
Spatial delivery of immune cues to lymph nodes to define therapeutic outcomes in cancer vaccination†
James I.
Andorko
a,
Shannon J.
Tsai
a,
Joshua M.
Gammon
a,
Sean T.
Carey
 a,
Xiangbin
Zeng
a,
Emily A.
Gosselin
a,
Camilla
Edwards
a,
Shrey A.
Shah
a,
Krystina L.
Hess
a and
Christopher M.
Jewell
*abcde
a,
Xiangbin
Zeng
a,
Emily A.
Gosselin
a,
Camilla
Edwards
a,
Shrey A.
Shah
a,
Krystina L.
Hess
a and
Christopher M.
Jewell
*abcde
aFischell Department of Bioengineering, University of Maryland, College Park, 8278 Paint Branch Drive, College Park, MD 20742, USA
bDepartment of Veterans Affairs, VA Maryland Health Care System, 10. N Green Street, Baltimore, MD 21201, USA
cRobert E. Fischell Institute for Biomedical Devices, 8278 Paint Branch Drive, College Park, MD 20742, USA. E-mail: cmjewell@umd.edu; Web: https://jewell.umd.edu Fax: +1 301-405-9953
dDepartment of Microbiology and Immunology, University of Maryland Medical School, 685 West Baltimore Street, HSF-I Suite 380, Baltimore, MD 21201, USA
eMarlene and Stewart Greenebaum Cancer Center, 22 S. Greene Street, Suite N9E17, Baltimore, MD 21201, USA
First published on 28th June 2022
Abstract
Recently approved cancer immunotherapies – including CAR-T cells and cancer vaccination, – show great promise. However, these technologies are hindered by the complexity and cost of isolating and engineering patient cells ex vivo. Lymph nodes (LNs) are key tissues that integrate immune signals to coordinate adaptive immunity. Directly controlling the signals and local environment in LNs could enable potent and safe immunotherapies without cell isolation, engineering, and reinfusion. Here we employ intra-LN (i.LN.) injection of immune signal-loaded biomaterial depots to directly control cancer vaccine deposition, revealing how the combination and geographic distribution of signals in and between LNs impact anti-tumor response. We show in healthy and diseased mice that relative proximity of antigen and adjuvant in LNs – and to tumors – defines unique local and systemic characteristics of innate and adaptive response. These factors ultimately control survival in mouse models of lymphoma and melanoma. Of note, with appropriate geographic signal distributions, a single i.LN. vaccine treatment confers near-complete survival to tumor challenge and re-challenge 100 days later, without additional treatments. These data inform design criteria for immunotherapies that leverage biomaterials for loco-regional LN therapy to generate responses that are systemic and specific, without systemically exposing patients to potent or immunotoxic drugs.
Introduction
Vaccines combat disease through molecularly-specific mechanisms that enable both selective and long-lasting outcomes. These technologies have also helped catalyze breakthrough cancer immunotherapies, such as chimeric antigen receptor (CAR) T cells,1–3 that are already providing remarkable impacts for certain patient groups. CAR T cell therapy involves isolation, engineering, expansion, and reinfusion of a patient's own immune cells. Another technology in cancer research is cancer vaccination. The first clinical example, Provenge, involves complex engineering of a patient's own antigen presenting cells (APCs) to generate an important, but modest therapeutic impact.4 Both of these cell-therapy examples are complex, requiring sophisticated manufacturing and regulatory regimens that create costs as high as $500![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 per treatment round.5 Thus, new, simpler immunotherapy approaches that provide this same type of vaccine-like specificity – without isolation, engineering, and reinfusion of patient cells – would be transformative.
000 per treatment round.5 Thus, new, simpler immunotherapy approaches that provide this same type of vaccine-like specificity – without isolation, engineering, and reinfusion of patient cells – would be transformative.
As with traditional vaccines, cancer vaccines are comprised of fragments of tumors or cancerous cells – termed “antigens”, mixed with stimulatory adjuvants in an attempt to drive strong, lasting responses against the tumor antigen. Adjuvants take a range of forms that mimic molecular patterns of pathogens such as toll-like receptor (TLR) agonists, insoluble particulates of aluminum salts, or physical moieties foreign to the host.6–9 However, anatomical geography also plays a crucial role in determining effectiveness,10,11 as vaccine and immunotherapy components must reach specialized immune tissues such as lymph nodes (LNs). These sites integrate combinations and concentrations of immune signals to coordinate appropriate antigen-specific responses. In LNs, APCs present antigen – both antigen that passively drains to LNs and antigen that is carried to LNs by APCs – to the resident T and B lymphocytes responsible for the specific nature of the response. For example, while traditional preventative vaccines seek long-lasting memory function, therapeutic vaccines might benefit from initially inducing rapid and intense effector function. More sophisticated insight into how immune signals are integrated in LNs to drive antigen specific responses tailored for immunotherapy would enable next generation therapeutic vaccines for cancer.12
Biomaterials offer a unique opportunity to engineer a delivery system with programmable parameters such as size (e.g., nano- or micro-scale), surface properties (e.g., charge, topography), and the release of included cargo over time (e.g., degradation, enzymatically-triggered).13 This distinctive control over parameters that biomaterials provide has motivated many pre-clinical approaches using carriers and scaffolds to direct cancer immunotherapy,14–18 and recently, in clinical trials.19 The central role of LNs to generate tumor-specific immune responses has motivated much of this work. The majority of strategies involve systemic injection or infusion of nanoparticles, for example, with chemical modifications introduced to engage natural host trafficking mechanisms to shuttle immune cues to LNs.20 For instance, polymeric particles or scaffold can be formulated to initially release cytokine to recruit immune cells of interest prior to releasing antigen and other immune cues to active these cells.21,22 In one such study, nanodiscs specifically designed to deliver cancer neoantigens alongside adjuvants were able to strongly enhance antigen-specific cytotoxic T cells after multiple subcutaneous injections.23 However, peripheral injections of immunotherapies are limited by the efficiency of targeting to LNs or tumors. Equally importantly, delivery of soluble potent immune drugs or nanoparticle therapies which need a higher overall dose are also limited by the toxicity or off-target effects. A portion of the injected dose of these therapies must reach LNs and be retained in these tissues at immunologically-relevant concentration and time scales without distribution or drainage to other tissues or organs to avoid off-target effects and toxicity.24,25 Thus, while there is no universal strategy to treat a disease as heterogeneous as cancer, these challenges have motivated great interest in alternative treatment schemes that do not require systemic or peripheral injection. The most basic example is the persistence of tumor resection as a treatment course in many cases. Newer concepts seek to exploit the geographic interplay between LNs, tumors, and the location of antigens, adjuvant or other signals.26–28 For example, compared with systemic injection, direct intra-tumoral injection and intradermal injection of immune checkpoint inhibitors can improve outcomes in pre-clinical models.29 Likewise, nanoparticle-based strategies are improved with similar loco-regional tumor injection approaches.30–32 In these processes, the route of antigen transport to and within LNs are important in determining outcomes.25 Another spatially-localized cancer immunotherapy is now testing a biomaterial scaffold for melanoma in clinical trials.19 For each patient, tumor cells are isolated and lysed, then scaffolds are loaded with lysate, adjuvants, and immune cell-recruiting molecules prior to implantation. The implanted scaffold will then recruit APCs to sample patient-derived antigen within the scaffold, and ideally migrate to LNs to generate tumor-specific adaptive responses. Together, these examples highlight the distinct potential of “geographic” immunotherapy in the context of solid tumors or other diseases with geographic localization.
Distinct form the tissue-restricted routes just mentioned, an intriguing injection route relevant to this geographic opportunity involves direct intra-LN (i.LN.) injection of immune signals to LNs using ultrasound guidance or lymphatic tracers. In the clinic, delivery of soluble antigen to LNs has been tested in immunotherapy contexts.33–40 By combining this approach with polymer depots, we have shown this antigen delivery strategy has promise to drive immune tolerance.41–43 While unlikely to be used as an everyday route of administration for vaccination again infectious diseases, in a therapeutic context, i.LN. injection of biomaterial depots creates a unique tool to directly study pro-immune signal integration during cancer immunotherapy: the ability to study responses and communication between injected LNs, non-injected LNs, and how these responses may vary when proximal or distal to tumors. This has significant implications for immunotherapy design, since during systemic administration, much broader biodistributions are achieved. Even using targeted particles that bias drainage to LNs, much of the material still reaches other sites. In particular, this level of precision is difficult to achieve using peripheral injections because most approaches rely on passive or active drainage of carriers and signals to LNs and again, are limited by the percentage of total dose delivered to the LN. Further, direct introduction of size-restricted degradable depots that are too large to drain from LNs after injection bypasses the need for lymphatic draining and APC targeting, creating potent therapeutic possibilities to generate tumor-specific immune cells without systemically exposing patients to potentially toxic immunotherapeutics. Thus, there is an opportunity to understand how juxtaposition of signals in a single LN using biomaterials alters the local environment of the treated LN, how localized these effects are with respect to other LNs, and the connections to systemic response. However, traditional vaccinology using systemic or peripheral injections does not reveal how local polymer depots injected to lymph nodes will function. For example, questions of how localized the signals will be retained in LNs after injection, how these cues will alter the local LN environment when released from the scaffolds, and the connection to systemic results remain unclear. This insight could help inform simple and potent immunotherapies that compare favorably to the cost of complex and expensive treatments such as CAR T cell therapies discussed above.
Toward the goals above, here we used geographic delivery of simple biomaterial systems to study how the relative proximity of antigen and adjuvant loaded in polymer depots impacts injected LNs and untreated LNs, and how communication between these sites determines efficacy in common pre-clinical cancer models. In particular, we used i.LN. injection to deliver widely-used signals with broad literature relevance to inguinal LNs of mice:44–46 ovalbumin (OVA) as an antigen, a molecular TLR agonist (PolyIC) as an adjuvant, and poly(lactic-co-glycolic acid) microparticle depots (PLGA MPs) as carriers. This platform mimics a key goal of robust adjuvant systems: adjuvant particles ready to be mixed with tumor antigens or patient-specific lysates. We show deposition of dose-matched treatments to a single LN, split equally in two LNs, or segregated with antigen and adjuvant in separate LNs results in vastly different local and systemic immunological responses. These changes include distinct innate immune cell activation profiles and antigen-specific T cell responses in LNs and blood. Ultimately, these geographic parameters led to large distinct efficacies during melanoma and lymphoma models, with a single treatment of the most potent signal distribution achieving near-complete efficacy during challenge and re-challenge over a 100 day period. Further, using this modular design, antigen could be readily exchanged for potential booster injections to facilitate antigen spreading. From a manufacturing and regulatory standpoint, the adjuvant particles could be normalized across a variety of products with different disease targets, further simplifying the overall workflow.
Experimental
Microparticle synthesis
Degradable MPs were synthesized via a double-emulsion, solvent evaporation technique as previously described.31,33,34 Briefly, to form lipid stabilized particles, 1,2-dioleoyl-sn-glycero-3-phosphocholine, 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[amino(polyethylene glycol)-2000], and 1,2-dioleoyl-3-trimethylammoniumpropane (Avanti Polar Lipids) were prepared at a 60:20:20 mol ratio and dried under nitrogen. For each particle batch, 80 mg of PLGA (Sigma) was dissolved with 5.15 μmol of lipids in 5 mL of dichloromethane. An inner aqueous phase containing 500 μL of water or 5 mg of PolyIC (Invivogen) in 500 μL of water was added to the organic phase containing polymer and lipid and sonicated for 30 seconds at 12 W to form the first emulsion. This emulsion was then added to 40 mL of water, homogenized for 3 min at 16000 rpm, and allowed to evaporate overnight while stirring to remove any excess organic solvent. After overnight stirring, particles were passed through a 40 μm cell strainer to remove any large aggregates and collected via centrifugation (5000g, 5 min, 4 °C). Supernatants were removed and particles were washed three times with 1 mL of water then suspended in water or PBS for animal studies, or lyophilized and stored at 4 °C prior to use. For preparation of fluorescently-labeled particles, 10 μL of DiO (Invitrogen) was added to the organic phase prior to forming the emulsion.
Microparticle characterization and cargo release
Particle diameter was determined using an LA-950 laser diffraction analyzer (Horiba). Zeta potential was measured using a Malvern Zetasizer Nano ZS90. The loading level of PolyIC was determined via UV/Vis spectrophotometry after hydrolyzing a known mass of lyophilized PolyIC MPs overnight in 0.2 M NaOH. Absorbance values were compared to standard curves of known masses of PolyIC to determine the mass of cargo per mass of polymer. MPs were imaged using a Hitachi SU-70 Schottky field emission gun scanning electron microscope after sputter coating lyophilized particles with gold. Cumulative release of PolyIC from MPs was characterized by suspending a known mass of MPs in media (RPMI 1640) and incubating at 37 °C. At given intervals, media was removed and replaced with fresh media. The removed media was analyzed via UV/Vis spectrophotometry and absorbance values were compared to standard curves of known masses of PolyIC in media.Mouse preparation and i.LN. vaccination
For each animal study, a small region of fur was removed from the lateral hind quarter of 6–10 weeks old C57BL6 mice (The Jackson Laboratory) by shaving the area and applying a mild depilatory. Tracer dye (1% w/v Evan's Blue) was injected subcutaneously on each side of the tail base as previously reported.31,33,34,36 After allowing for the tracer dye to drain to the inguinal LNs for visualization (approximately 16 hours), a 31G insulin needle was used to inject 10 μL containing the indicated treatment into inguinal LNs. To test how the localization of vaccine cargos to LNs impacted the local and systemic response MPs containing adjuvant (PolyIC) were mixed with soluble antigen (ovalbumin, OVA, Worthington) and introduced via i.LN. injection into one LN (‘One LN’), two LNs (‘Two LN’), or were split with the adjuvant MPs in one LN and the antigen in another LN (‘Split LN’). Within these studies, the overall dose of vaccine (adjuvant and antigen) delivered to the mouse was fixed while the dose delivered to an individual LN varied between treatments. For example, in the One LN treatment, 1 mg of PolyIC MPs containing ∼9 μg of PolyIC was co-injected with 50 μg of OVA suspended in 1× PBS to the left inguinal LN of a mouse. The right inguinal LN of the same mouse received only 1× PBS as an injection. In that same study, for a Two LN treatment, 0.5 mg of PolyIC MPs containing ∼4.5 μg of PolyIC was co-injected with 25 μg of OVA suspended in 1× PBS to the left and right inguinal LN of a mouse. In the Split LN treatment, 1 mg of PolyIC MPs containing ∼9 μg of PolyIC was injected into the left inguinal LN while 50 μg of OVA suspended in 1× PBS was delivered into the right inguinal LN. All studies involving mice were carried out in compliance with federal, state, and local laws and followed institutional guidelines, including the Guide for the Care and Use of Laboratory Animals and the Animal Welfare Act. All experiments were reviewed and approved by the University of Maryland's Institutional Animal Care and Use Committee (IACUC).LN and immune cargo imaging
For immune cargo and LN imaging, PolyIC was labeled with Cy5 per the manufacturer's protocol (Mirus Bio) and was encapsulated in MPs. FITC OVA (Thermofisher) was also used. 24 hours after i.LN. injection as described above, inguinal LNs were removed and frozen in OCT compound (Tissue-Tek). Frozen tissue was then sectioned at 6 μm thickness, air-dried for 2 hours, fixed in acetone, and washed in 1× PBS. LN sections were visualized using an Olympus IX83 fluorescent microscope and processed using ImageJ by comparing to an antibody isotype control. All images were equally adjusted across similar channels. Imaging of live and dissected whole mice was performed using a PerkinElmer IVIS Spectrum in vivo imaging system. i.LN. injections were performed as described above with FITC-labeled OVA and PLGA MPs synthesized with Cy5-labeled PolyIC. Animals were imaged live at 4 hours after injection then euthanized and dissected to image. Image analysis was performed using Living Image and quantitative measures were performed using region of interest (ROI) analysis of total radiant efficiency.Innate cell microparticle uptake and activation
To determine which innate cell subtypes were responsible for particle uptake, 1 mg of MPs labeled with DiO were introduced into the LNs of mice via i.LN. injection with either a One LN, Two LN, or Split LN treatment. 24 hours after treatment, mice were euthanized, the inguinal LNs were removed, and placed in 1× PBS. Tissues were processed into single cell suspensions by mechanical dissociation through a 40 μm strainer. After washing once with 100 μl of FACS buffer (1× PBS with 1% w/v bovine serum albumin, Sigma), cells were collected via centrifugation and blocked with Fc Block (anti-CD16/CD32, BD) for 10 minutes at room temperature to inhibit any nonspecific binding. Cells were then incubated for 30 minutes at room temperature with antibodies against cell surface markers including CD11b, CD8, F4/80, Ly6G, B220, and CD11c (all antibodies were from BD or Biolegend unless otherwise mentioned). Cells were then washed twice, suspended in FACS buffer, and particle uptake was quantified via flow cytometry (BD FACSCanto II) and analyzed using Flowjo (v.10, Treestar). To determine innate cell activation, mice were treated as above with PolyIC MP and OVA via i.LN. injection with either a One LN, Two LN, or Split LN treatment. Seven days after treatment, inguinal LNs were removed and stained against cell surface markers including CD11c, F4/80, CD40, CD80, CD86, I-A/I-E.Antigen-specific CD8+ T cell quantification via tetramer staining in blood and chemokine expression
At the indicated times after treatment of mouse LNs as above (e.g., Day 0, 7, 14, 21, 28), approximately 100 μL of blood was collected from anesthetized mice via submandibular bleeding. Red blood cells were removed by treating with 1 mL ACK lysis buffer (Lonza) for 5 minutes at room temperature. After pelleting cells by centrifugation (800g for 5 minutes at 4 °C) and aspirating the supernatant, cells were again treated with ACK lysis buffer, centrifuged, and the supernatant was removed. Following a 1 mL wash with FACS buffer, cells were blocked using Fc Block (anti-CD16/CD32, BD) for 10 minutes at room temperature to inhibit any non-specific binding. The cells were then stained with anti-SIINFEKL tetramer (H-2 Kb OVA, MBL International) for 30 minutes at room temperature followed by staining with antibodies for surface markers including CD4 and CD8 for 20 minutes at room temperature. After staining, cells were washed twice with 200 μL of FACS buffer, suspended in FACS buffer containing DAPI, and transferred to 5 mL flow cytometry tubes before being quantified on a BD FACSCanto II. The percentage of antigen-specific cytotoxic T cells (DAPI−, CD8+, tetramer+) were quantified. For experiments to determine chemokine expression, cells were prepared as above and stained with antibodies for CCR7 and CCR5. Chemokine expression was evaluated as overall number of positive cells or the percentage under CD8+ cells and CD8+, Tet+ cells.Antigen specificity and lymphocyte enumeration in LNs
At indicated terminal time points (e.g., Day 7 or 28 post vaccination), mice were euthanized, inguinal LNs were collected, placed in PBS, and processed into single cell suspensions by mechanical dissociation through a 40 μm strainer. Cells were washed once with 100 μL of FACS buffer then blocked with Fc Block for 10 minutes at room temperature. Cells was stained for lymphocyte populations and antigen-specific tetramer levels. First, 25 μL of anti-SIINFEKL tetramer was added and incubated for 30 min at room temperature followed by addition of 25 μL of antibodies against surface markers including CD4 and CD8 and incubated for 20 min at room temperature. Cells were then washed and evaluated, as above. The frequency of each cell population (percent of parent population) and number of counted cells per identical acquisition volume (80 μL) was evaluated.Investigation of adjuvant-involvement on split LN treatments and effect of inhibition of lymphocyte trafficking on antigen specificity
To determine the influence of adjuvant (PolyIC) delivery in the Split LN treatment group on antigen-specificity as quantified by SIINFEKL tetramer, mice were treated as described above for Split LN treatment with soluble OVA and either PolyIC MPs or with PBS. Antigen-specific CD8+ T cells in the blood and LNs were quantified using SIINFEKL tetramer as described above. Antigen-specific T cells were reported as a percentage of Live, CD8+ cells. In studies to determine the effect of lymphocyte trafficking on local and systemic antigen-specificity, FTY720 (Sigma) was administered daily at 1 mg kg−1via intraperitoneal injection one day prior to vaccination until the end of the experiment (7 days after vaccination). Following vaccination, antigen-specific CD8+ T cells in the blood and LNs were quantified using SIINFEKL tetramer as described above and reported as a percentage of Live, CD8+ cells and an overall number of cells from an identical acquisition volume (80 μL).Tumor inoculation, quantification, and rechallenge tumor studies
In preventative tumor studies, 7 days after treating mice with the indicated vaccines, mice were administered either 500000 B16-OVA cells (ATCC) or 1000000 E·G7-OVA cells (ATCC) in 100 μL of 1× PBS subcutaneously at the hind flank. Each day following inoculation, body weight was monitored and tumor burden was calculated as a product of two orthogonal diameters. Mice were euthanized according to IACUC-approved humane endpoints when the aggregate tumor burden exceeded 150 mm2. In the B16-OVA studies, mice that either did not establish or were able to clear the initial tumor were rechallenged with 100000 B16-OVA cells 13 weeks after initial tumor inoculation. The percentage of mice that either did not establish or cleared the secondary tumor were quantified as a percentage of the mice surviving the initial inoculation.
Statistical analysis
No pre-processing of data (transformation or removal of outliers) was performed prior statistical analysis. Sample sizes for each analysis are indicated in figure legends. Student's t tests were used in comparison of two groups. One-way ANOVA with a Tukey post-test was used to compare three or more groups, or two-way ANOVA for comparisons over time. In all cases, analyses were carried out with Graphpad Prism (version 8.4.3). Multiple comparisons of survival curve analysis was performed in Microsoft Excel as explained in GraphPad Prism using the Bonferroni method and the logrank p-values. Error bars in all panels represent the mean ± SEM and p-values ≤0.05 were considered significant with levels of significance were defined as *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001 unless otherwise noted. An absence of a symbol denotes non-significant findings and all statistical comparisons depicted are versus Sham unless otherwise noted.Results and discussion
Relative location of antigen and adjuvant loaded MPs determine distinct magnitudes of systemic response
We first used i.LN. delivery to confirm that the spatial juxtaposition of antigen and adjuvant that the immune system relies on can be selectively directed using local delivery of intra-LN depots. These particles were synthesized to be too large to drain from LNs, and thus diffusion limited.47 MPs exhibited characteristic cargo loading, size, surface charge, morphology, and release kinetics (ESI Fig. 1†). Throughout this work, mice were treated by administering a fixed total dose of OVA mixed with PolyIC MPs to either (i) a single inguinal LN (“One LN”), (ii) two inguinal LNs each receiving half the total dose (“Two LN”), or (iii) two inguinal LNs, with one LN receiving all of the antigen and one LN receiving all of the PolyIC MPs (“Split LN”) (Fig. 1a). To determine if immune signals are retained in treated LNs, mice were treated with both fluorescently-labeled PolyIC MPs and OVA. 24 hours later, histological analysis of treated LNs revealed deposition of both PolyIC MP (red) and OVA (green) into distinct LNs (Fig. 1b) compared to Sham treated LNs (ESI Fig. 2†). Of note, the soluble OVA was more diffuse than the PolyIC, which was encapsulated in MPs. Quantitative studies were subsequently performed using IVIS imaging in which mice were treated as in Fig. 1a with “One LN”, “Two LN”, or “Split LN” strategies. The geographic distribution of signals was then visualized after 4 hours (Fig. 1c). Similar to the histological results in Fig. 1b, images in Fig. 1c revealed localization to the expected LNs (e.g., “One LN”, “Split LN”) with more diffuse signal for soluble OVA. Quantification of each cohort using 3D image voxel analysis (Fig. 1d) revealed cargo in LNs at relative levels corresponding to the treatment scheme. For example, “Two LN” treatment of LNs where each node received half a total dose of immune cue exhibited approximately half of the signal of a node treated with a full dose. The observation in Fig. 1c of some diffusion in the OVA signal around the LN with the “Split” regimen could indicate possible association with MPs in vivo. While this was modest – since there was no significant increase in OVA levels in untreated nodes (Fig. 1d, “Split”), the interactions of free and particulate signals could be an interesting follow-on direction by functionalizing the surfaces of particles deposited in LNs.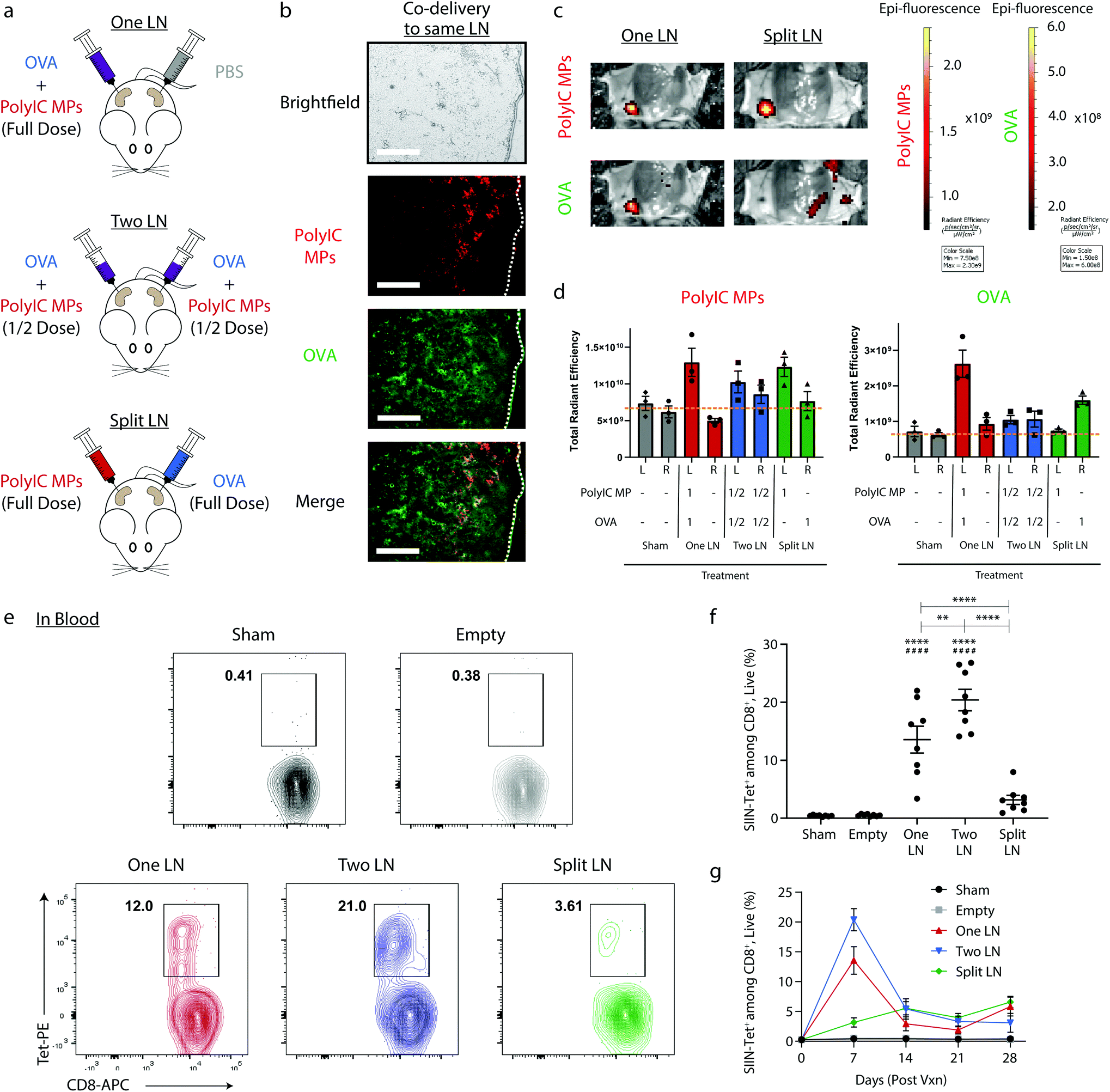 | ||
| Fig. 1 Immune cargos can be localized to different LNs, resulting in varied initial immune responses. (a) Schematic representation of treatment scheme where vaccine components are localized in overall matched doses given entirely to one LN (“One LN”, left), given in half doses to two inguinal LNs (“Two LN”, center), or split with the adjuvant-containing MPs in one LN and the antigen in the other (“Split LN”, right). (b) Representative fluorescent micrographs showing the colocalization of immune signals to LNs 24 hours after treatment via One LN injection. (PolyIC MP – red; FITC-OVA – green). Scale bar = 200 μm. (c) Representative IVIS images (left) of “One LN” and “Split LN” treated mice and (d) aggregate data for PolyIC MP deposition in LNs. Dashed gray line indicates the average total radiant efficiency for the “Sham” treatment group. n = 3 mice per group and errors bars represent SEM. (Using a One-way ANOVA with a Tukey post-test, * p < 0.05 when comparing left (L) to right (R) LN within the same treatment.) (e) Representative contour plots and (f) quantification of SIINFEKL-specific CD8+ T cells in blood 7 days after treatment as in (a). n = 4 per group for Day 14, 21, and 28 and errors bars represent SEM. (Using a One-way ANOVA with a Tukey post-test, * p < 0.05; ** p < 0.01; *** p < 0.001, ****p < 0.0001 compared to Sham treatment, #### p < 0.0001 compared to empty). (g) Evaluation of SIINFEKL-specific CD8+ T cells in the blood over time. N = 8 mice per group for Day 0 and 7. | ||
To investigate the influence of this geographic distribution on antigen-specific, adaptive immune response, blood was collected from mice 7 days after vaccination and MHC-I tetramer (SIINFEKL epitope, SIIN) was used to quantify the frequency of OVA-specific CD8+ T cells recognizing the SIINFEKL epitope fragment contained in OVA (Fig. 1e). Treating two LNs – each with half the total dose – resulted in the greatest frequency of OVA-specific CD8+ T cells after 7 days, with extremely strong responses indicated by an average frequency of 20.4%. Administration of the total dose to a single LN resulted in very strong (13.6%), but significantly lower responses compared to “Two LN” treatment (Fig. 1f). In contrast, the “Split LN” regimen generated a dramatically lower frequency 7 days after injection (3.16%) (Fig. 1f), confirming the importance of co-localizing antigen and adjuvant to generate potent immune responses. When evaluating the response over time, the largest differences between the percentage of SIIN-Tet+, CD8+ T cells were present 7 days after treatment (Fig. 1g). At 14 days post injection, the “Two LN” (Fig. 1g, blue) and “One LN” (Fig. 1g, red) treatments contracted, whereas the “Split LN” treatment regimen resulted in a slow, modest increase (Fig. 1g, green). The latter may suggest a low level of trafficking of adjuvant or antigen between nodes, an idea we assess directly in later studies. Overall, the results of Fig. 1 demonstrate intra-LN depots effectively localize signals to LNs, allowing rational control of where signals are localized and integrated.
Particle uptake and activation correlates with geographic signal distribution in LNs
We next directly studied the degree to which depots spatially-restrict the effects of adjuvant; this is clinically important since many cancer immunotherapeutics used to generate anti-tumor responses are also toxic.48 Mice were treated with OVA and fluorescent MPs using the “One LN”, “Two LN”, or “Split LN” regimens as above. When the total MP dose was administered to a single LN (i.e., “One LN” and right LN of “Split LN” regimen), approximately 10% of cells in the LN were positive for these large particles after 24 hours (Fig. 2a and b). In the case of the “Two LN” treatments, where half of the total dose of MPs was delivered to each LN, there was a corresponding decrease in the percentage of cells positive for particles by approximately half (Fig. 2a and b). No significant increases were observed in LNs of mice that were contralateral to a injected LN (Fig. 2a and b), further demonstrating the spatial control of this i.LN. injection approach. Assessment of the mean fluorescent intensity (MFI) also confirmed these biodistributions (ESI Fig. 3a†), further validating that the “mass balance” of the injected dose is almost complete, with a comparable number of cells taking up particles when integrating the portions of the total dose administered to each LN.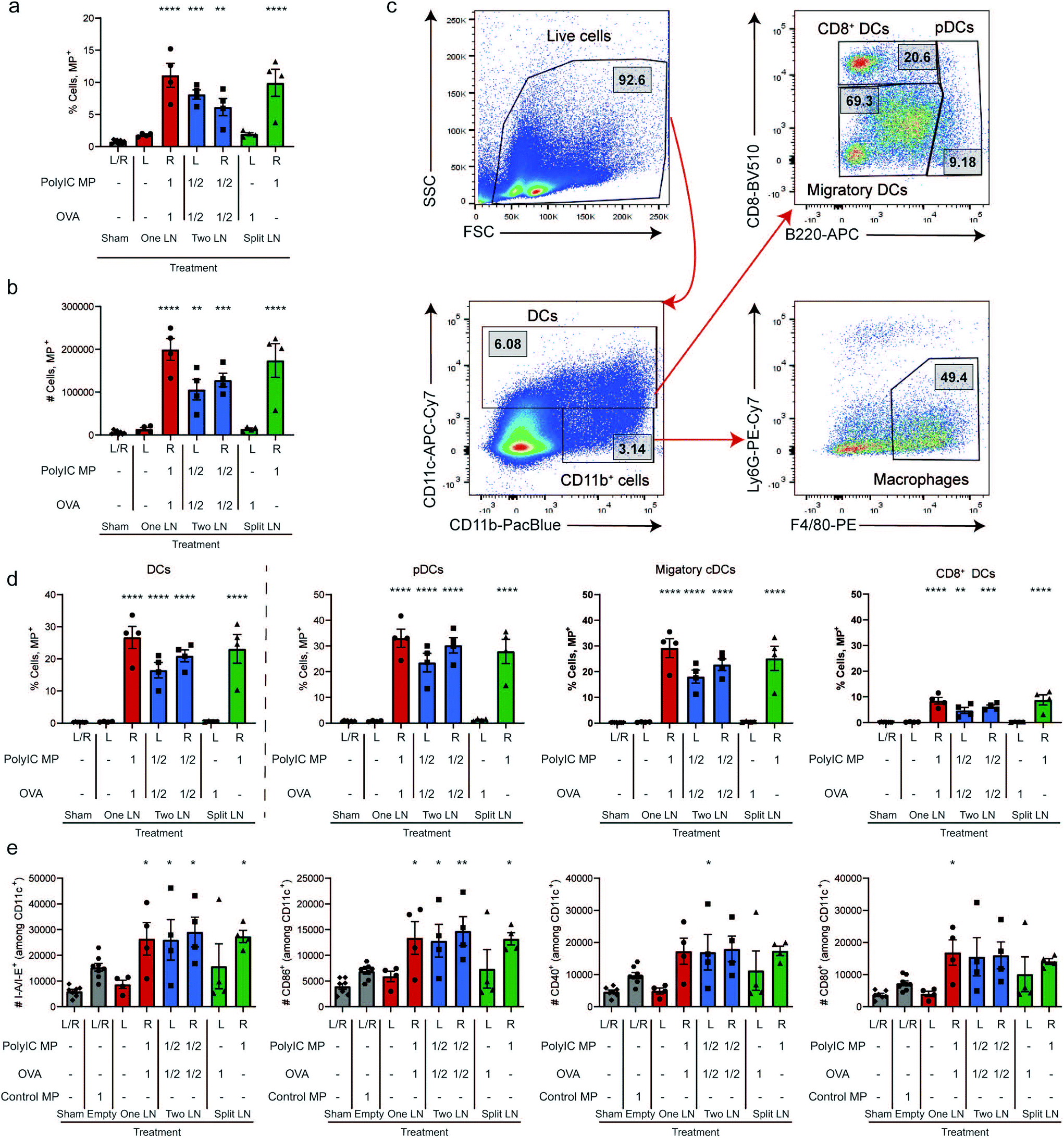 | ||
| Fig. 2 Rapid uptake of immune signals by innate immune cells within LNs leads to local activation. The overall percentage of cells (a) and number of cells (b) that were able to take up particles following “One LN”, “Two LN”, or “Split LN” treatment 24 hours after i.LN. treatment. (c) Representative gating scheme used to determine innate cell subpopulations including macrophages (CD11b+/CD11c−, F4/80+/Ly6G−) and DCs (CD11c+) which were further characterized as pDCs (CD11c+, B220+), CD8+ DCs (CD11c+, B220−/CD8+) or the remaining LN resident/migratory cDCs (CD11c+, B220−/CD8−), or. (d) Percentage of cells containing particles within DC parent (left) and subpopulations (right of dotted line). (e) Number of activated DCs (CD11c+) 7 days after treatment. For all panels, n = 4 mice per group and errors bars represent SEM. (Using a One-way ANOVA with a Tukey post-test, * p < 0.05; ** p < 0.01; *** p < 0.001, ****p < 0.0001 compared to Sham treatment). | ||
We next analyzed the MP uptake as a function of cell type, focusing on several important APC subsets: macrophages (CD11b+/CD11c−, F4/80+/Ly6G−) and dendritic cells (DCs; CD11c+), along with DC subsets of plasmacytoid DCs (pDCs; CD11c+, B220+), CD8+ DCs (CD11c+, B220−/CD8+), and the remaining LN resident/migratory conventional DCs (cDCs: CD11c+, B220−/CD8−); the gating scheme is shown in Fig. 2c. In LNs receiving particles, these studies revealed that 10–15% of macrophages and 20–25% of DCs were positive for MP signal (Fig. 2d, and ESI Fig. 3b†). As with the data for uptake by the total population of cells (Fig. 2a and b), the level of uptake in the “Two LN” regimen (where each LN receives half the dose) was generally lower than in LNs receiving the total dose of MPs (Fig. 2d). When assessing specific DC subsets, all DCs phagocytosed MPs, with pDCs and cDCs exhibiting similar levels of uptake; CD8+ DCs internalized fewer MPs, but with a similar trend across treated groups (Fig. 2d). This diminished uptake by CD8+ DCs may be due in part to their specialized endocytic compartments which are optimized for cross-presentation of antigen as well as the geographical location of these DC subsets within LNs.49 Next, we used key surface activation markers to determine if APC activation correlated to uptake levels. Compared to a control injection of Empty MPs which modestly increased activation, all LNs receiving PolyIC MP – regardless of the specific injection scheme – exhibited further increased DC activation, as indicated by elevated I-A/I-E (mouse MHCII) expression and expression of costimulatory markers (CD86, CD80, and CD40) (Fig. 2e, and ESI Fig. 3c†). Interestingly, activation was not observed in the Sham (PBS-treated) LNs, even when a full dose was injected contralaterally in the “One LN” regimen (Fig. 2e, left red bar); this underscores the precise spatial control in this platform. Analogous results were obtained when evaluating macrophages (ESI Fig. 3d†). In assessing the left LN of the “Split LN” regimen – which only received OVA, there was a slight trend toward increased activation in macrophages (ESI Fig. 3d†), and to a lesser extent, DCs (Fig. 2e). This suggests several possible explanations we investigate below, including drainage or transport of PolyIC from the right LN to the left LN, or migration of activated APCs between these sites.
Depot and relative signal localization dictate local expansion of antigen-specific T cells
In Fig. 1, we measured systemic antigen-specific response as a function of signal distribution. Fig. 2 revealed there are also distinct local differences in APC activation within LNs. Thus, we next assessed the level of antigen-specific T cells present directly in LNs using the same geographic injection schemes from above. Following injection (Day 0), LNs were isolated after 7 and 28 days (Fig. 3a), then the frequency of SIIN-Tet+, CD8+ cells was determined in both treated and untreated LNs using the gating scheme in Fig. 3b. 7 days after treatment, at the peak of T cell expansion and systemic responses observed in Fig. 1g, only LNs that received OVA (i.e., “One LN”, right; “Two LN”, right and left; “Split LN”, left) exhibited increased levels of antigen-specific cells (Fig. 3c, left plot). Interestingly, analysis of the MFIs among the CD8+, Tet+ cells revealed that any LN that received an immune signal (antigen or adjuvant), exhibited increased signal (Fig. 3d, left plot). Of note, the left LN from the “Split LN” treatment regimen (which received only OVA), contained a similarly high level of antigen-specific CD8+ T cells to those that received both OVA and PolyIC MPs. However, the APCs in these nodes were not strongly activated (Fig. 2e) and this same “Split LN” scheme did not result is significant antigen-specific T cells outside of the LN (i.e., circulating in blood) (Fig. 1e–g). Along these same lines, even though APCs in the right LN during the “Split LN” regimen were strongly activated (Fig. 2e, ESI Fig. 3d†), the percentage of antigen-specific T cell levels in this LN were very low due to the absence of antigen (Fig. 3c, left plot). This also indicates antigen, even though delivered here in a soluble form and not formulated in a MP, is not able to move or be carried from the left LN to the right LN over the period of 7 days. At longer times points (i.e., day 28 after injection), the antigen-specific T cell levels in the LNs contracted for all cases (Fig. 3c and d, right plots). Together, these observations suggest the T cells may expand in the LN during the “Split LN” scheme due to the presence of cognate antigen, but that these cells are not activated and cannot exit the LN to blood as functional antigen-specific T cells. To test these hypotheses directly, we next studied how the presence or absence of adjuvant in the LN influences local and systemic antigen-specific responses to antigen introduced to a contralateral LN (i.e., “Split LN” regimen).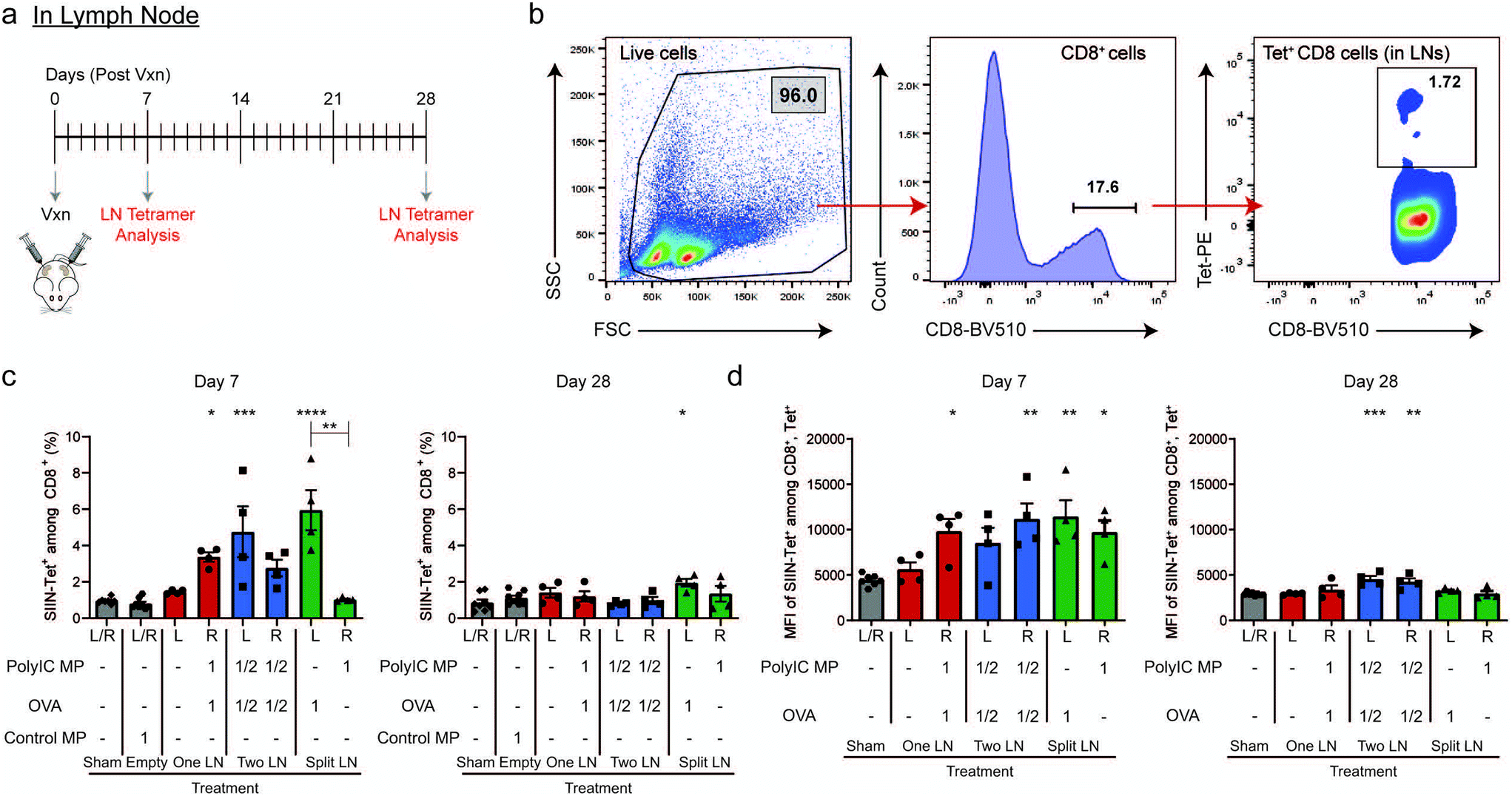 | ||
| Fig. 3 Vaccine location dictates the antigen-specific responses within the LN. (a) Schematic representation of vaccine treatment with experimental readouts and (b) representative gating scheme to determine CD8+, Tet+ cells in LNs. Evaluation of SIINFEKL-specific CD8+ T cells within the treated LNs of mice as treated in (a) at Day 7 (left) and Day 28 (right) post-treatment by percentage (c) and MFI (d) within the CD8+/Tet+ gate. For all panels, n = 4 mice per group and errors bars represent SEM. (Using a One-way ANOVA with a Tukey post-test, * p < 0.05; ** p < 0.01; *** p < 0.001, ****p < 0.0001 compared to Sham treatment). | ||
LN deposition of PolyIC MPs and cellular trafficking between LNs impact local and systemic antigen-specific responses
We next directly tested how the relative location of adjuvant depots controls priming of functional T cells and the ability of these lymphocytes to leave LNs. In these studies, mice received OVA in the left inguinal LN, and either PBS or PolyIC MPs in the contralateral (right) inguinal LN (Fig. 4a). The regimen using OVA and PBS (“Split LN (OVA only)”) increased systemic antigen-specific T cells approximately 2-fold compared to Sham but did not reach the level of statistical significance (p = 0.1494). However, treatment in the “Split LN” regimen with OVA and PolyIC MPs caused a statistically-significant increase in antigen-specific T cells in the blood by day 7; this represented an approximately 3-fold increase compared to the Sham baseline (Fig. 4b). Consistent with Fig. 1e and f, these responses were still much lower compared to antigen and adjuvant MPs delivered to the same LN. Assessment of the local concentration of OVA-specific T cells in LNs (Fig. 4c and d) revealed increases in the frequency and number of these populations in any (left) LN that received antigen, but not locally in contralateral (right) LNs that received either PBS or PolyIC MPs (Fig. 4c and d). These data (Fig. 4b–d) are consistent with the results of the “Split LN” regimen in Fig. 3c and d discussed above, suggesting that T cells in LNs receiving only antigen can expand, but are not activated and thus unable to exit LNs. Importantly, this result also suggests adjuvant contained in depots administered to LNs is largely localized to those sites. This capability could allow focused dosing of strong immunomodulatory therapeutic components to locally generate active, tumor-specific populations while limiting systemic toxicity.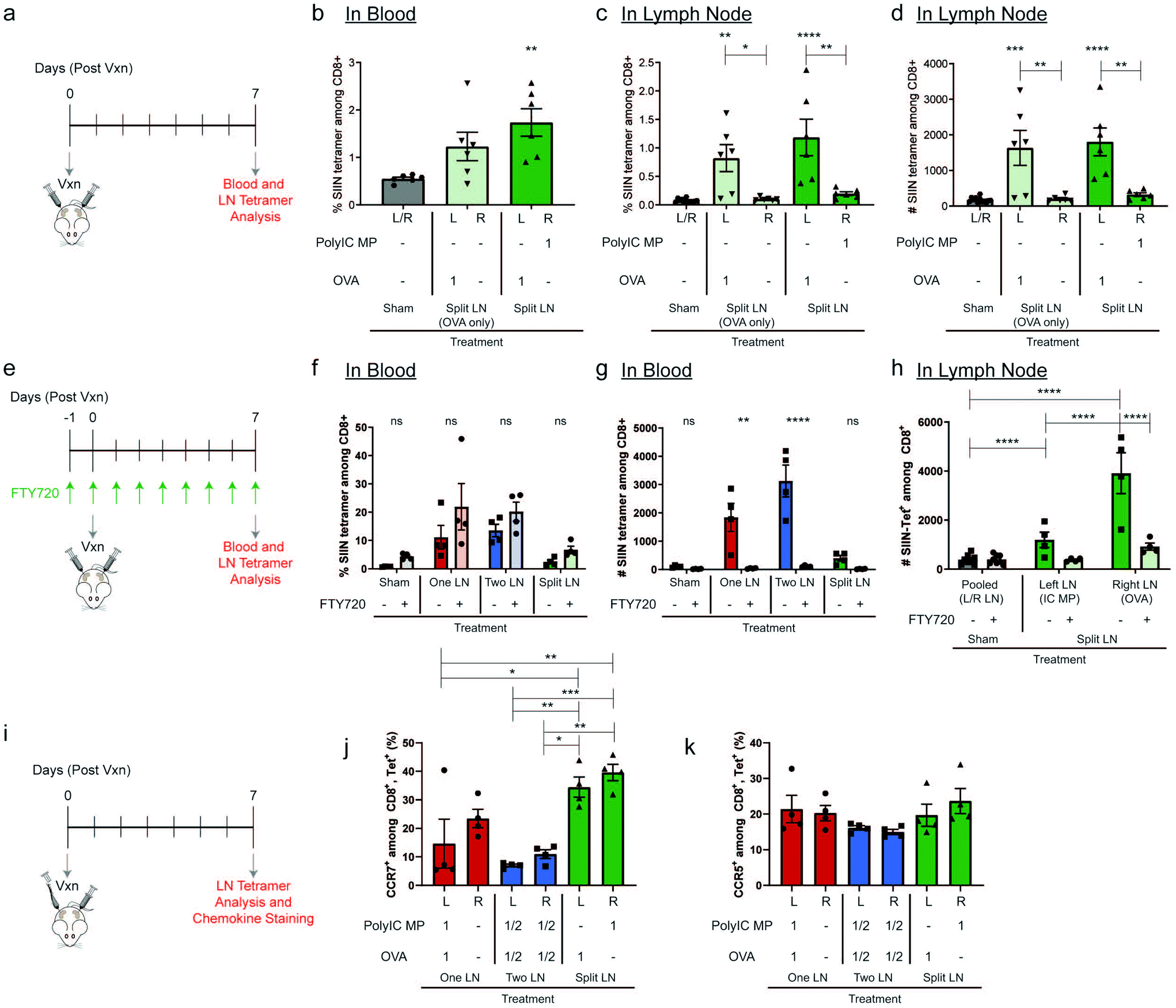 | ||
| Fig. 4 Split LN treatment relies on adjuvant treatment in LN to enhance antigen specificity and is dependent on cellular trafficking. (a) Schematic representation of treatment schedule and experimental readouts for studies comparing “Split LN” with OVA contralateral to PBS or PolyIC MPs. (b) Evaluation of SIINFEKL-specific CD8+ T cells in the blood following treatment in (a). Percentage (c) and number (d) of SIINFEKL-specific CD8+ T cells in the treated LNs following treatment as in (a). (e) Schematic representation of treatment schedule and experimental readouts for studies with FTY720 treatment to inhibit lymphocyte egress from lymphoid tissues. Percentage (f) and number (g) of SIINFEKL-specific CD8+ T cells in blood following treatment in (e). (h) Number of SIINFEKL-specific CD8+ T cells in LNs following treatment in (e). (i) Schematic representation of treatment schedule and experimental readouts for studies evaluating chemokine expression. Percentage of CCR7+ (j) and CCR5+ (k) among antigen-specific, CD8+ cells following i.LN. treatment; Sham treatment groups were omitted from this analysis among antigen-specific T cells since these animals did not receive a vaccine and did not exhibit measurable antigen-specific CD8+ cells. (panels: b–d) n = 6 mice per group and errors bars represent SEM. (Using a One-way ANOVA with a Tukey post-test, * p < 0.05; ** p < 0.01; *** p < 0.001, ****p < 0.0001 compared to Sham treatment). (panels: f–h) n = 4 mice per group and errors bars represent SEM. (Using a Two-way ANOVA with a Tukey post-test, * p < 0.05; ** p < 0.01; *** p < 0.001, ****p < 0.0001 compared to matched treatment without FTY720). (panels: j–k) n = 4 mice per group and errors bars represent SEM. (Using a One-way ANOVA with a Tukey post-test, * p < 0.05; ** p < 0.01; *** p < 0.001, ****p < 0.0001 as indicated by brackets). | ||
To directly investigate cellular trafficking, we administered daily systemic injections of a well-studied lymphocyte egress inhibitor, FTY720,50 beginning one day prior to local LN injection (Fig. 4e). We first validated the system by confirming FTY720 did not preferentially skew antigen-specific T cell generation for each injection scheme (Fig. 4f). Following treatment with FTY720, the percentage of antigen-specific T cells in blood did not significantly change, validating the ability to generate antigen-specific T cells. In contrast, the number of antigen-specific T cells in blood was greatly diminished (Fig. 4g), confirming the effectiveness of the FTY720 treatment in blocking LN egress. Using the “Split LN” regimen without FTY720, as with the earlier experiments, the OVA-treated LN exhibited local increases in antigen-specific T cells within LNs (Fig. 4h, “Split LN – Right LN (OVA)”, dark green), but not in blood (Fig. 4g); this further supports weak activation and an inability of these cells to migrate from the LN when adjuvant is restricted to other LNs using depots. In contrast, in mice receiving systemic FTY720, T cells in LNs receiving OVA (i.e., “Split – Right LN (OVA)” – light green) did not significantly expand in these tissues (Fig. 4h) or systemically in the blood (Fig. 4f and g). These results along with the data from Fig. 4c and d, further support that soluble PolyIC is not trafficking between nodes, regardless of whether FTY720 was administered. Therefore, one explanation for this finding is that FTY720 plays a role in limiting CD8+ expansion, while another is that this difference may indicate in the absence of FTY720, instead of soluble PolyIC draining between LNs, other cellular trafficking occurs. In this second scenario, activated lymphocytes or APCs from a treated LN egress from these tissues to contralateral LNs containing OVA, resulting in T cell expansion in these LNs. T cell migration and the methods through which these cells can enter and exit lymphatics is a highly investigated field51,52 and it has been established that T cells can migrate from one LN to another via the afferent lymphatics.53 Studies with additional treatment and inhibition regimens will allow these possibilities to be confirmed. This is analogous to the significant increases in systemic response observed when using PolyIC as a contralateral signal component compared with PBS (Fig. 4b). In all our studies, as one might expect, the generation of OVA-specific T cells was observed only in LNs that received OVA, not contralateral nodes receiving PolyIC MPs. Thus, these data further suggest free antigen did not drain significantly between nodes to promote antigen-specific T cell expansion in LNs treated only with PolyIC MPs.
To further probe the effect of cellular trafficking on contralateral LN activation, mice were treated using the “One LN”, “Two LN” and “Split LN” treatment scheme as above and chemokine expression on antigen-specific T cells within the LN was evaluated 7 days after injection (Fig. 4i, and ESI Fig. 4†). This chemokine staining was conducted to determine if immune cell homing signals, including C–C chemokine receptor type 5 and 7 (CCR5 and CCR7), are preferentially upregulated after injection. CCR7 is a receptor expressed on T cells to support homing to secondary lymphoid organs such as LNs, while CCR5 helps enable trafficking from LNs to peripheral tissues.54,55 In line with strong effector immune responses, the percentage of CCR7 expressed on antigen-specific CD8+ cells in LNs treated with both antigen and adjuvant decreased dramatically (Fig. 4j) when compared to other antigen-specific, CD8+ cells. “One LN” (Left LN) and “Two LN (Left and Right LN)” both decreased CCR7 expression by 2 to 3-fold, respectively, compared to LNs receiving individual or no immune cues. This shift in CCR7 enables primed lymphocytes to leave LNs to seek out targets they have been activated against. Interestingly, treatment of a LN with a single immune cue (OVA or PolyIC MPs individually in the “Split LN”) or PBS treatment in the “One LN” treatment (right LN), showed no reduction in CCR7; these treatments each had similar CCR7 expression levels. Within these studies, there were minimal changes to CCR5 regardless of treatment (Fig. 4k), suggesting future studies are needed to investigate the exact trafficking mechanisms of antigen-specific cells out of primed LNs and to areas of interest (e.g., homing marker profiles in blood, other LNs, tumors).
Distribution of antigen and adjuvant to LNs and tumors determines survival in murine melanoma models
Since the geographic distribution of signals resulted in dramatically different antigen-specific responses locally in LNs and systemically in blood, we next investigated these changes in a preventive cancer vaccination setting. Mice were vaccinated using the same “One LN”, “Two LN”, or “Split LN” treatment regimen as above and then inoculated subcutaneously with B16-OVA melanoma 7 days after treatment (Fig. 5a and b). In the case of the “One LN” and “Split LN” treatment regimens, we vaccinated mice in either tumor draining (tdLN) or non-tumor draining LN (non-tdLN) to test if vaccination proximity to the primary tumor influenced the resulting anti-tumor and antigen-specific response. In assessing tumor burden (Fig. 5c), the “Two LN” and either of the “One LN” regimens (tdLN or non-tdLN) generated immune responses that completely suppressed tumor growth. This efficacy resulted from a single injection and correlated to the significantly elevated percentage of antigen-specific T cells in the blood at the time of inoculation (ESI Fig. 5a†). Similar to trends seen in Fig. 1g, there was an increase in antigen-specific T cells in blood after 7 days which had a gradual contraction by day 28 (ESI Fig. 5b†).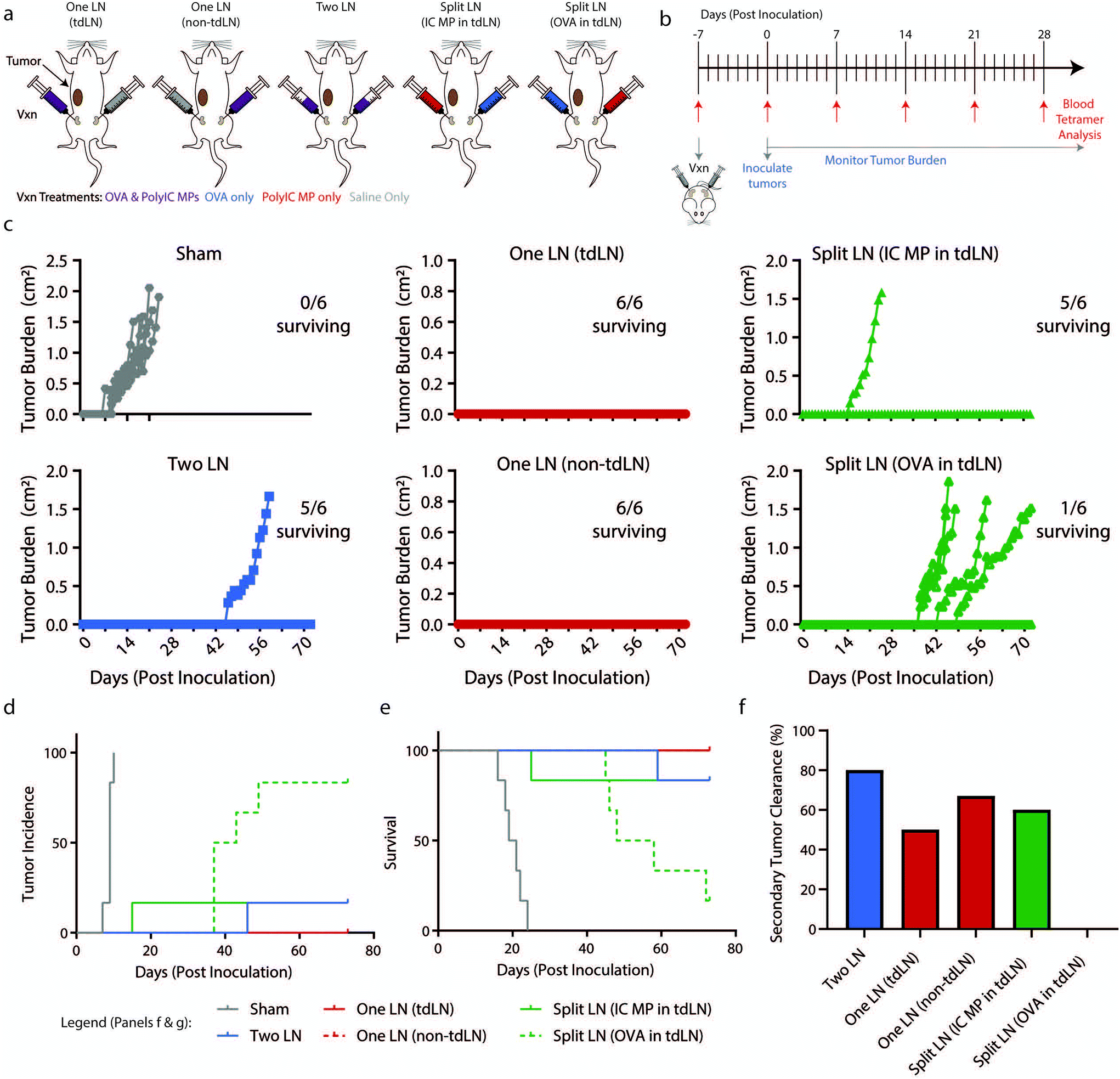 | ||
| Fig. 5 Geography of vaccination dictates survival following initial inoculation and rechallenge in a B16-OVA melanoma model. (a) Schematic representation of “Two LN”, “One LN”, or “Split LN” treatment and subsequent tumor inoculation in proximity to tumor-draining LNs (tdLN) and non-tumor-draining LNs (non-tdLN). (b) Schematic representation of treatment schedule and experimental readouts. (c) Traces of tumor volumes for individual mice with number of mice surviving indicated. (d) Incidence and (e) survival of mice following treatment in (b). (f) Percentage of mice which survived the initial inoculation that failed to grow a secondary tumor following a re-challenge on Day 91 (98 days after initial vaccination) with 100000 B16-OVA cells. (panels a–e) n = 6 mice per group. (panel d–e) Statistics indicated in ESI Table 1.† (panel h) Mice per group indicated in ESI Table 2.† | ||
When evaluating incidence (Fig. 5d) and survival (Fig. 5e), all treatment groups significantly differed from Sham treated LNs (ESI Table 1†). Intriguingly, the “Split LN” treatments resulted in divergent responses depending on which immune cue was delivered to the tdLN. When PolyIC MPs were delivered into the tdLN, tumor growth was suppressed (5/6 mice survived, Fig. 5c–e). However, when OVA was administered to the tdLN, most of the mice (5/6) developed tumors, albeit with a delayed onset of tumor formation. Thus, adjuvant depots localized to LNs draining tumors may create local environments able to generate potent antigen-specific tumor responses, while antigen sources (i.e., contralateral tumors or LNs) localized far from the LNs with adjuvant fails to provide protection. Along these lines, 14 days after vaccination the “Split LN” treatment with PolyIC MP in the tdLN exhibited increased antigen-specific cells in blood with (>5% Tet+), while treatment with OVA in the tdLN exhibited less than 5% Tet+ cells (ESI Fig. 5b†).
To investigate durability of tumor immunity, 13 weeks after the initial inoculation and well past the time of tumor growth and initial immune responses, surviving mice were rechallenged with 100000 B16-OVA cells. No additional vaccinations were performed, a design requiring mice to have established durable tumor-specific responses solely from the prime therapeutic vaccine 98 days earlier. At least 50% of the mice survived and cleared a secondary tumor in each of the treated groups (“Two LN”, “One LN (tdLN)”, “One LN (non-tdLN)”, “Split LN (IC MP-tdLN)”), except for the “Split LN” with OVA in the tdLN (Fig. 5f, and ESI Table 2†). In this group, the only mouse that did not succumb to the initial tumor did not survive a rechallenge. Together, the data from this rechallenge study supports the findings that adjuvant in tdLN ensures both tumor antigen and adjuvant are effectively presented. More broadly, these results indicate that potent, long-lasting anti-tumor responses can be achieved with a single treatment by creating appropriate relative signal geographies.
Co-delivery of antigen and adjuvant to LNs provides tumor protection and increased survival in a lymphoma model
To determine if findings from the melanoma model were generalizable to another pre-clinical model, “One LN”, “Two LN”, and “Split LN” treatments were applied to a lymphoma model in which OVA is constitutively expressed. Mice were treated in a preventative manner as in Fig. 5a with treatments in tdLN and non-tdLN, EG7-OVA cancer cells were implanted 7 days after vaccination, and mice were evaluated daily for tumor growth and via weekly blood draws (up to day 28 post inoculation) for antigen-specific T cell production (Fig. 6a). As with the findings from Fig. 1g and the previous melanoma experiment above, there were heightened antigen-specific T cell levels 7 days after treatment with all vaccine formulations (ESI Fig. 5c†). The “Two LN” or “One LN” treatment in either tdLN or non-tdLNs provided immune cues that generated anti-tumor immunity able to inhibit tumor growth and increase survival (17/18 mice surviving from these 3 groups) (Fig. 6b–d, and ESI Table 3†). However, in this lymphoma model, neither “Split LN” treatment (with PolyIC or OVA in the tdLN) was effective in protecting the mice from cancer with only 3/12 mice surviving in these treated groups (Fig. 6b–d). This suggests that compared to melanoma, even though the antigen-specific cells in circulation were heightened from “Split LN” treatment, these cells were not adequate to combat the lymphoma and provide protection, regardless of whether or not adjuvant was present in the tdLN.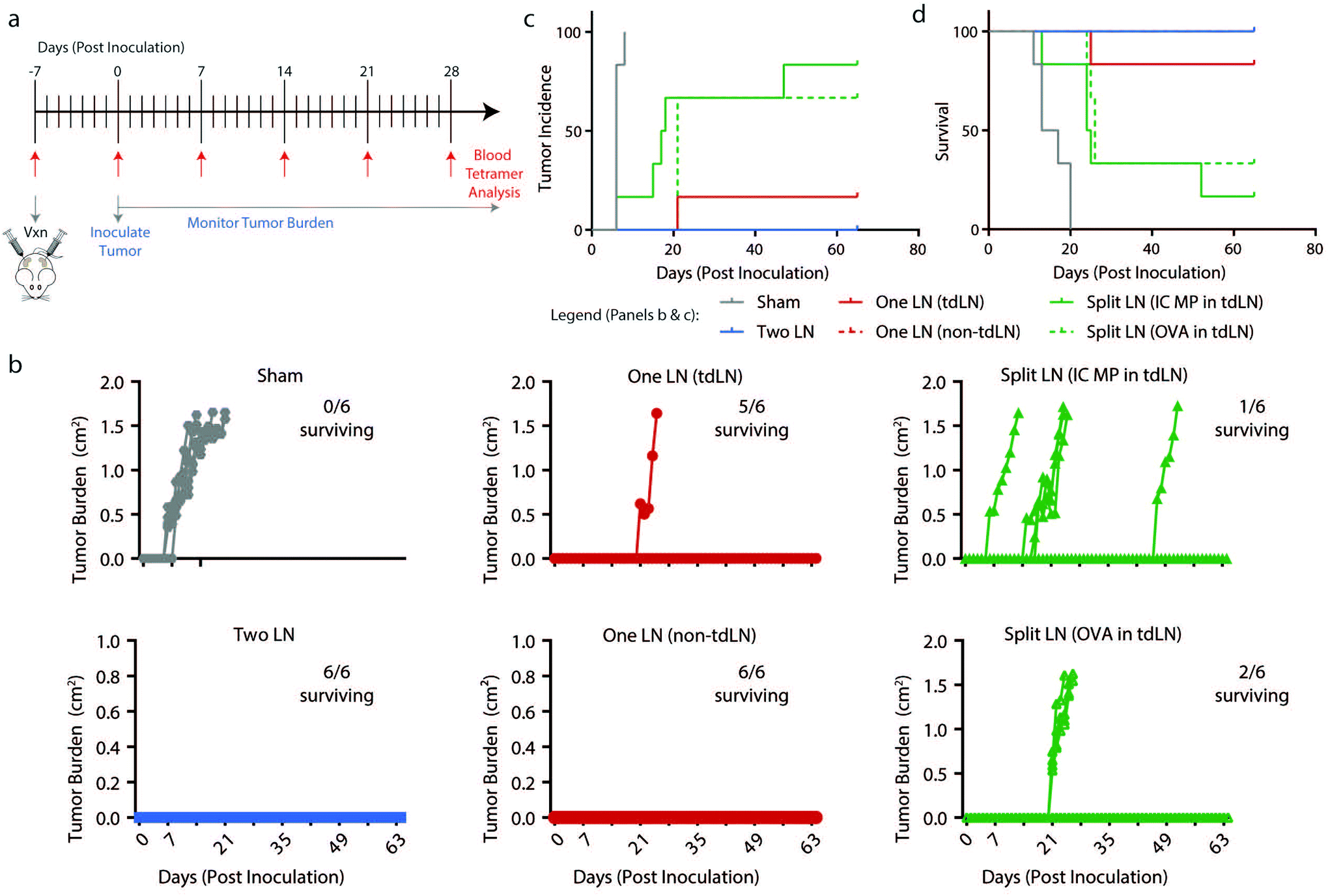 | ||
| Fig. 6 Presence of OVA and PolyIC MP within the same treated LNs decreases tumor burden, incidence, and increases survival in a lymphoma model. (a) Schematic representation of treatment schedule and experimental readouts. (b) Traces of tumor volumes for individual mice with number of mice surviving indicated. (c) Incidence and (d) survival of mice following treatment as in (a). (panels c–f) n = 6 mice per group and errors bars represent SEM. (panel c–d) Statistics indicated in ESI Table 3.† | ||
Conclusions
Prior to this study, i.LN. injection has been used in several therapeutic disease settings.41–43 However, there is tremendous opportunity to combine biomaterials with i.LN. injection as a tool to study immunological function and achieve disruptive therapeutic benefits. In particular, the use of larger, micron size particles deposited in these tissues creates diffusion limitations to spatially restrict signals, which could provide strong immune outcomes, restricting toxic immunomodulatory signals to specific sites. Intra-LN depots also provide some unique opportunities to precisely study the role of therapeutic design parameters, such as relative geography of where signals are deposited and the local signal concentrations, and the impact of these variables on kinetics on immune function. Using fixed total doses of widely studied immune cues – antigen (OVA) and adjuvant (PolyIC), we achieved precision localization of the injected cargo to LNs. This allowed isolation of the role of individual and combined immune signals on local and systemic immunity. When investigating the local, intranodal response to treatment, as expected, uptake of PolyIC MPs by innate cells, in particular DCs and macrophages, led to activation of these cells. Interestingly, regardless of whether receiving the full dose of PolyIC MPs (e.g., “One LN” treatment or “Split LN” treatment) or half the number of particles per LN (e.g., in each LN for “Two LN” treatment), activation levels were comparable. This may suggest a minimum threshold or particle concentration needed to drive tissue-wide activation of APCs. Using i.LN. delivery, titration studies could be performed to establish these minimal concentrations necessary for local APC activation. Likewise, introduction of a lower dose (i.e., 0.5×) into more LNs (i.e., two), drove greater systemic, antigen-specific responses. This may suggest another design rule for immunotherapies: localizing cargo to several LNs at relatively low doses, instead of high doses at a single node. We also observed in the potent regimens of “Two LN” and “One LN” that despite strong systemic response, the local levels of antigen-specific T cells were relatively low. This may reflect migration of primed cells out of these tissues and into the periphery. This highlights another interest area for further study, the kinetics of signal and cell trafficking from treated to untreated LNs. In conducting these studies, there are some important caveats. Of note, introducing polymer depots or scaffolds directly into LNs could influence stromal aspects of this local tissue environment – such as laminins involved in organizing microdomains important for immunity and tolerance.56–58 There are also physiochemical aspects to consider. For example, while PLGA is a widely used polymer with known degradation profiles, PLGA degradation results in lactic and glycolic acid. These products, which could alter local pH, might influence local cell or signaling processes, though we have not observed injection or carrier-induced effects. Taken together, combining biomaterial depots with local LN delivery creates potent immunotherapeutic opportunities, as well as to probe immune function. For example, by coupling these studies with immunological tools such as T cell receptor transgenic mice to study migration as a function of signal concentration and location. Finally, exploiting the unique capabilities of biomaterials to study immune function could provide significant insight relevant to a variety of vaccination and immunotherapy applications aimed at LN targeting, irrespective of the injection route.Author contributions
J.I.A. and C.M.J. conceptualized and formulated experimental design for the study. J.I.A., X.Z., S.J.T., J.M.G., S.T.C., C.E., S.A.S, E.A.G., and K.L.H. conducting the experimental procedures. X.Z., S.J.T., J.M.G., E.A.G., and K.L.H. assisted J.I.A. with flow cytometry studies. J.I.A., S.J.T., and S.T.C. prepared tissue for immunofluorescent staining, IVIS imaging, and conducted imaging. J.I.A., S.J.T., and S.T.C contributed to the analysis of the results. J.I.A., S.J.T., J.M.G., S.T.C., and C.M.J. contributed to the interpretation of results. J.I.A. and C.M.J. wrote the manuscript with input from all co-authors.Conflicts of interest
C.M.J. is an employee of the VA Maryland Health Care System. The views reported in this paper do not reflect the views of the Department of Veterans Affairs or the United States Government. C.M.J. has an equity position with Avidea Technologies. J.I.A. and C.M.J. are inventors on U.S. Patent No. 9610349. Issued April 4, 2017.Acknowledgements
This work was supported in part by NIH #R01EB026896, NSF CAREER Award #1351688, the Alliance for Cancer Gene Therapy #15051543, the Damon Runyon Foundation # DRR3415, and the Melanoma Research Alliance #348963. J. I. A. was supported in part as a Graduate Fellow of the American Association of Pharmaceutical Scientists Foundation and the University of Maryland Dean's Dissertation Fellowship. S. J. T. is a trainee of the NIH T32 Host–Pathogen Interaction Fellowship (# AI089621). J. M. G. was supported in part as a PhRMA Foundation Doctoral Fellow (Pharmaceutics Division). K. L. H. was a SMART Graduate Fellow funded by ASD/R&E, Defense – Wide/PE0601120D8Z National Defense Education Program (NDEP)/BA-1, Basic Research.References
- D. L. Porter, B. L. Levine, M. Kalos, A. Bagg and C. H. June, N. Engl. J. Med., 2011, 365, 725–733 CrossRef CAS PubMed.
- S. L. Maude, N. Frey, P. A. Shaw, R. Aplenc, D. M. Barrett, N. J. Bunin, A. Chew, V. E. Gonzalez, Z. Zheng, S. F. Lacey, Y. D. Mahnke, J. J. Melenhorst, S. R. Rheingold, A. Shen, D. T. Teachey, B. L. Levine, C. H. June, D. L. Porter and S. A. Grupp, N. Engl. J. Med., 2014, 371, 1507–1517 CrossRef PubMed.
- M. Klichinsky, M. Ruella, O. Shestova, X. M. Lu, A. Best, M. Zeeman, M. Schmierer, K. Gabrusiewicz, N. R. Anderson, N. E. Petty, K. D. Cummins, F. Shen, X. Shan, K. Veliz, K. Blouch, Y. Yashiro-Ohtani, S. S. Kenderian, M. Y. Kim, R. S. O’Connor, S. R. Wallace, M. S. Kozlowski, D. M. Marchione, M. Shestov, B. A. Garcia, C. H. June and S. Gill, Nat. Biotechnol., 2020, 38, 947–953 CrossRef CAS PubMed.
- J. Jou, K. J. Harrington, M. B. Zocca, E. Ehrnrooth and E. E. W. Cohen, Clin. Cancer Res., 2021, 27, 689–703 CrossRef CAS PubMed.
- A. K. Singh and J. P. McGuirk, Lancet Oncol., 2020, 21, e168–e178 CrossRef CAS PubMed.
- D. T. O'Hagan and C. B. Fox, Vaccine, 2015, 33, B14–B20 CrossRef PubMed.
- T. J. Moyer, A. C. Zmolek and D. J. Irvine, J. Clin. Invest., 2016, 126, 799–808 CrossRef PubMed.
- A. S. Cheung and D. J. Mooney, Nano Today, 2015, 10, 511–531 CrossRef CAS PubMed.
- C. J. H. Davitt and E. C. Lavelle, Adv. Drug Delivery Rev., 2015, 91, 52–69 CrossRef CAS PubMed.
- R. M. Zinkernagel, S. Ehl, P. Aichele, S. Oehen, T. Kündig and H. Hengartner, Immunol. Rev., 1997, 156, 199–209 CrossRef CAS PubMed.
- A. F. Ochsenbein, S. Sierro, B. Odermatt, M. Pericin, U. Karrer, J. Hermans, S. Hemmi, H. Hengartner and R. M. Zinkernagel, Nature, 2001, 411, 1058–1064 CrossRef CAS PubMed.
- E. Blass and P. A. Ott, Nat. Rev. Clin Oncol., 2021, 18, 215–229 CrossRef PubMed.
- J. I. Andorko and C. M. Jewell, Bioeng. Transl. Med., 2017, 2, 139–155 CrossRef PubMed.
- E. A. Gosselin, H. B. Eppler, J. S. Bromberg and C. M. Jewell, Nat. Mater., 2018, 17, 484–498 CrossRef CAS PubMed.
- H. B. Eppler and C. M. Jewell, Adv. Mater., 2020, 32, 26 CrossRef PubMed.
- A. J. Najibi and D. J. Mooney, Adv. Drug Delivery Rev., 2020, 161, 42–62 CrossRef PubMed.
- M. P. Manspeaker and S. N. Thomas, Adv. Drug Delivery Rev., 2020, 160, 19–35 CrossRef CAS PubMed.
- N. Gong, N. C. Sheppard, M. M. Billingsley, C. H. June and M. J. Mitchell, Nat. Nanotechnol., 2021, 16, 25–36 CrossRef CAS PubMed.
- F. S. Hodi, ClinicalTrials.org, 2017, https://clinicaltrials.gov/show/nct01753089.
- H. Liu, K. D. Moynihan, Y. Zheng, G. L. Szeto, A. V. Li, B. Huang, D. S. Van Egeren, C. Park and D. J. Irvine, Nature, 2014, 507, 519–522 CrossRef CAS PubMed.
- O. A. Ali, N. Huebsch, L. Cao, G. Dranoff and D. J. Mooney, Nat. Mater., 2009, 8, 151–158 CrossRef CAS PubMed.
- S. A. Bencherif, R. Warren Sands, O. A. Ali, W. A. Li, S. A. Lewin, T. M. Braschler, T. Y. Shih, C. S. Verbeke, D. Bhatta, G. Dranoff and D. J. Mooney, Nat. Commun., 2015, 6, 7556 CrossRef CAS PubMed.
- R. Kuai, L. J. Ochyl, K. S. Bahjat, A. Schwendeman and J. J. Moon, Nat. Mater., 2017, 16, 489–496 CrossRef CAS PubMed.
- D. J. Irvine and E. L. Dane, Nat. Rev. Immunol., 2020, 20, 321–334 CrossRef CAS PubMed.
- M. J. O’Melia, N. A. Rohner, M. P. Manspeaker, D. M. Francis, H. T. Kissick and S. N. Thomas, Sci. Adv., 2020, 6, 1–19 Search PubMed.
- R. S. Riley, C. H. June, R. Langer and M. J. Mitchell, Nat. Rev. Drug Discovery, 2019, 18, 175–196 CrossRef CAS PubMed.
- X. Duan, C. Chan and W. Lin, Angew. Chem., Int. Ed., 2019, 58, 670–680 CrossRef CAS PubMed.
- M. S. Goldberg, Nat. Rev. Cancer, 2019, 19, 587–602 CrossRef CAS PubMed.
- D. M. Francis, M. P. Manspeaker, A. Schudel, L. F. Sestito, M. J. O’Melia, H. T. Kissick, B. P. Pollack, E. K. Waller and S. N. Thomas, Sci. Transl. Med., 2020, 12, 563 Search PubMed.
- J. Kim, L. F. Sestito, S. Im, W. J. Kim and S. N. Thomas, Adv. Funct. Mater., 2020, 30, 1908788 CrossRef CAS PubMed.
- M. A. Aznar, L. Planelles, M. Perez-Olivares, C. Molina, S. Garasa, I. Etxeberría, G. Perez, I. Rodriguez, E. Bolaños, P. Lopez-Casas, M. E. Rodriguez-Ruiz, J. L. Perez-Gracia, I. Marquez-Rodas, A. Teijeira, M. Quintero and I. Melero, J. Immunother. Cancer, 2019, 7, 116 CrossRef PubMed.
- J. Xu, Q. Ma, Y. Zhang, Z. Fei, Y. Sun, Q. Fan, B. Liu, J. Bai, Y. Yu, J. Chu, J. Chen and C. Wang, Nat. Commun., 2022, 13, 110 CrossRef CAS PubMed.
- D. Weinfeld, U. Westin, L. Hellkvist, U.-H. Mellqvist, I. Jacobsson and L.-O. Cardell, Allergy, Asthma, Clin. Immunol., 2020, 16, 31 CrossRef CAS PubMed.
- S. H. Skaarup, J. M. Schmid, T. Skjold, O. Graumann and H. J. Hoffmann, J. Allergy Clin. Immunol., 2020, 147(3), 1011–1019 CrossRef PubMed.
- A. Ribas, J. S. Weber, B. Chmielowski, B. Comin-Anduix, D. Lu, M. Douek, N. Ragavendra, S. Raman, E. Seja, D. Rosario, S. Miles, D. C. Diamond, Z. Qiu, M. Obrocea and A. Bot, Clin. Cancer Res., 2011, 17, 2987–2996 CrossRef CAS PubMed.
- M. Adamina, R. Rosenthal, W. P. Weber, D. M. Frey, C. T. Viehl, M. Bolli, R. W. Huegli, A. L. Jacob, M. Heberer, D. Oertli, W. Marti, G. C. Spagnoli and P. Zajac, Mol. Ther., 2010, 18, 651–659 CrossRef CAS PubMed.
- G. Senti, B. M. Prinz Vavricka, I. Erdmann, M. I. Diaz, R. Markus, S. J. McCormack, J. J. Simard, B. Wüthrich, R. Crameri, N. Graf, P. Johansen and T. M. Kündig, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 17908–17912 CrossRef CAS PubMed.
- G. Senti, R. Crameri, D. Kuster, P. Johansen, J. M. Martinez-Gomez, N. Graf, M. Steiner, L. A. Hothorn, H. Grönlund, C. Tivig, A. Zaleska, O. Soyer, M. van Hage, C. A. Akdis, M. Akdis, H. Rose and T. M. Kündig, J. Allergy Clin. Immunol., 2012, 129, 1290–1296 CrossRef CAS PubMed.
- Y. Waeckerle-Men, N. Bruffaerts, Y. Liang, F. Jurion, P. Sander, T. M. Kündig, K. Huygen and P. Johansen, Vaccine, 2013, 31, 1057–1064 CrossRef CAS PubMed.
- P. Johansen, A. C. Haffner, E. Koch, K. Zepter, L. Erdmann, K. Maloy, J. J. Simard, T. Storni, G. Senti, A. Bot, B. Wuthrich and T. M. Kundig, Eur. J. Immunol., 2005, 35, 568–574 CrossRef CAS PubMed.
- J. I. Andorko, K. L. Hess and C. M. Jewell, AAPS J., 2015, 17, 323–338 CrossRef CAS PubMed.
- J. M. Gammon, L. H. Tostanoski, A. R. Adapa, Y. C. Chiu and C. M. Jewell, J. Controlled Release, 2015, 210, 169–178 CrossRef CAS PubMed.
- L. H. Tostanoski, Y. C. Chiu, J. M. Gammon, T. Simon, J. I. Andorko, J. S. Bromberg and C. M. Jewell, Cell Rep., 2016, 16, 2940–2952 CrossRef CAS PubMed.
- E. D. N. S. Abeyrathne, H. Y. Lee and D. U. Ahn, Poult. Sci., 2013, 92, 3292–3299 CrossRef CAS PubMed.
- M. Caskey, F. Lefebvre, A. Filali-Mouhim, M. J. Cameron, J.-P. Goulet, E. K. Haddad, G. Breton, C. Trumpfheller, S. Pollak, I. Shimeliovich, A. Duque-Alarcon, L. Pan, A. Nelkenbaum, A. M. Salazar, S. J. Schlesinger, R. M. Steinman and R. P. Sékaly, J. Exp. Med., 2011, 208, 2357–2366 CrossRef CAS PubMed.
- L. R. V. Antonelli, A. Gigliotti Rothfuchs, R. Gonçalves, E. Roffê, A. W. Cheever, A. Bafica, A. M. Salazar, C. G. Feng and A. Sher, J. Clin. Invest., 2010, 120, 1674–1682 CrossRef CAS PubMed.
- C. M. Jewell, S. C. B. Lopez and D. J. Irvine, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 15745–15750 CrossRef CAS PubMed.
- L. B. Kennedy and A. K. S. Salama, CA Cancer J. Clin., 2020, 70, 86–104 CrossRef PubMed.
- O. P. Joffre, E. Segura, A. Savina and S. Amigorena, Nat. Rev. Immunol., 2012, 12, 557–569 CrossRef CAS PubMed.
- V. Brinkmann, J. G. Cyster and T. Hla, Am. J. Transplant., 2004, 4, 1019–1025 CrossRef CAS PubMed.
- D. J. Fowell and M. Kim, Nat. Rev. Immunol., 2021, 21, 582–596 CrossRef CAS PubMed.
- V. Saxena, L. Li, C. Paluskievicz, V. Kasinath, A. Bean, R. Abdi, C. M. Jewell and J. S. Bromberg, Immunol. Rev., 2019, 292, 9–23 CrossRef CAS PubMed.
- D. Masopust and J. M. Schenkel, Nat. Rev. Immunol., 2013, 13, 309–320 CrossRef CAS PubMed.
- R. Förster, A. C. Davalos-Misslitz and A. Rot, Nat. Rev. Immunol., 2008, 8, 362–371 CrossRef PubMed.
- M. Oppermann, Cell. Signalling, 2004, 16, 1201–1210 CrossRef CAS PubMed.
- K. J. Warren, D. Iwami, D. G. Harris, J. S. Bromberg and B. E. Burrell, J. Clin. Invest., 2014, 124, 2204–2218 CrossRef CAS PubMed.
- T. Simon and J. S. Bromberg, Trends Immunol., 2017, 38, 858–871 CrossRef CAS PubMed.
- L. Li, M. W. Shirkey, T. Zhang, Y. Xiong, W. Piao, V. Saxena, C. Paluskievicz, Y. Lee, N. Toney, B. M. Cerel, Q. Li, T. Simon, K. D. Smith, K. L. Hippen, B. R. Blazar, R. Abdi and J. S. Bromberg, J. Clin. Invest., 2020, 130, 2602–2619 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2bm00403h |
| This journal is © The Royal Society of Chemistry 2022 |