
Multi-resistive pulse sensor microfluidic device†
Marcus
Pollard
,
Rushabh
Maugi
and
Mark
Platt
 *
*
School of Science, Loughborough University, Epinal Way, LE11 3TU, UK. E-mail: m.platt@lboro.ac.uk
First published on 25th February 2022
Abstract
Resistive pulse sensors have been used to characterise everything from whole cells to small molecules. Their integration into microfluidic devices has simplified sample handling whilst increasing throughput. Typically, these devices measure a limited size range, making them prone to blockages in complex sample matrixes. To prolong their life and facilitate their use, samples are often filtered or prepared to match the sample with the sensor diameter. Here, we advance our tuneable flow resistive pulse sensor which utilises additively manufactured parts. The sensor allows parts to be easily changed, washed and cleaned, its simplicity and versatility allow components from existing nanopore fabrication techniques such as glass pipettes to be integrated into a single device. This creates a multi-nanopore sensor that can simultaneously measure particles from 0.1 to 30 μm in diameter. The orientation and controlled fluid flow in the device allow the sensors to be placed in series, whereby smaller particles can be measured in the presence of larger ones without the risk of being blocked. We illustrate the concept of a multi-pore flow resistive pulse sensor, by combining an additively manufactured tuneable sensor, termed sensor 1, with a fixed nanopore sensor, termed sensor 2. Sensor 1 measures particles as small as 10 μm in diameter, whilst sensor 2 can be used to characterise particles as small as 100 nm, depending upon its dimensions. We illustrate the dual pore sensor by measuring 1 and 10 μm particles simultaneously.
Introduction
From its early applications in cell counting in the 1950′s, the Coulter Counter technology continues to be advanced and improved. Increasingly known as resistive pulse sensing/sensors (RPS), devices based on this principle have now been created using materials ranging from graphene,1–3 to polymers,4,5 silicon nitride6 and glass.7–11 The sensing process is simple, by monitoring the temporary changes in current caused by the translocation of an analyte through a narrow constriction, termed a sensing region or pore, RPS can characterise analytes according to size,12 concentration,13 shape14–17 and charge.18 The transport of an analyte through the pore is controlled by tuning the applied electric field, charge on the pore wall, electrophoretic mobility of the analyte, supporting electrolyte concentration and induced convection.19,20 To maintain sensitivity the pore size must be of comparable dimensions to the analyte.5 Therefore, whilst a smaller sensing region can detect smaller analytes, the drawback is an increased likelihood of blockages, and a decreased translocation frequency.Recent advances in the RPS sensors have utilised pores of different aspect ratios/shapes or specific electrode configurations to allow larger pores and sensing zones to be utilised.21–24 When integrated into microfluidic systems this technology is further advanced,25–29 allowing users to combine sample handling within the analytical system. A variety of innovations in this area, summarised in reviews elsewhere,30,31 have focused on different forms of fabrication, convection and applications. For example, electrophoresis can be used to drive the analyte through the pore. One example uses an applied electric field of 60 V to drive the sample flow for the detection of bacteria.32 Similar designs have coupled syringe pumps with lower voltages for the detection of yeast cells,28 polystyrene particles,21,33 DNA,33 red blood cells25 and algae.34 Another advantage of controlling convection and the design of the sensing zone is the ability to force the analyte through a series of RPS sensing regions. Zhang et al.25 presented such a device where five identical sensing regions were arranged in series. As the analyte travelled through the device it generated five pulses, allowing each analyte to be measured multiple times, whilst providing a regular signal pattern to be identified against random background fluctuations. The authors suggest that such a device could improve the signal-to-noise ratio by a factor of n1/2, where n is the number of sensing regions.
An emerging fabrication process for microfluidic systems is additive manufacturing (AM) or 3D printing.35–37 In the field of microfluidics the use of 3D printing and lab-on-a-chip fabrication has increased.35,37–39 Examples of RPS components integrated into AM are limited. This is likely due to the print resolution, although the use of two-photon-polymerisation (2PP) has now enabled some novel nanopore designs.40 We have previously used surface channels within AM devices, sealed with a polymer acetate film to overcome the resolution of AM.41 In a subsequent design, we extended the sensing dynamic range down to 5 μm using a re-sealable lid and tuneable PDMS gasket layer.15 However, this was insufficient to study nanomaterials.
To show how an AM-RPS device can be improved to cover a wide particle size range, here we present a beyond-state-of-the-art AM-RPS sensor design with advanced functionality by creating a system of fully integrated AM components. The motivation behind this work was to extend the detection range of the device, whilst making it easier to fabricate, reuse and be less prone to blockages. The final device creates a series of RPS sensors that can be independently tuned to an analyte size range of interest. We combine the additively manufactured tuneable sensor, termed sensor 1, with a fixed nanopore sensor, termed sensor 2 (see Fig. 1). The only restriction in design criteria is that sensor 1 must be tuned to a size range equal to or larger than sensor 2. We adhere to this restriction by using sensor 1 to measure particles >10 μm in size, whilst sensor 2 characterises particles as small as 100 nm. In the configurations shown in Fig. 1, the liquid always flows through sensor 1 and then, through and around sensor 2 (Fig. 1a). For nanomaterials to enter sensor 2, they must traverse the opening via a combination of electrophoresis (EP), osmosis (EO) or convection (Fig. 1b). These novel configurations facilitate the measurement of the broadest size range to date on a single device and represent the first integration of solid state nanopores into an AM-RPS. By controlling the fluid flow in the device several RPS sensors can be placed in series, whereby smaller particles can be measured in the presence or larger ones decreasing the risk of the pores being blocked.
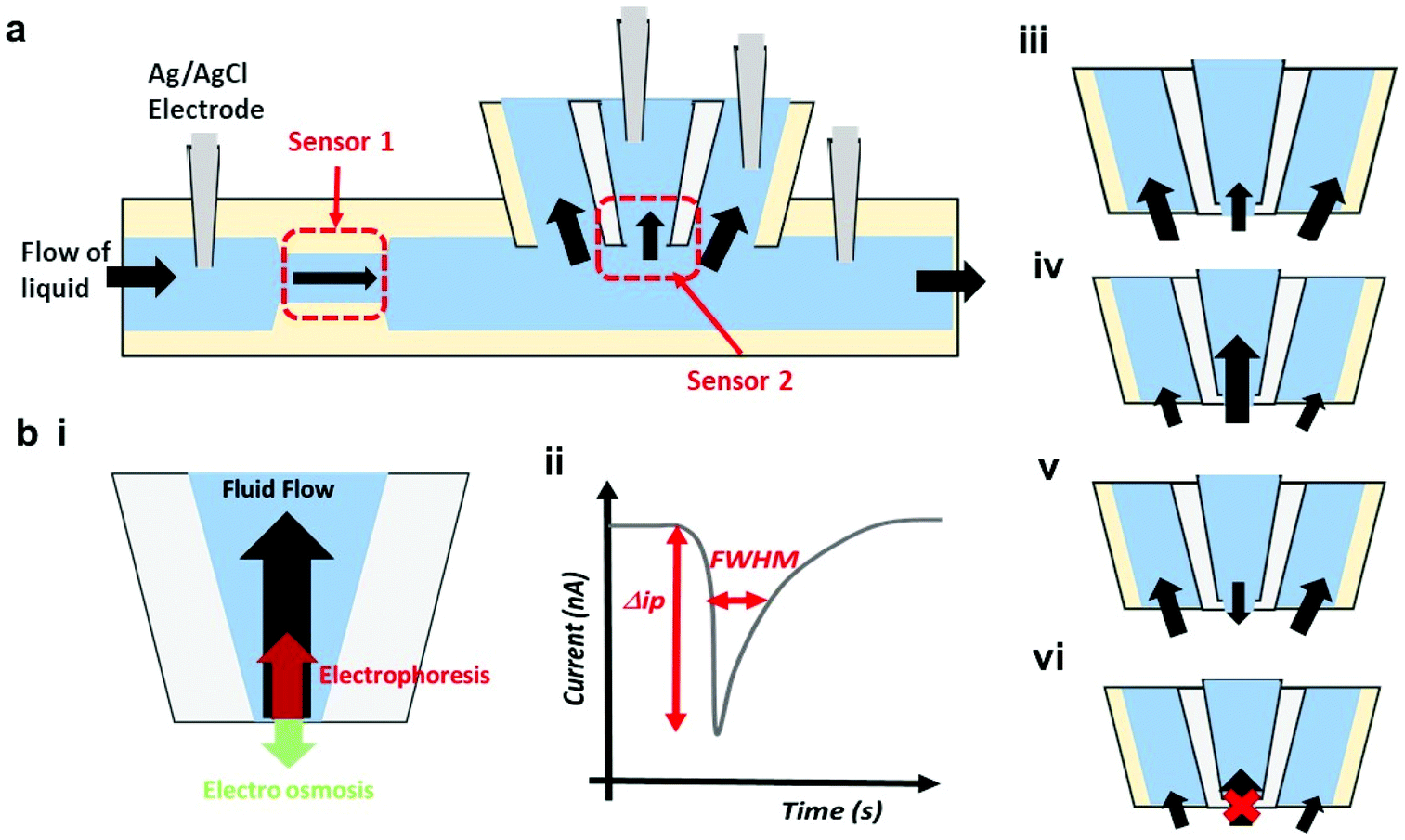 | ||
| Fig. 1 (a) Schematic of the dual pore setup showing the location of the sensors, electrodes and fluid flow. (b) Illustration of the forces acting upon the particles through sensor 2 (i), schematic of a blockade event or pulse in sensor 1 and 2, the pulse depth Δip is proportional the to the analyte volume, and the full width half maximum (FWHM) to the speed of the analyte through the sensing zone (ii). The black arrows in (iii)–(v) represent fluid flow where, by controlling the flow ratios, the flow through sensor 2 can be stopped (vi). | ||
Experimental methods
Chemicals and materials
Particles 100 nm – 2 μm were obtained from Izon Science Ltd, CPC100 (mean diameter 110 nm), CPC200 (mean diameter 200 nm) CPC2000, 2 micron carboxylated polystyrene calibration particles, 10 micron and 20 micron carboxlyated polystyrene calibration particles denoted as CP10M and CP20M were obtained from Izon Science Ltd 30 μm carboxylated polystyrene particles, Cat. No. 84135, ethanol >99.8%, acetone >97%, sulphuric acid 95–97% and 30% (w/w) perchloric acid were obtained from Sigma-Aldrich, potassium chloride obtained from Fisher Scientific UK, >99% cat no: P/4240/60, Acc Silicones QSil216 was obtained from RS components, catalogue no: 458–765, part no: QSil 216. Custom Faraday cages were purchased from Nanopore Solutions (nanoporesolutions.com), glass nanopore membranes (GNMs) from Electronic BioSciences USA and glass pipettes from World Precision Instruments (WPI) Europe. Silver wire >99.9% cat AG5487 was obtained from advent-rm. Dolomite mitos-P Pumps were used to supply the flow.Current amplifiers
Current amplifiers were supplied from Elements-ic, or were removed and repurposed from a Izon Qnano. Sampling frequencies were 50–60 kHz with a 5 kHz bandpass filter. Data analysis was performed within the data analysis module of clampfit. The two amplifiers were housed in Faraday cages, and controlled independently through the software supplied from the manufacturers.Device assembly
In order to assemble the device, the lid was either affixed to the base via 6 machine screws or using a clip (Fig. 2 and S1†). HPLC fittings were inserted into each screw thread to accommodate the inlet from the pumps, electrodes and the outlet. Once fully assembled, the device was placed into a custom-made Faraday cage and electrolyte solution was pumped into the device. The PDMS was poured onto the side of the lid with the ridge, such that the PDMS covered the entire lid surface and was <1 mm in thickness. Reference electrodes were made by wrapping silver wire around the tube before being thread through the screw thread. The wire and tube were then affixed in place using Araldite Adhesive. Flow rates were monitored by collecting the liquid from the outlets and weighing the solution over a period of time. For simplicity, the Ag/AgCl reference electrodes were incorporated into a HPLC style and sized, screw thread (Fig. 2a). Alongside the Ag wire, a tube was inserted into the screw thread to allow any trapped air bubbles to be removed during filling, and was usually sealed during operation, i.e., no flow was observed out of the reference electrode port during sensing. To simplify the number of parts and designs, the same sized screw thread was used to insert other parts and components. As shown in Fig. 2 (top right image), the screw thread was used for the inlet, outlets, reference electrodes and subsequent RPS materials. | ||
| Fig. 2 (a) The HPLC screw thread and mounted glass pipette and exit tube (i). Schematic of the different components – Inlet/outlet (ii), Ag/AgCl reference electrode (iii), and glass pipette and GNM holder with exit tube (iv). Illustrative example of all components in an assembled device. (b) Example base unit with screw ports (i), reverse side of base unit showing sensor 1 (ii), and with an assembled device and holder (iii). | ||
Device printing
The lid, screw threads and base of the device were printed using an Asiga Pico HD27 UV 3D printer with FORMlabs clear resin. Design files were converted from the CAD software (Siemens NX11) to STL and prepared for printing using Asiga Composer software. Once printed, the parts were cleaned and post cured using a UV light box.Polydimethylsiloxane (PDMS) gasket
The PDMS gaskets were formed by mixing parts A and B of QSil 216 clear liquid silicone in a 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio. The lid was placed into a Petri dish with the ridge oriented to the bottom. The uncured PDMS was then poured around the edge of the lid making sure that the whole lid was covered up to the ridge and no air pockets remained. The PDMS was then cured for 1 h at 70 °C or until set.
:1 ratio. The lid was placed into a Petri dish with the ridge oriented to the bottom. The uncured PDMS was then poured around the edge of the lid making sure that the whole lid was covered up to the ridge and no air pockets remained. The PDMS was then cured for 1 h at 70 °C or until set.
Optical imaging
Microscope images were captured using a Nixon Optiphot 2 optical microscope and optical images were captured using a DS 5 M camera with a DS-L1 camera control unit.Electrode fabrication
Electrodes were fabricated by inserting a section of 0.25 mm diameter silver wire (99.99% purity, Advent Research Materials) into a pipette tip. A small section of the wire was threaded through the narrow end of the pipette, glued in place using Araldite® Rapid epoxy resin and then left to dry.Results and discussion
The concept is built upon our previously published AM tuneable flow device15 as a core, referred to here as the “fluidic chip”. Exploiting the fluidic chip as the central part to build upon allowed other electrodes and RPS sensors to be added (Fig. 2). To facilitate ease of use, a custom holder for the device was created replacing the 6 screws that previously held the lid and base together, as shown in Fig. 2biii and S1.†Sensor 1
The base and lid had the same design as reported previously.15 Briefly, the fluidic chip was made of a base unit and lid, that when placed together created a RPS sensing zone, termed sensor 1. The base contained a surface channel and using this method, commercial stereolithography (SLA) printers and resins reproducibility produce channels ≥100 μm.41,42 Optical images of the channel are shown in Fig. 2bii. A PDMS gasket between the lid and base sealed the components preventing any leakage. The PDMS layer was typically 1 mm in thickness, the shape or dimensions (internal volume) of the sensing zone were controlled by compressing the PDMS layer, into the channel (Fig. S1c†).15 The screw in the back of the cell holder was created to tune the internal volume of sensor 1. It was hypothesised that turning a single screw on the back of the holder, would increased the force on the lid, compressing the PDMS into the channel (Fig. S1c†).To test the flow cell holder would seal the device in the first instance, the parts were assembled and 30 μm particles were flowed through. Fig. 3 shows the distribution of pulse magnitudes under three different screw tensions. Upon tightening the screw in the back of the holder, the blockage magnitude of the same sized particles increased.
 | ||
| Fig. 3 (a) Distribution of pulse magnitudes from 30 μm particles passing through sensor 1, under different screw tensions. (b) Example current trace from sensor 1. Particles at 3 × 105 particles per mL, 0.6V, 50 mM KCl. | ||
Sensor 2
To expand the sensing range of the device to smaller particles, an additional RPS sensor was needed, termed sensor 2. Within the literature, a range of materials have been tested for RPS applications, and some of the most common solid-state materials are SiN and SiO2.43 In keeping with our philosophy to create an easy to fabricate and assemble device, we opted to first create pores using pulled glass capillaries. A pipette was mounted into the screw thread shown in Fig. 2a. Alongside the pipettes we mounted an additional exit tube to allow the main liquid flow to exit out of the screw thread next to the pipette. In this setup the last port termed outlet in Fig. 4a was sealed closed. The samples during operation then passed through sensors 1 and out via the glass pipette screw thread. This enabled the samples to be drawn over the glass RPS sensing zone despite the recess from the main channel. This resulted in the liquid following out of the pipette and outlet flow simultaneously. | ||
| Fig. 4 (a) Schematic of the fluid chip and its components, connected to the outlet of sensor 2 was a syringe, where flow through the pipette could be enhanced (PLUS) or inhibited. (bi) Blockade magnitude distributions of 1μm particles passing through a 5 μm pore 1.5 V at 1 × 107 particles per mL, insert is a optical microscope image of the pipette taken before mounting into the screw thread. (bii) Example current trace from data in bi. | ||
Fig. 4 shows the schematic of the dual pore sensor, alongside example blockade distributions, and current traces for a 5 μm pipette, with 1 μm diameter particles in solution. Using a pipette of a different diameter allowed the sensing range of sensor 2 to be varied, by decreasing the pore diameter allows smaller particles to be detected, Fig. S2† illustrates this via the detection of 100 nm diameter particles. Whilst easy to make, the pulled pipettes have a thin diameter wall making them brittle and easy to break.
An alternative material is the glass nanopore membrane, GNM.44–46 The GNM's has a thicker wall diameter making them more robust to handle. This allowed the ends of the GNM's to be connected to a syringe that allowed liquid to be pulled from the main fluidic chamber through the RPS material, or vice versa, Fig. 4.
Fig. 5 shows the particle count rate for a 990 nm diameter GNM sensor 2 with 300 nm diameter particles in the sample liquid. Translocation events were observed instantly as the sample was flowed through the device. Where the flow rate through the main channel increased, the translocation frequency also increased, Fig. 5a. Halting the flow, resulted in a decrease in translocation events, but did not result in a loss of the signal altogether as the particles can move through the RPS sensors via a combination of EO and EP, grey line Fig. 5ai. This suggests the device could also be used as an injection loop to measure samples in the absence of flow. The velocity through the GNM, measured by the full width at half maximum (FWHM), also illustrates the effect of the flowing solution. In the absence of a flow, the particles traverse the sensor relatively slowly with a FWHM of 0.5 ms. Upon the addition of the flow, the FWHM decreases to 0.12 ms. The velocity does not decrease further as the flow rate goes from 0.5 to 0.75 mL min−1. The decrease in FWHM suggests the presence of some additional convection through the pore. The pulse magnitude decreases from 0.26 nA to 0.21 nA as the flow rate through the main channel increases, the cause for this is not clear, Fig. 5bii.
 | ||
| Fig. 5 (a) 990 nm GNM sensor 2 using 300 nm diameter particles, 1 × 109 particles per mL. 1.68V. Flow rates are measured at the outlet of sensor 1. (i) Translocation frequency. (ii) FWHM values, (iii) blockade magnitude histograms. (b) Translocation frequency of using 300 nm diameter particles through a 990 nm GNM sensor 2, 1 × 109 particles per mL. 1.68 V in the absence (Nano) and presence (Nano + Micro) of 10 μm particles 1 × 107 particles per mL. | ||
As the device works by drawing the sample across the RPS orifice and through the exit tube, akin to the reverse of a flow focusing chamber in flow cytometry, it was hypothesised that alongside the enhanced particle counting rate, the additional flow may help wash/clean and unblock the pore mouth if larger aggregates were present in solution. To test this hypothesis, a GNM was placed into the fluidic chamber, the solution filled with 300 nm particles, and the pulse frequencies were measured (Fig. 5c). Into this solution, 10 μm particles were also placed at a concentration of 1 × 107 particles per mL. The presence of the larger particles had no effect on ease, speed and consistency of particle translocations (Fig. 5b). The presence of the larger particle did not inhibit or change the measurement of the smaller ones.
In theory, the addition of a syringe to the back of sensor 2 allows the user to increase the fluid flow through the GNM, by pulling the syringe out, or prevent convection through the pore, by pushing the syringe down. The flow rate through the centre of the pipette was difficult to measure, given its small volume, and the flow rate ratios are not known here. It may be possible to control this further in more advanced systems and is the focus of future work. However, as the ratio of flow was changed to allow more liquid through the GMN, i.e., the syringe was pulled to draw liquid through the sensor, the pulse frequency also increased (Fig. 6). Conversely, when the liquid flow was reversed back from the GNM into the main channel, no translocation events were observed (no data available to plot in Fig. 6). As a comparison, the blockade size, frequency and translocation speed of the particles in the absence of any additional flow from the syringe are also shown in Fig. 6.
 | ||
| Fig. 6 (a) Distribution of blockade height and (b) translocation frequencies, (c) FWHM values using a 780 nm pore size in a GNM, 1.9 V in 50 mM KCl and a particle concentration of 200 nm particles 1 × 109 particles per mL. To create a PLUS liquid flow through the RPS, a syringe was withdrawn to value of 3 mL and held in a fixed position until after the experiments. To inhibit the flow through the RPS a syringe was depressed by a value of 1 mL. | ||
When two particle size populations, i.e. 10 and 1 μm, are mixed and present in solution at the same time, both particle sizes could be counted in the same device simultaneously using sensors 1 and 2. Fig. 7 shows the current trace and blockage magnitude distributions of the two sensors running simultaneously.
 | ||
| Fig. 7 (a) Current time trace of sensor 1 0.3V, (b) Current time trace of sensor 2, 5 μm diameter glass pipette 0.8V, (c) distribution of blockade heights from sensor 1 and 2. 10 μm particles at 1 × 103 particles per mL, 1 μm particles at 1 × 106 particles per mL. Flow rate of 0.5mL min−1 through sensor 1. | ||
The setup and process was tested across multiple runs, multiple assemblies over a numbers of days with several GNM. The data is tabulated in Table S1.† This ability to measure a wide range of particle sizes, or the properties of smaller ones without the need for sample preparation and filtration, may have many applications in biological and physical sciences.
Conclusions
In this article, we create a tuneable flow resistive pulse sensor which utilises additively manufactured parts. The sensor allows parts to be easily changed, washed and cleaned, its simplicity and versatility allow components from existing nanopore fabrication to be integrated into a single device. This creates a multi-nanopore sensor that can simultaneously measure particles an order of magnitude apart in diameter. The orientation and controlled fluid flow in the device allows the sensors to be placed in series, whereby smaller 200 nm particles can be measured in the presence of 10 μm particles without the risk of being blocked. Using GNM's further enhanced the ease of the device as they were robust and could be easily cleaned via sonication and reused. This allowed the same GNM to be used many times. The device here could greatly enhance the detection of microorganisms, characterise biological and inorganic nanomaterials with little or no sample preparation, and offers a platform that can be added to or run in series with, other RPS fluidic chips or complimentary analytical technologies.Conflicts of interest
The authors confirm no conflicts of interest.Acknowledgements
The authors would like to thanks Dr Micheal Scanlon, and Dr Angelika Holzinger for their advice and help during the work.References
- S. Nam, I. Choi, C. Fu, K. Kim, S. Hong, Y. Choi, A. Zettl and L. P. Lee, Nano Lett., 2014, 14, 5584–5589 CrossRef CAS PubMed.
- R. R. Henriquez, T. Ito, L. Sun and R. M. Crooks, Analyst, 2004, 129, 478–482 RSC.
- T. Ito, L. Sun, R. R. Henriquez and R. M. Crooks, Acc. Chem. Res., 2004, 37, 937–945 CrossRef CAS PubMed.
- G. S. Roberts, D. Kozak, W. Anderson, M. F. Broom, R. Vogel and M. Trau, Small, 2010, 6, 2653–2658 CrossRef CAS PubMed.
- E. Blundell, L. J. Mayne, E. R. Billinge and M. Platt, Anal. Methods, 2015, 7, 7055–7066 RSC.
- J. B. Heng, A. Aksimentiev, C. Ho, P. Marks, Y. V. Grinkova, S. Sligar, K. Schulten and G. Timp, Biophys. J., 2006, 90, 1098–1106 CrossRef CAS PubMed.
- W.-J. Lan, D. A. Holden, J. Liu and H. S. White, J. Phys. Chem. C, 2011, 115, 18445–18452 CrossRef CAS.
- M. Lemmer, M. S. Inkpen, K. Kornysheva, N. J. Long and T. Albrecht, Nat. Commun., 2016, 7, 12922 CrossRef CAS PubMed.
- G. Nguyen, S. Howorka and Z. S. Siwy, J. Membr. Biol., 2011, 239, 105–113 CrossRef CAS PubMed.
- H. S. White and A. Bund, Langmuir, 2008, 24, 2212–2218 CrossRef CAS PubMed.
- W.-J. J. Lan, C. Kubeil, J.-W. W. Xiong, A. Bund and H. S. White, J. Phys. Chem. C, 2014, 118, 2726–2734 CrossRef CAS.
- C. Bernabini, D. Holmes and H. Morgan, Lab Chip, 2011, 11, 407–412 RSC.
- A. D. Grabarek, D. Weinbuch, W. Jiskoot and A. Hawe, J. Pharm. Sci., 2019, 108, 563–573 CrossRef CAS PubMed.
- R. Maugi, P. Hauer, J. Bowen, E. Ashman, E. Hunsicker and M. Platt, Nanoscale, 2020, 12, 262–270 RSC.
- M. Pollard, E. Hunsicker and M. Platt, ACS Sens., 2020, 5, 2578–2586 CrossRef CAS PubMed.
- C. Chan, W. Mak, K. Cheung, K. Sin, C. Yu, T. Rainer and R. Renneberg, Annu. Rev. Anal. Chem., 2013, 6, 191–211 CrossRef CAS PubMed.
- N. A. W. Bell and U. F. Keyser, Nat. Nano, 2016, 11, 645–651 CrossRef CAS PubMed.
- E. L. C. J. Blundell, R. Vogel and M. Platt, Langmuir, 2016, 32, 1082–1090 CrossRef CAS PubMed.
- L. Mayne, C. Y. Lin, S. D. R. Christie, Z. S. Siwy and M. Platt, ACS Nano, 2018, 12, 4844–4852 CrossRef CAS PubMed.
- R. Vogel, G. Willmott, D. Kozak, G. S. Roberts, W. Anderson, L. Groenewegen, B. Glossop, A. Barnett, A. Turner and M. Trau, Anal. Chem., 2011, 83, 3499–3506 CrossRef CAS PubMed.
- R. Peng and D. Li, Talanta, 2018, 184, 418–428 CrossRef CAS PubMed.
- H. Yasaki, T. Yasui, T. Yanagida, N. Kaji, M. Kanai, K. Nagashima, T. Kawai and Y. Baba, J. Am. Chem. Soc., 2017, 139, 14137–14142 CrossRef CAS PubMed.
- P. Hinkle, T. M. Westerhof, Y. Qiu, D. J. Mallin, M. L. Wallace, E. L. Nelson, P. Taborek and Z. S. Siwy, Sci. Rep., 2017, 7, 10173 CrossRef PubMed.
- Z. Siwy, P. Apel, D. Dobrev, R. Neumann, R. Spohr, C. Trautmann and K. Voss, Nucl Instrum Methods Phys Res B, 2003, 208, 143–148 CrossRef CAS.
- W. Zhang, Y. Hu, G. Choi, S. Liang, M. Liu and W. Guan, Sens. Actuators, B, 2019, 296, 126615 CrossRef CAS.
- Z. Song, M. Li, B. Li, Y. Yan and Y. Song, Electrophoresis, 2019, 40, 897–905 CrossRef CAS PubMed.
- T. Zhou, Y. Song, Y. Yuan and D. Li, Anal. Chim. Acta, 2019, 1052, 113–123 CrossRef CAS PubMed.
- J. Sun, Y. Kang, E. M. Boczko and X. Jiang, Electroanalysis, 2013, 25, 1023–1028 CrossRef CAS.
- N. Khodaparastasgarabad, A. Mohebbi and C. Falamaki, Microsyst. Technol., 2019, 25, 3643–3653 CrossRef CAS.
- Z. Liu, J. Li, J. Yang, Y. Song, X. Pan and D. Li, Microfluid. Nanofluid., 2017, 21, 4 CrossRef.
- Y. Song, J. Zhang and D. Li, Micromachines, 2017, 8, 204 CrossRef PubMed.
- Y. Song, H. Zhang, C. H. Chon, S. Chen, X. Pan and D. Li, Anal. Chim. Acta, 2010, 681, 82–86 CrossRef CAS PubMed.
- R. Peng and D. Li, Nanoscale, 2017, 9, 5964–5974 RSC.
- Y. Song, J. Wang, J. Yang, Y. Wu, N. Li, N. Gong, X. Pan, Y. Sun and D. Li, Instrum. Sci. Technol., 2012, 40, 305–315 CrossRef CAS.
- T. Monaghan, M. J. Harding, R. A. Harris, R. J. Friel and S. D. R. Christie, Lab Chip, 2016, 16, 3362–3373 RSC.
- A. J. Capel, S. Edmondson, S. D. R. Christie, R. D. Goodridge, R. J. Bibb and M. Thurstans, Lab Chip, 2013, 13, 4583–4590 RSC.
- A. J. Capel, R. P. Rimington, M. P. Lewis and S. D. R. Christie, Nat. Rev. Chem., 2018, 2, 422–436 CrossRef.
- A. J. Capel, A. Wright, M. J. Harding, G. W. Weaver, Y. Li, R. A. Harris, S. Edmondson, R. D. Goodridge and S. R. D. Christie, Beilstein J. Org. Chem., 2017, 13, 111–119 CrossRef CAS PubMed.
- O. Okafor, A. Weilhard, J. A. Fernandes, E. Karjalainen, R. Goodridge and V. Sans, React. Chem. Eng., 2017, 2, 129–136 RSC.
- C. Liao, W. Anderson, F. Antaw and M. Trau, ACS Omega, 2019, 4, 1401–1409 CrossRef CAS PubMed.
- S. M. Hampson, M. Pollard, P. Hauer, H. Salway, S. D. R. Christie and M. Platt, Anal. Chem., 2019, 91, 2947–2954 CrossRef CAS PubMed.
- S. M. Hampson, W. Rowe, S. D. R. Christie and M. Platt, Sens. Actuators, B, 2018, 256, 1030–1037 CrossRef CAS.
- M. Pollard, R. Maugi, A. Holzinger, M. D. Scanlon and M. Platt, ChemRxiv. Cambridge.
- S. R. German, L. Luo, H. S. White and T. L. Mega, J. Phys. Chem. C, 2013, 117, 703–711 CrossRef CAS.
- W.-J. Lan and H. S. White, ACS Nano, 2012, 6, 1757–1765 CrossRef CAS PubMed.
- Y. Zhang, M. A. Edwards, S. R. German and H. S. White, J. Phys. Chem. C, 2016, 120, 20781–20788 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d2an00128d |
| This journal is © The Royal Society of Chemistry 2022 |