
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A convenient synthesis of ferrocene-(ethynylphenyl)thioacetates†
Troy L. R.
Bennett
and
Nicholas J.
Long
 *
*
Department of Chemistry, Imperial College London, London, W12 0BZ, UK. E-mail: n.long@imperial.ac.uk
First published on 19th October 2023
Abstract
Ferrocene is popular within the field of molecular electronics due to its well-defined electronic properties. However, where the conductance of highly-conjugated oligophenylethylenes has been widely studied, work on analogous ferrocenyl systems has been relatively rare, possibly due to difficulties associated with the synthesis of molecules containing terminal thioacetates, which are often used to bind molecules to metallic electrodes. Herein, a widely applicable synthetic methodology is demonstrated which can be used to synthesize a variety of conjugated ferrocene-alkyne systems terminated with thioacetates, including symmetric, asymmetric and multi-ferrocene systems. Conjugation of the ferrocene units to their terminal atoms is then shown through the use of both UV/Vis spectroscopy and cyclic voltammetry. This work paves the way for future studies and applications of conjugated ferrocene systems in the field of nanoelectronics.
Introduction
Since its discovery in the 1950s, ferrocene has fascinated the organometallic chemist, with compounds displaying high stability, interesting geometries (relating to interdependent rotation of the two cyclopentadienyl rings) and most prominently, exceptionally well-defined redox features.1 The electronic properties of ferrocene, in particular, have been studied fervently, to the point that it is now widely used as a common reference point for the study of other electroactive compounds.2 These electronic properties have made ferrocene an ideal candidate for studies in the field of molecular electronics.3A notable area of study within this field focusses on examining the conductivity of molecular systems, using techniques that have been developed to allow molecules to be trapped between two electrodes to study their conductive properties. However, where a wealth of research has focussed on the study of highly conductive oligo-phenylethylenes, which have been used as a building block in various organic and organometallic systems, investigations into analogous functionalised ferrocenes are relatively rare.4 This omission is surprising, considering the popularity of ferrocene in the wider field, and is made even more startling by the few studies that have been performed by e.g. the Sita and Wang groups, which suggest that these systems would be promising candidates in nanoelectronics, boasting near-perfect conduction, significantly higher than their organic counterparts.5
Aside from inherent rotational properties which can act to hinder the measurement of ‘linear’ 1,1′-disubstituted ferrocenes (i.e. where the two substituents point in opposite directions),3c,6 one potential reason for this relative lack of research could be attributed to difficulties in the synthesis of alkynyl-ferrocenes which can be strongly bound to an electrode surface. Thiols, and their thioacetate precursors, are widely applied in the field of molecular electronics for electrode binding, due to the strong bonds they can form with metallic electrodes.4a Unfortunately, where numerous methods exist for synthesising organic wires with terminal thiols (or thioacetates), these cannot be applied to alkynyl-ferrocenes.
For instance, it has previously been reported that (ethynylphenyl)thioacetates undergo a cyclo-oligomerisation when exposed to the palladium catalysts necessary for cross-coupling reactions.7 In organic systems, this can be compensated for by installing ethynyl groups, and subsequently coupling to iodo(arylthioacetates).4 However, this is not possible for ferrocene, due to the inherent instability of 1,1′-diethynylferrocene. Protecting group strategies have also proved ineffective, leading to either degradation or competitive side-reactions, associated with the electron-rich alkynes.8 Finally, though a handful of ferrocene-(alkynylphenyl)thioacetates have previously been prepared, via Stille couplings of ferrocene-ethynyl-SnBu3 units, these require many synthetic steps, utilise toxic reagents and are quite limited in scope.9 In order to synthesise more complicated derivatives, such as molecules containing unsymmetrical designs or complex geometries, a new synthetic route would need to be devised.
Herein the limitations of previously applied methods for synthesising ferrocenes with terminal thioacetate units are demonstrated, and a convenient and general method for the synthesis of this family of molecules, through use of a cyanoethyl-based sulphur protecting group strategy, is presented as an alternative. It is shown that this technique can be applied to create a wide range of both symmetric and asymmetrically substituted derivatives and can also be used to create multi-ferrocenyl systems, offering a valuable tool for further synthetic work. The optoelectronic properties of this new series of molecules were studied using a combination of both UV/Vis spectroscopy and cyclic voltammetry, showing that within these systems, the ferrocene units are effectively conjugated to their terminal aromatic substituents.
Results and discussion
Synthesis
Upon consideration of the synthesis of this class of molecules, it seemed reasonable to investigate the possibility of adapting previously attempted synthetic strategies. For instance, Engtrakul et al., have reported the synthesis of an asymmetric ferrocene-(ethynylphenyl)thioacetate, which they achieved by installing a terminal ethynyl group and subsequently cross-coupling this with 4-iodophenylthioacetate.5a,c In an attempt to mimic this, 1-ethynyl-1′-iodoferrocene (1) was synthesised via a 2-step procedure, first reacting TMSA with a slight excess of 1,1′-diiodoferrocene to give a monosubstituted derivative (1a), and then performing a basic deprotection of the TMS group using NaOH (Scheme 1A). The Sonogashira cross-coupling of this molecule with 4-iodophenylthioacetate was then attempted under standard conditions. Following flash-chromatography, this allowed for the desired product (2) to be isolated in good yield. Surprisingly though, it was found that by increasing the polarity of the eluent it was possible to isolate further fractions from the column, providing evidence for the formation of some interesting side-products (Scheme 1B).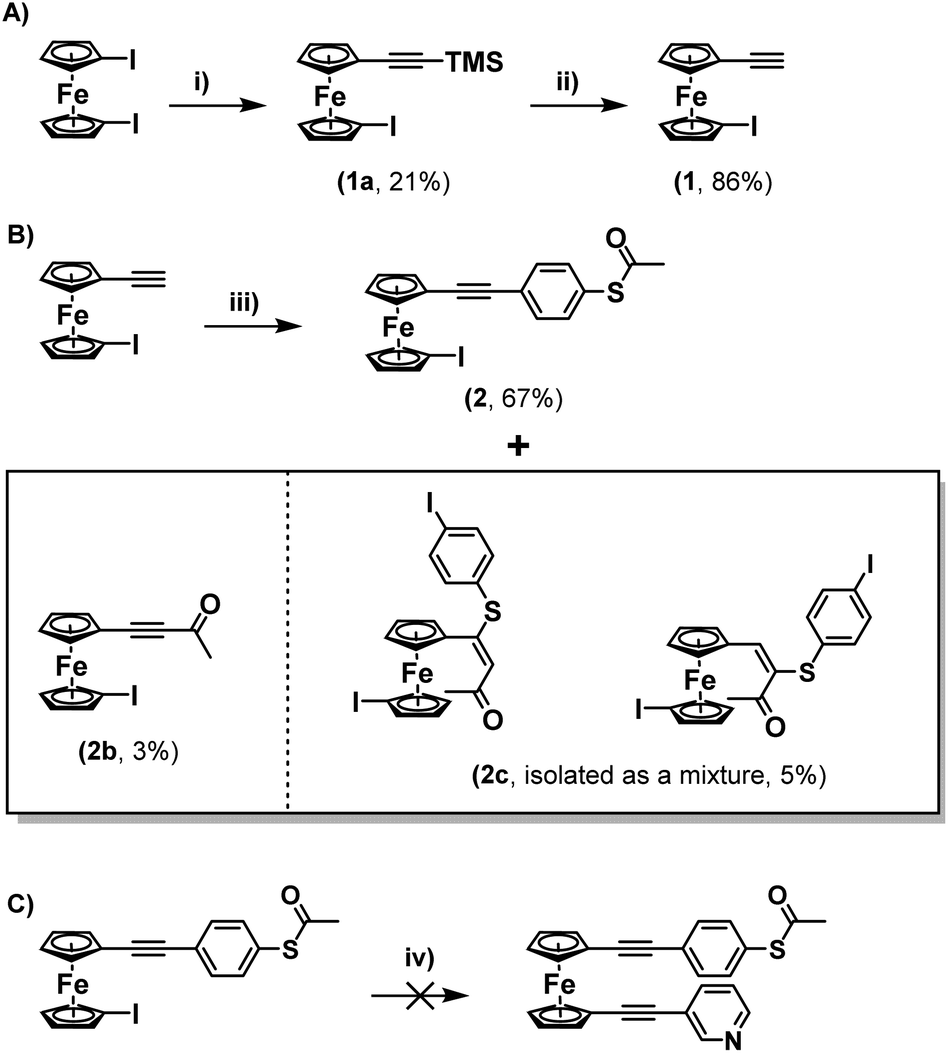 | ||
Scheme 1 (A) Synthesis of 1; (B) synthesis of 2 and its associated side products (2b and 2c) and (C) unsuccessful reaction attempted to functionalise 2 with a further alkyne substituent. Reaction conditions: (i) TMSA, CuI, Pd(P-tBu3)2, DIPA, RT, >16 h; (ii) NaOH, THF/MeOH (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), RT, 16 h; (iii) 4-iodophenylthioacetate, CuI, PdCl2(PPh3)2, DIPA, RT, >16 h; (iv) 3-ethynylpyridine, CuI, Pd(P-tBu3)2, DIPA, THF, RT, >16 h. :1), RT, 16 h; (iii) 4-iodophenylthioacetate, CuI, PdCl2(PPh3)2, DIPA, RT, >16 h; (iv) 3-ethynylpyridine, CuI, Pd(P-tBu3)2, DIPA, THF, RT, >16 h. | ||
The first of these gave a 1H-NMR spectrum with a singlet centred at 2.42 ppm, indicative of the presence of an acyl group, however there were no peaks in the aromatic region of the spectrum. Alongside the lack of an alkynyl-associated proton, this led to this molecule being identified as a terminal ynone (2b). This could likely have formed through oxidative addition across the S–Ac bond of 4-iodophenylthioacetate onto the palladium catalyst, followed by subsequent reductive elimination of the acyl group onto the terminal alkyne of 1.
Another fraction was isolated which seemingly contained a mixture of two species. This again presented peaks which could be associated with acyl groups (centred at 2.36 and 1.95 ppm respectively), however this time there were also peaks present in the aromatic region, as well as a distinct singlet for each of the two species, centred at 7.05 and 5.60 ppm respectively. The position of these peaks and their relative integrals, when compared to the other peaks in the spectrum, led to these being identified as alkenes (mixture 2c). These species were likely formed through conjugate addition of a previously formed ynone with a sulphur-containing species generated via removal of the acyl-group from 4-iodophenylthioacetate. This hypothesis aligns well with previous mechanistic work presented by Inkpen et al.7b The identities of both 2c and the ynone 2b were confirmed by high-resolution mass spectrometry (HRMS). 13C{1H} NMR was also useful in identifying 2b, however for the mixture of isomers 2c, limited sample quantities resulted in only one of the two molecules providing a full set of carbon signals (Fig. S10 of the ESI†). Regardless, when taken together: the 1H-NMR spectra, HRMS data, the similar polarities of the two species (as inferred from their co-elution during flash chromatography), and previous reports on similar systems, provide good evidence for the identity of 2c.7b
Regardless, with 2 in hand, an attempt was made to functionalise this further through Sonogashira cross-coupling, with 3-ethynylpyridine (Scheme 1C). Unfortunately, this led to the generation of a complex mixture of products which were not possible to separate. It is surmised that this may relate to competitive reactions relating to instability of the S–Ac bond under these conditions, as discussed above. In tandem with this work, the synthesis of a thioacetate-terminated biferrocene molecule was also investigated. For this 1,1′′′-diethynylbiferrocene (3) was synthesised by reaction of 1,1′′′-diiodobiferrocene with an excess of TMSA under standard Sonogashira cross-coupling conditions to give molecule 3a, followed by basic deprotection of the TMS group to give the desired diethynyl biferrocene (Scheme 2A).
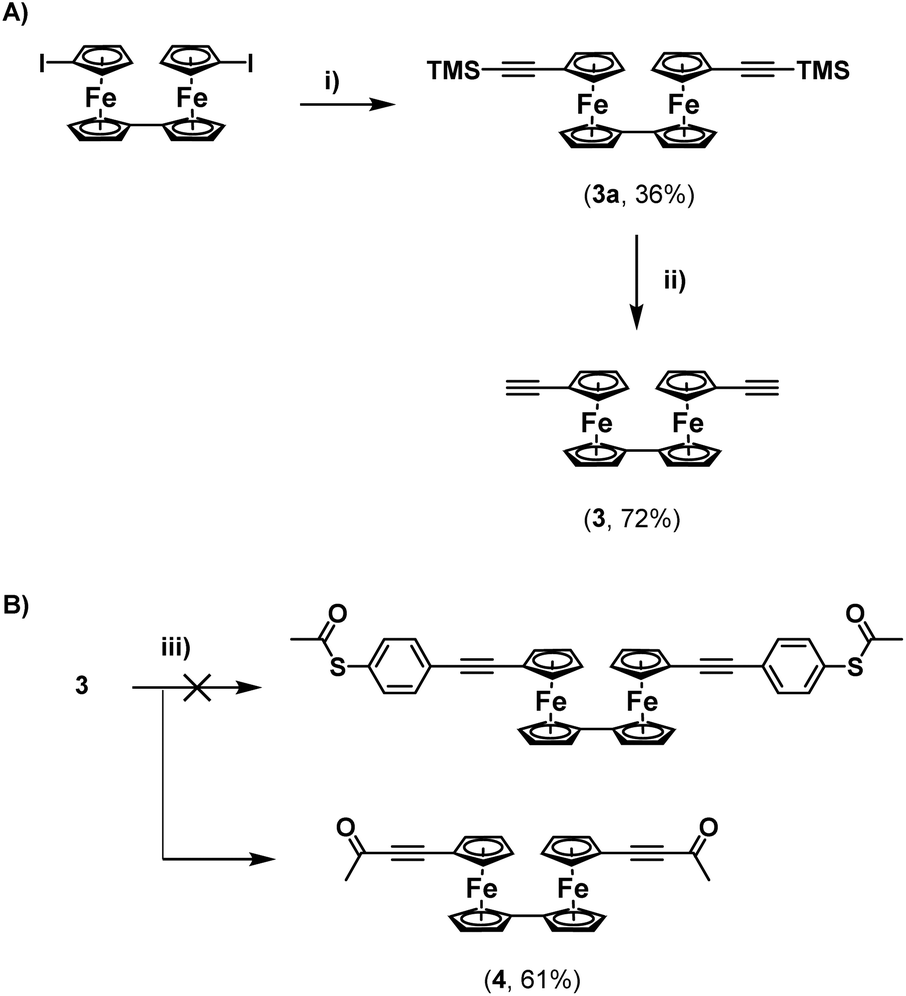 | ||
| Scheme 2 (A) Synthesis of 3 and (B) attempted synthesis of a biferrocene disubstituted with alkynyl(phenyl)thioacetates, which led to the competitive formation of a biferrocene di-ynone (4). Reaction conditions (i) TMSA, CuI, Pd(P-tBu3)2, DIPA, RT, >16 h; (ii) NaOH, THF/MeOH (1:1), RT, 16 h; (iii) 4-iodophenylthioacetate, CuI, PdCl2(PPh3)2, DIPA, RT, >16 h; (iv) 3-ethynylpyridine, CuI, Pd(P-tBu3)2, DIPA, RT, >16 h. | ||
Again, an attempt was made to functionalise this system through cross-coupling with 4-iodophenylthioacetate. Surprisingly, following flash-chromatography, the desired product was not obtained, and instead a material was isolated containing a singlet in its 1H-NMR spectrum located at 2.32 ppm, and with no peaks in the aromatic region of the spectrum. This again suggested the formation of a di-ynone species (4), which was further confirmed by 13C{1H}-NMR spectroscopy and HRMS (Scheme 2B). Again, this species was likely formed through oxidative addition across the S–Ac bond of the thioacetate onto the palladium catalyst, and subsequent reductive elimination onto the alkynes of the biferrocene species. Although interesting, and a potentially useful synthetic route towards a rare example of a biferrocene ynone which may have further synthetic applications,10 this was disappointing, and suggested that a protecting-group strategy would be required to synthesize terminal thioacetates, to compensate for their relative instability to cross-coupling conditions.
It has previously been shown that the commonly utilised tert-butyl protecting group would not be suitable for this synthesis, due to the acidic conditions required for its cleavage, combined with the instability of ferrocene when exposed to strong acids.7a,11 This ruled out many acid-labile protecting groups. The likely instability of the S–(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) group to basic conditions also rules out many base labile protecting groups. However, upon further consideration it was decided it may be possible to apply a protecting group strategy utilising a functional group with a lability to weakly basic conditions, namely a cyanoethyl group.
O) group to basic conditions also rules out many base labile protecting groups. However, upon further consideration it was decided it may be possible to apply a protecting group strategy utilising a functional group with a lability to weakly basic conditions, namely a cyanoethyl group.
For this, 4-(cyanoethylthio)ethynylbenzene was synthesised following a procedure laid out by Bryce et al.,12 and then subsequently reacted with 1,1′-diiodoferrocene and 1,1′′′-diiodobiferrocene, under previously optimised Sonogashira cross-coupling conditions (Scheme 3).7 These reactions proceeded efficiently, granting molecules 5a and 6a in high yields following purification by flash-chromatography. It was found that these systems could then be simply converted into their corresponding thioacetates, by first removing the cyanoethyl groups, through treatment with NaOMe, and subsequently adding acetic anhydride to ‘quench’ the free thiol, generating terminal thioacetates 5 and 6. Initial attempts to purify these systems were ineffective, with both silica and alumina causing some level of degradation. However, it was found that pre-treatment of the silica, by first preparing it as a slurry with hexane and a small amount of triethylamine (1%), decreased its acidity enough to allow for the isolation of 5 and 6 in high yields.
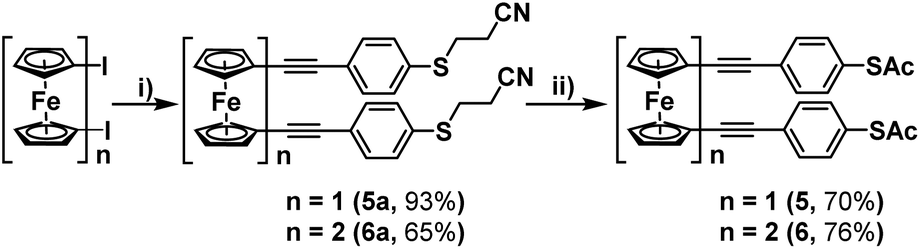 | ||
| Scheme 3 Synthesis of ferrocene-(ethynylphenyl)thioacetates 5 and 6. Reaction conditions: (i) 4-(cyanoethylthio)ethynylbenzene, CuI, Pd(PtBu3)2, DIPA, THF, 50 °C, >16 hours; (ii) NaOMe (25% in MeOH), THF, 30 min followed by Ac2O, 30 min. | ||
Spurred on by this success this methodology was explored further to examine whether it could be applied to synthesise asymmetric di-alkynyl ferrocenes. To this end, 4-(cyanoethylthio)ethnylbenzene was reacted with an excess of 1,1′-diiodoferrocene to form molecule 2a (Scheme 4). A series of terminal alkynes were then prepared (see ESI† for details), functionalised with a variety of different aryl groups. These were selected to investigate the tolerance of the ‘trans-protection’ step to different functional groups that may exhibit potential applications in molecular electronics, such as switching, electrode binding and on-surface functionalisation. These were then reacted with 2a to form a range of systems 7a–12a (from the following substituents: 4-pyridine (7a), 3-pyridine (8a), 6-(2,2′-bipyridine) (9a), pyrene (10a), 4-phenyldiazene (11a) and 4-benzaldehyde (12a)) (Scheme 4). Compounds 7a–12a were subsequently ‘trans-protected’, to form compounds 7–12. Most of these systems could be purified by flash chromatography alone, though in some cases a precipitation was also necessary, resulting in slightly reduced yields (see ESI† for details). It should be noted that the presence of a reactive aldehyde (in molecule 12a) also led to a reduction in yield.
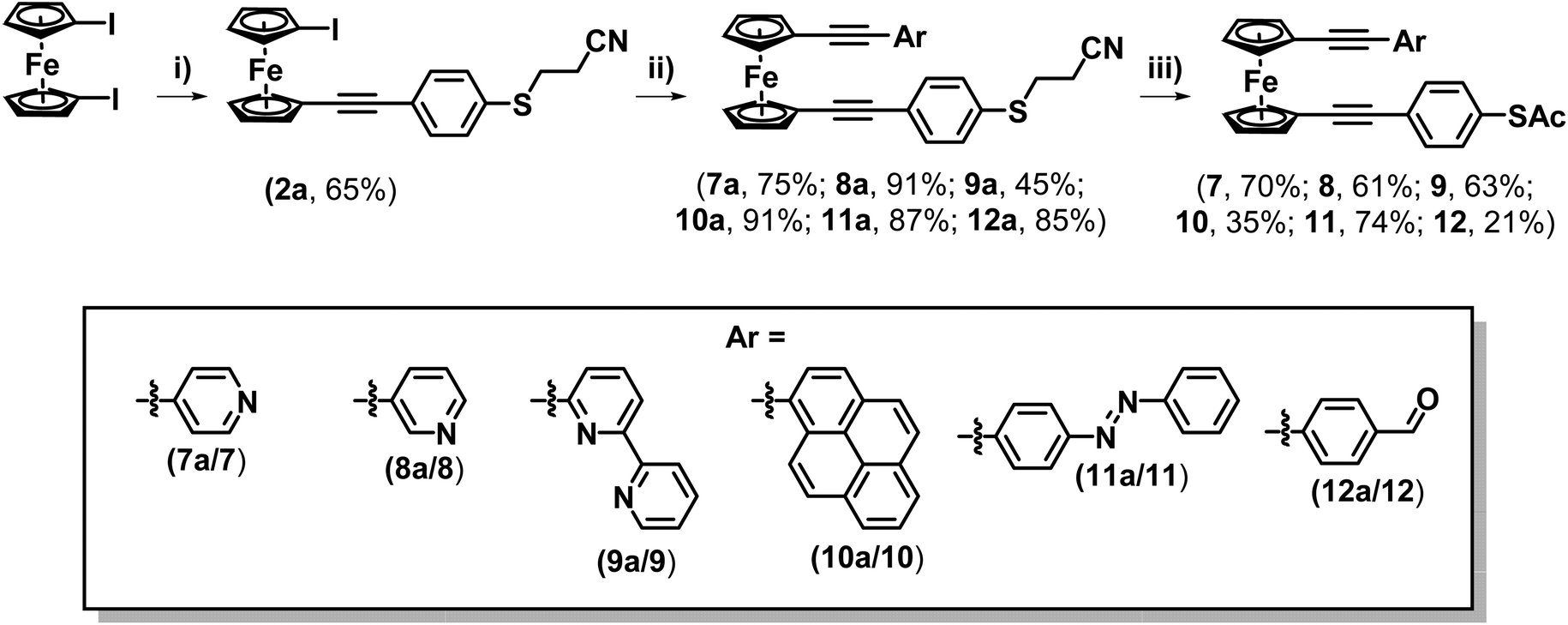 | ||
| Scheme 4 Synthetic route applied to form asymmetrically substituted ferrocene-(ethynylphenyl)thioacetates. Reaction conditions: (i) 4-(cyanoethylthio)ethynylbenzene, CuI, Pd(PtBu3)2, DIPA, THF, 50 °C, >16 h; (ii) terminal alkyne, CuI, Pd(PtBu3)2, DIPA, THF, 50 °C, 16 h; (iii) NaOMe (25% in MeOH), THF, 30 min, followed by Ac2O, 30 min. | ||
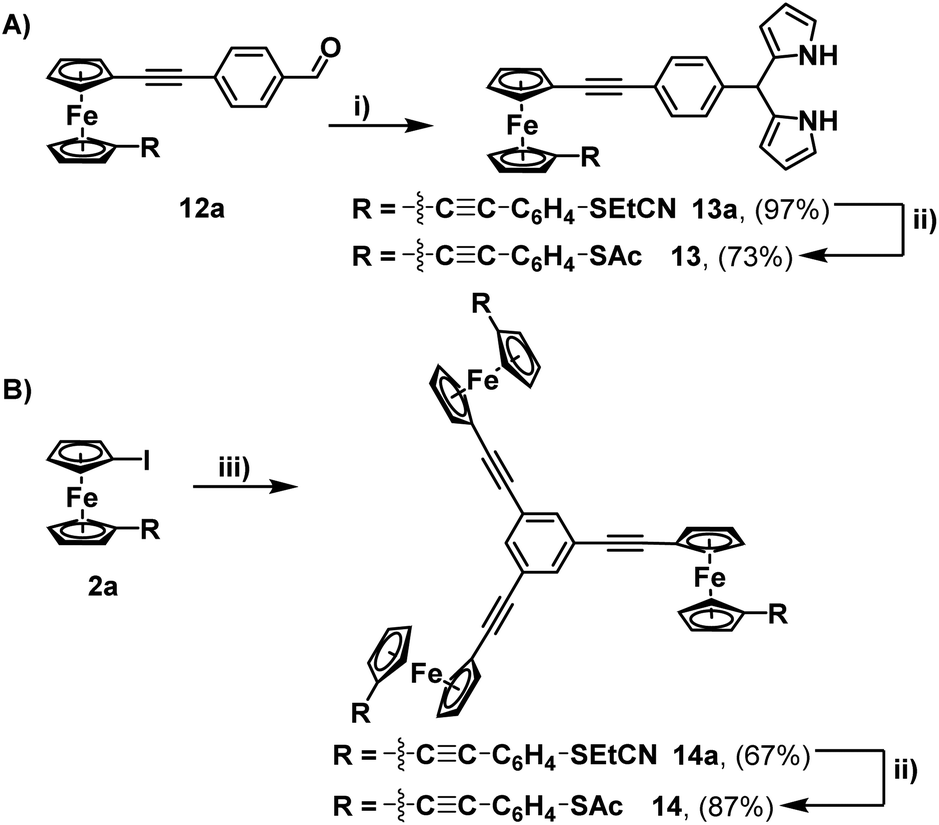
This aldehyde could, however, be trivially converted to a dipyrrole (13a), with no perturbation of the cyanoethyl group – hinting at the possibility for further modifications through non-basic reaction chemistry. To complete the synthesis, the feasibility of incorporating multiple units of 2a into a single molecule (beyond the simple covalently linked biferrocene 6a) was investigated. After a few attempts, it was found that dropwise addition of freshly prepared 1,3,5-triethynylbenzene to a solution of 2a allowed for the efficient isolation of a triferrocenyl-system (14a). Both 13a and 14a could then be converted to their thioacetate analogues (13 and 14) using the previously mentioned ‘trans-protection’ conditions (Scheme 5). All systems were fully characterised by 1H-NMR, 13C{1H}-NMR, high-resolution mass spectrometry and IR-spectroscopy demonstrating the expected results and providing strong evidence for the formation of the desired thioacetates. The 1H-NMR and IR analysis were particularly useful in judging the success of the reactions prior to purification, with both the cyanoethyl and thioacetate groups presenting distinct and characteristic peaks (Fig. 1).
 | ||
| Scheme 5 (A) Synthesis of 13 and (B) synthesis of 14. Reaction conditions: (i) pyrrole, TFA, RT, 4 hours; (ii) NaOMe (25% in MeOH), THF, 30 min followed by Ac2O 30 min; (iii) 1,3,5-triethynylbenzene, CuI, [Pd(CH3CN)2Cl2][PH(tBu3)2]BF4, DIPA, THF, RT, >16 h. | ||
 | ||
| Fig. 1 1H-NMR (CDCl3, 400 MHz, A) and IR (neat) (B) data of molecules 2a (red) and 2 (blue) that demonstrate indicative changes which occurred during the ‘trans-protection’ reactions. | ||
UV/Vis spectroscopy
UV/Vis spectroscopy allowed some initial conclusions on the electronic structure of these systems to be drawn. Previously reported analysis of unsubstituted ferrocene has shown that this displays a weak transition at approximately 442 nm, which is both Laporte and spin-forbidden.13 In these systems, introduction of the alkynyl ligands caused this peak to undergo a bathochromic shift to higher wavelengths (between 445–470 nm). Benchmarking this against previous literature results suggests that this is due to a reduction in the HOMO–LUMO gap, arising due to the electron withdrawing nature of the alkynyl substituents.14 However, within the family of thioacetates it becomes harder to draw clear conclusions on the relative electron-withdrawing nature of the substituents, due to the broadness of this peak.One general trend which can be identified from the data, however, is that moving from a cyanoethyl substituent to a thioacetate gives a consistent red-shift of the peak situated around 450 nm (with the only exception being the pair of molecules 8a/8). This may suggest a more pronounced electron-withdrawing effect within the thioacetate substituents. Trends in other ferrocene associated peaks are harder to quantify as these are masked by high absorptivity, most likely LMCT transitions, introduced by the alkynes, in the region of 230–350 nm. Despite this, however, it is clear that each ligand combination clearly presents its own distinct profile within this region (ESI, section 3†).
Cyclic voltammetry
Further electronic characterisation was performed using cyclic voltammetry. These experiments were conducted in dichloromethane solutions, with 0.1 M [nBu4][PF6] as the supporting electrolyte and are reported against an internal Fc/Fc+ standard, with relevant data reported in Table 1. All compounds displayed a single one-electron redox process, associated with an Fe2+/Fe3+ pair on the central ferrocene unit, except for compounds 6a/6, which showed two distinct one-electron processes (arising from separate Fe2+/Fe3+ pairs on the two ferrocenyl centres).15 All these processes were determined to be both chemically reversible, with ipa/ipc ≈ 1, and electrochemically reversible, with ΔE ≈ 59 mV. Scan-rate dependence experiments were also performed, by collecting voltammograms at several different scan-rates, demonstrating that all of these processes are also diffusion controlled, with ip ∝ (scan-rate)1/2, with the exception of 8a, which displayed a clear change in its redox profile over successive scans, possibly suggesting either molecular instability, or alternatively, deposition of the molecule onto the electrode when these specific measurement conditions were applied (see ESI section 2†).| Compound | E p/mV | E c/mV | E 1/2/mV | ΔE/mV | |ipa/ipc| |
|---|---|---|---|---|---|
| a This molecule showed significant instability over multiple scans, values reported are taken from a single scan at 100 mV s−1. All potentials are reported vs. the Fc/Fc+ redox pair. | |||||
| 2a | 245 | 178 | 211 | 66 | 1.03 |
| 2 | 252 | 186 | 219 | 66 | 0.98 |
| 5a | 194 | 125 | 160 | 69 | 0.97 |
| 5 | 218 | 144 | 181 | 74 | 1.04 |
| 6a | −11 | −79 | −45 | 68 | 0.99 |
| 338 | 269 | 303 | 69 | ||
| 6 | −2 | −60 | −31 | 58 | 1.01 |
| 350 | 292 | 321 | 58 | ||
| 7a | 234 | 169 | 206 | 75 | 1.01 |
| 7 | 258 | 186 | 221 | 72 | 0.91 |
| 8a | 223 | 148 | 185 | 75 | 0.98 |
| 8 | 229 | 166 | 197 | 63 | 1.00 |
| 9a | 229 | 152 | 190 | 77 | 0.97 |
| 9 | 233 | 163 | 198 | 70 | 0.98 |
| 10a | 167 | 101 | 133 | 66 | 0.97 |
| 10 | 176 | 117 | 146 | 59 | 0.97 |
| 11a | 199 | 132 | 165 | 67 | 0.99 |
| 11 | 208 | 143 | 175 | 65 | 0.96 |
| 12a | 227 | 148 | 187 | 78 | 0.98 |
| 12 | 233 | 160 | 196 | 73 | 0.97 |
| 13a | 180 | 117 | 149 | 63 | 0.95 |
| 13 | 196 | 133 | 165 | 64 | 1.05 |
| 14a | 207 | 135 | 171 | 72 | 1.02 |
| 14 | 223 | 158 | 190 | 65 | 0.92 |
All compounds displayed a redox potential (E1/2) which was positive when compared to ferrocene. Again, this can be attributed to the electron-withdrawing nature of the alkynyl substituents. This acts to extend the π-system of the ferrocene, leading to a decrease in the electron density at the redox-active iron-centre. Previously Bennett et al. reported a family of ferrocenes, terminated with various aryl-alkynes, which differed only in the position or type of heteroatom contained within their terminal aryl groups.16 It was found that these small differences exquisitely fine-tuned the redox-potential of the ferrocene unit, and this is further supported by the results reported here, with redox potential appearing to be directly correlated to the electron withdrawing or donating ability of the terminal aryl groups. This is evidenced by the fact that exchange of a cyanoethyl unit for a thioacetate causes a slight increase in the redox potential of each system, across the entire series of molecules (Fig. 2). This aligns well with simple-resonance analysis, with thioethers being better equipped to donate electrons to aromatic π-systems than thioacetates. Finally, though compound 14 includes three ferrocenyl centres, this only exhibits one redox process, indicating minimal communication between the ferrocene centres. This was expected, however, and has previously been observed in other multi-ferrocenyl compounds, bridged by a meta-substituted phenyl group.7b,17
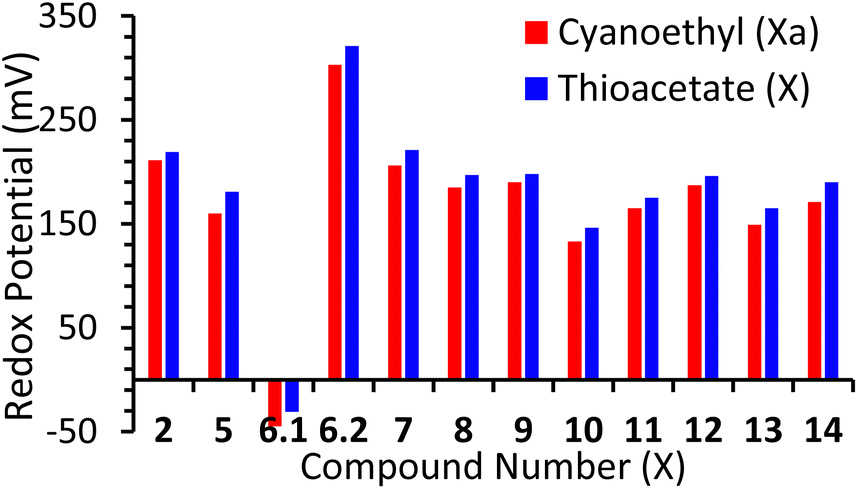 | ||
| Fig. 2 Redox potentials of all cyanoethyl/thioacetate compound's vs. Fc/Fc+. | ||
Conclusions
In summary, a convenient and robust synthetic route has been designed which allows for the facile fabrication of ferrocene alkyne compounds terminated with thioacetates. This strategy can tolerate a wide range of functional groups and can also be applied to synthesise multi-ferrocene systems, where each ferrocene has its own thioacetate substituent. Additionally, electronic characterisation of these systems demonstrated that the terminal substituents communicate well with their central ferrocene units. This work represents a critical step in the synthesis of conjugated ferrocene molecular wires with strong binding groups suitable for binding to widely used metallic electrodes. This work is amplified by recent works on asymmetric functionalisation of ferrocenes, optimisation of their cross-coupling efficiencies, and Butenschön's pioneering work on the synthesis of 1,1′-diaryl substituted ferrocenes.7,16,18Taken together, these works provide ripe opportunity for the design, synthesis and study of multitude ferrocene derivatives which may find use in a plethora of applications, extending far beyond the scope of simple conductance studies. The ability of these molecules to bind to metallic surfaces, and by extension, the potential for their use in the formation of self-assembled monolayers, could lead to their use as a component in the fabrication of electroactive inorganic–organic nanomaterials.19 These systems could also be used to fabricate multi-component systems using post-deposition modifications by taking advantage of the ability of the functional groups contained within these molecules (such as the nitrogen atoms present in molecules 7, 8, 9 and 13) to coordinate to other metals, with potential use in the formation of new electrochromic coatings.20
Finally, the rotational properties of these molecules are particularly interesting. Compound 14, for example, contains three ferrocene molecules, whose rotational flexibility could allow them to act as a hinge, conferring the possibility for this molecule to bind to a metal surface in a tripodal fashion. If realised, this unique geometry could potentially show interesting applications in the asymmetric transport of electricity and heat between different electrode materials.21 On-going work within the group is investigating whether this can be achieved within self-assembled monolayers.
Methodology
For full details of the experimental methodology, please refer to the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge funding from the EPSRC (studentship 2033057). N. J. L. is also grateful for a Royal Society Wolfson Research Merit Award.References
- (a) G. Wilkinson, M. Rosenblum, C. M. Whiting and R. B. Woodward, J. Am. Chem. Soc., 1952, 74, 2125 CrossRef CAS; (b) T. J. Kealy and P. L. Pauson, Nature, 1951, 168, 1039 CrossRef CAS; (c) K. Heinze and H. Lang, Organometallics, 2013, 32, 5623 CrossRef CAS; (d) D. Astruc, Eur. J. Inorg. Chem., 2017, 2017, 6 CrossRef CAS.
- (a) R. R. Gagne, C. A. Koval and G. C. Lisensky, Inorg. Chem., 1980, 19, 2854 CrossRef CAS; (b) N. Elgrishi, K. J. Rountree, B. D. McCarthy, E. S. Rountree, T. T. Eisenhart and J. L. Dempsey, J. Chem. Educ., 2018, 95, 197 CrossRef CAS.
- (a) A. C. Aragonès, N. Darwish, S. Ciampi, L. Jiang, R. Roesch, E. Ruiz, C. A. Nijhuis and I. Díez-Pérez, J. Am. Chem. Soc., 2019, 141, 240 CrossRef PubMed; (b) C. R. Peiris, Y. B. Vogel, A. P. Le Brun, A. C. Aragonès, M. L. Coote, I. Díez-Pérez, S. Ciampi and N. Darwish, J. Am. Chem. Soc., 2019, 141, 14788 CrossRef CAS PubMed; (c) Y. Yuan, J. F. Yan, D. Q. Lin, B. W. Mao and Y. F. Yuan, Chem. – Eur. J., 2018, 24, 3545 CrossRef CAS PubMed; (d) Y. Yang, J. Liu, H. Wen, J. Tian, J. Zheng, B. Schöllorn, C. Amatore, Z. Chen and Z. Tian, Nano Res., 2016, 9, 560 CrossRef CAS; (e) C. A. Nijuis, Z. Zhang, F. Adoah, C. Nickle, S. K. Karuppannan, L. Wang, L. Jiang, A. Tadich, B. Cowie, T. Salim, D. C. Qi, D. Thompson and E. Del Barco, Adv. Electron. Mater., 2023, 9, 2200637 CrossRef.
- (a) T. A. Su, M. Neupane, M. L. Steigerwald, L. Venkataraman and C. Nuckolls, Nat. Rev. Mater., 2016, 1, 1 Search PubMed; (b) M. L. Perrin, E. Burzurí and H. S. J. van der Zant, Chem. Soc. Rev., 2015, 44, 902 RSC; (c) S. J. Higgins and R. J. Nichols, Polyhedron, 2018, 140, 25 CrossRef CAS; (d) K. Wang, E. Meyhofer and P. Reddy, Adv. Funct. Mater., 2020, 30, 1904534 CrossRef CAS.
- (a) C. Engtrakul and L. R. Sita, Organometallics, 2008, 27, 927 CrossRef CAS; (b) S. A. Getty, C. Engtrakul, L. Wang, R. Liu, S. H. Ke, H. U. Baranger, W. Yang, M. S. Fuhrer and L. R. Sita, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 71, 1 CrossRef; (c) C. Engtrakul and L. R. Sita, Nano Lett., 2001, 1, 541 CrossRef CAS; (d) Q. Lu, C. Yao, X. Wang and F. Wang, J. Phys. Chem. C, 2012, 116, 17853 CrossRef CAS; (e) L. J. O'Driscoll and M. R. Bryce, Nanoscale, 2021, 13, 10668 RSC; (f) D. C. Milan, A. Vezzoli, I. J. Planje and P. J. Low, Dalton Trans., 2018, 47, 14125 RSC.
- (a) L. Q. Pei, J. R. Horley, J. W. Seng, X. Liu, Y. Q. Yeoh, M. X. Yu, X. H. Wu, A. D. Abell, J. F. Zheng, X. S. Zhou, J. Yu and S. Jin, ACS Appl. Mater. Interfaces, 2021, 13, 57646 CrossRef CAS PubMed; (b) L. A. Wilkinson, T. L. R. Bennett, I. M. Grace, J. Hamill, X. Wang, S. Au-Yong, A. Ismael, S. P. Jarvis, S. Hou, T. Albrecht, L. F. Cohen, C. Lambert, B. J. Robinson and N. J. Long, Chem. Sci., 2022, 13, 8380 RSC; (c) M. Camarasa-Gómez, D. Hernangómez-Pérez, M. S. Inkpen, G. Lovat, E. D. Fung, X. Roy, L. Venkataraman and F. Evers, Nano Lett., 2020, 20, 6381 CrossRef PubMed.
- (a) M. S. Inkpen, T. Albrecht and N. J. Long, Organometallics, 2013, 32, 6053 CrossRef CAS; (b) M. S. Inkpen, A. J. P. White, T. Albrecht and N. J. Long, Dalton Trans., 2014, 43, 15287 RSC.
- M. Vollmann and H. Butenschön, C. R. Chim., 2005, 8, 1282 CrossRef CAS.
- W. W. Zhang, M. Kondo, T. Fujita, K. Namiki, M. Murata and H. Nishihara, Molecules, 2010, 15, 150 CrossRef CAS PubMed.
- C. Nájera, L. K. Sydnes and M. Yus, Chem. Rev., 2019, 119, 11110 CrossRef PubMed.
- J. Ma, M. Vollmann, H. Menzel, S. Pohle and H. Butenschön, J. Inorg. Organomet. Polym. Mater., 2008, 18, 41 CrossRef CAS.
- C. Wang, M. R. Bryce, J. Gigon, G. J. Ashwell, I. Grace and C. J. Lambert, J. Org. Chem., 2008, 73, 4810 CrossRef CAS PubMed.
- Y. Yamaguchi, W. Ding, C. T. Sanderson, M. L. Borden, M. J. Morgan and C. Kutal, Coord. Chem. Rev., 2007, 251, 515 CrossRef CAS.
- (a) N. J. Long, J. A. Martin, R. Vilar, A. J. P. White, D. J. Williams and M. Younus, Organometallics, 1999, 18, 4261 CrossRef CAS; (b) A. Vacher, F. Barrière and D. Lorcy, Organometallics, 2013, 32, 6130 CrossRef CAS; (c) S. Bitter, M. Kunkel, L. Burkart, A. Mang, R. F. Winter and S. Polarz, ACS Omega, 2018, 3, 8854 CrossRef CAS PubMed.
- (a) M. J. Powers and T. J. Meyer, J. Am. Chem. Soc., 1977, 100, 4393 CrossRef; (b) D. Gottwald, Q. Yuan, M. Speck, J. Mahrholdt, M. Korb, K. Schreiter, S. Spange and H. Lang, J. Organomet. Chem., 2020, 923, 121447 CrossRef CAS; (c) S. A. Sheppard, T. L. R. Bennett and N. J. Long, Eur. J. Inorg. Chem., 2022, 2022, e202200055 CrossRef CAS.
- T. L. R. Bennett, L. A. Wilkinson, J. M. A. Lok, R. C. P. O'Toole and N. J. Long, Organometallics, 2021, 40, 1156 CrossRef CAS.
- (a) A. K. Diallo, C. Absalon, J. Ruiz and D. Astruc, J. Am. Chem. Soc., 2011, 133, 629 CrossRef CAS PubMed; (b) N. J. Long, A. J. Martin, A. J. P. White, D. J. Williams, M. Fontani, F. Laschi and P. Zanello, J. Chem. Soc., Dalton Trans., 2000, 19, 3387 RSC.
- (a) S. F. Schmiel and H. Butenschön, Eur. J. Org. Chem., 2021, 2388 CrossRef CAS; (b) I. Baumgardt and H. Butenschön, Eur. J. Org. Chem., 2010, 1076 CrossRef CAS.
- (a) Z. Ren, J. Yang, D. Qi, P. Sonar, L. Liu, Z. Lou, G. Shen and Z. Wei, Adv. Mater. Technol., 2021, 6, 2000889 CrossRef CAS; (b) C. Müller, L. Ouyang, A. Lund, K. Moth-Poulsen and M. M. Hamedi, Adv. Mater., 2021, 31, 1807286 CrossRef PubMed; (c) S. Y. Kim, S. J. Cho, S. E. Byeo, X. He and H. J. Yoon, Adv. Energy Mater., 2020, 10, 2002606 CrossRef CAS.
- (a) S. Shankar, M. Lahav and M. E. van der Boom, J. Am. Chem. Soc., 2015, 137, 4050 CrossRef CAS PubMed; (b) N. O. Laschuk, I. I. Ebralidze, E. B. Easton and O. V. Zenkina, ACS Appl. Mater. Interfaces, 2021, 13, 39573 CrossRef CAS PubMed.
- T. L. R. Bennett, M. Alshammari, S. Au-Yong, A. Almutlg, X. Wang, L. A. Wilkinson, T. Albrecht, S. P. Jarvis, L. F. Cohen, A. Ismael, C. J. Lambert, B. J. Robinson and N. J. Long, Chem. Sci., 2022, 13, 5176 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3dt02954a |
| This journal is © The Royal Society of Chemistry 2023 |