while refluxing in a three-neck round bottom flask sealed with rubber septa; water/volatiles vapor was evident after 5 minutes of heating and by 15 minutes, no vapor was visible. After 1 h, the flask was removed from heat and allowed to cool naturally to ambient temperature under N2 flow. A solution containing stoichiometric ethylenediamine to Zr in MeOH was prepared and then added to the flask using a syringe. The mixture was stirred magnetically for 24 h at ambient temperature. Rotary evaporation (60 rpm, 10−3 torr, 323 K) was utilized to remove MeOH and the final EDA-UiO-67(Zr) was immediately characterized via Fourier-transform infrared spectroscopy (FTIR).CO2 uptake experiments
Thermogravimetric analysis (TGA) on a PerkinElmer TGA 8000 was used to perform CO2 uptake experiments. In a typical experiment, 3–10 mg of each sample was placed in tared ceramic pans and massed prior to uptake measurements. Water and other volatile species were first removed by heating samples (5 K min−1) from 303 K to 383 K under 40 mL min−1 Ar flow for 3 h. The samples were subsequently cooled (5 K min−1) under 40 mL min−1 Ar until the adsorption temperature of 308 K was reached. Upon reaching 308 K, the flow was changed to 40 mL min−1 10% CO2 in He (for simulated flue gas) or 40 mL min−1 400 ppm CO2 in He (for DAC) and held for 1 h or 3 h, respectively, to allow CO2 uptake. After CO2 uptake, the samples were regenerated under 40 mL min−1 Ar for 2 h at 383 K (5 K min−1 ramp rate). Finally, samples were cooled (5 K min−1) back to 308 K, after which another CO2 uptake was performed; this uptake-regeneration cycling was performed 3 times for all materials unless otherwise specified.Material storage experiments
Freshly synthesized composite aminopolymer/UiO-67(Zr) materials were exposed to different storage environments (air, foil (in air), acetone, DCM, hexanes, MeCN, MeOH, toluene, and deionized water) for 7 days. In a typical experiment, 30 mg of each composite aminopolymer/UiO-67(Zr) sample and 3 mL of each liquid solvent was added to a plastic 10 mL centrifuge tube. Each tube was swirled to mix, capped, sealed with electrical tape to minimize volatile solvent losses, and allowed to rest stagnant for 7 days. After 7 days, all solids had settled at the bottom of each tube and the supernatant liquid was removed immediately prior to CO2 uptake experiments.Material characterization
X-ray diffraction (XRD) patterns were obtained on a Bruker D8 Discover diffractometer with Cu Kα radiation operating at 40 kV and 40 mA. Scanning electron microscopy (SEM) was performed on a Verios 460 Extreme High Resolution Scanning Electron Microscope. A Nicolet is50 spectrophotometer was utilized to obtain ex situ FTIR spectra; 13 mm diameter pellets of all samples were 2 wt% in KBr diluent and were pressed at 5000 lbs in a hydraulic press prior to spectra collection. 1H NMR was collected on a Bruker 500 MHz spectrometer with D2O solvent unless otherwise specified. All aminopolymer loadings were estimated with TGA by combustion under zero air from 383–1173 K. Finally, N2 adsorption–desorption isotherms (77 K) were collected on a Micromeritics 3flex physisorption unit and then utilized to calculate multipoint Brunauer–Emmett–Teller (BET) surface area and non-local density functional theory (NLDFT) pore size distributions. BET surface areas were calculated in accordance with Rouquerol consistency criterion,32 utilizing at least 5 points from relative pressure ranges between 0.01–0.15.Results and discussion
All synthesized UiO-67(Zr) are crystalline, exhibiting XRD patterns (Fig. S1, ESI†) consistent with both simulated UiO-67(Zr) and literature precedent.27,29 PPI weight loadings were varied from 20–50 wt% and all composite PPI/UiO-67(Zr) samples showed >100% increases in fresh (Fig. 2; cycle 1) dried adsorbent mass-normalized CO2 capacities relative to parent UiO-67(Zr) (0.31 ± 0.09 mmolCO2 gadsorbent−1) and neat unconfined PPI (0.26 mmolCO2 gadsorbent−1). Though the maximum observed CO2 capacity of 1.19 ± 0.02 mmolCO2 gadsorbent−1 for 40 wt% PPI/UiO-67(Zr) is lower relative to other aminopolymer–solid systems such as 45 wt% PEI/SBA-15 (2.2 mmolCO2 gadsorbent−1),33 this system is utilized to probe the impact of aminopolymer–support interactions, conformation, and stability, given the thermochemical robustness of the UiO-67(Zr) support. Notably, we first investigate the interplay between aminopolymer loadings and transport phenomena. The non-monotonic increase in mass-normalized CO2 capture as a function of PPI weight loadings suggests potential transport limitations at higher weight loadings. This is corroborated by monotonically increasing times to achieve 20% of equilibrium CO2 capacities (t20) as a function of PPI weight loadings (Fig. 2). With each successive CO2 uptake-regeneration cycle, there are volatile losses of lower molecular weight PPI oligomers in addition to desorbed CO2, as the mass lost in each regeneration cycle exceeds the mass of CO2 adsorbed in the preceding uptake step (Fig. S4b, ESI†). Despite these amine site losses, mass-normalized CO2 capacities for 30–50 wt% PPI/UiO-67(Zr) increase with each cycle, suggesting that prior to PPI liberation in regeneration cycles, CO2 diffusion to some internal amine sites is limited at higher PPI loadings. In contrast, 20 wt% PPI/UiO-67(Zr) demonstrates mild (<3%) decreases in mass-normalized CO2 capacities. As such, 20 wt% aminopolymer loadings are unlikely to suffer from pronounced CO2 diffusion limitations and are utilized for further characterization, uptake, and solvent environment stability studies to minimize confoundment by transport phenomena.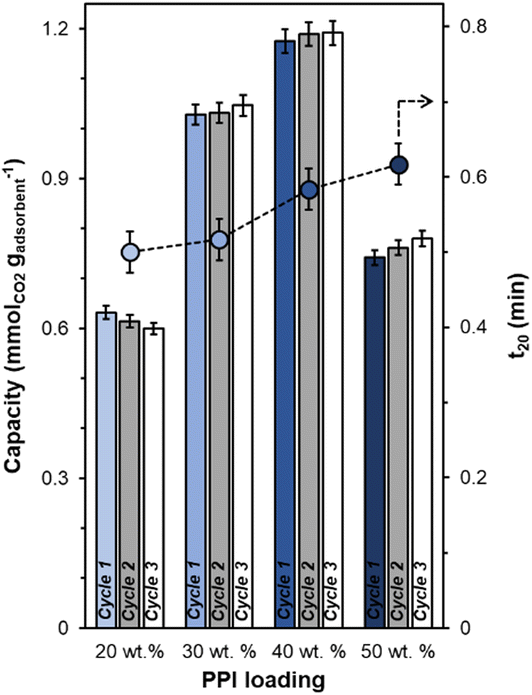 | ||
| Fig. 2 Mass-normalized CO2 capacities for varying weight percentages (20–50) of PPI supported on UiO-67(Zr) for three 1 h uptake cycles at 308 K from simulated flue gas with 2 h regeneration steps at 383 K under 40 mL min−1 Ar flow in between cycles. t20 (circles) are times to reach 20% of equilibrium CO2 uptakes. | ||
The incorporation of poly(ethylene imine) (PEI) and PPI into UiO-67(Zr) is first probed via N2 physisorption (Fig. 3) to determine relative BET surface areas and NLDFT pore size distributions (PSDs). Parent UiO-67(Zr) exhibits high (>2000 m2 g−1) BET surface areas and sharp PSDs at 1.0 nm and 2.4 nm, which are consistent with microporous tetrahedral and mesoporous octahedral cages, respectively.27 Both 20 wt% PPI/UiO-67(Zr) and 20 wt% PEI/UiO-67(Zr) are characterized by drastic reductions in BET surface areas (200–250 m2 g−1) that are commiserate with reductions in mesoporosity and no observed microporosity (Fig. 3(b)). These observations are consistent with internal PPI and PEI incorporation into UiO-67(Zr) pores, rather than dense agglomerates that exist exclusively on crystal surfaces.
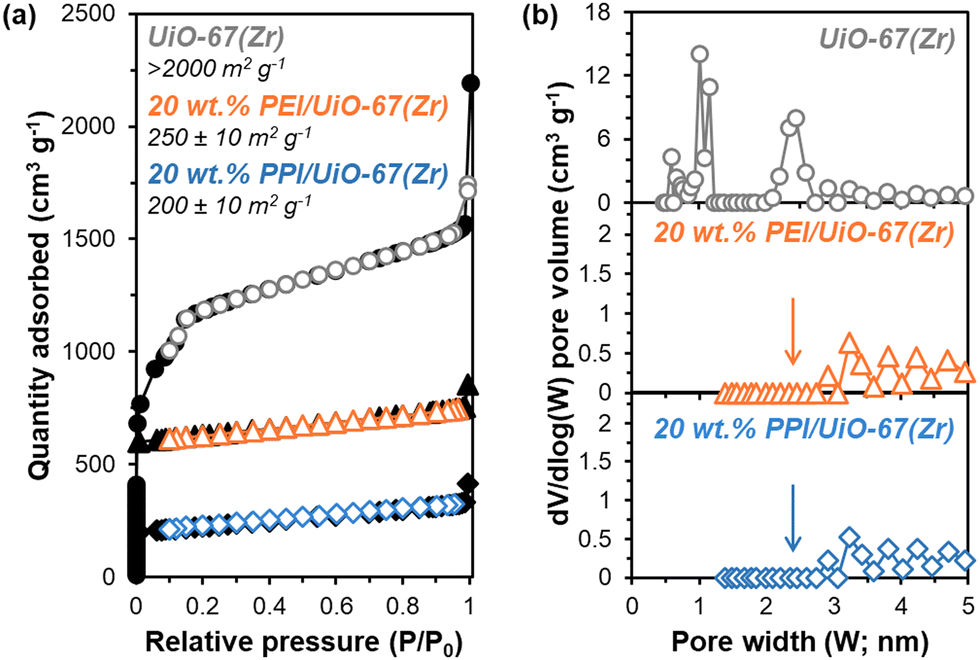 | ||
| Fig. 3 (a) Adsorption–desorption isotherms (77 K) and (b) NLDFT pore size distributions from N2 physisorption for UiO-67(Zr) (grey circles), 20 wt% PEI/UiO-67(Zr) (orange triangles), and 20 wt% PPI/UiO-67(Zr) (blue diamonds). Isotherms for 20 wt% PEI/UiO-67(Zr) and 20 wt% PPI/UiO-67(Zr) are shifted vertically for clarity. | ||
Fresh 20 wt% PPI/UiO-67(Zr) and 20 wt% PEI/UiO-67(Zr) demonstrate efficacy to increase mass-normalized CO2 adsorption compared to parent UiO-67(Zr) (0.31 ± 0.09 mmolCO2 gadsorbent−1), with similar uptakes for 20 wt% PEI/UiO-67(Zr) (0.58 ± 0.01 mmolCO2 gadsorbent−1) and 20 wt% PPI/UiO-67(Zr) (0.54 ± 0.01 mmolCO2 gadsorbent−1). Marginally lower initial CO2 uptakes for PPI compared to PEI observed here and for similar weight percentages in mesoporous SBA-157 are likely due to residual free acidic initiator (HBr) in synthesized PPI7 that outweigh the benefits of higher amine basicity, imposed by differing aliphatic chain lengths,24 for PPI compared to PEI. Moreover, secondary amines in commercial PEI are particularly susceptible to degradation in oxidative environments,12,21,22 resulting in decreased CO2 uptake capacities with time and uptake-regeneration cycles, while PPI has demonstrated enhanced oxidative resistance relative to PEI.7,8,23,24 Additionally, 20 wt% PPI/UiO-67(Zr) demonstrates cyclic stability under temporally realistic rapid CO2 uptake-regeneration cycles with statistically insignificant losses in mass-normalized CO2 capacities over 10 cycles (Fig. S10, ESI;† 15 min uptake at 308 K, 30 min desorption under Ar at 383 K). As such, we investigate the potential stabilization of aminopolymeric structures within UiO-67(Zr) voids against oxidative environments and explore alternative storage solutions to mitigate losses in CO2 uptake capacities with time.
Short-term storage experiments at ambient temperature and pressure are conducted over 7 days in a wide range of environments, including solvents that can be utilized at scale to store solid adsorbents as suspensions or cakes. After 7 days, 20 wt% PPI/UiO-67(Zr) and 20 wt% PEI/UiO-67(Zr) stored in various environments are evaluated for amine efficiencies (Fig. 4) that are determined from organic mass losses quantified via combustion TGA (Fig. S8, ESI†). Aminopolymer confinement within UiO-67(Zr) pores is first evaluated through comparison of fresh samples and samples stored in air for 7 days. Here, 20 wt% PPI/UiO-67(Zr) shows statistically identical amine efficiencies between the fresh sample (0.10 ± 0.03 mmolCO2 mmolN−1) and after 7 days in air (0.09 ± 0.03 mmolCO2 mmolN−1); both are notably higher than the amine efficiency of neat unconfined PPI (0.017 mmolCO2 mmolN−1). In contrast, though 20 wt% PEI/UiO-67(Zr) also shows a higher mass-normalized CO2 uptake (0.58 ± 0.01 mmolCO2 gadsorbent−1) and amine efficiency (0.113 ± 0.006 mmolCO2 mmolN−1) compared to neat unconfined PEI (0.10 mmolCO2 gadsorbent−1, 0.006 mmolCO2 mmolN−1), a modest 22% loss of amine efficiency from the fresh composite sample (0.113 ± 0.006 mmolCO2 mmolN−1) after 7 days in air (0.088 ± 0.006 mmolCO2 mmolN−1) is observed, partially due to the formation of amides through chain scission and dehydration reactions of hydroperoxides observed via1H NMR (Fig. S9, ESI†) and in literature.34 These observations are consistent with PPI's enhanced oxidative stability relative to PEI. However, confinement of PEI within UiO-67(Zr) confers mild stabilization against oxidative degradation, as approximately 32% of amines in branched PEI degrades in air at 298 K based on Arrhenius studies for the degradation of branched PEI (molecular weight of ∼3000 g mol−1);35 here, extracted pre-exponential factors and activation energies are utilized to calculate the PEI degradation rate constant at 298 K (6.2 × 10−7 s−1),35 which is then utilized to determine the first-order active amine center conversion to inactive degradation products after 7 days.
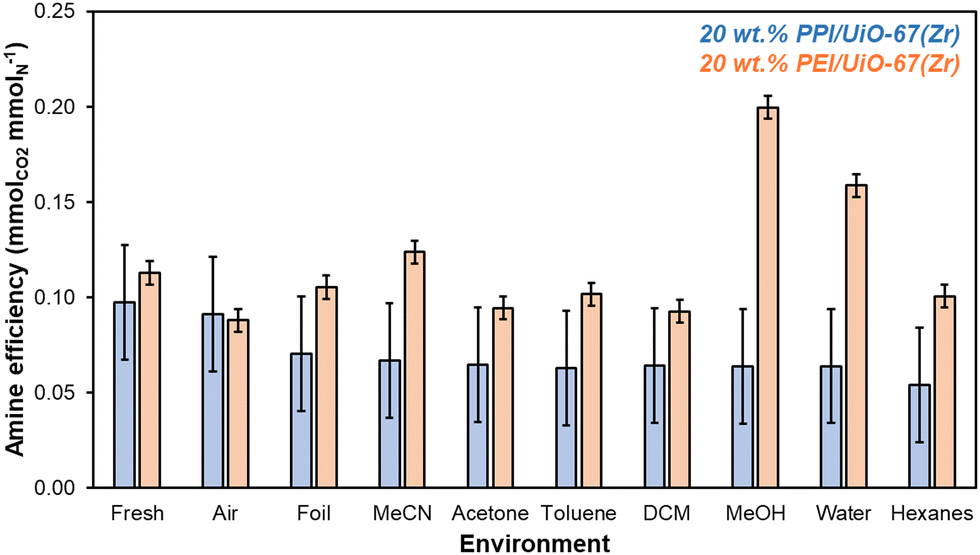 | ||
| Fig. 4 Amine efficiencies (mmolCO2 mmolN−1) determined from 1 h CO2 uptakes at 308 K from simulated flue gas for 20 wt% PPI/UiO-67(Zr) (blue) and 20 wt% PEI/UiO-67(Zr) (orange) after 7 days of exposure to each solvent environment. Amine contents are based on molecular weights of 400 g mol−1 for PPI (77% primary and secondary amines) and 800 g mol−1 for PEI (77% primary and secondary amines).36,37 | ||
PEI confinement within UiO-67(Zr) pores enhances amine oxidative stability in air relative to free PEI, though other storage methods may offer additional oxidative degradation resistance. Storage in air (humid or dry) is often the default storage method without consideration of the effect of light irradiation. After 7 days in ambient light conditions and in foil, 20 wt% PPI/UiO-67(Zr) amine efficiencies remain unchanged while 20 wt% PEI/UiO-67(Zr) retains its original fresh amine efficiency when shielded from light (0.111 ± 0.006 mmolCO2 mmolN−1) compared to its 22% loss relative to fresh amine efficiencies when stored in air and ambient light, in agreement with literature precedent reporting PEI degradation under oxidative UV irradiation conditions.34
Beyond gaseous storage conditions, liquid solvents of varying polarity, hydrogen-bonding capability, and size are evaluated for 20 wt% PPI/UiO-67(Zr) and 20 wt% PEI/UiO-67(Zr). 20 wt% PPI/UiO-67(Zr) demonstrates a high degree of chemical stability and resistance to solvent-induced polymer swelling or contraction, as all amine efficiencies after 7 days are statistically identical. As such, this discussion focuses on 20 wt% PEI/UiO-67(Zr). The effect of polarity is assessed through the comparison of water (1.000) and MeOH (0.762 relative to water) with a higher amine efficiency observed in MeOH for 20 wt% PEI/UiO-67(Zr), potentially due to greater polymer swelling through favorable interactions between MeOH's methyl moiety and PEI's aliphatic backbone; intramolecular interactions of PEI's hydrophobic aliphatic backbone may be favored in water, resulting in polymer contraction and impedance of CO2 access to internal amine adsorption sites. We note that despite similar aliphatic backbones in PEI and PPI, the apparent resistance to solvent-induced swelling or contraction observed for PPI suggests higher extents of cross-linking in as-synthesized PPI than in commercial PEI. Solvent hydrogen-bonding character is also potentially important for both interaction with UiO-67(Zr)'s hydroxyl moieties on the Zr6O4(OH)4 node and with aminopolymer N–H groups. MeOH is contrasted with acetone and MeCN to compare H-bonding acceptor/donor activity to exclusively H-bonding acceptor character to no appreciable H-bonding behavior, respectively. No trend between amine efficiency and H-bonding nature is observed, suggesting that any potential H-bonding stabilization of amine moieties during storage is minimal. Rather, polymer swelling or contraction in each solvent outweighs the impact of H-bonding. Notably, 20 wt% PEI/UiO-67(Zr) amine efficiencies in MeOH and MeCN are higher than in acetone, which is consistent with the rapid formation of a white, condensed mass in acetone that may initially hinder CO2 access to amine sites.
Since hydrogen bonding character has minimal impacts on amine efficiencies in 20 wt% PEI/UiO-67(Zr), the impact of different heteroatom moieties is examined through non-H-bonding DCM and MeCN. Here, amine efficiencies are lower in halogenated DCM due to possible reactions between primary, secondary, and tertiary amine sites with DCM to form a variety of products including aminals and chloromethyl quaternary chlorides that are not proficient CO2 adsorbents.38 Finally, hexanes and comparatively bulky toluene are contrasted to determine the impact of steric size. Though diffusion barriers are likely higher for toluene than for hexanes, neither 20 wt% PEI/UiO-67(Zr) nor 20 wt% PPI/UiO-67(Zr) are perturbed by higher or lower internal pore access for these two nonpolar solvents. Overall, 20 wt% PPI/UiO-67(Zr) is more stable than 20 wt% PEI/UiO-67(Zr) in air and shows enhanced stability in a wide swath of solvents. However, 20 wt% PEI/UiO-67(Zr) can be stabilized in some solvents with MeOH conferring the highest observed amine efficiency, potentially due to solvent-induced aminopolymer swelling that results in more facile amine access for CO2 adsorption. Therefore, storage as a cake or suspension rather than in air to preserve amine efficiencies over a longer period is a viable and scalable solution to stabilize both 20 wt% PPI/UiO-67(Zr) and 20 wt% PEI/UiO-67(Zr).
Beyond amine efficiencies, surface functionalities are probed for 20 wt% PPI/UiO-67(Zr) and 20 wt% PEI/UiO-67(Zr) through FTIR spectroscopy (Fig. 5) to assess if and how aminopolymers within UiO-67(Zr) pores are tethered and to identify potential chemical changes after 7 days in differing storage environments. N—H bending17 at 835 cm−1 is evident for all 20 wt% PPI/UiO-67(Zr) and 20 wt% PEI/UiO-67(Zr) after 7 days in all storage environments (Fig. 5), indicating aminopolymer retention within UiO-67(Zr). However, aminopolymer interactions with UiO-67(Zr) are limited to non-specific interactions due to steric hindrance, which is suggested by the absence of a C—N vibration at approximately 1050 cm−1 that is observed for ethylenediamine tethered to UiO-67(Zr) (EDA-UiO-67(Zr)); this band in EDA-UiO-67(Zr) (Fig. 5; grey) has been observed for grafted amines interacting with the μ3-OH (Fig. 1) in Zr oxide nodes.17,18 Characteristic UiO-67(Zr) peaks belonging to C–H, O–H, and Zr6O4(OH)4 nodes that are consistent with literature17,39 are not blue-shifted in any storage environment, which further implicates non-specific interactions between PEI/PPI and UiO-67(Zr). 20 wt% PPI/UiO-67(Zr) retains all characteristic bands for both UiO-67(Zr) and PPI after exposure to all storage environments, indicating minimal bulk changes in surface chemical functionality which agrees with its consistent amine efficiencies across storage environments. Though 20 wt% PEI/UiO-67(Zr) amine efficiencies vary in different storage environments, all samples retain bulk characteristic band positions, suggesting that amine efficiency differences are rooted in more subtle, localized phenomenon.
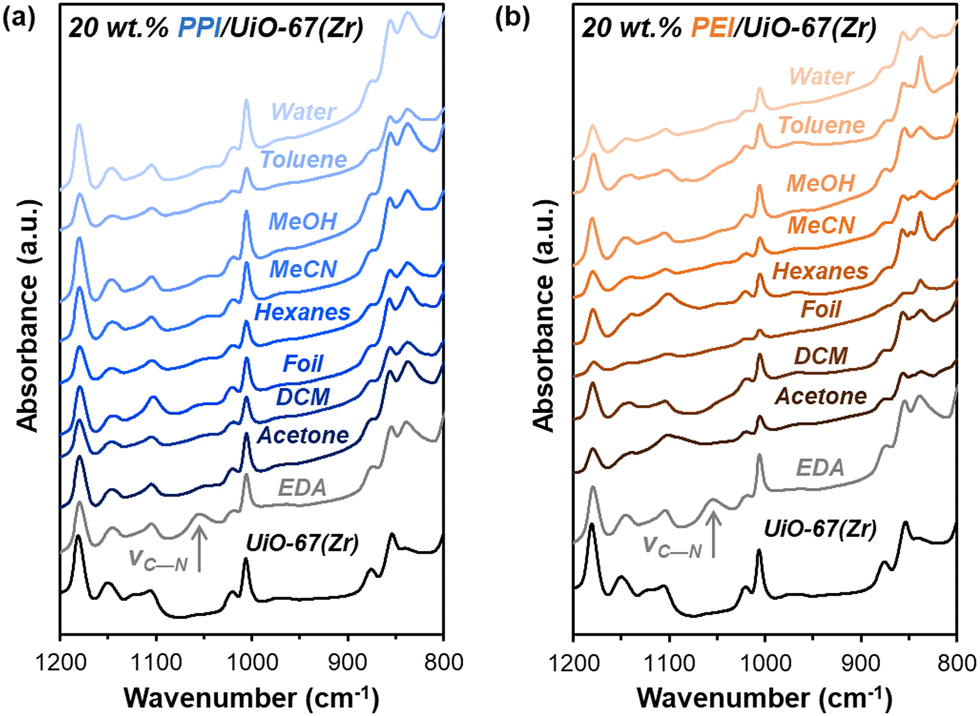 | ||
| Fig. 5 Ex situ FTIR spectra of fresh UiO-67(Zr) (black) and EDA-UiO-67(Zr) (grey) and (a) 20 wt% PPI/UiO-67(Zr) (blue) and (b) 20 wt% PEI/UiO-67(Zr) (orange) after 7 days in each solvent environment. All samples are 2 wt% in KBr diluent. | ||
Physicochemical changes in aminopolymer structure are probed through 1H NMR since parent UiO-67(Zr) mass-normalized CO2 uptakes and FTIR spectra are statistically identical across all storage environment conditions over 7 days (Fig. S3 and S6, ESI†) suggesting that changes in the MOF support are minimal. However, primary, secondary, and tertiary amine chemical shifts in PEI are not amenable to quantitative 1D 1H NMR and PPI contained in supernatant liquids recovered from 20 wt% PPI/UiO-67(Zr) samples after 7 days were present in concentrations too dilute for 1H NMR analysis, suggesting PPI retention in UiO-67(Zr) pores. Therefore, commercially available PEI and as-synthesized PPI are exposed to each storage condition in the absence of UiO-67(Zr) to probe qualitative aminopolymer aggregation behaviors for both PEI and PPI and quantitative primary, secondary, and tertiary amine distributions for PPI. Qualitatively, PPI is fully dissolvable in water and MeOH, but self-aggregates in all other liquid environments (Fig. S7b, ESI†), while PEI solubilizes in all solvents by the end of 7 days, except in DCM (Fig. S7a, ESI†). Formed PPI aggregates are all chromatically consistent (light yellow) except in acetone (tan) due to imine formation, but constant amine efficiencies across tested storage environments suggest that changes in moiety basicity due to reactions with the solvent or changes in aminopolymer conformation are not severe enough to alter measured overall amine efficiencies when supported in porous materials. PEI mixtures are all colorless and clear with the exception of acetone, which reacts with PEI to form yellow imine products,40 and DCM, which reacts with PEI to potentially form insoluble aminals and chloride species.38 Reactions of PEI amines with acetone and DCM yield lower amine efficiencies in these solvents relative to fresh samples or samples stored in other solvents. There is no literature precedent to our knowledge, however, for reactions with toluene or hexanes likely due to phase separation between these nonpolar solvents and aminopolymers. Quantitative primary, secondary, and tertiary amine distributions for PPI (Fig. 6) are similar for all solvents barring the most nonpolar solvents, toluene (0.099) and hexanes (0.009) that preferentially solvate aminopolymer species containing a higher tertiary amine fraction, which are inactive for CO2 adsorption under this system's dry conditions.41 This potentially indicates that more favorable interactions between tertiary amines and surrounding hexane or toluene molecules induce aminopolymer conformations that maximize the density of external tertiary moieties rather than active primary or secondary amines but this observation could also arise partially from solvent interactions with PPI that affect observed chemical shifts. The apparent aminopolymer conformational changes to favor external tertiary amine moieties aid in rationalizing lower amine efficiencies in toluene and hexanes than in MeOH, water, and MeCN for 20 wt% PEI/UiO-67(Zr).
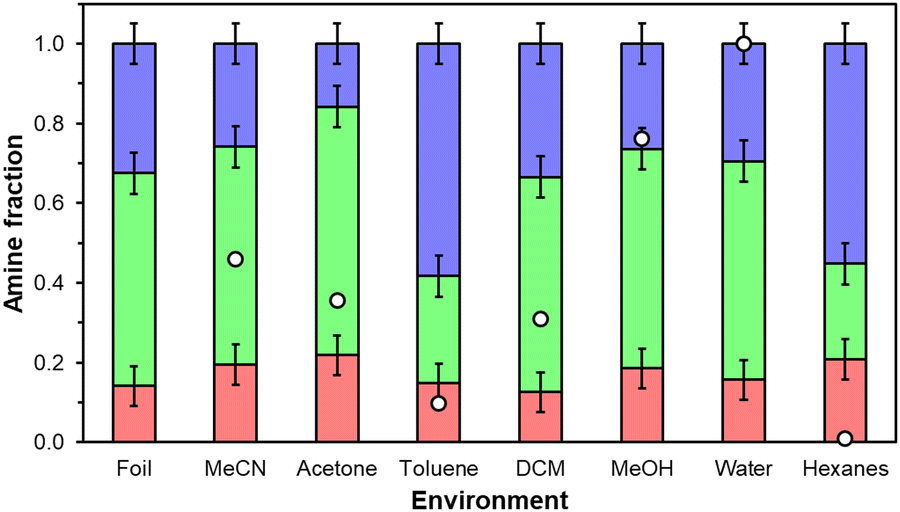 | ||
| Fig. 6 Primary (red), secondary (green), and tertiary (blue) amine distributions for as-synthesized poly(propylene imine) after exposure to different solvent environments of varying polarity (white circles) at ambient temperature (298 K) for 8 h. Distributions are determined via1H NMR in D2O solvent. | ||
Finally, though N2 physisorption adsorption–desorption isotherms and NLDFT pore size distributions suggest that PEI and PPI are both hosted within UiO-67(Zr) voids, it remains unclear whether non-specific interactions hinder aminopolymer migration during storage conditions. SEM images (Fig. 7(a), (b) and (e)) of as-synthesized UiO-67(Zr), 20 wt% PPI/UiO-67(Zr), and 20 wt% PEI/UiO-67(Zr) show the absence of large aminopolymer domains that would appear as films on the surface of UiO-67(Zr) micro-crystals of average diameter 0.60 ± 0.15 μm, supporting PEI and PPI internal incorporation into UiO-67(Zr). Based on amine efficiencies (Fig. 4), the storage environment that yielded the highest and lowest values are imaged to establish the starkest potential contrast in aminopolymer behavior. For 20 wt% PPI/UiO-67(Zr), air (Fig. 7(d)) confers the highest measured amine efficiency while hexanes yields the lowest (Fig. 7(c)); yet, no PPI aggregates were observed in either with comparable morphology and agglomeration of UiO-67(Zr) crystals in both air and hexanes. Similarly, 20 wt% PEI/UiO-67(Zr) stored in acetone (Fig. 7(f)) and MeOH (Fig. 7(g)) do not exhibit large aminopolymer domains and maintain bulk UiO-67(Zr) crystal morphologies. Therefore, PEI and PPI are not only incorporated within the UiO-67(Zr) framework, but also show limited ability to egress, which is promising for CO2 adsorption site retention during longer term storage, especially in air for PPI/UiO-67(Zr) and in MeOH for PEI/UiO-67(Zr).
 | ||
Fig. 7 SEM images at 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000× magnification of (a) UiO-67(Zr) with accompanying particle diameter distribution, (b) fresh 20 wt% PPI/UiO-67(Zr), (c) hexanes-20 wt% PPI/UiO-67(Zr), (d) air-20 wt% PPI/UiO-67(Zr), (e) fresh 20 wt% PEI/UiO-67(Zr), (f) acetone-20 wt% PEI/UiO-67(Zr), (g) MeOH-20 wt% PEI/UiO-67(Zr). 000× magnification of (a) UiO-67(Zr) with accompanying particle diameter distribution, (b) fresh 20 wt% PPI/UiO-67(Zr), (c) hexanes-20 wt% PPI/UiO-67(Zr), (d) air-20 wt% PPI/UiO-67(Zr), (e) fresh 20 wt% PEI/UiO-67(Zr), (f) acetone-20 wt% PEI/UiO-67(Zr), (g) MeOH-20 wt% PEI/UiO-67(Zr). | ||
Overall, 20 wt% PPI/UiO-67(Zr) and 20 wt% PEI/UiO-67(Zr) demonstrate activity for CO2 adsorption from concentrated 10% CO2 streams simulating point source emissions, but these materials also show efficacy for CO2 uptake from dilute streams (400 ppm CO2) representing direct air capture (DAC). Steady state (3 h) mass-normalized CO2 capacities at 308 K (Fig. 8) are similar between 20 wt% PPI/UiO-67(Zr) (0.42 ± 0.02 mmolCO2 gadsorbent−1) and 20 wt% PEI/UiO-67(Zr) (0.38 ± 0.02 mmolCO2 gadsorbent−1), consistent with trends observed for concentrated 10% CO2 streams. However, despite a 250× lower CO2 concentration in the DAC conditions, both materials retain >66% of the CO2 capacity and >68% of the amine efficiency observed under simulated flue gas conditions. As such, accessible amine sites in PEI and PPI demonstrate high affinity for CO2, even at dilute concentrations, and indicate the potential of these composite aminopolymer/UiO-67(Zr) adsorbent systems for practical air capture processes.
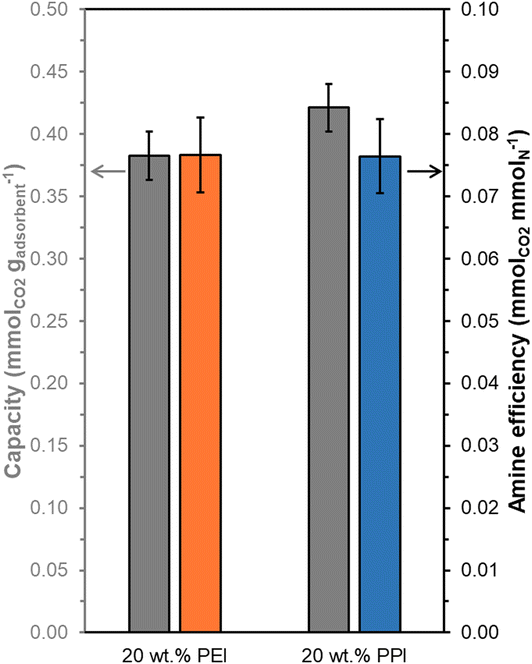 | ||
| Fig. 8 Amine efficiencies (orange, blue) and mass-normalized CO2 capacities (grey) at 308 K for 20 wt% PEI/UiO-67(Zr) (orange) and 20 wt% PPI/UiO-67(Zr) (blue) from 40 mL min−1 simulated air (400 ppm CO2 in He) after 3 h under 40 mL min−1 Ar at 383 K. | ||
Conclusions
Synthesized PPI and commercially available PEI are supported within nanoporous UiO-67(Zr) voids and tested for CO2 uptake from concentrated 10% CO2 gas streams to emulate flue gas compositions and from dilute 400 ppm CO2 streams for direct air capture applications. BET surface areas and NLDFT pore size distributions from N2 physisorption indicate that both PEI and PPI are contained within UiO-67(Zr) pores, but not tethered to Zr oxide nodes or the linker backbone. Repeated CO2 uptake-regeneration cycles for 20–50 wt% PPI/UiO-67(Zr) indicate that at higher aminopolymer weight loadings (30–50 wt%), CO2 mass transport limitations through the confined polymer become pronounced, with t20 values increasing monotonically with weight loading. Fresh mass-normalized CO2 uptake capacities for 20 wt% PEI/UiO-67(Zr) (0.58 ± 0.01 mmolCO2 gadsorbent−1) are similar to those for 20 wt% PPI/UiO-67(Zr) (0.54 ± 0.01 mmolCO2 gadsorbent−1), due to postulated residual acid modulator (HBr) presence in PPI that outweigh higher amine basicity benefits for PPI compared to PEI. The oxidative stability of 20 wt% PPI/UiO-67(Zr) and 20 wt% PEI/UiO-67(Zr) are investigated by exposing both composite materials to air at ambient temperatures and pressures for 7 days, with 20 wt% PPI/UiO-67(Zr) retaining its fresh amine efficiency and 20 wt% PEI/UiO-67(Zr) exhibiting a mild 22% loss in amine efficiency relative to its fresh value. Notably, PEI degradation in ambient air is reduced by 10% when confined within UiO-67(Zr) instead of as free polymer. Beyond PEI and PPI stabilization within UiO-67(Zr) confining voids, surrounding liquid solvent environments are explored to determine the impact of polarity, hydrogen bonding capability, and steric size on composite material stability and CO2 uptake efficacy. After 7 days of exposure to each solvent (water, MeOH, acetone, MeCN, DCM, hexanes, and toluene) at ambient temperatures and pressures, amine efficiencies are retained for 20 wt% PPI/UiO-67(Zr) in all solvents due to limited solvent-induced aminopolymer conformational changes. Surrounding solvent environments are more effectual for 20 wt% PEI/UiO-67(Zr), with storage in MeOH yielding the highest amine efficiency (0.200 ± 0.006 mmolCO2 mmolN−1) and acetone resulting in the lowest amine efficiency (0.094 ± 0.006 mmolCO2 mmolN−1) after 7 days due to solvent-induced PEI swelling and reaction to form less basic imine moieties, respectively. SEM images and 1H NMR spectra after storage for 7 days in air or liquid environments indicate no aminopolymer agglomeration or leaching, respectively, for 20 wt% PPI/UiO-67(Zr) and 20 wt% PEI/UiO-67(Zr). Finally, 20 wt% PPI/UiO-67(Zr) and 20 wt% PEI/UiO-67(Zr) demonstrate high CO2 affinity, retaining >66% of the CO2 capacity and >68% of the amine efficiency observed for flue gas conditions despite the 250× more dilute CO2 concentrations utilized to simulate air (400 ppm CO2), demonstrating the potential of these composite adsorbents for direct air capture applications. Overall, this work elucidates CO2 capture efficacies and material stabilities in different storage environments for PEI and PPI supported within UiO-67(Zr) and insight gleaned will have implications for solid adsorbent design and storage methods at both research and industrial scales.Author contributions
RAY and MLS wrote the manuscript. RAY, DRG, SC, JAV, and EJ synthesized and tested materials for CO2 uptake efficacy. RAY, SC, DRG, EJ, and JAV prepared FTIR pellets. RAY collected all FTIR spectra and XRD patterns. MRS obtained all SEM images. All authors discussed the presented data and contributed to the manuscript.Conflicts of interest
All authors declare no conflicts of interest.Acknowledgements
The authors acknowledge the use of Princeton's Imaging and Analysis Center, which is partially supported by the Princeton Center for Complex Materials, a National Science Foundation (NSF)-MRSEC program (DMR-1420541). We also appreciate the use of the Department of Chemistry's NMR facility at Princeton. Additional funding support includes the Peter B. Lewis Fund for Student Innovation in Energy and the Environment, the Lidow Independent Work/Senior Thesis Fund, and the DuPont Senior Thesis Fellowship Grant in memory of Michelle Goudie ’93.References
- L. A. Darunte, A. D. Oetomo, K. S. Walton, D. S. Sholl and C. W. Jones, ACS Sustainable Chem. Eng., 2016, 4, 5761–5768 CrossRef CAS
.
-
B. Kirtman, S. B. Power, A. J. Adedoyin, G. J. Boer, R. Bojariu, I. Camilloni, F. Doblas-Reyes, A. M. Fiore, M. Kimoto, G. Meehl, M. Prather, A. Sarr, C. Schär, R. Sutton, G. J. van Oldenborgh, G. Vecchi and H. J. Wang, Clim. Chang. 2013 Phys. Sci. Basis Work. Gr. I Contrib. to Fifth Assess. Rep. Intergov. Panel Clim. Chang., 2013, vol. 9781107057, pp.953–1028 Search PubMed
- United States Environmental Protection Agency, Inventory of U.S. Greenhouse Gas Emissions and Sinks: 1990–2019, 2021.
- A. B. Rao and E. S. Rubin, Environ. Sci. Technol., 2002, 36, 4467–4475 CrossRef CAS
- K. Goto, K. Yogo and T. Higashii, Appl. Energy, 2013, 111, 710–720 CrossRef CAS
- K. Li, J. Jiang, F. Yan, S. Tian and X. Chen, Appl. Energy, 2014, 136, 750–755 CrossRef CAS
- M. L. Sarazen and C. W. Jones, Macromolecules, 2017, 50, 9135–9143 CrossRef CAS
- M. L. Sarazen, M. A. Sakwa-Novak, E. W. Ping and C. W. Jones, ACS Sustainable Chem. Eng., 2019, 7, 7338–7345 CrossRef
- W. Zhang, H. Liu, C. Sun, T. C. Drage and C. E. Snape, Chem. Eng. J., 2014, 251, 293–303 CrossRef
- X. Xu, C. Song, J. M. Andresen, B. G. Miller and A. W. Scaroni, Energy Fuels, 2002, 16, 1463–1469 CrossRef
- C. Chen, J. Kim and W. S. Ahn, Korean J. Chem. Eng., 2014, 31, 1919–1934 CrossRef
- S. Bali, T. T. Chen, W. Chaikittisilp and C. W. Jones, Energy Fuels, 2013, 27, 1547–1554 CrossRef
- G. Rim, F. Kong, M. Song, C. Rosu, P. Priyadarshini, R. P. Lively and C. W. Jones, JACS Au, 2022, 2, 380–393 CrossRef PubMed
- J. Zhu, L. Wu, Z. Bu, S. Jie and B. G. Li, ACS Omega, 2019, 4, 3188–3197 CrossRef CAS PubMed
- A. Sinha, L. A. Darunte, C. W. Jones, M. J. Realff and Y. Kawajiri, Ind. Eng. Chem. Res., 2017, 56, 750–764 CrossRef CAS
- E. S. Sanz-Pérez, C. R. Murdock, S. A. Didas and C. W. Jones, Chem. Rev., 2016, 116, 11840–11876 CrossRef PubMed
- N. Erfaninia, R. Tayebee, M. Dusek and M. M. Amini, Appl. Organomet. Chem., 2018, 32, 1–10 CrossRef
- G. Y. Cha, H. Chun, D. Y. Hong, J. Kim, K. H. Cho, U. H. Lee, J. S. Chang, S. G. Ryu, H. W. Lee, S. J. Kim, B. Han and Y. K. Hwang, J. Hazard. Mater., 2020, 398, 122857 CrossRef CAS PubMed
- E. S. Sanz-Pérez, M. Olivares-Marín, A. Arencibia, R. Sanz, G. Calleja and M. M. Maroto-Valer, Int. J. Greenhouse Gas Control, 2013, 17, 366–375 CrossRef
- T. C. Drage, A. Arenillas, K. M. Smith and C. E. Snape, Microporous Mesoporous Mater., 2008, 116, 504–512 CrossRef CAS
- P. Bollini, S. Choi, J. H. Drese and C. W. Jones, Energy Fuels, 2011, 25, 2416–2425 CrossRef CAS
- G. Calleja, R. Sanz, A. Arencibia and E. S. Sanz-Pérez, Top. Catal., 2011, 54, 135–145 CrossRef CAS
- S. H. Pang, R. P. Lively and C. W. Jones, ChemSusChem, 2018, 11, 2628–2637 CrossRef CAS
- S. H. Pang, L. C. Lee, M. A. Sakwa-Novak, R. P. Lively and C. W. Jones, J. Am. Chem. Soc., 2017, 139, 3627–3630 CrossRef CAS
- T. Ghanbari, F. Abnisa and W. M. A. Wan Daud, Sci. Total Environ., 2020, 707, 135090 CrossRef CAS
- Q. Liu, L. Ning, S. Zheng, M. Tao, Y. Shi and Y. He, Sci. Rep., 2013, 3, 1–6 CAS
- M. J. Katz, Z. J. Brown, Y. J. Colón, P. W. Siu, K. A. Scheidt, R. Q. Snurr, J. T. Hupp and O. K. Farha, Chem. Commun., 2013, 49, 9449–9451 RSC
- H. Duo, Y. Wang, L. Wang, X. Lu and X. Liang, J. Sep. Sci., 2018, 41, 4149–4158 CrossRef PubMed
- J. B. Decoste, G. W. Peterson, H. Jasuja, T. G. Glover, Y. G. Huang and K. S. Walton, J. Mater. Chem. A, 2013, 1, 5642–5650 RSC
- M. Younas, M. Rezakazemi, M. Daud, M. B. Wazir, S. Ahmad, N. Ullah, Inamuddin and S. Ramakrishna, Prog. Energy Combust. Sci., 2020, 80, 100849 CrossRef
- D. K. Yoo and S. H. Jhung, J. Mater. Chem. A, 2022, 67, 8856–8865 RSC
- D. A. Gómez-Gualdrón, P. Z. Moghadam, J. T. Hupp, O. K. Farha and R. Q. Snurr, J. Am. Chem. Soc., 2016, 138, 215–224 CrossRef PubMed
- S. Choi, J. H. Drese, P. M. Eisenberger and C. W. Jones, Environ. Sci. Technol., 2011, 45, 2420–2427 CrossRef PubMed
- S. A. Idris, O. A. Mkhatresh and F. Heatley, Polym. Int., 2006, 55, 1040–1048 CrossRef CAS
- V. V. Nedel’ko, B. L. Korsunskii, F. I. Dubovitskii and G. L. Gromova, Polym. Sci. U.S.S.R., 1975, 17, 1697–1703 CrossRef
- A. Holewinski, M. A. Sakwa-Novak and C. W. Jones, J. Am. Chem. Soc., 2015, 137, 11749–11759 CrossRef CAS
- K. Grenda, A. Idström, L. Evenäs, M. Persson, K. Holmberg and R. Bordes, J. Appl. Polym. Sci., 2022, 139, 51657 CrossRef CAS
- J. E. Mills, C. A. Maryanoff, R. M. Cosgrove, L. Scott and D. F. McComsey, Org. Prep. Proced. Int., 1984, 16, 97–114 CrossRef CAS
- S. Chavan, J. G. Vitillo, D. Gianolio, O. Zavorotynska, B. Civalleri, S. Jakobsen, M. H. Nilsen, L. Valenzano, C. Lamberti, K. P. Lillerud and S. Bordiga, Phys. Chem. Chem. Phys., 2012, 14, 1614–1626 RSC
- V. G. Zaikin, R. S. Borisov, N. Y. Polovkov and L. N. Kulikova, J. Anal. Chem., 2012, 67, 1001–1004 CrossRef CAS
- L. Yu, M. Kanezashi, H. Nagasawa and T. Tsuru, Appl. Sci., 2018, 8, 1032 CrossRef
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ma01020h |
| This journal is © The Royal Society of Chemistry 2023 |