
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Non-innocent cyanido ligands: tetracyanidoferrate(–II) as carbonyl copycat†
Franziska
Jach‡
 *ab,
Theresa
Block
c,
Yurii
Prots
a,
Marcus
Schmidt
a,
Matej
Bobnar§
a,
Rainer
Pöttgen
c,
Michael
Ruck
ab and
Peter
Höhn
a
*ab,
Theresa
Block
c,
Yurii
Prots
a,
Marcus
Schmidt
a,
Matej
Bobnar§
a,
Rainer
Pöttgen
c,
Michael
Ruck
ab and
Peter
Höhn
a
aMax-Planck-Institute for Chemical Physics of Solids, Nöthnitzer Straße 40, 01187 Dresden, Germany. E-mail: Franziska.Jach@tu-dresden.de
bFaculty of Chemistry and Food Chemistry, Technische Universität Dresden, 01062 Dresden, Germany
cInstitut für Anorganische und Analytische Chemie, Westfälische Wilhelms-Universität Münster, Corrensstraße 30, 48149 Münster, Germany
First published on 6th April 2022
Abstract
While a negative oxidation state occurs rarely for metals in general, this is commonly known for metal carbonyl anions, i.e. carbonyl metalates. Although CO and CN− are isoelectronic, cyanidometalates usually do not exhibit metal centers with negative oxidation states. However, we report on the electron-rich tetrahedral tetracyanidoferrate(–II) anion [Fe(CN)4]6−, which was stabilized in (Sr3N)2[Fe(CN)4] (space group R3c, a = 702.12(2) pm, c = 4155.5(2) pm). Microcrystalline powders were synthesized by a solid-state route, single crystals were obtained from Na metal flux. In comparison to classical cyanidometalates, C–N distances are longer and stretching frequencies are lower as indicated by X-ray diffraction, IR and Raman spectroscopy. Weak C–N, strong Fe–C bonds as well as the anion geometry resemble the isoelectronic tetrahedral carbonyl ferrate [Fe(CO)4]2−. 57Fe Mössbauer spectroscopic measurements reveal a negative isomer shift in agreement with substantially delocalized d electrons due to strong π back-bonding. These results point to a very similar bonding situation of both 18e tetracyanido and tetracarbonyl ferrates including non-innocent redox-active ligands and a d10 closed shell configuration on iron. Hereby, new tetracyanidoferrate(–II) provides a missing link for a more in-depth understanding of the chemical bonding trends of highly-reduced cyanidometalates in the quest for even higher reduced transition metals in this exceptional class of compounds.
Introduction
The possibility of metal anions has fascinated chemists for over 100 years.1 Systematic studies of transition metals with negative oxidation states started in the 1930s with metal carbonyl anions such as [FeII(en)3][Fe–II(CO)4] (en = ethylenediamine), although its ionic nature was not revealed until later.2–4 During the following decades, carbonyl metalates were applied as starting materials and catalysts in (metal)organic syntheses.5,6 Additionally, a quest for even further reduced metalates resulted in extreme examples such as [Co(CO)3]3− and [Cr(CO)4]4−.1,7–10Although the cyanide anion is isoelectronic to carbon monoxide, respective cyanidometalates typically differ significantly from carbonyl metalates with regard to chemical bonding, properties and applications. The σ-donor function of the typically redox-inactive CN ligand allows for stabilization of relatively high oxidation states of the transition metal. In contrast, negative oxidation states for carbonyl metalates are achieved by strong π back-bonding shifting electron density towards the non-innocent, redox-active CO ligands.11 These differences are best investigated by IR and Raman spectroscopy: stretching frequencies of the CN ligand ν(CN) are similar for complexes and simple salts (typically 2000–2200 cm−1).12,13 For carbonyl metalates, ν(CO) frequencies are much lower in comparison to gaseous CO (2143 cm−1),14e.g. 1614 cm−1 for [Co(CO)3]3−.8,9
However, exotic examples of highly reduced cyanido complexes have been reported, e.g. [Co(CN)3]6− and [Fe(CN)3]7−.15,16 These metalates mimic the bonding situation of carbonyl metalates including unusual non-innocent CN ligands exhibiting very low ν(CN), such as 1641–1696 cm−1 for Ba3[Co(CN)3].15 Quantum chemical calculations revealed a closed shell d10 configuration for the Co and Fe centers and strong π back-bonding yielding intermediate-valent CN1.67− ligands due to occupied CN ppπ* antibonding orbitals.15,16
Taking into account the striking similarities between isoelectronic [Co(CO)3]3− and [Co(CN)3]6−, it can be anticipated, that further highly reduced cyanido copycats of metal carbonyl anions exist. In this work, we present the tetracyanidoferrate(–II) anion stabilized in (Sr3N)2[Fe(CN)4] which is isoelectronic to Hieber's famous prototype [Fe(CO)4]2−.
Results and discussion
Synthesis and thermal stability
Air and moisture sensitive, nearly single-phase micro-crystalline powders of (Sr3N)2[Fe(CN)4] were synthesized using pelletized mixtures of Sr2N, iron powder, graphite, and NaN3 in the molar ratio of Sr![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Fe:C:N = 6:1:4:6 at 1070 K in Ar atmosphere. The synthesis is very sensitive to the nitrogen partial pressure: higher nitrogen contents yield thermodynamically more stable carbodiimides such as SrCN2.17 Despite reasonable efforts, small contaminations of elemental iron and Sr4N[C2N][CN2]18 could not be avoided (see ESI†). Transparent yellow hexagonal plate-like crystals were grown from a slowly cooled solution of starting materials in Na flux.
:Fe:C:N = 6:1:4:6 at 1070 K in Ar atmosphere. The synthesis is very sensitive to the nitrogen partial pressure: higher nitrogen contents yield thermodynamically more stable carbodiimides such as SrCN2.17 Despite reasonable efforts, small contaminations of elemental iron and Sr4N[C2N][CN2]18 could not be avoided (see ESI†). Transparent yellow hexagonal plate-like crystals were grown from a slowly cooled solution of starting materials in Na flux.
The compound rearranges to Sr3.5[Fe(CN)3]16 upon annealing at 970 K, while the latter structure is stable up to ca. 1200 K in Ar, as indicated by thermal analysis (see Fig. S1, ESI†). This reveals the high thermal stability of highly reduced cyanidometalates.
Crystal structure
The trigonal crystal structure of (Sr3N)2[Fe(CN)4] (space group R3c, no. 161, a = 702.12(2) pm, c = 4155.5(2) pm, 300 K) contains layers of edge-sharing (NSr6/2)-octahedra forming a honeycomb motif in the ab plane (Fig. 1). Slightly distorted tetrahedral cyanidoferrate anions [Fe(CN)4]6− are positioned in between these layers with one CN-ligand rising into the honeycomb void.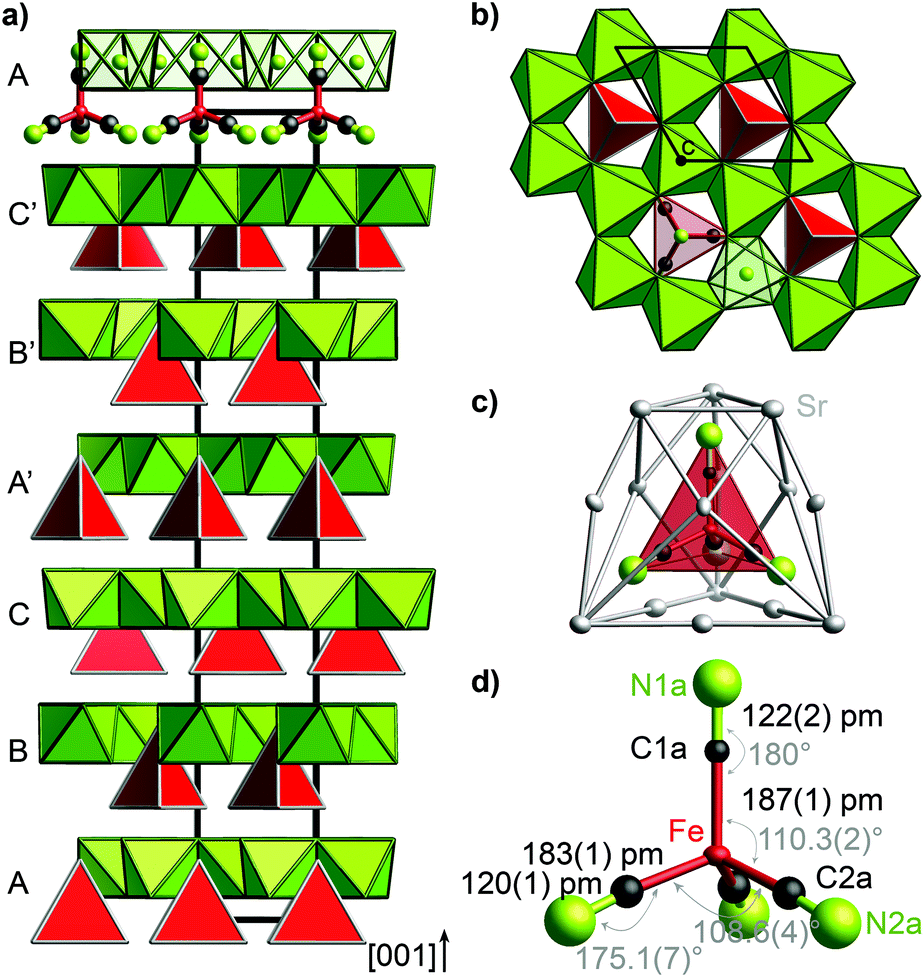 | ||
| Fig. 1 Crystal structure of (Sr3N)2[Fe(CN)4] (a) layers of (NSr6/2) octahedra (green) and [Fe(CN)4]6− tetrahedra (red) exhibit an ABCA′B′C′ stacking sequence in [001] direction. (b) Honeycomb motif of (NSr6/2) octahedra (green) in the ab plane. [Fe(CN)4]6− tetrahedra (red) rise into the honeycomb void. (c) Cage of 15 Sr2+ cations surrounding the [Fe(CN)4]6− tetrahedron. (d) Tetrahedral anion [Fe(CN)4]6− with bond lengths (black) and angles (gray). Ellipsoids in c and d are set at 99% probability. For simplicity reasons, the ideal structure without stacking faults is shown in this figure. | ||
The uniform orientation of the [Fe(CN)4]6− tetrahedra grounds the polarity of the structure. The double layers of (NSr6/2)-octahedra and [Fe(CN)4]6− tetrahedra are stacked along c with shifted honeycomb voids and tetrahedra from one layer to the next. The stacking sequence ABCA′B′C′ thereby yields the large cell parameter c. The nearest neighbors of the cyanidoferrate themselves form a cage of 15 Sr2+ cations (Fig. 1c). The differing environment for the CN ligands results in small deviations from tetrahedral symmetry for [Fe(CN)4]6− with respect to C–N distances and bond angles (Fig. 1d). In addition to this description, stacking faults are revealed by diffuse scattering along c* and additional peaks in the difference Fourier map. For details see Fig. S2–S5 (ESI†).
Nitridometalates such as (Ca3N)2[FeN3] exhibit a related crystal structure in space group P63/mcm.19 In contrast to the title compound, trigonal planar [FeN3]6− anions are located in between honeycomb layers of (NCa6/2) octahedra forming a much simpler stacking sequence.
IR and Raman spectroscopy
IR and Raman spectra of (Sr3N)2[Fe(CN)4] exhibit three distinct regions of sharp bands (Fig. 2): the ν(CN) valence band region, a region of ν(FeC) and δ(FeCN) deformation modes and so-called lattice modes below 300 cm−1. A tetrahedral molecule of point group Td containing nine atoms exhibits 21 vibrations: 2A1 + 2E + F1 + 4F2.20,21 In accordance to this point group and the isoelectronic tetrahedral molecular anion [Fe(CO)4]2−,20,22 two Raman and one IR mode are observed for ν(CN) at similar frequencies for both anions (see Table 1). In contrast, ν(FeC) and δ(FeCN) modes are split as a result of the symmetry reduction for [Fe(CN)4]6− in the solid state structure (site symmetry C3). An unambiguous assignment of ν(FeC) and δ(FeCN) modes within that region is not possible without further investigations. For molecular site group analysis, see ESI.†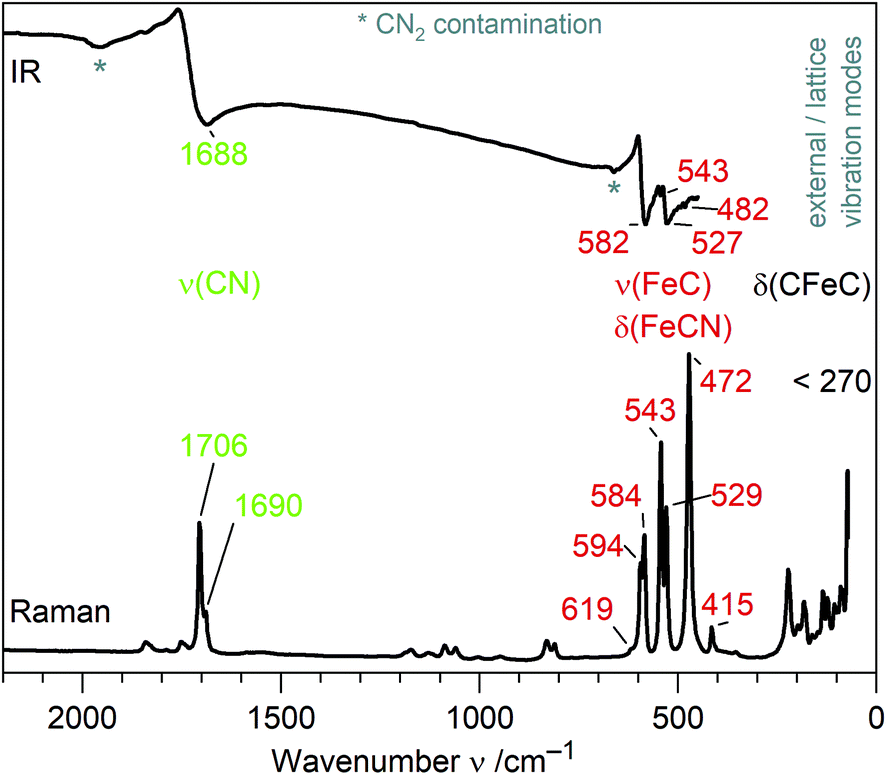 | ||
| Fig. 2 IR and Raman spectra of (Sr3N)2[Fe(CN)4] with assigned molecular vibrations of [Fe(CN)4]6−. | ||
| Ligand | Anion | Compound | Geometry | d(C–N) | ν(CN)/ν(CO) | d(M–C) | ν(MC) & δ(MCN)/δ(MCO) | Ref. |
|---|---|---|---|---|---|---|---|---|
| a Italic: IR data; regular: Raman data. b dien = diethylenetriamine. c Xyl = 2,6-dimethylphenyl. d QuinH = quinuclidinium. e Calculated values. | ||||||||
| CN | [Fe(CN)4]6− | (Sr3N)2[Fe(CN)4] | Tetrahedral | 120(1), 122(2) | 1688; 1690, 1706 | 183(1), 187(1) | 482–582; 415–619 | This work |
| [Fe(CN)3]7− | LiSr3[Fe(CN)3] | Trigonal-planar | 126.7(3) | 1490; 1581 | 178.6(4) | 557–664; 464–611 | 16 | |
| [Co(CN)3]6− | Ba3[Co(CN)3] | Trigonal-planar | 123.5(5) | 1641; 1696 | 176.9(3) | 542, 569; 420–582 | 15 and 27 | |
| [Fe(CN)4]2− | [Ni(dien)2][Fe(CN)4]b | Square-planar | 114–116 | 2077, 2117 | 185–186 | 23 | ||
| [Fe(CN)6]3− | Rb2Na[Fe(CN)6] | Octahedral | 114, 115 | 2122 | 194 | 394, 523 | 25 | |
| [Fe(CN)6]4− | Ba(NH4)2[Fe(CN)6]3H2O | Octahedral | 115 | 2041; 2060, 2095 | 191 | <585 | 28 | |
| [Fe(CNXyl)4]2− | [K([2.2.2]cryptand)]2 [Fe(CNXyl)4]c | Tetrahedral | 123–124 | 1675 | 176–177 | 24 | ||
| NaCN | 116 | 2088 | 29 | |||||
| CO | [Fe(CO)4]2− | A 2[Fe(CO)4] (A = K, Na) | Tetrahedral | 118 | 1729–1786; 1788 | 175 | 555, 646; 464–785 | 22, 30–32 |
| [Co(CO)4]− | A[Co(CO)4] (A = QuinH, Na)d | Tetrahedral | 115 | 1886; 1883, 1918 | 176–178 | 530, 555; 439–715 | 22, 30 and 33 | |
| [Co(CO)3]3− | Na3[Co(CO)3] | Trigonal-planar | 126e | 1614 | 177e | 8 and 9 | ||
| CO | CO (gas) | 113 | 2143 | 14 and 29 | ||||
Comparison of [Fe(CN)4]6− and related metalates
To the best of our knowledge, [Fe(CN)4]6− is the first homoleptic 18e cyanidoferrate with four ligands. The only other four-coordinated cyanidoferrate is 14e square-planar [Fe(CN)4]2−.23 On the other hand, 18e tetraisocyanidometalates were reported, such as [Fe(CNXyl)4]2− (Xyl = 2,6-dimethylphenyl).24In contrast to simple cyanide salts and classical cyanidometalates such as [Fe(CN)4]2− or [Fe(CN)6]3−,23,25 C–N bond lengths in [Fe(CN)4]6− are unusually long and ν(CN) valence frequencies are unusually low pointing towards severely weakened C–N bonds (Table 1). This is accompanied by much stronger Fe–C bonds than in [Fe(CN)6]3− evidenced by shorter distances and higher ν(FeC)/δ(FeCN) frequencies. These observations are in good accordance with the textbook description of π back-bonding: electrons are shifted from metal d orbitals to antibonding π* orbitals of the CN ligand, which strengthens the Fe–C bond and weakens the C–N bond. High values of δ(FeCN) vibrations likewise point to a stiffer Fe–C–N unit due to increased π bonding.26 Similar trends have been discussed for the complexes [Fe(CN)6]3− and [Fe(CN)6]4−, with the higher charged metalate exhibiting more pronounced π back-bonding.11,12 However, changes for hexacyanidoferrates are small in comparison to the tetracyanidoferrates [Fe(CN)4]2− and [Fe(CN)4]6− (Table 1). Instead of classical cyanidoferrates, chemical bonding in [Fe(CN)4]6− resembles the situation in isoelectronic and likewise tetrahedral [Fe(CO)4]2− evidenced by similar values for bond lengths and stretching frequencies.
With regard to the previously reported highly reduced cyanidometalates, a qualitatively similar bonding situation can be assumed for all 18e complexes [Co(CN)3]6−, [Fe(CN)3]7−, and [Fe(CN)4]6− based on interatomic distances and vibrational frequencies. Quantitatively, C–N bonds are stronger (shorter d(C–N), higher ν(CN)) and Fe–C bonds are weaker for [Fe(CN)4]6− in comparison to [Fe(CN)3]7− pointing to somewhat less π back-bonding for the tetracyanidoferrate. This is reasonable since the lesser charge of [Fe(CN)4]6− is distributed over more ligands which is equivalent to less π back-bonding in contrast to [Fe(CN)3]7−. A similar trend is observed for the pair [Co(CO)3]3− and [Co(CO)4]− (Table 1).
The isoelectronic [Fe(CNXyl)4]2− exhibits somewhat longer C–N distances and lower ν(CN) frequencies than [Fe(CN)4]6− due to the bridging CN groups instead of terminal cyanido ligands.
Physical properties
(Sr3N)2[Fe(CN)4] exhibits electrically insulating behavior consistent with mainly ionic interactions between Sr2+, N3−, and complex anions [Fe(CN)4]6−. This accords well with reported properties of similar reduced cyanidometalates such as Sr3[Co(CN)3] and Ba3.5[Fe(CN)3].15,16 Unfortunately, the conclusive examination of magnetic susceptibility data of (Sr3N)2[Fe(CN)4] was hindered by too large contaminations of elemental Fe from synthesis.57Fe Mössbauer spectroscopy
To get a closer look into the valence state of the iron atoms in (Sr3N)2[Fe(CN)4], 57Fe Mössbauer spectroscopic measurements were conducted at 293 and 78 K (Fig. 3). Both spectra could be well reproduced with a superposition of two sub-signals in a ratio of 63(1):37(1) at 293 (top) and 59(1):41(1) at 78 K (bottom), respectively. Both spectra show similar fitting parameters (Table 2) for the particular simulated sub-spectra, hence we will focus on the 293 K data. The blue sub-signal can be ascribed to residual elemental iron from the synthesis, since the isomer shift and the hyperfine field splitting accord well with the reported data (δ = −0.1 mm s−1, BHf = 33.1 T for elemental Fe);34,35 these iron atoms in cubic environment show no quadrupole splitting. The green sub-spectrum with an isomer shift of δ = −0.227(2) mm s−1 and a quadrupole splitting parameter of ΔEQ = 0.373(2) mm s−1 can be assigned to the iron atoms in the tetrahedral cyanidoferrate(–II) units of (Sr3N)2[Fe(CN)4]. In 57Fe Mössbauer spectroscopy, a more negative isomer shift indicates a higher s-electron density at the iron nucleus. The d-electrons shield the s-electrons, which leads to a lower delocalization and a higher s-electron density at the nucleus with a decreasing number of d-electrons. As a result we observe more negative shifts for compounds with iron atoms in highly positive oxidation states like K2Fe+VIO4 with an isomer shift of δ = −1.013 mm s−1 (the original value in the literature is δ = −1.262 mm s−1 referred to a 57Co/Pt source).36,37 For iron in the formal oxidation state –II we would hence assume (just by extrapolation) a positive isomer shift at first sight. Unlike this assumption we observe a negative isomer shift for the iron atoms in the [Fe(CN)4]6− unit. A similar shift is reported for the isoelectric anion [Fe(CO)4]2− in Na2[Fe(CO)4] by Erickson and Fairhall.38 Na2[Fe(CO)4] shows an isomer shift of δ = −0.251 mm s−1 relative to elemental iron which corresponds to an isomer shift of δ = −0.351 mm s−1 relative to a 57Co/Rh source.35,36
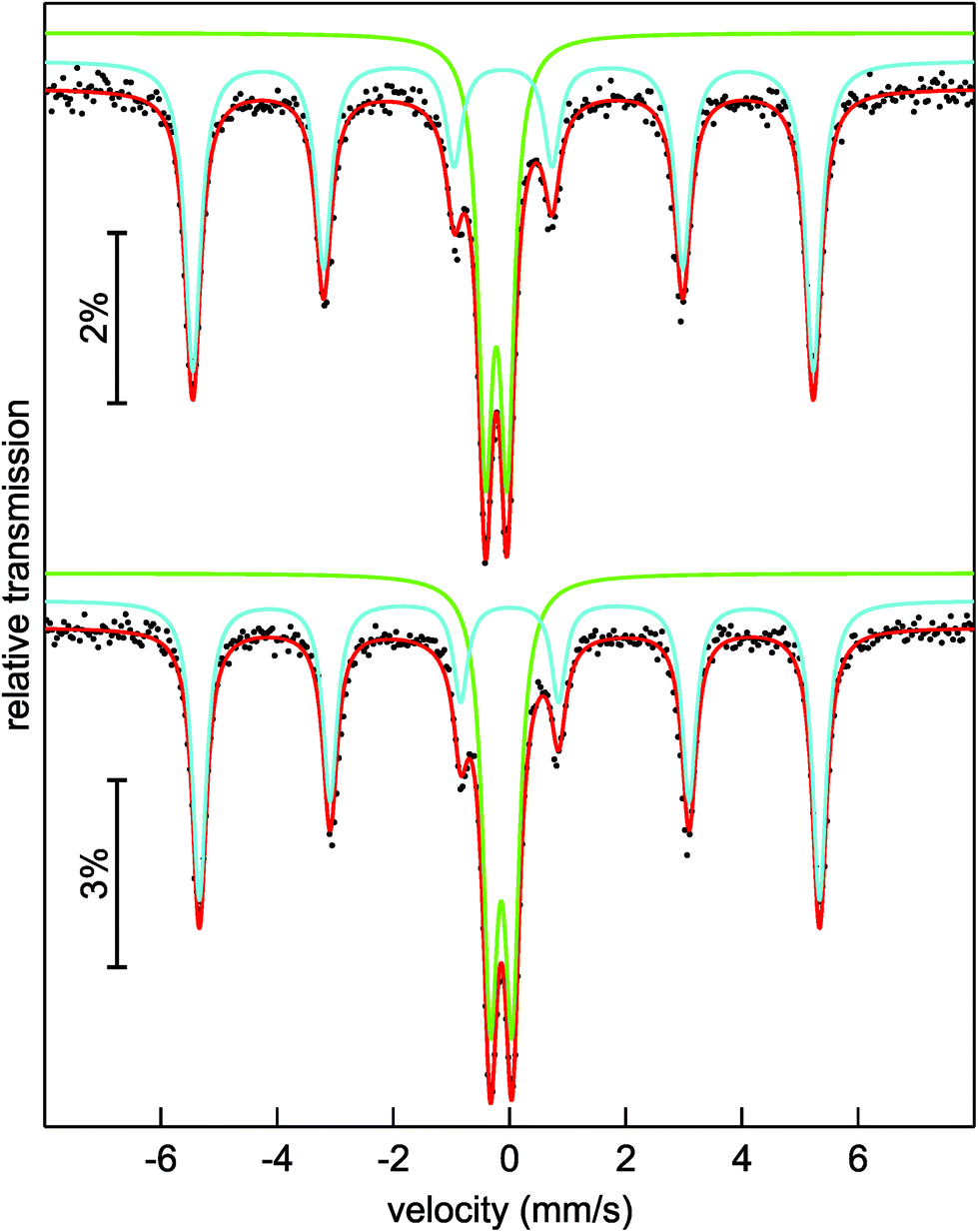 | ||
| Fig. 3 Experimental (black dots) and simulated (lines) 57Fe Mössbauer spectra of (Sr3N)2[Fe(CN)4] at 293 K (top) and 78 K (bottom). | ||
| T/K | δ (mm s−1) | ΔEQ (mm s−1) | Γ (mm s−1) | B Hf (T) | Area (%) |
|---|---|---|---|---|---|
| *Parameter was kept fixed during fitting procedure. |
|||||
| 293 | −0.110(1) | 0* | 0.328(5) | 33.12(1) | 63(1) |
| −0.227(2) | 0.373(2) | 0.297(4) | — | 37(1) | |
| 78 | 0.001(1) | 0* | 0.298(4) | 33.10(1) | 59(1) |
| −0.143(1) | 0.374(2) | 0.307(3) | — | 41(1) | |
Erickson and Fairhall assigned the observation of the negative isomer shift to the strong π back-bonding between the carbonyl ligands and the iron d-orbitals, leading to substantial delocalization of the d-electrons which results in a reduced shielding of the s-electrons and therefore a higher s-electron density at the nucleus. This is underlined by the observed long C–N distances within the [Fe(CN)4]6− anion and the low valence vibration frequencies ν(CN). The bonding situation in (Sr3N)2[Fe(CN)4] is thus very similar to Fe(CO)42− anion in Na2[Fe(CO)4].38 The less negative isomer shift for [Fe(CN)4]6− in comparison to the [Fe(CO)4]2− anion can be ascribed to the ligands. The carbonyl ligand is a stronger π-acceptor than cyanide which leads to a weaker π back-bonding and a weaker delocalization of the d-electrons in [Fe(CN)4]6−.
The slightly distorted coordination tetrahedron of [Fe(CN)4]6− leads to weak quadrupole splitting (Table 2). The small increase of the isomer shifts at 78 K of around 0.1 mm s−1 is caused by the second order Doppler shift which describes a temperature dependent contribution to the total isomer shift and which depends on the mean square of velocity of the nucleus.35,36 The experimental line widths Γ (Table 2) are in the usual range for 57Fe Mössbauer signals.
Conclusions
Tetracyanidoferrate(–II) extends the growing class of highly reduced cyanidometalates to tetrahedral complexes. Judging from bond lengths, vibrational frequencies, as well as Mössbauer spectroscopic measurements, chemical bonding in the isolated [Fe(CN)4]6− anion corroborates well to the isoelectronic tetrahedral [Fe(CO)4]2−. Thus, a chemical bonding scheme consisting of strong π back-bonding between non-innocent ligands and a d10 metal center can be assumed for [Fe(CN)4]6− too, as previously confirmed for the famous carbonyl metalate prototype.9,39,40 The tetrahedral anion geometry of [Fe(CN)4]6− corroborates well with that picture: considering cyanide as a pseudohalide, the title complex resembles other tetrahedral d10 halide complexes such as [ZnCl4]2− and [AlCl4]− anions.41,42 Although the qualitative bonding scheme seems similar to previously known reduced cyanidometalates, it differs on a quantitative level of π back-bonding strength as observed by differences of interatomic distances and vibrational frequencies, e.g. in comparison to the [Fe(CN)3]7− anion.16Crystal structures of highly reduced cyanidometalates resemble those known from ternary nitridometalates: the structure of (Sr3N)2[Fe(CN)4] shows similarities to (Ca3N)2[FeN3],19 while LiSr3[Fe(CN)3] and Ba3[Co(CN)3] are closely related to Ba3[FeN3].43
Thus, the new tetracyanidoferrate(–II) provides a missing link for a more in-depth understanding of the trends regarding crystal chemistry and chemical bonding of electron-rich cyanidometalates. Bearing these concepts in mind will enable the future extension of highly charged cyanidometalates beyond group 8 and 9 – following the quest for even higher charged metalates. In view of the numerous applications of carbonyl metalates, similar usages can be anticipated for reduced cyanidometalates.
Author contributions
F.J., P.H., and M.R. planned and carried out the material synthesis. F.J., P.H., Yu.P., and M.R. planned and carried out the single-crystal X-ray diffraction measurements and analyzed the data. Yu.P. collected synchrotron powder diffraction data, while F.J. analyzed these. F.J. recorded and analyzed IR and Raman spectra. Th.B. and R.P. recorded and analyzed Mössbauer spectra. M.S. performed thermal analyses. M.B. performed physical property measurements. All authors contributed in writing the manuscript. M.R., P.H., and R.P. supervised the project.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge Dr H. Borrmann and S. Hückmann for X-ray powder diffraction data collection, K. Zechel and G. Yildirim for help with sample preparation, S. Scharsach for thermal analyses, as well as U. Schmidt and A. Völzke for chemical analyses. We acknowledge ALBA Barcelona, Spain, for using the high resolution powder diffraction facilities at BL04-MSPD beamline. We thank the German Research Foundation (DFG) for financial support (project-id 422042965). Open Access funding provided by the Max Planck Society.Notes and references
- J. E. Ellis, Inorg. Chem., 2006, 45, 3167–3186 CrossRef CAS PubMed ; and references therein.
- W. Hieber and F. Leutert, Ber. Dtsch. Chem. Ges., 1931, 64, 2832–2839 CrossRef.
- F. Feigl and P. Krumholz, Z. Anorg. Allg. Chem., 1933, 215, 242–248 CrossRef CAS.
- W. Hieber, J. Sedlmeier and R. Werner, Chem. Ber., 1957, 90, 278–286 CrossRef CAS.
- J.-J. Brunet, Chem. Rev., 1990, 90, 1041–1059 CrossRef CAS.
- W. Beck, Angew. Chem., 1991, 103, 173–174 ( Angew. Chem., Int. Ed. Engl. , 1991 , 30 , 168–169 ) CrossRef CAS.
- J. E. Ellis, Organometallics, 2003, 22, 3322–3338 CrossRef CAS.
- J. E. Ellis, P. T. Barger, M. L. Winzenburg and G. F. Warnock, J. Organomet. Chem., 1990, 383, 521–530 CrossRef CAS.
- Z. Chen, Y. Deng, J. Bian, L. Li and G. Xu, J. Mol. Struct.: THEOCHEM, 1998, 434, 155–161 CrossRef CAS.
- J. T. Lin, G. P. Hagen and J. E. Ellis, J. Am. Chem. Soc., 1983, 105, 2296–2303 CrossRef CAS.
- C. Janiak, T. M. Klapötke and H.-J. Meyer, Moderne Anorganische Chemie, ed. E. Riedel, de Gruyter, Berlin, 2nd edn, 2003 Search PubMed.
- A. G. Sharpe, The Chemistry of Cyano Complexes of Transition Metals, Academic Press, London, 1976 Search PubMed.
- K. R. Dunbar and R. A. Heintz, Prog. Inorg. Chem., 1997, 45, 283–391 CAS.
- D. H. Rank, D. P. Eastman, B. S. Rao and T. A. Wiggins, J. Opt. Soc. Am., 1961, 51, 929–936 CrossRef CAS.
- P. Höhn, F. Jach, B. Karabiyik, Yu. Prots, S. Agrestini, F. R. Wagner, M. Ruck, L. H. Tjeng and R. Kniep, Angew. Chem., 2011, 123, 9533–9536 ( Angew. Chem., Int. Ed. , 2011 , 50 , 9361–9364 ) CrossRef.
- F. Jach, F. R. Wagner, Z. H. Amber, M. Rüsing, J. Hunger, Yu. Prots, M. Kaiser, M. Bobnar, A. Jesche, L. M. Eng, M. Ruck and P. Höhn, Angew. Chem., 2021, 133, 16015–16021 ( Angew. Chem., Int. Ed. , 2021 , 60 , 15879–15885 ) CrossRef.
- U. Berger and W. Schnick, J. Alloys Compd., 1994, 206, 179–184 CrossRef CAS.
- F. Jach, P. Höhn, Yu. Prots and M. Ruck, Eur. J. Inorg. Chem., 2019, 1207–1211 CrossRef CAS.
- G. Cordier, R. Kniep, P. Höhn and A. Rabenau, Z. Anorg. Allg. Chem., 1990, 591, 58–66 CrossRef CAS.
- J. Weidlein, U. Müller and K. Dehnicke, Schwingungsspektroskopie, Thieme, Stuttgart, 1988 Search PubMed.
- D. L. Rousseau, R. P. Bauman and S. P. S. Porto, J. Raman Spectrosc., 1981, 10, 253–290 CrossRef CAS.
- H. Stammreich, K. Kawai, Y. Tavares, P. Krumholz, J. Behmoiras and S. Bril, J. Chem. Phys., 1960, 32, 1482–1487 CrossRef CAS.
- H. Bie, J. Yu, D. Chu, J. Xu, J. Lu, Y. Sun, X. Cui, Y. Li and X. Zhang, Pol. J. Chem., 2004, 78, 1969–1978 CAS.
- W. W. Brennessel and J. E. Ellis, Angew. Chem., 2007, 119, 604–606 ( Angew. Chem., Int. Ed. , 2007 , 46 , 598–600 ) CrossRef.
- S. Peschel, W. Paulus and D. Babel, Z. Naturforsch., B: J. Chem. Sci., 1996, 51, 826–831 CrossRef CAS.
- V. Jonas and W. Thiel, Organometallics, 1998, 17, 353–360 CrossRef CAS.
- F. Jach, Dissertation, TU Dresden, 2018.
- L. M. Córdoba, G. A. Echeverría, O. E. Piro and M. I. Gómez, J. Therm. Anal. Calorim., 2015, 120, 1827–1834 CrossRef.
- A. Holleman and E. Wiberg, Lehrbuch der Anorganischen Chemie, de Gruyter, Berlin, 1995 Search PubMed.
- W. F. Edgell, J. Huff, J. Thomas, H. Lehman, C. Angell and G. Asato, J. Am. Chem. Soc., 1960, 82, 1254–1255 CrossRef CAS.
- R. G. Teller, R. G. Finke, J. P. Collman, H. B. Chin and R. Bau, J. Am. Chem. Soc., 1977, 99, 1104–1111 CrossRef CAS.
- J. E. Ellis, Adv. Organomet. Chem., 1990, 31, 1–51 CrossRef CAS.
- L. Brammer, J. C. Mareque Rivas and D. Zhao, Inorg. Chem., 1998, 37, 5512–5518 CrossRef CAS PubMed.
- D. Barb, Grundlagen und Anwendungen der Mössbauerspektroskopie, Editura Academiei Republicii Socialiste România and Akademie-Verlag, Bukarest and Berlin, 1980 Search PubMed.
- P. Gütlich, E. Bill and A. X. Trautwein, Mössbauer Spectroscopy and Transition Metal Chemistry: Fundamentals and Applications, Springer-Verlag Berlin Heidelberg, Berlin and Heidelberg, 2011 Search PubMed.
- G. K. Shenoy and F. E. Wagner, Mössbauer Isomer Shifts, North Holland Publishing Company, Amsterdam, 1978 Search PubMed.
- E. Fluck, W. Kerler and W. Neuwirth, Angew. Chem., 1963, 75, 461–472 ( Angew. Chem., Int. Ed. , 1963 , 2 , 277–287 ) CrossRef.
- N. E. Erickson and A. W. Fairhall, Inorg. Chem., 1965, 4, 1320–1322 CrossRef CAS.
- D. Tiana, E. Francisco, M. A. Blanco, P. Macchi, A. Sironi and A. Martín Pendás, J. Chem. Theory Comput., 2010, 6, 1064–1074 CrossRef CAS.
- H. G. Cutforth and P. W. Selwood, J. Am. Chem. Soc., 1943, 65, 2414–2415 CrossRef CAS.
- H. Matsunaga, J. Phys. Soc. Jpn., 1982, 51, 864–872 CrossRef CAS.
- G. Mairesse, P. Barbier and J.-P. Wignacourt, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1979, 35, 1573–1580 CrossRef.
- P. Höhn, R. Kniep and A. Rabenau, Z. Kristallogr., 1991, 196, 153–158 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. CSD 2127618 for (Sr3N)2[Fe(CN)4]. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2dt00833e |
| ‡ Current address: Fraunhofer Institute for Integrated Systems and Device Technology IISB, Schottkystraße 10, 91058 Erlangen, Germany. |
| § Current address: J. Stefan Institute, Jamova 39, 1000 Ljubljana, Slovenia. |
| This journal is © The Royal Society of Chemistry 2022 |