
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBinding affinity of aniline-substituted dodecaborates to cyclodextrins†‡
Tarek
Marei§
 a,
Mahmoud K.
Al-Joumhawy§
a,
Mohammad A.
Alnajjar§
a,
Werner M.
Nau
a,
Khaleel I.
Assaf
b and
Detlef
Gabel
*a
a,
Mahmoud K.
Al-Joumhawy§
a,
Mohammad A.
Alnajjar§
a,
Werner M.
Nau
a,
Khaleel I.
Assaf
b and
Detlef
Gabel
*a
aDepartment of Life Sciences and Chemistry, Jacobs University Bremen, Bremen D-28759, Germany. E-mail: d.gabel@jacobs-university.de
bDepartment of Chemistry, Faculty of Science, Al-Balqa Applied University, Al-Salt 19117, Jordan
First published on 18th January 2022
Abstract
A new set of hybrid guest molecules bearing organic and inorganic residues have been studied for their recognition by cyclodextrins in aqueous solution. The guest molecules consist of nitroanilines linked through their amino group to the dodecahydrido-closo-dodecaborate cluster B12H122−, which serves as an anchor group. They show sizable affinity to cyclodextrins, and unexpected photophysical properties, with a very strong and low-energy charge-transfer band. The dodecaborate cluster increases the pKa of the anilines by 5.0 to 5.7 pH units, and the deprotonated forms of the o- and p-nitroaniline derivatives show strong charge transfer absorption bands in the visible part of the spectrum.
Boron cluster chemistry has seen tremendous growth in the last decades with respect to the synthesis of new derivatives and their applications in various research fields, ranging from materials science to medicinal chemistry.1–8 The properties of ionic icosahedral boron clusters such as B12H122− are different from those of organic anionic molecules. They are well soluble in water, but their interaction with water is influenced by the distribution of the charges within the entire cluster, and the hydridic character of the H atoms.9 The two effects combine in enabling these compounds to dissolve well in water, yet bind to hydrophobic surfaces. Recently, boron cluster anions have been recognized as strong cavity binders to macrocyclic receptors, in particular cyclodextrins (CDs, Fig. 1a).10–17 For example, the binding affinities of closo-dodecaborate anions (B12X122−, X = H, Cl, Br, I) exceed those of hydrophobic guest molecules.15 The high affinity was traced back to the chaotropic nature of the dodecaborate anions, classifying boron clusters as superchaotropic anions.15,18 Through the investigation of several boron-based clusters, it transpires that dihydrogen bonds, dispersion interactions, and the desolvation of the anionic clusters govern the trend in affinities in aqueous solution. Their high chemical and thermal stability as well as the lack of metabolization in the body add additional incentives to pursue their utilization in both material applications and drugs.
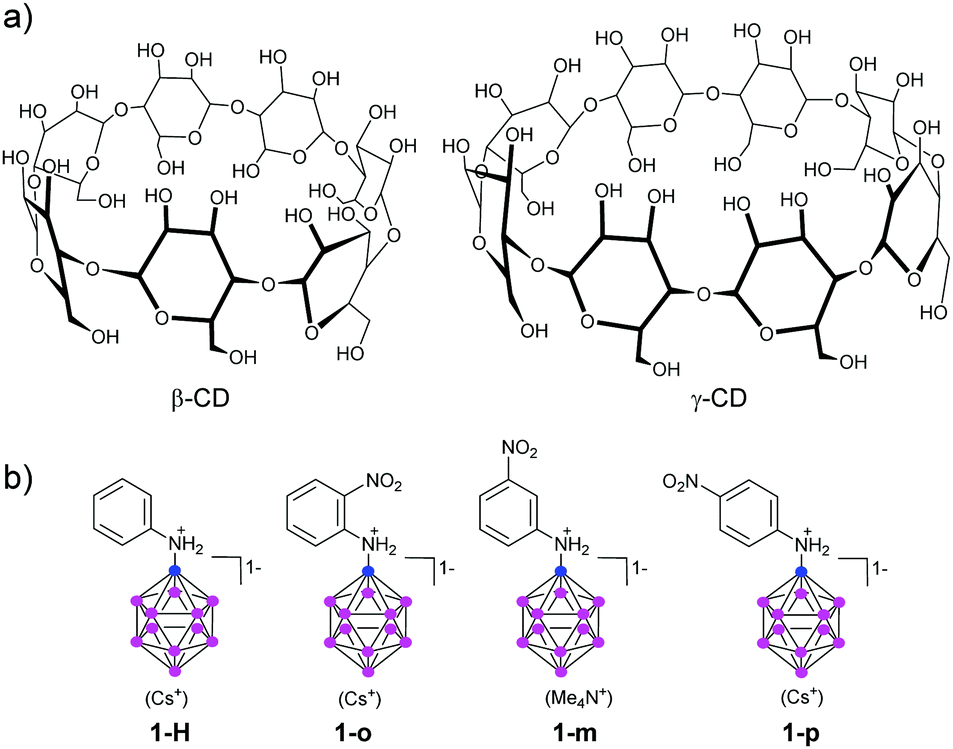 | ||
| Fig. 1 Molecular structure of cyclodextrins (CDs), the parent aniline-substituted dodecaborate (1-H), and its nitroaniline derivatives (o-, m-, p-, designated as 1-o, 1-m, and 1-p). | ||
Recently, one of the major problems in connecting boron clusters to atoms such as C and N has found new solutions through the use of Pd-catalysed cross-coupling reactions with B12H11I2−.19–21 This allows one to study the properties of series of similar compounds where chemical changes are small and well-defined. We had reported the synthesis of a number of aniline derivatives, where the dodecaborate cluster is linked directly to the aniline nitrogen;21 we had used a Buchwald–Hartwig type reaction with iodododecaborate, for which an improved synthesis method had been found.22 With this tool, it is now possible to explore properties and potential applications of such inorganic–organic hybrid molecules.
We picked the three nitroaniline derivatives of dodecaborate (o-, m-, p-, designated as 1-o, 1-m, and 1-p, respectively, Fig. 1b) the synthesis of which has been described by us before,21 together with the parent aniline derivative 1-H, and investigated the influence of the boron cluster on the HOMO–LUMO gap (as estimated by UV-Visible spectroscopy), the pKa values of the aniline N atom, and the binding to CDs of different sizes. We hoped to gain insight into how binding and physical–chemical properties of the derivatives vary with respect to their structure. The nitro group as electron acceptor and the unprotonated aminododecaborate as electron donor should result in a strong push–pull effect, with a considerable red shift of the absorption bands. This shift should disappear when the amine becomes protonated, or when the acceptor is absent. In addition, due to lack of π conjugation between the substituents, a nitro group in the meta position with respect to the amine substituent should display a relatively smaller influence on the HOMO–LUMO gap.
For these experiments, water-soluble salts are required. We could precipitate the Cs salts of the nitroaniline derivatives by dissolving the cross-coupling mixture21 in methanol after evaporation of DMSO and adding a methanolic solution of CsF. The product precipitated, and was found to be of high purity without requiring chromatography. Solely for the parent aniline derivative 1-H column chromatography was necessary to remove other dodecaborate by-products.
The pKa values of nitroanilines are known to be much lower than those of the parent (unsubstituted) aniline (Table 1). The pKa values for the nitroaniline boron clusters and the non-nitrated derivative are, however, much higher, by about 5 units (1-o: 5.4, 1-m: 7.8, 1-p: 6.1, 1-H: 9.9), than those of the corresponding free anilines (−0.3, 2.5, 1.0, and 4.9, see Table 1). The considerable increase in basicity of the N atom reflects the high electron-donating power and the charge of the cluster, strongly favoring the protonated form. Of interest is the observation that the m-nitroaniline derivative (1-m) has a pKa value close to physiological pH, and the pKa of 1-H is comparable to that of aliphatic amines. Such knowledge will be of importance when drugs would be constructed with such substituents. In terms of the Hammett and Pytela constants σp and σi, often used to describe the electronic influence on aromatic systems, the N-dodecaborate derivatives are much stronger electron donors than any of the commonly used substituents. This is borne out also in N-dodecaborate amides described previously,23 where the benzamide was isolated in its protonated form.
| 1-H | 1-o | 1-m | 1-p | |
|---|---|---|---|---|
| a From ref. 24 b Error in data is ca. 20%. c Excess CD was added to ensure a high degree of complexation (>90%). d nd: No reliable determination possible, see note.25 | ||||
| Parent anilinea | 4.9 | −0.3 | 2.5 | 1.0 |
| Boron-substituted aniline | 9.9 | 5.4 | 7.8 | 6.1 |
| ΔpKab | 5.0 | 5.7 | 5.3 | 5.1 |
| Complexed with β-CDc | ndd | 4.4 | 7.2 | 4.8 |
| Complexed with γ-CDc | ndd | 5.4 | 8.7 | 5.6 |
In the spectra of the non-protonated forms of 1-o, 1-m, and 1-p (see Fig. 2), we found charge-transfer (CT) absorption bands which were much more red-shifted than those described for m- and p-nitroaniline.26 Thus, for p-nitroaniline in water, the maximum absorption due to the CT band is around 360 nm, whereas the dodecaborate analogue 1-p has its maximum at 450 nm. The change in the CT band corresponds to the high electron-donating power of dodecaborate, which is empirically known from its chemical reactions. There is no influence of the protonation state on the CT band of 1-H, and its long-wavelength absorbance at 290 nm stays constant with pH. For 1-m, as expected, we found a greatly reduced CT band in comparison to 1-o and 1-p, due to lack of resonance effects in this position.
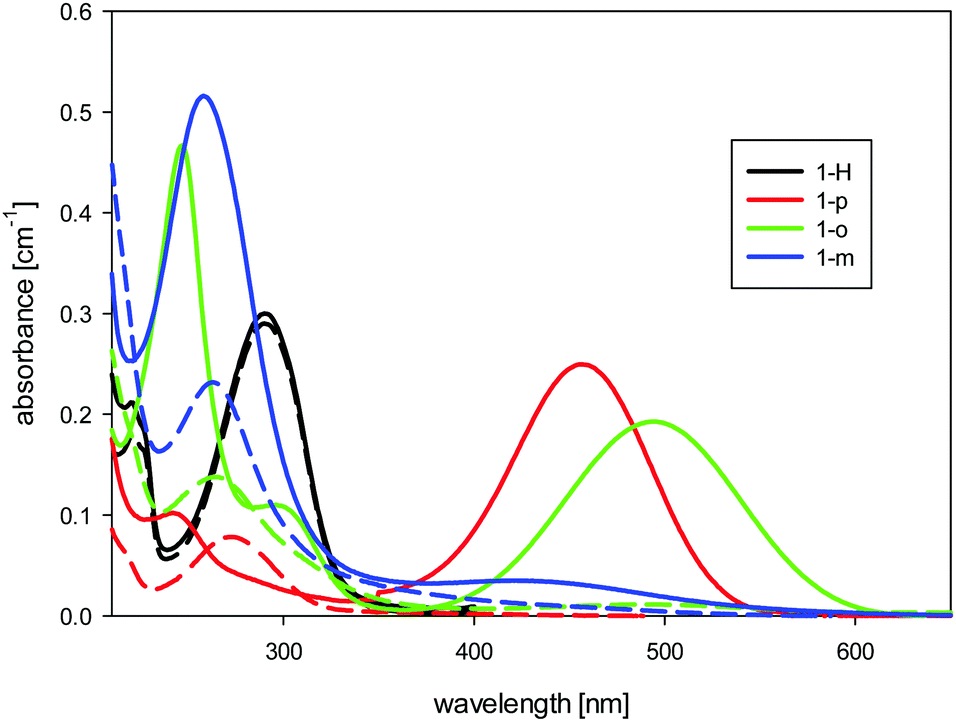 | ||
| Fig. 2 UV-Visible absorption spectra of the dodecaborate-substituted anilines in water (concentration 10 μM) at high (solid line) and low (dashed line) pH values. | ||
We knew from previous experiments that monosubstituted boron clusters can bind more strongly to CDs than the non-substituted parent clusters.14 Therefore, we expected the nitroaniline derivatives to show a higher affinity than B12H122−. With structurally related substituents, we hoped to see how much the physical–chemical properties and the binding affinity to CDs of different sizes could be related to the substitution pattern.
At high pH, i.e., when the guests are in their deprotonated forms, the nitroaniline derivatives bind well, with millimolar or higher affinities, to both β-CD and γ-CD (see Table 2).25 Guest molecules undergo a complexation-induced pKa shift upon complexation by CDs, and this results generally in a decrease of the pKa values of ammonium ions/amines in their CD-complexed states.27–29 The underlying reason is that CDs tend to bind hydrophobic neutral amines more strongly in their hydrophobic cavity.30 For β-CD complexation of the nitroaniline derivatives, the shift is in the expected direction (pKa values lower than the uncomplexed compounds) (Table 1), with the deprotonated form as the favored form for binding. As the net charge is decreased by deprotonation (from −1 to −2), a higher pKa would have been expected if the net charge was the dominant driving force in complexation. As, however, a pronounced hydration of the positively charged N atoms has been found in molecular dynamics simulations,9 binding to β-CD would require ammonium-solvating water to be removed, with an energetic penalty. Upon complexation with γ-CD, the pKa values for 1-o and 1-p are influenced less than for 1-m; for the latter, an increase (rather than an expected decrease) of 0.9 pH units is found.
| Derivative | CD | K ass /103 M−1 | −ΔHb | TΔSb | −ΔGb |
|---|---|---|---|---|---|
| a Values without parentheses pertain to the deprotonated form (determined by ITC at pH 9), values in parentheses to the protonated form (extrapolated from the pKa shifts in Table 1 for 1-o, 1-m, and 1-p, and determined by ITC at pH 3 for 1-H). b All in kcal mol−1, T = 298 K. c By NMR, from ref. 15 d nd: No reliable determination possible, see note.25 | |||||
| B12H122− | βc | 0.2 | |||
| γc | 2.0 | ||||
| 1-H | β | ndd (31 ± 6) | (11 ± 3) | (5.0) | (6.0) |
| γ | ndd (5.9 ± 3.4) | (7.5 ± 0.7) | (2.4) | (5.1) | |
| 1-o | β | 5.7 ± 2.0 (0.6) | 1.2 ± 0.7 | 3.9 | 5.1 |
| γ | 15 ± 4 (15) | 2.9 ± 0.1 | 2.9 | 5.8 | |
| 1-m | β | 18 ± 6 (4.5) | 2.8 ± 0.1 | 3.0 | 5.8 |
| γ | 9.6 ± 3.6 (76) | 1.2 ± 0.1 | 4.2 | 5.4 | |
| 1-p | β | 19 ± 5 (1.0) | 2.0 ± 0.3 | 3.8 | 5.8 |
| γ | 7.6 ± 0.7 (2.4) | 4.3 ± 0.6 | 1.0 | 5.3 | |
As we knew the pKa values of both the free and bound dodecaborate anilines (Table 1), and since the binding constants of the unprotonated forms could be directly measured by isothermal titration calorimetry (ITC, Table S2 and ESI‡), one can estimate the association constants of the protonated forms through the known thermodynamic cycle. These binding constants are also found in Table 2 (values in parentheses). Binding to β-CD is stronger than binding to γ-CD for equivalent protonation states. For γ-CD, Kass does not show a clear dependence between the two protonation states.
While being unprotonated, in their dianionic forms, the three nitroaniline derivatives bind quite strongly (Kass around 104 M−1) to γ-CD, more strongly than the unsubstituted parent cluster. Binding to β-CD of all derivatives in both protonation states investigated here is stronger than the binding of the parent B12H122−.25 For B12H122− and β-CD, we had found an association constant of only 2 × 102 M−1.15 The binding of B12H122− to γ-CD is one order of magnitude stronger (2.0 × 103 M−1). This demonstrates that monosubstitution has a synergetic effect on the binding. Binding is driven about equally by enthalpic and entropic contributions, which suggests an interplay between chaotropic cluster binding (enthalpically favorable) and hydrophobic binding (entropically driven).18 The enthalpy–entropy compensation which we had observed before for other derivatives15 is also found for the compounds investigated here (thermodynamic data from ITC in Table 2).
In the UV/Vis spectra, the binding of the different derivatives to both CDs leads only to small bathochromic shifts. This is in contrast to one of the two dodecaborate-substituted nitrobenzodiazoles described earlier,14 where larger shifts were found. In the case of the anilines, the shifts were too small to be used for a reliable determination of the association constants by UV/Vis titrations, such that the ITC affinity data could not be readily verified by an independent technique. However, the complexation of the nitroaniline derivatives could be further structurally verified by using NMR spectroscopy (Fig. 3). The formation of host–guest inclusion complexes could be monitored through the complexation-induced chemical shifts, and in particular for the signals of the CD hosts.14,15 For example, the complexation of 1-m with β-CD resulted in an up-field shift of H1 and H2, as well as a down-field shift of the H3 proton, which is located inside the cavity. The resonances of the aromatic protons of the anilines also experienced shifts upon complexation. Similar effects were observed for the complexation of 1-p with β-CD. For γ-CD, very pronounced shifts were observed for the complexation of 1-m and 1-p (Fig. 3), which might indicate that the guest protrudes more deeply into the larger cavity of γ-CD. Overall, the 1H NMR experiments suggest that the complexation of the nitroaniline derivatives occurs through the boron cluster, as also anticipated from our previous study.13–15 To further support this conclusion, 2D-ROESY experiments (see ESI‡) were conducted, which did not reveal any cross-peaks between the aromatic protons of the anilines and the CD protons (see Fig. 4 for modelled structures). Furthermore, theoretical calculations indicated that the inclusion complexation between the CDs and the inorganic cluster is preferred over that for the organic part (Fig. 4).
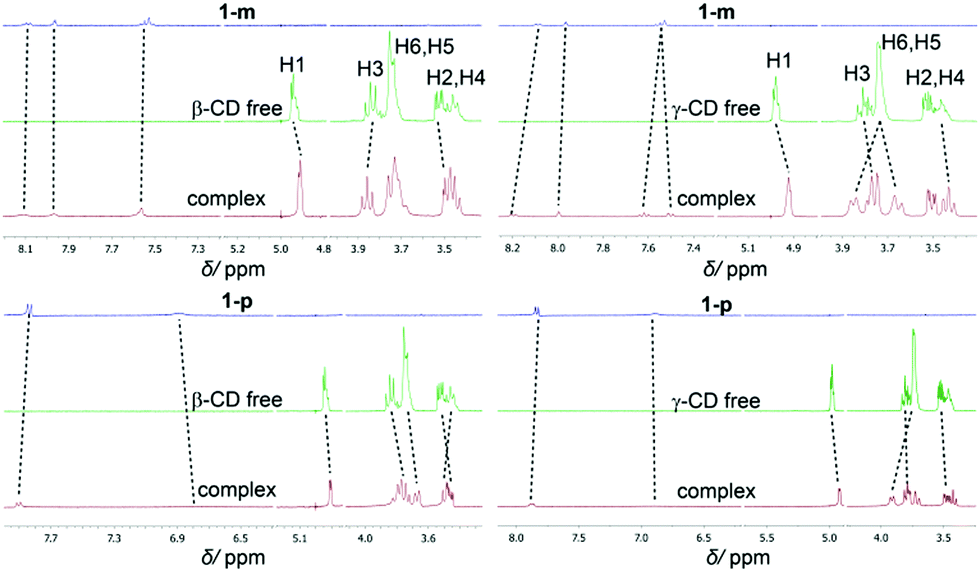 | ||
| Fig. 3 Shifts of 1H-NMR resonances upon binding of 1-m (top) and 1-p (bottom) to β-(left) and γ-CD (right). | ||
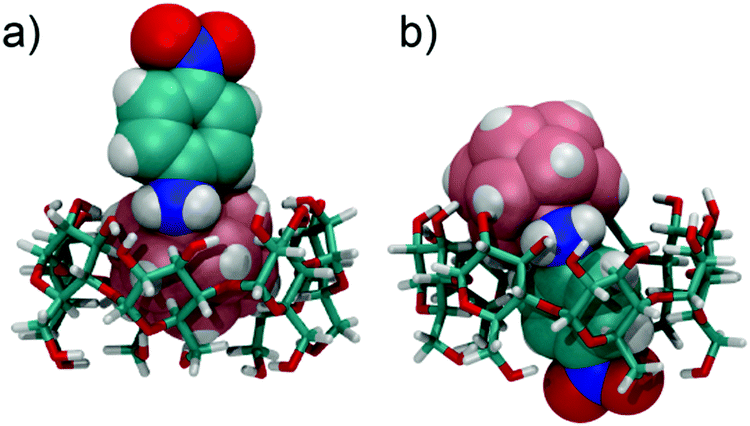 | ||
| Fig. 4 Binding of 1-p, in its protonated state, to β-CD. (a) “inorganic head-first”; (b) “organic feet-first”; structures were optimized by using the semi-empirical method PM3. | ||
In conclusion, we have found that the aniline-substituted dodecaborates are more basic than the parent anilines, with pKa values shifting such that N-protonation under physiological pH values must be considered. In comparison to non-boronated anilines, the charge-transfer bands of the ortho- and para-substituted derivatives are shifted to considerably longer visible wavelengths. The compounds complex strongly (mM and higher affinities) with both β- and γ-CD.
The authors are grateful to the DFG for grants GA 250/55-1, NA 686/15-1, and NA 686/8-2.
Conflicts of interest
There are no conflicts to declare.Notes and references
- E. Justus, K. Rischka, J. F. Wishart, K. Werner and D. Gabel, Chem. – Eur. J., 2008, 14, 1918–1923 CrossRef CAS PubMed.
- T. Kondo, Sci. Technol. Adv. Mater., 2017, 18, 780–804 CrossRef PubMed.
- E. Hey-Hawkins and C. Viñas, Teixidor, boron-based compounds: potential and emerging applications in medicine, John Wiley & Sons, 2018 Search PubMed.
- B. P. Dash, R. Satapathy, J. A. Maguire and N. S. Hosmane, New J. Chem., 2011, 35, 1955–1972 RSC.
- D. Gabel, Pure Appl. Chem., 2015, 87, 173–179 CAS.
- P. Řezáčová, J. Pokorná, J. Brynda, M. Kožíšek, P. Cígler, M. Lepšík, J. Fanfrlík, J. Řezáč, K. Grantz Šašková, I. Sieglová, J. Plešek, V. Šícha, B. Grüner, H. Oberwinkler, J. Sedláček’, H.-G. Kräusslich, P. Hobza, V. Král and J. Konvalinka, J. Med. Chem., 2009, 52, 7132–7141 CrossRef PubMed.
- L. He, H.-W. Li, H. Nakajima, N. Tumanov, Y. Filinchuk, S.-J. Hwang, M. Sharma, H. Hagemann and E. Akiba, Chem. Mater., 2015, 27, 5483–5486 CrossRef CAS.
- Y. Zhang, L. Yang, L. Wang, S. Duttwyler and H. Xing, Angew. Chem., Int. Ed., 2019, 58, 8145–8150 CrossRef CAS PubMed.
- K. Karki, D. Gabel and D. Roccatano, Inorg. Chem., 2012, 51, 4894–4896 CrossRef CAS PubMed.
- S. El Anwar, K. I. Assaf, B. Begaj, M. A. Samsonov, Z. Růžičková, J. Holub, D. Bavol, W. M. Nau, D. Gabel and B. Grűner, Chem. Commun., 2019, 55, 13669–13672 RSC.
- K. I. Assaf, B. Begaj, A. Frank, M. Nilam, A. S. Mougharbel, U. Kortz, J. Nekvinda, B. Grüner, D. Gabel and W. M. Nau, J. Org. Chem., 2019, 84, 11790–11798 CrossRef CAS PubMed.
- K. I. Assaf, A. Hennig, S. Peng, D.-S. Guo, D. Gabel and W. M. Nau, Chem. Commun., 2017, 53, 4616–4619 RSC.
- K. I. Assaf, D. Gabel, W. Zimmermann and W. M. Nau, Org. Biomol. Chem., 2016, 14, 7702–7706 RSC.
- K. I. Assaf, O. Suckova, N. Al Danaf, V. von Glasenapp, D. Gabel and W. M. Nau, Org. Lett., 2016, 18, 932–935 CrossRef CAS PubMed.
- K. I. Assaf, M. S. Ural, F. Pan, T. Georgiev, S. Simova, K. Rissanen, D. Gabel and W. M. Nau, Angew. Chem., Int. Ed., 2015, 54, 6852–6856 CrossRef CAS PubMed.
- K. I. Assaf, J. Holub, E. Bernhardt, J. M. Oliva-Enrich, M. I. Fernández Pérez, M. Canle, J. A. Santaballa, J. Fanfrlík, D. Hnyk and W. M. Nau, ChemPhysChem, 2020, 21, 971–976 CrossRef CAS PubMed.
- S. M. Eyrilmez, E. Bernhardt, J. Z. Dávalos, M. Lepšík, P. Hobza, K. I. Assaf, W. M. Nau, J. Holub, J. M. Oliva-Enrich, J. Fanfrlík and D. Hnyk, Phys. Chem. Chem. Phys., 2017, 19, 11748–11752 RSC.
- K. I. Assaf and W. M. Nau, Angew. Chem., Int. Ed., 2018, 57, 13968–13981 CrossRef CAS PubMed.
- T. Peymann, C. B. Knobler and M. F. Hawthorne, Inorg. Chem., 1998, 37, 1544–1548 CrossRef CAS.
- A. Himmelspach, M. Finze, A. Vöge and D. Gabel, Z. Anorg. Allg. Chem., 2012, 638, 512–519 CrossRef CAS.
- M. K. Al-Joumhawy, T. Marei, A. Shmalko, P. Cendoya, J. La Borde and D. Gabel, Chem. Commun., 2021, 57, 10007–10010 RSC.
- M. Al-Joumhawy, P. Cendoya, A. Shmalko, T. Marei and D. Gabel, J. Organomet. Chem., 2021, 949, 121967 CrossRef CAS.
- S. Hoffmann, E. Justus, M. Ratajski, E. Lork and D. Gabel, J. Organomet. Chem., 2005, 690, 2757–2760 CrossRef CAS.
- CRC Handbook of Chemistry and Physics, ed. J. R. Rumble, CRC Press, 101st edn, 2020 Search PubMed.
- For 1-H, because of its high pKa value of 9.9, partial deprotonation of CDs, particularly β-CD (ref. 30), and the possible interplay between deprotonation of CDs and 1-H might make the measured values less certain, such that we omit them from the discussion.
- F. Bureš, RSC Adv., 2014, 4, 58826–58851 RSC.
- I. Ghosh and W. M. Nau, Adv. Drug Delivery Rev., 2012, 64, 764–783 CrossRef CAS PubMed.
- A. I. Lazar, J. Rohacova and W. M. Nau, J. Phys. Chem. B, 2017, 121, 11390–11398 CrossRef CAS PubMed.
- J. Mohanty, A. C. Bhasikuttan, W. M. Nau and H. Pal, J. Phys. Chem. B, 2006, 110, 5132–5138 CrossRef CAS PubMed.
- X. Zhang, G. Gramlich, X. Wang and W. M. Nau, J. Am. Chem. Soc., 2002, 124, 254–263 CrossRef CAS PubMed.
Footnotes |
| † Dedicated to Clara Viñas and Francesc Teixidor on the occasion of their 70th birthdays. |
| ‡ >Electronic supplementary information (ESI) available. See DOI: 10.1039/d1cc06524f |
| § These authors contributed equally to the work. |
| This journal is © The Royal Society of Chemistry 2022 |