
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Recent advances in photorelease complexes for therapeutic applications
Andrew C.
Benniston
 *a and
Lingli
Zeng
b
*a and
Lingli
Zeng
b
aChemistry-School of Natural & Environmental Sciences, Newcastle University, Newcastle upon Tyne, NE7 1RU, UK. E-mail: andrew.benniston@ncl.ac.uk
bGuangdong Provincial Key Laboratory of Malignant Tumor Epigenetics and Gene Regulation, Guangdong-Hong Kong Joint Laboratory for RNA Medicine, Medical Research Center, Sun Yat-Sen Memorial Hospital, Sun Yat-Sen University, Guangzhou 510120, China
First published on 16th February 2022
Abstract
Photorelease complexes represent a class of agents for which UV-visible light triggers the expulsion of a specfic molecule that is intrinsically part of the inner coordination sphere or held in close proximity to the metal centre. The reaction does not occur in the ground-state complex and requires a photon, but an additional agent may be present that facilitates the release process. In this context, the perspective article covers recent papers from the past five years (2017–2021) on metal-based complexes containing ligands that are expelled under light activation. In addition, the examples primarily focus on ligands with potential biological activity and have specfic therapeutic applications. Some examples include NO, CO, Cl−, peptides, pharmacophores and redox-active compounds.
 Andrew C. Benniston | Andy Benniston has spent a vast majority of his academic career at Newcastle University, after a spell at Glasgow University where he started as a lecturer. He is a professor in chemistry and has held several positions including Head of Chemistry, Deputy Head of School of Natural and Environmental Sciences and now the Associate Dean of Strategic Projects. His work has covered many different aspects including coordination chemistry of macrocycles, the photophysical properties of organic and inorganic compounds and artificial photosynthesis. He has published over 160 papers and co-written several review articles. |
 Lingli Zeng | Lingli Zeng is currently a postdoctoral researcher at the Sun Yat-sen University, Sun Yat-Sen Memorial Hospital. She obtained her PhD degree from Newcastle University in May 2020 under the supervision of Prof. Andrew Benniston. Her research interests include synthesis and applications of light-activated molecular systems, multimodel bioimaging, tumor diagnosis, drug delivery with nanosystems and the related disciplines. She has several publications in the fields of organic molecules, coordination compounds and nanomaterials for detection and tumor therapy. |
1. Introduction
Light–matter interaction is critical for sustaining the vast majority of life on the planet Earth, providing the initial ignition to facilitate chemical and physical processes spanning from the ability to detect objects (e.g., rhodopsin photoisomerization)1 to mass flora production (e.g., H2O and CO2 redox reactions).2–4 The lessons learnt from the study of such natural processes provide the blueprint to design new compounds for which light is the decisive trigger to promote a physicochemical response. The general field of photodynamic therapy (PDT)5 is one such example, designed for a variety of treatments such as tumour removal and requiring a specialised photoactive compound (PAC). The common feature is the active agent (e.g., singlet oxygen, 1O2) is produced by a secondary sphere interaction following light activation of the PAC.6,7 Specifically, ground state oxygen (i.e., 3O2) must firstly diffuse to the PAC and undergo a triplet–triplet energy transfer process.8,9 However, also within the broad envelope of PDT is the class of PACs for which light stimulation directly affects their core components to result in the ejection of a molecule.10 The released molecule may have a direct therapeutic effect or play a key role in various physiological processes.11 Since light can be highly focused and directed to a specific site the photorelease process can offer both spatial and temporal control for a therapy. There are of course downsides to the applicability of the method, since light does not penetrate far into dense objects, and light scattering and its absorption by other coloured compounds is difficult to avoid.12Metal-based complexes dominate as examples of PACs especially those utilising polypyridyl ligands (e.g., 2,2′-bipyridyl) coordinated to a ruthenium(II) metal centre.13,14 One reason for their popularity stems from the rich and well understood basic photophysics for the complexes.15 They are highly coloured because of light absorption associated with a metal-to-ligand charge-transfer (MLCT) band. The presence of the MLCT absorption band more importantly gives a suitable handle to photo-excite the complex and promote secondary processes. In this context, therefore, the concept of photorelease complexes is well established and they have been extensively studied for several years.16 It is more recent that their application for therapeutics has grown in stature and forms the basis of this article. Examples are sampled from the past five years and several previously published reviews are highlighted for further detailed reading.17
2. Basic photophysics and photorelease mechanism
In order to appreciate the photorelease process, the basic photophysics of two archetypical complexes is highlighted; namely, [Ru(bipy)3]2+ (Rubipy) and [Ru(terpy)2]2+ (Ruterpy), where bipy = 2,2′-bipyridyl and terpy = 2,2′;6′,2′′-terpyridine (Fig. 1). Even though both complexes are six coordinate and use similar donor groups, the precise geometry at the metal centre plays a major role in determining their photophysical properties. The poor bite angle for the terpy ligands in Ruterpy essentially means that the ligand-field splitting is weaker when compared to the Rubipy complex.18 The importance of this effect becomes very clear when discussing the relative energies of electronic states within the two complexes (vide infra).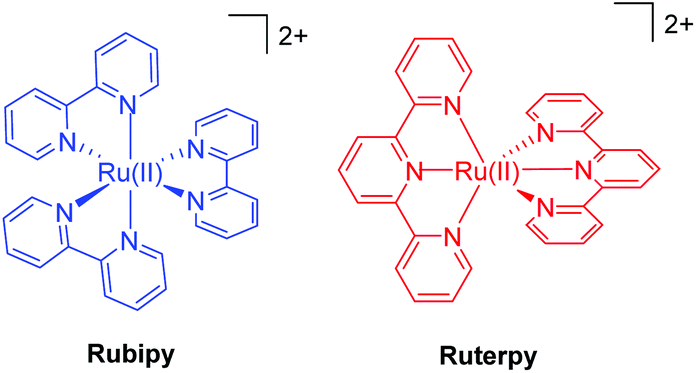 | ||
| Fig. 1 Illustrations of the two archetypical complexes based on ruthenium(II) and the polypyridyl ligands 2,2′-bipyridyl (left) and 2,2′;6′,2′′-terpyridine (right). | ||
As mentioned previously the prominent ground-state absorption profile in the visible region for both complexes is associated with a 1MLCT state, formed by electron migration from the ruthenium metal ion to an empty π* orbital on the polypyridyl ligand. The state can be written as Ru(III)-L˙−, where L = bipy or terpy. It is worth noting that this representation helps explain why the excited complex can act as an oxidant (i.e., Ru(III) + e− → Ru(II) or a reductant (L˙− → L + e−).19,20 Extremely rapid intersystem crossing (<30 fs) results in efficient population of the triplet state (3MLCT) from which processes for deactivation of the excited state originate (e.g., luminescence).21,22
The secondary feature of the complexes is their d–d states, and the presence of the eg* anti-bonding orbitals. The relative positioning of the 3MLCT and the d–d states is especially critical in comprehending the lifetime of the 3MLCT and the timescale available for a secondary process (Fig. 2). Room temperature emission from the 3MLCT state for Rubipy![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 23 in fluid solution is readily observed but the same is not true for Ruterpy. Likewise the excited state lifetime for Rubipy24 is in the microsecond region but only a few nanoseconds for Ruterpy.25 The disparity in properties is explained by inspection of the Ruterpy potential energy surfaces and the closeness in energy of the 3MLCT and d–d states (Fig. 2). Efficient electron crossover into the d–d state results in rapid deactivation back to the ground state, facilitated by the favourable conical intersection (yellow line). The case for Rubipy is somewhat different since the 3MLCT and d–d states are not so strongly coupled, requiring thermal population for deactivation. A point to note is the anti-bonding character of the d–d state and the fact that an electron will reside in it after the cross over process (vide supra). Hence, elongation of the Ru–N bond is expected,26–28 leading to a weakening of the coordination bond. Despite what may appear to be a clear pathway for ligand loss both Rubipy and Ruterpy are stable in solution to light stimulation. There is a clear chelate effect, explained that even if one pyridine became detached the other anchoring ring would still hold the ligand in place.29,30 A bond elongation process, by population of the anti-bonding metal-centred d-orbital, would appear to be more significant in a photorelease mechanism for mono-dentate ligands. The full ejection of a chelate after excitation may require a secondary agent, utilising the excited state redox properties of a complex. The excited state lifetime is more critical in this scenario since a bi-molecular reaction is now required.31 The alternative to release a chelate is to introduce a steric effect, modify its electronic property or change the type of donor atom.32
23 in fluid solution is readily observed but the same is not true for Ruterpy. Likewise the excited state lifetime for Rubipy24 is in the microsecond region but only a few nanoseconds for Ruterpy.25 The disparity in properties is explained by inspection of the Ruterpy potential energy surfaces and the closeness in energy of the 3MLCT and d–d states (Fig. 2). Efficient electron crossover into the d–d state results in rapid deactivation back to the ground state, facilitated by the favourable conical intersection (yellow line). The case for Rubipy is somewhat different since the 3MLCT and d–d states are not so strongly coupled, requiring thermal population for deactivation. A point to note is the anti-bonding character of the d–d state and the fact that an electron will reside in it after the cross over process (vide supra). Hence, elongation of the Ru–N bond is expected,26–28 leading to a weakening of the coordination bond. Despite what may appear to be a clear pathway for ligand loss both Rubipy and Ruterpy are stable in solution to light stimulation. There is a clear chelate effect, explained that even if one pyridine became detached the other anchoring ring would still hold the ligand in place.29,30 A bond elongation process, by population of the anti-bonding metal-centred d-orbital, would appear to be more significant in a photorelease mechanism for mono-dentate ligands. The full ejection of a chelate after excitation may require a secondary agent, utilising the excited state redox properties of a complex. The excited state lifetime is more critical in this scenario since a bi-molecular reaction is now required.31 The alternative to release a chelate is to introduce a steric effect, modify its electronic property or change the type of donor atom.32
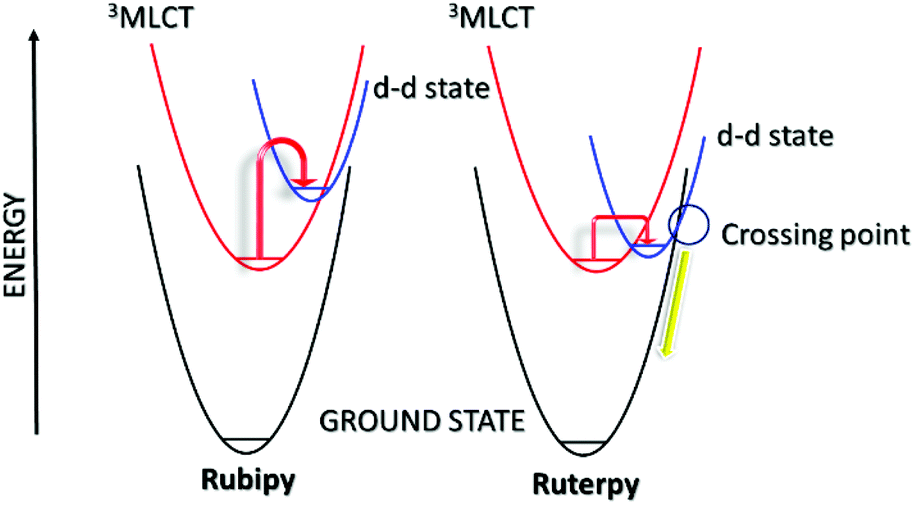 | ||
| Fig. 2 Simplified potential energy surface diagrams for Rubipy (left) and Ruterpy (right). Note: the first-excited singlet 1MLCT surface is omitted for clarity and the difference in energy between the potential energy surfaces are only qualitative. | ||
Hopefully it is evident that exchange of a single terpy ligand in Ruterpy or one/two bipy ligand(s) in Rubipy for other donors, opens up the possibility for developing photorelease complexes. It is also reasonable to consider mixed bipy-terpy complexes or those where a pyridine is swapped for a different aromatic subunit. A further point to note is the capacity of the agent to be released to be an electron acceptor. In relation to Fig. 2 this introduces a further MLCT potential energy surface to consider with possible dissociative character and multiple surface crossing points. The following sections represent several different classes of photorelease agents based on the ruthenium(II) polypyridyl core. Other examples represent disparate types of complexes but still requiring light as the trigger.
3. Poly- and pyridine-based ligand dissociation
For most biological applications the photoreaction should ideally take place in aqueous solution. However, complexes invariably behave differently when dissolved in disparate solvents, and they will have influence on the ligand dissociation rate. To demonstrate this concept, Khnayzer and co-workers studied [Ru(bipy)2(dmphen)]2+ (dmphen = 2,9-dimethyl-1,10-phenanthroline) (RU1a). In acetonitrile the sterically constrained dmphen ligand is released much faster (half-life ∼5 min) compared to water (half-life ∼25 min). This observation was attributed to the better solubility of dmphen in acetonitrile.33 Interestingly the complex was investigated against the ML-2 acute myeloid leukemia (AML) cancer cell line upon irradiation in water. The dmphen appears to be the cytotoxic agent rather than the [Ru(bipy)2(OH2)]2+ ion produced during the dissociation process. Other work by Khnayzer and co-workers focused on the more constrained complex RU1b. The photophysics and photochemistry results showed that the complex can dissociate both bipyridine and the bathocuproine ligands.34 However, not all the cytotoxic effects are caused by the ligand that is released from the light activation. For instance, to mimic the antitumor mechanism of cisplatin, Bonnet et al. demonstrated for the three ruthenium-based complexes [Ru(bipy)2(dmbpy)]2+ (RU2a), [Ru(bipy)2(mtmp)]2+ (RU2b), and [Ru(Ph2phen)2(mtmp)]2+ (RU2c), it is the ruthenium bis-aqua photoproduct of [Ru(Ph2phen)2(mtmp)]2+ instead of the ligand that kills the cancer cells.35 Antitumor activity is always limited by the amount of a drug that accumulates in a cell. To improve cell uptake and nuclear accumulation levels, Zhou and co-workers obtained four pyridine-2-sulfonate (py-SO3−) ligand-based Ru(II) complexes (RU3a–RU3d). Under visible light the py-SO3− ligand dissociated and the remaining ruthenium bipyridine or phenanthroline precursor bound to DNA in what is termed photoactivated chemotherapy (PACT).36 The mechanism of PACT is very classical in that the vacant coordination sites on ruthenium complex combine with base pairs of DNA and restrict its replication.Periphery ligands with functional groups may also influence the properties of photorelease complexes. For example, Zhou and co-workers prepared pyridine-based ruthenium(II) complexes containing an additional fluorinated dppz (dipyridophenazine) ligand (RU4). The complexes undergo pyridine loss upon 470 nm irradiation coupled to DNA covalent binding. Compared to the parent complex, RU4a, the other complexes (RU4b–RU4d) displayed enhanced phototoxicity against tumor cell lines HeLa and SKOV-3 but diminished dark cytotoxicity, which is ideal for PACT. Meanwhile, in the normal cell line L-O2 the complexes exhibit less toxicity.37 Follow up work tested the complexes for antibacterial activity against methicillin-resistant Staphylococcus aureus (MRSA), vancomycin-resistant Enterococcus (VRE), and Escherichia coli (E. coli). Compared to the parent un-fluorinated complex, the strong electronegativity of fluorine atom stabilizes the chemical structure, increases thermal stability and gives extra hydrogen bond interactions, expanding the application scope from antitumor to antibacterial.38 A similar periphery ligand effect is demonstrated by the pyrene-based complex RU5, where the one pyridine of the terpy group is non-coordinating. Generally, a terpy ligand when fully coordinated is non photolabile. However, the uncoordinated pyrene-based terpy weakens the ligand-field and it is released under irradiation. The pyrene modification enables two-photon excited synergistic PDT and PACT activity, as well as “turn-on” fluorescence after ligand dissociation.39
The final example from Fig. 3 represents a complex (RU6) designed to photorelease a pharmacophore for application as a metalloenzyme inhibitor.40 The key component is the gallic acid portion of RU6 which is a recognized inhibitor of the N-terminal domain (PAN) of the PA polymerase for the influenza virus. Basic experiments demonstrated that the pyridine–gallic acid conjugate was released from RU6 following irradiation (λirr = 450 nm) in water. An enzymatic activity evaluation confirmed that the photoreleased conjugate was able to inhibit the PAN endonuclease activity.
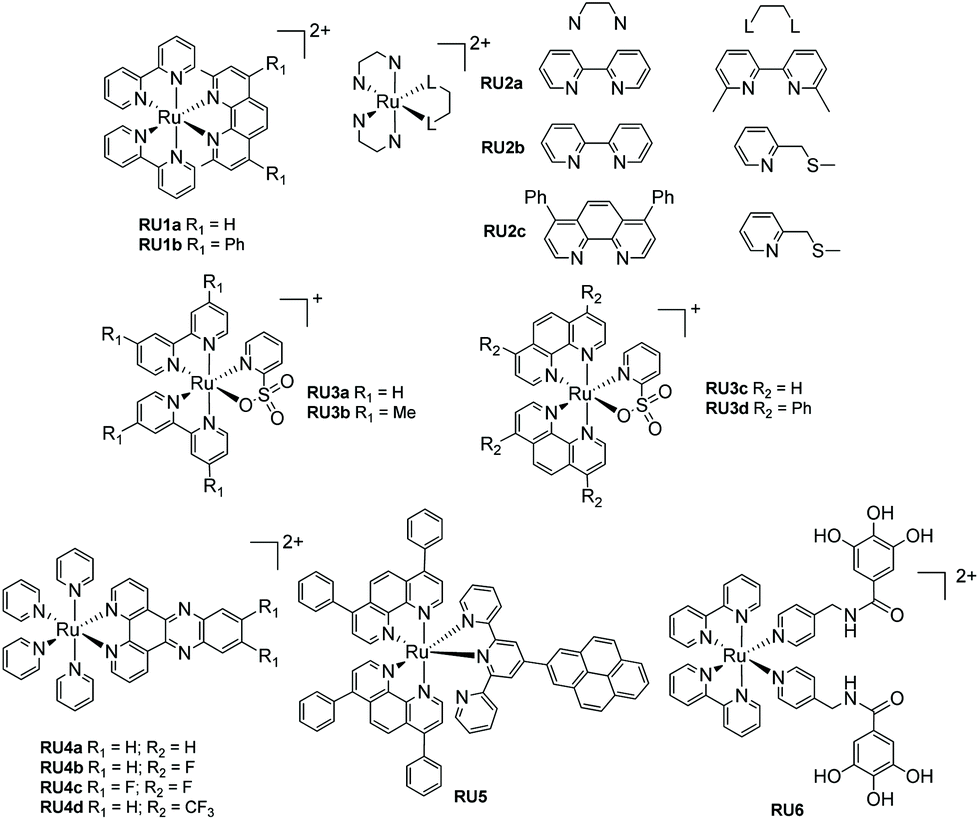 | ||
| Fig. 3 Examples of ruthenium(II) polypyridyl complexes that undergo ligand loss upon irradiation in water. | ||
Generally, the photorelease of tridentate chelates is more difficult than bidentate chelates. The example RU7 (Fig. 4) represents an interesting complex comprising a thioether-based terpy ligand (btap). White light irradiation of a solution of RU7 in MeCN resulted in alterations in the absorption spectrum consistent with formation of the solvate complex and ejection of btap.10
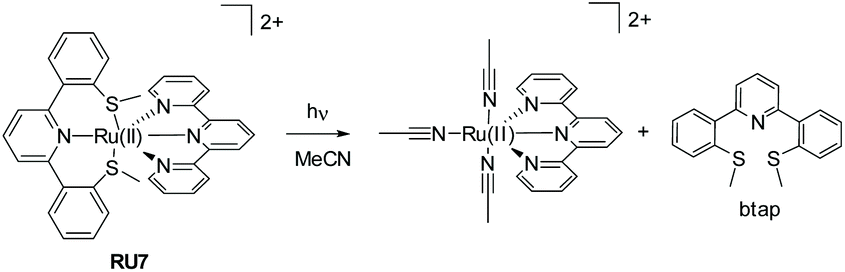 | ||
| Fig. 4 Example of a mixed-ligand ruthenium(II) complex and the breakdown products formed under while light irradiation. | ||
4. Nitric oxide (NO) photorelease complexes
The discovery that nitric oxide (NO˙) plays an important role in mammalian biological regulation and immunology, has aroused great interest in the chemistry and biochemistry of nitric oxide and its derivatives.41 Also of interest are strategies for delivering NO˙ on demand to biological targets.42 In this context ruthenium(II) nitrosyl complexes [(L)Ru(II)(NO+)], where L = any ligand(s), have been especially targeted since they often have low toxicity, good chemical stability, but are capable of releasing NO˙ upon irradiation (eqn (1)).43| [(L)Ru(II)(NO+)] + solvent + hν → [(L)Ru(III)(solvent)] + NO˙ | (1) |
It is worth noting the change in oxidation state of the ruthenium centre in the process of nitric oxide release (i.e., +2 to +3). Interestingly DFT theoretical calculations (CASPT2) using trans-[Ru(Cl)(NO)(py)4]2+, where py = pyridine, supports that NO˙ photorelease is a sequential two-photon process coupled with partial photoisomerization of the nitric oxide ligand.44 This two-photon mechanism should certainly be considered for the ruthenium(II) polypyridyl nitrosyl examples illustrated in Fig. 5. The photorelease properties of complexes RU8a and RU8b were studied in DMSO by Malfant and co-workers,45 both showing extreme sensitivity to traces of water and an equilibrium between the nitrosyl and nitro adduct. A similar set of complexes (RU8c–RU8e) had been previously studied by the same group highlighting alterations in the NO˙ releasing efficiency by swapping from fluorenyl to carbazole subunits.46 The presence of the tertiary amine in the carbazole (RU8d, RU8e) introduced additional charge-transfer transitions toward the Ru-NO fragment. The observed enhancement of the two-photon absorption property for the complexes did not however manifest in an increase in the quantum yield for the NO˙ release. This result is intriguing in the context of the theoretical calculations concept, demonstrating that the nitric oxide release mechanism may be very sensitive to electronic effects. The examples for the two series RU9a–RU9d and RU10a–RU10d were developed to investigate two effects on the photorelease of nitric oxide.47 The first is the influence of the nature of the substituent on the 4′-positon of the terpy ligand, and the second is the change in the chloride position at the ruthenium metal centre (i.e., cis vs. trans). The quantum yield (ϕNO) for the photorelease of NO˙ was measured in MeCN for all the compounds. Across the series the ϕNO for the cis isomer was greater than the trans isomer. A simple explanation for the difference is the electron donating chloride trans to the NO˙ facilitates its release. No correlation between ϕNO and the electron donating or withdrawing effect of the 4′-substituent was observed. One point to note is the same solvate photoproduct was produced for both the cis and trans isomers.
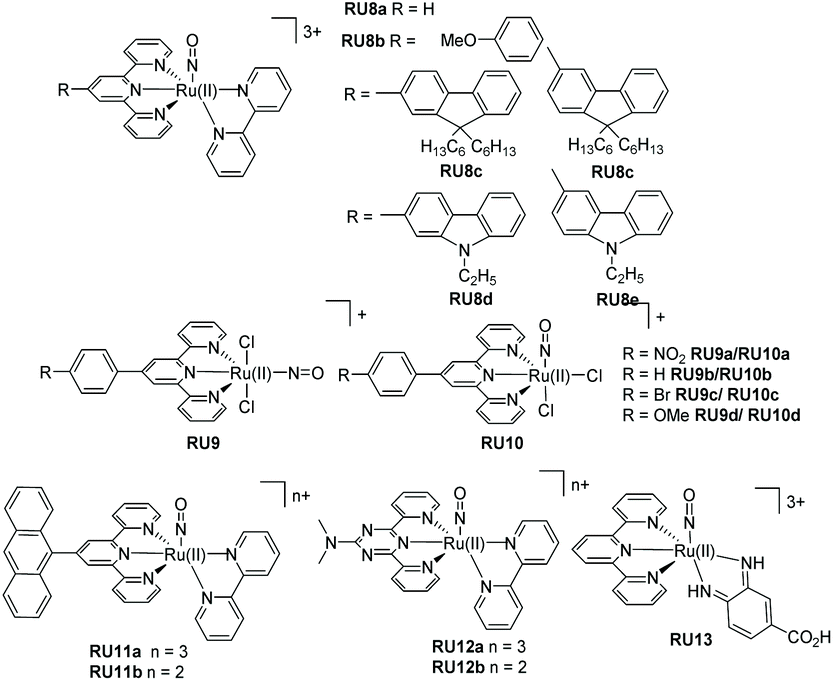 | ||
| Fig. 5 Examples of NO photoreleasing agents based on ruthenium(II) terpyridine nitrosyl complexes. | ||
The final examples shown in Fig. 5 are from the group of Maji and co-workers.16 In each case the tri-positive and di-positive complexes were prepared, the latter by chemical reduction of the bound nitric oxide ligand (i.e., NO+ to NO˙) The anthracene-based complexes RU11a–RU11b were specifically targeted for the treatment of prostate cancer. The rate of nitric oxide photorelease in MeCN for RU11b is ca. 4 fold greater than RU11a, under irradiation with a xenon light source (200 W). Presumably the change is related to a weakening of the Ru–N bond in the RU11a complex. The photo-cyctotoxicity of both complexes was determined against a VCaP human prostate cancer cell line. They demonstrated a reduction in VCaP cells after 6 hours of irradiation with white light (400–700 nm). The complexes RU12a and RU12b contain the nitrogen-rich ligand N,N-dimethyl-4,6-di(pyridine-2-yl)-1,3,5-triazin-2-amine.48 As noted in the previous examples the photorelease of nitric oxide is faster in the complex containing the reduced ligand; the difference in rates being ca. 5. For a very specific application the complex RU13 was developed for treating skin diseases.49 Specifically, the complex's penetration into skin was studied after the application of iontophoresis; the low level application of constant low-density electric current through an electrolyte and the skin. Compared to control experiments NO˙ release into skin was achieved using the complex.
The additional examples shown in Fig. 6 highlight bis-bidentate chelate ruthenium(II) complexes, with the two remaining coordination sites occupied by a NO+ and another mono-dentate ligand. The complex RU14 developed by Lopes et al.50 was synthesised from its nitro precursor for testing if the complex was a vasodilator. Photorelease of NO˙ was confirmed by irradiation of an aqueous solutions of RU14 (pH 2) at 365, 453 and 505 nm. A lower quantum yield of NO˙ release was noted when the long-wavelength light was employed. The two complexes RU15a and RU15b, again studied by the group of Lopes,51 used the phenanthroline chelate and a thiocarbonyl subunit. As well as studying the nitric oxide release process further work studied their DNA cleaving properties in the presence and absence of blue light. Whereas complex RU15a promoted DNA damage both in the dark and the light, the same was not true for RU15b which required blue light activation. The complex RU16 containing 2,2′-biquinoline (biq) is especially interesting since on its own poor NO˙ release was observed following white light irradiation in MeCN. However, significant enhancement occurred if the dichloride analogue [Ru(II)(biq)2Cl2] was used as a co-sensitizer. It was hypothesised that a supramolecular dimer formed in solution, and from this species the NO˙ was released.52 The more elaborate adduct RU17 combines nitric oxide release with potential other free radical formation from the porphyrin group.53
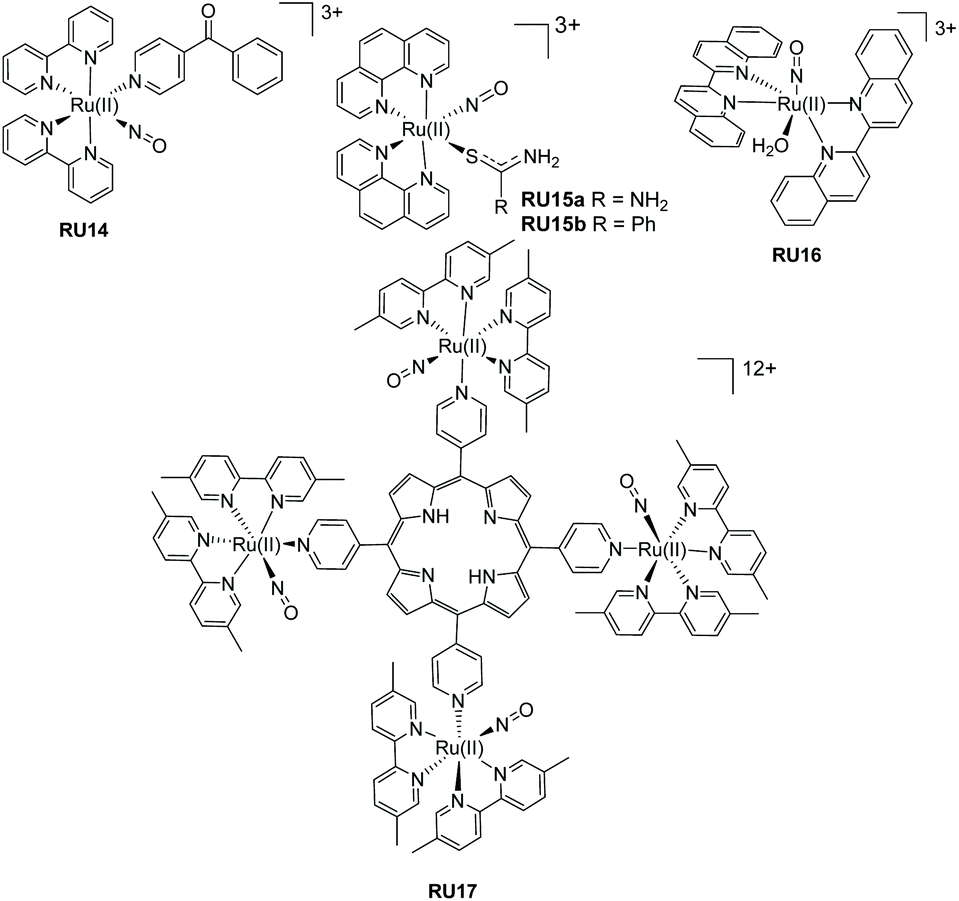 | ||
| Fig. 6 Ruthenium(II) nitrosyl complexes based around bipyridine, biquinoline and phenanthroline ligands. | ||
It is not exclusively necessary to construct nitric oxide photorelease complexes based around the ruthenium(II) polypyridyl core. The example shown in Fig. 7 is a disparate system developed by Kumar et al.; a cyclometalated derivative coupled to an azobenzene moiety.54 The ruthenium(III) precursor when treated with sodium nitrite and acid produced the nitrosyl complex RU17, noting that the aromatic ring was also nitrated in the reaction. The complex when irradiated in DCM with white light resulted in loss of the NO˙, but it is a reversible process as evidenced by spectroscopic measurements. The photoisomerization from trans to cis at the azobenzene unit is probably the driving force behind the NO˙ ejection process. The released nitric oxide could be trapped by reduced myoglobin in a phosphate buffer. Other measurements using fluorescent activated cell sorting analysis confirmed NO˙ release resulted in A549 human breast cancer cell death.
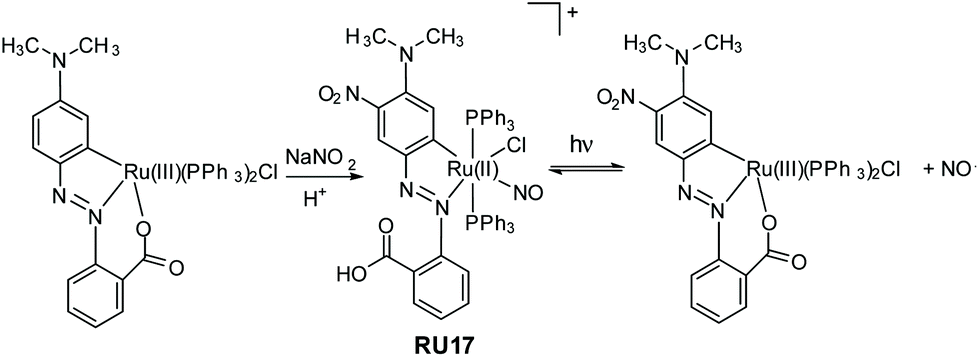 | ||
| Fig. 7 Example of a ruthenium(II) cyclometalated nitrosyl complex prepared by Kumar and co-workers and the reversible loss and binding of nitric oxide under while light irradiation. | ||
It should be noted that the photoinduced release of NO˙ molecules from pure organic components linked to a photosensitizer has also been explored by several groups. For instance, Thomsen et al. reported the room temperature release of NO from a cupferron O-alkylated with an anthracene derivative.55 Nakagawa and his colleagues also conducted in-depth research in this direction using a hindered nitrobenzene derivative.56
5. Photorelease of other agents
The previous examples are classical as that much of the early worked on photodissociation used similar examples. This next section delves into complexes that release other molecules. It is broken down into two parts termed direct and indirect to represent the different characteristics of the photorelease process. In the direct reaction the agent to be released is directly attached to the metal centre as discussed previously, whereas in the indirect examples the agent is held in close proximity, but both still require a photon as the trigger. Other metal ions besides ruthenium are also considered.5.1 Direct
Recognising that CO is isoelectronic with NO+ it is not too surprising that studies have focused on its photorelease.57 Carbon monoxide though poisonous in excess, is still one of the most important information molecules in the animal body and helps to regulate various physiological processes.58 In addition, as a diagnostic reagent, CO plays a role in the treatment of inflammation and cell protection.59 However, the real-time monitoring and study of CO in organisms are easily disturbed by the background. In the past decades, photoactive CO-releasing molecules (PhotoCORMs) have aroused scientists’ interests because the CO molecule could be involved in immune and anti-inflammatory responses, as well as in vaso-relaxation.60A sample of recent PhotoCORMS are depicted in Fig. 8, again relying on the use of polypyridyl, triazole-based and thiazole-based compounds as the ancillary ligands. The complex RU18 containing the triazamacrocycle was studied in depth by Slep and co-workers61 using both steady-state and ultrafast transient absorption spectroscopic techniques. Steady-state photoinitiated CO release conducted in both water and MeCN showed that the solvento species was produced in both cases. This simple picture hides a more complicated process which was addressed by the ultrafast spectroscopic studies. The diagram shown in Fig. 9 represents the proposed species involved from the single occupied molecular orbitals (SOMOs) after excitation and their subsequent roles.
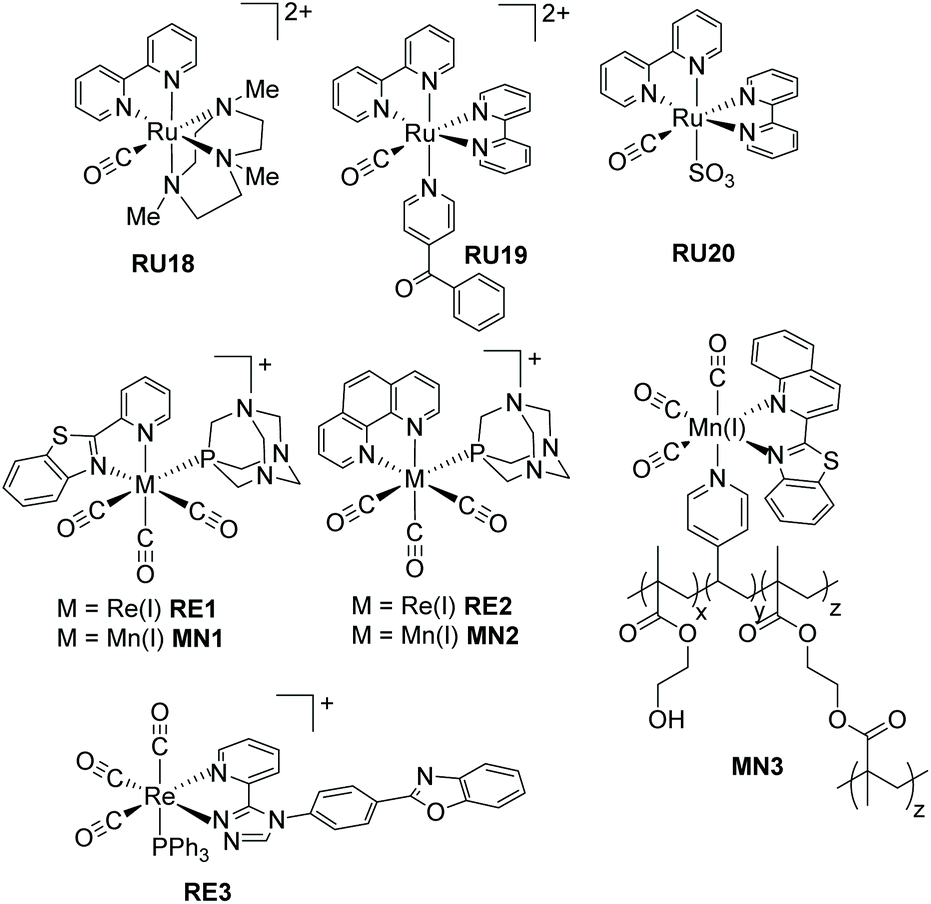 | ||
| Fig. 8 Illustrations of PhotoCORMS based on ruthenium, rhenium and manganese complexes. | ||
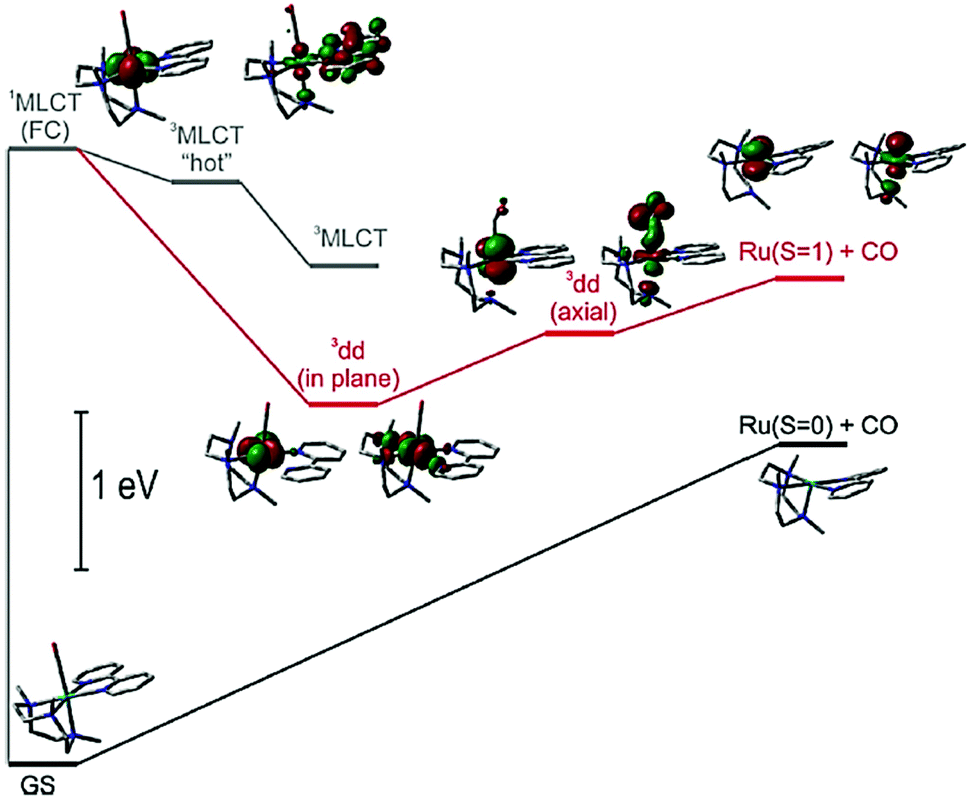 | ||
| Fig. 9 Basic energy diagram representing the SOMOs of the states involved in the photophysics and photochemistry of RU18. Taken from ref. 61. Copyright permission © 2020 American Chemical Society. | ||
The complexes RU19 and RU20 represent examples based on the “Ru(bipy)2CO” core, but where the final ligands in the coordination sphere are very different. The softer S donor is introduced in the second example. The irradiation of RU19 in water with long-wavelength light (λirr = 453 nm) resulted in selective loss of the benzoyl pyridine ligand and formation of the aqua complex [Ru(bipy)2(CO)H2O]2+. A similar experiment performed using light of λirr = 365 nm firstly formed the same complex, which lost the CO ligand to finally generate [Ru(bipy)2(H2O)2]2+. The quantum yield (ϕCO) for CO loss was found to be 0.041 ± 0.004.62 It is worth noting that irradiation of RU20 in water (λirr = 350 nm) resulted in only CO loss and a ϕCO = 0.301 ± 0.003.63 These separate studies by two different groups would seem to suggest that secondary soft donors may enhance CO release in PhotoCORMS based around the “Ru(bipy)2CO” core.
The remainder of the examples shown in Fig. 8 move way from ruthenium and focus on the other d6 ions manganese(I) and rhenium(I). Three CO ligands are present and one of the other ancillary ligands is a polyaromatic. The complexes RE1–RE2 and MN1–MN2 were specifically designed to be water soluble by the addition of the 1,3,5-triaza-7-phosphasadamantane (PTA) to the coordination sphere.64 A further aim was to produce photoCORMS that worked under low-power and broad-band visible light irradiation. Although MN1 displayed no fluorescence in solution the release of CO under broad-band white light low-power (15 mW cm−2) irradiation resulted in strong fluorescence centred at 400 nm. Interestingly the same was no seen for the complex MN2; the rate of CO loss was, in addition, slower under identical conditions. The loss of CO from the rhenium complexes RE1 and RE2 worked only under low power UV-A light (λirr = 360 nm, 5 mW cm−2) and the dissociation rates for CO were comparable for the two complexes. Additional experiments focused on the luminescent complex RE1 and demonstrated its cellular internalization in MDA-MB-231 (human breast cancer) cells. The manganese complexes MN1 and MN2 also exhibited dose-dependent eradication of MDA-MB-231 cells following irradiation and CO dissociation. The group of Mascharak developed the manganese-based photoCORM concept by its incorporation into a polymer (MN3).65 The polymer was used to release CO under low-power broadband visible light (<1 mW cm−2) using a fibre optics technology approach. A fibre optical device was used to deliver CO to a suspension of human colorectal adenocarcinoma (HT-29) cells.
The final complex RE3 incorporates the extended ligand based around pyridinetriazole and phenylbenzoxazole, with a triphenylphosphine to aid in the CO dissociation process.66 Irradiation of a solution of RE3 in MeCN at λirr = 350 nm resulted in absorption alterations consistent with CO loss and formation of the solvento complex. A 1H NMR study coupled to a mass spectrometry experiment confirmed the loss of only one CO during the photolysis. The relatively strong emission observed from RE3 diminished as the single CO was replaced by MeCN, consistent with previous findings on similar complexes. One drawback of RE3 is its poor solubility in water and its self-aggregation.
5.2 Indirect
A series of fine examples of indirect release agents was designed and produced by Meyer and co-workers,67 based on a supramolecular halide photorelease concept (Fig. 10). The early work by the group focused on RU21 and RU22, noting that the halide binding site was kept constant but the electron donating/withdrawing effect at the two bipy ligands was changed. The design element behind this was to alter the direction of the excited state dipole moment. For complex RU21 the MLCT dipole would point toward the halide but for RU22 it would be directed away. The ground state binding constant towards chloride for both complexes in DCM was found to be ca. 4 × 106 M−1. 1H NMR experiments were consistent with the chloride binding to the R2 side chains, rather than it sitting randomly in the second coordination sphere. Light excitation of RU21 was shown to eject the chloride out the supramolecular cage. The binding constant was lowered by a factor of 20 in the excited state, meaning that around 45% of the chloride dissociated. In contrast, light excitation of RU22 resulted in a 45 fold increase in the excited state binding constant. The ejection of the chloride can be rationalized in terms of a coulombic repulsion concept. The electron in the MLCT state for RU21 will sit exclusively on the bipy containing the amide binding sites, and the repulsive electrostatic ion–ion repulsion essentially pushes out the chloride. Follow up work by the same group further studied the halide photorelease concept using RU23 to RU27 with chloride and bromide.67,68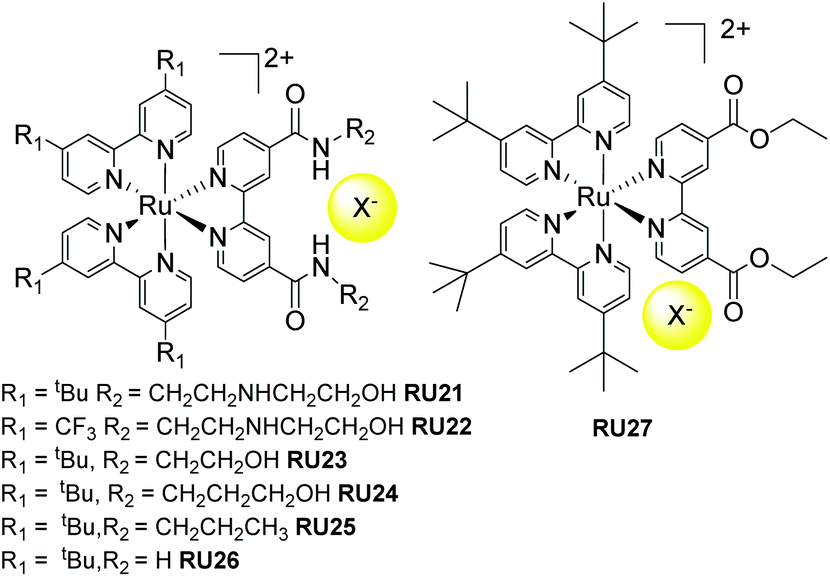 | ||
| Fig. 10 Chloride photorelease complexes based on secondary supramolecular interactions with the amide functionalised bipy ligand. | ||
6. Miscellaneous examples
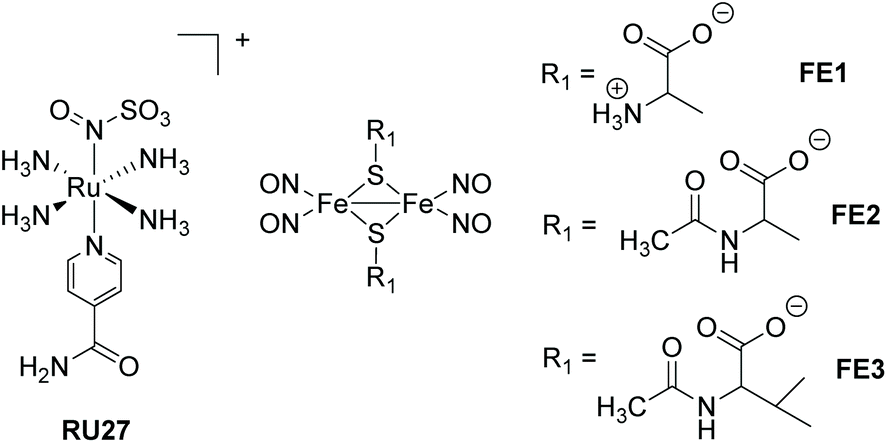
In this section other direct photorelease complexes are noted that lack the presence of extended polyaromatic ligands at the metal centre. The use of the “Fe2S2” core is also discussed which is a disparate feature for NO˙ release agents (Fig. 11). In addition, an illustration of light controlled release of therapeutic proteins from a cobalamin-based conjugate is considered in the context of protein therapeutics (Fig. 12). The ruthenium(II) nitrosulphito complex RU27 is an interesting example produced by the reaction of trans-[Ru(NH3)4(isn)NO], where isn = isonicotinimide, with sulfite.69 Previous to the work by Roveda and co-workers only four other similar complexes were known and their photochemistry was unexplored. The irradiation of RU27 in phosphate buffered water (λirr = 355/410 nm) resulted in loss of the N(O)SO3− anion, which further decomposed to give NO˙ and the sulfur trioxide anion radical (SO3˙−). There was no indication from the photolysis that a Ru3+ complex was formed, and so the final complex was assigned to [Ru(NH3)4(H2O)(isn)]2+. This dual release of two radicals may have further applications, especially considering the rich solution chemistry of the sulfite radical (eqn (2)–(4)).| SO3˙− + O2 → −O3SOO˙ | (2) |
| −O3SOO˙ + SO32− → SO4˙− + SO42− | (3) |
| SO4˙− + SO32− → SO42− + SO3˙− | (4) |
 | ||
| Fig. 11 Examples of other types of photorelease complexes. | ||
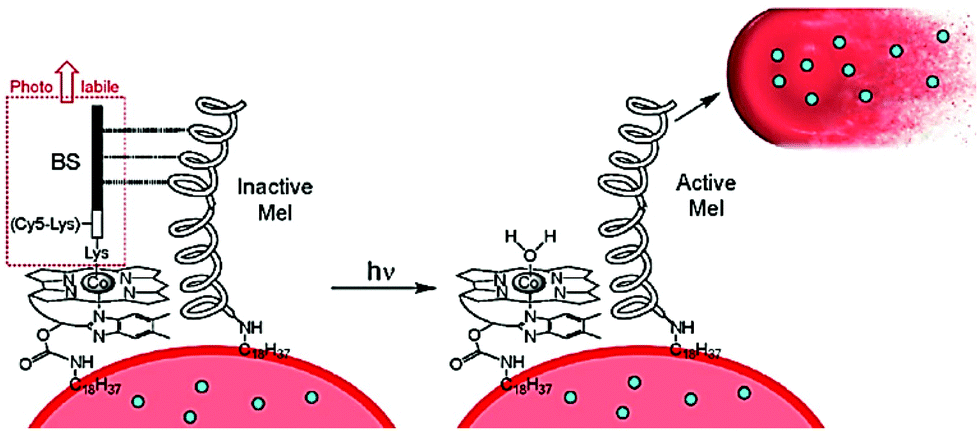 | ||
| Fig. 12 A basic representation for the concept behind a photoactivated protein therapeutic, where ejection of the BS peptide unit activates the proximal protein to facilitate the loss of therapeutic proteins (blue dots) from the red blood cell (RBC). Taken from ref. 78. Copyright © 2020 American Chemical Society. | ||
Roussin's red ester is a dimer nitrosyl-iron complex with the formula [Fe(μ-S)2(NO)4]2−, and has been extensively studied as a NO˙ release agent.70,71 Recent work by Lim et al. focused on the cysteine-derived complexes FE1–FE3 which displayed excellent water solubility, suitable to complete an in-depth study into their photodissociation dynamics using femtosecond time-resolved infrared spectroscopy.72 The thermal stability of the complexes FE2 and FE3 in water is around five times greater than the simple cysteine derivative FE1. Probing the excited state for FE1–FE3 revealed that they released one NO˙ to form [Fe2((μ-RS)2(NO)3], or relaxed to the ground state without photodeligation via an electronically excited intermediate state. The loss of the single NO˙ is extremely fast (<0.3 ps). Although [Fe2((μ-RS)2(NO)3] combined with the solvent a small fraction also underwent geminate recombination with the NO˙. The solvent complex was also shown to bimolecularly recombine with NO˙ (k = 1.3–1.6 × 108 M−1 s−1).
6.1 Amino acid and peptide release
The release of an amino acid or a peptide chain can be involved in regulating tumour cells apoptosis. For example, Rohrabaugh, Jr. et al. produced a new Ru(II) complex [Ru(terpy)(dppn)(Cbz-Leu-NHCH2CN)]2+, where dppn = benzo[i]dipyrido[3,2-a:2′,3′-c]phenazine to release the cathepsin K inhibitor Cbz-Leu-NHCH2CN ligand. And the resulting [Ru(terpy)(dppn)(OH2)]2+ was able to further generate 1O2 for tumour cells apoptosis.73 In the field of neurophysiology, scientists have made great efforts in modulating neural activities. Amino acids are also one of the most common neurotransmitters. Etchenique and co-workers developed the ruthenium-based caged 5-hydroxytryptamine (5HT) complex [Ru(bipy)2(PMe3)(5HT)](PF6)2 for modulating the excitability of mouse prefrontal principal neurons.74 The similar derivative [Ru(bipy)2(PMe3)Arg](PF6)2, where Arg = arginine, could be photoactivated by blue or green light via one-photon excitation. The reduced L-arginine was able to elicit feeding response in the freshwater cnidarian Hydra vulgaris.75Protein therapeutics is a growing area of research representing a class of drugs that are both selective and potent in their application.76,77 However, deficiencies are their short circulatory lifetimes and side reactions at healthy tissue. One strategy is to keep the protein masked up to the point of action, and the use of red blood cells (RBCs) as carriers is a promising approach. The challenge is to control the release of proteins at the desired target. To this end the use of a photorelease process was demonstrated by the group of Lawrence as outlined in the simple picture of Fig. 12.78 The concept is based on the well-established photochemistry of cobalamin (Cbl), in which the Co–C bond of methyl and adenosyl derivatives can be homolytically cleaved under light illumination (330–550 nm range). The working of the functionalized RBC of Fig. 12 relies on the release of the photolabile BS protein following white light illumination. The inactive melittin (Mel) protein is activated following this process, which then triggers the release of therapeutic proteins from the RBS. The successful application of photoresponsive thrombin loaded RBCs was demonstrated in vivo using FVB mice, noting that thrombin is capable of promoting clots in blood.
7. External agent assisted photorelease complexes
In the previous sections the function of the complexes only required input from light and no other agent was needed to facilitate the process. In this section the tenet of using the excited state-state redox chemistry to facilitate ligand loss is explored. The concept is not especially original79 but the application for therapeutics is based on recognising that biological systems often contain oxidants and reductants.80 It is proposed that the excited-state photochemistry could disturb their operating concentrations and result in loss of a ligand with detrimental/beneficial effects. Anthraquinone compounds fit the profile of possessing interesting therapeutic applications,81,82 have a rich redox chemistry and can act as ligands to metal ions.83 One problem arises from the low reduction potential for an anthraquinone which is manifested in a MLCT band of long-wavelength. At first this seems a positive feature for irradiation of a sample with low-energy white light, but has a negative effect of shortening the excited-state lifetime into the picosecond region.84 Referring back to the picture of Fig. 1 the low-energy MLCT state introduced strongly couples to the ground state; non-radiative deactivation is highly efficient because of the energy-gap law.85 The ultrashort excited lifetime would appear to preclude an effective bimolecular reaction with an oxidant/reductant from promoting efficient ligand dissociation. Despite this problem the two examples (RU28–RU29) shown in Fig. 13, with excited state lifetimes of only 5–7 ps, were shown to release the anthraquinone ligand in the presence of hydrogen peroxide.86,87 The decomposition of RU29 would appear to be promoted by an autocatalytic process, which helps explain why it can readily eject the anthraquinone. To date no biological experiments were carried out to ascertain if such complexes would have a therapeutic application. However, it is worth noting the triple functioning capability of the anthraquinone-based complex RU30 as a PACT, photoredox catalyst and a PDT agent.88 The ruthenium(III) centre and anthraquinones radical anion produced by RU30 upon irradiation, and analogues to that created in RU28 and RU29, will oxidize NADH/NADPH to form O2˙− which is cytotoxic. The field of study for these type of complexes for therapeutic applications is clearly still at an early development stage.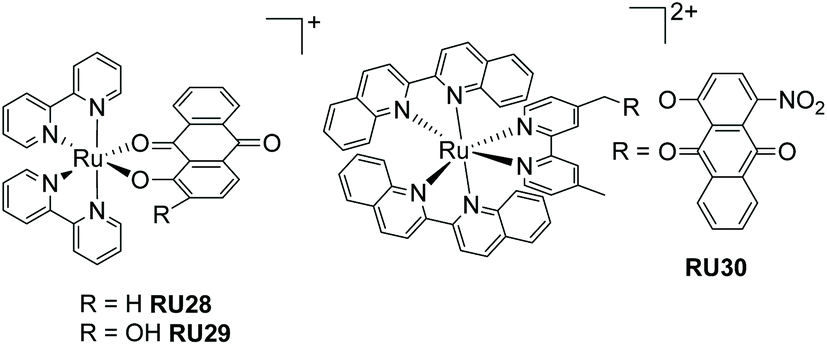 | ||
| Fig. 13 Two anthraquinone-based ruthenium(II) complexes that decompose under light irradiation in the presence of hydrogen peroxide (left), and a ruthenium(II) bis-biquinoline complex appended with an anthraquinone. | ||
8. Conclusions
This review has attempted to capture the recent advances made in photorelease complexes for therapeutic treatments. The application of complexes that eject CO or NO is certainly well established and there are numerous examples. More recent ultrafast time-resolved studies coupled to high-level DFT calculations have helped shed more light on the multifaceted nature of the dissociation process, and have pointed towards ways in which to improve future compounds. In comparison, the indirect release of an agent such as a halide or agent-assisted ligand photodissociation is still to be fully developed. Mitochondria targeting therapy is an area of research growing in popularity,89,90 and may be the ideal target since they contain a plethora of redoxactive species. One could envisage disrupting the balance of reagents following the photoactivation of a drug.Author contributions
This article was a joint project with equal contributions from the authors toward writing and literature surveys.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Newcastle University for financial support.Notes and references
- Y. Hontani, M. Broser, M. Luck, J. Weißenborn, M. Kloz, P. Hegemann and J. T. M. Kennis, J. Am. Chem. Soc., 2020, 142, 11464–11473 CrossRef CAS PubMed.
- S. K. Lee, M. Kondo, M. Okamura, T. Enomoto, G. Nakamura and S. Masaoka, J. Am. Chem. Soc., 2018, 140, 16899–16903 CrossRef CAS PubMed.
- M. A. Hoque, M. Gil-Sepulcre, A. de Aguirre, J. A. A. W. Elemans, D. Moonshiram, R. Matheu, Y. Shi, J. Benet-Buchholz, X. Sala, M. Malfois, E. Solano, J. Lim, A. Garzón-Manjón, C. Scheu, M. Lanza, F. Maseras, C. Gimbert-Suriñach and A. Llobet, Nat. Chem., 2020, 12, 1060–1066 CrossRef CAS PubMed.
- R. R. Rao, M. J. Kolb, L. Giordano, A. F. Pedersen, Y. Katayama, J. Hwang, A. Mehta, H. You, J. R. Lunger, H. Zhou, N. B. Halck, T. Vegge, I. Chorkendorff, I. E. L. Stephens and Y. Shao-Horn, Nat. Catal., 2020, 3, 516–525 CrossRef CAS.
- F. Heinemann, J. Karges and G. Gasser, Acc. Chem. Res., 2017, 50, 2727–2736 CrossRef CAS PubMed.
- P. Kumar, P. Singh, S. Saren, S. Pakira, S. Sivakumar and A. K. Patra, Dalton Trans., 2021, 50, 8196–8217 RSC.
- N. Soliman, L. K. McKenzie, J. Karges, E. Bertrand, M. Tharaud, M. Jakubaszek, V. Guérineau, B. Goud, M. Hollenstein, G. Gasser and C. M. Thomas, Chem. Sci., 2020, 11, 2657–2663 RSC.
- S. Cerfontaine, L. Troian-Gautier, S. A. M. Wehlin, F. Loiseau, E. Cauët and B. Elias, Dalton Trans., 2020, 49, 8096–8106 RSC.
- V. Balzani, P. Ceroni, A. Credi and M. Venturi, Coord. Chem. Rev., 2021, 433, 213758 CrossRef CAS.
- A. Awada, F. Loiseau and D. Jouvenot, Eur. J. Inorg. Chem., 2021, 2021, 4539–4542 CrossRef CAS.
- H. Li, C. Xie, R. Lan, S. Zha, C.-F. Chan, W.-Y. Wong, K.-L. Ho, B. D. Chan, Y. Luo and J.-X. Zhang, J. Med. Chem., 2017, 60, 8923–8932 CrossRef CAS PubMed.
- R. Bensasson, C. Salet and V. Balzani, J. Am. Chem. Soc., 1976, 98, 3722–3724 CrossRef CAS.
- G.-B. Jiang, W.-Y. Zhang, M. He, Y.-Y. Gu, L. Bai, Y.-J. Wang, Q.-Y. Yi and F. Du, J. Inorg. Biochem., 2020, 208, 111104 CrossRef CAS PubMed.
- S. Li, G. Xu, Y. Zhu, J. Zhao and S. Gou, Dalton Trans., 2020, 49, 9454–9463 RSC.
- J. G. Vos and J. M. Kelly, Dalton Trans., 2006, 4869–4883 RSC.
- B. Giri, T. Saini, S. Kumbhakar, K. K. Selvan, A. Muley, A. Misra and S. Maji, Dalton Trans., 2020, 49, 10772–10785 RSC.
- J. Shum, P. K.-K. Leung and K. K.-W. Lo, Inorg. Chem., 2019, 8, 2231–2247 CrossRef PubMed.
- M. T. Rupp, N. Shevchenko, G. S. Hanan and D. G. Kurth, Coord. Chem. Rev., 2021, 446, 214127 CrossRef CAS.
- E. Cuéllar, L. Pastor, G. García-Herbosa, J. Nganga, A. M. Angeles-Boza, A. Diez-Varga, T. Torroba, J. M. Martín-Alvarez, D. Miguel and F. Villafañe, Inorg. Chem., 2020, 60, 692–704 CrossRef PubMed.
- D. Oyama, T. Kanno and T. Takase, Dalton Trans., 2021, 50, 7759–7767 RSC.
- V. Vorobyev, D. S. Budkina and A. N. Tarnovsky, J. Phys. Chem. Lett., 2020, 11, 4639–4643 CrossRef CAS PubMed.
- A. Cannizzo, F. van Mourik, W. Gawelda, G. Zgrablic, C. Bressler and M. Chergui, Angew. Chem., 2006, 118, 3246–3248 CrossRef.
- A. C. Bhasikuttan, M. Suzuki, S. Nakashima and T. Okada, J. Am. Chem. Soc., 2002, 124, 8398–8405 CrossRef CAS PubMed.
- T. P. Cheshire, M. K. Brennaman, P. G. Giokas, D. F. Zigler, A. M. Moran, J. M. Papanikolas, G. J. Meyer, T. J. Meyer and F. A. Houle, J. Phys. Chem. B, 2020, 124, 5971–5985 CrossRef CAS PubMed.
- D. Luo, T. Zuo, J. Zheng, Z.-H. Long, X.-Z. Wang, Y.-L. Huang, X.-P. Zhou and D. Li, Mater. Chem. Front., 2021, 5, 2777–2782 RSC.
- A. Soupart, F. Alary, J.-L. Heully, P. I. P. Elliott and I. M. Dixon, Inorg. Chem., 2020, 59, 14679–14695 CrossRef CAS PubMed.
- A. Soupart, F. Alary, J.-L. Heully, P. I. P. Elliott and I. M. Dixon, Inorg. Chem., 2018, 57, 3192–3196 CrossRef CAS PubMed.
- M. Asahara, H. Kurimoto, M. Nakamizu, S. Hattori and K. Shinozaki, Phys. Chem. Chem. Phys., 2020, 22, 6361–6369 RSC.
- M. S. Meijer and S. Bonnet, Inorg. Chem., 2019, 58, 11689–11698 CrossRef CAS PubMed.
- M. Hirahara, H. Nakano, K. Uchida, R. Yamamoto and Y. Umemura, Inorg. Chem., 2020, 59, 11273–11286 CrossRef CAS PubMed.
- A. Soupart, F. Alary, J.-L. Heully, P. I. P. Elliott and I. M. Dixon, Coord. Chem. Rev., 2020, 408, 213184 CrossRef CAS.
- D. Lazić, A. Scheurer, D. Ćoćić, J. Milovanović, A. Arsenijević, B. Stojanović, N. Arsenijević, M. Milovanović and A. R. Simović, Dalton Trans., 2021, 50, 7686–7701 RSC.
- D. F. Azar, H. Audi, S. Farhat, M. El-Sibai, R. J. Abi-Habib and R. S. Khnayzer, Dalton Trans., 2017, 46, 11529–11532 RSC.
- S. Mehanna, N. Mansour, H. Audi, K. Bodman-Smith, M. A. Mroueh, R. I. Taleb, C. F. Daher and R. S. Khnayzer, RSC Adv., 2019, 9, 17254–17265 RSC.
- J.-A. Cuello-Garibo, M. S. Meijer and S. Bonnet, Chem. Commun., 2017, 53, 6768–6771 RSC.
- N. Tian, Y. Feng, W. Sun, J. Lu, S. Lu, Y. Yao, C. Li, X. Wang and Q. Zhou, Dalton Trans., 2019, 48, 6492–6500 RSC.
- R. Boerhan, W. Sun, N. Tian, Y. Wang, J. Lu, C. Li, X. Cheng, X. Wang and Q. Zhou, Dalton Trans., 2019, 48, 12177–12185 RSC.
- W. Sun, R. Boerhan, N. Tian, Y. Feng, J. Lu, X. Wang and Q. Zhou, RSC Adv., 2020, 10, 25364–25369 RSC.
- Z. Jin, S. Qi, X. Guo, Y. Jian, Y. Hou, C. Li, X. Wang and Q. Zhou, Chem. Commun., 2021, 57, 3259–3262 RSC.
- J. Karges, R. W. Stokes and S. M. Cohen, Dalton Trans., 2021, 50, 2757–2765 RSC.
- A. A. Mikhailov, D. V. Khantakova, V. A. Nichiporenko, E. M. Glebov, V. P. Grivin, V. F. Plyusnin, V. V. Yanshole, D. V. Petrova, G. A. Kostin and I. R. Grin, Metallomics, 2019, 11, 1999–2009 CrossRef CAS PubMed.
- B. Giri, S. Kumbhakar, K. K. Selvan, A. Muley and S. Maji, Inorg. Chim. Acta, 2020, 502, 119360 CrossRef CAS.
- A. Enriquez-Cabrera, I. Sasaki, V. Bukhanko, M. Tassé, S. Mallet-Ladeira, P. G. Lacroix, R. M. Barba-Barba, G. Ramos-Ortiz, N. Farfán, Z. Voitenko and I. Malfant, Eur. J. Inorg. Chem., 2017, 2017, 1446–1456 CrossRef CAS.
- F. Talotta, L. González and M. Boggio-Pasqua, Molecules, 2020, 25, 2613 CrossRef CAS PubMed.
- N. Marchenko, P. G. Lacroix, V. Bukhanko, M. Tasse, C. Duhayon, M. Boggio-Pasqua and I. Malfant, Molecules, 2020, 25, 2205 CrossRef CAS PubMed.
- A. Enriquez-Cabrera, P. G. Lacroix, I. Sasaki, S. Mallet-Ladeira, N. Farfán, R. M. Barba-Barba, G. Ramos-Ortiz and I. Malfant, Eur. J. Inorg. Chem., 2018, 2018, 531–543 CrossRef CAS.
- S. Amabilino, M. Tasse, P. G. Lacroix, S. Mallet-Ladeira, V. Pimienta, J. Akl, I. Sasaki and I. Malfant, New J. Chem., 2017, 41, 7371–7383 RSC.
- B. Giri, S. Kumbhakar, K. K. Selvan, A. Muley and S. Maji, New J. Chem., 2020, 44, 18732–18744 RSC.
- D. C. A. S. De Santana, K. Dias, J. G. Souza, A. T. Ogunjimi, M. C. Souza, R. S. Silva and R. F. V. Lopez, Molecules, 2017, 22, 104 CrossRef PubMed.
- A. P. de Sousa, A. F. Fernandes, I. A. Paz, N. R. F. Nascimento, J. Ellena, E. H. S. Sousa, L. G. F. Lopes and A. K. M. Holanda, J. Braz. Chem. Soc., 2017, 28, 2117–2129 CAS.
- C. D. S. Silva, I. A. Paz, F. D. Abreu, A. P. de Sousa, C. P. Veríssimo, N. R. F. Nascimento, T. F. Paulo, D. Zampieri, M. N. Eberlin, A. C. S. Gondim, L. C. Andrade, I. M. M. Carvalho, E. H. S. Sousa and L. G. F. Lopes, J. Inorg. Biochem., 2018, 182, 83–91 CrossRef CAS PubMed.
- D. B. G. Mateus, A. P. L. Batista, R. L. Rodrigues and S. Nikolaou, J. Braz. Chem. Soc., 2020, 31, 2319–2330 CAS.
- M. I. F. Barbosa, G. G. Parra, R. S. Correa, R. N. Sampaio, L. N. Magno, R. C. Silva, A. C. Doriguetto, J. Ellena, N. M. B. Neto, A. A. Batista and P. J. Gonçalvese, J. Photochem. Photobiol., A, 2017, 338, 152–160 CrossRef CAS.
- R. Kumar, A. Yadav, A. Ratnam, S. Kumar, M. Bala, D. Sur, S. Narang, U. P. Singh, P. K. Mandal and K. Ghosh, Eur. J. Inorg. Chem., 2017, 2017, 5334–5343 CrossRef CAS.
- H. Thomsen, N. Marino, S. Conoci, S. Sortino and M. B. Ericson, Sci. Rep., 2018, 8, 1–8 CAS.
- K. Hishikawa, H. Nakagawa, T. Furuta, K. Fukuhara, H. Tsumoto, T. Suzuki and N. Miyata, J. Am. Chem. Soc., 2009, 131, 7488–7489 CrossRef CAS PubMed.
- Z. Yuan, X. Yang, Y. Ye, R. Tripathi and B. Wang, Anal. Chem., 2021, 93, 5317–5326 CrossRef CAS PubMed.
- A. Ismailova, D. Kuter, D. S. Bohle and I. S. Butler, Bioinorg. Chem. Appl., 2018, 2018, 2018–2041 Search PubMed.
- Y. Zhou, W. Yu, J. Cao and H. Gao, Biomaterials, 2020, 255, 120193 CrossRef CAS PubMed.
- B. Pauwels, C. Boydens, L. Vanden Daele and J. Van de Voorde, J. Pharm. Pharmacol., 2016, 68, 293–304 CrossRef CAS PubMed.
- N. Levin, J. P. Marcolongo, A. Cadranel and L. D. Slep, Inorg. Chem., 2020, 59, 12075–12085 CrossRef CAS PubMed.
- A. P. de Sousa, E. M. Carvalho, J. Ellena, E. H. S. Sousa, J. R. de Sousa, L. G. F. Lopes, P. C. Ford and A. K. M. Holanda, J. Inorg. Biochem., 2017, 173, 144–151 CrossRef CAS PubMed.
- A. P. de Sousa, J. S. do Nascimento, A. P. Ayala, B. P. Bezerra, E. H. S. Sousa, L. G. F. Lopes and A. K. M. Holanda, Polyhedron, 2019, 167, 111–118 CrossRef CAS.
- I. Chakraborty, S. J. Carrington, G. Roseman and P. K. Mascharak, Inorg. Chem., 2017, 56, 1534–1545 CrossRef CAS PubMed.
- M. N. Pinto, I. Chakraborty, C. Sandoval and P. K. Mascharak, J. Controlled Release, 2017, 264, 192–202 CrossRef CAS PubMed.
- A. D. H. Mejias, A. Poirot, M. Rmili, N. Leygue, M. Wolff, N. Saffon-Merceron, E. Benoist and S. Fery-Forgues, Dalton Trans., 2021, 50, 1313–1323 RSC.
- M. D. Turlington, L. Troian-Gautier, R. N. Sampaio, E. E. Beauvilliers and G. J. Meyer, Inorg. Chem., 2018, 57, 5624–5631 CrossRef CAS PubMed.
- M. D. Turlington, L. Troian-Gautier, R. N. Sampaio, E. E. Beauvilliers and G. J. Meyer, Inorg. Chem., 2019, 58, 3316–3328 CrossRef CAS PubMed.
- A. C. Roveda, W. G. Santos, M. L. Souza, C. N. Adelson, F. S. Gonçalves, E. E. Castellano, C. Garino, D. W. Franco and D. R. Cardoso, Dalton Trans., 2019, 48, 10812–10823 RSC.
- H. Yoon, S. Park and M. Lim, J. Phys. Chem. Lett., 2020, 11, 3198–3202 CrossRef CAS PubMed.
- D. C. Pectol, S. Khan, R. B. Chupik, M. Elsabahy, K. L. Wooley, M. Y. Darensbourg and S.-M. Lim, Mol. Pharmaceutics, 2019, 16, 3178–3187 CrossRef CAS PubMed.
- H. Yoon, S. Park and M. Lim, ACS Omega, 2021, 6, 27158–27169 CrossRef CAS PubMed.
- T. N. Rohrabaugh, K. A. Collins, C. Xue, J. K. White, J. J. Kodanko and C. Turro, Dalton Trans., 2018, 47, 11851–11858 RSC.
- R. Cabrera, O. Filevich, B. García-Acosta, J. Athilingam, K. J. Bender, K. E. Poskanzer and R. Etchenique, ACS Chem. Neurosci., 2017, 8, 1036–1042 CrossRef CAS PubMed.
- Y. R. Pérez and R. Etchenique, Photochem. Photobiol. Sci., 2019, 18, 208–212 CrossRef PubMed.
- V. Ramu, S. Aute, N. Taye, R. Guha, M. G. Walker, D. Mogare, A. Parulekar, J. A. Thomas, S. Chattopadhyay and A. Das, Dalton Trans., 2017, 46, 6634–6644 RSC.
- Z. Zhao, K. Qiu, J. Liu, X. Hao and J. Wang, Chem. Commun., 2020, 56, 12542–12545 RSC.
- B. M. Vickerman, C. P. O'Banion, X. Tan and D. S. Lawrence, ACS Cent. Sci., 2020, 7, 93–103 CrossRef PubMed.
- L. Ma, Q. Zhao, X. Zhang and X. Chen, Chem. Phys. Lett., 2019, 730, 253–258 CrossRef CAS.
- A. Notaro, A. Frei, R. Rubbiani, M. Jakubaszek, U. Basu, S. Koch, C. Mari, M. Dotou, O. Blacque, J. Gouyon, F. Bedioui, N. Rotthowe, R. F. Winter, B. Goud, S. Ferrari, M. Tharaud, M. Řezáčová, J. Humajová, P. Tomšík and G. Gasser, J. Med. Chem., 2020, 63, 5568–5584 CrossRef CAS PubMed.
- W. Tian, C. Wang, D. Li and H. Hou, Future Med. Chem., 2020, 12, 627–644 CAS.
- S. Siddamurthi, G. Gutti, S. Jana, A. Kumar and S. K. Singh, Future Med. Chem., 2020, 12, 1037–1069 CrossRef CAS PubMed.
- A. Dey, J. Dana, S. Aute, A. Das and H. N. Ghosh, Photochem. Photobiol. Sci., 2019, 18, 2430–2441 CrossRef CAS PubMed.
- C. B. Larsen, G. A. Farrow, L. D. Smith, M. V. Appleby, D. Chekulaev, J. A. Weinstein and O. S. Wenger, Inorg. Chem., 2020, 59, 10430–10438 CrossRef CAS PubMed.
- D. Cullinane, K. S. Gkika, A. Byrne and T. E. Keyes, J. Inorg. Biochem., 2020, 207, 111032 CrossRef CAS PubMed.
- L. Zeng, D. Sirbu, P. G. Waddell, N. V. Tkachenko, M. R. Probert and A. C. Benniston, Dalton Trans., 2020, 49, 13243–13252 RSC.
- L. Zeng, D. Sirbu, N. V. Tkachenko and A. C. Benniston, Dalton Trans., 2021, 50, 7640–7646 RSC.
- C. Zhang, X. Guo, X. Da, Z. Wang, X. Wang and Q. Zhou, Dalton Trans., 2021, 50, 10845–10852 RSC.
- L. Xie, L. Wang, R. Guan, L. Ji and H. Chao, J. Inorg. Biochem., 2021, 217, 111380 CrossRef CAS PubMed.
- S. Chakrabortty, B. K. Agrawalla, A. Stumper, N. M. Vegi, S. Fischer, C. Reichardt, M. Kögler, B. Dietzek, M. Feuring-Buske, C. Buske, S. Rau and T. Weil, J. Am. Chem. Soc., 2017, 139, 2512–2519 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2022 |