
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLayered 2D PtX2 (X = S, Se, Te) for the electrocatalytic HER in comparison with Mo/WX2 and Pt/C: are we missing the bigger picture?
Sengeni
Anantharaj
 *ab and
Suguru
Noda
ab
*ab and
Suguru
Noda
ab
aDepartment of Applied Chemistry, School of Advanced Science and Engineering, Waseda University, 3-4-1 Okubo, Shinjuku-ku, Tokyo 169-8555, Japan. E-mail: ananth@aoni.waseda.jp; anantharaj1402@gmail.com
bWaseda Research Institute for Science and Engineering, Waseda University, 3-4-1 Okubo, Shinjuku-ku, Tokyo 169-8555, Japan
First published on 7th March 2022
Abstract
The hydrogen evolution reaction (HER) is best catalyzed by metallic Pt with the lowest overpotential, Tafel slope, and highest exchange current density. But its scarcity made us look for abundant alternatives which come at the price of poor activity and stability. Hence, all non-Pt HER electrocatalysts are compared with Pt (often Pt/C). In such cases, a closer activity to that of Pt is usually appreciated for non-Pt materials. In contrast, when an HER electrocatalyst is made of Pt, it is expected to surpass the activity of Pt/C as it has the same Pt. A familiar example of this kind is the dilution of Pt without any compromise in activity. The recently evolved layered dichalcogenides of Pt (PtX2) do not satisfy this expectation as they not only perform poorer than Pt/C but also against other familiar MX2 HER electrocatalysts such as MoX2 and WX2. However, the studies that engaged in the evaluation of the HER activity of PtX2 were quite useful in deducing the structure–activity relationship and mechanism which are clearly inevitable pieces of knowledge needed in all kinds of electrocatalysis. Though PtX2 are poorer HER catalysts in their pristine form, structural engineering and other activation methods made them as active as Pt/C while a few were better than Pt/C. This perspective is dedicated to presenting the recent progress in the area of the PtX2 catalyzed HER in comparison with MoX2 and WX2 while highlighting the opportunities and challenges. Particularly, how we are missing the bigger picture in designing a Pt-based HER electrocatalyst and the ways in which poorly active PtX2 can be made into a superior one to Pt/C are critically discussed.
 Sengeni Anantharaj | Dr Sengeni Anantharaj obtained his PhD in 2018 from CSIR-CECRI, Karaikudi, Tamil Nadu, India, following which he worked with Prof. Noda as a JSPS postdoc fellow from January 2019 to March 2021. Currently, he is an Assistant Professor (Junior Researcher) at RISE, Waseda University since April 2021. He was the first ‘ECS India Section S. K. Rangarajan Graduate Student Award’ winner of the year 2017 which was presented to him by The Electrochemical Society, USA, for his noteworthy works in the area of electrocatalysis and materials electrochemistry during his PhD. He is an electrochemist by passion and a (teaching) researcher by profession. He places his interest on everything where there is a transfer of electrons that results in the transformation of matter, especially at the electrode–electrolyte interface. |
 Suguru Noda | Prof. Suguru Noda received his PhD in 1999 from The University of Tokyo, Japan, became an assistant professor and associate professor there, and then joined Waseda University in 2012 as a full professor. He is a chemical engineer conducting research in the field of materials processes. He has recently been focusing on the practical production of carbon and silicon nanomaterials such as carbon nanotubes and silicon films/nanoparticles, and applying these materials to energy and electronic devices including rechargeable batteries. |
Broader contextEnergy conversion electrocatalysis ranging from water electrolysis and fuel cells to recently attention-grabbing CO2 and N2 electrolysis depends mainly on the development of highly efficient electrocatalysts in terms of activity, stability, and selectivity. In that context, it has always been a custom to compare all the newly developed catalysts with the state-of-the-art of the field. The same is also true for the electrocatalytic hydrogen evolution reaction (HER) where Pt is the state-of-the-art. The ultimate goals in the HER are no Pt, low-Pt, and better kinetics in a medium where Pt alone cannot perform better. The recent evolution of PtX2 as HER electrocatalysts has concerned the community as they do not perform better than Pt/C despite the fact that they are also made of Pt. A material that takes a huge amount of time, energy, and resources to make but performs poorer than Pt is not as encouraging as the advancements being made with other HER electrocatalysts. Hence, it is essential to ensure that the developments happening in the field of HER electrocatalysis are focused on the ultimate goals and the bigger picture of the field rather than being driven by the fancy structural properties of a particular class of materials (i.e., PtX2). |
Introduction
The hydrogen evolution reaction (HER) is the cathodic half-cell reaction of water electrolysis that governs the H2 generation efficiency significantly alongside the anodic oxygen evolution reaction (OER).1–6 Though the kinetic complexity of the OER attracted significant attention earlier, the HER has also gained equal attention in recent years as the significance of replacing or lowering the amount of Pt used and improving the poor kinetics of Pt in the alkaline HER are realized.7–14 In general, catalysts that have little or no Pt suffer from poor activity and kinetics in all media. On the other hand, Pt-based HER catalysts suffer from poor kinetics when the concentration of protons available in the solution is very low or nil (i.e., in highly alkaline solutions).8,10,15 In such cases, the proton adsorption and discharge step (Volmer step) is coupled with water dissociation. Recently, several advancements have been made to overcome this particular issue in the alkaline HER among which heterostructuring Pt with a metal hydroxide co-catalyst and doping Ru in limited proportions are the most efficient ones.7,8,16 Materials that contain Pt and Ru are the only known HER electrocatalysts to have an onset potential of 0.0 V vs. RHE and demand low overpotentials under benchmarking conditions with a few exceptional metal phosphides of Fe and Co.6 Promising alternatives to Pt are chalcogenides, phosphides, nitrides, and carbides of transition metals that include but are not limited to Ni, Co, Fe, Cu, Mn, W, Mo, Rh, and Re.3,6,17 Particularly, phosphides of Fe and its alloys show very close HER activity to those of Pt and Pt-based HER electrocatalysts.18–22 However, it is difficult to prepare phosphides, carbides, and nitrides and they always require high pressure and temperature conditions with an inert atmosphere.14,23–26 On the other hand, chalcogenides have always been obtained under relatively milder conditions and often show appreciable HER activity in terms of an overpotential at 10 mA cm−2 (apparent activity) that is just 50 to 80 mV higher than that of metal phosphides and 80 to 150 mV higher than those of Pt and Ru-based catalysts.1,9,27 Chalcogenides of first-row transition metals (mainly of Ni, Co, Fe, and Cu) are of different stoichiometries and catalyze the HER distinctly depending on the metal to chalcogen ratio.3,28 However, these metals rarely form 2D layered structures as their electronic configurations are usually not fit for the formation of octahedral and trigonal prismatic phases. Layered 2D transition metal chalcogenides (TMDs), on the other hand, are the most preferred form of crystallization for many transition metal cations in their tetravalent state including W, Mo, and Pt. These materials are of recent interest in the area of energy conversion electrocatalysis because of their fascinating physical and chemical properties and their structural resemblance to graphene.29–32 Among them, WX2 and MoX2 are reported in vast counts in the literature for their synthesis, and optical, electronic, spintronic, magnetic, and electrocatalytic properties.33–38 Tetravalent cations of both Mo and W have just two electrons in their d-orbitals which tend to degenerate in a trigonal prismatic fashion and always result in a semiconducting 2H phase under ordinary synthetic conditions.29 On the other hand, the tetravalent cation of Pt having six d-electrons degenerates into octahedral symmetry in which the t2g level can be completely filled. Hence, PtX2 always and almost crystallizes in the metallic 1T phase.33,39 TMDs of W, Mo, and Pt have been reported for the HER recently in literature more often now than ever mainly because of the recent revolution in the area of materials science and technology. MoX2 and WX2 are certainly worth reporting for the HER as they possess better activity than metallic Mo and W.40–42 In contrast, pristine PtX2 can never perform better than its metallic counterpart Pt in HER electrocatalysis.39 In that context, it is meaningless to toil with PtX2 just to have a poorer HER electrocatalyst when we actually have the state-of-the-art metallic Pt. However, other advancements (i.e., recent understanding of how PtX2 works in HER electrocatalysis in comparison with other MX2 materials and the use of PtX2 as a good starting material or a pre-catalyst for making an excellent HER electrocatalyst) made in this area are worthy of a critical discussion. Thus, this can lead to further advancements in the area of HER electrocatalysis and associated catalyst design strategies. This perspective comes with such a critical opinionated discussion on the recent developments in the area of PtX2 design and application to HER electrocatalysis.TMDs: an overview
TMDs are a unique class of materials formed by transition metals when they combine chemically with the chalcogenide anions with a general formula of MX2 in which M is a tetravalent transition metal cation and X is a chalcogenide dianion.38 Any transition metal that can have an electronically stable 4+ oxidation state can form TMDs. TMDs resemble 2D graphene in their structure and tend to have resonating properties.30,39,43–49 Very common transition metals with a stable 4+ oxidation state and a capability of forming TMDs are W, Mo, Ti, Cr, Mn, Re, Pd, and Pt. Among them, TMDs of Mo and W are the most frequently used HER electrocatalysts. TMDs formed by these stable tetravalent cations of transition metals commonly crystallize in trigonal prismatic (2H) and octahedral (1T) lattices (Fig. 1a and b) of which the 2H-phase is semiconducting and the 1T-phase is metallic. Other than these two phases, a few M4+ ions from the transition metal series are known to form the semiconducting 3R phase of the same trigonal prismatic lattice system to that of the 2H phase but with a different point of symmetry under non-standard conditions.50 In recent years, the relationship between the d-electron population and the degeneracy of electronic states upon forming TMDs is correlated. Tetravalent transition metal cations with incompletely filled d-orbitals (e.g. W4+ and Mo4+ with four d-electrons) degenerate into e′ (dxz,yz), a1 (dz2), and e (dx2−y2,xy) sub-levels forming the trigonal prismatic structure so as to have a high crystal field stabilization. On the other hand, tetravalent cations with more than four d-electrons degenerate into eg (dz2,x2−y2) and t2g (dxy,yz,xz) sub-levels forming the octahedral structure.30 A familiar example of this kind is Pt4+ with six d-electrons.39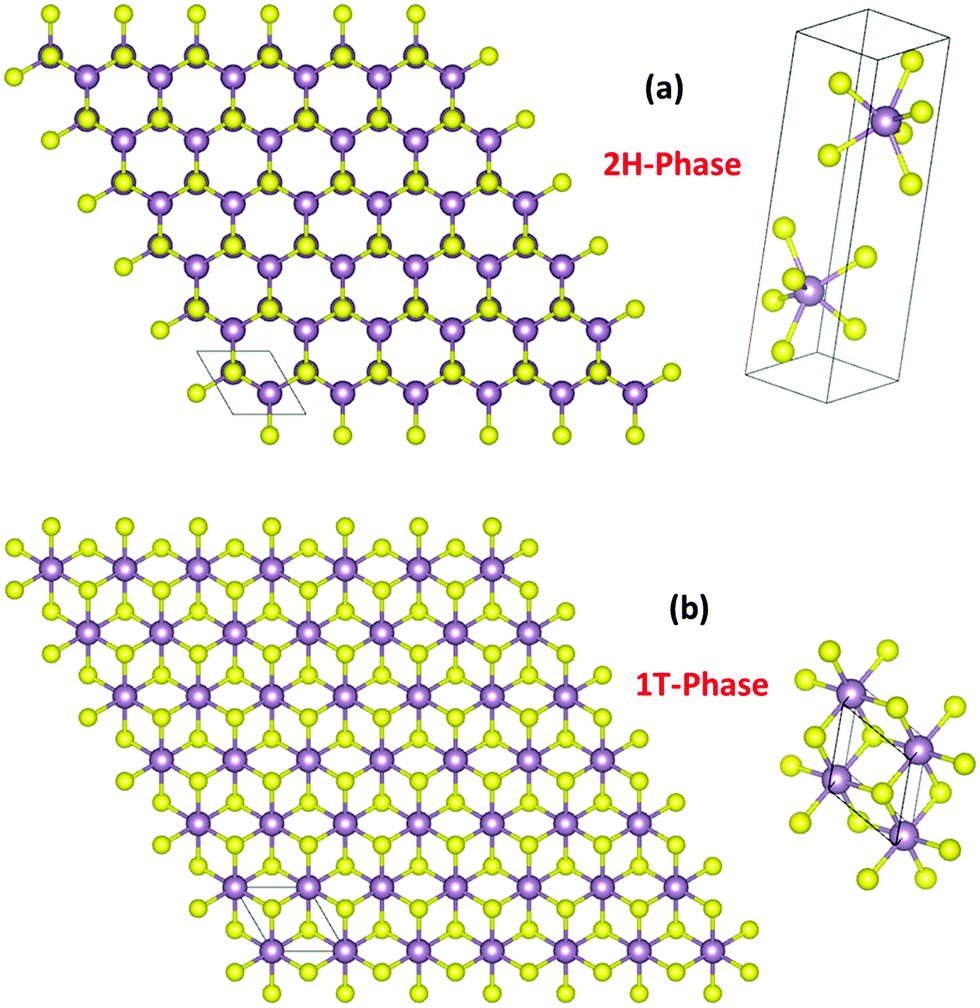 | ||
| Fig. 1 2H (a) and 1T (b) phases formed commonly by TMDs with the unit cells showing trigonal prismatic and octahedral arrangements of atoms, respectively. | ||
When Pt4+ undergoes electronic degeneration to result in an octahedral geometry, the t2g sub-level gets completely filled which in turn results in high crystal field stabilization. Between the 2H and 1T phases of TMDs, the latter was found to be outperforming the former as it is metallic in nature, thus, offers a better charge transfer during the HER. On the other hand, the 2H phase of MoX2 and WX2 in a few cases was found to transform into the 1T phase during the HER as Mo4+ and W4+ cations are reduced electrochemically in the potential window of the HER.49,51 However, it is not shown that such a transformation is possible with other MX2 catalysts under similar conditions. As far as the synthesis is concerned, high-quality oxygen impurity-free TMDs are obtained only via physical methods in a vacuum. Vapor deposition, sputtering, and laser ablation are the ones prominently used in this area.52–54 Though aqueous routes are known to impart a considerable quantity of oxygen impurities, their effect on HER activity is poorly understood. Hence, irrespective of the method by which the TMDs are synthesized and the lattice system in which they crystallized into, their HER activity tends not to vary much unless other modes of material engineering strategies are deployed to enhance the activities additionally. The most common material engineering strategies used with TMDs are amorphization, making defects and vacancies, and chemically opening up the basal planes by rupturing M–X bonds and creating more edges.41,55–57 Other than these, compositing, heterostructuring, anion exchange, and doping have also been shown to enhance the HER activity of these TMDs as these strategies are known to bring out HER-favouring changes in their electronic structures.58–61 The electronic structure of a catalyst is crucial in HER electrocatalysis as it reveals the density of states (DOS) at different applied potentials. In general, materials with a significant DOS intensity at and around 0 eV are better for the HER as the reversible potential of HER is 0.0 V vs. RHE. Having a high DOS intensity at 0.0 eV ensures a better charge transfer at and around the reversible potential of the HER which is the most important requirement of all for an electrocatalyst.62
Unlike many other energy conversion electrocatalytic reactions, the HER does not require the catalytic site to undergo a self-redox cycle to catalyse the evolution of H2. All that happens in the HER is adsorption of proton (water dissociation coupled-proton adsorption in alkali), discharge, and delivery of H2 and hence, the DOS matters the most besides the energy of interactions of proton/water and M–H intermediates. Fig. 2 shows the electronic structure of a few commonly used TMDs for the HER in comparison with Pt and PtO2.63 Among them, Pt is the better HER electrocatalyst than all and the same is witnessed by its electronic structure and high DOS intensity at 0.0 eV. For materials with a weak and too strong proton/water adsorption free energy change, the overpotential (work required to evolve H2) is considerably higher. Such materials, even when supported by HER-favouring DOS intensities will perform poorly because of their opposing energy of interaction with reaction intermediates. Familiar examples for this kind are Ru and Ni, these two metals have HER-favouring DOS intensities at and around 0.0 eV but they form strong metal hydride bonds prohibiting the easier delivery of H2. Particularly, Ru has the same onset as that of Pt because of their similar DOS maps yet the exchange current density of Ru remains a few orders lower than that of Pt because of its nature of forming strong hydride bonds.7,64 Hence, for designing an efficient HER electrocatalyst, it is essential to pay attention to both the electronic structure and the energies of the interaction of intermediates involved.
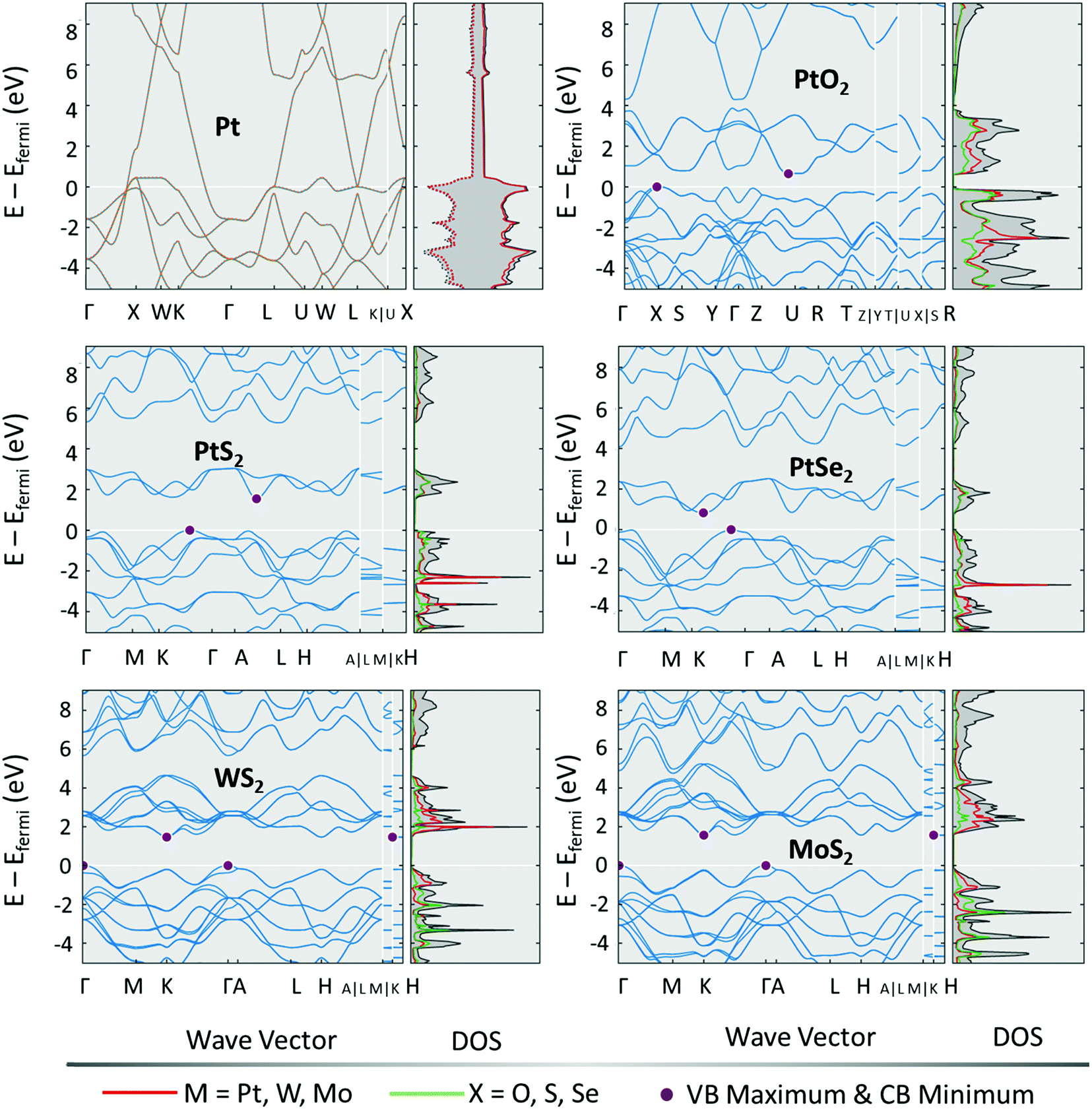 | ||
| Fig. 2 Electronic band structures with DOS for Pt, PtO2, PtS2, PtSe2, WS2, and MoS2. Reproduced with permission from ref. 64 copyright (2012, materials project). | ||
Things to go after in the HER
The HER is the cathodic half-cell reaction of water electrolysis which is relatively simpler than its counterpart, the anodic OER in terms of mechanism. Even though the OER is the one that takes up a huge amount of energy due to its sluggish kinetics, understanding the mechanism of the HER is also inevitable in order to design an efficient electrocatalyst. There are only two elementary steps in the HER, namely, adsorption of proton/water and subsequent discharge (i.e., the Volmer step: H+/H2O + e− + S → S–H (+OH−) that results in a hydridic intermediate (S–H)) and the evolution of H2 either by repeating the Volmer step via the hydridic intermediate (i.e., the Heyrovsky step: ) or via chemical release of H2 from two adjacent hydridic intermediates (i.e., the Tafel step: 2S–H → 2S + H2↑). In theory, the HER is the reaction that occurs at 0.0 V vs. RHE on the surface of a standard hydrogen electrode (Pt(s)|1 M [H+]/H2(g) 1 atm) at pH 0 and against RHE under all conditions.5 There are only two other elements (Ru and Rh) which can have an onset potential of 0.0 V vs. RHE for the HER.7,65 However, their exchange current density is too low when compared to that of Pt because of their strong adsorption tendency towards H and OH adatoms that result during the HER. Hence, Pt is still the best of all.64 However, there have been notable developments in recent years in the area of catalyst design. In the last five years, many non-Pt catalysts have been reported to have excellent HER activity in both acid and alkali and there are several reviews on the same.6,9 Hence, further research and development in this area of energy conversion have recently attracted greater attention.
) or via chemical release of H2 from two adjacent hydridic intermediates (i.e., the Tafel step: 2S–H → 2S + H2↑). In theory, the HER is the reaction that occurs at 0.0 V vs. RHE on the surface of a standard hydrogen electrode (Pt(s)|1 M [H+]/H2(g) 1 atm) at pH 0 and against RHE under all conditions.5 There are only two other elements (Ru and Rh) which can have an onset potential of 0.0 V vs. RHE for the HER.7,65 However, their exchange current density is too low when compared to that of Pt because of their strong adsorption tendency towards H and OH adatoms that result during the HER. Hence, Pt is still the best of all.64 However, there have been notable developments in recent years in the area of catalyst design. In the last five years, many non-Pt catalysts have been reported to have excellent HER activity in both acid and alkali and there are several reviews on the same.6,9 Hence, further research and development in this area of energy conversion have recently attracted greater attention.
As for every electrocatalyst, HER electrocatalysts are also screened for their activity, stability, and selectivity.66 In extreme pH conditions, there is neither a catalyst-confined self-redox reaction nor an electrolyte-based competing reaction. Hence, selectivity is not an issue with most of the HER electrocatalysts. Stability, on the other hand, is tested by both potentiodynamic sweeping techniques and controlled current/potential voltammetry and for most of the metal-based HER electrocatalysts, it is quite good. However, the stability of most of the alkaline HER electrocatalysts is poorer where the water dissociation coupled Volmer step (proton adsorption and discharge) and highly alkaline environment cause anion exchange at the surface leading to the formation of metal hydroxide containing heterostructures.8,67 It has recently been understood that a metal hydroxide interface is essential to realize better HER activity in alkaline medium even with Pt and Ru which are known for their exceptionally lower overpotentials at benchmarking conditions.7 As stability and selectivity are almost of no concern in the HER, activity is given greater attention. In general, the activity of any electrocatalyst can be classified into apparent activity and intrinsic activity in which the former is the performance of the catalyst in practical conditions while the latter is the true activity of the catalyst free from the effects of iR drop, surface area, and mass.68,69 The intrinsic activity forms the base for further optimization and structural engineering to realize a better apparent activity under practical conditions. The apparent activity of an HER electrocatalyst is given by the overpotential at a fixed current density which is usually 10 mA cm−2, exchange current density, and mass activity whereas intrinsic activity is given only by turnover frequency (TOF) and specific activity at a given overpotential. Both the TOF and specific activity require the knowledge of the real surface area, the exact number of active sites, and faradaic efficiency (FE) for precise determination.69 Between the TOF and specific activity, the former is a straightforward measure of the intrinsic activity of an electrocatalyst. We have detailed the ways in which all these activity markers can be precisely obtained in our earlier reviews and perspectives.66,68 In addition to these three primary evaluation parameters (activity, selectivity, and stability), mechanistic evaluation by Tafel analysis is also inevitable. In HER electrocatalysis, Tafel analysis provides vital information on the mechanism and relative information on the kinetics. However, the use of potentiodynamic polarization curves for Tafel analysis has significantly led to the unintended falsification of data in the literature. To get precise values of the Tafel slope and exchange current density, it is strongly advised to use steady-state responses that are corrected for 100% iR drop. More detailed discussion on the appropriate ways of Tafel analysis can be found in our recent viewpoint article.70 In general, a better HER electrocatalyst is anticipated to have lower overpotentials and higher TOF, exchange current density, stability, and selectivity.
HER activity trends in PtX2
Layered dichalcogenides of Pt4+ ions with a d6 electronic configuration always and almost prefer to degenerate into octahedral symmetry (i.e., 1T phase) unlike W4+ and Mo4+ ions. This 1T phase of TMDs is metallic and electronically highly conductive. Hence, it is obvious to anticipate that PtX2 will perform better than MoX2 and WX2 for the HER. However, in practice, no such superior activity to that of MoX2 and WX2 is generally witnessed with PtX2 especially when the latter is in its pristine form. Instead, most of the time, the activity reported for PtX2 is only as good as those reported for MoX2 and WX2. However, there are exceptions where PtX2 was shown to have better HER activity. In such exceptional cases, other factors (to be discussed in the latter part) have contributed more to the overall activity enhancement than the intrinsic activity of pristine PtX2.PtX2 with poorer HER activity than Pt or Pt/C
A general theoretical account on the HER activity of diselenide and disulphide of Pt was first given by Tsai and co-workers71 along with a bunch of other metals including W and Mo. In this study, both 1T and 2H phases with basal plane termination and edge-site termination were examined. Since Pt4+ ion can only form the 1T phase, for PtS2 and PtSe2, calculations were done only on the basal plane terminated and edge-terminated 1T phase. The Gibbs free energy changes associated with H-adsorption on the chalcogen site (ΔHX) and on Pt (ΔH) of PtS2 and PtSe2 are as follows. For the basal plane, ΔH values are 1.35 and 1.44 eV, respectively. For the edge-site, the values (of ΔH) are −0.08 and −0.02 eV, respectively. For H-adsorption on the X site (ΔHX), the basal plane of PtS2 and PtSe2 showed 0.34 and −0.18 eV, respectively, whereas for the edge-site, both of them showed significantly higher negative values (−0.84 and −0.93 eV, respectively). These values are plotted against one another in Fig. 3a and b along with the values of other TMDs. In general, for a material to be more active in any electrocatalytic reaction, the Gibbs free energy change associated with the adsorption of an intermediate should be moderate. For the HER, the values should be closer to 0.0 eV in theory. A very high positive value for H-adsorption implies poor H-adsorption while a very high negative value implies spontaneous adsorption and the formation of a strong M–H bond. Both would require more energy to release the adsorbed H atoms as H2 molecules. From the values provided above, it is explicit that only undercoordinated Pt atoms at the edge-site are appreciably active for the HER while the same Pt site in the basal plane and X sites in both the basal plane and edge-site are either too weak or too strong for H-adsorption. This information right away implies that PtX2 cannot be as efficient as Pt or Pt/C for the HER. The same trend was supported by the simulated DOSs of PtS2 and PtSe2 (Fig. 3c) which when compared with the DOSs of other TMDs studied together in this report were found to be having similar patterns and no population of electrons at around 0.0 eV. Soon after this study, Chia and co-workers39 reported the monotonic dependence of HER activity of PtX2 on the size of the chalcogen present in it. They found that the increasing size of the chalcogen increases the metallicity of PtX2 as one goes from S to Te in the chalcogen group. | ||
| Fig. 3 (a and b) Plots of Gibbs free energy of H-adsorption on the metal site (ΔH) and on the chalcogen site (ΔHX) for various TMDs including W, Mo, and Pt for the semiconducting 2H phase and metallic 1T phase. (c) Projected DOS of various TMDs in comparison with that of W, Mo, and Pt showing no significant electron population for PtS2 and PtSe2 at around 0.0 eV. Reproduced with permission from ref. 71 (Copyright 2015, Elsevier). | ||
Interestingly, the experimental results also resonated with their theoretical predictions (Fig. 4). However, none of this dichalcogenide was able to perform HER effectively when compared to Pt/C. This raises the question ‘is it really meaningful to apply PtX2 for the HER when we have a better performing catalyst of the same element (i.e., Pt in the form of Pt/C)?’. Similar calculations but with significant changes in simulation parameters reported by Mir and co-workers,72 Liu and co-workers,73 and Huang and co-workers74 have all concluded that PtX2 can never catalyse the HER as efficiently as Pt/C. Very recently, Ma and Shen75 also reported a similar trend even with vacancy engineering and Pd doping. Apart from these studies, there have been many experimental works as well. Particularly, the number of reports on PtSe2 is higher despite it being shown to be poorer than PtTe2. Lin and co-workers76 reported the magnetron sputtering-assisted selenization of the Pt film to form PtSe2 which showed poorer HER activity than Pt under identical conditions (Fig. 5a). Specifically, PtSe2 delivered −225 mA cm−2 at −0.8 V vs. RHE whereas Pt delivered −510 mA cm−2 at the same potential.
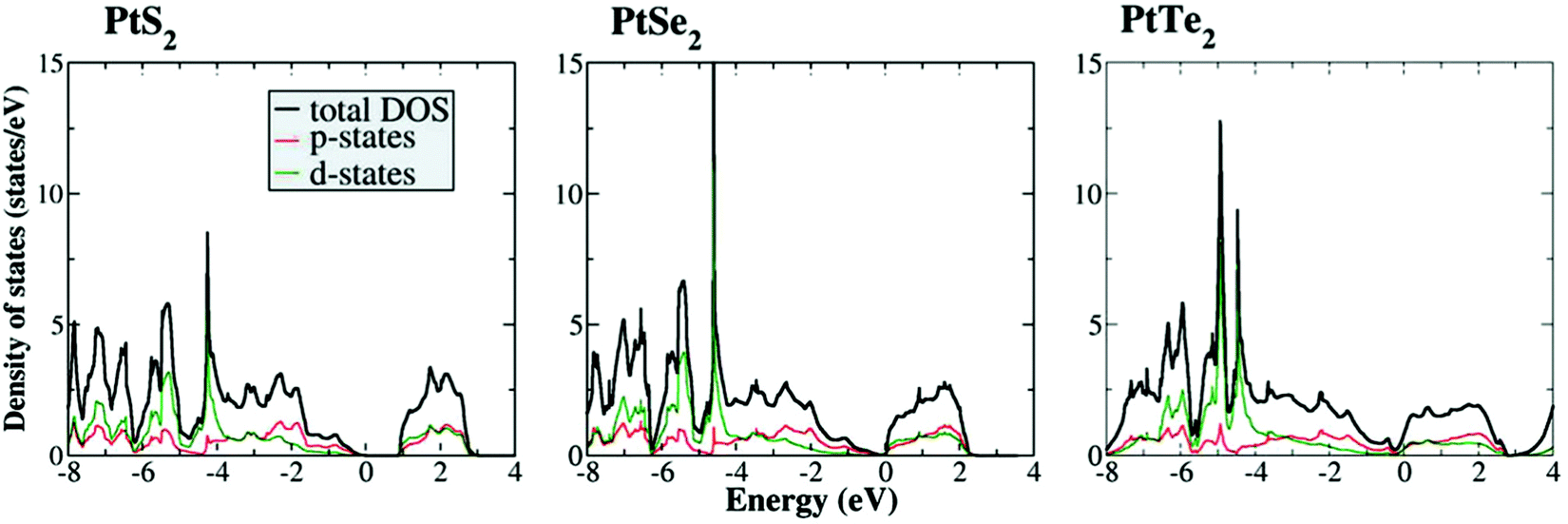 | ||
| Fig. 4 Calculated DOSs of the p and d-states of PtX2 (X = S, Se, and Te) showing an increasing metallic character and improved electron population at 0.0 eV with the increasing size of the chalcogen. Reproduced with permission from ref. 39 (Copyright 2016, Wiley). | ||
 | ||
| Fig. 5 (a) PtSe2 with increasing HER activity with the increasing thickness. Reproduced from ref. 76 (Copyright 2017, Elsevier). (b) PtSe2 thin films with the increasing HER activity with thickness until it reached 51.1 nm (PtSe2@CC-3) and activity reversal thereafter. Reproduced with permission from ref. 77 (Copyright 2021, Royal Society of Chemistry). Notably, none of these films showed any comparable activity to that of Pt or Pt/C under identical conditions. | ||
Zhang and co-workers77 have recently reported a two-step approach to grow edge-rich PtSe2 on carbon cloth (CC) substrate in which Pt sputtering follows the selenization in an inert atmosphere at high temperature (Fig. 5b). When the thickness of the PtSe2 film was varied as 10.7, 24.8, 51.1, and 100.2 nm (labelled as PtSe2@CC-1, PtSe2@CC-2, PtSe2@CC-3, and PtSe2@CC-4 in Fig. 5b), the HER activity was found to increase with the increasing thickness until 51.1 nm and began lowering afterward. Notably, none of these PtSe2 films matched the activity of Pt under identical conditions. When both PtX2 and Pt/C have the same precious metal (i.e., Pt), preparing and applying PtX2 for the HER appear to be meaningless as no PtX2 can have a better HER activity than Pt/C.
However, valuable insights brought out by these studies on the role of layer numbers, the importance of having an edge-rich surface, the role of vacancies and defects, and the film thickness are something that deserves to be appreciated as these results are invaluable in understanding the patterns and trends of HER activity for a variety of TMDs. PtTe2, on the other hand, is relatively less frequently reported in the literature though it was shown to be the best of PtX2 for the HER earlier by Chia and co-workers.39 Also, PtTe2 is mostly reported with one or more additional metals. When there are additional metal sites, telluride of Pt has always and almost outperformed Pt/C. Classic examples of this kind are PtPdRuTe reported by Liu and co-workers78 and the Pt/PtTe2/NiCoTe2 heterostructure reported by Yi and co-workers.79 Similarly, PtS2/TiC reported by Jeong and co-workers80 and PtSe2/Pt reported by Wang and co-workers81 have performed as have done by Pt/C. Otherwise, none of the reported PtX2 were able to surpass the activity exerted by Pt or Pt/C in the HER.
Role of layer numbers and defects/vacancies on the HER activity of PtX2
As introduced above, when there are one or more metals besides Pt in a PtX2, the observed HER activity is always better than Pt or Pt/C. However, there are instances when such high HER activity was realized just with PtX2. In such cases, increasing the number of layers of PtX2 and creating defects/vacancies that would lead to the formation of metallic Pt clusters are behind this high HER activity. In general, WX2 and MoX2 are shown to exhibit enhancement in HER activity when the layer number is decreased by exfoliation as it leads to an increased active surface area. Hence, there have been many attempts of making and studying monolayers of MoX2 and WX2 to understand the origin of their HER activity.82–85 The results have unanimously supported the fact that creation of more edges was the reason for enhanced HER electrocatalysis with MoX2 and WX2.86–90 In sharp contrast to this trend, PtX2 have shown decreasing activity with the decreasing layer numbers despite the fact that it also belongs to the family of isostructural layered 2D TMDs. This was quite intriguing and led to a few notable and dedicated works that studied the effect of layer numbers of PtX2 and related electronic properties.Villaos and co-workers91 provided the theoretically calculated band structures for the 1T phase of bulk PtX2 in comparison with the band structures of the same with increasing layer numbers from 1L to 10L of which the band structures of bulk 1T phases of PtS2, PtSe2, and PtTe2 are shown together with the band structures of 1T PtTe2 of layer numbers 1 to 10 in Fig. 6a-e. Evidently, the bulk phases of PtX2 are more metallic in nature and the metallicity increases with the increasing size of the chalcogen as shown by Chia and co-workers39 as well. The crossover of Fermi level over 0.0 eV was witnessed with the bulk 1T phases of PtSe2 and PtTe2 but not with PtS2. This implies that no matter how many layers PtS2 has, it will continue to have poor electronic conductivity which also accounts for its poor HER performance. Increasing layer numbers, on the other hand, showed the same trend for all three of them. Particularly, the crossover of the Fermi level over 0.0 eV was witnessed when there were four or more layers with PtSe2 whereas for PtTe2 this crossover was witnessed right away with the addition of the second layer (2L) which continues to increase with the increasing layers. In addition to that, clear overlaps of the conduction band and valence band were also witnessed with the increasing number of layers in PtTe2. This increasing metallicity is the main reason why PtX2 have an opposite activity trend to that of WX2 and MoX2 with the increasing number of layers. In the meantime, Hu and co-workers92 have shown that with the increasing number of layers of 1T PtSe2, HER activity increases and edged closer to Pt/C when there were 20 layers (Fig. 7a–h). This observation aligns well with the predictions made by Villaos and co-workers and it was once again proven that PtX2 is not like WX2 or MoX2 when it comes to the electrocatalytic HER. Even though the edge-sites remain to be the active sites with PtX2 just like in WX2 and MoX2, increasing metallicity with the increasing layer numbers makes it behave entirely differently. With the increasing metallicity, the Pt-like character of PtX2 does also increase leading to enhanced HER activity. This once again urges us to ask ‘is it really meaningful to apply PtX2 for the HER when it is clear that Pt or Pt/C is better in every aspect?’.
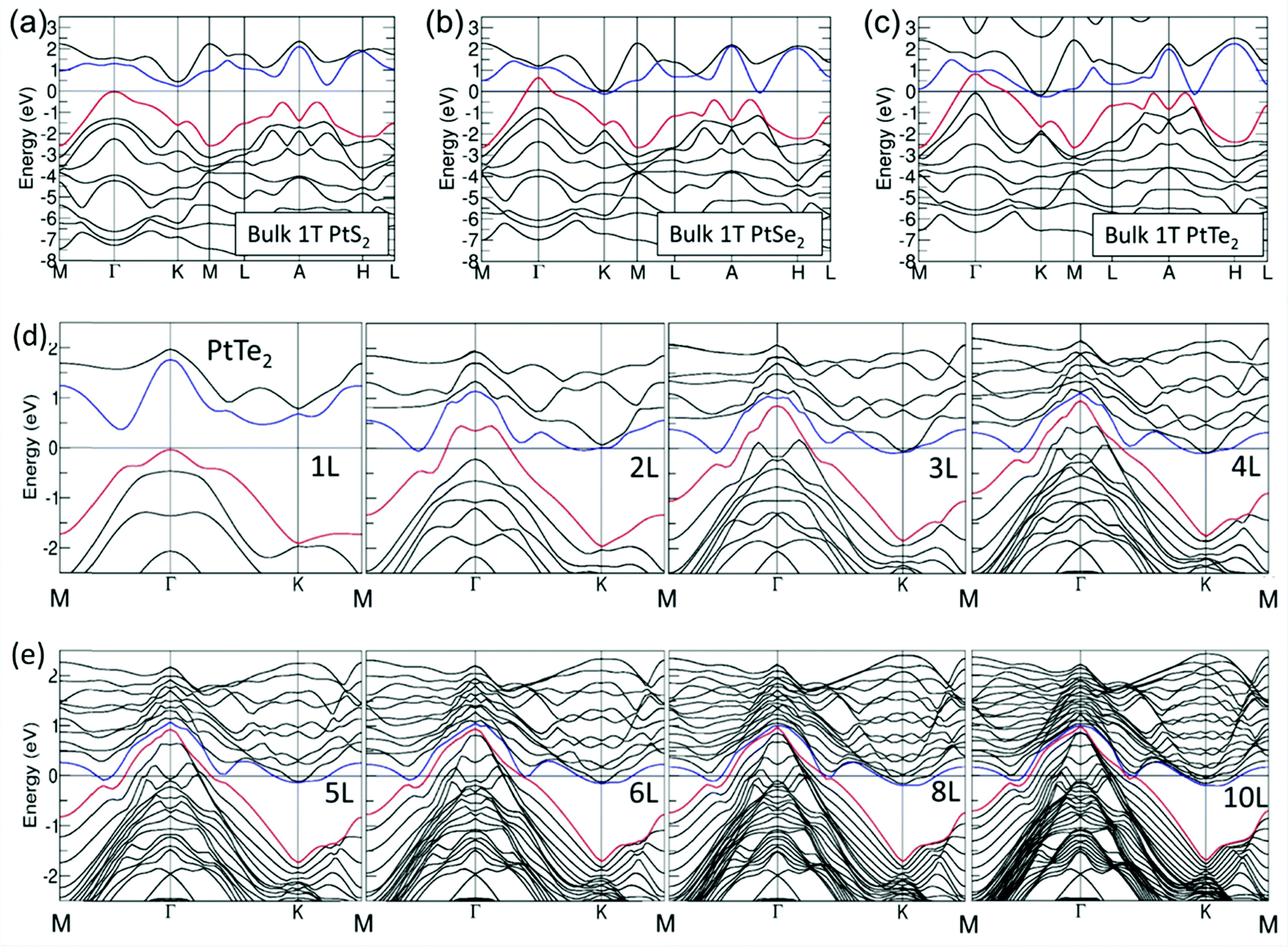 | ||
| Fig. 6 Electronic band structures of bulk 1T PtS2 (a), bulk 1T PtSe2 (b), and 1T PtTe2 (c) with the electronic band structures of 1T PtTe2 of 1–4 layers (d) and 5, 6, 8, and 10 layers (e) showing increasing metallicity and the overlap of conduction and valence bands with increasing layer numbers. Reproduced with permission from ref. 91 (Copyright 2019, Nature). | ||
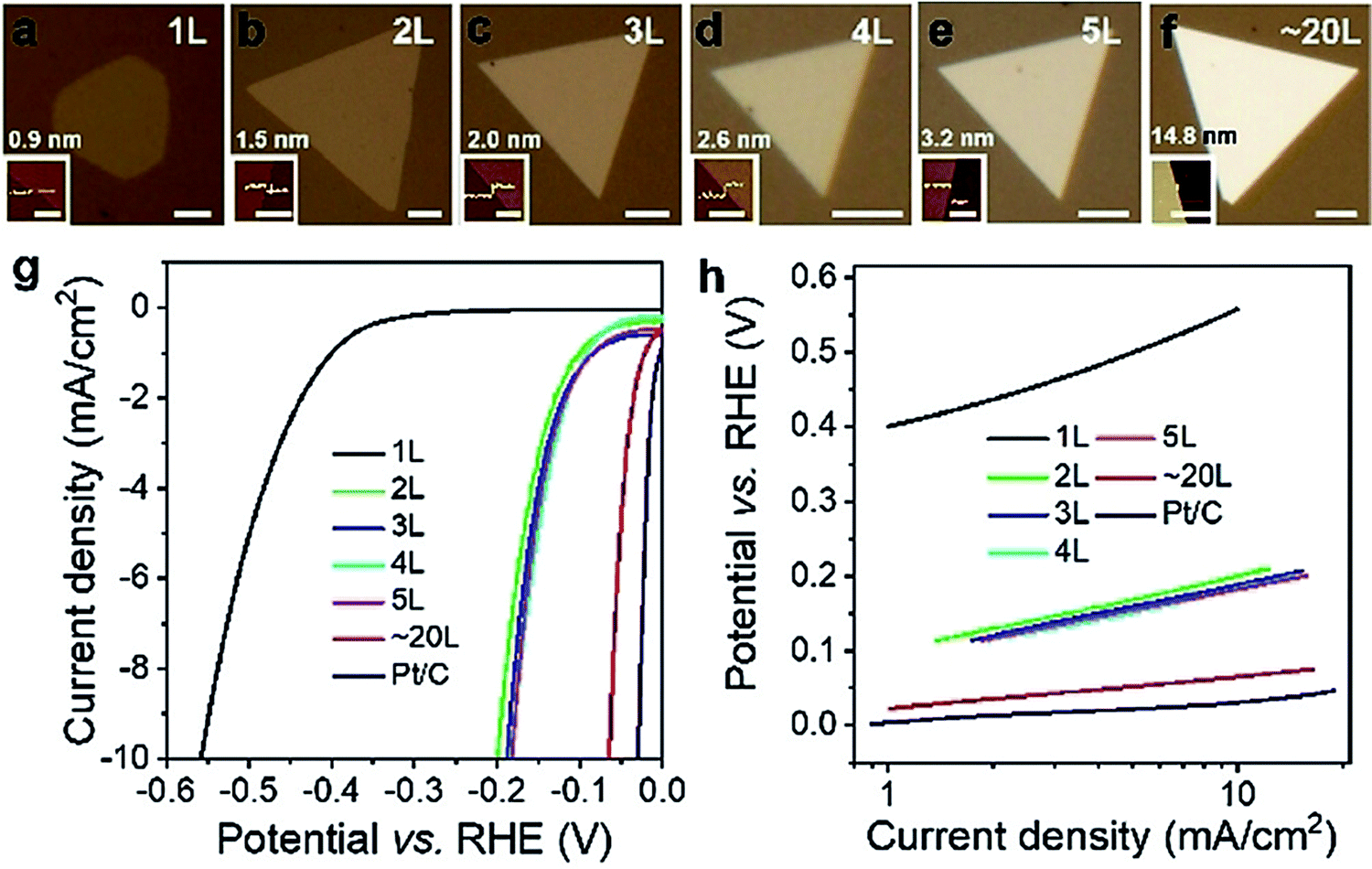 | ||
| Fig. 7 (a–f) Optical images of PtSe2 flakes grown using a chemical vapor transfer technique. (g and h) The corresponding HER LSVs and Tafel lines, respectively, showing better HER activity with the increasing number of layers. Reproduced with permission from ref. 92 (Copyright 2019, Wiley). | ||
Defects or vacancies, in contrast, were sometimes found to bring out parallel activity to that of Pt/C. However, the origin of this high HER activity was not the PtX2 sheets (basal plane or edge-site) but the undercoordinated Pt atoms and Pt clusters that were formed as a result of inducing defects and vacancies. These undercoordinated Pt atoms and Pt clusters are much like Pt or Pt/C in HER activity. Li and co-workers93 have recently shown how creating ordered Te vacancies in PtTe2 could make it perform better in the HER than Pt/C (Fig. 8a–f). To induce such ordered vacancies, they used the two-step top-down approach in which exfoliation followed the heat treatment.
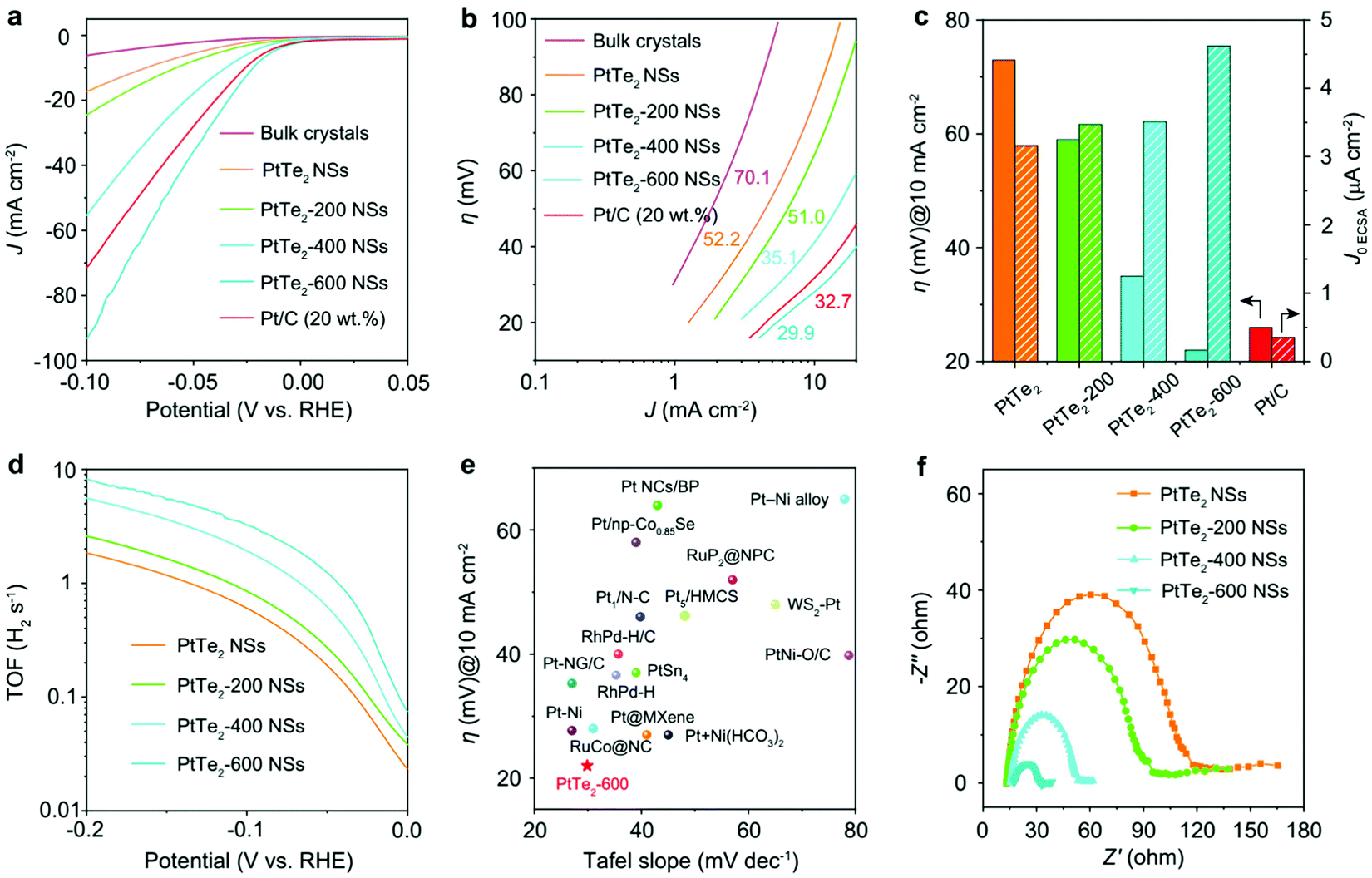 | ||
| Fig. 8 (a) HER LSVs of PtTe2 as bulk crystals and as nanosheets with and without heat treatment at 200, 400, and 600 °C. (b) Corresponding Tafel lines. (c) Histogram of the overpotential at 10 mA cm−2 and exchange current density for the same catalysts. (d) Calculated TOF at all overpotentials. (e) Plot of overpotential at 10 mA cm-2 against the Tafel slope. (f) Nyquist plots of the same ascertaining the activity trend observed in LSVs. Reproduced with permission from ref. 93 (Copyright 2021, The Authors (published by Nature)). | ||
The one with ordered single atom Te vacancies created by heating at 600 °C after exfoliation performed better than Pt/C. Detailed XPS and XANES investigations suggested that PtTe2 with ordered Te vacancies was almost identical to metallic Pt. Besides, as PtTe2 with ordered Te vacancies also had an additional boost from the undercoordinated Pt atoms from the edge-sites, it was able to surpass the HER activity of Pt/C at all overpotentials. Intriguingly, PtTe2 with ordered Te vacancies did also show higher TOF and excellent cycling and potentiostatic electrolysis stabilities which were superior to Pt/C. Ping and co-workers,94 in their recent work, demonstrated that 5–20 layers of 1T PtSe2 flakes synthesized by their chemical vapor transport method can be engineered by irradiating the flakes with a mild Ar-plasma (5–15 W) to have atomic vacancies (both Pt and Te) and Pt clusters. These PtSe2 flakes with atomic vacancies and Pt clusters were found to perform as well as Pt. From this study, it was also revealed that the increasing time of plasma irradiation increased its HER activity. Advanced spectroscopic and microscopic analyses carried out in this work have explicitly shown that there was metallic Pt after Ar-plasma treatment (Fig. 9a–j). Hence, it can safely be concluded here that no matter how fancy the structural and electronic properties of PtX2 sound, they will always be poorer electrocatalytic interfaces for the HER unless otherwise there will be some special pre-treatments (such as defect engineering and increasing the number of layers) enhancing their Pt-like characteristics. Table 1 summarizes the recent PtX2 HER electrocatalysts reported in the literature in the ascending order of their overpotentials at benchmarking conditions (i.e., apparent activity).
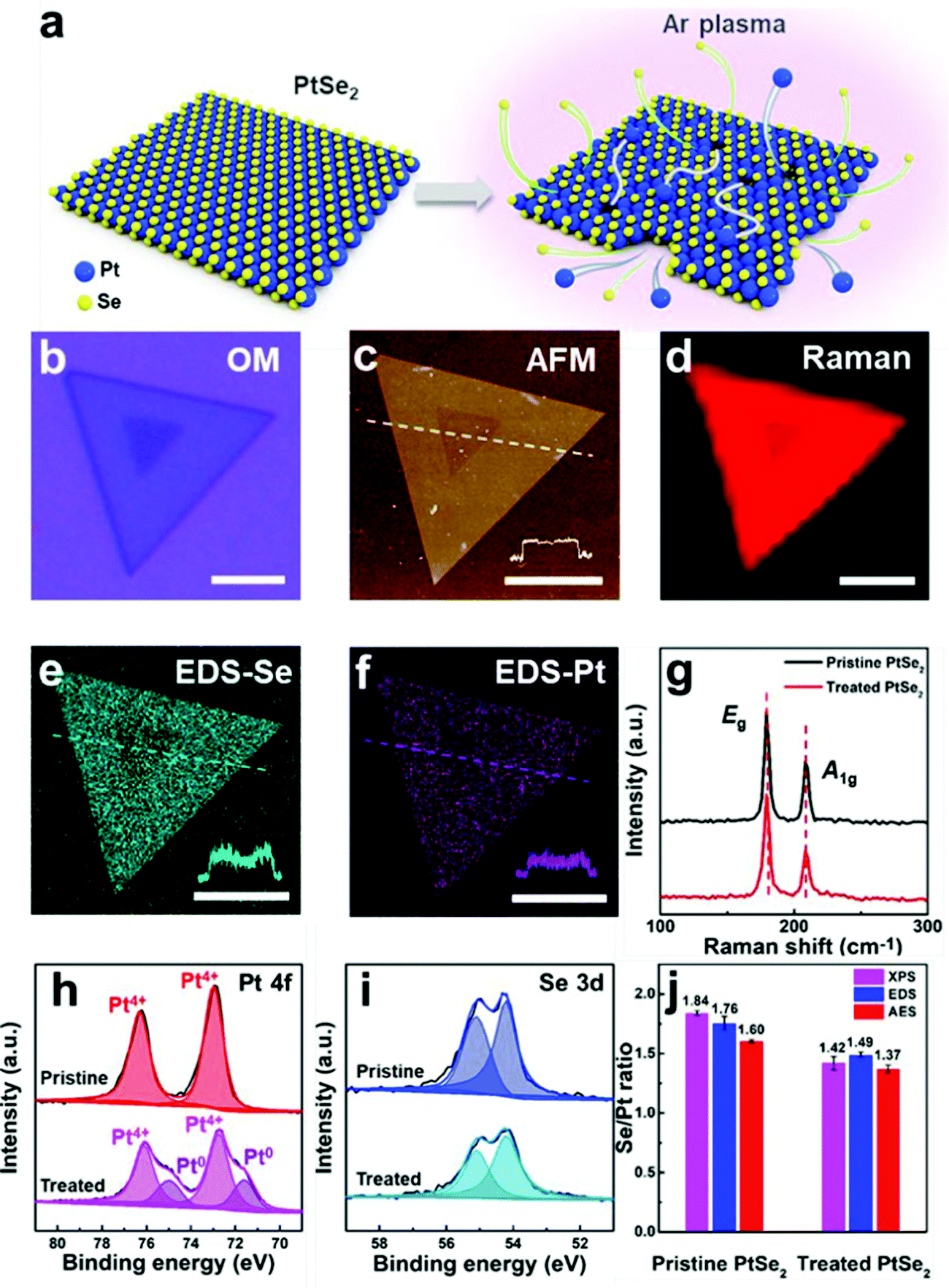 | ||
| Fig. 9 (a) Graphic showing the Ar-plasma etching induced creation of atomic vacancies and Pt clusters on PtSe2. (b–d) The optical image, AFM images, and Raman map of a single PtSe2 flake after etching. (e and f) EDS map of Se and Pt. (g) Raman spectrum of the same. (h and i) XPS narrow scans of Pt 4f and Se 3d levels of Pt and Se in PtSe2 before and after Ar-plasma treatment. (j) Histogram showing the Se/Pt ratio of PtSe2 before and after Ar-plasma treatment as revealed by XPS, EDS, and AES showing the increase in Pt content after treatment. Reproduced with permission from ref. 94 (Copyright 2021, American Chemical Society). | ||
| Catalyst | Medium | η 10/mV | Tafel slope/mV dec−1 | Loading/mg cm−2 | Ref. |
|---|---|---|---|---|---|
| Note: N/A implies that the corresponding data are not available in cited reports. | |||||
| PtSe2 with vacancies and Pt clusters | 0.5 M H2SO4 | 11 | 48 | N/A | Ping et al.94 |
| PtTe2 with Te vacancy | 0.5 M H2SO4 | 22 | 29.9 | 0.809 | Li et al.93 |
| Pt/PtTe2/NiCoTe2/NPFC HFSs | 0.5 M H2SO4 | 34 | 81 | N/A | Yi et al.79 |
| PtPdRuTe | 0.5 M H2SO4 | 39 | 32 | 0.285 | Liu et al.78 |
| PtSe2/Pt | 0.5 M H2SO4 | 42 | 53 | N/A | Wang et al.81 |
| PtS2 QDs on TiC | 0.5 M H2SO4 | 55 | 60 | N/A | Jeong et al.80 |
| PtSe2 20L | 0.5 M H2SO4 | 60 | 41 | N/A | Hu et al.92 |
| Pt3Bi2S2 | 0.5 M H2SO4 | 61 | 51 | N/A | Fang et al.95 |
| 1TPtSe2/CC | 0.5 M H2SO4 | 177 | 67 | N/A | Zhang et al.77 |
| PtSe2 2L | 0.5 M H2SO4 | 200 | 92 | N/A | Hu et al.92 |
| PtSe2 film (76.0 nm) | 0.5 M H2SO4 | 280 | 47 | N/A | Lin et al.76 |
| PtSe2 film (57.6 nm) | 0.5 M H2SO4 | 320 | 63 | N/A | Lin et al.76 |
| PtSe2 film (38.0 nm) | 0.5 M H2SO4 | 380 | 47 | N/A | Lin et al.76 |
| PtSe2 film (19.0 nm) | 0.5 M H2SO4 | 450 | 60 | N/A | Lin et al.76 |
| PtSe2 film (11.4 nm) | 0.5 M H2SO4 | 510 | 32 | N/A | Lin et al.76 |
| PtTe2 | 0.5 M H2SO4 | 540 | 110 | N/A | Chia et al.39 |
| PtSe2 1L | 0.5 M H2SO4 | 550 | 140 | N/A | Hu et al.92 |
| PtSe2 film (7.6 nm) | 0.5 M H2SO4 | 575 | 43 | N/A | Lin et al.76 |
| PtSe2 film (3.8 nm) | 0.5 M H2SO4 | 590 | 50 | N/A | Lin et al.76 |
| PtSe2 film (1.9 nm) | 0.5 M H2SO4 | 615 | 60 | N/A | Lin et al.76 |
| PtSe2 | 0.5 M H2SO4 | 630 | 132 | N/A | Chia et al.39 |
| PtS2 | 0.5 M H2SO4 | 860 | 216 | N/A | Chia et al.39 |
| PtPdRuTe | 1.0 M KOH | 22 | 22 | 0.285 | Liu et al.78 |
| Pt/PtTe2/NiCoTe2/NPFC HFSs | 1.0 M KOH | 43 | 161 | N/A | Yi et al.79 |
| Pt/PtTe2/NiCoTe2/NPFC HFSs | 1.0 M PBS | 36 | 80 | N/A | Yi et al.79 |
From Table 1, it is once again witnessed that PtX2 (also other Pt-based chalcogenides) are poorer catalysts for the HER when compared to Pt/C unless otherwise they have additional metals, Pt heterophase, and vacancies that can result in Pt-like undercoordinated Pt sites, and metallic Pt clusters. Hence, it is apparent that to design and apply a Pt-based HER electrocatalyst (PtX2) that would never have a comparable performance to that of Pt is of least practical meaning.
It is understood from the reports on PtX2 that all the attempts made to make PtX2 an efficient HER catalysts were mainly due to its structural resemblance to two of the most widely studied TMDs in HER electrocatalysis (which are WX2 and MoX2). A quick literature survey on the recent HER reports using WX2 and MoX2 as electrocatalysts revealed the following key points on their activity trends. Both MoX2 and WX2 deliver the benchmarking current density (apparent activity) at 10 mA cm−2 in the overpotential range of 150 to 300 V and always and almost follow the Volmer–Heyrovský mechanism with a Tafel slope ranging from 60 to 120 mV dec−1 in both acid and alkaline solutions. The Tafel slope values tend to be closer to 120 mV dec−1 in alkaline medium as the rate-determining step is usually the water dissociation coupled proton adsorption and discharge unlike the simple proton adsorption and discharge in acidic conditions. Due to the coupled water dissociation in the Volmer step in alkaline conditions, it is also common to witness abnormal Tafel slopes (>120 mV dec−1). In order to compare the performance of PtX2 catalysts listed in Table 1, a collection of recent MoX2 and WX2 catalysts are benchmarked based on their reported apparent activity in Table 2. It is advised here that the readers should be mindful of the effects of surface area, loading, and screening conditions all of which have significant influences on apparent activity.
| Catalyst | Medium | η10/mV | Tafel slope/mV dec−1 | Loading/mg cm−2 | Ref. |
|---|---|---|---|---|---|
| Note: N/A implies that the corresponding data are not available in cited reports. | |||||
| MoX2 HER electrocatalysts | |||||
| 1T MoTe2 | 0.5 M H2SO4 | 73 | 46.3 | N/A | He et al.96 |
| MoSe2–WS2 | 0.5 M H2SO4 | 75 | 60 | N/A | Vikraman et al.61 |
| 1T-2H MoSe2/graphene | 0.5 M H2SO4 | 98 | 49 | N/A | Deng et al.97 |
| N-Doped MoS2 | 0.5 M H2SO4 | 108 | 37 | 0.5 | Bolar et al.98 |
| MoS2–WSe2 | 0.5 M H2SO4 | 116 | 76 | N/A | Vikraman et al.99 |
| Graphene wrapped MoS2 | 0.5 M H2SO4 | 118 | 73 | N/A | Nguyen et al.100 |
| MoSe2 with vacancies | 0.5 M H2SO4 | 125 | 35 | N/A | Xia et al.101 |
| MoS2–WS2 heterostructure | 0.5 M H2SO4 | 129 | 72 | N/A | Vikraman et al.102 |
| Se-Rich MoSe2 | 0.5 M H2SO4 | 130 | 46 | N/A | Kwon et al.47 |
| MoS2–WTe2 | 0.5 M H2SO4 | 140 | 40 | 1.2 | Zhou et al.84 |
| MoSe2–MoO2 | 0.5 M H2SO4 | 142 | 48.9 | N/A | Jian et al.103 |
| 3D MoS2/graphene | 0.5 M H2SO4 | 143 | 71 | 0.5 | Meng et al.104 |
| Disordered 1T MoSe2 | 0.5 M H2SO4 | 152 | 52 | 0.14 | Yin et al.105 |
| MoS2 Moiré superlattice | 0.5 M H2SO4 | 153 | 73 | N/A | Jiang et al.106 |
| MOS2 nanomesh | 0.5 M H2SO4 | 160 | 46 | N/A | Yin et al.107 |
| MoSe2–MoS2 | 0.5 M H2SO4 | 162 | 61 | N/A | Li et al.108 |
| Electroactivated MoTe2 | 0.5 M H2SO4 | 178 | 116 | N/A | McGlynn et al.109 |
| 2D MoS2–MoSe2 thin sheets | 0.5 M H2SO4 | 186 | 71 | N/A | Sharma et al.110 |
| Pores-rich MoS2 | 0.5 M H2SO4 | 190 | 163 | N/A | Zhou et al.111 |
| Deformed MoS2 | 0.5 M H2SO4 | 191 | 64 | 0.13 | Chen et al.112 |
| Amorphous MoS2 | 0.5 M H2SO4 | 210 | 42 | N/A | Wu et al.113 |
| WS2–MoS2@CNT | 0.5 M H2SO4 | 212 | 50 | 0.3 | Thangasamy et al.114 |
| MoSe2 nanoflowers | 0.5 M H2SO4 | 220 | 61 | N/A | Masurkar et al.115 |
| 1T′ MoTe2/CC | 0.5 M H2SO4 | 220 | 127 | N/A | Lu et al.116 |
| MoSe2–CNT | 0.5 M H2SO4 | 245 | 49 | 0.112 | Maity et al.117 |
| 1T′-2H MoS2 edges | 0.5 M H2SO4 | 290 | 83 | N/A | Zhang et al.52 |
| Et2N-Ph–MoS2 | 0.5 M H2SO4 | 348 | 75 | N/A | Benson et al.32 |
| MoSeTe | 0.5 M H2SO4 | 410 | 62 | 0.001 | Kosmala et al.48 |
| MoTe2 | 0.5 M H2SO4 | 460 | 67 | N/A | McManus et al.118 |
| 1T′ MoTe2 | 0.5 M H2SO4 | 530 | 177 | N/A | Zhuang et al.119 |
| 1T MoS2 with defects | 1.0 M KOH | 90 | 100 | 1 | Anjum et al.120 |
| N-Doped MoS2 | 1.0 M KOH | 141 | 48 | 0.5 | Bolar et al.98 |
| 1T-2H MoS2 heterostructure | 1.0 M KOH | 260 | 65 | N/A | Wang et al.54 |
| WX2 HER electrocatalysts | |||||
| WS2 with S-vacancy | 0.5 M H2SO4 | 116 | 37.9 | N/A | Zhu et al.121 |
| WSe2 3D dendrite | 0.5 M H2SO4 | 175 | 80 | N/A | Zou et al.87 |
| WS2–graphene | 0.5 M H2SO4 | 180 | 76 | N/A | Le et al.122 |
| WSe2 films | 0.5 M H2SO4 | 189 | 72 | N/A | Li et al.123 |
| WS2 with dominant 1T phase | 0.5 M H2SO4 | 200 | 50.4 | N/A | Liu et al.124 |
| Trigonal WS2–CNT | 0.5 M H2SO4 | 205 | 84 | N/A | Guo et al.125 |
| WS2–CNT | 0.5 M H2SO4 | 210 | 59.7 | N/A | Wang et al.126 |
| Te doped WS2 | 0.5 M H2SO4 | 210 | 94 | N/A | Pan et al.127 |
| WSe2 monolayer | 0.5 M H2SO4 | 245 | 76 | N/A | Sun et al.128 |
| Sputtered WS2 | 0.5 M H2SO4 | 250 | 126.3 | N/A | Nam et al.129 |
| WSe2 nanosheets | 0.5 M H2SO4 | 275 | 78 | N/A | Wang et al.88 |
| WS2–graphdyine | 0.5 M H2SO4 | 275 | 54 | 0.29 | Yao et al.130 |
| WS2–graphite protected | 0.5 M H2SO4 | 280 | 47.9 | N/A | Yang et al.131 |
| WTe2 edges | 0.5 M H2SO4 | 325 | 96 | N/A | Ling et al.132 |
| 1T-WS2 | 0.5 M H2SO4 | 350 | 95 | N/A | Kim et al.133 |
| WS2–WO3 | 0.5 M H2SO4 | 380 | 50 | 0.14 | Shang et al.134 |
| WSe2–rGO | 0.5 M H2SO4 | 390 | 85 | N/A | Liu et al.135 |
| Edge-engineered WS2 | 0.5 M H2SO4 | 390 | 122 | N/A | Shirazi et al.136 |
| WSe2–S2−/Na+ | 0.5 M H2SO4 | 400 | 97 | 1 | Kim et al.137 |
| Semimetallic WTe2 | 0.5 M H2SO4 | 450 | 57 | N/A | Hong et al.138 |
| Te-Vacant WTe2 | 0.5 M H2SO4 | 550 | 159 | N/A | Kwon et al.139 |
| Selected state-of-the-art in HER electrocatalysts | |||||
| Rh2P | 0.5 M H2SO4 | 14 | 32 | N/A | Yang et al.140 |
| Ru–NC | 1.0 M KOH | 17 | 32 | 0.24 | Liu et al.141 |
| Rh2P | 0.5 M H2SO4 | 17 | 35 | 0.114 | Zhao et al.142 |
| NiFe2O4–Ru–Ni | 1.0 M KOH | 18 | 27 | 0.1 | Niu et al.143 |
| Pt | 0.5 M H2SO4 | 24 | 31 | N/A | Yang et al.140 |
| Pt/C–Ni foam | 1.0 M KOH | 25 | 99 | 0.1 | Niu et al.143 |
| Pt/C | 1.0 M KOH | 25 | 23 | 0.285 | Luo et al.144 |
| Ru–C | 1.0 M KOH | 27 | 33 | 0.285 | Luo et al.144 |
| Pt/C | 0.5 M H2SO4 | 27 | 32 | 0.114 | Zhao et al.142 |
| NiFe LDH–Ru | 1.0 M KOH | 29 | 31 | N/A | Chen et al.145 |
| Rh2P | 1.0 M KOH | 30 | 50 | N/A | Yang et al.140 |
| Pt/C | 1.0 M KOH | 31 | 32 | N/A | Chen et al.145 |
| Pt/C | 1.0 M KOH | 32 | 32 | 0.24 | Liu et al.141 |
| Ru–NC | 1.0 M KOH | 32 | 64 | N/A | Wang et al.146 |
| Pt | 1.0 M PBS | 32 | 51 | N/A | Yang et al.140 |
| Pt/C | 1.0 M KOH | 34 | N/A | N/A | Wang et al.146 |
| Rh2P | 1.0 M PBS | 38 | 46 | N/A | Yang et al.140 |
| Pt | 1.0 M KOH | 58 | 77 | N/A | Yang et al.140 |
From Table 2, it is evident that MoX2 and WX2 have average overpotentials of 200 and 286 mV, respectively. Apparently, these merits are far better than those of metallic Mo and W, and hence, designing and screening MoX2 and WX2 for the HER in comparison with Pt/C is truly purposeful and makes sense unlike comparing PtX2 with Pt/C. Also, when the activity trends of Mo/WX2 are compared with that of PtX2, the poorer HER activity of pristine PtX2 (with no defects, vacancies, metallic single atoms, and cluster of Pt, etc.,) is explicitly evidenced. Besides, a list of recent state-of-the-art HER electrocatalysts listed in the second part of Table 2 suggest that pristine PtX2 are nowhere near good enough to be compared with the state-of-the-art despite being made of Pt. This implies that working hard on a material (in its pristine form) that will never perform better than its metallic counterpart (i.e., Pt) is simply a waste of time and resources. On the other hand, the structural engineering strategies (vacancy engineering, creating Pt clusters and single atoms, and increasing the layer numbers) reported recently show some that these PtX2 materials have some potential to actually become better than Pt/C. However, the question is ‘at what cost?’. One has to be mindful of the time, energy, and resources spent in creating a material that would have a comparable activity (most of the time) or a slightly better activity than Pt/C and must ask ‘Is it worth it?’.
Are we missing the bigger picture here?
Undoubtedly, any effort put forward by researchers to find highly efficient electrocatalysts for energy conversion reactions including the HER of water electrolysis deserves to be appreciated. Since Pt is the superior catalyst of all for the HER in almost all pH conditions, all the other materials made of metals other than Pt are conventionally compared with Pt/C under identical working conditions to judge them for their suitability as an alternate to Pt.9 Similarly, there is also huge interest among researchers to lower the overall Pt content instead of replacing it completely so that one would not have to compromise activity and energy-efficiency. Nanostructuring-assisted high surface area Pt particles synthesis and their subsequent stabilization on a suitable support/substrate is an active research area.147–149 In such cases, these low-Pt nanocatalysts are compared with Pt/C (20 wt%) to assess their superiority. In electrocatalytic water splitting, these two objectives are important as they address the issue of the scarcity of Pt. However, when a catalyst made of Pt (such as PtX2) is incapable of surpassing the parent Pt or Pt/C in HER electrocatalytic activity, putting so much time and resources in this area is simply meaningless. From the above discussion and Tables 1 and 2, it could have been witnessed that even MoX2 and WX2 deliver better HER performances than pristine PtX2. To get a clear picture of the HER activity trends of MoX2, WX2, and PtX2 in comparison with Pt/C, the average overpotential at 10 mA cm−2 required by all these materials (an average of the values from cited reports and there could significant deviation when just a single study is considered) are plotted as a histogram (Fig. 10). From Fig. 10, it is once again proven that working with PtX2 for the purpose of applying it to HER when we have a better catalyst comprised of the same Pt (Pt/C) for the same is essentially meaningless. Moreover, even when PtX2 with undercoordinated Pt sites, Pt clusters, and other metals are put to the job of the HER, their average overpotential at 10 mA cm−2 remains still higher than that of Pt/C. Since we cannot completely criticise the advantages of PtX2 that it may have in HER electrocatalysis, we looked into other facts in which PtX2 can actually be better than Pt. The main advantage with PtX2 (that too only with the monolayers of PtX2) is the 100% accessibility of Pt sites unlike the solid Pt electrodes. Hence, if the activity is reported in mA Pt−1, PtX2 can surely deliver better activity than Pt as only the surface sites are available for the HER with the latter one. To show this, we took the data reported by Chia and co-workers and converted it into activity per Pt site (Fig. 11a–c). The number of Pt sites was calculated from the lattice parameters of Pt, PtS2, PtSe2, and PtTe2. | ||
| Fig. 10 Histogram of the average overpotential at 10 mA cm−2 (apparent activity) required by MoX2, WX2, pristine PtX2, and Pt/C pH 0 with MoX2 in pH 14 (green bar) showing the superiority of Pt/C over PtX2. The asterisk symbol indicates that these PtX2 catalysts had assistance from other contributors like undercoordinated Pt-sites, more layer numbers, vacancies leading to the formation of Pt clusters, other highly active materials, and having an additional Pt heterointerface. Each ηavg.10 value is the average one calculated using the data from multiple reports in Tables 1 and 2. | ||
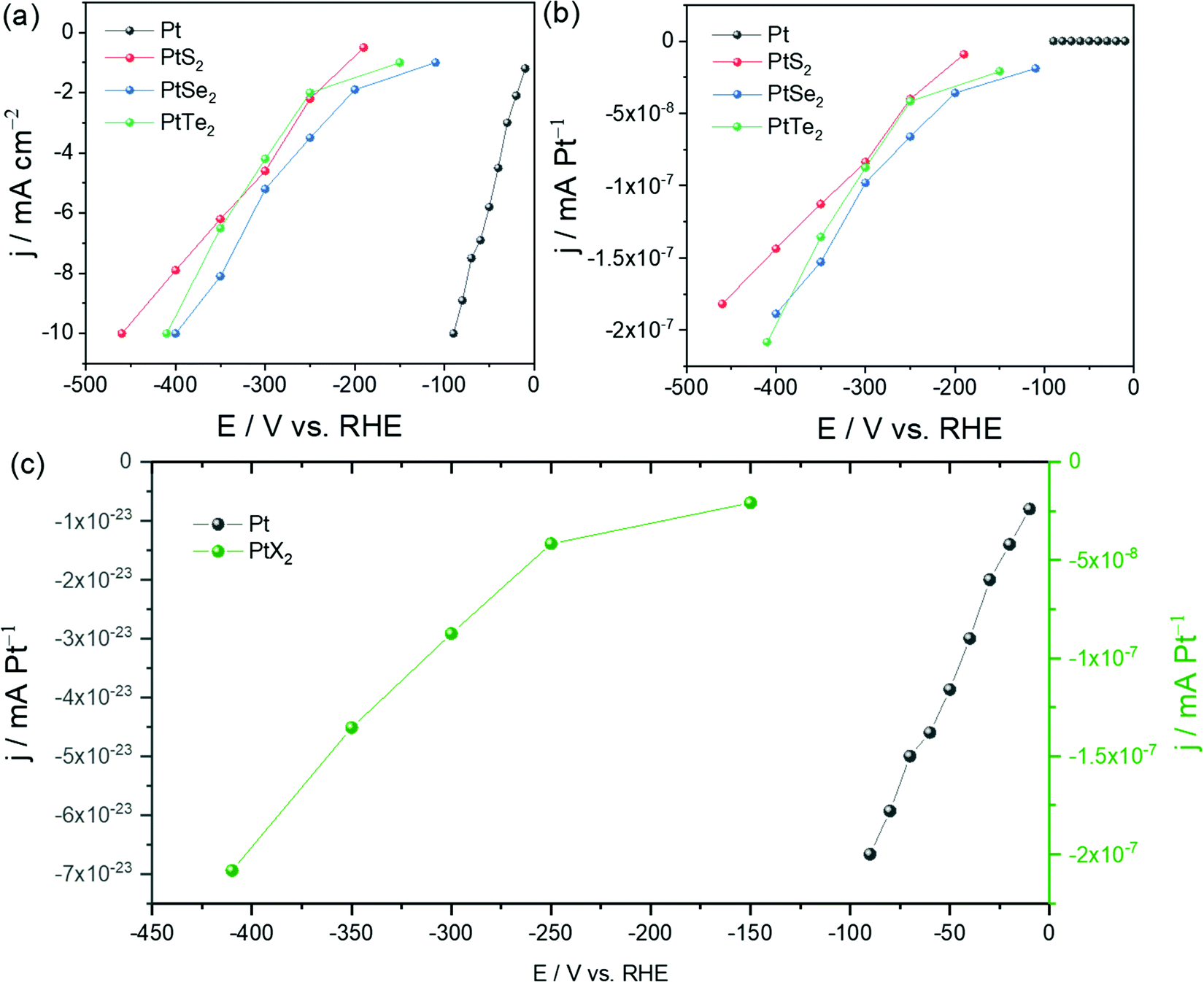 | ||
| Fig. 11 (a) Activity of PtX2 catalysts in comparison with Pt as reported by Chia and co-workers.39 (b) The activity of the same catalysts normalized by the number of Pt Sites. (c) Number of Pt sites normalized activity of PtTe2 and Pt showing several orders of magnitudes of difference but with the same onset potentials implying that Pt is still superior to PtX2 in intrinsic activity (so is in apparent activity). Note: The values of activity were manually read from the figures given by the original study by Chia and co-workers39 and there could be some deviations in the data points. Hence, the readers are solicited to act on their own discretion. | ||
Fig. 11a shows the as-adopted data with activity normalized by geometrical area. Fig. 11b on the other hand shows the activity normalized by the number of Pt sites. Clearly, PtX2 delivered several orders of magnitudes of activity (in mA Pt−1) higher than Pt as shown in Fig. 11c. However, one should also note here that irrespective of the high activity per Pt sites, the onset potential remains the same indicating that Pt has the upper hand when it comes to practical electrolysis. Hence, the higher activity per Pt site shown in Fig. 11b and c is useless unless the same can be capitalized into apparent activity. Other than these observations, the following are also the reasons why PtX2 could never outperform Pt in practical electrolysis of water in its pristine form.
• 100% accessibility of Pt sites is possible with only monolayer PtX2. However, the experimental studies suggest that monolayer PtX2 (in sharp contrast to that of Mo/WX2) catalyze the HER poorly compared with those with higher layer numbers. This is mainly because all the Pt atoms accessed in monolayer PtX2 are actually Pt4+ ions. The density of states (DOS) of Pt4+ ions are quite different from those of Pt0 and do not possess a HER favoring band structure (please refer to our discussion on the role of DOS on HER activity).
• Only PtX2 catalysts with vacancies and defects that led to the formation of metallic Pt single atoms and clusters performed better and possessed a comparable activity to that of Pt/C. This indicates that having metallic Pt is more important than ensuring 100% accessibility in the form of Pt4+.
• Because of such differences in DOS, pristine PtX2 have always had a higher onset overpotential (even higher than Mo/WX2) for the HER than Pt/C. This basically undermines the goals of HER electrocatalysis. In other words, there is no meaning in ensuring 100% accessibility when we need to trade off the onset overpotential to a greater extent.
From the above discussion, we can say ‘We Are Missing the Bigger Picture Here’. However, it should not be left without mentioning that the reports published on the HER activity trends of PtX2 catalysts have helped us to understand the structure–activity relationship in PtX2 as well as in other MX2 catalysts. Hence, it is not that just that PtX2 is a poor catalyst for HER, it is totally useless. It could be useful and actually be better than Pt in other areas of applications. Hence, it is concluded here that in HER electrocatalysis, the bigger picture (finding an alternate with comparable activity to Pt and lowering the total Pt content without compromising its HER activity) should not be missed.
Conclusions and outlook
HER electrocatalysis is an important area of water electrolysis research that focuses mainly on three goals. (1) Replacing Pt entirely with a non-Pt catalyst for HER, with a non-Pt catalyst, (2) lowering the total Pt content without making any compromise in activity, and (3) improving the poor kinetics of Pt in alkaline conditions. With all these goals, any progress made is conventionally compared with that of Pt/C under identical experimental conditions as it is the state-of-the-art to date. In studies where Pt-based catalysts are engineered structurally to lower the total Pt content and improve its kinetics, they are expected to surpass the commercial Pt/C in terms of both activity and HER kinetics. With PtX2, it appears that this basic requisite has been forgotten. Pristine PtX2 with an average overpotential of 476 mV (calculated from the works cited in this study and could vary notably but not extensively depending on the studies that may emerge in the future) at 10 mA cm−2 is nowhere near the performance delivered by Pt/C which is basically undermining the whole objective of designing and applying a material for the HER electrocatalyst. On the other hand, PtX2 that had assistance from other factors listed above performed better yet still poorer than Pt/C on average. Hence, it is time that we stop and question ourselves ‘Are we doing it right?’. However, it is unanimously agreed here that new knowledge added by the studies on the activity trend of PtX2 and the correlations that were made with the activity trends of other MX2 materials in comparison with PtX2 is invaluable. Similarly, since a monolayer PtX2 ensures 100% accessibility to all the Pt sites and PtX2 with more layers to a notable extent than one can access with Pt/C, the activity reported per Pt atom could actually add weightage to PtX2-based HER catalysts over the same reported for Pt/C. However, it should be reminded here that all the sites that are accessed with PtX2 are tetravalent Pt cations (Pt4+) and not metallic Pt. These two have very distinct electronic structures and DOS population at and around 0.0 eV which suggests that only metallic Pt has an appropriate electronic structure for facile HER electrocatalysis on its surface. Hence, it may not matter how much percentage of Pt we can access as long as the accessed Pt site is chemically different from metallic Pt and will always have a poorer HER activity (as evidenced from the higher onset overpotentials of PtX2 in the HER). The huge anticipation laid on PtX2 to have a better HER performance was mainly because it is isostructural to Mo/WX2 HER electrocatalysts. However, Mo/WX2 are very different in their electronic properties linked to HER activity and have an inverse trend of activity with the increasing number of layers. This is one of many reasons why the theoretical predictions about PtX2 failed in experimental studies. This implies that both theoretical and experimental studies should go hand in hand in order to identify, justify, and resolve any hurdles that may be encountered in the way of developing a better catalyst while being complementary to one another.Despite the humongous issues pointed out in this perspective, we will not recommend abandoning PtX2 completely. Instead, these graphene-like and structurally intriguing PtX2 can be used as synergistic supports and pre-catalysts. It has been shown that electrochemical cycling and potentiostatic/galvanostatic electrolysis can activate PtX2 towards the HER by lowering the demanded overpotential significantly. Key insights of such studies indicate that such electroactivation results in the formation of metallic Pt clusters and nanoparticles on the PtX2 matrix while leaving multiple vacancies behind which in turn resulted in undercoordinated metallic Pt-like Pt atomic sites. This can be used as a potential way to enhance the activity of PtX2 so that it could outperform Pt/C as demonstrated by a few already. However, high activity realized just by (single atom) vacancy engineering cannot be a long-term solution, as these vacancies are highly prone to the restructuring of the local environment around it particularly when the working condition is reductive in nature. Other options that are available with PtX2 are heterostructuring and doping metallic Pt clusters and particles so that one could lower the total content yet can have a better performance than Pt/C. Other than this application (HER)-oriented prospects, PtX2 can become a useful model catalyst to study the structure–activity relationship and elucidating mechanism of the HER in comparison with other MX2 HER electrocatalysts. If not, spending time and resources just to create a poorly active HER electrocatalyst (PtX2) out of the state-of-the-art (Pt) is unfortunately and undeniably meaningless.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported by the Grant-in-Aid for Researchers of Research Institute for Science and Engineering, Waseda University, Japan.References
- M.-R. Gao, Y.-F. Xu, J. Jiang and S.-H. Yu, Chem. Soc. Rev., 2013, 42, 2986–3017 RSC
.
- Y. Jiao, Y. Zheng, M. Jaroniec and S. Z. Qiao, Chem. Soc. Rev., 2015, 44, 2060–2086 RSC
- S. Anantharaj, S. R. Ede, K. Sakthikumar, K. Karthick, S. Mishra and S. Kundu, ACS Catal., 2016, 6, 8069–8097 CrossRef CAS
- S. Anantharaj and V. Aravindan, Adv. Energy Mater., 2019, 2, 1902666 Search PubMed
- S. Anantharaj and S. Noda, Small, 2020, 16, 1905779 CrossRef CAS PubMed
- Y. Shi and B. Zhang, Chem. Soc. Rev., 2016, 45, 1529–1541 RSC
- S. Anantharaj, J. Mater. Chem. A, 2021, 9, 6710–6731 RSC
- S. Anantharaj, S. Noda, V. R. R. Jothi, S. C. Yi, M. Driess and P. W. Menezes, Angew. Chem., Int. Ed., 2021, 60(35), 18981–19006, DOI:10.1002/anie.202015738
- S. Anantharaj, S. Kundu and S. Noda, J. Mater. Chem. A, 2020, 4174–4192 RSC
- N. Dubouis and A. Grimaud, Chem. Sci., 2019, 10, 9165–9181 RSC
- M. Miao, J. Pan, T. He, Y. Yan, B. Y. Xia and X. Wang, Chem. – Eur. J., 2017, 23, 10947–10961 CrossRef CAS PubMed
- W. Luo, Y. Wang and C. Cheng, Mater. Today Phys., 2020, 15, 100274 CrossRef
- Y. Yan, B. Xia, Z. Xu and X. Wang, ACS Catal., 2014, 4, 1693–1705 CrossRef CAS
- P. Xiao, W. Chen and X. Wang, Adv. Energy Mater., 2015, 5, 1500985 CrossRef
- D. Strmcnik, P. P. Lopes, B. Genorio, V. R. Stamenkovic and N. M. Markovic, Nano Energy, 2016, 29, 29–36 CrossRef CAS
- Y. Zheng, Y. Jiao, A. Vasileff and S. Z. Qiao, Angew. Chem., Int. Ed., 2018, 57, 7568–7579 CrossRef CAS PubMed
- J. Wang, W. Cui, Q. Liu, Z. Xing, A. M. Asiri and X. Sun, Adv. Mater., 2016, 28, 215–230 CrossRef CAS PubMed
- Q. Liu, Z. Pu, A. M. Asiri and X. Sun, Electrochim. Acta, 2014, 149, 324–329 CrossRef CAS
- J. Tian, Q. Liu, Y. Liang, Z. Xing, A. M. Asiri and X. Sun, ACS Appl. Mater. Interfaces, 2014, 6, 20579–20584 CrossRef CAS PubMed
- P. Jiang, Q. Liu, Y. Liang, J. Tian, A. M. Asiri and X. Sun, Angew. Chem., Int. Ed., 2014, 53, 12855–12859 CrossRef CAS PubMed
- J. Tian, Q. Liu, Y. Liang, Z. Xing, A. M. Asiri and X. Sun, ACS Appl. Mater. Interfaces, 2014, 6, 20579–20584 CrossRef CAS PubMed
- Y. Liang, Q. Liu, A. M. Asiri, X. Sun and Y. Luo, ACS Catal., 2014, 4, 4065–4069 CrossRef CAS
- J. Park, B. Koo, K. Y. Yoon, Y. Hwang, M. Kang, J. G. Park and T. Hyeon, J. Am. Chem. Soc., 2005, 127, 8433–8440 CrossRef CAS PubMed
- J. F. Callejas, C. G. Read, C. W. Roske, N. S. Lewis and R. E. Schaak, Chem. Mater., 2016, 28(17), 6017–6044, DOI:10.1021/acs.chemmater.6b02148
- J. Kibsgaard, C. Tsai, K. Chan, J. D. Benck, J. K. Nørskov, F. Abild-Pedersen and T. F. Jaramillo, Energy Environ. Sci., 2015, 8, 3022–3029 RSC
- A. E. Henkes, Y. Vasquez and R. E. Schaak, J. Am. Chem. Soc., 2007, 129, 1896–1897 CrossRef CAS PubMed
- S. Anantharaj, S. R. Ede, K. Sakthikumar, K. Karthick, S. Mishra and S. Kundu, ACS Catal., 2016, 6, 8069–8097 CrossRef CAS
- S. Anantharaj, E. Subhashini, K. C. Swaathini, T. S. Amarnath, S. Chatterjee, K. Karthick and S. Kundu, Appl. Surf. Sci., 2019, 487, 1152–1158 CrossRef CAS
- X. Chia, Z. Sofer, J. Luxa and M. Pumera, ACS Appl. Mater. Interfaces, 2017, 9, 25587–25599 CrossRef CAS PubMed
- J. Luxa, P. Vosecký, V. Mazánek, D. Sedmidubský, M. Pumera, P. Lazar and Z. Sofer, ACS Catal., 2017, 7, 5706–5716 CrossRef CAS
- D. Voiry, J. Yang and M. Chhowalla, Adv. Mater., 2016, 28, 6197–6206 CrossRef CAS PubMed
- E. E. Benson, H. Zhang, S. A. Schuman, S. U. Nanayakkara, N. D. Bronstein, S. Ferrere, J. L. Blackburn and E. M. Miller, J. Am. Chem. Soc., 2018, 140, 441–450 CrossRef CAS PubMed
- W. L. Tao, Y. Q. Zhao, Z. Y. Zeng, X. R. Chen and H. Y. Geng, ACS Appl. Mater. Interfaces, 2021, 13, 8700–8709 CrossRef CAS PubMed
- X.-W. Tong, J.-J. Wang, J.-X. Li, X.-F. Hu, D. Wu and L.-B. Luo, Sens. Actuators, A, 2021, 322, 112625 CrossRef CAS
- Y. Shan, T. Li and L. Liu, Phys. E, 2020, 116, 113741 CrossRef CAS
- J. Mangin and P. Veber, J. Cryst. Growth, 2008, 310, 3077–3083 CrossRef CAS
- D. Wang, W. Ju, D. Kang, T. Li and H. Li, Vacuum, 2021, 183, 109859 CrossRef CAS
- N. Rohaizad, C. C. Mayorga-Martinez, M. Fojtů, N. M. Latiff and M. Pumera, Chem. Soc. Rev., 2021, 50, 619–657 RSC
- X. Chia, A. Adriano, P. Lazar, Z. Sofer, J. Luxa and M. Pumera, Adv. Funct. Mater., 2016, 26, 4306–4318 CrossRef CAS
- J. D. Benck, T. R. Hellstern, J. Kibsgaard, P. Chakthranont and T. F. Jaramillo, ACS Catal., 2014, 4, 3957–3971 CrossRef CAS
- Q. Ding, B. Song, P. Xu and S. Jin, Chem, 2016, 1, 699–726 CAS
- J. Theerthagiri, R. A. Senthil, B. Senthilkumar, A. Reddy Polu, J. Madhavan and M. Ashokkumar, J. Solid State Chem., 2017, 252, 43–71 CrossRef CAS
- P. M. Pataniya, D. Late and C. K. Sumesh, ACS Appl. Energy Mater., 2021, 4, 755–762 CrossRef CAS
- J. Seok, J. H. Lee, S. Cho, B. Ji, H. W. Kim, M. Kwon, D. Kim, Y. M. Kim, S. H. Oh, S. W. Kim, Y. H. Lee, Y. W. Son and H. Yang, 2D Mater., 2017, 4, 025061, DOI:10.1088/2053-1583/aa659d
- X. Han, X. Tong, X. Liu, A. Chen, X. Wen, N. Yang and X. Y. Guo, ACS Catal., 2018, 8, 1828–1836 CrossRef CAS
- R. K. Biroju, D. Das, R. Sharma, S. Pal, L. P. L. Mawlong, K. Bhorkar, P. K. Giri, A. K. Singh and T. N. Narayanan, ACS Energy Lett., 2017, 2, 1355–1361 CrossRef CAS
- I. S. Kwon, I. H. Kwak, T. T. Debela, H. G. Abbas, Y. C. Park, J. P. Ahn, J. Park and H. S. Kang, ACS Nano, 2020, 14, 6295–6304 CrossRef CAS PubMed
- T. Kosmala, H. Coy Diaz, H.-P. Komsa, Y. Ma, A. V. Krasheninnikov, M. Batzill and S. Agnoli, Adv. Energy Mater., 2018, 8, 1800031 CrossRef
- S. Deng, F. Yang, Q. Zhang, Y. Zhong, Y. Zeng, S. Lin, X. Wang, X. Lu, C. Wang, L. Gu, X. Xia and J. Tu, Adv. Mater., 2018, 30, 1802223 CrossRef PubMed
- R. J. Toh, Z. Sofer, J. Luxa, D. Sedmidubský and M. Pumera, Chem. Commun., 2017, 53, 3054–3057 RSC
- S. Deng, M. Luo, C. Ai, Y. Zhang, B. Liu, L. Huang, Z. Jiang, Q. Zhang, L. Gu, S. Lin, X. Wang, L. Yu, J. Wen, J. Wang, G. Pan, X. Xia and J. Tu, Angew. Chem., Int. Ed., 2019, 58, 16289–16296 CrossRef CAS PubMed
- J. Zhang, J. Wu, H. Guo, W. Chen, J. Yuan, U. Martinez, G. Gupta, A. Mohite, P. M. Ajayan and J. Lou, Adv. Mater., 2017, 29, 1701955 CrossRef PubMed
- J. M. Woods, Y. Jung, Y. Xie, W. Liu, Y. Liu, H. Wang and J. J. Cha, ACS Nano, 2016, 10, 2004–2009 CrossRef CAS PubMed
- S. Wang, D. Zhang, B. Li, C. Zhang, Z. Du, H. Yin, X. Bi and S. Yang, Adv. Energy Mater., 2018, 8, 1801345 CrossRef
- W. Zhou, M. Chen, M. Guo, A. Hong, T. Yu, X. Luo, C. Yuan, W. Lei and S. Wang, Nano Lett., 2020, 20, 2923–2930 CrossRef CAS PubMed
- J. Zhang, X. Xu, L. Yang, D. Cheng and D. Cao, Small Methods, 2019, 3, 1900653 CrossRef CAS
- J. D. Benck, Z. Chen, L. Y. Kuritzky, A. J. Forman and T. F. Jaramillo, ACS Catal., 2012, 2, 1916–1923 CrossRef CAS
- F. Wang, P. He, Y. Li, T. A. Shifa, Y. Deng, K. Liu, Q. Wang, F. Wang, Y. Wen, Z. Wang, X. Zhan, L. Sun and J. He, Adv. Funct. Mater., 2017, 27, 1605802 CrossRef
- Y. Jing, X. Mu, C. Xie, H. Liu, R. Yan, H. Dai, C. Liu and X. D. Zhang, Int. J. Hydrogen Energy, 2019, 44, 809–818 CrossRef CAS
- J. Wu, T. Chen, C. Zhu, J. Du, L. Huang, J. Yan, D. Cai, C. Guan and C. Pan, ACS Sustainable Chem. Eng., 2020, 8, 4474–4480 CrossRef CAS
- D. Vikraman, S. Hussain, S. A. Patil, L. Truong, A. A. Arbab, S. H. Jeong, S. H. Chun, J. Jung and H. S. Kim, ACS Appl. Mater. Interfaces, 2021, 13, 5061–5072 CrossRef CAS PubMed
- M. S. Shawkat, J. Gil, S. S. Han, T. J. Ko, M. Wang, D. Dev, J. Kwon, G. H. Lee, K. H. Oh, H. S. Chung, T. Roy, Y. Jung and Y. Jung, ACS Appl. Mater. Interfaces, 2020, 12, 14341–14351 CrossRef CAS PubMed
- K. A. Persson, B. Waldwick, P. Lazic and G. Ceder, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 85, 235438 CrossRef
- J. N. Hansen, H. Prats, K. K. Toudahl, N. Mørch Secher, K. Chan, J. Kibsgaard and I. Chorkendorff, ACS Energy Lett., 2021, 6, 1175–1180 CrossRef CAS PubMed
- Y. Cheng, S. Lu, F. Liao, L. Liu, Y. Li and M. Shao, Adv. Funct. Mater., 2017, 27, 1700359 CrossRef
- S. Anantharaj, S. R. Ede, K. Karthick, S. Sam Sankar, K. Sangeetha, P. E. Karthik, S. Kundu, E. K. Pitchiah and S. Kundu, Energy Environ. Sci., 2018, 11, 744–771 RSC
- M. Gong, D. Y. Wang, C. C. Chen, B. J. Hwang and H. Dai, Nano Res., 2016, 9, 28–46 CrossRef CAS
- S. Anantharaj and S. Kundu, ACS Energy Lett., 2019, 4, 1260–1264 CrossRef CAS
- S. Anantharaj, P. E. Karthik and S. Noda, Angew. Chem., Int. Ed., 2021, 60, 23051–23067 CrossRef CAS PubMed
- S. Anantharaj, S. Noda, M. Driess and P. W. Menezes, ACS Energy Lett., 2021, 6, 1607–1611 CrossRef CAS
- C. Tsai, K. Chan, J. K. Nørskov and F. Abild-Pedersen, Surf. Sci., 2015, 640, 133–140 CrossRef CAS
- S. H. Mir, S. Chakraborty, J. Wärnå, S. Narayan, P. C. Jha, P. K. Jha and R. Ahuja, Catal. Sci. Technol., 2017, 7, 687–692 RSC
- J. Liu, H. Xu, J. Yan, J. Huang, Y. Song, J. Deng, J. Wu, C. Ding, X. Wu, S. Yuan and H. Li, J. Mater. Chem. A, 2019, 7, 18906–18914 RSC
- H. Huang, X. Fan, D. J. Singh and W. Zheng, ACS Omega, 2018, 3, 10058–10065 CrossRef CAS PubMed
- L. J. Ma and H. Shen, Appl. Surf. Sci., 2021, 545, 149013 CrossRef CAS
- S. Lin, Y. Liu, Z. Hu, W. Lu, C. H. Mak, L. Zeng, J. Zhao, Y. Li, F. Yan, Y. H. Tsang, X. Zhang and S. P. Lau, Nano Energy, 2017, 42, 26–33 CrossRef CAS
- L. Zhang, D. Kong, Q. Zhuang, M. Wang, T. Zhang, J. Zang, W. Shen, T. Xu, D. Wu, Y. Tian, Y. Wang, X. Li and X. Huang, J. Mater. Chem. C, 2021, 9, 9524–9531 RSC
- S. Liu, X. Mu, W. Li, M. Lv, B. Chen, C. Chen and S. Mu, Nano Energy, 2019, 61, 346–351 CrossRef CAS
- M. Yi, S. Hu, B. B. Lu, N. Li, Z. Zhu, X. Huang, M. Wang and J. Zhang, J. Alloys Compd., 2021, 884, 161042, DOI:10.1016/j.jallcom.2021.161042
- S. Jeong, H. D. Mai, T. K. Nguyen, J. S. Youn, K. H. Nam, C. M. Park and K. J. Jeon, Appl. Catal., B, 2021, 293, 120227 CrossRef CAS
- Z. Wang, B. Xiao, Z. Lin, Y. Xu, Y. Lin, F. Meng, Q. Zhang, L. Gu, B. Fang, S. Guo and W. Zhong, Angew. Chem., 2021, 60(43), 23388–23393, DOI:10.1002/ange.202110335
- D. Voiry, H. Yamaguchi, J. Li, R. Silva, D. C. B. Alves, T. Fujita, M. Chen, T. Asefa, V. B. Shenoy, G. Eda and M. Chhowalla, Nat. Mater., 2013, 12, 850–855 CrossRef CAS PubMed
- D. Escalera-López, R. Griffin, M. Isaacs, K. Wilson, R. E. Palmer and N. V. Rees, Appl. Mater. Today, 2018, 11, 70–81 CrossRef
- Y. Zhou, J. V. Pondick, J. L. Silva, J. M. Woods, D. J. Hynek, G. Matthews, X. Shen, Q. Feng, W. Liu, Z. Lu, Z. Liang, B. Brena, Z. Cai, M. Wu, L. Jiao, S. Hu, H. Wang, C. M. Araujo and J. J. Cha, Small, 2019, 15, 1900078 CrossRef PubMed
- D. Xu, P. Xu, Y. Zhu, W. Peng, Y. Li, G. Zhang, F. Zhang, T. E. Mallouk and X. Fan, ACS Appl. Mater. Interfaces, 2018, 10, 2810–2818 CrossRef CAS PubMed
- X. Zhou, Y. Liu, H. Ju, B. Pan, J. Zhu, T. Ding, C. Wang and Q. Yang, Chem. Mater., 2016, 28, 1838–1846 CrossRef CAS
- M. Zou, J. Zhang, H. Zhu, M. Du, Q. Wang, M. Zhang and X. Zhang, J. Mater. Chem. A, 2015, 3, 12149–12153 RSC
- X. Wang, Y. Chen, B. Zheng, F. Qi, J. He, Q. Li, P. Li and W. Zhang, J. Alloys Compd., 2017, 691, 698–704 CrossRef CAS
- X. Zhang, L. Li, Y. Guo, D. Liu and T. You, J. Colloid Interface Sci., 2016, 472, 69–75 CrossRef CAS PubMed
- J. Xie, H. Qu, J. Xin, X. Zhang, G. Cui, X. Zhang, J. Bao, B. Tang and Y. Xie, Nano Res., 2017, 10, 1178–1188 CrossRef CAS
- R. A. B. Villaos, C. P. Crisostomo, Z.-Q. Huang, S.-M. Huang, A. A. B. Padama, M. A. Albao, H. Lin and F.-C. Chuang, npj 2D Mater. Appl., 2019, 3, 1–8 CrossRef CAS
- D. Hu, T. Zhao, X. Ping, H. Zheng, L. Xing, X. Liu, J. Zheng, L. Sun, L. Gu, C. Tao, D. Wang and L. Jiao, Angew. Chem., 2019, 131, 7051–7055 CrossRef
- X. Li, Y. Fang, J. Wang, H. Fang, S. Xi, X. Zhao, D. Xu, H. Xu, W. Yu, X. Hai, C. Chen, C. Yao, H. B. Tao, A. G. R. Howe, S. J. Pennycook, B. Liu, J. Lu and C. Su, Nat. Commun., 2021, 12, 2351 CrossRef CAS PubMed
- X. Ping, D. Liang, Y. Wu, X. Yan, S. Zhou, D. Hu, X. Pan, P. Lu and L. Jiao, Nano Lett., 2021, 21, 3857–3863 CrossRef CAS PubMed
- Y. Fang, S. Wang, G. Lin, X. Wang and F. Huang, Chem. Commun., 2021, 57, 7946–7949 RSC
- Y. He, M. Boubeche, Y. Zhou, D. Yan, L. Zeng, X. Wang, K. Yan and H. Luo, J. Phys. Mater., 2020, 4, 014001 CrossRef
- S. Deng, Y. Zhong, Y. Zeng, Y. Wang, Z. Yao, F. Yang, S. Lin, X. Wang, X. Lu, X. Xia and J. Tu, Adv. Mater., 2017, 29, 1700748 CrossRef PubMed
- S. Bolar, S. Shit, N. C. Murmu and T. Kuila, ChemElectroChem, 2020, 7, 336–346 CrossRef CAS
- D. Vikraman, S. Hussain, L. Truong, K. Karuppasamy, H. J. Kim, T. Maiyalagan, S. H. Chun, J. Jung and H. S. Kim, Appl. Surf. Sci., 2019, 480, 611–620 CrossRef CAS
- V. T. Nguyen, P. A. Le, Y. C. Hsu and K. H. Wei, ACS Appl. Mater. Interfaces, 2020, 12, 11533–11542 CrossRef CAS PubMed
- B. Xia, T. Wang, X. Jiang, T. Zhang, J. Li, W. Xiao, P. Xi, D. Gao, D. Xue and J. Ding, ACS Energy Lett., 2018, 3, 2167–2172 CrossRef CAS
- D. Vikraman, S. Hussain, K. Akbar, L. Truong, A. Kathalingam, S. H. Chun, J. Jung, H. J. Park and H. S. Kim, ACS Sustainable Chem. Eng., 2018, 6, 8400–8409 CrossRef CAS
- C. Jian, Q. Cai, W. Hong, J. Li and W. Liu, Small, 2018, 14, 1703798 CrossRef PubMed
- X. Meng, L. Yu, C. Ma, B. Nan, R. Si, Y. Tu, J. Deng, D. Deng and X. Bao, Nano Energy, 2019, 61, 611–616 CrossRef CAS
- Y. Yin, Y. Zhang, T. Gao, T. Yao, X. Zhang, J. Han, X. Wang, Z. Zhang, P. Xu, P. Zhang, X. Cao, B. Song and S. Jin, Adv. Mater., 2017, 29, 1700311 CrossRef PubMed
- Z. Jiang, W. Zhou, A. Hong, M. Guo, X. Luo and C. Yuan, ACS Energy Lett., 2019, 2830–2835 CrossRef CAS
- Y. Li, K. Yin, L. Wang, X. Lu, Y. Zhang, Y. Liu, D. Yan, Y. Song and S. Luo, Appl. Catal., B, 2018, 239, 537–544 CrossRef CAS
- S. Li, W. Zang, X. Liu, S. J. Pennycook, Z. Kou, C. Yang, C. Guan and J. Wang, Chem. Eng. J., 2019, 359, 1419–1426 CrossRef CAS
- J. C. McGlynn, T. Dankwort, L. Kienle, N. A. G. Bandeira, J. P. Fraser, E. K. Gibson, I. Cascallana-Matías, K. Kamarás, M. D. Symes, H. N. Miras and A. Y. Ganin, Nat. Commun., 2019, 10, 4916 CrossRef PubMed
- M. D. Sharma, C. Mahala and M. Basu, Inorg. Chem., 2020, 59, 4377–4388 CrossRef CAS PubMed
- Q. Zhou, X. Luo, Y. Li, Y. Nan, H. Deng, E. Ou and W. Xu, Int. J. Hydrogen Energy, 2020, 45, 433–442 CrossRef CAS
- Y. Chen, A. Lu, P. Lu, X. Yang, C. Jiang, M. Mariano, B. Kaehr, O. Lin, A. Taylor, I. D. Sharp, L. Li, S. S. Chou and V. Tung, Adv. Mater., 2017, 29, 1703863 CrossRef PubMed
- L. Wu, A. Longo, N. Y. Dzade, A. Sharma, M. M. R. M. Hendrix, A. A. Bol, N. H. de Leeuw, E. J. M. Hensen and J. P. Hofmann, ChemSusChem, 2019, 12, 4383–4389 CrossRef CAS PubMed
- P. Thangasamy, S. Oh, S. Nam and I. K. Oh, Carbon, 2020, 158, 216–225 CrossRef CAS
- N. Masurkar, N. K. Thangavel and L. M. R. Arava, ACS Appl. Mater. Interfaces, 2018, 10, 27771–27779 CrossRef CAS PubMed
- D. Lu, X. Ren, L. Ren, W. Xue, S. Liu, Y. Liu, Q. Chen, X. Qi and J. Zhong, ACS Appl. Energy Mater., 2020, 3, 3212–3219 CrossRef CAS
- S. Maity, B. Das, M. Samanta, B. K. Das, S. Ghosh and K. K. Chattopadhyay, ACS Appl. Energy Mater., 2020, 3, 5067–5076 CrossRef CAS
- J. B. McManus, G. Cunningham, N. McEvoy, C. P. Cullen, F. Gity, M. Schmidt, D. McAteer, D. Mullarkey, I. V. Shvets, P. K. Hurley, T. Hallam and G. S. Duesberg, ACS Appl. Energy Mater., 2019, 2, 521–530 CrossRef CAS
- P. Zhuang, Y. Sun, P. Dong, W. Smith, Z. Sun, Y. Ge, Y. Pei, Z. Cao, P. M. Ajayan, J. Shen and M. Ye, Adv. Funct. Mater., 2019, 29, 1901290 CrossRef
- M. A. R. Anjum, H. Y. Jeong, M. H. Lee, H. S. Shin and J. S. Lee, Adv. Mater., 2018, 30, 1707105 CrossRef PubMed
- Q. Zhu, W. Chen, H. Cheng, Z. Lu and H. Pan, ChemCatChem, 2019, 11, 2667–2675 CrossRef CAS
- P. A. Le, V. T. Nguyen, V. Q. Le, Y. C. Lu, S. Y. Huang, S. K. Sahoo, Y. H. Chu and K. H. Wei, ACS Appl. Energy Mater., 2021, 4, 5143–5154 CrossRef CAS
- H. Li, J. Zou, S. Xie, X. Leng, D. Gao, H. Yang and X. Mao, J. Alloys Compd., 2017, 725, 884–890 CrossRef CAS
- Z. Liu, N. Li, C. Su, H. Zhao, L. Xu, Z. Yin, J. Li and Y. Du, Nano Energy, 2018, 50, 176–181 CrossRef CAS
- X. Guo, J. Ji, Q. Jiang, L. Zhang, Z. Ao, X. Fan, S. Wang, Y. Li, F. Zhang, G. Zhang and W. Peng, ACS Appl. Mater. Interfaces, 2017, 9, 30591–30598 CrossRef CAS PubMed
- X. Wang, Y. Chen, F. Qi, B. Zheng, J. He, Q. Li, P. Li, W. Zhang and Y. Li, Electrochem. Commun., 2016, 72, 74–78 CrossRef CAS
- Y. Pan, F. Zheng, X. Wang, H. Qin, E. Liu, J. Sha, N. Zhao, P. Zhang and L. Ma, J. Catal., 2020, 382, 204–211 CrossRef CAS
- Y. Sun, X. Zhang, B. Mao and M. Cao, Chem. Commun., 2016, 52, 14266–14269 RSC
- J. H. Nam, M. J. Jang, H. Y. Jang, W. Park, X. Wang, S. M. Choi and B. Cho, J. Energy Chem., 2020, 47, 107–111 CrossRef
- Y. Yao, Z. Jin, Y. Chen, Z. Gao, J. Yan, H. Liu, J. Wang, Y. Li and S. (Frank) Liu, Carbon, 2018, 129, 228–235 CrossRef CAS
- M. Yang, Z. Sun and Y. H. Hu, J. Phys. Chem. C, 2019, 123, 12142–12148 CrossRef CAS
- N. Ling, S. Zheng, Y. Lee, M. Zhao, E. Kim, S. Cho and H. Yang, APL Mater., 2021, 9, 061108 CrossRef CAS
- H. Kim, V. Kanade, M. Kim, K. S. Kim, B. An, H. Seok, H. Yoo, L. E. Chaney, S. Kim, C. Yang, G. Y. Yeom, D. Whang, J. Lee and T. Kim, Small, 2020, 16, 1905000 CrossRef CAS PubMed
- X. Shang, Y. Rao, S. S. Lu, B. Dong, L. M. Zhang, X. H. Liu, X. Li, Y. R. Liu, Y. M. Chai and C. G. Liu, Mater. Chem. Phys., 2017, 197, 123–128 CrossRef CAS
- J. Li, P. Liu, Y. Qu, T. Liao and B. Xiang, Int. J. Hydrogen Energy, 2018, 43, 2601–2609 CrossRef CAS
- S. Balasubramanyam, M. Shirazi, M. A. Bloodgood, L. Wu, M. A. Verheijen, V. Vandalon, W. M. M. Kessels, J. P. Hofmann and A. A. Bol, Chem. Mater., 2019, 31, 5104–5115 CrossRef CAS PubMed
- M. Kim, G. H. Park, S. Seo, V. Q. Bui, Y. Cho, Y. Hong, Y. Kawazoe and H. Lee, ACS Appl. Mater. Interfaces, 2021, 13, 11403–11413 CrossRef CAS PubMed
- J. Li, M. Hong, L. Sun, W. Zhang, H. Shu and H. Chang, ACS Appl. Mater. Interfaces, 2018, 10, 458–467 CrossRef CAS PubMed
- H. Kwon, B. Ji, D. Bae, J. H. Lee, H. J. Park, D. H. Kim, Y. M. Kim, Y. W. Son, H. Yang and S. Cho, Appl. Surf. Sci., 2020, 515, 145972 CrossRef CAS
- F. Yang, Y. Zhao, Y. Du, Y. Chen, G. Cheng, S. Chen and W. Luo, Adv. Energy Mater., 2018, 8, 1703489 CrossRef
- J. Liu, G. Ding, J. Yu, X. Liu, X. Zhang, J. Guo, J. Zhang, W. Ren and R. Che, J. Mater. Chem. A, 2019, 7, 18072–18080 RSC
- Y. Zhao, N. Jia, X. R. Wu, F. M. Li, P. Chen, P. J. Jin, S. Yin and Y. Chen, Appl. Catal., B, 2020, 270, 118880 CrossRef CAS
- S. Niu, S. Li, J. Hu, Y. Li, Y. Du, X. Han and P. Xu, Chem. Commun., 2019, 55, 14649–14652 RSC
- D. Luo, B. Zhou, Z. Li, X. Qin, Y. Wen, D. Shi, Q. Lu, M. Yang, H. Zhou and Y. Liu, J. Mater. Chem. A, 2018, 6, 2311–2317 RSC
- G. Chen, T. Wang, J. Zhang, P. Liu, H. Sun, X. Zhuang, M. Chen and X. Feng, Adv. Mater., 2018, 30, 1706279 CrossRef PubMed
- J. Wang, Z. Wei, S. Mao, H. Li and Y. Wang, Energy Environ. Sci., 2018, 11, 800–806 RSC
- S. Anantharaj, P. E. Karthik, B. Subramanian, S. Kundu, S. Balasubramanian and S. Kundu, ACS Catal., 2016, 6, 4660–4672 CrossRef CAS
- Y. Zhang, J. Yan, X. Ren, L. Pang, H. Chen and S. (Frank) Liu, Int. J. Hydrogen Energy, 2017, 42, 5472–5477 CrossRef CAS
- S. Anantharaj, K. Karthick, M. Venkatesh, T. V. S. V. Simha, A. S. Salunke, L. Ma, H. Liang and S. Kundu, Nano Energy, 2017, 39, 30–43 CrossRef CAS
| This journal is © The Royal Society of Chemistry 2022 |