
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePAM-less conditional DNA substrates leverage trans-cleavage of CRISPR-Cas12a for versatile live-cell biosensing†
Siyu
Chen
,
Rujia
Wang
,
Shuang
Peng
,
Shiyi
Xie
,
Chunyang
Lei
 *,
Yan
Huang
and
Zhou
Nie
*
*,
Yan
Huang
and
Zhou
Nie
*
State Key Laboratory of Chemo/Bio-Sensing and Chemometrics, College of Chemistry and Chemical Engineering, Hunan Provincial Key Laboratory of Biomacromolecular Chemical Biology, Hunan University, Changsha 410082, P. R. China. E-mail: niezhou.hnu@gmail.com; cylei@hnu.edu.cn
First published on 17th January 2022
Abstract
The CRISPR-Cas system has been repurposed as a powerful live-cell imaging tool, but its utility is limited to genomic loci and mRNA imaging in living cells. Here, we demonstrated the potential of the CRISPR-Cas system as a generalizable live-cell biosensing tool by extending its applicability to monitor diverse intracellular biomolecules. In this work, we engineered a CRISPR-Cas12a system with a generalized stimulus-responsive switch mechanism based on PAM-less conditional DNA substrates (pcDNAs). The pcDNAs with stimulus-responsiveness toward a trigger were constructed from the DNA substrates featuring no requirement of a protospacer-adjacent motif (PAM) and a bubble structure. With further leveraging the trans-cleavage activity of CRISPR-Cas12a for signal reporting, we established a versatile CRISPR-based live-cell biosensing system. This system enabled the sensitive sensing of various intracellular biomolecules, such as telomerase, ATP, and microRNA-21, making it a helpful tool for basic biochemical research and disease diagnostics.
Introduction
The visualization and detection of important biomolecules in living cells can directly reveal their cellular spatiotemporal distribution and dynamics, contributing to unveil their functions in biochemical reaction networks.1,2 Bioimaging tools, including fluorescent protein-based biosensors and nucleic acid-based probes,3,4 have been developed for monitoring a variety of biomolecules with high sensitivity and specificity. Despite these advancements, high-performance biosensing and bioimaging tools remain highly desirable in fundamental biomedical research, disease diagnosis, and drug screening. The clustered regularly interspaced short palindromic repeat (CRISPR)/CRISPR-associated protein (Cas) system enables precise sequence recognition under the guidance of RNA,5–9 which rationally integrates the advantages of proteins and nucleic acids, thus highlighting its potential in bioimaging.10,11 For instance, by fusing the nuclease-deficient Cas protein (dCas) with fluorescent tags or engineering guide RNA (gRNA) with fluorescent labels, the CRISPR-Cas system has been repurposed as a powerful imaging tool for tracking the dynamics of genomic loci containing protospacer-adjacent motifs (PAMs) and RNA in living cells, which contribute to gain better insights into the dynamics of nuclear organization,12–18 gene regulation and viral infections.19,20 Given the significance of this progress, we were intrigued to find whether the CRISPR-Cas system can be deployed as a universal live-cell biosensing tool for a variety of intracellular biomolecules, rather than just for the nucleic acid targets of interest.To this purpose, we need to engineer the RNA-guided nucleic acid binding and nuclease activation functions of the CRISPR-Cas system with a generalized stimulus-responsive switch mechanism. By engineering the CRISPR-Cas components with responsiveness toward a stimulus, several studies have realized conditional regulation of CRISPR-Cas functions in gene editing.21–29 However, the complexity and diversity in the tertiary structures of the Cas protein and guide RNA make this strategy still face challenges when extending to other analytes. Given the programmability and scalability of manipulating the secondary structure of DNA,30 engineering the substrate DNA sequence with a generalized responsive mechanism can be a viable solution to address this limitation. The DNA-targeting class II type V Cas effectors (e.g., Cas12a and Cas12b) should be suitable candidates,31 because promiscuous degradation of endogenous RNAs by the RNA-targeting Cas proteins could result in cell apoptosis.32,33 According to mechanism studies on the CRISPR-Cas12a system, unwinding of the seed region by the Cas12a/gRNA complex after PAM recognition is a crucial pre-step for the subsequent R-loop formation and further allosteric activation of nuclease activity.34–36 A recent study found that Cas12a/gRNA complexes can process unwound DNA substrates without the requirement of PAM recognition.37 Collectively, we reasoned that the strict PAM dependence of the CRISPR-Cas12a system on a DNA substrate could be circumvented if an artificially unwound seed region was constructed. In such a scenario, a new type of DNA substrate without PAM requirement can be presented, allowing us to design a generalized stimulus-responsive DNA substrate by rationally taking advantage of the flexibility of the pre-unwound seed region in molecular engineering.
Here, we engineered the CRISPR-Cas12a system with a generalizable stimulus-responsive mechanism and deployed it to monitor various biomolecules in living cells. Specifically, we described a versatile strategy to eliminate the strict PAM preference of the CRISPR-Cas12a system on the DNA substrate by introducing an artificial bubble structure in the seed region. Then, we constructed a PAM-less conditional DNA substrate (pcDNA) whose activity is dependent on the presence of a biomolecule trigger (Scheme 1). Benefiting from the programmability and modularity of base-pairing, pcDNAs for different analytes can be readily built via fine-tuning the integrated DNA module. By further coupling the trans-cleavage of CRISPR-Cas12a with a high turnover for signal reporting, conditional CRISPR-Cas12a systems that enable sensing of different types of biomolecules in living cells were established, without involving any genetic manipulations or chemical modifications to the CRISPR-Cas components. The versatility of pcDNA-based CRISPR-Cas12a systems was demonstrated by the sensitive sensing of several biomolecules in living cells, including intracellular enzymes, small molecules, and microRNAs. This work explores the potential of Cas12a for live-cell sensing by integrating pcDNA with Cas12a's nuclease activity, presenting a novel paradigm for the development of high-performance CRISPR-Cas biosensing systems.
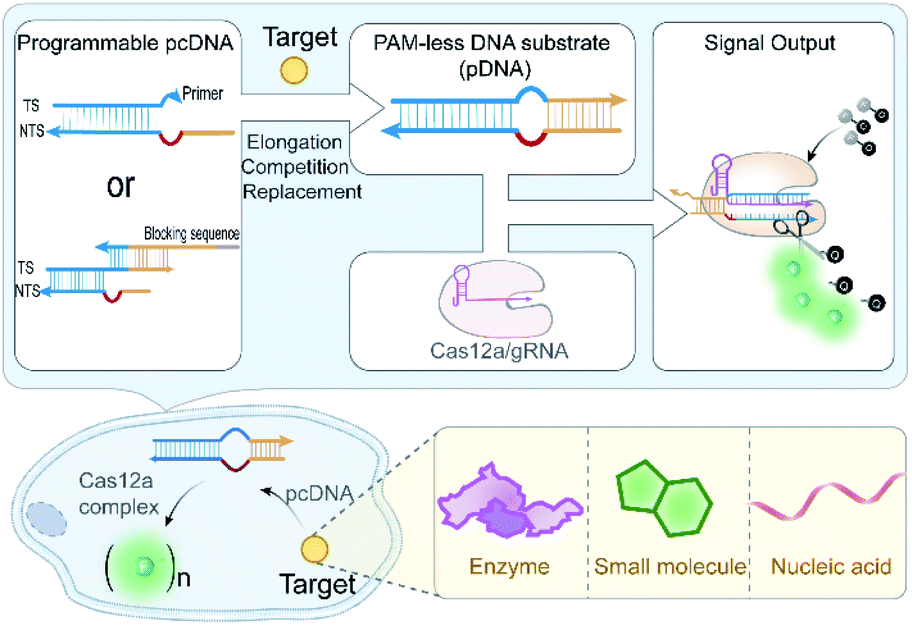 | ||
| Scheme 1 Schematic illustration of the pcDNA-based CRISPR-Cas12a systems. Different types of intracellular targets convert the corresponding pcDNA into a PAM-less dsDNA substrate (pDNA) that can activate the nuclease activities of Cas12a to generate signals for in vitro and live-cell biosensing. | ||
Results and discussion
Construction of PAM-less DNA substrates
We first sought to fabricate the PAM-less DNA substrates (pDNAs) that can be recognized and processed by using the CRISPR-Cas12a system in this work. Structural studies have revealed that the Cas12a/gRNA complex recognizes the PAM, unwinds the seed region in the DNA substrate, and facilitates the formation of the R-loop between the target strand (TS) and the spacer in gRNA, resulting in allosterically activating the cis- and trans-cleavage activities of Cas12a.34 Therefore, the seed region unwinding is the connecting step between PAM recognition and R-loop formation. Moreover, when the complementary sequence of a spacer in a DNA substrate was completely unwound, the PAM is dispensable for activating both the cis- and trans-cleavage activities of Cas12a.35,36 The single-molecule fluorescence study also confirmed this phenomenon, revealing that the PAM is required only for R-loop initiation during the target searching and cleavage process of Cas12a.37 In our recent study, even the unwinding of only one site in the seed region could accelerate the trans-cleavage rate of Cas12a.38 Inspired by this progress, we speculated that unwinding of the seed region might allow the seed segment in gRNA to hybridize with TS directly without requiring the assistance of the PAM recognition process (Fig. 1a).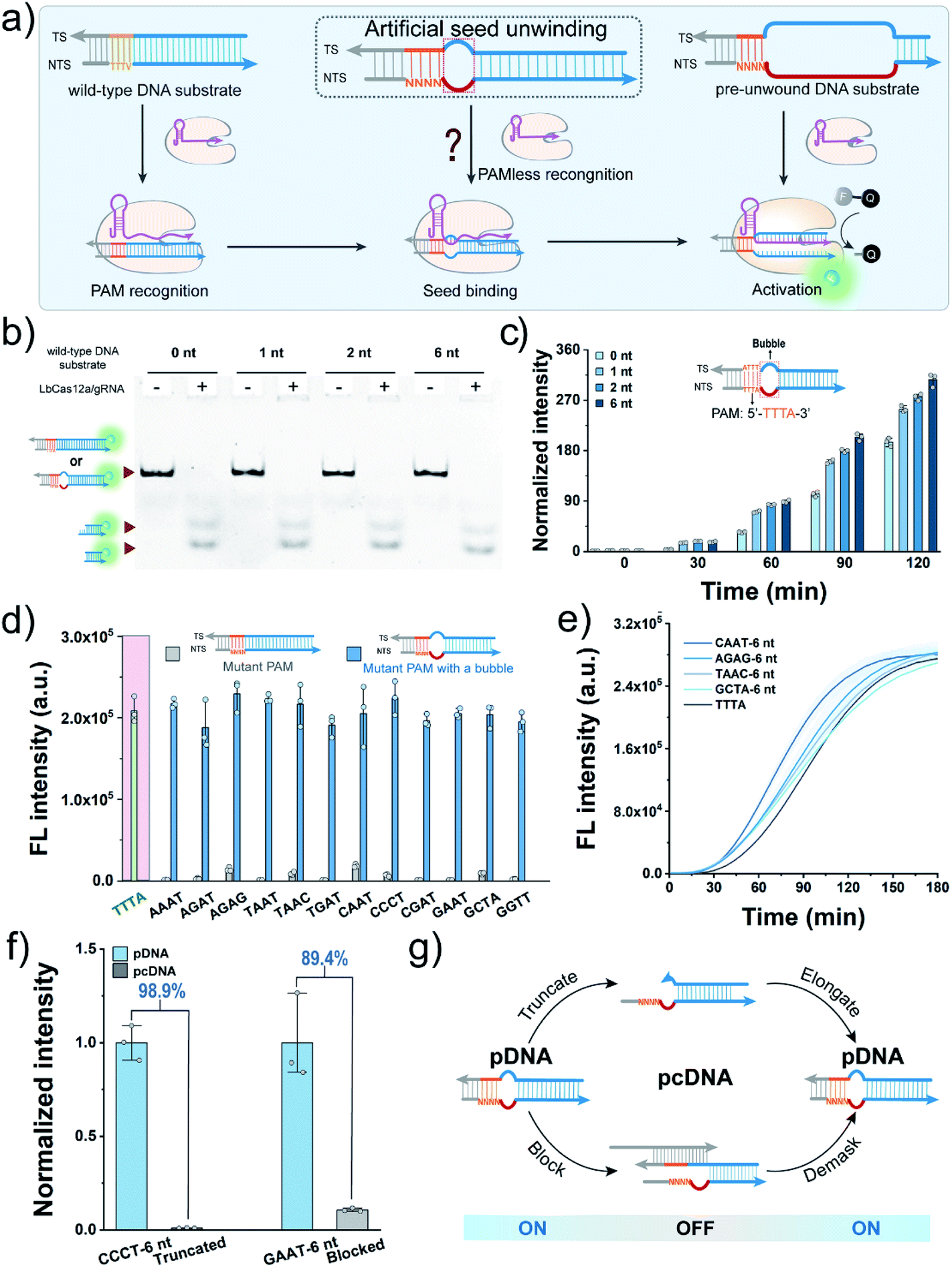 | ||
| Fig. 1 Construction of pDNAs. (a) Schematic diagram of the recognition of pDNA by Cas12a. V represents G, C or A, and N refers to any of the four canonical nucleotides. (b) Native PAGE analysis of cis-cleavage of wild-type DNA substrates with different nonmatched sites by LbCas12a. (c) Fluorescence signals of FQ reporter cleavage by Cas12a induced by the dsDNA substrates containing different nonmatched sites in the seed region. (d) Evaluation of the activation of LbCas12a by the substrates with a mutant PAM and their counterparts with a 6 nt bubble in the seed region using the FQ reporters. (e) Fluorescence kinetic analysis of trans-cleavage of FQ reporters by Cas12a activated by the bubble DNA substrates. (f) Normalized trans-cleavage signals induced by pDNA and truncated/blocked pcDNA. (g) Blueprint of constructing pcDNAs with a switch mechanism by modulating the formation of a bubble structure in the seed region. LbCas12a, 100 nM; gRNA, 100 nM; wild-type DNA substrate, pDNA or pcDNA, 50 nM; FQ reporter, 1.0 μM. FL, fluorescence; a.u., arbitrary units. | ||
Here, Cas12a from the Lachnospiraceae bacterium (LbCas12a) was chosen as an example to test this assumption (Fig. S1†). LbCas12a, a widely used Cas protein in CRISPR-based diagnostics,39–42 mainly recognizes the DNA substrates containing the 5′–TTTV–3′ PAM.31 In this work, a series of DNA substrates with different nonmatched sites (1-, 2- and 6 nt bubbles) in the seed region were prepared and processed by LbCas12a/gRNA complexes, and the reaction products were analysed by gel shift assay. All the DNA substrates with a bubble structure were efficiently cis-cleaved by LbCa12a (Fig. 1b), suggesting that LbCas12a can recognize and process these bubble DNA substrates. Next, we investigated whether these bubble DNA substrates could trigger the trans-cleavage activity of LbCas12a using a fluorescent ssDNA reporter (FAM-TTATT-BHQ1, FQ reporter).39 As shown in Fig. 1c, all the bubble DNA substrates induced a significant fluorescence signal, and the fluorescence intensity had a positive relationship with the number of nonmatched sites. A further increment in nonmatched sites did not increase the trans-cleavage signals (Fig. S2†). Using the DNA substrate with a 6 nt bubble as the starting sequence, we designed a panel of counterpart sequences with a mutant PAM that was reported to be unrecognizable by LbCas12a.31 These sequences could also activate the trans-cleavage activity of LbCas12a (Fig. 1d and S3†). Moreover, several sequences had a faster trans-cleavage dynamic over the wild-type DNA substrate (Fig. 1e and S4†), which might contribute to a better detection sensitivity in biosensing.43 Taken together, we can conclude that introducing a bubble structure into the seed region is able to eliminate the PAM preference of LbCas12a toward the DNA substrates.
Engineering pDNA with a switch mechanism
To engineer pDNA with a stimulus-responsiveness, we need to devise it with both switch-on and switch-off mechanisms. First, we sought to switch off its function by undermining the integrity of the PAM region in pDNA. Toward this end, we introduced different numbers of nonmatched sites into the PAM, and tested their abilities to activate the trans-cleavage activity of LbCas12a. After incubating these sequences with LbCas12a/gRNA complexes and FQ reporters, strong fluorescent signals comparable to the control group were observed (Fig. S5†), which suggested that PAM integrity is dispensable for the pDNAs. Therefore, the bubble structure might perform a crucial role in the maintenance of pDNAs' function. To verify this hypothesis, we created a truncated pDNA by removing the bubble seed region from the TS and assessed its function based on the trans-cleavage signal. A sharp decline of 98.9% in the fluorescent signal revealed that the truncated counterpart lost the ability to activate the LbCas12a's nuclease activity (Fig. 1f). Moreover, the pDNA could be efficiently blocked by hybridizing the bubble seed region and its 3′-flank with a complementary strand (declined by 89.4%, Fig. 1f). Together, these results indicate that we can switch off the function of pDNA by destroying the bubble structure.Given that the seed region is indispensable to maintain the function of pDNAs, it is reasonable to envisage a blueprint for constructing pcDNAs by modulating the formation of the bubble structure in the seed region (Fig. 1g). Specifically, we can restore the activity of the truncated pDNA via enzymatic chain elongation or demask the blocked pDNA in combination with dynamic nucleic acid hybridization.44,45 In this scenario, diverse pcDNAs are expected to be constructed for the sensing of different biomolecules. The advantages of no PAM requirement and the bubble structure confer the pDNAs with better flexibility and freedom in designing a stimulus-responsiveness compared to the wild-type DNA substrates.
Cas12a-mediated telomerase activity assay
Telomerase activation is observed in most human tumours, and the immortality conferred by telomerase plays a key role in cancer development.46–48 As a proof-of-concept, we attempted to construct pcDNATel with telomerase-responsive capacity and deployed it to develop Cas12a-based telomerase assay (Fig. 2a). The advantages of the bubble structure and no PAM requirement allow us to implement the construction of pcDNATel based on a truncated pDNA using a 20 nt primer sequence of human telomerase as the TS. The NTS in pcDNATel is composed of the complementary sequence of two telomeric repeats (12 nt, orange), a 6 nt linker sequence (red), and a partially complementary region of the telomerase primer (14 nt, blue). In the presence of human telomerase, tandem telomeric repeats (AGGGTT)n are enzymatically appended to the 3′-terminus of the telomerase primer,46 leading to the generation of the duplex DNA containing a bubble structure. Using gRNA targeting the telomerase primer sequence, the elongation products can function as the pDNA to activate the nuclease activity of LbCas12a. Consequently, telomerase activity is converted and amplified into a detectable fluorescent signal via the efficient trans-cleavage of FQ reporters.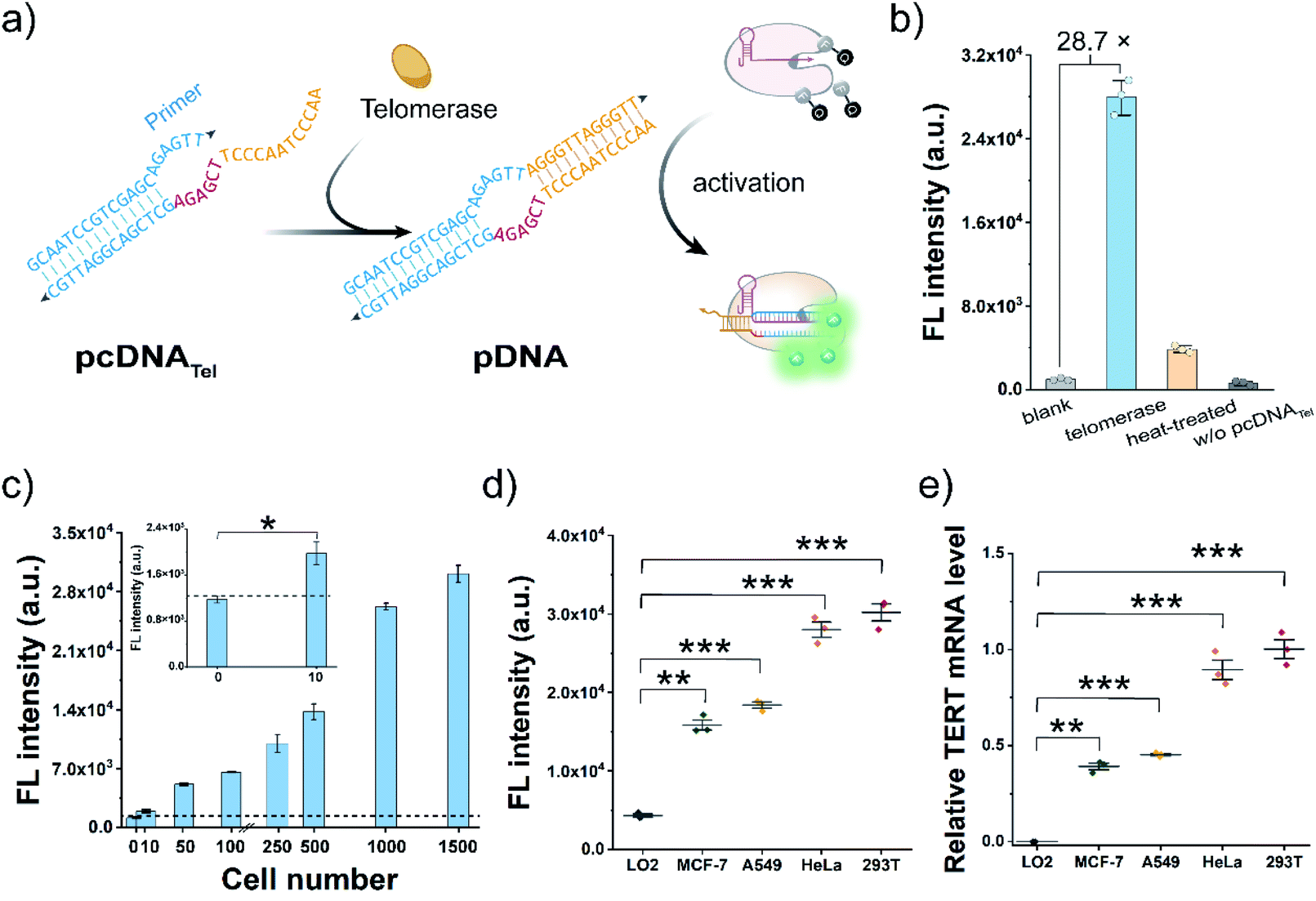 | ||
| Fig. 2 The pcDNA-based Cas12a assay for the analysis of telomerase activity. (a) Scheme of pcDNA-based Cas12a assay for the detection of telomerase activity. (b) In vitro fluorescent detection of the telomerase activity in the extracts from 1000 HeLa cells in the pcDNA-based Cas12a assay. The blank sample contains the same components except the cell extract; the w/o pcDNATel sample contains all the components except pcDNATel. (c) Fluorescence responses of pcDNA-based Cas12a assay in response to telomerase from different numbers of HeLa cells. The black dot line represents the fluorescence threshold value, followed by the 3σ principle for the distinction of telomerase in cell lysates. The inset shows the enlarged view of fluorescence intensity of 0 and 10 HeLa cells. Data represent means ± SD. *P < 0.05 and Student's test. (d) Fluorescence signals induced by telomerase from different types of human cell lines for 1000 cells. (e) qRT-PCR result of the relative TERT mRNA expression levels in different cells. Data represent means ± SD. **P < 0.01, ***P < 0.001, and ANOVA followed by Tukey's post hoc test. | ||
To verify the feasibility of this design, we extracted telomerase from HeLa cells and incubated it with the pcDNATel in the buffer solution containing dNTP mixtures. Elongation products with slower migration rates were detected in the gel shift assay (Fig. S6†). Then, the elongation products were mixed with LbCas12a/gRNA complexes and FQ reporters to produce detectable signals. As shown in Fig. 2b, a strong fluorescence signal that is 28.7-times that of the control without telomerase was observed. When the telomerase was treated with different concentrations of 3′–azido–3′-deoxythymidine (AZT, a telomerase inhibitor),49 a dose-dependent decrease in the trans-cleavage signal was observed (Fig. S7†). Therefore, the telomerase-catalysed elongation reaction can efficiently and specifically trigger the trans-cleavage activity of LbCas12a. In this assay, the highest fluorescence signal was obtained with an elongation time of 1.0 h (Fig. S8†), which might be attributed to the fact that over-elongated products could serve as the competitive substrates to retard the trans-cleavage rate of FQ reporters. Under this optimal condition, fluorescence response dynamics of the elongation products at different concentrations of telomerase were measured (Fig. 2c). The fluorescence signal had a positive correlation with the number of HeLa cells over the range of 10–1500 cells, and the lowest detection cell number was as low as 8 cells (3σ/slope). Next, to assess the potential utility of this assay in clinical diagnosis, we chose three other typical human tumour cell lines (MCF-7, A549 and 293T) and a normal cell line (LO2) as the models to test the cellular telomerase activity. The fluorescence signals of the tumour cell line groups were significantly higher than that of the LO2 cell line group (Fig. 2d and S9a†), which are consistent with the telomerase activities determined by telomeric repeat amplification protocol (TRAP) assay and the mRNA levels of human telomerase reverse transcriptase (TERT) quantified by qRT-PCR (Fig. 2e and S9b†). Collectively, by rationally designing a telomeric elongation-responsive pcDNA, we have established Cas12a-based assay for the sensitive detection of telomerase activities.
Cas12a-mediated live-cell detection of telomerase activity
Having realized the analysis of telomerase activities in vitro, we next sought to employ this pcDNA-based Cas12a assay to monitor telomerase activity in living cells (Fig. 3a). Although the trans-cleavage activity of Cas12a has been widely employed for signal reporting in CRISPR-based biosensing and diagnostics,39–42,50–52 whether it can function in living cells for biosensing remains to be verified. To this end, we first tested the trans-cleavage effect of LbCas12a in the cytoplasm using the FQ reporters. Before that, Mg2+, a key cofactor of Cas12a, was confirmed to be sufficient to maintain the nuclease activity of Cas12a under the intracellular Mg2+ concentration in the range of 0.5–1.0 mM![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 53,54 (Fig. S10a†). Moreover, the FQ reporters were validated to be relatively stable in complex biological samples, contributing to gain a low background signal (Fig. S10b and c†). The FQ reporters, wild-type DNA substrates and LbCas12a/gRNA complexes were stepwise transfected into different cell lines using a commercial transfection reagent. The cells were imaged by confocal laser scanning microscopy (CLSM), and bright fluorescent spots were detected in various cell lines, including HeLa, A549, MCF-7, and LO2 (Fig. 3b and S11†), suggesting that LbCas12a can be activated to trans-cleave the FQ reporters effectively in the cytoplasm. The slight effects on cell viabilities determined by the standard CCK8 assay implied the low cytotoxicity of the Cas12-based signal reporting system (Fig. S12†). Collectively, these data have indicated the promising potential of Cas12a's trans-cleavage effect in live-cell biosensing.
53,54 (Fig. S10a†). Moreover, the FQ reporters were validated to be relatively stable in complex biological samples, contributing to gain a low background signal (Fig. S10b and c†). The FQ reporters, wild-type DNA substrates and LbCas12a/gRNA complexes were stepwise transfected into different cell lines using a commercial transfection reagent. The cells were imaged by confocal laser scanning microscopy (CLSM), and bright fluorescent spots were detected in various cell lines, including HeLa, A549, MCF-7, and LO2 (Fig. 3b and S11†), suggesting that LbCas12a can be activated to trans-cleave the FQ reporters effectively in the cytoplasm. The slight effects on cell viabilities determined by the standard CCK8 assay implied the low cytotoxicity of the Cas12-based signal reporting system (Fig. S12†). Collectively, these data have indicated the promising potential of Cas12a's trans-cleavage effect in live-cell biosensing.
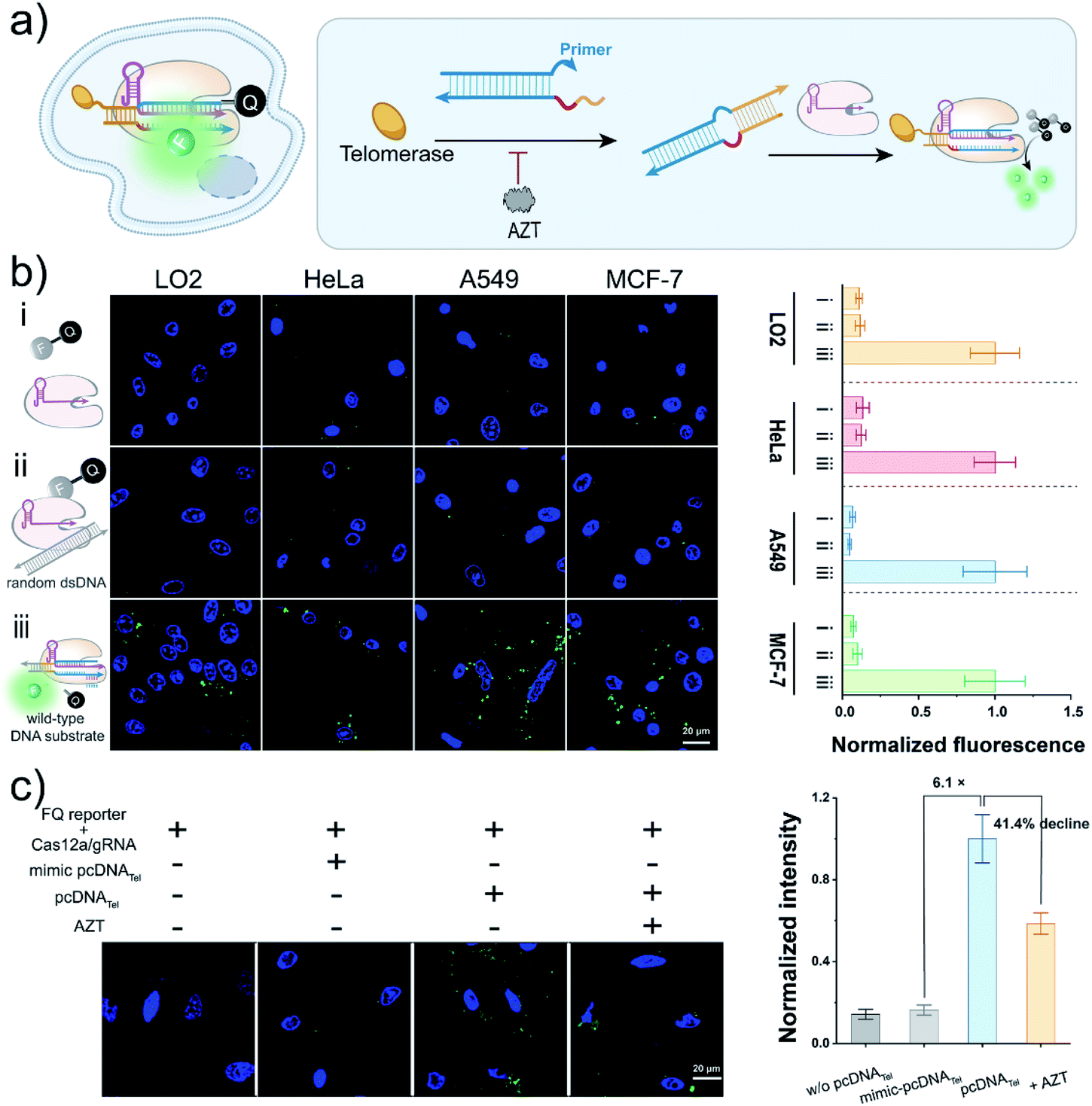 | ||
| Fig. 3 The pcDNA-based Cas12a assay for the live-cell sensing of telomerase activity. (a) Scheme of pcDNA-based Cas12a assay for sensing of the telomerase activity in living cells. (b) CLSM images (left) and the corresponding fluorescence signals (right) of the trans-cleavage activity of LbCas12a in different cell lines with indicated treatments. (i) LbCas12a/gRNA and FQ reporter; (ii) LbCas12a/gRNA, FQ reporter and random dsDNA; (iii) LbCas12a/gRNA, FQ reporter and a wild-type DNA substrate. Fluorescence intensity was quantified from per condition using ImageJ software. Data represent means ± SD (n = 81, 63, 63, 44, 52, 46, 53, 51, 52, 74, 58, and 45 cells, from top to bottom). (c) The CLSM images (left) and the corresponding fluorescence signals (right) of the Cas12a assay for sensing of telomerase activity in HeLa cells. LbCas12a, 50 nM; gRNA, 50 nM; wild-type DNA substrate, random dsDNA or pcDNA, 100 nM; FQ reporter, 1.0 μM; AZT, 200 μM. Scale bar, 20 μm. Data represent means ± SD (n = 41, 44, 42, 57 cells, from left to right). | ||
To test the usability of the pcDNA-based Cas12a assay for the live-cell sensing of telomerase activity, we transfected pcDNATel, LbCas12a/gRNA complexes and FQ reporters into HeLa cell lines, followed by CLSM analysis at different times (Fig. S13†). As shown in Fig. 3c and S14,† strong fluorescence signals were detected in the HeLa cells, which were 6.1 times over the control group using a mimic-pcDNATel without telomerase-responsive capacity. The fluorescence signal declined by 41.4% when the cells were pre-treated with AZT (Fig. 3c and S15†), indicating that the fluorescence signal was derived from the elongation catalysed by intracellular telomerase. Therefore, the rational integration of pcDNA and the trans-cleavage activity of LbCas12a have enabled the specific biosensing of telomerase activity in living cells.
Cas12a-mediated sensing of ATP
The bubble structure and no PAM requirement in the pDNA provides a high degree of freedom to design pcDNA by integrating with functional nucleic acids and toehold-mediated strand exchange reactions.45 Thus, we next sought to explore the versability of pcDNA in sensing different biomolecules. To validate the applicability of pcDNA in the sensing of small biomolecules, we chose ATP that plays an essential regulatory and integral role in cell metabolism as the model target,55 and fabricated pcDNAATP with ATP-responsiveness. In the pcDNAATP, an ATP aptamer containing a 7 nt tail at the 3′ terminus is introduced to block the function of a pDNA through complementary base pairing (Fig. 4a). In the presence of ATP, the specific binding between ATP and its aptamer results in the release of the pDNA, which subsequently triggers the activation of LbCas12a's nuclease activity, thereby leading to the generation of fluorescence signals via trans-cleavage of FQ reporters. Thus, both the bubble structure and no PAM requirement are indispensable for the integration of ATP aptamer to build the pcDNAATP. The structure of pcDNAATP was theoretically predicted by using NUPACK (Fig. S16†). The fabrication of pcDNAATP and its responsiveness toward ATP were confirmed by the gel shift assay (Fig. 4b). Then, ATP was incubated with pcDNAATP, and the reaction products were mixed with LbCas12a/gRNA complexes and FQ reporters. An intense fluorescence signal that is about 8.5-times the control without ATP was obtained (Fig. S17†). After optimizing the concentration of pcDNAATP, the ratio of the fluorescence without ATP was obtained (Fig. S17†). After optimizing the concentration of pcDNAATP, the ratio of the fluorescence response was further improved to 9.8 (Fig. S18†). By determining the fluorescence responses toward ATP at different concentrations, the calibration curve for ATP detection was found to be linear over a concentration range of 2.5–800 μM with a limit of detection of 0.47 μM (3σ/slope, Fig. 4c). Because of the high specificity of the aptamer, this pcDNA-based Cas12a assay showed good selectivity toward ATP (Fig. 4d). Next, we attempted to investigate the feasibility of the pcDNA-based Cas12a assay in live-cell biosensing of intracellular ATP. The mixture of pcDNAATP, LbCas12a/gRNA complexes, and FQ reporters was transfected into A549 cell lines. The CLSM image showed a strong fluorescence signal, which is 5.5-fold over the control group using a mimic pcDNAATP without responsiveness toward ATP (Fig. 4e and S19†). Moreover, when the mitochondrial ATP synthesis was inhibited by oligomycin,56 the fluorescence signal declined by 62.4%. Overall, these results have demonstrated that Cas12a-based sensing of ATP both in vitro and live-cell have been achieved by rationally designing pcDNAATP and leveraging the trans-cleavage activity for signal reporting.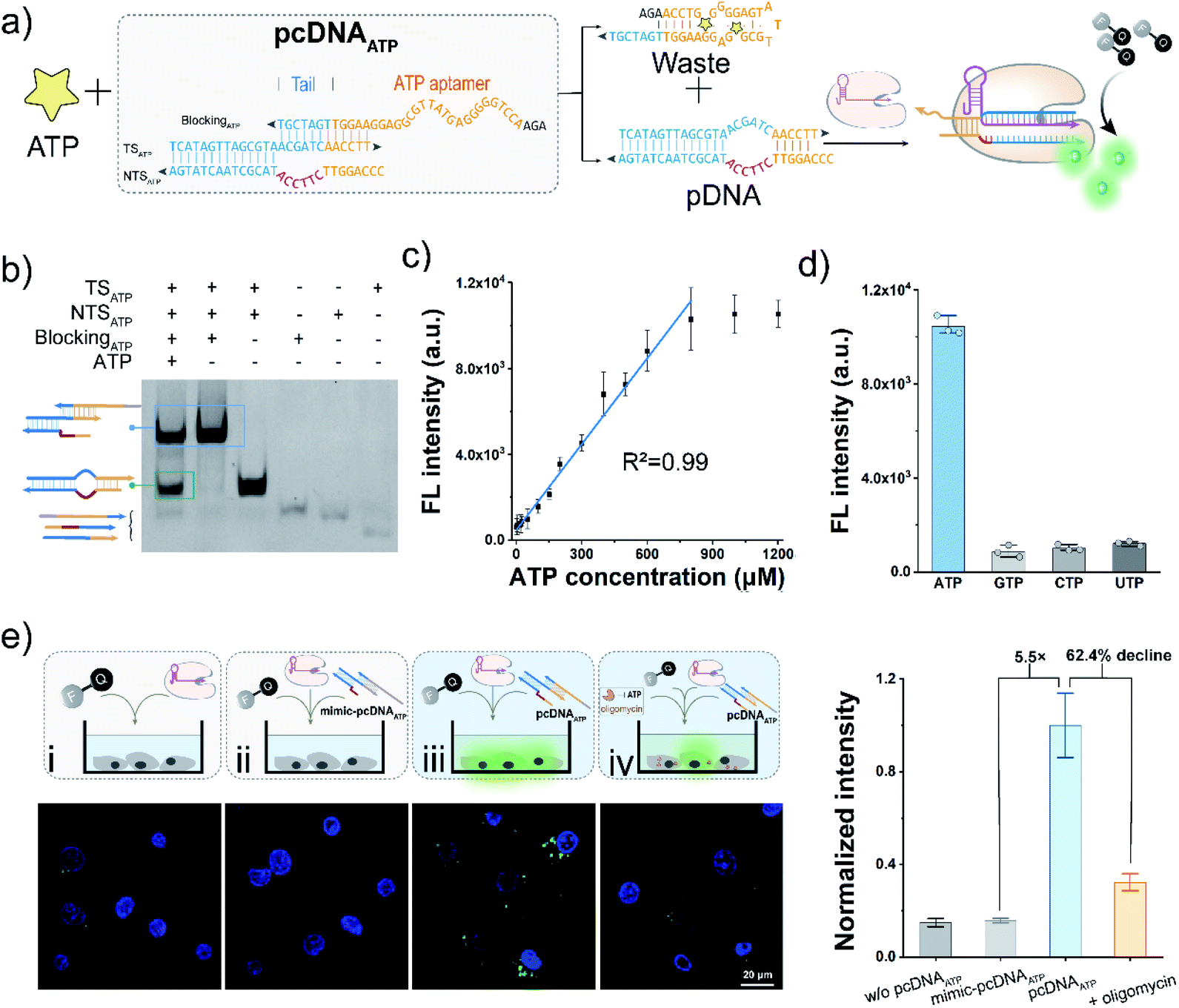 | ||
| Fig. 4 The pcDNA-based Cas12a assay for the sensing of ATP. (a) Schematic diagram of ATP-sensing using the pcDNAATP. (b) Native PAGE analysis of the responsiveness of pcDNAATP toward ATP. (c) Calibration curve of the fluorescence intensity as a function of the ATP concentration. pcDNAATP, 375 nM. (d) Fluorescence signals of ATP and other nucleoside triphosphates. The concentration of each nucleoside triphosphate is 1.0 mM. Data represent means ± SD (n = 3). (e) Sensing of ATP in A549 cells. The corresponding fluorescence signals of ATP in A549 cells with different treatments are shown on the right. Scale bar, 20 μm. Data represent means ± SD (n = 97, 91, 67, and 95 cells, from left to right). | ||
Cas12a-mediated sensing of miRNA
To further extend the applicability of the pcDNA-based Cas12a assay, we tested its potential in the live-cell sensing of the disease-associated miRNAs. The miRNA-21 is a known oncogenic miRNA, which has been identified as upregulated in almost all kinds of cancers and involved in tumorigenesis.57 Here, taking miRNA-21 as the model miRNA biomarker, pcDNAmiRNA was devised to enable the specific response toward miRNA-21. Analogously, the advantages of the bubble structure and no PAM requirement allow us to construct pcDNAmiRNA based on a blocked pDNA. Similar to the pcDNAATP, the complementary sequence of miRNA-21 conjugating a 6 nt tail at the 3′ terminus is introduced to block the functionality of a pDNA in the pcDNAmiRNA (Fig. 5a). In principle, miRNA-21 hybridizes with the toehold in pcDNAmiRNA and initiates the strand exchange reaction, thereby restoring the function of the pDNA. Therefore, by simply designing pcDNAmiRNA, the existence of miRNA-21 can be converted into the promoted nuclease activity of LbCas12a, which can generate amplified fluorescence signals through the trans-cleavage of FQ reporters. As shown in Fig. 5b, pcDNAmiRNA only showed a very slight background fluorescence signal when incubated with LbCas12a/gRNA complexes and FQ reporters. In contrast, a significant fluorescence response was observed in the presence of miRNA-21. The response of pcDNAmiRNA toward miRNA-21 was also validated by gel shift assay (Fig. S20†). Analogously pcDNAmiRNA was designed and fabricated to detect miRNA-122 (Fig. S21†). Notably, the parallel pcDNAmiRNA only responded to their corresponding miRNA targets with a negligible crosstalk (Fig. 5c). Under the optimal conditions, the fluorescence response in this assay showed a linear relationship between miRNA-21 concentration in the range of 1–250 nM with a limit of detection of 0.25 nM (Fig. 5d). Next, the feasibility of the pcDNA-based Cas12a assay in probing the endogenous miRNAs in living cells was further investigated. Toward this end, the components of this assay were co-transfected into A549 cells and LO2 cells, respectively. As shown in Fig. 5e and S22,† bright fluorescent spots were observed in the cytoplasm of A549 cells transfected with all the components, accomplished by the weak background fluorescence in the control group without pcDNAmiRNA. At the same time, no significant fluorescence signal was detected in the LO2 cells, suggesting the low expression of miRNA21. These results agreed with the higher expression level of miRNA-21 in cancerous A549 cells determined by qRT-PCR (inset of Fig. 5f). Taken together, these data have proved that the pcDNA-based Cas12a assay can be extended for the sensitive sensing of endogenous miRNAs in living cells.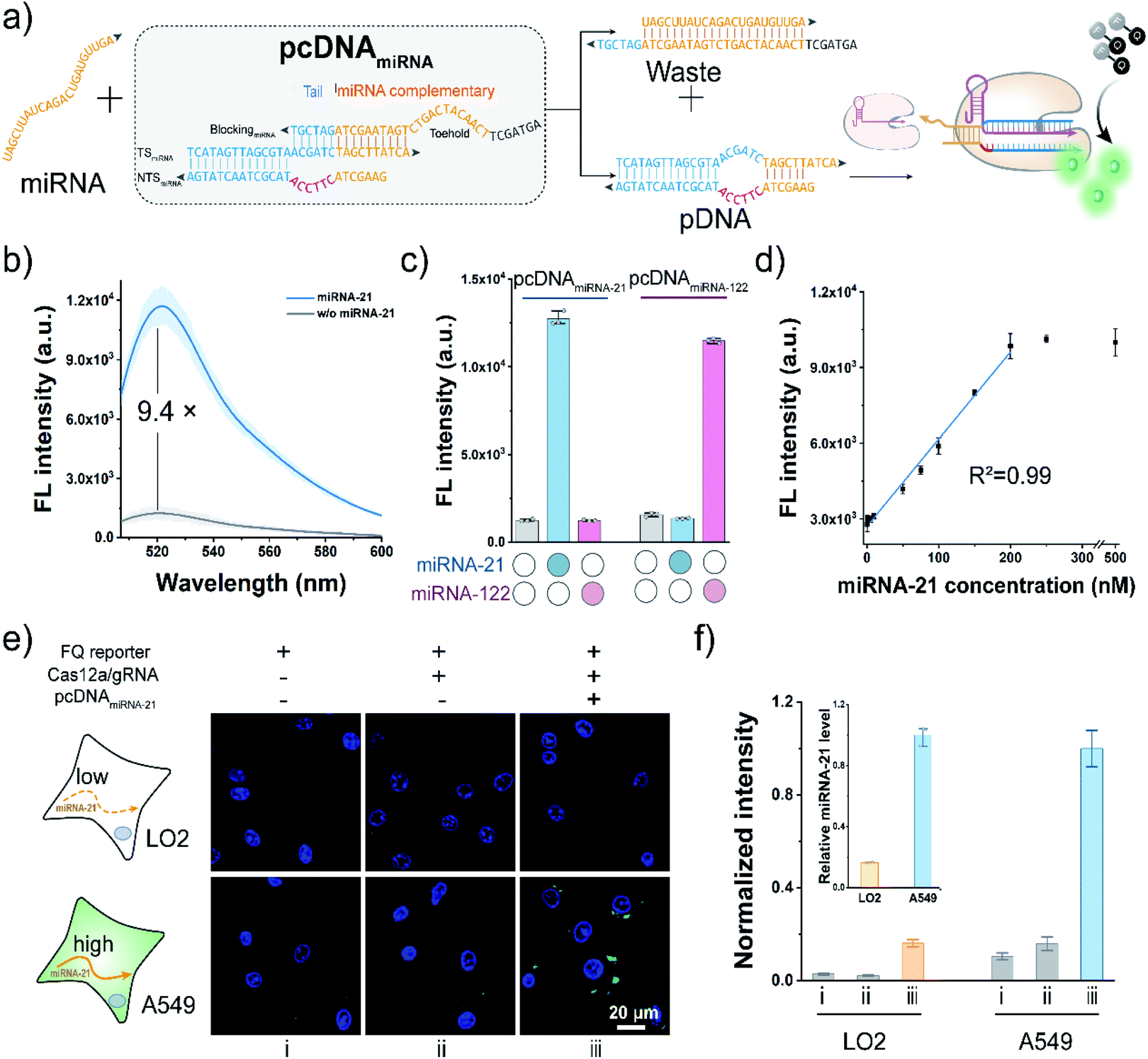 | ||
| Fig. 5 The pcDNA-based Cas12a assay for the sensing of miRNA. (a) Schematic diagram of the sensing of miRNA based on pcDNAmiRNA. (b) Fluorescence emission spectra of the pcDNA-based Cas12a assay in response to miRNA-21. pcDNAmiRNA, 100 nM. miRNA-21, 500 nM. (c) Fluorescence signals of miRNA-21 and miRNA-122 in the orthogonal assay. The concentration of each miRNA is 500 nM. (d) Calibration curve of the observed fluorescence intensity as a function of the miRNA-21 concentration. (e) Sensing of endogenous miRNA-21 in living cells using the pcDNA-based Cas12a assay. (f) Fluorescence signals of each cell line with different treatments. The inset shows the qRT-PCR result of the relative miRNA-21 levels in LO2 and A549 cells. Scale bar, 20 μm. Data represent means ± SD (n = 81, 78, 67, 49, 57, and 82 cells, from left to right; n = 3 for qRT-PCR). | ||
Conclusions
The CRISPR-Cas system has been repurposed as a powerful tool for imaging genomic loci, mRNA, and their dynamics in living cells, due to its RNA-guided precise nucleic acid sequence recognition capacity. Here, we demonstrated that the CRISPR-Cas system presents a generic live-cell biosensing tool by extending its applicability to the monitoring of telomerase, ATP and miRNA in living cells. Based on the structural and mechanistic studies, we envisioned eliminating the PAM preference of the CRISPR-Cas12a system toward DNA substrates by introducing an artificial unwound seed region. After a comprehensive investigation, we validated this assumption and developed a series of pDNAs containing a 6 nt bubble in the seed region. These pDNAs can activate both the cis- and trans-cleavage activities of LbCas12a even better than the corresponding wild-type counterparts. Further studies revealed that the bubble structure is crucial to maintain the function of the pcDNAs, which allows us to switch off their function by truncating or blocking the TS in these DNAs. By integrating with enzymatic chain elongations or functional nucleic acids, we constructed the pcDNA whose activities depend on the presence of the corresponding target biomolecules, including telomerase, ATP and miRNA-21. Moreover, we demonstrated the potential of Cas12a's trans-cleavage effect in live-cell biosensing for the first time. Finally, sensitive detection of these biomolecules in living cells was achieved by leveraging the trans-cleavage effect for signal reporting, expanding the applicability of CRISPR-Cas system in live-cell biosensing.This work constructed pcDNAs and leveraged the trans-cleavage activity of CRISPR-Cas12a for versatile live-cell sensing, which possesses several merits: (i) no PAM requirement and a bubble structure in the pcDNAs enable great flexibility and high degree of freedom in designing stimulus-responsiveness; (ii) scalability can be readily implemented by coupling with diverse functional nucleic acid motifs; (iii) engineering on a DNA substrate represents simplicity and low cost in methodology; (iv) high turnovers of trans-cleavage allow for amplified signal reporting. In this live-cell sensing system, the reaction kinetics relies on the diffusion of each component for random collisions and interactions in the cytoplasm, leading to a compromise in detection speed and sensitivity. Considering that biochemical reactions can be accelerated by concentrating the reactants together, trapping the components of the sensing system in confined spaces via DNA technology or liquid–liquid phase separation could be a promising solution to mitigate this limitation.58–61
In summary, this work has exploited the potential of the CRISPR-Cas system in live-cell biosensing by developing a facile and generalizable strategy. This strategy engineers the DNA substrates with a generalizable stimulus-responsive mechanism and employs the trans-cleavage activity of CRISPR-Cas12a for signal reporting. Such a rational combination enables the versatile sensing of different types of biomolecules in living cells. Taking advantage of the programmability of Watson–Crick base pairing and the diversity of functional nucleic acids, this pcDNA-based Cas12a live-cell biosensing tool can be expanded into a generic biosensing system for the monitoring of intracellular biomolecules in the future. Therefore, this work presents a novel paradigm for developing high-performance CRISPR-Cas biosensing systems, exhibiting great potential in basic biochemical research and clinical diagnostics.
Data availability
All data required to evaluate the conclusions are present in the paper and/or ESI.†Author contributions
S. C., C. L. and Z. N. conceived the original idea. S. C. and S. P. designed the experiments. S. C., R. W. and S. X. performed most of the assays. S. C. and C. L. performed data analysis and wrote the manuscript. C. L., Y. H. and Z. N. commented and revised the paper. Z. N. and C. L. supervised the project.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Key R&D Program of China (No. 2021YFA0910100), and the National Natural Science Foundation of China (No. 22034002, 21725503, 21974038, and 22074034).Notes and references
- D. J. Stephens and V. J. Allan, Science, 2003, 300, 82–86 CrossRef CAS PubMed.
- V. Ntziachristos, Nat. Methods, 2010, 7, 603–614 CrossRef CAS PubMed.
- R. N. Day and M. W. Davidson, Chem. Soc. Rev., 2009, 38, 2887–2921 RSC.
- K. Quan, C. Yi, X. Yang, X. He, J. Huang and K. Wang, TrAC, Trends Anal. Chem., 2020, 124, 115784 CrossRef CAS.
- S. J. J. Brouns, M. M. Jore, E. R. Westra, R. J. H. Slijkhuis, A. P. L. Snijders, M. J. Dickman, K. S. Makarova, E. V. Koonin and J. van der Oost, Science, 2008, 321, 960–964 CrossRef CAS PubMed.
- K. S. Makarova, Y. I. Wolf, O. S. Alkhnbashi, F. Costa, S. A. Shah, S. J. Saunders, R. Barrangou, S. J. J. Brouns, E. Charpentier, D. H. Haft, P. Horvath, S. Moineau, F. J. M. Mojica, R. M. Terns, M. P. Terns, M. F. White, A. F. Yakunin, R. A. Garrett, J. van der Oost, R. Backofen and E. V. Koonin, Nat. Rev. Microbiol., 2015, 13, 722–736 CrossRef CAS PubMed.
- S. Shmakov, O. O. Abudayyeh, K. S. Makarova, Y. I. Wolf, J. S. Gootengerg, E. Semenova, L. Minakhin, H. Joung, S. Konermann, K. Severinov, F. Zhang and E. V. Koonin, Mol. Cell, 2015, 60, 385–397 CrossRef CAS PubMed.
- T. Yamano, H. Nishimasu, B. Zetsche, H. Hirano, I. M. Slaymaker, Y. Li, I. Fedorova, T. Nakane, K. S. Makarova, E. V. Koonin, R. Ishitani, F. Zhang and O. Nureki, Cell, 2016, 165, 959–962 CrossRef PubMed.
- A. Pickar-Oliver and C. A. Gersbach, Nat. Rev. Mol. Cell Biol., 2019, 20, 490–507 CrossRef CAS PubMed.
- S. C. Knight, R. Tjian and J. A. Doudna, Angew. Chem., Int. Ed., 2018, 57, 4329–4337 CrossRef CAS PubMed.
- X. Wu, S. Mao, Y. Ying, C. J. Krueger and A. K. Chen, Genomics, Proteomics Bioinf., 2019, 17, 119–128 CrossRef PubMed.
- B. Chen, L. A. Gilbert, B. A. Cimini, J. Schnitzbauer, W. Zhang, G.-W. Li, J. Park, E. H. Blackburn, J. S. Weissman, L. S. Qi and B. Huang, Cell, 2013, 155, 1479–1491 CrossRef CAS PubMed.
- X. Chen, D. Zhang, N. Su, B. Bao, X. Xie, F. Zuo, L. Yang, H. Wang, L. Jiang, Q. Lin, M. Fang, N. Li, X. Hua, Z. Chen, C. Bao, J. Xu, W. Du, L. Zhang, Y. Zhao, L. Zhu, J. Loscalzo and Y. Yang, Nat. Biotechnol., 2019, 37, 1287–1293 CrossRef CAS PubMed.
- S. Wang, Y. Hao, L. Zhang, F. Wang, J. Li, L. Wang and C. Fan, CCS Chem., 2019, 1, 278–285 CAS.
- D. A. Nelles, M. Y. Fang, M. R. O'Connell, J. L. Xu, S. J. Markmiller and J. A. Doudna, Cell, 2016, 165, 488–496 CrossRef CAS PubMed.
- O. O. Abudayyeh, J. S. Gootenberg, P. Essletzbichler, S. Han, J. Joung, J. J. belanto, V. Verdine, D. B. T. Cox, M. J. Kellner, A. Regev, E. S. Lander, D. F. Voytas, A. Y. Ting and F. Zhang, Nature, 2017, 550, 280–284 CrossRef PubMed.
- L.-Z. Yang, Y. Wang, S.-Q. Li, R.-W. Yao, P.-F. Luan, H. Wu, G. G. Carmichael and L.-L. Chen, Mol. Cell, 2019, 76, 1–17 CrossRef PubMed.
- K. Zhang, R. Deng, X. Teng, Y. Li, Y. Sun, X. Ren and J. Li, J. Am. Chem. Soc., 2018, 140, 11293–11301 CrossRef CAS PubMed.
- H. Wang, M. Nakamura, T. R. Abbott, D. Zhao, K. Luo, C. Yu, C. M. Nguyen, A. Lo, T. P. Daley, M. L. Russa, Y. Liu and L. S. Qi, Science, 2019, 365, 1301–1305 CrossRef CAS PubMed.
- Y.-B. Yang, Y.-D. Tang, Y. Hu, F. Yu, J.-Y. Xiong, M.-X. Sun, C. Lyu, J.-M. Peng, Z.-J. Tian, X.-H. Cai and T.-Q. An, Nano Lett., 2020, 20, 1417–1427 CrossRef CAS PubMed.
- Y. Nihongaki, T. Otabe, Y. Ueda and M. Sato, Nat. Chem. Biol., 2019, 15, 882–888 CrossRef CAS PubMed.
- B. L. Oakes, C. Fellmann, H. Rishi, K. L. Taylor, S. M. Ren, D. C. Nadler, R. Yokoo, A. P. Arkin, J. A. Doudna and D. F. Savage, Cell, 2019, 176, 254–267 CrossRef CAS PubMed.
- J. Hemphill, E. K. Borchardt, K. Brown, A. Asokan and A. Deiters, J. Am. Chem. Soc., 2015, 137, 5642–5645 CrossRef CAS PubMed.
- M. H. Hanewich-Hollatz, Z. Chen, L. M. Hochrein, J. Huang and N. A. Pierce, ACS Cent. Sci., 2019, 5, 1241–1249 CrossRef CAS PubMed.
- S.-R. Wang, L.-Y. Wu, H.-Y. Huang, W. Xiong, J. Liu, L. Wei, P. Yin, T. Tian and X. Zhou, Nat. Commun., 2020, 11, 91 CrossRef PubMed.
- W. Tang, J. H. Hu and D. R. Liu, Nat. Commun., 2017, 8, 15939 CrossRef CAS PubMed.
- H. Yin, C.-Q. Song, S. Suresh, Q. Wu, S. Walsh, H. Rhym, E. Mintzer, M. F. Bolukbasi, L. J. Zhu, K. Kauffman, H. Mou, A. Oberholzer, J. Ding, S.-Y. Kwan, R. L. Bogorad, T. Zatsepin, V. Koteliansky, S. A. Wolfe, W. Xue, R. Langer and D. G. Anderson, Nat. Biotechnol., 2017, 35, 1179–1187 CrossRef CAS PubMed.
- Y. Liu, R. S. Zou, S. He, Y. Nihongaki, X. Li, S. Razavi, B. Wu and T. Ha, Science, 2020, 368, 1265–1269 CrossRef CAS PubMed.
- L. Oesinghaus and F. C. Simmel, Nat. Commun., 2019, 10, 2092 CrossRef PubMed.
- Y.-J. Chen, B. Groves, R. A. Muscat and G. Seelig, Nat. Nanotechnol., 2015, 10, 748–760 CrossRef CAS PubMed.
- B. Zetsche, J. S. Gootenberg, O. O. Abudayyeh, I. M. Slaymaker, K. S. Makarova, P. Essletzbichler, S. E. Volz, J. Joung, J. van der Oost, A. Regev, E. V. Koonin and F. Zhang, Cell, 2015, 163, 759–771 CrossRef CAS PubMed.
- O. O. Abudayyeh, J. S. Gootenberg, S. Konermann, J. Joung, I. M. Slaymaker, D. B. T. Cox, S. Shmakov, K. S. Makarova, E. Semenova, L. Minakhin, K. Severinov, A. Regev, E. S. Lander, E. V. Koonin and F. Zhang, Science, 2016, 353, aaf5573 CrossRef PubMed.
- K. Kiga, X.-E. Tan, R. Ibarra-Chávez, S. Watanabe, Y. Aiba, Y. Li, F.-Y. Sato'o, T. Sasahara, B. Cui, M. Kawauchi, T. Boonsir, K. Thitiananpakorn, Y. Taki, A. H. Azam, M. Suzuki, J. R. Penadés and L. Cui, Nat. Commun., 2020, 11, 2934 CrossRef CAS PubMed.
- D. Dong, K. Ren, X. Qiu, J. Zheng, M. Guo, X. Guan, H. Liu, N. Li, B. Zhang, D. Yang, C. Ma, S. Wang, D. Wu, Y. Ma, S. Fan, J. Wang, N. Gao and Z. Huang, Nature, 2016, 532, 522–526 CrossRef CAS PubMed.
- S. Stella, P. Mesa, J. Thomsen, B. Paul, P. Alcón, S. B. Jensen, B. Saligram, M. E. Moses, N. S. Hatzakis and G. Montoya, Cell, 2018, 175, 1–16 CrossRef PubMed.
- D. C. Swarts and M. Jinek, Mol. Cell, 2019, 73, 589–600 CrossRef CAS PubMed.
- Y. Jeon, Y. H. Choi, Y. Jang, J. Yu, J. Goo, G. Lee, Y. K. Jeong, S. H. Lee, I.-S. Kim, J.-S. Kim, C. Jeong, S. Lee and S. Bae, Nat. Commun., 2018, 9, 2777 CrossRef PubMed.
- S. Peng, Z. Tan, S. Chen, C. Lei and Z. Nie, Chem. Sci., 2020, 11, 7362–7368 RSC.
- J. S. Chen, E. Ma, L. B. Harrington, M. D. Costa, X. Tian, J. M. Palefsky and J. A. Doudna, Science, 2018, 360, 436–439 CrossRef CAS PubMed.
- S.-Y. Li, Q.-X. Cheng, J.-M. Wang, X.-Y. Li, Z.-L. Zhang, S. Gao, R.-B. Cao, G.-P. Zhao and J. Wang, Cell Discovery, 2018, 4, 20 CrossRef PubMed.
- Y. Dai, R. A. Somoza, L. Wang, J. F. Welter, Y. Li, A. I. Caplan and C. C. Liu, Angew. Chem., Int. Ed., 2019, 58, 17399–17405 CrossRef CAS PubMed.
- K. Shi, S. Xie, R. Tian, S. Wang, Q. Lu, D. Gao, C. Lei, H. Zhu and Z. Nie, Sci. Adv., 2021, 7, eabc7802 CrossRef CAS PubMed.
- H. Yue, B. Shu, T. Tian, E. Xiong, M. Huang, D. Zhu, J. Sun, Q. Liu, S. Wang, Y. Li and X. Zhou, Nano Lett., 2021, 21, 4643–4653 CrossRef CAS PubMed.
- Y. Dong, C. Yao, Y. Zhu, L. Yang, D. Luo and D. Yang, Chem. Rev., 2020, 120, 9420–9481 CrossRef CAS PubMed.
- D. Y. Zhang and G. Seelig, Nat. Chem., 2011, 3, 103–113 CrossRef CAS PubMed.
- G. E. Ghanim, A. J. Fountain, A.-M. M. van Roon, R. Rangan, R. Das, K. Collins and T. H. D. Nguyen, Nature, 2021, 593, 449–453 CrossRef CAS PubMed.
- K. Collins, Nat. Rev. Mol. Cell Biol., 2006, 7, 484–494 CrossRef CAS PubMed.
- R. Qian, L. Ding, L. Yan, M. Lin and H. Ju, J. Am. Chem. Soc., 2014, 136, 8205–8208 CrossRef CAS PubMed.
- S. M. Melana, J. F. Holland and B. G.-T. Pogo, Clin. Cancer Res., 1998, 4, 693–696 CAS.
- J. P. Broughton, X. Deng, G. Yu, C. L. Fasching, V. Servellita, J. Singh, X. Miao, J. A. Streithorst, A. Granados, A. Sotomayor-Gonzalez, K. Zorn, A. Gopez, E. Hsu, W. Gu, S. Miller, C.-Y. Pan, H. Guevara, D. A. Wadford, J. S. Chen and C. Y. Chiu, Nat. Biotechnol., 2020, 38, 870–874 CrossRef CAS PubMed.
- M. Liang, Z. Li, W. Wang, J. Liu, L. Liu, G. Zhu, L. Karthik, M. Wang, K.-F. Wang, Z. Wang, J. Yu, Y. Shuai, J. Yu, L. Zhang, Z. Yang, C. Li, Q. Zhang, T. Shi, L. Zhou, F. Xie, H. Dai, X. Liu, J. Zhang, G. Liu, Y. Zhuo, B. Zhang, C. Liu, S. Li, X. Xia, Y. Tong, Y. Liu, G. Alterovitz, G.-Y. Tan and L.-X. Zhang, Nat. Commun., 2019, 10, 3672 CrossRef PubMed.
- Y. Xiong, J. Zhang, Z. Yang, Q. Mou, Y. Ma, Y. Xiong and Y. Lu, J. Am. Chem. Soc., 2020, 142, 207–213 CrossRef CAS PubMed.
- F. I. Wolf and A. Cittadini, Mol. Aspects Med., 2003, 24, 3–9 CrossRef CAS PubMed.
- A. Tomita, M. Zhang, F. Jin, W. Zhuang, H. Takeda, T. Maruyama, M. Osawa, K. ichi Hashimoto, H. Kawasaki, K. Ito, N. Dohmae, R. Ishitani, I. Shimada, Z. Yan, M. Hattori and O. Nureki, Nat. Commun., 2017, 8, 148 CrossRef PubMed.
- C. Ma, C. Lin, Y. Wang and X. Chen, TrAC, Trends Anal. Chem., 2016, 77, 226–241 CrossRef CAS.
- J. Symersky, D. Osowski, D. E. Walters and D. M. Mueller, Proc. Natl. Acad. Sci. U.S.A., 2012, 109, 13961–13965 CrossRef CAS PubMed.
- H. Dong, J. Lei, L. Ding, Y. Wen, H. Ju and X. Zhang, Chem. Rev., 2013, 113, 6207–6233 CrossRef CAS PubMed.
- N. C. Seeman and H. F. Sleiman, Nat. Rev. Mater., 2017, 3, 17068 CrossRef.
- A. Joesaar, S. Yang, B. Bögels, A. van der Linden, P. Pieters, B. V. V. S. Pavan Kumar, N. Dalchau, A. Phillips, S. Mann and T. F. A. de Greef, Nat. Nanotechnol., 2019, 14, 369–378 CrossRef CAS PubMed.
- S. Boeynaems, S. Alberti, N. L. Fawzi, T. Mittag, M. Polymenidou, F. Rousseau, J. Schymkowitz, J. Shorter, B. Wolozin, L. V. D. Bosch, P. Tompa and M. Fuxreiter, Trends Cell Biol., 2018, 28, 420–435 CrossRef CAS PubMed.
- D. Bracha, M. T. Walls and C. P. Brangwynne, Nat. Biotechnol., 2019, 37, 1435–1445 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sc05558e |
| This journal is © The Royal Society of Chemistry 2022 |