
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRed-shifted activity-based sensors for ethylene via direct conjugation of fluorophore to metal–carbene†
Nicholas J.
Dacon
,
Nathan B.
Wu
and
Brian W.
Michel
 *
*
Department of Chemistry and Biochemistry, University of Denver, Denver, CO 80210, USA. E-mail: Brian.Michel@du.edu
First published on 25th September 2023
Abstract
A number of Activity-Based Sensors (ABS) for relatively unreactive small molecules, such as ethylene, necessitates a transition metal for reaction under ambient conditions. Olefin metathesis has emerged as one of the primary strategies to achieve ethylene detection, and other transition metals are used for similarly challenging-to-detect analytes. However, limited studies exist investigating how fluorophore-metal attachment impacts photophysical properties of such ABS. Two new probes were prepared with the chelating benzlidene Ru–ligand directly conjugated to a BODIPY fluorophore and the photophysical properties of the new conjugated ABS were evaluated.
Introduction
The simplest alkene ethylene (IUPAC name ethene) has a range of important and interesting roles in biological systems. Ethylene is an important plant hormone well-known for its role in the ripening of climacteric fruit, but it also has broad signalling responsibilities affecting the expression of a range of genes.1–6 Additionally, ethylene is produced in mammalian systems. Peroxidation of unsaturated fatty acids and subsequent radical fragmentation pathways produce ethylene,7 which can be detected in exhaled breath, making it an intriguing biomarker of oxidative stress.8Molecular detection of ethylene has largely focused on roles in plant systems inspiring several elegant strategies due to the lack of main group reactivity under mild conditions.9,10 To detect this simple alkene, transition metals have emerged as an obvious choice owing to the wealth of coordination chemistry and reactivity accessible under ambient conditions due to additional bonding interactions such as pi-back bonding.11 Approaches reported for molecular ethylene detection can be broadly categorized as coordination-based sensors12–16 or activity-based sensors (ABS)17–24 with the latter emerging as an attractive approach for selective ethylene detection in complex environments.9,25,26 In 2018, our group reported a fluorescent tagged Hoveyda–Grubbs-type complex that provided sensitive and selective detection of ethylene (Fig. 1(A)).18 Since, additional reports have demonstrated olefin metathesis as a viable strategy for ethylene detection along with a recent report using migratory insertion into a cyclorhodinated complex.23 Beyond ethylene detection, transition metal-based ethylene probes represent examples of transition-metal based ABS, which are gaining traction for the detection of small molecule analytes that lack reactive functionality.27–35
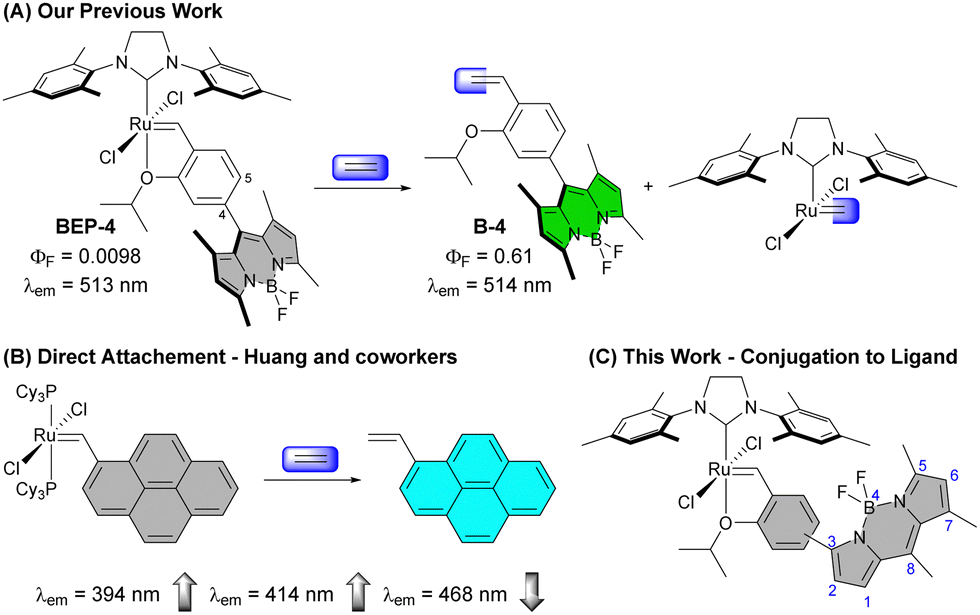 | ||
| Fig. 1 (A) Previously reported probes BEP-4 and BEP-5 (not shown) demonstrated a large (∼113-fold) increase in luminescence to ethylene with minimal shift in the wavelength of emission. (B) The Grubbs 1st generation Ru complex derived from 1-vinylpyrene was reported by Huang and coworkers with 20-fold increase in luminescence. (C) The work described here evaluates modified BODIPY ethylene probes with the chelating benzylidene ligand conjugated to the extended fluorophore π system – Con-BEPs. | ||
To improve sensitivity for ethylene, it is desirable to increase signal response to ethylene while minimizing background signal. One effective approach has been to “protect” the ruthenium complex in hydrophobic binding pockets of proteins such as albumin, which is an intriguing strategy with a range of potential applications.20,22,36 Alternatively, we have been interested in using ligand modulation37 about the reactive ruthenium species to improve signal and reactivity. More broadly transition metal-based ABS are often quenched prior to analyte detection by the presence of the metal. Therefore, it is important to understand how different means of connecting the reactive ligand to the fluorophore impact the optical properties of the ABS.
A Grubbs 1st generation-based probe reported by Huang and co-workers has the Ru directly bound to a fluorescent pyrene core (Fig. 1(B)).19 This probe shows an interesting modulation of emission peaks upon reaction with ethylene with a decrease in intensity of a small peak at 468 nm and increase in the peaks at 394 and 414 nm. Other Ru- and Rh-based ethylene probes reported to date show only the expected increase in emission intensity upon reaction with ethylene.
Results and discussion
Synthesis of Con-BEP-4 and Con-BEP-5
The substitution pattern on the BODIPY of BEP-4, with methyl groups at the 1 and 7 positions, restrict rotation of aryl adjacent π systems. In the context of BEP-4 (or BEP-5, not shown) this suggests that quenching occurs via a through space process (i.e. PET or FRET). We hypothesized that conjugation of the chelating ligand to the fluorophore would result in a red-shifted probe with potentially interesting modulation of the optical properties and instruct future design of transition metal containing ABS. Herein we report the synthesis and characterization of two new conjugated BEPs (Con-BEPs), one of which displays large turn-on coupled with a hypsochromic shift of the emission peak relative to the ethylene adduct.We initially sought to prepare two complexes with the chelating isopropoxy styrene at the 3-position of the BODIPY analogous to BEP-4 and BEP-5. To achieve this, we envisioned a convergent route (Scheme 1) coupling the known asymmetric chloro-BODIPY 138 and the pinacol boronate esters 2 and 3, which are readily prepared from the commercially available bromo salicyladehydes. Pd-mediated cross coupling provided the ligands Con-B-4 and Con-B-5 in unoptimized 31% and 16% yields, respectively, providing sufficient material for preparing the desired Ru-complexes. Con-BEP-4 and Con-BEP-5 were prepared using the indenylidene monopyridine complex (M310) and Amberlyst-15 resin.39,40 In our hands, we have found this to be the most reliable method for preparing high purity complexes. This procedure is touted for operational simplicity and not requiring column chromatography; however, the presence of even a small amount highly fluorescent impurity results in a higher baseline signal and reduced turn-on for ethylene detection. As a result, we have found synthesis and chromatographic purification of the final complex in a nitrogen filled glove box beneficial. This provides isolated complex with the largest observed turn-on, although batch to batch variability was still observed. It should be noted that these complexes are bench stable in the solid state after purification.
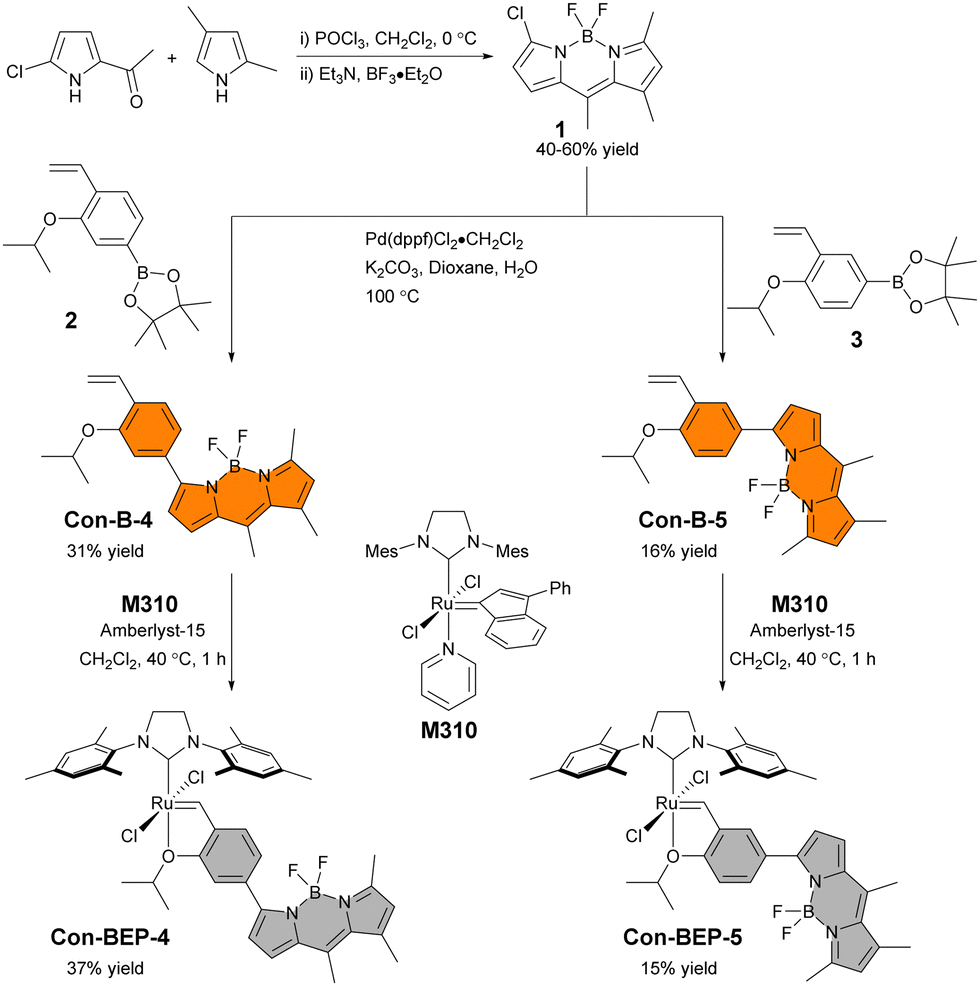 | ||
| Scheme 1 Synthetic routes to Con-BEP-4 and Con-BEP-5. | ||
Photophysical characterization and structure
The optical properties of the quenched Con-BEPs were investigated along with their respective precursors (Fig. 2(A)). Both Ru complexes, Con-BEP-5 and Con-BEP-4, were efficiently quenched and the corresponding ethylene adducts, Con-B-5 and Con-B-4 are strongly emissive. As expected, all four compounds were red shifted as compared to BEP-4 and the further separation from the primary absorption of HG2 supports an electron transfer quenching process over an energy transfer (Fig. 2(B)).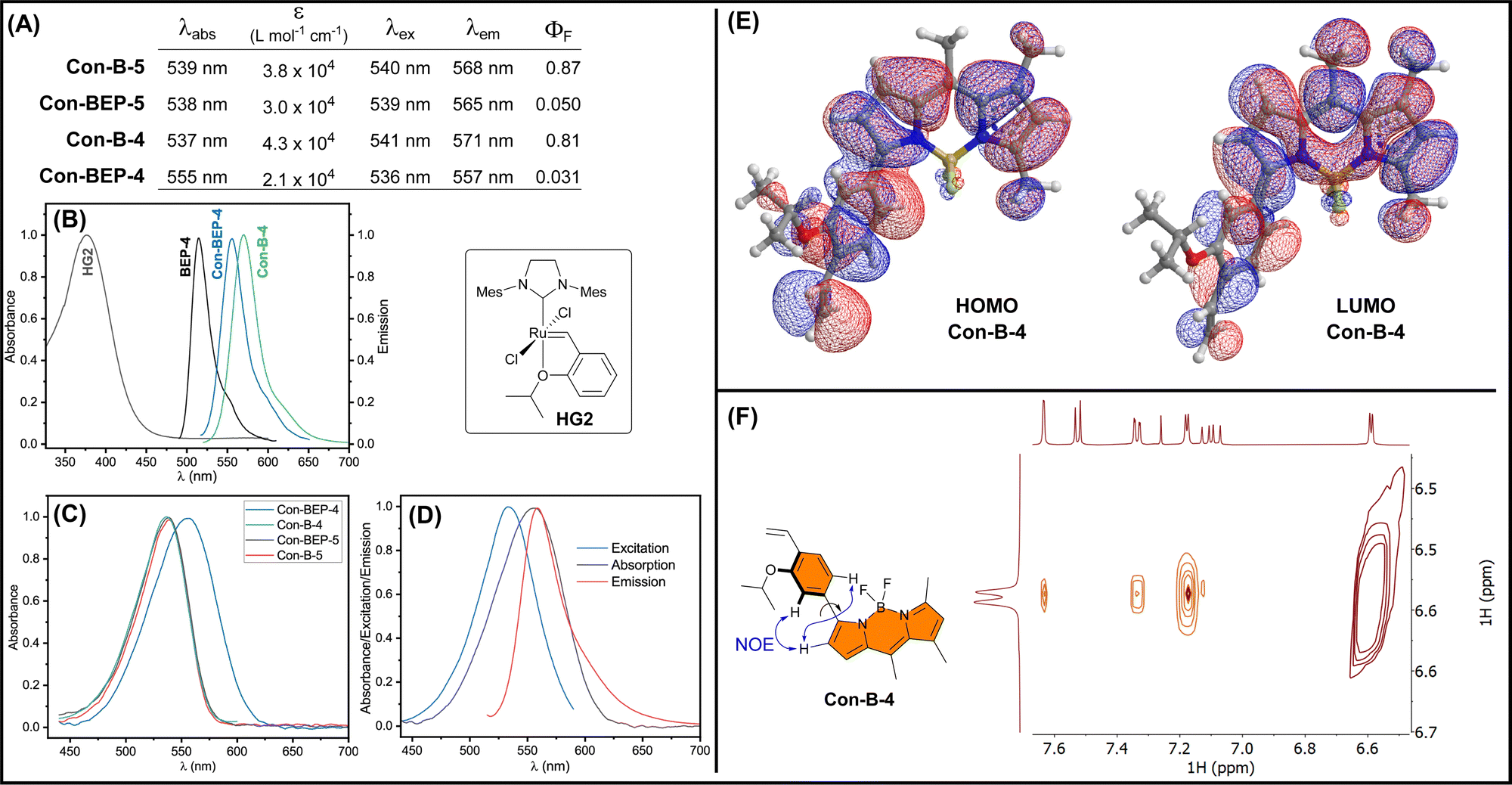 | ||
| Fig. 2 (A) Summary of the photophysical properties of Con-BEP-5 and Con-BEP-4 along with the ethylene adducts measured in PhMe at RT. Molar absorptivity (ε) measured at the respective λabs maxima. (B) Comparison of the absorption spectrum of HG2 (gray) with the emission spectra for BEP-4 (black), Con-BEP-4 (blue), and Con-B-4 (green) showing minimal spectral overlap. (C) Absorption spectra for the Con-BEPs and ethylene adducts: Con-B-4/5. (D) Excitation, absorption, and emission spectra for Con-BEP-4. (E) Calculated molecular orbitals for Con-B-4. (F) 2D NOESY spectrum for Con-B-4. | ||
Con-BEP-4 displays some additional noteworthy photophysical features including a hypsochromic shift of the emission peak relative to Con-B-4, which is potentially valuable effect that presumably arises from the positioning of the Ru para to the BODIPY. Further, comparison of the absorbance spectrum showed a surprising broadening and bathochromic shift for Con-BEP-4, which is not observed for the other three compounds (Fig. 2(C)). This puts the λabs almost on top of the λem, yet the excitation spectrum does not overlay the absorption (Fig. 2(D)). This interesting series of observations may explain the more quenched fluorescence observed for Con-BEP-4 and we suspect it is related to non-emissive absorption arising from extended conjugation to the benzylidene carbon.
Although the red-shifted emission supports extended conjugation of the fluorophore pi-system, we sought additional information on the structural implications of incorporating the aryl unit at the 3-position of the BODIPY. DFT calculations were performed b3lyp/6-311+G(d), which identified an energy minimized structure with a dihedral angle of 38° between the BODIPY and aryl units (Fig. 2(E)). Forcing a more planar structure with a dihedral angle of 8° results in a 2 kcal mol−1 penalty (see ESI†). In both conformations the HOMO (and HOMO−1) for both structures extends across the BODIPY and aryl moieties. To gain insight into the solution structure, a NOESY spectrum of Con-B-4 was acquired (Fig. 2(F)). Notably, through space correlations were observed between the proximal pyrrole H and both ortho H's of the arene unit suggesting a range of conformations are readily accessible about the BODIPY-aryl bond.
Response to ethylene
Next, the fluorescence response of Con-BEP-5 and Con-BEP-4 to ethylene was evaluated (Fig. 3(A)). Con-BEP-5 showed an ∼15-fold increase in integrated emission over 60 min and an endpoint turn-on of 22-fold (Fig. S14, ESI†). Notably Con-BEP-4 had a much larger increase in fluorescence upon exposure to excess ethylene along with an ∼14 nm bathochromic shift of λem (Fig. 3(B)). Integrating the emission peak showed up to a 123-fold turn-on in 60 min and an endpoint turn-on of 160-fold (Fig. 3(C)). However, by taking advantage of the shift in the emission peak and monitoring turn-on at the wavelengths of maximal difference (∼580–581 nm), a 179-fold turn-on was observed in 60 min with an endpoint turn-on of 235-fold!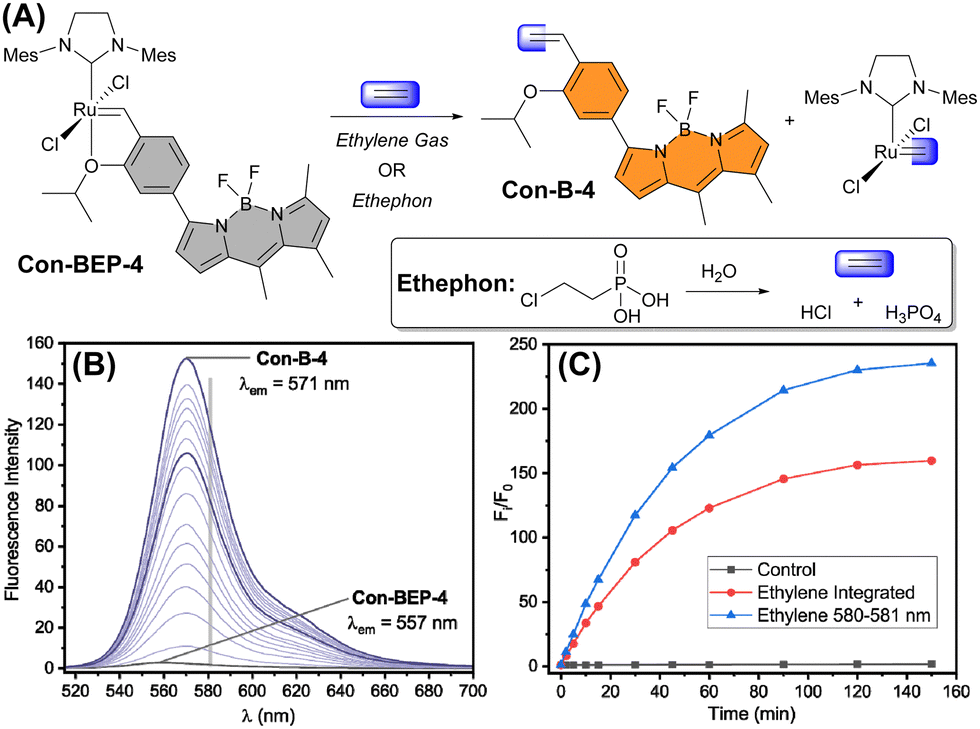 | ||
| Fig. 3 (A) Reaction of Con-BEP-4 with ethylene gas or ethylene from ethephon hydrolysis. (B) Emission spectra of Con-BEP-4 exposed to ethylene. Timepoints represented are t = 0, 5, 10, 15, 20, 25, 30, 40, 50, 60, 75, 90, 105, 120, 150, and 180 min. Gray bar at 581 nm represents wavelength with maximum increase in emission intensity. (C) Normalized emission intensity of Con-BEP-4 control sample or treated with ethylene (integrated emission and 580–581 nm). | ||
The kinetic profiles for both probes were measured by adding probe to a saturated solution of ethylene in toluene at 25 °C. The increase in fluorescence intensity was monitored under these pseudo-first-order conditions as a measure of product formation (see ESI† for full details). The observed rate constants (kobs) for Con-BEP-5 (kobs = 4.1 × 10−4 ± 0.3 s−1) and Con-BEP-4 (kobs = 3.1 × 10−4 ± 0.3 s−1) were of similar magnitude, which is not surprising given the minimal structural changes to the coordination environment about Ru.
Since direct application of ethylene gas is often impractical in agricultural settings, ethephon (2-chlorophosphonic acid) is used widely to expose plants to a controlled release of ethylene. Due to limited aqueous solubility, similar challenges are presented in accurately dosing ethylene in laboratory settings such as the cell studies described below. Therefore, we sought to demonstrate that ethylene released from slow hydrolysis of ethephon could be detected. Con-BEP-4 was converted to Con-B-4 under biphasic conditions (PBS/PhMe), which was readily assessed by NMR (Fig. S18, ESI†). Specifically, the observation of the terminal alkene hydrogens from Con-B-4 can only be observed from metathesis and not from probe decomposition. In a similar experiment, conversion of Con-BEP-4 to Con-B-4 was achieved in the presence of cell growth media (Fig. S19, ESI†).
Cell studies
Inspired by these intriguing findings Con-BEP-4 was evaluated for detection of ethylene in live A549 cells. Cells were first treated with varying amounts the ethylene releasing molecule, ethephon, followed by washing, and treating with Con-BEP-4. Compared to BEP-4, the red-shifted λex allows for flexible image acquisition parameters. Images were acquired with two different laser sources and observation windows. Exciting with a 561 nm laser and observing from 578–602 nm showed good turn-on and a dose dependent response to increased concentrations of ethylene (via ethephon hydrolysis, Fig. 4(A)–(C) and (G)). Additional images acquired independently with a 488 nm laser source and observing over a wider range of wavelengths, 539–601 nm, showed a larger mean fluorescence response (Fig. 4(D)–(G)). It is possible that the larger response under the later imaging conditions arises from the larger observation window or excitation efficiencies at different wavelengths. However, the later explanation seems unlikely when comparing the laser lines overlayed on the respective excitation spectra of Con-BEP-4 and Con-B-4 (Fig. 4(H)). Given that an intermediate laser closer to the excitation maxima was not available, these results highlight the utility of Con-BEP-4, and similar fluorophores, for varied experimental and microscope setups.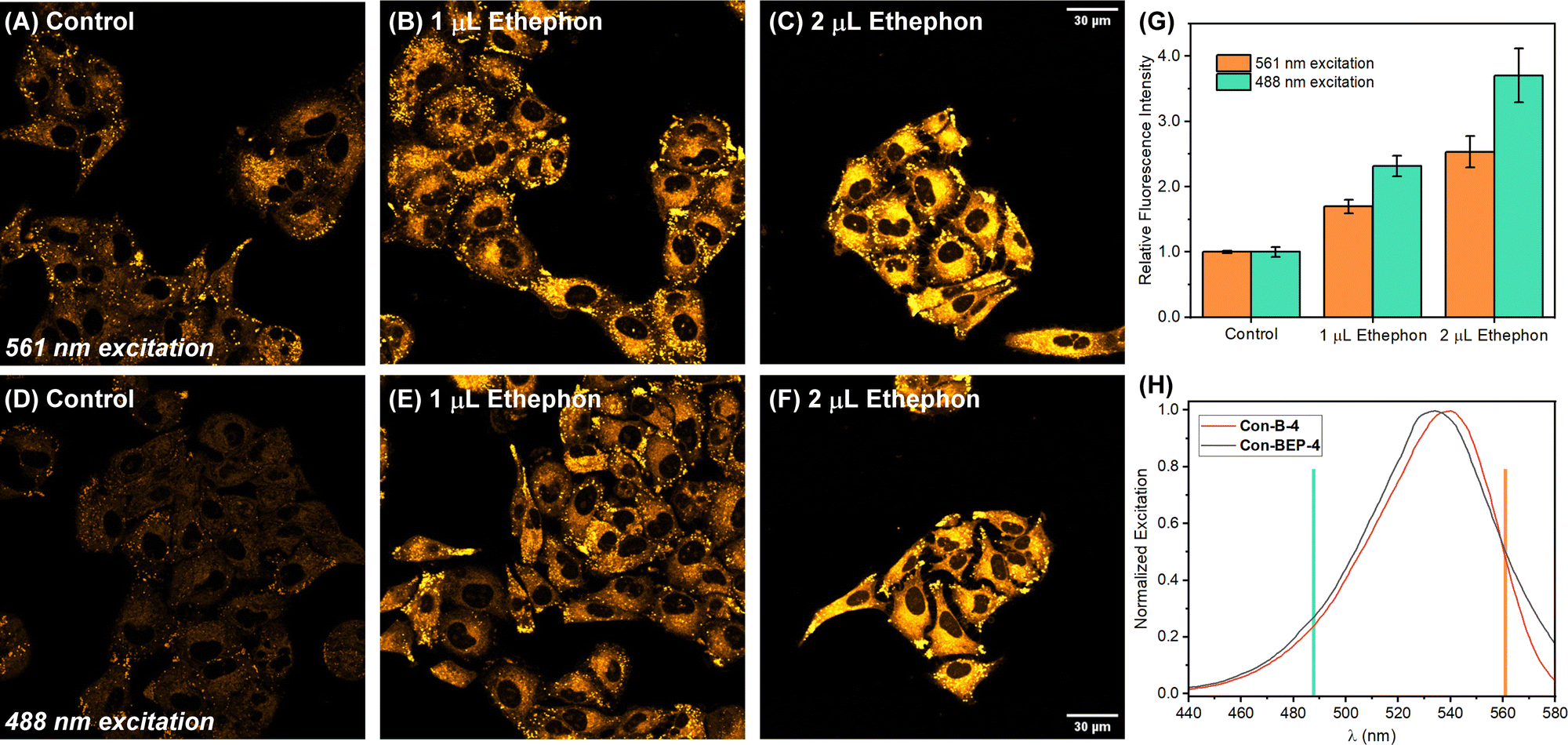 | ||
| Fig. 4 Representative confocal microscopy images of A549 cells treated with varying amounts of ethephon followed by Con-BEP-4. (A)–(F) Cells were pre-treated with 0, 1, or 2 μL of ethephon (40% solution) in 400 μL DMEM for 2 h at 37 °C. Cells were washed with PBS and stained with 1 μM Con-BEP-4 in PBS (0.5% DMSO) for 1 h at 37 °C. Images acquired exciting with a (A)–(C) 561 nm laser or a (D)–(F) 488 nm laser. (H) Mean fluorescence intensity measured across the entire field. Error bars denote ±SD. Statistical analysis was performed using a two-tailed test and all measurements are statistically significant with respect to the control, varied ethephon concentrations, and excitation with different lasers (p < 0.01). (I) Excitation scans of Con-BEP-4 and the ethylene adduct Con-B-4 with the respective laser lines indicated. | ||
Finally, the potential for cytotoxicity was evaluated. Cells were exposed to varied concentrations of Con-BEP-4, Con-B-4, Con-BEP-5, and Con-B-5 under analogues conditions to those used for live-cell imaging. Cell viability was assessed by an MTT assay and no significant toxicity was observed for the concentrations evaluated (Fig. S21, ESI†).
Conclusions
The direct conjugation of the Hoveyda–Grubbs-type chelating ligand to BODIPY fluorophores at the 3-position results in the expected, red-shifted ethylene probes that may be useful for cellular imaging studies, particularly in mammalian systems. Both Con-BEP-4 and Con-BEP-5 are efficiently quenched, despite poor overlap with the absorption peak of HG2. This suggests an electron transfer mechanism as the primary means of quenching and not an energy transfer process. The positioning of the Ru-benzylidene para to the BODIPY in Con-BEP-4 resulted in an interesting hypsochromic shift of ∼14 nm relative to ethylene adduct Con-B-4 and this effect is not observed for this isomeric Con-BEP-5. This shift coupled with the large increase in fluorescence upon reaction with ethylene allows for increased observed turn-on by judicious selection of excitation and observed emission wavelengths. This observation has potential implications in the design of future ethylene probes and more broadly in other transition metal-based ABS. Additionally, imaging studies in live cells demonstrate dose dependent ethylene detection and flexibility in choice of excitation sources treated with varying amounts of ethephon followed by Con-BEP-4.Materials and methods
Synthesis of Con-BEP-4 and Con-BEP-5
All reactions utilizing air- or moisture-sensitive reagents were performed in dried glassware under an atmosphere of dry N2. When dry solvent was used the solvent was passed over activated alumina. Other reagents were purchased and used without further purification unless otherwise stated. Silica gel (RediSep Rf gold) was used for column chromatography. Grubbs second generation catalyst, ethylene gas, and all other reagents were purchased from commercial sources and used as received. 1H and 13C NMR spectra for characterization of new compounds were collected in CDCl3 at 25 °C at the NMR Facility at the University of Denver using a Bruker Avance III 500 MHz instrument. All chemical shifts are reported in parts per million and referenced to the residual solvent peak from CDCl3 (δ = 7.26 ppm, 1H; 77.16 ppm, 13C). Splitting patterns are indicated as follows: s, singlet; d, doublet; t, triplet; dd, doublet of doublets. Glovebox experiments were conducted in a nitrogen-filled glovebox at less than 10 ppm O2 level and 1 ppm H2O. High-Resolution Mass spectrometry data were obtained using a Shimadzu LCMS-9030 Quadrupole iRef TOF in the indicated ionization mode (ESI or APCI).![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) solution was added to the flask via syringe and the solution was degassed for 20 minutes. The flask was added to a 100 °C pre-heated oil bath and stirred for 4.5 hours until completion. The solution was extracted with ethyl acetate (3 × 50 mL) and washed with water and brine (3 × 100 mL), dried with sodium sulfate, and concentrated. Purification by flash chromatography with hexanes/CH2Cl2 gradient of 40% → 70% to yield Con-B-4 as an orange solid (61.9 mg, 0.157 mmol, 31%). 1H NMR (500 MHz, CDCl3): δ 7.61 (s, 1H), 7.52 (d, J = 8.2 Hz, 1H), 7.33 (d, J = 8.0 Hz, 1H), 7.19 (d, J = 4.2 Hz, 1H), 7.09 (dd, J = 17.8, 11.2 Hz, 1H), 6.59 (d, J = 4.2 Hz, 1H), 6.14 (s, 1H), 5.80 (d, J = 17.8 Hz, 1H), 5.27 (d, J = 11.2 Hz, 1H), 4.68 (sept, J = 6.2 Hz, 1H), 2.59 (s, 3H), 2.53 (s, 3H), 2.43 (s, 3H), 1.40 (d, J = 6.0 Hz, 6H). 13C NMR (126 MHz, CDCl3): δ 158.6, 154.9, 154.4, 143.8, 140.7, 136.3, 133.5, 131.9, 128.1, 126.2, 125.1, 122.7, 121.4 (t, JCF = 2.7 Hz), 118.07, 118.05, 114.9 (t, JCF = 6.1 Hz), 114.6, 70.8, 22.3, 16.9, 16.5, 15.0. HRMS (APCI+) m/z: calculated for C23H26BF2N2O (M + H+) 395.21008; found 395.20529, C23H25BF2N2ONa (M + Na+) 417.19202; found 417.18717.
:1 → 2:1 → 1:1) to afford Con-BEP-4 as purple solid (22.5 mg, 26.2 mmol) in 37% yield. 1H NMR (500 MHz, CDCl3) δ 16.52 (s, 1H), 7.57 (s, 1H), 7.29 (d, J = 7.9 Hz, 1H), 7.08 (d, J = 11.8 Hz, 6H), 6.99–6.94 (m, 1H), 6.56 (d, J = 4.3 Hz, 1H), 6.14 (s, 1H), 4.99 (m, 1H), 4.18 (s, 4H), 2.56–2.36 (m, 27H), 1.32 (d, J = 6.0 Hz, 6H). 13C NMR (126 MHz, CDCl3) δ 294.3 (from HSQC), 211.7, 159.6, 153.9, 152.1, 145.0, 144.3, 140.5, 139.0, 136.7, 133.9, 129.5, 129.2, 128.4, 125.5, 124.9, 123.8, 123.3, 122.0, 117.9, 114.2, 75.6, 66.0, 37.3, 32.1, 29.9, 22.9, 21.2, 17.0, 16.5, 15.4, 15.2, 14.3. HRMS (APCI+) m/z: calculated for C43H49BF2N4ORu (M-Cl2 + H+) 789.30837; found 789.30242.
:water (5:1) solution was added to the flask via syringe and the solution was degassed for 20 minutes. The flask was added to a 100 °C pre-heated oil bath and stirred for 4.5 hours until completion. The solution was extracted with ethyl acetate (3 × 50 mL) and washed with water and brine (3 × 100 mL), dried with sodium sulfate and concentrated under reduced pressure to yield an orange solid. Purification by column chromatography with hexanes/CH2Cl2 gradient of 40% → 70% to yield Con-B-5 as an orange solid (34.1 mg, 86.5 μmol, 16%) 1H NMR (500 MHz, CDCl3) δ 7.98 (d, J = 2.4 Hz, 1H), 7.84 (dd, J = 8.7, 2.4 Hz, 1H), 7.19 (d, J = 4.3 Hz, 1H), 7.09 (dd, J = 17.7, 11.1 Hz, 1H), 6.95 (d, J = 8.7 Hz, 1H), 6.56 (d, J = 4.1 Hz, 1H), 6.12 (s, 1H), 5.83 (dd, J = 17.8, 1.6 Hz, 1H), 5.27 (dd, J = 11.1, 1.5Hz, 1H), 4.63 (sept, J = 6.1 Hz, 1H), 2.56 (s, 3H), 2.54 (s, 3H), 2.41 (s, 3H), 1.39 (d, J = 6.1 Hz, 6H). 13C NMR (126 MHz, CDCl3) δ 157.6, 156.1, 155.1, 143.1, 140.3, 136.2, 133.2, 131.9, 129.9 (t, JCF = 5.0 Hz), 128.0 (t, JCF = 3.4 Hz), 127.3, 125.5, 125.4, 122.4, 118.1, 114.6, 113.0, 70.7, 29.8, 22.4, 16.8, 16.5, 15.0. HRMS (APCI+) m/z: calculated for C23H26BF2N2O (M + H+) 395.21008; found 395.20524, C23H25BF2N2ONa (M + Na+) 417.19202; found 417.18711.
:1 → 2:1 → 1:1) to afford Con-BEP-5 as a red solid (8.9 mg, 0.0104 mmol) in 15% yield. 1H NMR (500 MHz, CDCl3) δ 16.50 (s, 1H), 8.22 (dd, J = 8.6, 2.3 Hz, 1H), 7.34 (d, J = 2.3 Hz, 1H), 7.19 (d, J = 4.0 Hz, 1H), 7.07 (s, 4H), 6.88 (d, J = 8.8 Hz, 1H), 6.43 (d, J = 4.5 Hz, 1H), 6.13 (s, 1H), 4.93 (sept, J = 6.2 Hz, 1H), 4.18 (s, 4H), 2.59 (s, 3H), 2.53 (s, 3H), 2.48 (s, 12H), 2.44 (s, 3H), 2.38 (s, 6H), 1.30 (d, J = 6.1 Hz, 6H). 13C NMR (126 MHz, CDCl3) δ 296.8 (from HSQC), 211.6, 158.3, 153.5, 153.0, 145.1, 143.5, 140.5, 139.1, 136.2, 133.4, 130.8, 129.5, 129.2, 128.4, 127.6, 125.3, 123.8, 122.6, 118.1, 112.8, 75.7, 51.6, 37.2, 32.1, 29.8, 29.5, 22.8, 21.3, 21.2, 16.9, 16.5, 14.3. HRMS (APCI+) m/z: calculated for C43H50BF2N4ORu (M + H+) 789.30837; found 789.30209.
:1) solution was added to the flask via syringe and the solution was degassed for 20 minutes. The flask was added to a 100 °C pre-heated oil bath and stirred for 4.5 hours until completion. The solution was extracted with ethyl acetate (3 × 50 mL) and washed with water and brine (3 × 100 mL), dried with sodium sulfate, and concentrated. Purification by flash chromatography with hexanes/CH2Cl2 gradient of 40% → 70% to yield Con-B-4 as an orange solid (61.9 mg, 0.157 mmol, 31%). 1H NMR (500 MHz, CDCl3): δ 7.61 (s, 1H), 7.52 (d, J = 8.2 Hz, 1H), 7.33 (d, J = 8.0 Hz, 1H), 7.19 (d, J = 4.2 Hz, 1H), 7.09 (dd, J = 17.8, 11.2 Hz, 1H), 6.59 (d, J = 4.2 Hz, 1H), 6.14 (s, 1H), 5.80 (d, J = 17.8 Hz, 1H), 5.27 (d, J = 11.2 Hz, 1H), 4.68 (sept, J = 6.2 Hz, 1H), 2.59 (s, 3H), 2.53 (s, 3H), 2.43 (s, 3H), 1.40 (d, J = 6.0 Hz, 6H). 13C NMR (126 MHz, CDCl3): δ 158.6, 154.9, 154.4, 143.8, 140.7, 136.3, 133.5, 131.9, 128.1, 126.2, 125.1, 122.7, 121.4 (t, JCF = 2.7 Hz), 118.07, 118.05, 114.9 (t, JCF = 6.1 Hz), 114.6, 70.8, 22.3, 16.9, 16.5, 15.0. HRMS (APCI+) m/z: calculated for C23H26BF2N2O (M + H+) 395.21008; found 395.20529, C23H25BF2N2ONa (M + Na+) 417.19202; found 417.18717.
:1 → 2:1 → 1:1) to afford Con-BEP-4 as purple solid (22.5 mg, 26.2 mmol) in 37% yield. 1H NMR (500 MHz, CDCl3) δ 16.52 (s, 1H), 7.57 (s, 1H), 7.29 (d, J = 7.9 Hz, 1H), 7.08 (d, J = 11.8 Hz, 6H), 6.99–6.94 (m, 1H), 6.56 (d, J = 4.3 Hz, 1H), 6.14 (s, 1H), 4.99 (m, 1H), 4.18 (s, 4H), 2.56–2.36 (m, 27H), 1.32 (d, J = 6.0 Hz, 6H). 13C NMR (126 MHz, CDCl3) δ 294.3 (from HSQC), 211.7, 159.6, 153.9, 152.1, 145.0, 144.3, 140.5, 139.0, 136.7, 133.9, 129.5, 129.2, 128.4, 125.5, 124.9, 123.8, 123.3, 122.0, 117.9, 114.2, 75.6, 66.0, 37.3, 32.1, 29.9, 22.9, 21.2, 17.0, 16.5, 15.4, 15.2, 14.3. HRMS (APCI+) m/z: calculated for C43H49BF2N4ORu (M-Cl2 + H+) 789.30837; found 789.30242.
:water (5:1) solution was added to the flask via syringe and the solution was degassed for 20 minutes. The flask was added to a 100 °C pre-heated oil bath and stirred for 4.5 hours until completion. The solution was extracted with ethyl acetate (3 × 50 mL) and washed with water and brine (3 × 100 mL), dried with sodium sulfate and concentrated under reduced pressure to yield an orange solid. Purification by column chromatography with hexanes/CH2Cl2 gradient of 40% → 70% to yield Con-B-5 as an orange solid (34.1 mg, 86.5 μmol, 16%) 1H NMR (500 MHz, CDCl3) δ 7.98 (d, J = 2.4 Hz, 1H), 7.84 (dd, J = 8.7, 2.4 Hz, 1H), 7.19 (d, J = 4.3 Hz, 1H), 7.09 (dd, J = 17.7, 11.1 Hz, 1H), 6.95 (d, J = 8.7 Hz, 1H), 6.56 (d, J = 4.1 Hz, 1H), 6.12 (s, 1H), 5.83 (dd, J = 17.8, 1.6 Hz, 1H), 5.27 (dd, J = 11.1, 1.5Hz, 1H), 4.63 (sept, J = 6.1 Hz, 1H), 2.56 (s, 3H), 2.54 (s, 3H), 2.41 (s, 3H), 1.39 (d, J = 6.1 Hz, 6H). 13C NMR (126 MHz, CDCl3) δ 157.6, 156.1, 155.1, 143.1, 140.3, 136.2, 133.2, 131.9, 129.9 (t, JCF = 5.0 Hz), 128.0 (t, JCF = 3.4 Hz), 127.3, 125.5, 125.4, 122.4, 118.1, 114.6, 113.0, 70.7, 29.8, 22.4, 16.8, 16.5, 15.0. HRMS (APCI+) m/z: calculated for C23H26BF2N2O (M + H+) 395.21008; found 395.20524, C23H25BF2N2ONa (M + Na+) 417.19202; found 417.18711.
:1 → 2:1 → 1:1) to afford Con-BEP-5 as a red solid (8.9 mg, 0.0104 mmol) in 15% yield. 1H NMR (500 MHz, CDCl3) δ 16.50 (s, 1H), 8.22 (dd, J = 8.6, 2.3 Hz, 1H), 7.34 (d, J = 2.3 Hz, 1H), 7.19 (d, J = 4.0 Hz, 1H), 7.07 (s, 4H), 6.88 (d, J = 8.8 Hz, 1H), 6.43 (d, J = 4.5 Hz, 1H), 6.13 (s, 1H), 4.93 (sept, J = 6.2 Hz, 1H), 4.18 (s, 4H), 2.59 (s, 3H), 2.53 (s, 3H), 2.48 (s, 12H), 2.44 (s, 3H), 2.38 (s, 6H), 1.30 (d, J = 6.1 Hz, 6H). 13C NMR (126 MHz, CDCl3) δ 296.8 (from HSQC), 211.6, 158.3, 153.5, 153.0, 145.1, 143.5, 140.5, 139.1, 136.2, 133.4, 130.8, 129.5, 129.2, 128.4, 127.6, 125.3, 123.8, 122.6, 118.1, 112.8, 75.7, 51.6, 37.2, 32.1, 29.8, 29.5, 22.8, 21.3, 21.2, 16.9, 16.5, 14.3. HRMS (APCI+) m/z: calculated for C43H50BF2N4ORu (M + H+) 789.30837; found 789.30209.
Response to ethylene
:1 hexanes/ethyl acetate to provide orange crystals of Con-B-4 in approximately quantitative yield.
Live cell imaging studies
A549 cells were cultured in Dulbecco's Modified Eagle Medium supplemented with 10% fetal bovine serum and 1% penicillin–streptomycin in LabTek 8-well dishes (400 μL per well). At approximately 80% confluency cells were 0, 1, or 2 μL treated with 40% aqueous solution of ethephon (AK Scientific) and the media was immediately titrated with a 1 M NaOH solution back to neutral pH as indicated by the phenol red in DMEM (∼7–10 μL 1 M NaOH per μL ethephon solution). The cells were incubated for 1–3 h at 37 °C in 5% CO2. After the defined period of DMEM (or DMEM + Ethephon) solutions were removed, cells were washed with pre-warmed PBS (1 × 400 μL), and then stained with Con-BEP-4 in prewarmed PBS (1 μM, 0.5% DMSO). The cells were again incubated for 30–60 min at 37 °C in 5% CO2, then removed for imaging. Control cells were treated in the same fashion, except no ethephon was added. Confocal images of A549 cell were acquired on a Flowview 3000 microscope with a 60× objective using the FV315-SW confocal control software. Con-BEP-4 (or the resulting ethylene adduct – Con-B-4) were excited using either a 488 nm laser (2% Laser power, HV 500, Gain 2×, offset 3%, observe 578–602 nm) or a 561 nm laser (3% Laser power, HV 550, Gain 2×, offset 3%, 539–601 nm). DIC images were obtained with the 640 nm laser. At least three representative fields of cells within the same well were imaged. Image analysis and quantification was performed using ImageJ (National Institutes of Health) with the OlympusViewer plugin. The threshold was appropriately adjusted to the same level for all images being compared and mean fluorescence intensity was measured across the entire field using the measure function.Cytotoxicity assay
A549 cells were cultured in DMEM supplemented with 10% fetal bovine serum and 1% penicillin–streptomycin in 96-well plates (∼104 cells per well). Cell culture media was removed, and cells were treated with Con-BEP-4, Con-B-4, Con-BEP-5, or Con-B-5 (0.2–2 μM) dissolved in Opti-MEM (1X) with 0.5% DMSO (100 μL per well) for 1 hour. After incubation the wells were aspirated, and fresh Opti-MEM was added to the wells with 10 μL of MTT reagent in PBS and incubated for 3 hours. After incubation the wells were aspirated and 100 μL of DMSO was added to the wells. Absorbance readings at 570 nm were measured on a plate reader (Tecan Infinite M200 Pro). The normalized absorbance was averaged over a minimum of four replicates for each condition evaluated (Fig. S21, ESI†).Author contributions
NJD, NBW, and BWM designed and performed experiments. NJD and BWM analysed data and wrote the manuscript. BWM conceived and supervised the research.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Prof. Andrei G. Kutateladze (University of Denver) is acknowledged for assistance with computational work. Tyler D. Ball and Prof. Sunil Kumar (University of Denver) are acknowledged for assistance with cytotoxicity assays. Prof. Todd A. Wells (University of Denver) is thanked for helpful discussions. This material is based upon work supported by the National Science Foundation under Grant CHE-1900482. Research reported in this publication was supported by NIGMS R35GM150937.References
- J. J. Kieber, Annu. Rev. Plant Physiol. Plant Mol. Biol., 1997, 48, 277–296 CrossRef CAS PubMed.
- K. L. C. Wang, H. Li and J. R. Ecker, Plant Cell, 2002, 14, S131–S151 CrossRef CAS PubMed.
- J. M. Alonso and A. N. Stepanova, Science, 2004, 306, 1513–1515 CrossRef CAS PubMed.
- S. Janssen, K. Schmitt, M. Blanke, M. L. Bauersfeld, J. Wollenstein and W. Lang, Philos. Trans. R. Soc., A, 2014, 372, 20130311 CrossRef CAS PubMed.
- F. Caprioli and L. Quercia, Sens. Actuators, B, 2014, 203, 187–196 CrossRef CAS.
- K. M. Light, J. A. Wisniewski, W. A. Vinyard and M. T. Kieber-Emmons, JBIC, J. Biol. Inorg. Chem., 2016, 21, 715–728 CrossRef CAS PubMed.
- E. E. Dumelin and A. L. Tappel, Lipids, 1977, 12, 894 CrossRef CAS PubMed.
- L. M. Paardekooper, G. van den Bogaart, M. Kox, I. Dingjan, A. H. Neerincx, M. B. Bendix, M. ter Beest, F. J. M. Harren, T. Risby, P. Pickkers, N. Marczin and S. M. Cristescu, Sci. Rep., 2017, 7, 6889 CrossRef PubMed.
- K. H. Jensen and B. W. Michel, Analysis Sensing, 2023, 3, e202200058 CrossRef CAS PubMed.
- X. Yang, Q. Zhang, S. Zhang, M. Lai, X. Ji, Y. Ye, H. Li and M. Zhao, Coord. Chem. Rev., 2023, 487, 215154 CrossRef CAS.
- J. Chatt, L. A. Duncanson and L. M. Venanzi, J. Chem. Soc., 1955, 4456–4460 RSC.
- O. Green, N. A. Smith, A. B. Ellis and J. N. Burstyn, J. Am. Chem. Soc., 2004, 126, 5952–5953 CrossRef CAS PubMed.
- M. Santiago Cintrón, O. Green and J. N. Burstyn, Inorg. Chem., 2012, 51, 2737–2746 CrossRef PubMed.
- B. Esser and T. M. Swager, Angew. Chem., Int. Ed., 2010, 49, 8872–8875 CrossRef CAS PubMed , S8872/1-S8872/15.
- B. Esser, J. M. Schnorr and T. M. Swager, Angew. Chem., Int. Ed., 2012, 51, 5752–5756 CrossRef CAS PubMed , S5752/1-S5752/13.
- Y. Hitomi, T. Nagai and M. Kodera, Chem. Commun., 2012, 48, 10392–10394 RSC.
- P. Cabanillas-Galan, L. Farmer, T. Hagan, M. Nieuwenhuyzen, S. L. James and M. C. Lagunas, Inorg. Chem., 2008, 47, 9035–9041 CrossRef CAS PubMed.
- S. N. W. Toussaint, R. T. Calkins, S. Lee and B. W. Michel, J. Am. Chem. Soc., 2018, 140, 13151–13155 CrossRef CAS PubMed.
- M. Sun, X. Yang, Y. Zhang, S. Wang, M. W. Wong, R. Ni and D. Huang, J. Agric. Food Chem., 2019, 67, 507–513 CrossRef CAS PubMed.
- K. Vong, S. Eda, Y. Kadota, I. Nasibullin, T. Wakatake, S. Yokoshima, K. Shirasu and K. Tanaka, Nat. Commun., 2019, 10, 5746 CrossRef CAS PubMed.
- D. Fong, S.-X. Luo, R. S. Andre and T. M. Swager, ACS Cent. Sci., 2020, 6, 507–512 CrossRef CAS PubMed.
- M. Wu, C. Yin, L. Fu, T. Liu, M. Jiang, Q. Sun, L. Chen and N. Niu, Chem. Eng. J., 2022, 435, 135045 CrossRef CAS.
- Y. Chen, W. Yan, D. Guo, Y. Li, J. Li, H. Liu, L. Wei, N. Yu, B. Wang, Y. Zheng, M. Jing, J. Zhao and Y. Ye, Angew. Chem., Int. Ed., 2021, 60, 21934–21942 CrossRef CAS PubMed.
- Y. Yang, H. Bian, Z. Jia and P. Tu, ACS Omega, 2023, 8, 15350–15359 CrossRef CAS PubMed.
- J. Ohata, K. J. Bruemmer and C. J. Chang, Acc. Chem. Res., 2019, 52, 2841–2848 CrossRef CAS PubMed.
- K. J. Bruemmer, S. W. M. Crossley and C. J. Chang, Angew. Chem., Int. Ed., 2020, 59, 13734–13762 CrossRef CAS PubMed.
- M. H. Lim, B. A. Wong, W. H. Pitcock, D. Mokshagundam, M.-H. Baik and S. J. Lippard, J. Am. Chem. Soc., 2006, 128, 14364–14373 CrossRef CAS PubMed.
- M. H. Lim, D. Xu and S. J. Lippard, Nat. Chem. Biol., 2006, 2, 375–380 CrossRef CAS PubMed.
- B. W. Michel, A. R. Lippert and C. J. Chang, J. Am. Chem. Soc., 2012, 134, 15668–15671 CrossRef CAS PubMed.
- M. Strianese and C. Pellecchia, Coord. Chem. Rev., 2016, 318, 16–28 CrossRef CAS.
- D. A. Iovan, S. Jia and C. J. Chang, Inorg. Chem., 2019, 58(20), 13546–13560 CrossRef CAS PubMed.
- B. J. Bezner, L. S. Ryan and A. R. Lippert, Anal. Chem., 2020, 92, 309–326 CrossRef CAS PubMed.
- J. Alday, A. Mazzeo and S. Suarez, Inorg. Chim. Acta, 2020, 510, 119696 CrossRef CAS.
- R. R. Walvoord, M. R. Schneider and B. W. Michel, in Carbon Monoxide in Drug Discovery: Basics, Pharmacology, and Therapeutic Potential, ed. B. Wang and O. Wang, John Wiley & Sons, Hoboken, NJ, 2022, pp. 321–344 Search PubMed.
- X. Yang, Z. Yuan, W. Lu, C. Yang, M. Wang, R. Tripathi, Z. Fultz, C. Tan and B. Wang, J. Am. Chem. Soc., 2023, 145, 78–88 CrossRef CAS PubMed.
- D. F. Sauer, T. Himiyama, K. Tachikawa, K. Fukumoto, A. Onoda, E. Mizohata, T. Inoue, M. Bocola, U. Schwaneberg, T. Hayashi and J. Okuda, ACS Catal., 2015, 5, 7519–7522 CrossRef CAS.
- J. Morstein, D. Höfler, K. Ueno, J. W. Jurss, R. R. Walvoord, K. J. Bruemmer, S. P. Rezgui, T. F. Brewer, M. Saitoe, B. W. Michel and C. J. Chang, J. Am. Chem. Soc., 2020, 142, 15917–15930 CrossRef CAS PubMed.
- V. Leen, E. Braeken, K. Luckermans, C. Jackers, M. V. der Auweraer, N. Boens and W. Dehaen, Chem. Commun., 2009, 4515–4517 RSC.
- S. Monsaert, R. Drozdzak, V. Dragutan, I. Dragutan and F. Verpoort, Eur. J. Inorg. Chem., 2008, 432–440 CrossRef CAS.
- C. A. Urbina-Blanco, A. Leitgeb, C. Slugovc, X. Bantreil, H. Clavier, A. M. Z. Slawin and S. P. Nolan, Chem. – Eur. J., 2011, 17, 5045–5053 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Additional experimental details, photophysical characterization data, supplemental figures, and NMR spectra. See DOI: https://doi.org/10.1039/d3cb00079f |
| This journal is © The Royal Society of Chemistry 2023 |