
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAssessing formic and acetic acid emissions and chemistry in western U.S. wildfire smoke: implications for atmospheric modeling†
Wade
Permar
 *a,
Catherine
Wielgasz
a,
Lixu
Jin
a,
Xin
Chen
b,
Matthew M.
Coggon
c,
Lauren A.
Garofalo
d,
Georgios I.
Gkatzelis
e,
Damien
Ketcherside
a,
Dylan B.
Millet
b,
Brett B.
Palm
f,
Qiaoyun
Peng
g,
Michael A.
Robinson
c,
Joel A.
Thornton
g,
Patrick
Veres
c,
Carsten
Warneke
c,
Robert J.
Yokelson
a,
Emily V.
Fischer
h and
Lu
Hu
a
*a,
Catherine
Wielgasz
a,
Lixu
Jin
a,
Xin
Chen
b,
Matthew M.
Coggon
c,
Lauren A.
Garofalo
d,
Georgios I.
Gkatzelis
e,
Damien
Ketcherside
a,
Dylan B.
Millet
b,
Brett B.
Palm
f,
Qiaoyun
Peng
g,
Michael A.
Robinson
c,
Joel A.
Thornton
g,
Patrick
Veres
c,
Carsten
Warneke
c,
Robert J.
Yokelson
a,
Emily V.
Fischer
h and
Lu
Hu
a
aDepartment of Chemistry and Biochemistry, University of Montana, Missoula, MT, USA. E-mail: wade.permar@umontana.edu
bDepartment of Soil, Water, and Climate, University of Minnesota, St. Paul, MN, USA
cChemical Sciences Laboratory, National Oceanic and Atmospheric Administration, Boulder, CO, USA
dDepartment of Chemistry, Colorado State University, Fort Collins, CO, USA
eInstitute of Energy and Climate Research, IEK-8: Troposphere, Forschungszentrum Jülich GmbH, Jülich, Germany
fAtmospheric Chemistry Observations & Modeling Laboratory, National Center for Atmospheric Research, Boulder, CO, USA
gDepartment of Atmospheric Sciences, University of Washington, Seattle, WA, USA
hDepartment of Atmospheric Science, Colorado State University, Fort Collins, CO, USA
First published on 4th September 2023
Abstract
Formic acid (FA) and acetic acid (AA), two of the most abundant organic acids in the atmosphere, are typically underestimated by atmospheric models. Here we investigate their emissions, chemistry, and measurement uncertainties in biomass burning smoke sampled during the WE-CAN and FIREX-AQ aircraft campaigns. Our observed FA emission ratios (ERs) and emission factors (EFs) were generally higher than the 75th percentile of literature values, with little dependence on fuel type or combustion efficiency. Rapid in-plume FA production was observed (2.7 ppb ppmCO−1 h−1), representing up to ∼20% of the total emitted reactive organic carbon being converted to FA within half a day. AA ERs and EFs showed good agreement with the literature, with little or no secondary production observed within <8 hours of plume aging. Observed FA and AA trends in the near-field were not captured by a box model using the explicit Master Chemical Mechanism nor simplified GEOS-Chem chemistry, even after tripling the model's initial VOC concentrations. Consequently, the GEOS-Chem chemical transport model underestimates both acids in the western U.S. by a factor of >4. This is likely due to missing secondary chemistry in biomass burning smoke and/or coniferous forest biogenic emissions. This work highlights uncertainties in measurements (up to 100%) and even large unknowns in the chemical formation of organic acids in polluted environments, both of which need to be addressed to better understand their global budget.
Environmental significanceFormic and acetic acid are the most abundant organic acids in the troposphere, playing an important role in regulating cloud droplet and aerosol pH levels, aqueous-phase chemistry, and gas–aerosol partitioning. However, their underestimation by atmospheric models in regions with significant biomass burning highlights an incomplete understanding of their emissions and production. Using measurements from the WE-CAN and FIREX-AQ aircraft campaigns, this study finds rapid formic acid production in wildfire plumes with higher emissions than previously reported. Moreover, current modeling approaches underestimate both acids across the western U.S. during wildfire season, due to missing secondary chemistry. Addressing these uncertainties is essential for advancing model development and improving our understanding of how biomass burning emissions impact regional air quality and atmospheric chemistry. |
1. Introduction
Formic acid (FA) and acetic acid (AA) are the two most prevalent organic acids in the troposphere, affecting aqueous-phase chemistry1 and gas–aerosol partitioning2 by regulating pH levels in cloud droplets and aerosols.3–6 Multiple studies have shown that various models continuously underestimate both FA and AA abundance compared to ground, airborne, and satellite observations. This low model bias is most pronounced in biogenic source regions,7,8 including United States (U.S.) deciduous forests,9,10 boreal forests,11 tropical forests,6 and in the Arctic tundra.12 Additionally, models typically fail to capture FA and AA enhancements in plumes from mixed anthropogenic sources13,14 and in biomass burning (BB) impacted regions,15–17 indicating potential missing primary and/or secondary sources in smoke. In this work, we investigate emissions, secondary productions, and model representations of FA and AA in the western U.S. during two wildfire seasons, using measurements made during the WE-CAN (Western Wildfire Experiment for Cloud Chemistry, Aerosol Absorption, and Nitrogen) and FIREX-AQ (Fire Influence on Regional to Global Environments and Air Quality) field campaigns.FA and AA are two of the most abundantly emitted volatile organic compounds (VOCs) from BB, accounting for 16% of the average VOC emissions by mass in western U.S. wildfires.18 As smoke plumes age, substantial secondary production of FA and AA may occur,19–23 resulting in these compounds together being one of the largest OH sinks in smoke aged more than 3 days, accounting for up to ∼25% of plume OH reactivity.24 Similarly, FA and AA can account for up to 15% of the VOC OH reactivity in urban atmospheres as well as the clean free troposphere in the western U.S. during wildfire season.24
Globally, top-down estimates suggest FA sources could reach 100–120 Tg y−1,6 which is two to three times higher than the sum of its known sources.6,8,16 Of this, photochemical production from biogenic sources has been estimated to contribute up to 90% of the global FA budget.6 Biomass burning may account for up to 16 Tg y−1 (∼13–16%) of FA globally,16 though such estimates for BB are mostly based on direct emissions. The secondary production of FA from BB precursors is poorly known due in part to the high uncertainty in BB emissions and a large amount of reactive BB precursors, such as furan containing species, not being implemented in current chemical transport models (CTMs).24 Similarly, global AA sources have been estimated using a bottom-up approach to be 85 Tg y−1,8 which is likely a lower bound.7 Despite BB being a major source of AA,23–26 the contribution of BB to the global AA budget is rarely discussed in the literature and is not well constrained.
The primary sinks of atmospheric FA and AA include wet and dry deposition, photochemical oxidation by OH radicals, and the irreversible uptake on dust resulting in atmospheric lifetimes of 2–4 days for FA and ∼2 days for AA.6–8,27 Consequently, their relatively short atmospheric lifetimes coupled with the localized and seasonal nature of fires likely means BB alone cannot close the global FA and AA budgets.8 However, in regions heavily impacted by BB it is likely that fires play an important role in their regional abundance and a more detailed understanding of their emissions and chemistry in wildfire smoke is needed.
As the two simplest organic acids, FA and AA may be produced from the oxidation of many different VOCs and are known photochemical products of isoprene, terminal alkenes, monoterpenes, glycolaldehyde, aromatics, acetone, and acetaldehyde.7,8,14,28–31 Heterogeneous formation of FA in aerosols and cloud droplets has also been identified as a potential major source, which, when included in the global chemistry – climate model ECHAM5/MESSy (EMAC), has been found to largely reconcile the global FA budget.32–34 However, regional discrepancies remain. For example, FA abundances were still underestimated in boreal forested regions, likely due to low emissions of FA precursors from BB.32
Analytical challenges measuring FA and AA,35 along with an incomplete understanding of chemical processes in smoke,36 has made it difficult to accurately model their evolution in BB plumes.16,37 For example, it has been well documented that the GEOS-Chem CTM underpredicts FA and AA abundances. Missing secondary production from biogenic precursors is thought to be one of the most significant reasons for the low model bias,7 though in some ecosystems there may still be missing primary emissions and/or in-canopy sources.11,38 The overall model sink may also be too large.39 By updating the model chemistry to reflect photochemical FA production from alkynes, monoterpenes, isoprene, methyl peroxy radical (CH3O2), ozonolysis of terminal alkenes, keto–enol tautomerization, and phototautomerization of acetaldehyde,7,8,40 Chen et al.17 were able to improve GEOS-Chem representation of the remote free troposphere relative to observations during the Atmospheric Tomography Mission (ATOM) field campagin. Despite the updated chemistry, the model underestimated the median FA![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :CO ratio by a factor of >2 and the 95th percentile by a factor of >4,17 suggesting that there are still significant missing secondary sources in smoke.
:CO ratio by a factor of >2 and the 95th percentile by a factor of >4,17 suggesting that there are still significant missing secondary sources in smoke.
In this work, we examine FA and AA emissions and chemistry in wildfire smoke to better understand the role of BB in their regional budgets. Using observations from the WE-CAN and FIREX-AQ aircraft campaigns, we first assess FA measurements made by two commonly used chemical ionization mass spectrometers, PTR-ToF and I− CIMS (proton-transfer-reaction time-of-flight mass spectrometer and iodide adduct chemical-ionization mass spectrometer). Emissions for FA and AA are then compared with literature values before examining their chemistry during WE-CAN in five pseudo-Lagrangian sampled smoke plumes. Finally, we assess GEOS-Chem representation of both acids across two fire seasons, first using observations made during the WE-CAN field campaign and then FIREX-AQ as an additional test for year-to-year variability and regional representativeness.
2. Methods
2.1. WE-CAN and FIREX-AQ campaign overviews and sampling approach
Comprehensive gas and aerosol measurements were made in wildfire smoke plumes across seven western U.S. states from 24 July to 13 September 2018 during the WE-CAN aircraft campaign (https://www.eol.ucar.edu/field_projects/we-can). In situ smoke plume sampling was carried out aboard the NSF/NCAR C-130 research aircraft based out of Boise, ID, typically between 14:00 and 19:00 local time when burning conditions were most active. Fig. S1† depicts the C-130 flight tracks during WE-CAN, colored by the observed formic and acetic acid mixing ratios. Upon arriving at a fire, the C-130 would typically sample fire emissions by flying perpendicular transects through the plume, as near to the source as was allowed by firefighting operations and plane safety constraints. To investigate plume aging, most plumes were subsequently sampled using a pseudo-Lagrangian approach where perpendicular transects were performed in a stepwise pattern starting near a fire and continuing as far downwind as possible (seen as the zig-zag flight pattern in Fig. S1†). In total, WE-CAN sampled more than 22 hours of wildfire smoke, including 31 emission transects of 24 unique fires18 and 1.2 hours of smoke estimated to have aged >3 days, along with 4.8 hours of the clean free troposphere.24The FIREX-AQ aircraft campaign sampled BB plumes across the western and southeastern U.S. from 22 July to 5 September 2019 (https://csl.noaa.gov/projects/firex-aq) following a similar sampling approach as WE-CAN.41 In this work, we separate FIREX-AQ data into its western and southeastern U.S. portions (FIREX-AQ-W and FIREX-AQ-SE), delimitated by the 105th meridian west, for a more accurate regional comparison. This allows us to assess FA and AA representation in the GEOS-Chem CTM across multiple fire seasons and regions. As the total VOC emissions in the western U.S. during the 2018 WE-CAN campaign were ∼10× higher, with ∼2× more area burned, than during the 2019 FIREX-AQ campaign (190 GgC vs. 20 GgC, 3.5 × 106 ha vs. 1.9 × 106 ha),42,43 these two datasets provide complementary representation of a wide range of seasonal fire activity allowing the model to be assessed under varying real-world conditions.
2.2. Measurements of formic acid, acetic acid, and organic aerosol
FA and AA were both measured by two different proton-transfer-reaction time-of-flight mass spectrometers (PTR-ToF) and iodide adduct chemical-ionization mass spectrometers (I− CIMS) during the WE-CAN and FIREX-AQ campaigns. The PTR-ToF18 and I− CIMS44–47 operated during WE-CAN and FIREX-AQ48–50 and referenced in this work have been extensively described by the cited literature, while here we include those details most relevant to their measurements of FA and AA.Organic aerosol (OA) was measured by high-resolution aerosol mass spectrometry (HR-AMS; Aerodyne Inc.), described in detail by Garofalo et al.51 During WE-CAN, the HR-AMS measured OA with 5 seconds time resolution, vacuum aerodynamic diameter of ∼70–1000 nm, and uncertainty of 35%. In this work we primarily use the fractional component of OA attributed to the CO2+ ion (f44), an OA oxidation marker.52,53 For plume transects, f44 averages are weighted by the measured OA mass.54
Calibrating FA and AA is analytically challenging due to their instability in gas standards and known humidity-dependent sensitivities in PTR-ToF measurements.55 To overcome these challenges, humidity-dependent FA and AA sensitivities were determined in the laboratory post-campaign using a commercial liquid calibration unit (LCU; Ionicon Analytik). Analytical grade FA and AA were volatilized in the LCU and dynamically diluted into zero air where the humidity was varied within the range observed during WE-CAN as determined by the internal humidity proxy of H2O·H3O+ to H3O+ ([m/z 39]/[m/z 21], 0–6%).55,56 The resulting calibration curves for FA and AA sensitivities as a function of the percent [m/z 39]/[m/z 21] are shown in Fig. S2† and applied to all WE-CAN FA and AA PTR-ToF measurements. Similar calibrations and humidity corrections were applied to PTR-ToF measurements during FIREX-AQ. Over the 0–6% [m/z 39]/[m/z 21] range, sensitivities for both species were observed to decrease with increased humidity, ranging ∼9–4 ncps per ppb, similar to the sensitivity change reported by Baasandorj et al.55 During WE-CAN the PTR-ToF FA and AA uncertainties are conservatively estimated as 50%, mostly due to 40% potential instrument drift between WE-CAN and the laboratory calibrations as determined from the observed instrument sensitivity change of other gas standards. The detection limits are 1.0 ppb for FA and 0.5 ppb for AA for 1 Hz measurements, defined as 3σ for the inflight instrument zeros.
In PTR-ToF, FA (HCOOH) and AA (CH3COOH) are detected at their protonated masses, m/z 47.013 and m/z 61.028 respectively. The corresponding mass resolution during WE-CAN is 2120 m/Δm at m/z 47 and 3060 m/Δm at m/z 61, where Δm is the full width at half maximum for the ion peak. FA has three major potential interfering ions: dimethyl ether (DME, m/z 47.077), ethanol (m/z 47.050), and N2H3O+ (m/z 47.024).25,55 The mass resolution during WE-CAN was high enough to separate DME and ethanol signals from FA, with ethanol abundance also expected to be ∼4× lower than FA in BB smoke with an instrumental sensitivity ∼10× lower than FA.25 The N2H3O+ signal, which was not fully resolved from FA, was observed to stay constant regardless of emission source strength throughout the campaign and was therefore classified and corrected as instrumental background. Consequently, we treat the m/z 47 signal as being primarily FA in agreement with previous literature.55–57
Potential interferences of AA in PTR-TOF measurements include 2-propanol and n-propanol (m/z 61.065), peroxyacetic acid (PAA) fragments (m/z 61.028), ethyl acetate fragments (m/z 61.028), methyl formate (m/z 61.028), and glycolaldehyde (m/z 61.028).25,55,57–61 Propanol was resolved from AA during WE-CAN, while PAA fragments, ethyl acetate fragments, methyl formate, and glycolaldehyde are all isomeric with AA. PAA is formed by the reaction of CH3C(O)O2 radicals with HO2, which may be important in low NOx conditions55 but is ∼100× less abundant than AA in fresh BB smoke (∼20× less abundant after 1.5 hours aging)22 making its fragment unlikely to be a significantly contributor to m/z 61.25 Ethyl acetate is used in coatings, adhesives, cosmetics, and as a process solvent,62 resulting in it being most prevalent in anthropogenically polluted areas, while it has not been reported in significant quantities in BB emissions.25,63 For methyl formate, a small peak can be seen by GC-MS during the FIREX-AQ laboratory burning experiment, but FTIR comparison suggests its contribution is negligible.25 Due to interference from these isomers being minimal in BB smoke, we do not attempt to correct for their presence, and assume m/z 61 to be predominantly AA and glycolaldehyde in wildfire emissions.64
Based on the FIREX-AQ Missoula fire laboratory burning experiments, the m/z 61 signal is on average 67% AA and 33% glycolaldehyde (±45% of value) in fresh BB emissions.25,65 However, the glycolaldehyde contribution in aged smoke is not well described. As glycolaldehyde's atmospheric lifetime of 1 day66 is approximately half of that of AA,8 it is likely that m/z 61 becomes more predominantly AA in aged air masses, though glycolaldehyde production could offset its loss. Given that the relative contribution of glycolaldehyde to m/z 61 was not constrained during WE-CAN, we do not attempt to correct for the potential glycolaldehyde interference and apply only the humidity dependent AA sensitivity to m/z 61. Though we treat and discuss the PTR-ToF m/z 61 as AA in this work, the reported values reflect the combined AA and glycolaldehyde isomers and therefore likely represent an upper bound for AA.
The I− CIMS employed a fast-zeroing approach described in Palm et al.46 where background concentrations were found by sampling ultra-high purity N2 into the IMR for 6 seconds every minute. The fast zeros were used to determine the background-subtracted signal by isolating the effects of adsorption and desorption of ‘sticky’ molecules on the internal IMR surfaces. This zero occurred both in and out of smoke plumes to account for the changes in background signal with varying sampled concentrations. A full inlet zero was also performed for 10 seconds every 20 minutes to determine the combined background signal from inlet tubing plus IMR surfaces, which confirmed that the dominant source of the background was from the IMR and not inlet tubing.46
The I− CIMS detects FA as a cluster with iodide at m/z 172.911. For WE-CAN, FA was calibrated in the laboratory prior to the campaign by flowing pure air over heated permeation tubes with gravimetrically determined permeation rates. Although I− CIMS measures AA, due to its low sensitivity and apparent interference from an unknown compound during WE-CAN, we only report AA from PTR-ToF. Recent work has shown that one potential source of uncertainty for the I− CIMS FA measurement is that its sensitivity to FA decreases with increasing IMR temperature.49 The I− CIMS deployed during WE-CAN did not directly regulate temperature in the IMR. During smoke sampling periods the C-130 cabin temperatures measured near the I− CIMS ranged from 20 to 32 °C (10th and 90th percentiles: 22–26 °C). Robinson et al.49 showed that a 10 °C change in IMR temperature could correspond to a 50% change in sensitivity, though differences in pressures and tuning between instruments makes applying this uncertainty to the WE-CAN deployment highly uncertain. It is also unlikely that the IMR temperature fluctuated as widely as the cabin temperature, though a lack of data makes it difficult to constrain the actual IMR temperature during WE-CAN or during the laboratory calibrations. Consequently, we conservatively estimate the I− CIMS FA measurement to be a likely upper bound, with 60% uncertainty and 30 ppt detection limit for 1 Hz data, based on calibration uncertainties and potential variation in IMR temperature.
For the I− CIMS, ambient air was sampled at 6 slpm through a mass-flow-controlled PFA inlet (70 cm length, 6.4 mm I. D.) maintained at 40 °C. A pressure control region upstream of a critical orifice at the entrance to the IMR was maintained at 140 mbar, and thus a constant flow of 1.2 slpm ambient air entered the IMR to mix with the 1 slpm ion source flow. Similar to the I− CIMS deployed during WE-CAN, the IMR was humidified to minimize instrument humidity dependence. The instrument background signal was determined in flight by overflowing the inlet with scrubbed ambient air for 30 seconds every 10 minutes through a port located 2 cm downstream of the inlet entrance.48 IMR temperature was not controlled during FIREX-AQ, but a post-campaign temperature correction was applied to the data as described in Robinson et al.49 The instrument uncertainty for FA is 15% ± 30 ppt with a 3σ detection limit of 6 ppt.
2.3. GEOS-Chem chemical transport model
GEOS-Chem nested grid simulations (version 12.1.1)68,69 over North America were run for the WE-CAN and FIREX-AQ periods using the model conditions described in Chen et al.17 Simulations were carried out using Goddard Earth Observation System Forward Processing (GEOS-FP) assimilated meteorology data with detailed HOx, NOx, VOC, ozone, halogen, and aerosol chemistry. Model runs were conducted at 0.25° × 0.3125° (∼25 km) resolution with time steps of 5 minutes (transport/convection) and 10 minutes (chemistry/emission). Emissions follow Chen et al.17 with the notable exception that we use Global Fire Assimilation System version 1.2 (GFAS) BB emissions with FA and AA emission ratios updated based on Permar et al.,18 which in turn reflect the WE-CAN averages discussed in Section 7. GEOS-Chem was subsequently sampled along both campaign flight tracks for comparison to the observations.The GEOS-Chem simulations also reflect updated FA chemistry including photochemical FA production based on OH initiated oxidation of alkynes, monoterpenes, isoprene, and CH3O2, ozonolysis of terminal alkenes (e.g. ethene and propene), keto–enol tautomerization,7,8 and phototautomerization of acetaldehyde.40 The model does not included the aerosol chemistry proposed by Franco et al.32 Based on these updates, Chen et al.17 found that GEOS-Chem accurately simulated FA concentrations in the remote free troposphere during the ATom aircraft campaign, indicating that GEOS-Chem is not missing any significant FA sources in the remote free troposphere. The model was found to significantly underestimate FA mixing ratios in 1–10 days aged plumes attributed to both anthropogenic and BB sources. In this work, we investigate how well GEOS-Chem, with the Chen et al.17 treatment of FA and AA chemistry, represents these acids in the western U.S. under heavily smoke impacted conditions.
3. Formic acid measurement intercomparison
Formic acid is analytically challenging to measure due to its ‘stickiness’ in sample inlets and its humidity/temperature dependent sensitivities in PTR-ToF35,55 and I− CIMS.44,49Fig. 1 shows the 1 Hz time series and cumulative mixing ratios of FA measured by PTR-ToF and I− CIMS during five plume transects (<20 km downwind) of the Taylor Creek (TC) fire sampled during WE-CAN (Research Flight #3). When corrected for inlet residence times, the two measurements show good temporal agreement, capturing the real-time plume variability. However, the PTR-ToF consistently measures ∼2× lower maximum FA concentrations than the I− CIMS during the plume transects likely representing sample retention in the inlet, a baseline offset due to background correction differences, and/or calibration errors.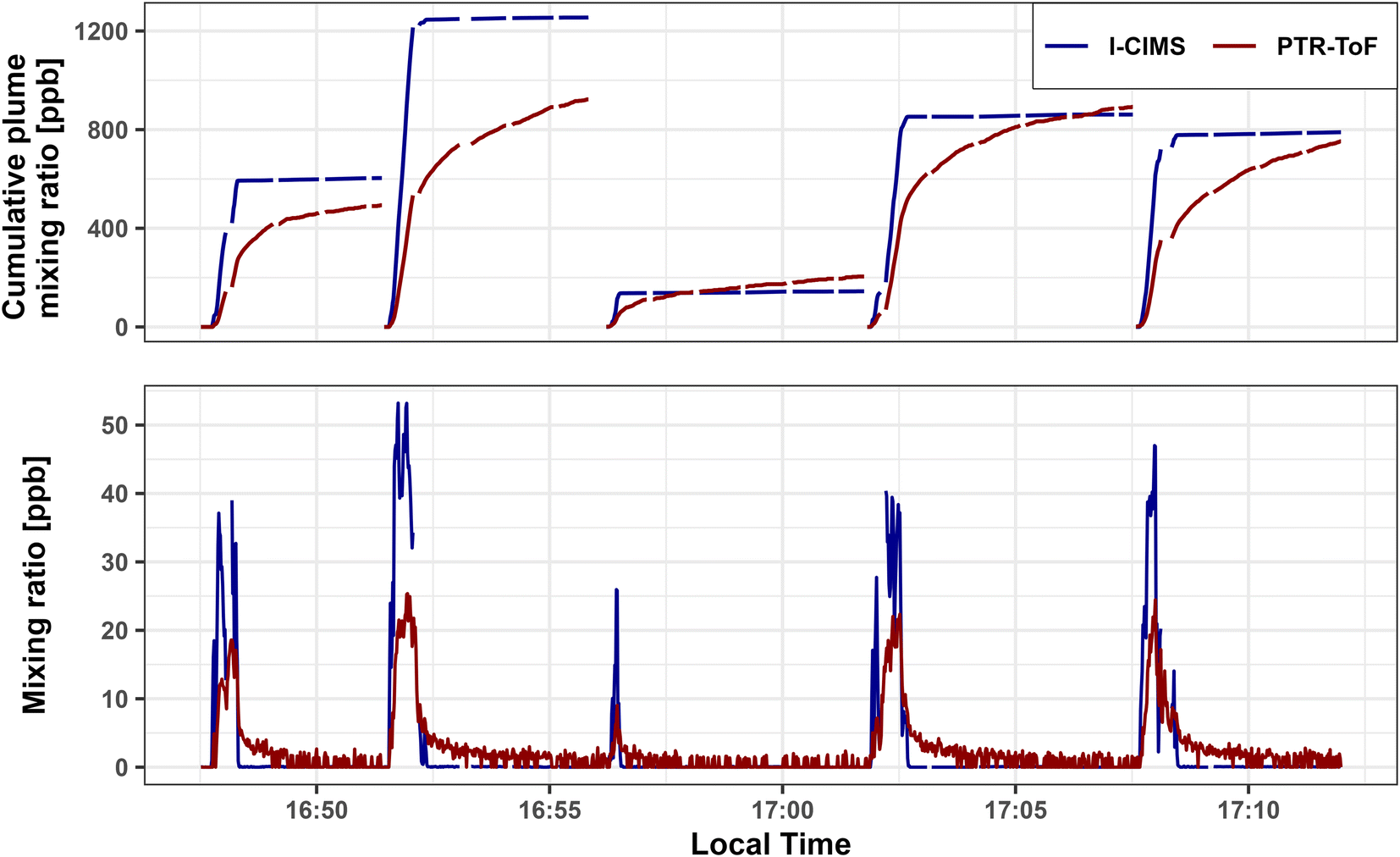 | ||
| Fig. 1 Time series of 1 Hz PTR-ToF and I− CIMS formic acid mixing ratios (bottom panel) and cumulative mixing ratios for each plume through the following background period (top panel) during 5 plume transects made <20 km downwind from the Taylor Creek Fire, OR during WE-CAN. | ||
The TC fire was sampled shortly after injection into the free troposphere with little to no regional smoke impacts, resulting in clearly defined plume edges that can be seen in Fig. 1 by the rapid FA enhancement upon entry into the smoke. However, when exiting the plumes, the PTR-ToF trace shows a distinct tail indicative of FA being initially retained in the inlet before flushing out in the 60–90 seconds after returning to background air. This is further illustrated by the upper panel in Fig. 1, where the cumulative mixing ratios for each plume through the subsequent background sampling periods are shown for both instruments. For all plumes shown in the figure I− CIMS and PTR-ToF integrated FA mixing ratios agree within <50% after accounting for residual FA in the inlet. This indicates that the two measurements agree within their stated uncertainty given sufficient time to recapture FA from the PTR-ToF inlet. However, Plumes 2 and 3 also demonstrate how FA may wash out of the inlet and increase the signal in subsequent transects. Due to most other sampling periods having either more poorly defined plume edges, elevated background signals from regional smoke, and/or not having enough time between consecutive transects, we are unable to accurately extend this analysis to other fires. It is likely though that inlet retention decreases the maximum PTR-ToF measured FA in most plumes sampled during WE-CAN.
The lack of a similar inlet artifact in the I− CIMS measurement is likely explained by a few characteristics. First, inlet sizes and materials differ slightly between the two instruments. The I− CIMS inlet is significantly shorter than the one used by the PTR-ToF, resulting in a shorter residence time in I− CIMS (<0.7 vs. 2 s). The I− CIMS inlet was also comprised of only PTFE, while the PTR-ToF used PFA and PEEK tubing. Second, the I− CIMS fast zeroing strategy (seen as the data gaps in the I− CIMS trace in Fig. 1) results in a FA background-subtracted signal that minimizes the effects of adsorption and desorption from walls and surfaces in the instrument.46 Consequently, this points to the importance of the instrument's inlet configuration and background correction procedures for the most accurate measurement of FA in environments with rapid concentration changes.
Although inlet retention explains a large part of the disagreement of FA measured by the two instruments while sampling smoke plumes with high, rapidly changing concentrations, the average flight integrated I− CIMS (120 ± 61 ppm) to PTR-ToF (66 ± 28 ppm) formic acid ratio of 2.1 and total least squares regression (TLS; slope = 2.06, r2 = 0.82), indicates that the I− CIMS measured ∼2× more FA than the PTR-ToF across all research flights (Fig. S3†). Although the exact reason for this disagreement is unknown, it likely stems from both a baseline offset and calibration uncertainty. For example, the PTR-ToF inflight zeros may have contained residual FA due to desorption from the instrument/inlet surfaces and/or incomplete oxidation in the catalyst-generated zero air. Consequently, an excessive background signal was subtracted, resulting in the mixing ratios being biased slightly low, especially when sampling relatively clean air.
The FA sensitivity in I− CIMS is also strongly dependent on IMR temperature,49 which was not directly controlled or logged during WE-CAN. The IMR used during WE-CAN46 was different than the IMR used in Robinson et al.,49 with each also operated at different pressures (100 mbar vs. 40 mbar). Although variability in IMR temperature may influence the I− CIMS FA sensitivity during WE-CAN, instrument differences likely change the temperature dependence between studies in ways that have not been tested. Consequently, the full extent of temperature effects on the I− CIMS sensitivity during WE-CAN is unknown and future work should focus on controlling IMR temperature while further characterizing the sensitivity dependence on temperature under different instrument conditions.
We repeat a similar analysis with FIREX-AQ FA measurements and find that in contrast to WE-CAN, the PTR-ToF generally measured slightly lower FA mixing ratios than the I− CIMS (TLS slope = 0.87, r2 = 0.78) for all FIREX-AQ data at altitudes below ∼4.9 km, with the two agreeing well within their stated instrument uncertainty. When compared between regions, the FIREX-AQ-W FA measurements shows better agreement (slope = 0.89, r2 = 0.82) than FIREX-AQ-SE (slope = 0.69, r2 = 0.67), possibly representing uncertainty in the instrument sensitivity due to the higher humidity typical of the southeastern U.S. relative to the western U.S. (Fig. S3†). Additionally, neither instrument shows significant inlet retention, with FA mixing ratios for both instruments generally returning to background levels at the same rate after exiting plumes (Fig. S4†).
The observed disagreement between the two instruments during WE-CAN is likely due to a combination of factors including uncertainty in the FA sensitivity due to its humidity and temperature dependence, potential instrument drift between the laboratory calibrations and field measurements, inlet losses, and differences in background correction procedures. FIREX-AQ FA measurements largely corrected for these issues by using a shorter PTR-ToF inlet along with correcting for the I− CIMS IMR temperature variations. Consequently, to improve future FA measurements made by I− CIMS and PTR-ToF special attention should be given to shortening inlet residence times and minimizing sensitivity dependencies on temperature and humidity.
In this work, we primarily use I− CIMS FA measurements for most analysis and discussion due to its lower detection limits and lack of apparent inlet artifacts. This is likely an upper bound for WE-CAN data and therefore we include PTR-ToF FA observations to further constrain the measurement uncertainty where appropriate. We note that despite the high uncertainty in the FA measurements (up to 100%), model underestimates discussed in Section 6 are much greater than the measurement uncertainty.
4. Emissions of formic and acetic acid from WE-CAN sampled fires
Emission ratios (ERs) and emission factors (EFs) were calculated for 31 WE-CAN emission transects of 24 individual fires as described in Permar et al.18 Here, emission transects are defined as the nearest transect of a well-defined smoke plume that is traceable to a single emission source sampled 27–130 minutes downwind from the fire, as calculated by wind speeds measured aboard the C-130 and fire locations reported by the U.S. Forest Service.18 Although these transects represent the freshest smoke sampled during the campaign, this is sufficient time for substantial secondary formation to have occurred.19,47,70,71 Consequently, FA and AA ERs and EFs during WE-CAN represent their combined production and loss before being sampled by the research aircraft, which may be more appropriate for the spatial and temporal resolution of many CTMs.72 Normalized excess mixing ratios (NEMRs) were calculated using the background corrected plume integrated mixing ratios of a VOC to CO (ppb VOC ppmCO−1) for each emission and subsequent downwind plume transect. ERs were used to calculate EFs, expressed as grams of VOC emitted per kilogram of burned fuel, using the carbon mass balance method23,73 with the total emitted carbon as the sum of CO2, CO, CH4, organic carbon, black carbon, and 161 VOCs.18 ERs and EFs for 16 fires sampled during FIREX-AQ are from Gkatzelis et al.,50 with estimated ages of 10–150 minutes. Fuels burned during WE-CAN and FIREX-AQ were primarily those characteristic of mixed conifer forests.Literature values were compiled from 16 different papers, reporting 330 FA and 289 AA ERs and EFs.74 Approximately half of the EFs were also recorded and retrieved from the Smoke Emissions Reference Application (SERA), which may include some additional values recalculated to match their fuel types.75 Average FA and AA ERs and EFs for the literature described in this work are summarized in Table S1† and represent a variety of burned fuels. Pre 2007 FA data measured by Fourier transform infrared spectroscopy (FTIR) have also been corrected by a factor of 2.1 following Yokelson et al.76 Similar to vegetation classifications in global BB emissions inventories, we broadly categorize these ERs and EFs as conifer forest (147 FA and 122 AA EFs), mixed hardwood forest (17, 23), shrubland (53, 38), grassland (36, 31), crop residue (46, 47), and organic soil/peat (31, 28). Table S1† also includes modified combustion efficiency (MCE) when available, instrumentation used, region of fuels burned, and whether the data are from a laboratory or field study.19,20,23,25,26,35,77–80
Fig. 2 shows FA and AA ERs for each of the 24 fires sampled during WE-CAN (green points), 16 fires sampled during FIREX-AQ (blue points), and literature values for all fuel types in our review (box-and-whisker plots). During WE-CAN, the average formic acid ER calculated from I− CIMS data was found to be 9.5 ± 4.2 ppb ppmCO−1 (Table 1), which is 3.5 times higher than the literature average of 2.7 ± 2.6 ppb ppmCO−1 calculated from 168 data points reported for 10 of the 16 studies in Table S1.† We note that although FA ERs calculated from PTR-ToF measurements (average 6.6 ± 2.5 ppb ppmCO−1) are slightly lower than those from I− CIMS, both are generally higher than the 75th percentile of literature values. Similarly, though FA ER during FIREX-AQ (average 3.31 ± 2.0 ppb ppmCO−1) are lower than during WE-CAN, half are still above the 75th percentile of the literature. Additionally, constraining ERs to only western U.S. fuels has little effect on this comparison as discussed in more detail below.
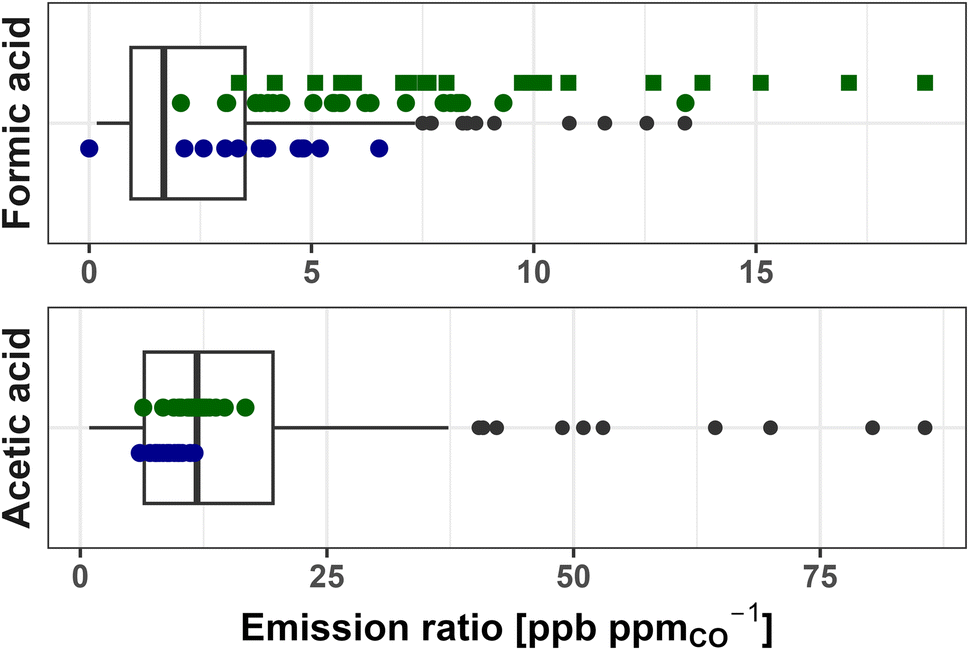 | ||
| Fig. 2 Emission ratios of formic and acetic acid for literature values (box-and-whisker), WE-CAN PTR-ToF observations (green points), I− CIMS FA (green squares), and FIREX-AQ PTR-ToF (blue points). The box and whisker plots reported include literature ERs from all studies in Table S1,† representing a variety of fuels (204 data points for formic acid and 196 for acetic acid). Boxes represent the 25th and 75th percentiles, with vertical lines as median, whiskers as 1.5× the interquartile range, and black points as >1.5 × interquartile range of literature values. | ||
| WE-CAN | FIREX-AQ | Literature average | Recommended average | ||
|---|---|---|---|---|---|
| Formic acid | ER ± 1σ (range) | 9.5 ± 4.2 (3.4–18.8) | 3.3 ± 2.0 (0–6.5) | 2.7 ± 2.6 (0.17–13.4) | 3.5 ± 3.4 |
| EF ± 1σ (range) | 1.5 ± 0.60 (0.55–2.5) | 0.60 ± 0.42 (0–1.6) | 0.35 ± 0.48 (0.002–4.2) | 0.42 ± 0.56 | |
| n. obs | 20 | 16 | 168 ERs, 330 EFs | 204, 366 | |
| Eq. with MCE | y = −4.8x + 5.8 | y = −6.4x + 6.3 | y = −9.7x + 9.4 | ||
| r 2 = 0.03 | r 2 = 0.35 | r 2 = 0.31 | |||
| Acetic acid | ER ± 1σ (range) | 11.5 ± 2.1 (6.4–16.7) | 8.9 ± 1.5 (6.0–11.7) | 15.5 ± 14.2 (0.9–85.6) | 14.5 ± 12.8 |
| EF ± 1σ (range) | 2.4 ± 6.1 (1.2–3.3) | 2.1 ± 0.63 (1.1–3.4) | 2.5 ± 2.6 (0.14–14.0) | 2.0 ± 1.9 | |
| n. obs | 24 | 16 | 156 ERs, 289 EFs | 196, 254 | |
| Eq. with MCE | y = −20.7x + 21 | y = −20.2x + 20.8 | y = −19.9x + 20.5 | ||
| r 2 = 0.52 | r 2 = 0.14 | r 2 = 0.15 | |||
One possible explanation for the higher ERs observed during WE-CAN and FIREX-AQ is that a significant amount of FA has been produced in the plumes prior to being intercepted by the C-130 (Section 5). To approximate how much FA may have been formed before being sampled, we estimate t0 emission ratios from the least squares regression of WE-CAN NEMRs vs. physical age for three of the five pseudo-Lagrangian sampled smoke plumes discussed in Section 5. Assuming a constant production rate, projected FA NEMRs from I− CIMS measurements at t0 range from 5.7–7.4 ppb ppmCO−1, which is still approximately 2–3 times higher than the literature average. Consequently, while many of the FA ERs measured during WE-CAN likely reflect some plume aging (which, although hard to quantify, may also be the case in literature values), near-field production alone is not enough to explain the disagreement. Given that WE-CAN ERs calculated using both I− CIMS (9.5 ppb ppmCO−1) and PTR-ToF (6.6 ppb ppmCO−1) measurements agree within their stated uncertainty, it is likely that the ERs observed during WE-CAN generally represent higher FA emissions from the wildfires sampled that season than the literature average (Fig. S6†). As FIREX-AQ FA ERs are also generally higher than the literature, this may in part reflect the bias of these two datasets towards sampling relatively large wildfires, which could produce different FA emissions than laboratory burns and the smaller fires predominantly represented in the literature. We recommend that future studies report their estimated aging when reporting FA ERs.
In contrast, AA ERs measured during WE-CAN and FIREX-AQ mostly fall within the 25th–75th percentiles of literature values (Fig. 2 and Table 1), with good agreement between their averages (WE-CAN 11.5 ± 2.1 ppb ppmCO−1, FIREX-AQ 8.9 ± 1.5 ppb ppmCO−1, literature 15.5 ± 14.2 ppb ppmCO−1). We note that by treating the PTR-ToF m/z 61 as being primarily AA (Section 2.2.1), the WE-CAN and FIREX-AQ ERs and EFs likely represent an upper bound, though still agreeing well with literature values if assuming potentially 30% glycolaldehyde interference.
Fig. S5† shows that FA EFs follow the same trend as the ERs, with the WE-CAN average EF of 1.5 ± 0.60 g kg−1 (PTR-ToF = 0.96 ± 0.39 g kg−1) and FIREX-AQ average EF of 0.6 ± 0.42 g kg−1 approximately 5 and 2 times higher than the literature average of 0.35 ± 0.48 g kg−1 Similarly, both campaign AA EFs are within the 25th–75th percentile of literature values, with good agreement between their averages (WE-CAN 2.4 ± 6.1 g kg−1, FIREX-AQ 2.1 ± 6.3 g kg−1, literature 2.5 ± 2.6 g kg−1).
To examine if the observed organic acid emission variability is related to burning condition, we compare the derived EFs from WE-CAN and literature coniferous forests to the modified combustion efficiency, which is a simple proxy used to describe the degree of flaming versus smoldering combustion in a fire. MCE is defined as,
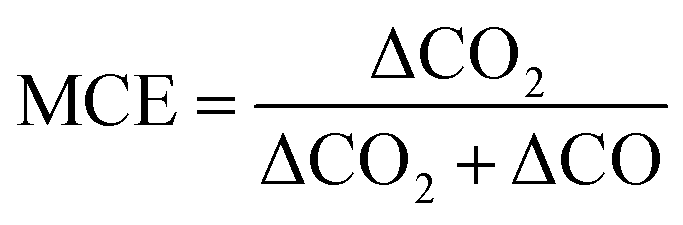 | (1) |
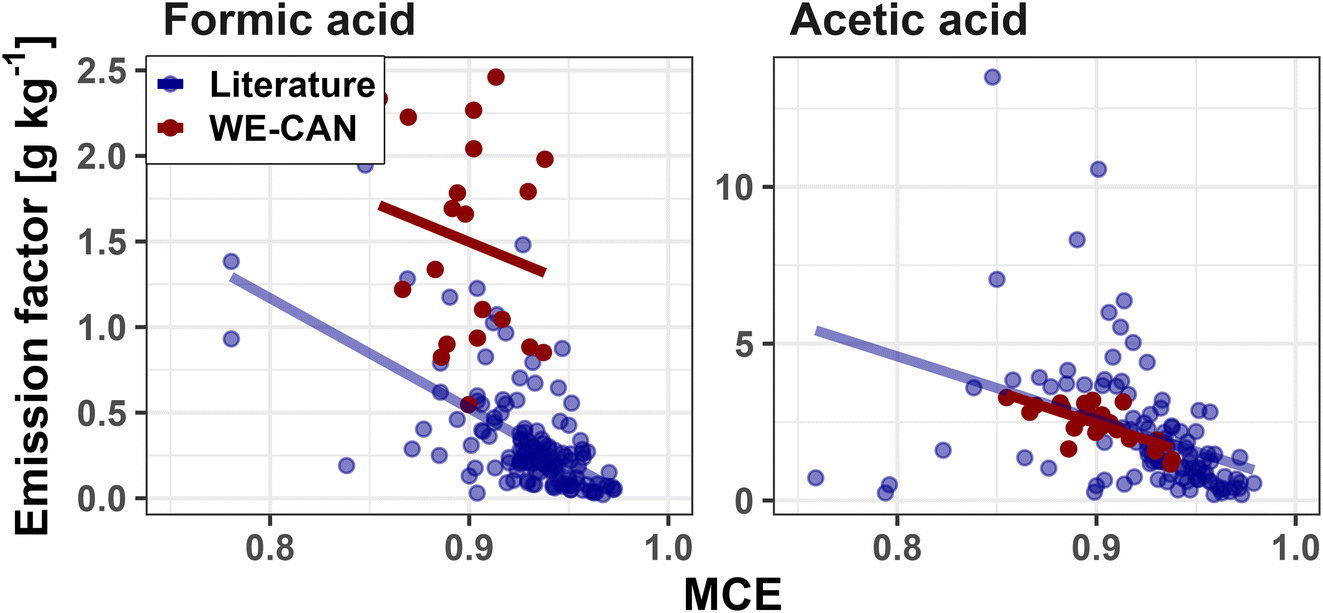 | ||
| Fig. 3 Correlations of FA (left) and AA (right) EFs versus MCE for both WE-CAN (red points) and literature reported coniferous forest values (blue points). The least squares regression for each group is shown in corresponding colors. For FA, the line of best fit for WE-CAN data is y = −4.8x + 5.8 (r2 = 0.03) and y = −6.4x + 6.3 (r2 = 0.35) for literature values. For AA, the line of best fit for WE-CAN data is y = −20.7x + 21.1 (r2 = 0.52) and y = −20.2x + 20.8 (r2 = 0.14) for literature values. Detailed statistics including PTR-ToF FA are shown in Table 1. | ||
To determine if the type of fuel burned influenced FA or AA emissions, we compare WE-CAN and literature EFs between the six fuel categories described above. For each organic acid we use a Tukey's range test to evaluate if the 95% confidence interval (CI) of emission factors for each fuel type overlap. For FA, the Tukey range test p-values are >0.05 for comparisons between all fuel types except with shrubland, indicating an overlap in the 95% CI for most fuels. This suggests that FA EFs for shrubland, mainly consisting of chaparral vegetation types in our literature review, have statistically significant differences from the other 5 fuel categories. Alternatively, no statistical difference was found between any of the other categories. Coupled with the lack of correlation with MCE, this suggests that a single FA ER of 3.5 ± 3.4 ppb ppmCO−1 and EF of 0.42 ± 0.56 g kg−1 (average of WE-CAN, FIREX-AQ, and literature values ±1σ, Table 1) best describe most BB emissions, though a fuel-specific EF for shrubland fuels (0.11 ± 0.09 g kg−1) may be more accurate.
AA EFs between coniferous forests, mixed hardwood forests, shrubland, and grassland similarly show no statistically significant fuel related difference, and an average ER of 14.5 ± 12.8 ppb ppmCO−1 and EF of 2.0 ± 1.9 g kg−1 may best describe most fuel types. However, organic soil/peat and crop residue both have p-values <0.05 when compared to the other four fuels, suggesting that MCE and fuel dependent EFs may be needed to best describe AA EFs. Given that AA shows some dependence on MCE, it is possible that the differences between crop residue and organic soil/peat EFs compared to EFs for the other fuel categories is in part due to combustion efficiency. For example, organic soil/peat combustion is generally dominated by smoldering (MCE = 0.68–0.92 in this work), which would result in higher EFs (Fig. 3). Box plots of FA and AA EFs for each fuel category are shown in Fig. S6.†
5. Near-field acid production during WE-CAN
FA and AA concentrations varied widely during WE-CAN with maximum mixing ratios of 98 ppb and 89 ppb, respectively. The highest FA NEMR of 71 ppb ppmCO−1 was observed in smoke aged ∼13 hours. ERs were not measured for this fire; however, this is ∼7 × higher than campaign average ER (9.5 ± 4.2 ppb ppmCO−1), and 4× higher than the maximum ER (18.8 ppb ppmCO−1). This suggests a maximum FA production rate of 4.0–4.7 ppb ppmCO−1 h−1 in aged smoke sampled during WE-CAN. This NEMR is approximately half of the maximum observed during ATom in smoke sampled off the African coast estimated to have been aged 1–10 days (140 ppb ppmCO−1), though this latter value is similar to many other plumes intercepted during that campaign.17Fig. 4 shows FA and AA NEMRs as a function of smoke plume age for 5 fires with more than 10 plume transects sampled in a pseudo-Lagrangian fashion during WE-CAN, while NEMRs for all sampled plumes are shown in Fig. S7† for reference. In the first 8 hours of plume aging FA is rapidly produced at an average rate of 2.7 ppb ppmCO−1 h−1. This is in good agreement with FA production seen in other studies including 2.6–3.3 ppb ppmCO−1 h−1 in smoke from Alaskan boreal forest fires,20 1.6 ppb ppmCO−1 h−1 from BB in the Yucatan, Mexico,22 and 0.9 ppb ppmCO−1 h−1 in chaparral fires in California.19 Given that a majority of the EFs/ERs in our literature review are from laboratory burns, with the few field sampled fires being small enough that aircraft could often sample directly over the source, they represent smoke with little to no aging. As WE-CAN and FIREX-AQ emissions are estimated to have been sampled 10–150 minutes downwind from the fires, we hypothesize that the higher FA EFs and ERs discussed in Section 3 are partially due to the rapid FA production observed during the campaigns.
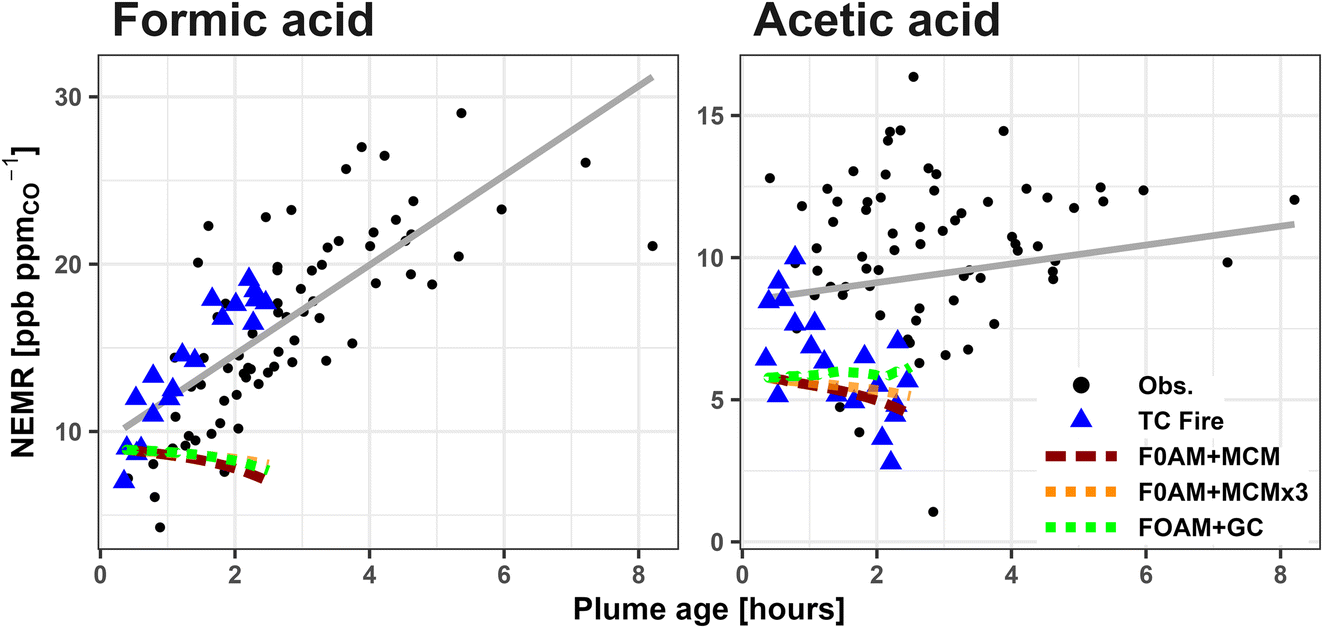 | ||
| Fig. 4 NEMRs of FA and AA for 5 research flights with more than 10 pseudo-Lagrangian transects. Blue triangles highlight plume transects of the Taylor Creek (TC) fire and correspond to the red dashed F0AM + MCM, orange F0AM + MCM × 3, and green F0AM + GC predicted NEMRs for the same fire. Black points correspond to the other 4 fires. Least squares regression lines for the aggregated data are shown in gray. During the first 8 hours of plume aging FA NEMR increased on average 2.7 ppb ppmCO−1 per hour (r2 = 0.58, intercept = 9.3 ppb ppmCO−1), while AA has a statistically insignificant increase of 0.3 ppb ppmCO−1 per hour (r2 = 0.03, intercept = 8.4 ppb ppmCO−1). | ||
During WE-CAN, AA NEMRs remain relatively constant in the first 8 hours of plume aging, increasing by a statistically insignificant 0.3 ppb ppmCO−1 h−1 (p = 0.13, r2 = 0.03, Fig. 4). Additionally, the maximum AA NEMR was observed to be 17 ppb ppmCO−1 in the same ∼13 hours aged plume discussed above, which is within 3σ of the campaign average ER. The extent that AA is produced in BB plumes is not well understood. For example, no net AA production has similarly been observed in smoke aged 1.4 hours over the Mexican Yucatán Peninsula22 nor inferred in BB plumes measured across Alaska and western Canada.82 However, multiple other studies have observed rapid AA production in the first few hours of plume aging including: 2.3 ppb ppmCO−1 h−1 in smoke from a Californian chaparral fire aged 4.5 hours,19 1.5 ppb ppmCO−1 h−1 in 1 hour aged smoke from southeast U.S. prescribed agricultural burning,35 1.5–2.0 ppb ppmCO−1 h−1 in smoke aged <1 hour from African Savanah fires,26 and 7.2 ppb ppmCO−1 h−1 in smoke aged 1 hour from Alaskan boreal forest fires.20 Future work is needed to better characterize AA production, especially in smoke that has aged more than half a day.
One potential explanation for the lack of observed AA production during WE-CAN is that the removal of glycolaldehyde offsets the formation of AA. This is possible because AA measured by PTR-ToF may be ∼30% glycolaldehyde in fresh emissions,25,65 both species have similar sensitivities in PTR-MS,55 and glycolaldehyde is ∼20× more reactive than AA with OH (kOH 1.1 × 10−11 cm3 molecule−1 s−1vs. 7.4 × 10−13 cm3 molecule−1 s−1; NIST chemical kinetics database average). Additionally, of the studies that observed AA production listed above, only Müller et al.35 used a PTR-ToF while the others used FTIR, which does not have isomeric interferences for AA. To test this hypothesis, we attribute only 66% of the PTR-ToF m/z 61 to AA to calculate a corrected campaign average ER of 7.6 ppb ppmCO−1. Assuming there is negligible glycolaldehyde formation downwind, such that the PTR-ToF is only measuring AA in aged smoke, the maximum AA production rate over ∼13 hours of plume aging would be 0.7 ppb ppmCO−1 h−1. Although this production rate is still lower than in the literature, the 30% reduction in the campaign average AA ER brings it outside of the observed NEMR variance for most aged smoke samples. This suggests that AA production could be statistically significant if these assumptions are true, and more detailed characterization of the glycolaldehyde interference in aged smoke is needed.
It is also possible that most of the AA formation happens in the first hours of plume aging, which is then averaged out in our analysis of plumes that are aged 8 hours during WE-CAN. For example, in one plume, Goode et al.20 observed an AA increase of 11 ppb ppmCO−1 in the first 1.5 hours of plume aging followed by no net production relative to emissions in 2.8 hours old smoke. However, limiting WE-CAN observations to those with <2 hours aging in Fig. 4 still results in a non-statistically significant NEMR-vs-age slope of 0.23 ppb ppmCO−1 h−1 (p = 0.80, r2 = 0.002). Similarly, if each of the 5 fires in Fig. 4 are treated individually, only two have statistically significant correlations (p = 0.02) with plume age, both with negative slopes (−0.2 and −0.3 ppb ppmCO−1 h−1). Given the relatively long atmospheric lifetime of AA (∼2–3 days),8,39 it is unlikely that a significant amount was removed from the plume in the 8 hours of aging shown here. Consequently, these results suggest that most of the observed AA in the near-field during WE-CAN is from primary emissions, though photochemical production may still be an important source in some fires and over longer plume aging times which should be investigated further.
Previous work has used the Framework for 0-D modeling (F0AM)83 to simulate the Taylor Creek (TC) fire sampled during WE-CAN due to it being a well isolated plume with pseudo-Lagrangian samples performed just after injection into the free troposphere.24,47,51,84–87 Here, we use the same F0AM model run as originally described in Peng et al.87 with updated VOC emissions per Permar et al.24 Briefly, F0AM was initialized using 49 VOCs, plus NO, NO2, HONO, O3, and CO (Table S2†). For VOCs measured by PTR-ToF, potential interfering isomers, including glycolaldehyde, were removed so that the model was initialized based on the proportion of the mass attributed only to the given species following Koss et al.25 Physical parameters such as photolysis frequencies, temperature, and pressure were constrained to measured values at each model step with a dilution correction factor based on CO observations. Model chemistry was simulated using the explicit and updated Master Chemical Mechanism (F0AM + MCM) including recently developed furans and phenolic chemistry,88,89 with an additional sensitivity test run using 3× VOC initial values for all gases except FA and AA (F0AM + MCM × 3). We also run the same box model driven with the GEOS-Chem chemical mechanisms (F0AM + GC) to test if recent updates by Chen et al.17 significantly impact the modeled FA production in fresh smoke.
Fig. 4 shows that neither F0AM + MCM, F0AM + MCM × 3, nor F0AM + GC can reproduce the rapid FA formation observed in the TC plume, with modeled FA instead decreasing slightly with plume age. The MCM predicted loss rate for FA in the base run is ∼0.3 ppb h−1 by reaction with OH, while there is minimal production (∼0.01 ppb h−1) from C4H6O3 and CH2OO Criegee intermediates. Coupled with the MCM being insensitive to increased initial values, this represents more FA being removed in the model than is being produced and indicates that both the MCM and GC are missing a substantial amount of secondary FA production in BB smoke.
The total emitted VOC carbon during WE-CAN averaged 367.3 ± 29.6 ppbC ppmCO−1.18 As the average FA enhancement shown in Fig. 4 is 21 ppb ppmCO−1, approximately 5.7% of the VOC carbon oxidation would need to go to FA to explain the observed production. Additionally, the maximum observed FA NEMR of 71 ppb ppmCO−1 after ∼13 hours of plume aging represents a 61 ppb ppmCO−1 enhancement relative to the campaign average ER, indicating that up to 17% of the total emitted reactive organic carbon could be converted to FA within half a day.
AA NEMRs in the TC plume are highly variable, likely representing changes in fire emissions or sampling different parts of the plume. Fig. 4 shows that F0AM + MCM and F0AM + GC generally have good agreement with the observed AA NEMRs for that fire, though poorly represents the general trend for the other 4 fires. Similar to FA, AA is also mainly lost in the MCM through reaction with OH at ∼0.4 ppb h−1, with negligible production (<0.01 ppb h−1) from CH3CHOO Criegee intermediates and CH3C(O)O2 peroxy acetyl radicals.
A current lack of understanding of the major FA and AA precursors is one of the largest hurdles to accurately modeling their evolution in smoke. To evaluate potential VOC precursors, NEMRs for both acids measured in the same 5 wildfires as described above were compared to NEMRs of 152 VOCs measured during WE-CAN using least squares regression. FA was found to have statistically significant negative correlations (p-value <0.05, r2 > 0.10) with 94 VOCs. Over the 8 hours of plume aging the oxidation of these 94 species collectively accounts for 127 ppbC ppmCO−1 that is reacted away. This indicates that those species lose 6× more carbon than is needed to account for the observed FA production, though the exact chemical pathways are often unknown. For example, C3H4O2 (methyl glyoxal + acrylic acid), styrene, and furanoid compounds such as 3-methylfuran and furfural are among the species with strongest correlations to FA in fresh smoke during WE-CAN (Fig. 5; r2 > 0.40). As furanoid compounds could be important dicarbonyl precursors,90,91 FA production from these VOCs may follow a similar mechanism as has been proposed for ketene–enol and dicarbonyl oxidation from aromatic compounds.92 However, the updated MCM does not currently incorporate this specific mechanism, nor does it show any chemical production of FA from furanoid or aromatic compounds.88,93,94 Although, the GEOS-Chem mechanism predicts ∼25% of the FA production in the Taylor Creek fire is from aromatics following recent mechanism/model developments,92,95 furanoid compounds, styrene, and methyl glyoxal/acrylic acid are currently not implemented.
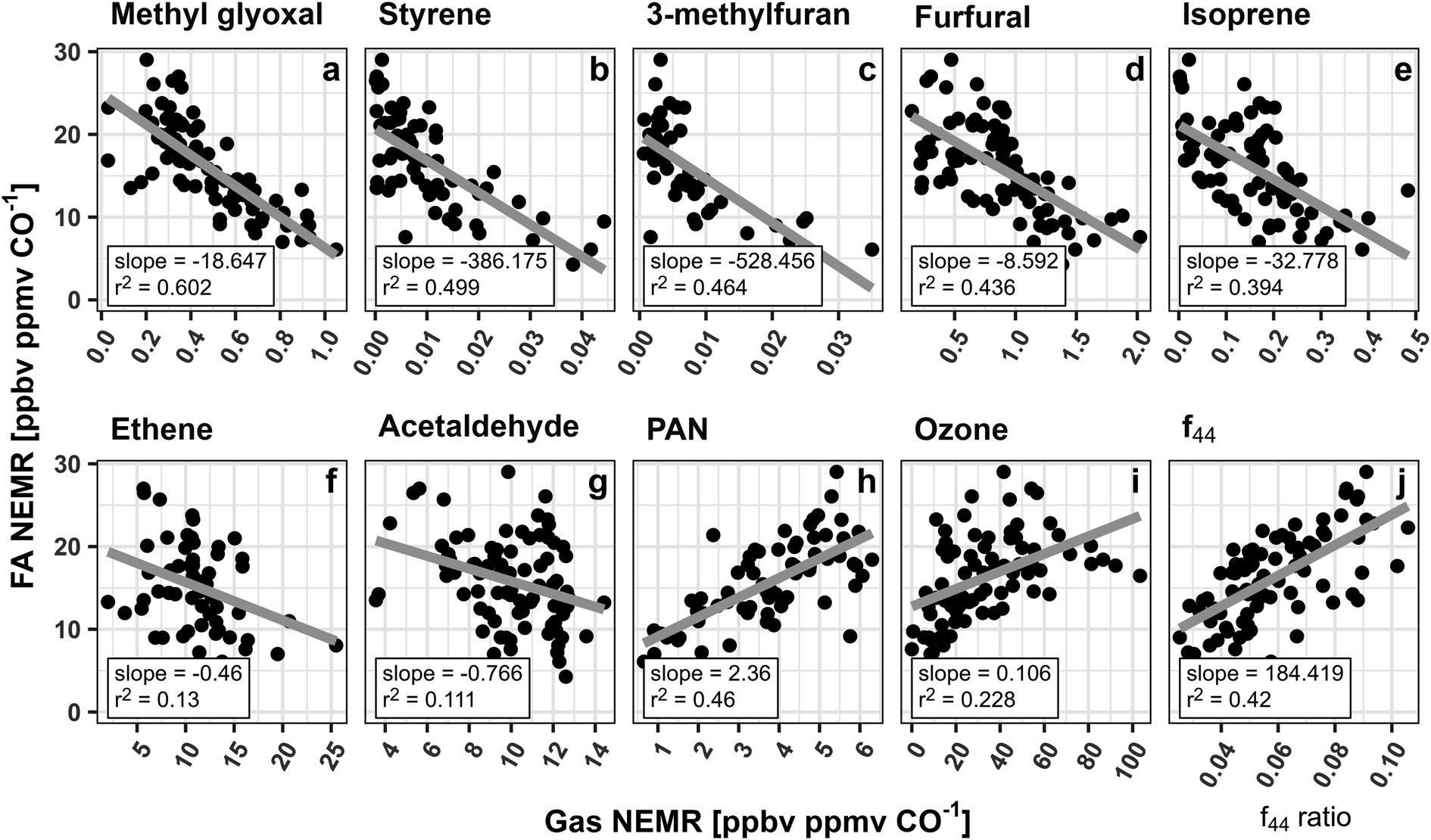 | ||
| Fig. 5 FA NEMRs compared to various gas phase species NEMRs and aerosol f44 ratios measured in 5 smoke plumes with more than 10 pseudo-Lagrangian plume transects. Slope and r2 for the least squares regression of each species are shown at the bottom of each panel, while the gray lines represent the best fit. Panels (a)–(c) show the three VOCs with the strongest correlation to FA. Panels (d) and (e) show two of the largest OH radical sinks (ranked by OH reactivity from individual VOC)24 that are highly correlated with FA in wildfire emissions. Panels (f), and (g) show known FA precursors, while quantities plotted in (h)–(j) are representative of the overall plume oxidation. Note, methyl glyoxal is measured with acrylic acid (C3H4O2). PAN = peroxyacetyl nitrate. f44 = ratio of m/z 44 to the total signal in the aerosol component spectrum with higher ratios indicating more aged organic aerosol and higher O:C. | ||
Fig. 5 similarly shows that FA is well correlated (r2 > 0.4) with isoprene, ethene, and acetaldehyde, consistent with the current understanding of these species being known FA precursors. Additionally, FA was found to have a strong correlation with peroxyacetyl nitrate (PAN; r2 = 0.46) and a modest correlation with ozone (r2 = 0.23), further indicating that FA production follows the overall plume gas phase oxidization. While the correlations of FA NEMRs with the VOCs in Fig. 5 do not directly indicate that they are FA precursors in smoke plumes, when coupled with FA being well corelated to PAN, ozone, and 94 different VOCs, they do demonstrate that FA is likely being produced through the oxidation of many different species, most of which are currently not well studied in the literature.
Heterogenous formation is also likely to be an important FA source in smoke via a multiphase pathway where methanediol (HOCH2OH) is off gassed from aerosols and is rapidly oxidized by OH to form HCOOH.32–34 The WE-CAN dataset does not have sufficient data to fully examine how this pathway may contribute to the FA production observed during the campaign. Instead, we explore whether the FA NEMRs show dependence on OA aging during WE-CAN in Fig. 5j by comparing FA NEMRs with the OA oxidation marker f44.52,53f44 is the fractional component of OA attributed to the CO2+ ion which is ascribed to fragments of acids or acid-derived species.96 Consequently, f44 is generally well correlated with the OA elemental O:C ratio,97 where both increase as the bulk aerosol becomes more oxidized. During WE-CAN f44 was found to increase with smoke plume age, while the dilution-adjusted OA mass generally remained unchanged over ∼8 hours of plume aging.51Fig. 5j shows that FA NEMRs are positively correlated with f44 (r2 = 0.42) as well as with the OA O:C ratio (r2 = 0.32, not shown). This suggests that FA production follows the bulk aerosol oxidation during WE-CAN. Additionally, the increasing OA oxidation with the constant downwind dilution-adjusted OA mass reported by Garofalo et al.51 requires a balance between evaporation and condensation of semivolatile species. This indicates that FA could be formed as part of this OA mass balance and more detailed laboratory and field studies are needed to better understand this potentially significant FA formation pathway in BB smoke.
Similar analysis with AA is shown in Fig. S8,† with AA NEMRs plotted against a similar grouping of gases as in Fig. 5. The three species with the strongest correlation against AA are shown in Fig. S8a–c:† C3H6O2 (hydroxyacetone + methyl acetate + ethyl formate; r2 = 0.62), C5H8O3 (5-hydroxymethyl tetrahydro 2-furanone; r2 = 0.48), and methyl propionate (r2 = 0.45). Like AA, NEMRs for these three species are not well correlated with the physical plume age. Fig. S8† also shows that AA has only modest correlation with the reactive VOCs furfural and isoprene as well as with acetaldehyde and ozone (r2 = 0.14–0.32) but is poorly correlated with ethene and PAN (r2 < 0.1). The fact that (a) AA is most strongly correlated with other VOCs whose NEMRs remain mostly unchanged with plume age and (b) has poor negative correlations with the plume oxidation indicators such as PAN, ozone, and f44, further supports the observation that little AA is produced and instead is mainly from primary emissions in the WE-CAN sample wildfire plumes.
6. GEOS-Chem representation of FA and AA during WE-CAN and FIREX-AQ
Global chemical transport models typically have difficulty simulating formic and acetic acid mixing ratios, particularly in the presence of BB smoke. Section 5 suggests that the GEOS-Chem chemistry underestimates a significant amount of secondary production of FA in fresh smoke. Here we investigate how the GEOS-Chem CTM, with the most recent updates for FA implemented by Chen et al.17 (Section 2.3), represents FA and AA in different environments sampled during WE-CAN, FIREX-AQ-W, and FIREX-AQ-SE. All WE-CAN and FIREX-AQ measurements have also been averaged to 5 minutes to match the model resolution. GEOS-Chem was sampled along the plane flight tracks at the time of each corresponding research flight.Fig. 6 shows that GEOS-Chem generally underestimates the vertical distribution of FA observed during WE-CAN (−92%; normalized mean bias to I− CIMS, NMB) and in the middle to lower troposphere (>450 hPa or below ∼7.2 km above sea level) during FIREX-AQ-W (−76% NMB) and FIREX-AQ-SE (−37%). This corresponds to the model underestimating the average measured FA by nearly a factor of 13 during the WE-CAN deployment, while also underestimating FA by a factor of 4 and 2 in the lower altitude FIREX-AQ-W and FIREX-AQ-SE samples. However, GEOS-Chem does significantly better simulating FA in the middle to upper troposphere (<450 hPa; −27% NMB in FIREX-AQ-W), consistent with findings by Chen et al.17 Interestingly, GEOS-Chem overestimates FA mixing ratios compared to I− CIMS measurements at higher altitudes (<450 hPa) in the southeastern U.S. (213%), though the measured FA is reaching the stated I− CIMS detection limit (∼30 ppt). Fig. S9† shows a similar underestimation for acetic acid mixing ratios with NMB ranging −92% to −99% in both high and low altitude WE-CAN and FIREX-AQ-W samples, and slightly better agreement (−80%) with lower altitude FIREX-AQ-SE periods. We note that this significant underestimate of FA by GEOS-Chem holds true regardless of the high uncertainty in FA measured during the WE-CAN deployment as the difference between the modeled and measured values is much greater than the instrument uncertainty. Similarly, the model underestimate of AA is much greater than what can be explained by the potential interference of glycolaldehyde measured at m/z 61.
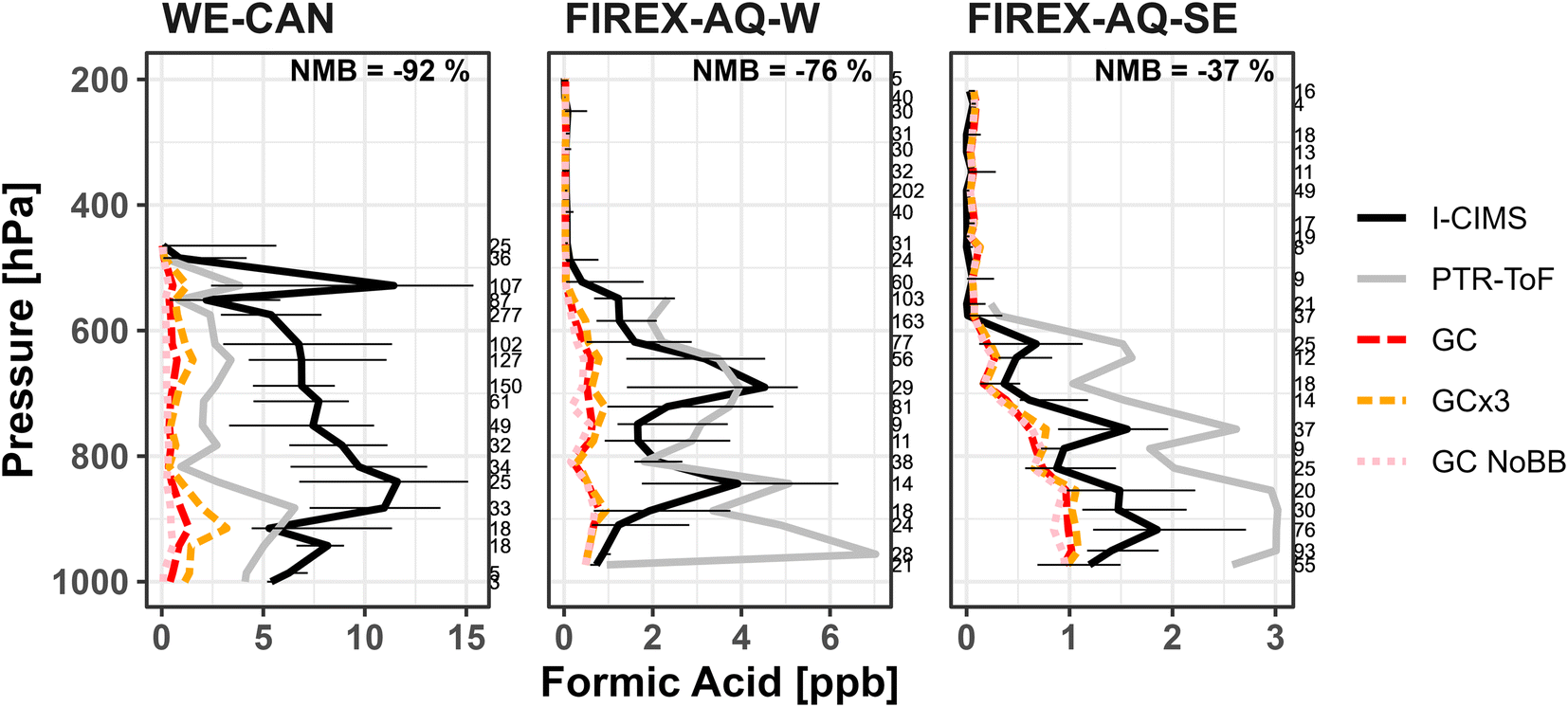 | ||
| Fig. 6 Vertical profiles of the median formic acid mixing ratios measured during the WE-CAN and FIREX-AQ field campaigns, binned at every 33 hPa. Black and gray lines correspond to the measurements made by I− CIMS and PTR-ToF, with error bars representing the 25th and 75th percentile of I− CIMS measurements at each pressure bin. Red dashed lines correspond to GEOS-Chem with GFAS BB emissions (GC), orange dashed lines represent GEOS-Chem with 3× GFAS BB emissions (GC × 3), and the pink dotted lines show GEOS-Chem with BB emissions turned off (GC NoBB). The number of samples in each pressure bin are shown on the right of the plots, while the normalized mean bias (NMB) to the I− CIMS measurement for lower altitude observations (>450 hPa) are shown at the top. | ||
There are a few possible explanations for why GEOS-Chem underestimates FA and AA during the two campaigns, including: incorrect or missing emissions, sampling bias, and/or missing secondary chemistry from BB (Section 5) and biogenic precursors. Recent model developments have improved the GEOS-Chem representation of the free troposphere,17 chemistry, and biogenic sources.7 Subsequently, we hypothesize that missing secondary production from BB and specific biogenic precursors in the western U.S. are likely key reasons for GEOS-Chem underestimating FA mixing ratios during WE-CAN and FIREX-AQ-W. The exact reason for the underestimation of AA mixing ratios is uncertain, though we speculate it may in part be due to the model sink for AA being too large39 and/or secondary production in BB smoke aged over greater processing times than discussed in Section 5 (i.e. >8 hours).
6.1. Model BB emissions and sampling bias
Recent work has shown that commonly used global emission inventories, including GFAS, GFED4, QFED, and FINNv1.5, underestimate BB emissions by a factor of three or more in the western U.S. when compared to aircraft and ground-based measurements.42,98 Jin et al.42 attributes this mostly to the significant underestimation of the dry biomass burned in the BB emission inventories. To explore if underestimated BB emissions can explain the low FA and AA model bias, GEOS-Chem was also initiated with 3× GFAS BB emissions as a sensitivity test, in which the BB VOC and CO emissions are tripled from the base run. Fig. S10† shows that GEOS-Chem with base GFAS emissions underestimate CO during WE-CAN, FIREX-AQ-W, and FIREX-AQ-SE. Model representation in the western U.S. is improved by the 3× GFAS model run, in good agreement with Jin et al.42 Similarly, though benzene and acetone are better represented by the base model in this work, the 3× GFAS emission simulation further improves their model agreement. Despite this, Fig. 6 and S9† show that increasing BB emissions by a factor of 3 only slightly increases the model FA and AA mixing ratios, decreasing the NMB by ∼5% in all cases. Given (1) that tripling BB emissions has minimal impact on the modeled FA or AA, (2) that the GFAS inventory is not missing the location/timing of the fires sampled during both campaigns,42 and (3) that FA and AA BB emissions were implemented per observed ERs,18 underestimation of primary BB emissions of either acid or their known precursors in GEOS-Chem alone cannot account for the low model bias. This reflects that the contribution of primary BB emissions to ambient FA during WE-CAN and FIREX-AQ in the western US is small. Additionally, the 3× GFAS run also increases emissions for all BB implemented species,24 thus pointing to missing secondary formation pathways from either implemented and/or unknown precursors in the model (Section 5). Given the lack of evidence for near-field AA production during WE-CAN, the model being largely insensitive to a 3-fold increase in AA emissions suggests that AA production in BB plumes aged greater than the 8 hours observed during WE-CAN may still be significant and/or the overall model sink is too large.The WE-CAN and FIREX-AQ aircraft campaigns were focused on sampling and tracking BB smoke whenever possible. As GEOS-Chem was run at 0.25° × 0.3125° (∼25 km) resolution, the low model bias may in part also reflect the dilution of narrow smoke plumes over the model grid. Though some error is inherent in the model comparisons due to this sampling bias, using GEOS-Chem run with the same WE-CAN and FIREX-AQ datasets, Jin et al.42 demonstrated that the model also had difficulty simulating smoke impacts at longer term ground measurement sites across the western U.S. This is indicative that the low model biases cannot be explained by the model resolution alone. Similarly, Jin et al.42 showed that fire detection products across emission inventories did well capturing the large fires sampled during WE-CAN and that GEOS-Chem is fairly insensitive to plume injection heights for the averaged WE-CAN profiles, likely due to efficient vertical mixing during the summer months.99,100 However, because of these issues when comparing fire plumes sampled by aircraft to global CTMs, the GEOS-Chem evaluation here further focuses on the campaign averages across two different years, in smoke impacted, no/low smoke, and clean free troposphere environments.
6.2. Representation in different environments
To investigate potential model deficiencies over broad regions, we further examine the model performance in different environments sampled during the campaigns as described in our previous work.24 Here, smoke-impacted sampling periods for both campaigns are defined as those with hydrogen cyanide (HCN) > 250 ppt and acetonitrile (CH3CN) > 200 ppt, while periods below this threshold are discussed as low/no-smoke. However, due to widespread regional smoke during the fire season, the low/no-smoke samples likely still represent some BB influence. In addition to this coarse filter, clean free troposphere samples were also defined for both campaigns based on HCN < 250 ppt, CH3CN < 150 ppt, and pressure < 624 hPa (∼4 km above sea level, representing the maximum boundary layer height as determined from vertical temperature profiles).
Fig. 7 shows the vertical profiles for the median observed and modeled FA in the three different environments. We find GEOS-Chem underestimates the median FA mixing ratio most significantly in smoke impacted samples, doing slightly better during low/no smoke periods in the western U.S. Alternatively, GEOS-Chem does well simulating FA mixing ratios in the free troposphere during all three periods, in good agreement with Chen et al.17 This is particularly evident in FIREX-AQ-W free troposphere samples, which agree nearly 1:1 with the model. Similarly, the model also does very well simulating median FA mixing ratios in the low/no smoke southeast U.S. samples (NMB −36%). As this profile reflects minimal smoke impact during the period, it suggests that the model is accurately simulating FA from biogenic sources in the southeast U.S., as reflected in recent model developments including production from stabilized Criegee intermediates and acetaldehyde tautomerization as implemented by Millet et al.7 and Chen et al.17
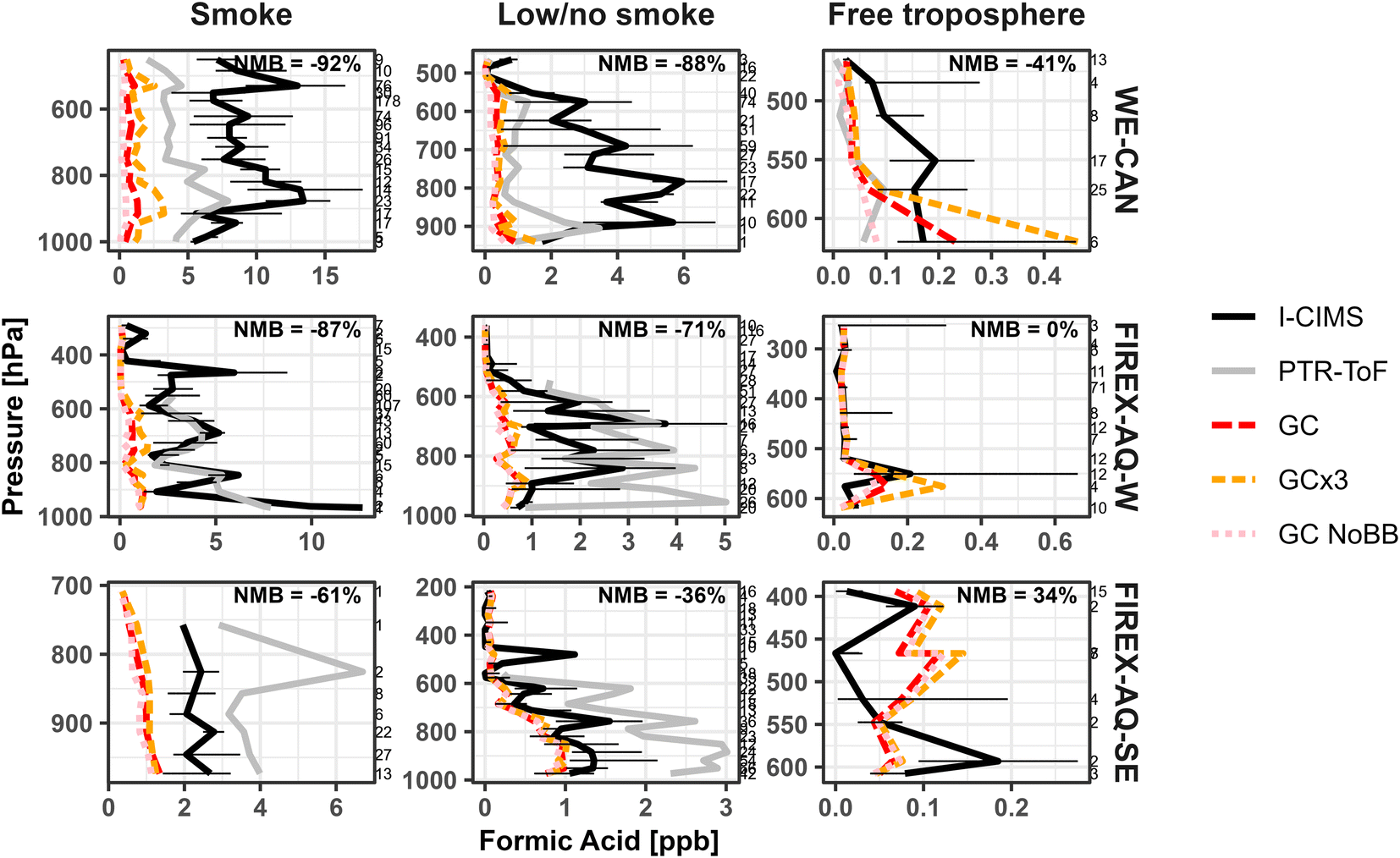 | ||
| Fig. 7 Vertical profiles of the median formic acid mixing ratios measured during the WE-CAN field campaign for smoke impacted, low/no smoke, and free troposphere sampling periods. Pressures are binned at every 33 hPa. Black and gray lines correspond to the measurements made by I− CIMS and PTR-ToF. Red dashed lines correspond to GEOS-Chem with GFAS BB emissions (GC), orange dashed lines are GEOS-Chem with 3× GFAS BB emissions (GC × 3), and the pink dotted lines are GEOS-Chem with BB emissions turned off (GC NoBB). Error bars are the 25th and 75th percentile of the I− CIMS measurement at each pressure bin. | ||
Although GEOS-Chem does better simulating FA during low/no smoke samples than in smoke in the western U.S., the improvement is only modest with NMB decreasing by <10%. This may in part reflect the widespread smoke impacts in the western U.S. during fire season, where a pool of longer-lived oxygenated species could persist in the region.24 However, it also suggests that the model may be missing a FA source or secondary chemistry from biogenic precursors unique to coniferous forests,11 which are likely different than those most responsible for FA in the southeastern U.S. For example, isoprene oxidation is thought to be one of the main contributors to FA formation above deciduous forests,7 while in coniferous forests emissions are typically dominated by monoterpenes and 2-methyl-3-buten-2-ol (MBO),101 whose potential contribution to FA formation is unclear.
To explore the regional sources of FA using the two campaign datasets, Fig. 8 shows how FA correlates with CO, methanol, acetone, and MVK + MACR (methyl vinyl ketone and methacrolein) in the three regions and environments shown in Fig. 7. The plot of FA vs. CO shows two distinct populations between the smoke and low/no smoke environments. As CO is mainly from BB in the WE-CAN and FIREX-AQ datasets, the correlation of FA with CO in smoke samples indicates FA coming from BB sources, while the spread likely represents FA enhancement relative to primary emissions. In low/no smoke samples, the FA:CO slope is steeper than in the smoke samples, suggesting a FA source that is independent of the combustion process thus pointing to photochemical origin.
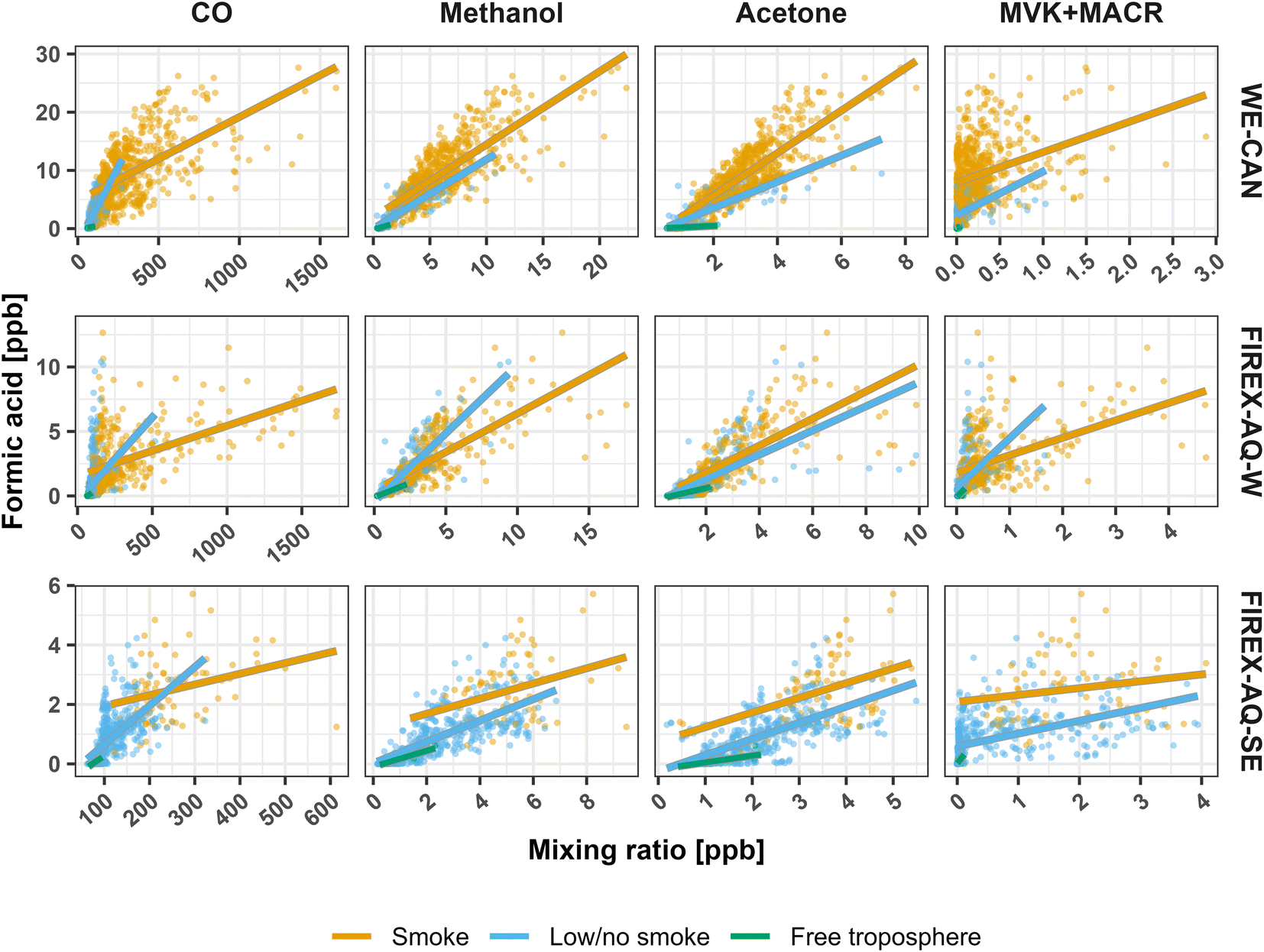 | ||
| Fig. 8 Correlations of FA with CO, methanol, acetone, and MVK + MACR (methyl vinyl ketone and methacrolein) in WE-CAN and FIREX-AQ observations. Orange points represent smoke-impacted data, blue points indicate low/no smoke impact, and green points show clean free troposphere measurements (see main texts for definitions). The data have been averaged to 5 minutes. Lines show the least squares regression corresponding to each set of colored points. Note that acetone is also measured with its isomer propanal. | ||
Interestingly, Fig. 8 also shows that FA is well correlated with both methanol (r2 = 0.55–0.75) and acetone (0.42–0.72), with generally similar slopes in both smoke and low/no smoke samples (methanol = 0.5–0.9, acetone = 0.2–0.4) during WE-CAN and FIREX-AQ-W periods. In contrast to FA, neither methanol nor acetone measured during WE-CAN show net production in the 5 smoke plumes and ∼8 hours of aging discussed in Section 5 (Fig. S11†). This suggests that their correlations in Fig. 8 are not due to near-field production in BB, though enhancement in much more aged plumes (>2 days) has been observed.102,103 As methanol and acetone are known to be major primary emissions and secondary products from biogenic sources,12,31,102,104–106 we hypothesize that their strong correlation with FA during WE-CAN and FIREX-AQ indicates that a portion of the observed FA may be of biogenic origin, though the long atmospheric lifetimes of all three species (>2 days) likely also play a role in why they are well correlated with each other.
As isoprene is known to be the major FA precursor in deciduous forests, Fig. 8 also shows FA vs. MVK + MACR, an important isoprene oxidation product. During WE-CAN and FIREX-AQ, FA has a weak positive correlation with MVK + MACR in most environments (r2 = 0.11–0.24), further indicating that some of the observed FA is indeed related to biogenic species. Some of the agreement between the two is also likely due to both being primary BB emissions,18 while the large spread in the correlations also points to MVK + MACR being lost as the plumes age (Fig. S11†). Consequently, the model underestimate of FA in the western U.S. is likely due to both missing secondary chemistry from BB and biogenic sources, pointing to a need for more detailed studies of FA production from both BB and coniferous forest emissions.
A similar trend can be seen for acetic acid in Fig. S12,† where GEOS-Chem underestimates AA in both smoke-impacted and low/no smoke environments during all three sampling periods, with NMB improving by <10% between smoke low/no smoke conditions. Additionally, AA is better captured by the model in the clean free troposphere during WE-CAN (NMB −44%), though the disparity is larger for both portions of FIREX-AQ (NMB −92%). Fig. S13† shows that AA is well correlated with CO across all WE-CAN and FIREX-AQ-W samples, with a slope in the range of reported ERs (WE-CAN slope = 15.8 ppb ppmCO−1, r2 = 0.84; FIREX-AQ-W slope = 11.0 ppb ppmCO−1, r2 = 0.92). This further indicates that AA and CO in the western U.S. come from the same source, likely BB. Given the lack of evidence for production of AA in the fresh BB plumes sampled during WE-CAN (Section 5), near-field production is unlikely to explain the low model bias, though production in plumes aged longer than those sampled during WE-CAN is still possible. Additionally, the underrepresentation cannot be accounted for by BB emission alone for two reasons: (1) the AA (and FA) emission ratio in the model was implemented using the WE-CAN observations per Permar et al.18 and (2) the GEOS-Chem + 3 × GFAS, which should account for the underestimated BB primary emissions per Jin et al.42 only slightly increases the modeled AA. Consequently, the exact reason behind the low model bias for AA is unknown, though it may be due to too large of a model sink and/or missing secondary production from long lived biogenic and BB precursors.
7. Conclusions
Using detailed formic acid and acetic acid measurements made during the WE-CAN and FIREX-AQ aircraft campaigns, we assess their emissions, chemistry, and model representation in the western and southeastern U.S. FA measured by two commonly used mass spectrometers, PTR-ToF and I− CIMS, was found to have high measurement uncertainty during the WE-CAN deployment (up to 100%) due to its humidity and temperature dependent sensitivities, inlet artifacts, and instrument baseline issues. However, FA measured by two different PTR-ToF and I− CIMS instruments during the FIREX-AQ campaign were found to agree within their measurement uncertainty. Accuracy for the FA measurement could be greatly improved by reducing inlet losses via shorter sampling lines, increased flow rate, and/or reduced sampling line diameter, thus reducing sample residence time in the instrument. In addition, regulating reaction chamber temperatures and humidity while performing more frequent humidity dependent calibrations and instrument zeros are key to improving the FA measurement quality in both instrument types. Despite the high uncertainty in FA measured during WE-CAN, model underestimates of FA mixing ratios were found to be much greater than the measurement uncertainty.During WE-CAN FA ERs and EFs were found to be 9.5 ± 4.2 (1σ) ppb ppmCO−1 and 1.5 ± 0.60 g kg−1 respectively, which are 3.5 times higher than literature values. In FIREX-AQ, AA EFs and ERs agree better with the literature; however, they are still often higher than the 75th percentile of literature values. As FA was found to have little to no dependence on MCE or fuel type. The exact reason for this discrepancy is currently unknown, though may reflect differences in emissions between the larger wildfires in this work and those from laboratory BB studies and the smaller fires typically reported in the literature. It also reflects some extent of early plume production, as the WE-CAN flights sampled 27–130 minutes downwind from the source. However, extrapolating FA NEMRs measured downwind from these fires to t0 does little to close the gap between WE-CAN and literature ERs and EFs.
Analysis of 5 smoke plumes sampled in a pseudo-Lagrangian fashion finds that FA is rapidly produced at 2.7 ppb ppmCO−1 h−1 during the first 8 hours of plume aging, in good agreement with previous studies. However, F0AM run with explicit MCM or simplified GEOS-Chem chemistry was unable to capture the observed production due to missing secondary sources. Observed FA production was found to have statistically significant correlations (p-value <0.05) with 94 VOCs measured during WE-CAN. The oxidation of these 94 species collectively accounts for 127 ppbC ppmCO−1 that is reacted away over the 8 hours of plume aging. This indicates that those species could lose 6× more carbon than is needed to account for the observed FA production, though the exact chemical pathways are often unknown.
AA ERs and EFs were found to fall within the 25th and 75th percentiles of the literature-reported values, exhibiting a modest negative dependence on MCE and some fuel types. In contrast to some previous studies, AA was not found to have any statistically significant production during the first 8 hours of plume aging during WE-CAN, with downwind NEMRs generally in the range of observed ERs. Consequently, most of the observed AA in the nearfield is likely from primary emissions, though photochemical production may still be important for certain fires/fuels and in more aged smoke.
GEOS-Chem simulations with updated FA and AA chemistry and emissions were performed for the WE-CAN and FIREX-AQ campaigns and compared to field observations. For both campaigns, FA and AA were found to be biased low in the model by ∼90%. The model does slightly better simulating FA mixing ratios in no/low smoke impacted western U.S. samples, and significantly better in no/low smoke periods over southeast U.S. forests. It is likely that much of the low model bias for FA is due to missing secondary production from both BB and coniferous forest biogenic sources. The factors leading the underestimate of AA are unknown, but may reflect too large of a model sink and secondary production in smoke aged longer than observed during WE-CAN.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was supported by the U.S. National Science Foundation (AGS # 2144896, EPSCoR # 2242802). The 2018 WE-CAN field campaign was supported by NSF through grants AGS # 1650275 (U of Montana), # 1650786 (Colorado State U), # 1650288 (U of Colorado at Boulder), # 1650493 (U of Wyoming), # 1652688 (U of Washington), # 1748266 (U of Montana), and the National Oceanic and Atmospheric Administration (award # NA17OAR4310010, Colorado State U). Dr Dylan B. Millet acknowledges additional support from NOAA (#NA22OAR4310200). This material was also based upon work supported by the NCAR, which is a major facility sponsored by the NSF under cooperative agreement no. 1852977. The WE-CAN data were collected using NSF's Lower Atmosphere Observing Facilities, which are managed and operated by NCAR's Earth Observing Laboratory. The authors thank Dr Delphine K. Farmer for use of the AMS measurements made during WE-CAN and acknowledge high-performance computing resources and support from Cheyenne (DOI: 10.5065/D6RX99HX) provided by the NCAR Computational and Information Systems Laboratory, sponsored by the NSF, and the U of Montana's Griz Shared Computing Cluster (GSCC).References
- D. J. Jacob, Chemistry of OH in remote clouds and its role in the production of formic acid and peroxymonosulfate, J. Geophys. Res.: Atmos., 1986, 91, 9807–9826 CrossRef CAS.
- S. Metzger, N. Mihalopoulos and J. Lelieveld, Importance of mineral cations and organics in gas–aerosol partitioning of reactive nitrogen compounds: case study based on MINOS results, Atmos. Chem. Phys., 2006, 6, 2549–2567 CrossRef CAS.
- M. O. Andreae, R. W. Talbot, T. W. Andreae and R. C. Harriss, Formic and acetic acid over the central Amazon region, Brazil: 1. Dry season, J. Geophys. Res.: Atmos., 1988, 93, 1616–1624 CrossRef CAS.
- J. N. Galloway, G. E. Likens, W. C. Keene and J. M. Miller, The composition of precipitation in remote areas of the world, J. Geophys. Res.: Oceans, 1982, 87, 8771–8786 CrossRef CAS.
- W. C. Keene, J. N. Galloway and J. D. Holden Jr., Measurement of weak organic acidity in precipitation from remote areas of the world, J. Geophys. Res.: Oceans, 1983, 88, 5122–5130 CrossRef CAS.
- T. Stavrakou, J.-F. Müller, J. Peeters, A. Razavi, L. Clarisse, C. Clerbaux, P.-F. Coheur, D. Hurtmans, M. De Mazière, C. Vigouroux, N. M. Deutscher, D. W. T. Griffith, N. Jones and C. Paton-Walsh, Satellite evidence for a large source of formic acid from boreal and tropical forests, Nat. Geosci., 2012, 5, 26–30 CrossRef CAS.
- D. B. Millet, M. Baasandorj, D. K. Farmer, J. A. Thornton, K. Baumann, P. Brophy, S. Chaliyakunnel, J. A. de Gouw, M. Graus, L. Hu, A. Koss, B. H. Lee, F. D. Lopez-Hilfiker, J. A. Neuman, F. Paulot, J. Peischl, I. B. Pollack, T. B. Ryerson, C. Warneke, B. J. Williams and J. Xu, A large and ubiquitous source of atmospheric formic acid, Atmos. Chem. Phys., 2015, 15, 6283–6304 CrossRef CAS.
- F. Paulot, D. Wunch, J. D. Crounse, G. C. Toon, D. B. Millet, P. F. DeCarlo, C. Vigouroux, N. M. Deutscher, G. González Abad, J. Notholt, T. Warneke, J. W. Hannigan, C. Warneke, J. A. de Gouw, E. J. Dunlea, M. De Mazière, D. W. T. Griffith, P. Bernath, J. L. Jimenez and P. O. Wennberg, Importance of secondary sources in the atmospheric budgets of formic and acetic acids, Atmos. Chem. Phys., 2011, 11, 1989–2013 CrossRef CAS PubMed.
- H. D. Alwe, D. B. Millet, X. Chen, J. D. Raff, Z. C. Payne and K. Fledderman, Oxidation of Volatile Organic Compounds as the Major Source of Formic Acid in a Mixed Forest Canopy, Geophys. Res. Lett., 2019, 46, 2940–2948 CrossRef CAS PubMed.
- T. B. Nguyen, J. D. Crounse, A. P. Teng, J. M. S. Clair, F. Paulot, G. M. Wolfe and P. O. Wennberg, Rapid deposition of oxidized biogenic compounds to a temperate forest, Proc. Natl. Acad. Sci., 2015, 112, E392–E401 CAS.
- S. Schobesberger, F. D. Lopez-Hilfiker, D. Taipale, D. B. Millet, E. L. D'Ambro, P. Rantala, I. Mammarella, P. Zhou, G. M. Wolfe, B. H. Lee, M. Boy and J. A. Thornton, High upward fluxes of formic acid from a boreal forest canopy, Geophys. Res. Lett., 2016, 43, 9342–9351 CrossRef.
- V. Selimovic, D. Ketcherside, S. Chaliyakunnel, C. Wielgasz, W. Permar, H. Angot, D. B. Millet, A. Fried, D. Helmig and L. Hu, Atmospheric biogenic volatile organic compounds in the Alaskan Arctic tundra: constraints from measurements at Toolik Field Station, Atmos. Chem. Phys., 2022, 22, 14037–14058 CrossRef CAS PubMed.
- M. Le Breton, M. R. McGillen, J. B. A. Muller, A. Bacak, D. E. Shallcross, P. Xiao, L. G. Huey, D. Tanner, H. Coe and C. J. Percival, Airborne observations of formic acid using a chemical ionization mass spectrometer, Atmos. Meas. Tech., 2012, 5, 3029–3039 CrossRef CAS.
- B. Yuan, P. R. Veres, C. Warneke, J. M. Roberts, J. B. Gilman, A. Koss, P. M. Edwards, M. Graus, W. C. Kuster, S.-M. Li, R. J. Wild, S. S. Brown, W. P. Dubé, B. M. Lerner, E. J. Williams, J. E. Johnson, P. K. Quinn, T. S. Bates, B. Lefer, P. L. Hayes, J. L. Jimenez, R. J. Weber, R. Zamora, B. Ervens, D. B. Millet, B. Rappenglück and J. A. de Gouw, Investigation of secondary formation of formic acid: urban environment vs. oil and gas producing region, Atmos. Chem. Phys., 2015, 15, 1975–1993 CrossRef CAS.
- K. E. Cady-Pereira, S. Chaliyakunnel, M. W. Shephard, D. B. Millet, M. Luo and K. C. Wells, HCOOH measurements from space: TES retrieval algorithm and observed global distribution, Atmos. Meas. Tech., 2014, 7, 2297–2311 CrossRef CAS PubMed.
- S. Chaliyakunnel, D. B. Millet, K. C. Wells, K. E. Cady-Pereira and M. W. Shephard, A Large Underestimate of Formic Acid from Tropical Fires: Constraints from Space-Borne Measurements, Environ. Sci. Technol., 2016, 50, 5631–5640 CrossRef CAS PubMed.
- X. Chen, D. B. Millet, J. A. Neuman, P. R. Veres, E. A. Ray, R. Commane, B. C. Daube, K. McKain, J. P. Schwarz, J. M. Katich, K. D. Froyd, G. P. Schill, M. J. Kim, J. D. Crounse, H. M. Allen, E. C. Apel, R. S. Hornbrook, D. R. Blake, B. A. Nault, P. Campuzano-Jost, J. L. Jimenez and J. E. Dibb, HCOOH in the Remote Atmosphere: Constraints from Atmospheric Tomography (ATom) Airborne Observations, ACS Earth Space Chem., 2021, 5, 1436–1454 CrossRef CAS PubMed.
- W. Permar, Q. Wang, V. Selimovic, C. Wielgasz, R. J. Yokelson, R. S. Hornbrook, A. J. Hills, E. C. Apel, I.-T. Ku, Y. Zhou, B. C. Sive, A. P. Sullivan, J. L. Collett Jr., T. L. Campos, B. B. Palm, Q. Peng, J. A. Thornton, L. A. Garofalo, D. K. Farmer, S. M. Kreidenweis, E. J. T. Levin, P. J. DeMott, F. Flocke, E. V. Fischer and L. Hu, Emissions of Trace Organic Gases From Western U.S. Wildfires Based on WE-CAN Aircraft Measurements, J. Geophys. Res.: Atmos., 2021, 126, e2020JD033838 CrossRef CAS.
- S. K. Akagi, J. S. Craven, J. W. Taylor, G. R. McMeeking, R. J. Yokelson, I. R. Burling, S. P. Urbanski, C. E. Wold, J. H. Seinfeld, H. Coe, M. J. Alvarado and D. R. Weise, Evolution of trace gases and particles emitted by a chaparral fire in California, Atmos. Chem. Phys., 2012, 12, 1397–1421 CrossRef CAS.
- J. G. Goode, R. J. Yokelson, D. E. Ward, R. A. Susott, R. E. Babbitt, M. A. Davies and W. M. Hao, Measurements of excess O3, CO2, CO, CH4, C2H4, C2H2, HCN, NO, NH3, HCOOH, CH3COOH, HCHO, and CH3OH in 1997 Alaskan biomass burning plumes by airborne Fourier transform infrared spectroscopy (AFTIR), J. Geophys. Res.: Atmos., 2000, 105, 22147–22166 CrossRef CAS.
- M. Grutter, N. Glatthor, G. P. Stiller, H. Fischer, U. Grabowski, M. Höpfner, S. Kellmann, A. Linden and T. von Clarmann, Global distribution and variability of formic acid as observed by MIPAS-ENVISAT, J. Geophys. Res.: Atmos., 2010, 115, D10303 CrossRef.
- R. J. Yokelson, J. D. Crounse, P. F. DeCarlo, T. Karl, S. Urbanski, E. Atlas, T. Campos, Y. Shinozuka, V. Kapustin, A. D. Clarke, A. Weinheimer, D. J. Knapp, D. D. Montzka, J. Holloway, P. Weibring, F. Flocke, W. Zheng, D. Toohey, P. O. Wennberg, C. Wiedinmyer, L. Mauldin, A. Fried, D. Richter, J. Walega, J. L. Jimenez, K. Adachi, P. R. Buseck, S. R. Hall and R. Shetter, Emissions from biomass burning in the Yucatan, Atmos. Chem. Phys., 2009, 9, 5785–5812 CrossRef CAS.
- R. J. Yokelson, J. G. Goode, D. E. Ward, R. A. Susott, R. E. Babbitt, D. D. Wade, I. Bertschi, D. W. T. Griffith and W. M. Hao, Emissions of formaldehyde, acetic acid, methanol, and other trace gases from biomass fires in North Carolina measured by airborne Fourier transform infrared spectroscopy, J. Geophys. Res.: Atmos., 1999, 104, 30109–30125 CrossRef CAS.
- W. Permar, L. Jin, Q. Peng, K. O'Dell, E. Lill, V. Selimovic, R. J. Yokelson, R. S. Hornbrook, A. J. Hills, E. C. Apel, I.-T. Ku, Y. Zhou, B. C. Sive, A. P. Sullivan, J. L. Collett, B. B. Palm, J. A. Thornton, F. Flocke, E. V. Fischer and L. Hu, Atmospheric OH reactivity in the western United States determined from comprehensive gas-phase measurements during WE-CAN, Environ. Sci.: Atmos., 2023, 3, 97–114 CAS.
- A. R. Koss, K. Sekimoto, J. B. Gilman, V. Selimovic, M. M. Coggon, K. J. Zarzana, B. Yuan, B. M. Lerner, S. S. Brown, J. L. Jimenez, J. Krechmer, J. M. Roberts, C. Warneke, R. J. Yokelson and J. de Gouw, Non-methane organic gas emissions from biomass burning: identification, quantification, and emission factors from PTR-ToF during the FIREX 2016 laboratory experiment, Atmos. Chem. Phys., 2018, 18, 3299–3319 CrossRef CAS.
- R. J. Yokelson, I. T. Bertschi, T. J. Christian, P. V. Hobbs, D. E. Ward and W. M. Hao, Trace gas measurements in nascent, aged, and cloud-processed smoke from African savanna fires by airborne Fourier transform infrared spectroscopy (AFTIR), J. Geophys. Res.: Atmos., 2003, 108(D13), 8478 CrossRef.
- A. Chebbi and P. Carlier, Carboxylic acids in the troposphere, occurrence, sources, and sinks: a review, Atmos. Environ., 1996, 30, 4233–4249 CrossRef CAS.
- J.-F. Müller, T. Stavrakou and J. Peeters, Chemistry and deposition in the Model of Atmospheric composition at Global and Regional scales using Inversion Techniques for Trace gas Emissions (MAGRITTE v1.1) – Part 1: Chemical mechanism, Geosci. Model Dev., 2019, 12, 2307–2356 CrossRef.
- U. Baltensperger, M. Kalberer, J. Dommen, D. Paulsen, M. R. Alfarra, H. Coe, R. Fisseha, A. Gascho, M. Gysel, S. Nyeki, M. Sax, M. Steinbacher, A. S. H. Prevot, S. Sjögren, E. Weingartner and R. Zenobi, Secondary organic aerosols from anthropogenic and biogenic precursors, Faraday Discuss., 2005, 130, 265–278 RSC.
- K. P. Wyche, P. S. Monks, A. M. Ellis, R. L. Cordell, A. E. Parker, C. Whyte, A. Metzger, J. Dommen, J. Duplissy, A. S. H. Prevot, U. Baltensperger, A. R. Rickard and F. Wulfert, Gas phase precursors to anthropogenic secondary organic aerosol: detailed observations of 1,3,5-trimethylbenzene photooxidation, Atmos. Chem. Phys., 2009, 9, 635–665 CrossRef CAS.
- S. Wang, E. C. Apel, R. H. Schwantes, K. H. Bates, D. J. Jacob, E. V. Fischer, R. S. Hornbrook, A. J. Hills, L. K. Emmons, L. L. Pan, S. Honomichl, S. Tilmes, J.-F. Lamarque, M. Yang, C. A. Marandino, E. S. Saltzman, W. de Bruyn, S. Kameyama, H. Tanimoto, Y. Omori, S. R. Hall, K. Ullmann, T. B. Ryerson, C. R. Thompson, J. Peischl, B. C. Daube, R. Commane, K. McKain, C. Sweeney, A. B. Thames, D. O. Miller, W. H. Brune, G. S. Diskin, J. P. DiGangi and S. C. Wofsy, Global Atmospheric Budget of Acetone: Air–Sea Exchange and the Contribution to Hydroxyl Radicals, J. Geophys. Res.: Atmos., 2020, 125, e2020JD032553 CrossRef CAS.
- B. Franco, T. Blumenstock, C. Cho, L. Clarisse, C. Clerbaux, P.-F. Coheur, M. De Mazière, I. De Smedt, H.-P. Dorn, T. Emmerichs, H. Fuchs, G. Gkatzelis, D. W. T. Griffith, S. Gromov, J. W. Hannigan, F. Hase, T. Hohaus, N. Jones, A. Kerkweg, A. Kiendler-Scharr, E. Lutsch, E. Mahieu, A. Novelli, I. Ortega, C. Paton-Walsh, M. Pommier, A. Pozzer, D. Reimer, S. Rosanka, R. Sander, M. Schneider, K. Strong, R. Tillmann, M. Van Roozendael, L. Vereecken, C. Vigouroux, A. Wahner and D. Taraborrelli, Ubiquitous atmospheric production of organic acids mediated by cloud droplets, Nature, 2021, 593, 233–237 CrossRef CAS PubMed.
- J. D. Cope, K. A. Abellar, K. H. Bates, X. Fu and T. B. Nguyen, Aqueous Photochemistry of 2-Methyltetrol and Erythritol as Sources of Formic Acid and Acetic Acid in the Atmosphere, ACS Earth Space Chem., 2021, 5, 1265–1277 CrossRef CAS.
- Y. Jiang, M. Xia, Z. Wang, P. Zheng, Y. Chen and T. Wang, Photochemical aging of aerosols contributes significantly to the production of atmospheric formic acid, EGUsphere [preprint], 2023, DOI:10.5194/egusphere-2023-1140.
- M. Müller, B. E. Anderson, A. J. Beyersdorf, J. H. Crawford, G. S. Diskin, P. Eichler, A. Fried, F. N. Keutsch, T. Mikoviny, K. L. Thornhill, J. G. Walega, A. J. Weinheimer, M. Yang, R. J. Yokelson and A. Wisthaler, In situ measurements and modeling of reactive trace gases in a small biomass burning plume, Atmos. Chem. Phys., 2016, 16, 3813–3824 CrossRef.
- S. K. Akagi, R. J. Yokelson, I. R. Burling, S. Meinardi, I. Simpson, D. R. Blake, G. R. McMeeking, A. Sullivan, T. Lee, S. Kreidenweis, S. Urbanski, J. Reardon, D. W. T. Griffith, T. J. Johnson and D. R. Weise, Measurements of reactive trace gases and variable O3 formation rates in some South Carolina biomass burning plumes, Atmos. Chem. Phys., 2013, 13, 1141–1165 CrossRef.
- J. Trentmann, R. J. Yokelson, P. V. Hobbs, T. Winterrath, T. J. Christian, M. O. Andreae and S. A. Mason, An analysis of the chemical processes in the smoke plume from a savanna fire, J. Geophys. Res.: Atmos., 2005, 110, D12301 CrossRef.
- S. R. Fulgham, P. Brophy, M. Link, J. Ortega, I. Pollack and D. K. Farmer, Seasonal Flux Measurements over a Colorado Pine Forest Demonstrate a Persistent Source of Organic Acids, ACS Earth Space Chem., 2019, 3, 2017–2032 CrossRef CAS.
- X. Lee, D. Huang, Q. Liu, X. Liu, H. Zhou, Q. Wang and Y. Ma, Underrated primary biogenic origin and lifetime of atmospheric formic and acetic acid, Sci. Rep., 2021, 11, 7176 CrossRef CAS PubMed.
- M. F. Shaw, B. Sztáray, L. K. Whalley, D. E. Heard, D. B. Millet, M. J. T. Jordan, D. L. Osborn and S. H. Kable, Photo-tautomerization of acetaldehyde as a photochemical source of formic acid in the troposphere, Nat. Commun., 2018, 9, 2584 CrossRef PubMed.
- C. Warneke, J. P. Schwarz, J. Dibb, O. Kalashnikova, G. Frost, J. Al-Saad, S. S. Brown, W. A. Brewer, A. Soja, F. C. Seidel, R. A. Washenfelder, E. B. Wiggins, R. H. Moore, B. E. Anderson, C. Jordan, T. I. Yacovitch, S. C. Herndon, S. Liu, T. Kuwayama, D. Jaffe, N. Johnston, V. Selimovic, R. Yokelson, D. M. Giles, B. N. Holben, P. Goloub, I. Popovici, M. Trainer, A. Kumar, R. B. Pierce, D. Fahey, J. Roberts, E. M. Gargulinski, D. A. Peterson, X. Ye, L. H. Thapa, P. E. Saide, C. H. Fite, C. D. Holmes, S. Wang, M. M. Coggon, Z. C. J. Decker, C. E. Stockwell, L. Xu, G. Gkatzelis, K. Aikin, B. Lefer, J. Kaspari, D. Griffin, L. Zeng, R. Weber, M. Hastings, J. Chai, G. M. Wolfe, T. F. Hanisco, J. Liao, P. Campuzano Jost, H. Guo, J. L. Jimenez, J. Crawford and T. F.-A. S. Team, Fire Influence on Regional to Global Environments and Air Quality (FIREX-AQ), J. Geophys. Res.: Atmos., 2023, 128, e2022JD037758 CrossRef CAS.
- L. Jin, W. Permar, V. Selimovic, D. Ketcherside, R. J. Yokelson, R. S. Hornbrook, E. C. Apel, I.-T. Ku, J. L. Collett Jr., A. P. Sullivan, D. A. Jaffe, J. R. Pierce, A. Fried, M. M. Coggon, G. I. Gkatzelis, C. Warneke, E. V. Fischer and L. Hu, Constraining emissions of volatile organic compounds from western US wildfires with WE-CAN and FIREX-AQ airborne observations, Atmos. Chem. Phys., 2023, 23, 5969–5991 CrossRef CAS.
- NIFC, https://www.nifc.gov/fire-information/statistics/wildfires, (accessed 7 February 2023).
- B. H. Lee, F. D. Lopez-Hilfiker, C. Mohr, T. Kurtén, D. R. Worsnop and J. A. Thornton, An Iodide-Adduct High-Resolution Time-of-Flight Chemical-Ionization Mass Spectrometer: Application to Atmospheric Inorganic and Organic Compounds, Environ. Sci. Technol., 2014, 48, 6309–6317 CrossRef CAS PubMed.
- B. H. Lee, F. D. Lopez-Hilfiker, P. R. Veres, E. E. McDuffie, D. L. Fibiger, T. L. Sparks, C. J. Ebben, J. R. Green, J. C. Schroder, P. Campuzano-Jost, S. Iyer, E. L. D'Ambro, S. Schobesberger, S. S. Brown, P. J. Wooldridge, R. C. Cohen, M. N. Fiddler, S. Bililign, J. L. Jimenez, T. Kurtén, A. J. Weinheimer, L. Jaegle and J. A. Thornton, Flight Deployment of a High-Resolution Time-of-Flight Chemical Ionization Mass Spectrometer: Observations of Reactive Halogen and Nitrogen Oxide Species, J. Geophys. Res.: Atmos., 2018, 123, 7670–7686 CrossRef CAS.
- B. B. Palm, X. Liu, J. L. Jimenez and J. A. Thornton, Performance of a new coaxial ion–molecule reaction region for low-pressure chemical ionization mass spectrometry with reduced instrument wall interactions, Atmos. Meas. Tech., 2019, 12, 5829–5844 CrossRef CAS.
- Q. Peng, B. B. Palm, K. E. Melander, B. H. Lee, S. R. Hall, K. Ullmann, T. Campos, A. J. Weinheimer, E. C. Apel, R. S. Hornbrook, A. J. Hills, D. D. Montzka, F. Flocke, L. Hu, W. Permar, C. Wielgasz, J. Lindaas, I. B. Pollack, E. V. Fischer, T. H. Bertram and J. A. Thornton, HONO Emissions from Western U.S. Wildfires Provide Dominant Radical Source in Fresh Wildfire Smoke, Environ. Sci. Technol., 2020, 54, 5954–5963 CrossRef CAS PubMed.
- I. Bourgeois, J. Peischl, J. A. Neuman, S. S. Brown, H. M. Allen, P. Campuzano-Jost, M. M. Coggon, J. P. DiGangi, G. S. Diskin, J. B. Gilman, G. I. Gkatzelis, H. Guo, H. A. Halliday, T. F. Hanisco, C. D. Holmes, L. G. Huey, J. L. Jimenez, A. D. Lamplugh, Y. R. Lee, J. Lindaas, R. H. Moore, B. A. Nault, J. B. Nowak, D. Pagonis, P. S. Rickly, M. A. Robinson, A. W. Rollins, V. Selimovic, J. M. St. Clair, D. Tanner, K. T. Vasquez, P. R. Veres, C. Warneke, P. O. Wennberg, R. A. Washenfelder, E. B. Wiggins, C. C. Womack, L. Xu, K. J. Zarzana and T. B. Ryerson, Comparison of airborne measurements of NO, NO2, HONO, NOy, and CO during FIREX-AQ, Atmos. Meas. Tech., 2022, 15, 4901–4930 CrossRef CAS.
- M. A. Robinson, J. A. Neuman, L. G. Huey, J. M. Roberts, S. S. Brown and P. R. Veres, Temperature-dependent sensitivity of iodide chemical ionization mass spectrometers, Atmos. Meas. Tech., 2022, 15, 4295–4305 CrossRef CAS.
- G. I. Gkatzelis, M. M. Coggon, C. E. Stockwell, R. S. Hornbrook, H. Allen, E. C. Apel, K. Ball, M. M. Bela, D. R. Blake, I. Bourgeois, S. S. Brown, P. Campuzano-Jost, J. M. St. Clair, J. H. Crawford, J. D. Crounse, D. A. Day, J. DiGangi, G. Diskin, A. Fried, J. Gilman, H. Guo, J. W. Hair, H. A. Halliday, T. F. Hanisco, R. Hannun, A. Hills, G. Huey, J. L. Jimenez, J. M. Katich, A. Lamplugh, Y. R. Lee, J. Liao, J. Lindaas, S. A. McKeen, T. Mikoviny, B. A. Nault, J. A. Neuman, J. B. Nowak, D. Pagonis, J. Peischl, A. E. Perring, F. Piel, P. S. Rickly, M. A. Robinson, A. W. Rollins, T. B. Ryerson, M. K. Schueneman, R. H. Schwantes, J. P. Schwarz, K. Sekimoto, V. Selimovic, T. Shingler, D. J. Tanner, L. Tomsche, K. Vasquez, P. R. Veres, R. Washenfelder, P. Weibring, P. O. Wennberg, A. Wisthaler, G. Wolfe, C. Womack, L. Xu, R. Yokelson and C. Warneke, Parameterizations of US wildfire and prescribed fire emission ratios and emission factors based on FIREX-AQ aircraft measurements, EGUsphere [preprint], 2023, DOI:10.5194/egusphere-2023-1439.
- L. A. Garofalo, M. A. Pothier, E. J. T. Levin, T. Campos, S. M. Kreidenweis and D. K. Farmer, Emission and Evolution of Submicron Organic Aerosol in Smoke from Wildfires in the Western United States, ACS Earth Space Chem., 2019, 3, 1237–1247 CrossRef CAS.
- M. J. Cubison, A. M. Ortega, P. L. Hayes, D. K. Farmer, D. Day, M. J. Lechner, W. H. Brune, E. Apel, G. S. Diskin, J. A. Fisher, H. E. Fuelberg, A. Hecobian, D. J. Knapp, T. Mikoviny, D. Riemer, G. W. Sachse, W. Sessions, R. J. Weber, A. J. Weinheimer, A. Wisthaler and J. L. Jimenez, Effects of aging on organic aerosol from open biomass burning smoke in aircraft and laboratory studies, Atmos. Chem. Phys., 2011, 11, 12049–12064 CrossRef.
- J. L. Jimenez, M. R. Canagaratna, N. M. Donahue, A. S. H. Prevot, Q. Zhang, J. H. Kroll, P. F. DeCarlo, J. D. Allan, H. Coe, N. L. Ng, A. C. Aiken, K. S. Docherty, I. M. Ulbrich, A. P. Grieshop, A. L. Robinson, J. Duplissy, J. D. Smith, K. R. Wilson, V. A. Lanz, C. Hueglin, Y. L. Sun, J. Tian, A. Laaksonen, T. Raatikainen, J. Rautiainen, P. Vaattovaara, M. Ehn, M. Kulmala, J. M. Tomlinson, D. R. Collins, M. J. Cubison, E. J. Dunlea, J. A. Huffman, T. B. Onasch, M. R. Alfarra, P. I. Williams, K. Bower, Y. Kondo, J. Schneider, F. Drewnick, S. Borrmann, S. Weimer, K. Demerjian, D. Salcedo, L. Cottrell, R. Griffin, A. Takami, T. Miyoshi, S. Hatakeyama, A. Shimono, J. Y. Sun, Y. M. Zhang, K. Dzepina, J. R. Kimmel, D. Sueper, J. T. Jayne, S. C. Herndon, A. M. Trimborn, L. R. Williams, E. C. Wood, A. M. Middlebrook, C. E. Kolb, U. Baltensperger and D. R. Worsnop, Evolution of Organic Aerosols in the Atmosphere, Science, 2009, 326, 1525–1529 CrossRef CAS PubMed.
- J. L. Jimenez, P. Campuzano-Jost, D. A. Day, B. A. Nault, H. Guo, J. C. Schroder and M. J. Cubison, Frequently Asked AMS Questions for AMS Data Users, https://cires1.colorado.edu/jimenez-group/wiki/index.php/FAQ_for_AMS_Data_Users, (accessed 10 June 2023) Search PubMed.
- M. Baasandorj, D. B. Millet, L. Hu, D. Mitroo and B. J. Williams, Measuring acetic and formic acid by proton-transfer-reaction mass spectrometry: sensitivity, humidity dependence, and quantifying interferences, Atmos. Meas. Tech., 2015, 8, 1303–1321 CrossRef CAS.
- J. A. de Gouw and C. Warneke, Measurements of volatile organic compounds in the earth's atmosphere using proton-transfer-reaction mass spectrometry, Mass Spectrom. Rev., 2007, 26, 223–257 CrossRef CAS PubMed.
- B. Yuan, A. R. Koss, C. Warneke, M. Coggon, K. Sekimoto and J. A. de Gouw, Proton-Transfer-Reaction Mass Spectrometry: Applications in Atmospheric Sciences, Chem. Rev., 2017, 117, 13187–13229 CrossRef CAS PubMed.
- E. C. Fortner, J. Zheng, R. Zhang, W. Berk Knighton, R. M. Volkamer, P. Sheehy, L. Molina and M. André, Measurements of Volatile Organic Compounds Using Proton Transfer Reaction – Mass Spectrometry during the MILAGRO 2006 Campaign, Atmos. Chem. Phys., 2009, 9, 467–481 CrossRef CAS.
- J. B. Gilman, B. M. Lerner, W. C. Kuster, P. D. Goldan, C. Warneke, P. R. Veres, J. M. Roberts, J. A. de Gouw, I. R. Burling and R. J. Yokelson, Biomass burning emissions and potential air quality impacts of volatile organic compounds and other trace gases from fuels common in the US, Atmos. Chem. Phys., 2015, 15, 13915–13938 CrossRef CAS.
- P. Španěl, A. M. Diskin, T. Wang and D. Smith, A SIFT study of the reactions of H3O+, NO+ and O2+ with hydrogen peroxide and peroxyacetic acid, Int. J. Mass Spectrom., 2003, 228, 269–283 CrossRef.
- R. J. Yokelson, R. Susott, D. E. Ward, J. Reardon and D. W. T. Griffith, Emissions from smoldering combustion of biomass measured by open-path Fourier transform infrared spectroscopy, J. Geophys. Res.: Atmos., 1997, 102, 18865–18877 CrossRef CAS.
- D. J. Marino, in Encyclopedia of Toxicology, ed. P. Wexler, Elsevier, New York, 2nd edn, 2005, pp. 277–279 Search PubMed.
- L. E. Hatch, W. Luo, J. F. Pankow, R. J. Yokelson, C. E. Stockwell and K. C. Barsanti, Identification and quantification of gaseous organic compounds emitted from biomass burning using two-dimensional gas chromatography-time-of-flight mass spectrometry, Atmos. Chem. Phys., 2015, 15, 1865–1899 CrossRef CAS.
- T. J. Christian, B. Kleiss, R. J. Yokelson, R. Holzinger, P. J. Crutzen, W. M. Hao, B. H. Saharjo and D. E. Ward, Comprehensive laboratory measurements of biomass-burning emissions: 1. Emissions from Indonesian, African, and other fuels, J. Geophys. Res.: Atmos., 2003, 108(D23), 4719 CrossRef.
- V. Selimovic, R. J. Yokelson, C. Warneke, J. M. Roberts, J. de Gouw, J. Reardon and D. W. T. Griffith, Aerosol optical properties and trace gas emissions by PAX and OP-FTIR for laboratory-simulated western US wildfires during FIREX, Atmos. Chem. Phys., 2018, 18, 2929–2948 CrossRef CAS.
- C. Bacher, G. S. Tyndall and J. J. Orlando, The Atmospheric Chemistry of Glycolaldehyde, J. Atmos. Chem., 2001, 39, 171–189 CrossRef CAS.
- P. Veres, J. B. Gilman, J. M. Roberts, W. C. Kuster, C. Warneke, I. R. Burling and J. de Gouw, Development and validation of a portable gas phase standard generation and calibration system for volatile organic compounds, Atmos. Meas. Tech., 2010, 3, 683–691 CrossRef CAS.
- I. Bey, D. J. Jacob, R. M. Yantosca, J. A. Logan, B. D. Field, A. M. Fiore, Q. Li, H. Y. Liu, L. J. Mickley and M. G. Schultz, Global modeling of tropospheric chemistry with assimilated meteorology: model description and evaluation, J. Geophys. Res.: Atmos., 2001, 106, 23073–23095 CrossRef CAS.
- The International GEOS-Chem User Community, 2018 Search PubMed.
- P. V. Hobbs, P. Sinha, R. J. Yokelson, T. J. Christian, D. R. Blake, S. Gao, T. W. Kirchstetter, T. Novakov and P. Pilewskie, Evolution of gases and particles from a savanna fire in South Africa, J. Geophys. Res.: Atmos., 2003, 108, 8485 Search PubMed.
- J. Lindaas, I. B. Pollack, L. A. Garofalo, M. A. Pothier, D. K. Farmer, S. M. Kreidenweis, T. L. Campos, F. Flocke, A. J. Weinheimer, D. D. Montzka, G. S. Tyndall, B. B. Palm, Q. Peng, J. A. Thornton, W. Permar, C. Wielgasz, L. Hu, R. D. Ottmar, J. C. Restaino, A. T. Hudak, I.-T. Ku, Y. Zhou, B. C. Sive, A. Sullivan, J. L. Collett Jr. and E. V. Fischer, Emissions of Reactive Nitrogen From Western U.S. Wildfires During Summer 2018, J. Geophys. Res.: Atmos., 2021, 126, e2020JD032657 CrossRef CAS.
- C. R. Lonsdale, M. J. Alvarado, A. L. Hodshire, E. Ramnarine and J. R. Pierce, Simulating the forest fire plume dispersion, chemistry, and aerosol formation using SAM-ASP version 1.0, Geosci. Model Dev., 2020, 13, 4579–4593 CrossRef CAS.
- R. J. Yokelson, D. W. T. Griffith and D. E. Ward, Open-path Fourier transform infrared studies of large-scale laboratory biomass fires, J. Geophys. Res.: Atmos., 1996, 101, 21067–21080 CrossRef CAS.
- C. Wielgasz, Investigation of Emissions and Chemistry of Formic Acid And Acetic Acid in Wildfire Smoke, Graduate Student theses, Dissertartions, & Professional Papers, Universtity of Montana, 2021, https://scholarworks.umt.edu/etd/11676.
- S. J. Prichard, S. M. O'Neill, P. Eagle, A. G. Andreu, B. Drye, J. Dubowy, S. Urbanski and T. M. Strand, Wildland fire emission factors in North America: synthesis of existing data, measurement needs and management applications, Int. J. Wildland Fire, 2020, 29, 132 CrossRef.
- R. J. Yokelson, S. K. Akagi, D. W. T. Griffith and T. J. Johnson, Interactive comment on Exceptional emissions of NH3 and HCOOH in the 2010 Russian wildfires by Y. R'Honi et al., Atmos. Chem. Phys. Discuss., 2013, 12, C11864–C11868 Search PubMed.
- C. E. Stockwell, P. R. Veres, J. Williams and R. J. Yokelson, Characterization of biomass burning emissions from cooking fires, peat, crop residue, and other fuels with high-resolution proton-transfer-reaction time-of-flight mass spectrometry, Atmos. Chem. Phys., 2015, 15, 845–865 CrossRef.
- I. Bertschi, R. J. Yokelson, D. E. Ward, R. E. Babbitt, R. A. Susott, J. G. Goode and W. M. Hao, Trace gas and particle emissions from fires in large diameter and belowground biomass fuels, J. Geophys. Res.: Atmos., 2003, 108(D13), 8472 Search PubMed.
- T. J. Christian, B. Kleiss, R. J. Yokelson, R. Holzinger, P. J. Crutzen, W. M. Hao, B. H. Saharjo and D. E. Ward, Comprehensive laboratory measurements of biomass-burning emissions: 1. Emissions from Indonesian, African, and other fuels, J. Geophys. Res.: Atmos., 2003, 108(D23), 4719 CrossRef.
- L. M. McKenzie, W. M. Hao, G. N. Richards and D. E. Ward, Measurement and Modeling of Air Toxins from Smoldering Combustion of Biomass, Environ. Sci. Technol., 1995, 29, 2047–2054 CrossRef CAS PubMed.
- S. K. Akagi, R. J. Yokelson, C. Wiedinmyer, M. J. Alvarado, J. S. Reid, T. Karl, J. D. Crounse and P. O. Wennberg, Emission factors for open and domestic biomass burning for use in atmospheric models, Atmos. Chem. Phys., 2011, 11, 4039–4072 CrossRef CAS.
- J. A. de Gouw, C. Warneke, A. Stohl, A. G. Wollny, C. A. Brock, O. R. Cooper, J. S. Holloway, M. Trainer, F. C. Fehsenfeld, E. L. Atlas, S. G. Donnelly, V. Stroud and A. Lueb, Volatile organic compounds composition of merged and aged forest fire plumes from Alaska and western Canada, J. Geophys. Res.: Atmos., 2006, 111, D10303 CrossRef.
- G. M. Wolfe, M. R. Marvin, S. J. Roberts, K. R. Travis and J. Liao, The Framework for 0-D Atmospheric Modeling (F0AM) v3.1, Geosci. Model Dev., 2016, 9, 3309–3319 CrossRef.
- J. Lindaas, I. B. Pollack, J. J. Calahorrano, K. O'Dell, L. A. Garofalo, M. A. Pothier, D. K. Farmer, S. M. Kreidenweis, T. Campos, F. Flocke, A. J. Weinheimer, D. D. Montzka, G. S. Tyndall, E. C. Apel, A. J. Hills, R. S. Hornbrook, B. B. Palm, Q. Peng, J. A. Thornton, W. Permar, C. Wielgasz, L. Hu, J. R. Pierce, J. L. Collett Jr., A. P. Sullivan and E. V. Fischer, Empirical Insights Into the Fate of Ammonia in Western U.S. Wildfire Smoke Plumes, J. Geophys. Res.: Atmos., 2021, 126, e2020JD033730 CrossRef CAS.
- B. B. Palm, Q. Peng, S. R. Hall, K. Ullmann, T. L. Campos, A. Weinheimer, D. Montzka, G. Tyndall, W. Permar, L. Hu, F. Flocke, E. V. Fischer and J. A. Thornton, Spatially Resolved Photochemistry Impacts Emissions Estimates in Fresh Wildfire Plumes, Geophys. Res. Lett., 2021, 48, e2021GL095443 CrossRef CAS.
- B. B. Palm, Q. Peng, C. D. Fredrickson, B. H. Lee, L. A. Garofalo, M. A. Pothier, S. M. Kreidenweis, D. K. Farmer, R. P. Pokhrel, Y. Shen, S. M. Murphy, W. Permar, L. Hu, T. L. Campos, S. R. Hall, K. Ullmann, X. Zhang, F. Flocke, E. V. Fischer and J. A. Thornton, Quantification of organic aerosol and brown carbon evolution in fresh wildfire plumes, Proc. Natl. Acad. Sci., 2020, 117, 29469–29477 CrossRef CAS PubMed.
- Q. Peng, B. B. Palm, C. D. Fredrickson, B. H. Lee, S. R. Hall, K. Ullmann, T. Campos, A. J. Weinheimer, E. C. Apel, F. Flocke, W. Permar, L. Hu, L. A. Garofalo, M. A. Pothier, D. K. Farmer, I.-T. Ku, A. P. Sullivan, J. L. Collett, E. Fischer and J. A. Thornton, Observations and Modeling of NOx Photochemistry and Fate in Fresh Wildfire Plumes, ACS Earth Space Chem., 2021, 5, 2652–2667 CrossRef CAS.
- M. M. Coggon, C. Y. Lim, A. R. Koss, K. Sekimoto, B. Yuan, J. B. Gilman, D. H. Hagan, V. Selimovic, K. J. Zarzana, S. S. Brown, J. M. Roberts, M. Müller, R. Yokelson, A. Wisthaler, J. E. Krechmer, J. L. Jimenez, C. Cappa, J. H. Kroll, J. de Gouw and C. Warneke, OH chemistry of non-methane organic gases (NMOGs) emitted from laboratory and ambient biomass burning smoke: evaluating the influence of furans and oxygenated aromatics on ozone and secondary NMOG formation, Atmos. Chem. Phys., 2019, 19, 14875–14899 CrossRef CAS.
- Z. C. J. Decker, K. J. Zarzana, M. Coggon, K.-E. Min, I. Pollack, T. B. Ryerson, J. Peischl, P. Edwards, W. P. Dubé, M. Z. Markovic, J. M. Roberts, P. R. Veres, M. Graus, C. Warneke, J. de Gouw, L. E. Hatch, K. C. Barsanti and S. S. Brown, Nighttime Chemical Transformation in Biomass Burning Plumes: A Box Model Analysis Initialized with Aircraft Observations, Environ. Sci. Technol., 2019, 53, 2529–2538 CrossRef CAS PubMed.
- S. M. Aschmann, N. Nishino, J. Arey and R. Atkinson, Products of the OH Radical-Initiated Reactions of Furan, 2- and 3-Methylfuran, and 2,3- and 2,5-Dimethylfuran in the Presence of NO, J. Phys. Chem. A, 2014, 118, 457–466 CrossRef CAS PubMed.
- Y. Yuan, X. Zhao, S. Wang and L. Wang, Atmospheric Oxidation of Furan and Methyl-Substituted Furans Initiated by Hydroxyl Radicals, J. Phys. Chem. A, 2017, 121, 9306–9319 CrossRef CAS PubMed.
- S. Wang, M. J. Newland, W. Deng, A. R. Rickard, J. F. Hamilton, A. Muñoz, M. Ródenas, M. M. Vázquez, L. Wang and X. Wang, Aromatic Photo-oxidation, A New Source of Atmospheric Acidity, Environ. Sci. Technol., 2020, 54, 7798–7806 CrossRef CAS PubMed.
- Leeds MCM, https://mcm.leeds.ac.uk/MCM, (accessed 5 May 2023).
- J. Jiang, W. P. L. Carter, D. R. Cocker and K. C. Barsanti, Development and Evaluation of a Detailed Mechanism for Gas-Phase Atmospheric Reactions of Furans, ACS Earth Space Chem., 2020, 4, 1254–1268 CrossRef CAS.
- K. H. Bates, D. J. Jacob, K. Li, P. D. Ivatt, M. J. Evans, Y. Yan and J. Lin, Development and evaluation of a new compact mechanism for aromatic oxidation in atmospheric models, Atmos. Chem. Phys., 2021, 21, 18351–18374 CrossRef CAS.
- M. R. Canagaratna, J. L. Jimenez, J. H. Kroll, Q. Chen, S. H. Kessler, P. Massoli, L. Hildebrandt Ruiz, E. Fortner, L. R. Williams, K. R. Wilson, J. D. Surratt, N. M. Donahue, J. T. Jayne and D. R. Worsnop, Elemental ratio measurements of organic compounds using aerosol mass spectrometry: characterization, improved calibration, and implications, Atmos. Chem. Phys., 2015, 15, 253–272 CrossRef.
- A. C. Aiken, P. F. DeCarlo, J. H. Kroll, D. R. Worsnop, J. A. Huffman, K. S. Docherty, I. M. Ulbrich, C. Mohr, J. R. Kimmel, D. Sueper, Y. Sun, Q. Zhang, A. Trimborn, M. Northway, P. J. Ziemann, M. R. Canagaratna, T. B. Onasch, M. R. Alfarra, A. S. H. Prevot, J. Dommen, J. Duplissy, A. Metzger, U. Baltensperger and J. L. Jimenez, O/C and OM/OC Ratios of Primary, Secondary, and Ambient Organic Aerosols with High-Resolution Time-of-Flight Aerosol Mass Spectrometry, Environ. Sci. Technol., 2008, 42, 4478–4485 CrossRef CAS PubMed.
- C. E. Stockwell, M. M. Bela, M. M. Coggon, G. I. Gkatzelis, E. Wiggins, E. M. Gargulinski, T. Shingler, M. Fenn, D. Griffin, C. D. Holmes, X. Ye, P. E. Saide, I. Bourgeois, J. Peischl, C. C. Womack, R. A. Washenfelder, P. R. Veres, J. A. Neuman, J. B. Gilman, A. Lamplugh, R. H. Schwantes, S. A. McKeen, A. Wisthaler, F. Piel, H. Guo, P. Campuzano-Jost, J. L. Jimenez, A. Fried, T. F. Hanisco, L. G. Huey, A. Perring, J. M. Katich, G. S. Diskin, J. B. Nowak, T. P. Bui, H. S. Halliday, J. P. DiGangi, G. Pereira, E. P. James, R. Ahmadov, C. A. McLinden, A. J. Soja, R. H. Moore, J. W. Hair and C. Warneke, Airborne Emission Rate Measurements Validate Remote Sensing Observations and Emission Inventories of Western U.S. Wildfires, Environ. Sci. Technol., 2022, 56, 7564–7577 CrossRef CAS PubMed.
- Y. Chen, Q. Li, J. T. Randerson, E. A. Lyons, R. A. Kahn, D. L. Nelson and D. J. Diner, The sensitivity of CO and aerosol transport to the temporal and vertical distribution of North American boreal fire emissions, Atmos. Chem. Phys., 2009, 6559–6580 CrossRef CAS.
- Y. Jian and T.-M. Fu, Injection heights of springtime biomass-burning plumes over peninsular Southeast Asia and their impacts on long-range pollutant transport, Atmos. Chem. Phys., 2014, 14, 3977–3989 CrossRef.
- J. F. Hunter, D. A. Day, B. B. Palm, R. L. N. Yatavelli, A. W. H. Chan, L. Kaser, L. Cappellin, P. L. Hayes, E. S. Cross, A. J. Carrasquillo, P. Campuzano-Jost, H. Stark, Y. Zhao, T. Hohaus, J. N. Smith, A. Hansel, T. Karl, A. H. Goldstein, A. Guenther, D. R. Worsnop, J. A. Thornton, C. L. Heald, J. L. Jimenez and J. H. Kroll, Comprehensive characterization of atmospheric organic carbon at a forested site, Nat. Geosci., 2017, 10, 748–753 CrossRef CAS.
- K. H. Bates, D. J. Jacob, S. Wang, R. S. Hornbrook, E. C. Apel, M. J. Kim, D. B. Millet, K. C. Wells, X. Chen, J. F. Brewer, E. A. Ray, R. Commane, G. S. Diskin and S. C. Wofsy, The Global Budget of Atmospheric Methanol: New Constraints on Secondary, Oceanic, and Terrestrial Sources, J. Geophys. Res.: Atmos., 2021, 126, e2020JD033439 CrossRef CAS.
- R. Holzinger, J. Williams, G. Salisbury, T. Klüpfel, M. de Reus, M. Traub, P. J. Crutzen and J. Lelieveld, Oxygenated compounds in aged biomass burning plumes over the Eastern Mediterranean: evidence for strong secondary production of methanol and acetone, Atmos. Chem. Phys., 2005, 5, 39–46 CrossRef CAS.
- L. Hu, D. B. Millet, S. Y. Kim, K. C. Wells, T. J. Griffis, E. V. Fischer, D. Helmig, J. Hueber and A. J. Curtis, North American acetone sources determined from tall tower measurements and inverse modeling, Atmos. Chem. Phys., 2013, 13, 3379–3392 CrossRef CAS PubMed.
- L. Hu, D. B. Millet, M. J. Mohr, K. C. Wells, T. J. Griffis and D. Helmig, Sources and seasonality of atmospheric methanol based on tall tower measurements in the US Upper Midwest, Atmos. Chem. Phys., 2011, 11, 11145–11156 CrossRef CAS.
- K. C. Wells, D. B. Millet, L. Hu, K. E. Cady-Pereira, Y. Xiao, M. W. Shephard, C. L. Clerbaux, L. Clarisse, P.-F. Coheur, E. C. Apel, J. de Gouw, C. Warneke, H. B. Singh, A. H. Goldstein and B. C. Sive, Tropospheric methanol observations from space: retrieval evaluation and constraints on the seasonality of biogenic emissions, Atmos. Chem. Phys., 2012, 12, 5897–5912 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ea00098b |
| This journal is © The Royal Society of Chemistry 2023 |