
On-demand shape transformation of polymer vesicles via site-specific isomerization of hydrazone photoswitches in monodisperse hydrophobic oligomers†
Valene
Wang
,
Jiwon
Kim
 ,
Junyoung
Kim
,
Seul Woo
Lee
and
Kyoung Taek
Kim
*
,
Junyoung
Kim
,
Seul Woo
Lee
and
Kyoung Taek
Kim
*
Department of Chemistry, Seoul National University, Seoul 08826, Korea. E-mail: ktkim72@snu.ac.kr
First published on 16th August 2021
Abstract
The shape control of nanostructures formed by the solution self-assembly of block copolymers is of significance for drug delivery. In particular, site-specific perturbation resulting in the conformational change of the hydrophobic block has attracted considerable attention because of the possibility of creating polymer vesicles capable of releasing cargo molecules on demand by responding to specific stimuli. Herein, we report the synthesis of amphiphilic block copolymers based on the monodisperse molecular-weight oligo(phenyllactic acid) (OPLA) having hydrazone photoswitches at specific locations. Upon light irradiation, the photoswitch undergoes E–Z isomerization, resulting in the conformational change of the OPLA block. Polymer vesicles composed of these block copolymers exhibited reversible shape transformation upon irradiation with UV or visible light due to the E–Z isomerization of the photoswitch. Furthermore, the location and the number of hydrazone photoswitches in the monodisperse OPLA block are the decisive factors for the reversible shape transformation of the polymer vesicles from an isotropic to an anisotropic morphology.
Introduction
Stimuli-responsive polymer vesicles (polymersomes) have been extensively investigated as intelligent vehicles for on-demand drug delivery in response to chemical, physical, and biological stimuli.1–6 When the compartmentalizing membranes of polymersomes are exposed to external stimuli, the polymersomes release the guest molecules encapsulated in their cavity via molecular transport through the bilayer membrane consisting of self-assembled block copolymers (BCPs). Generally, the transmembrane transport of guest molecules requires pore generation in the polymersome membranes, which occurs through the solubility switching of the BCP building blocks or pore generators embedded in the membrane; this leads to the disassembly or perforation of the membranes under specific external stimuli such as pH, temperature, biomolecules and light.7–10 However, pore generation based on these mechanisms can cause permanent perforation of the membranes or complete disassembly of the polymersomes.11–13 Therefore, the development of polymersomes that can reversibly release or retain the encapsulated cargo molecules under external stimuli has attracted recent interest.14In this regard, the photo-triggered release of guests from polymersomes composed of photo-responsive BCPs has been extensively investigated because these polymersomes enable reversible mass transport through the bilayers without the permanent disruption of the vesicular membrane.15 The permeability of these polymersomes can be changed by inducing the conformational change of the membrane-forming BCPs in response to a certain light wavelength.16 To achieve the light-triggered conformational change of polymers, photoisomerizable azobenzene groups are routinely introduced as photoswitches in BCPs. Recently, Thayumanavan et al. took an alternative approach known as interface-induced dynamic facilitation, which promotes propagation across a glassy polymeric membrane via the configurational switching of a single azobenzene unit.17
Most photo-responsive BCPs are synthesized by controlled polymerization methods involving the introduction of photo-responsive units as pendent groups18–32 on the polymer backbone or at the junction interfacing the constituting polymer blocks.17,33–36 However, studying the effect of the location of the photoswitch in polymeric systems on their conformational change using traditional polymerization techniques is challenging. We envisioned that the synthesis of discrete hydrophobic blocks using a convergent approach would allow a more sophisticated control over the position of the photoswitch incorporated in the hydrophobic core and thus help understand the relationship between the structure and actuation of the self-assembled BCPs.
Herein, we report the synthesis of BCPs composed of hydrophilic polyethylene glycol (PEG) blocks and discrete oligo(phenyllactic acid) (OPLA) blocks with hydrazone-based photoswitches at specific positions. To determine the effect of the light-induced configurational switching of the photoswitches, they were embedded in two different positions within the BCP chains: (1) in the middle of the hydrophobic OPLA chains, and (2) at the junction of the hydrophilic and hydrophobic blocks. Moreover, to investigate the effect of the number of photoswitches in the BCP backbone on the extent of the shape transformations of the self-assembled nanostructures, a BCP containing multiple photoswitches was synthesized. The shape transformation of the self-assembled nanostructures of these BCPs were triggered by the E/Z isomerization of the hydrazone-based photoswitches upon irradiation with a light source (λ = 405 nm or 365 nm), which changed the conformation of the discrete OPLA blocks. The polymersomes exhibited reversible shape transformation via the E/Z isomerization of the hydrazone photoswitch embedded in the discrete hydrophobic block. The effect of the conformational change of the hydrophobic block on the shape transformation of the BCP-based polymersomes was even more pronounced when the number of hydrazone switches was increased. The present study highlights the importance of the perfect control of the position of a versatile hydrazone photoswitch in BCPs for the configurational change at the nanoscale to be translated to microscopic actuation of the BCP membrane, i.e., shape transformation (Fig. 1). The elucidation of the on-demand actuation of our system can open new avenues for the tailoring of polymeric morphologies for versatile applications.
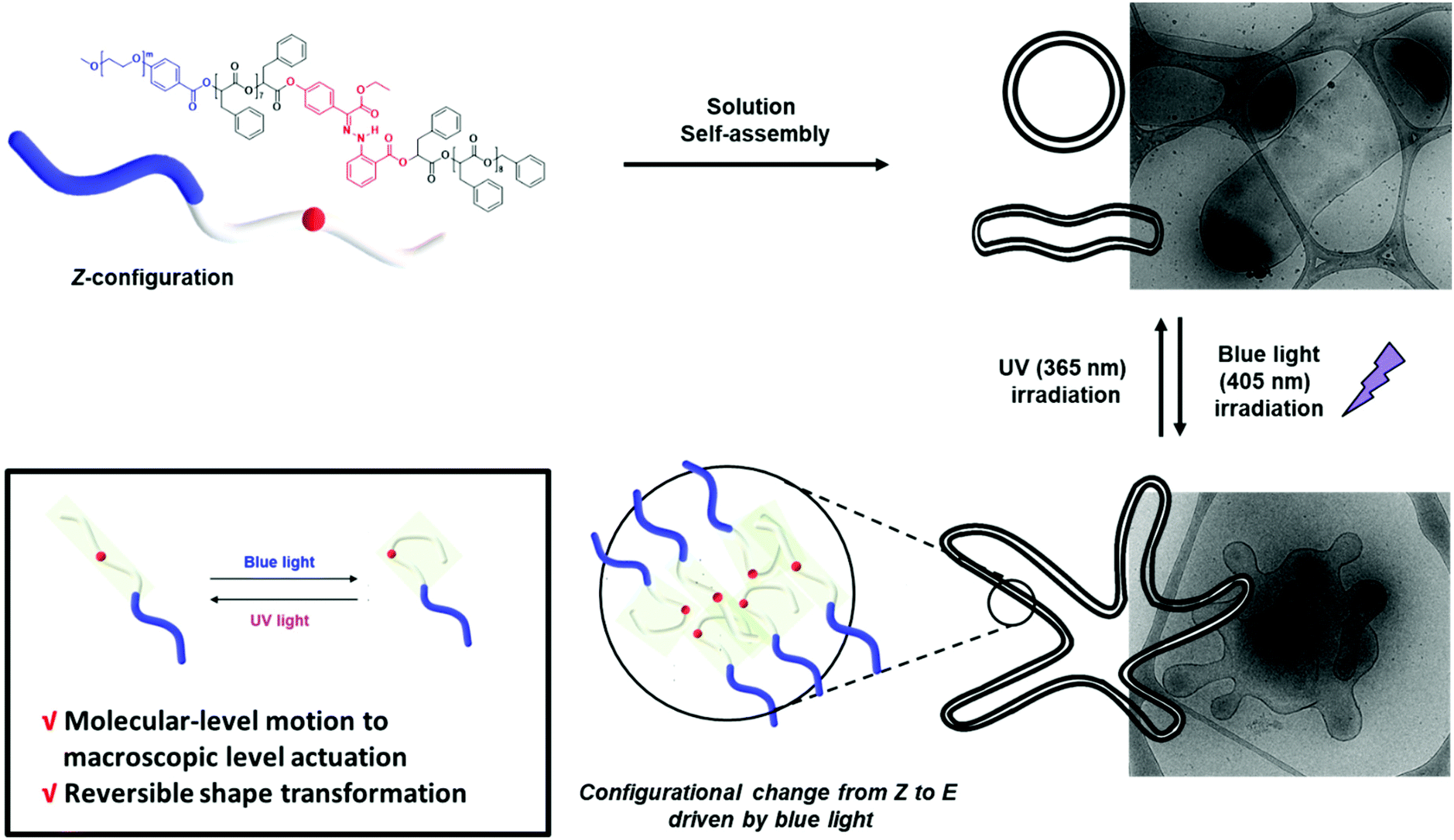 | ||
| Fig. 1 Schematic representation of the reversible shape transformation of self-assembled structures of PEG-b-[OPLA] containing hydrazone-based photoswitches via configurational switching of the system. | ||
Results and discussion
Effect of the position of hydrazone photoswitches in the BCP backbone
Hydrazone-based photoswitches contain the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N–NH functional group, renders the system virtually bistable (thermal relaxation (τ1/2) up to 5300 years).37,38 This characteristic of hydrazone photoswitches can be ideal for permanently locking the actuation of dynamic molecules for the on-demand sustained release of guest molecules.39 The hydrazone-based photoswitch (H1) was synthesized using a previously reported procedure (Scheme S1 and Fig. S1†).40
N–NH functional group, renders the system virtually bistable (thermal relaxation (τ1/2) up to 5300 years).37,38 This characteristic of hydrazone photoswitches can be ideal for permanently locking the actuation of dynamic molecules for the on-demand sustained release of guest molecules.39 The hydrazone-based photoswitch (H1) was synthesized using a previously reported procedure (Scheme S1 and Fig. S1†).40
To prepare discrete hydrophobic blocks, OPLAs of different molecular weights were synthesized by iterative convergent methods (Schemes 1 and S2†).41,42 The orthogonally protected dimer of phenyllactic acid, TBDMS-OPLA2-Bz, was deprotected to prepare HO-OPLA2-Bz and TBDMS-OPLA2-COOH by desilylation with BF3 and hydrogenation, respectively. The deprotected OPLA2 compounds were coupled by esterification using N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide (EDC) as a coupling agent. The resulting TBDMS-OPLA4-Bz was used for further convergence to obtain OPLA8 and OPLA16 (Schemes 1 and S3†). The discrete OPLAs were purified by column chromatography, and their purity was determined by NMR spectroscopy and matrix-assisted laser desorption/ionization time-of-flight (MALDI-TOF) mass spectrometry (Fig. S3–S6†).
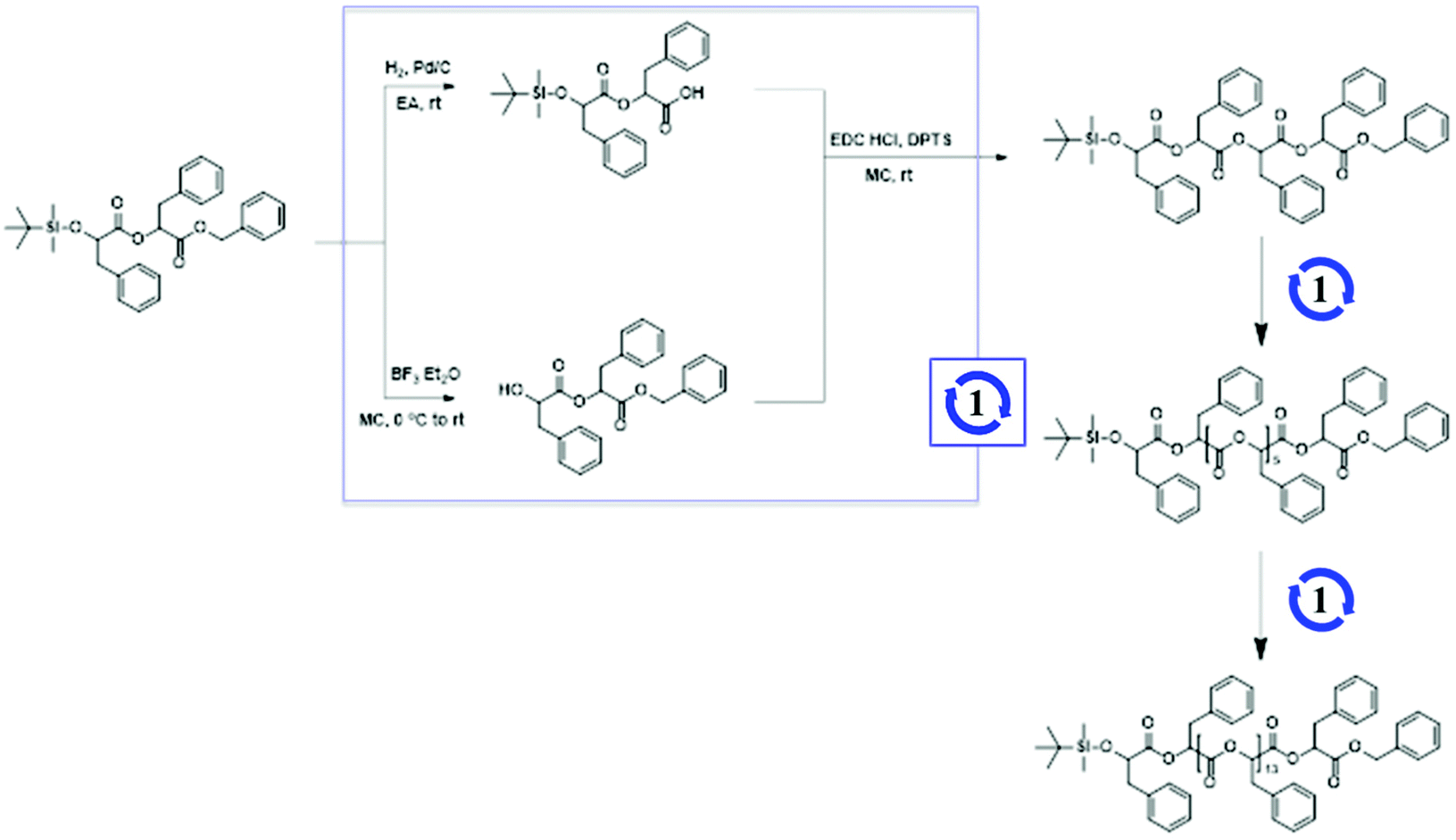 | ||
| Scheme 1 Synthesis of discrete oligo(phenyl lactic acid)s (OPLAs). | ||
Using the discrete OPLAs as building blocks, we synthesized discrete hydrophobic blocks by introducing a photoswitch, H1, at the desired position in the polymer backbone. We first synthesized discrete OPLA17 by coupling two OPLA8 units using H1 as a linker. To connect the OPLAs via the H1 linker, a single unit of phenyllactic acid was used as a spacer between H1 and the OPLAs. To introduce H1 in the mid-position of the OPLA block, OH-OPLA8 was reacted with H1-PLA1-COOH using EDC as a coupling agent. After acetal deprotection with PPTS, the resulting OH-H1-OPLA9 was esterified with OPLA8-COOH to obtain a monodisperse hydrophobic block OPLA8-H1-OPLA9 with the photoswitch in the middle of the oligomer backbone (Schemes 2, S5 and Fig. S7, S8†). To introduce H1 at the junction of the hydrophilic and OPLA blocks, H1-OPLA17 was synthesized by the esterification of H1-PLA1-COOH and OH-OPLA16 (Scheme S6†). The synthesized hydrophobic block was characterized by 1H NMR spectroscopy and MALDI-TOF mass spectrometry (Fig. 2A, B, S11A and S11B†).
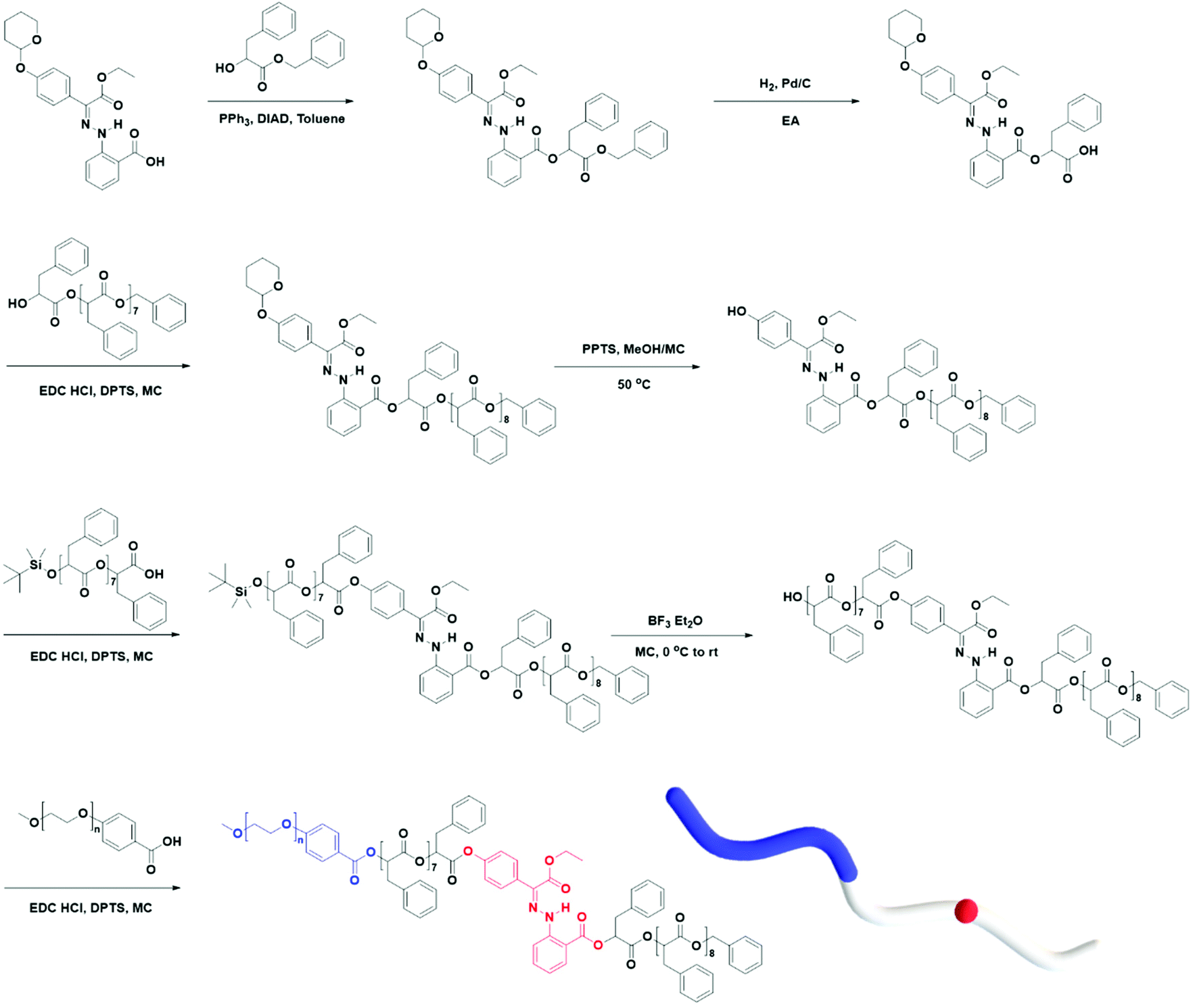 | ||
| Scheme 2 Synthesis of BCP having hydrazone-based photo-switches (PEG-b-[OPLA8-H1-OPLA9]). | ||
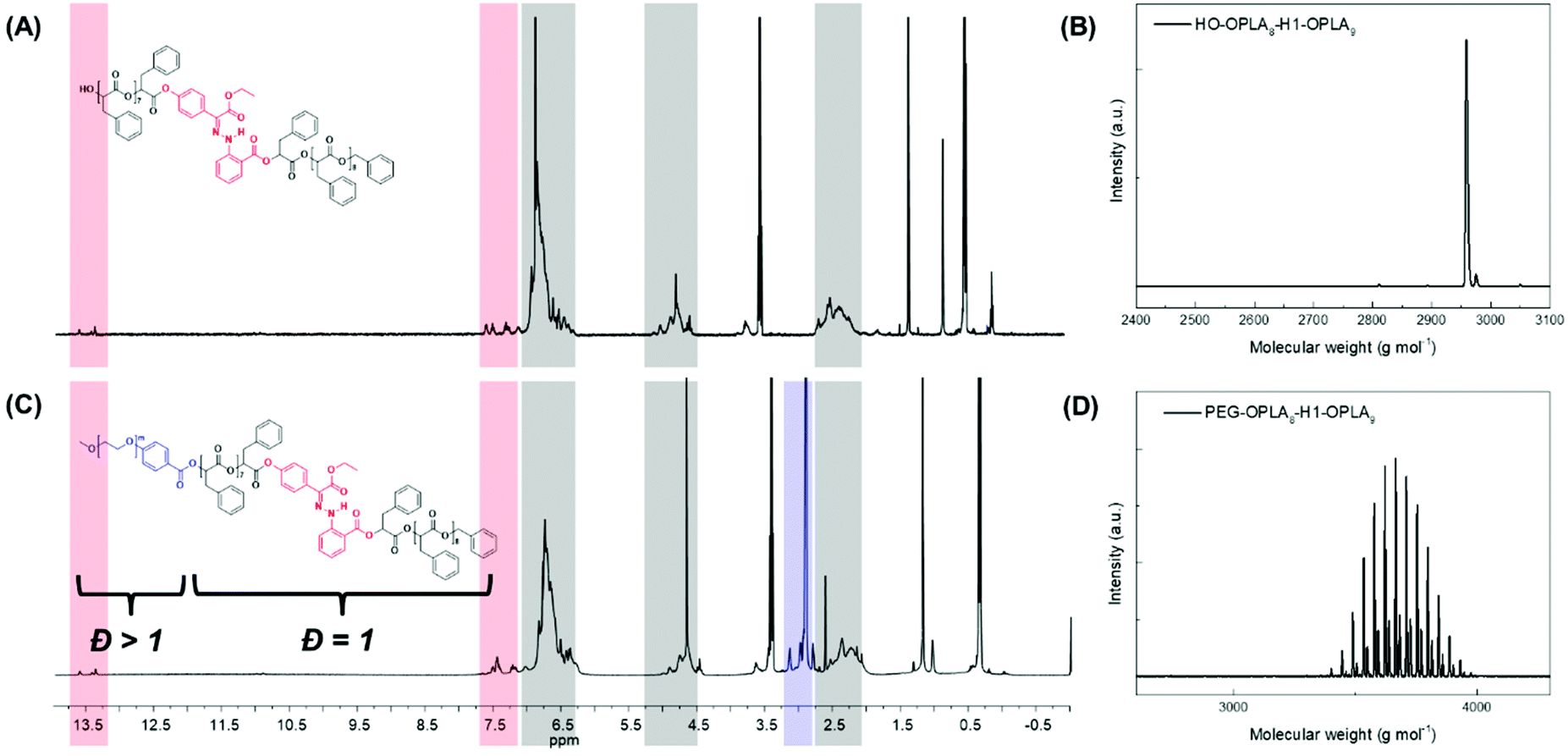 | ||
| Fig. 2 (A and C) 1H NMR spectra in CDCl3 and (B and D) MALDI-TOF spectra of monodisperse hydrophobic block (HO-OPLA8-H1-OPLA9) and amphiphilic block copolymer (PEG550-b-[OPLA8-H1-OPLA9]), respectively. | ||
The synthesis of BCPs composed of a discrete hydrophobic block H1 at different positions and a hydrophilic PEG block of different molecular weights (Mn = 550 and 1000) was confirmed by 1H NMR spectroscopy and MALDI-TOF spectrometry (Fig. 2C, D, S9, S10, S11C, S11D and S12†); the results are summarized in Table 1.
| Sample | Name | Position of H1 | M n (g mol−1)a of hydrophobic block | M n (g mol−1)a of hydrophilic block | Đ of BCPs |
f
PEG![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) b (%) b (%) |
|---|---|---|---|---|---|---|
| a Number average molecular weight (Mn) and molecular weight distribution (Đ) determined by GPC using polystyrene (PS) standards. Elution was performed with THF at a flow rate of 1 mL min−1 at 30 °C. b Molecular weight fraction of the PEG domain relative to the amphiphilic block copolymer. | ||||||
| 1 | PEG550-b-[OPLA8-H1-OPLA9] | Center | 2958 | 550 | 1.02 | 15.7 |
| 2 | PEG550-b-[H1-OPLA17] | Junction | 2958 | 550 | 1.02 | 15.7 |
| 3 | PEG1000-b-[OPLA8-H1-OPLA9] | Center | 2958 | 1000 | 1.02 | 25.2 |
| 4 | PEG1000-b-[H1-OPLA17] | Junction | 2958 | 1000 | 1.02 | 25.2 |
| 5 | PEG1000-b-[OPLA4-H1-OPLA8-H1-OPLA4] | Center | 3100 | 1000 | 1.02 | 24.4 |
The 1H NMR spectroscopic analysis of the as-prepared H1 in DMSO revealed that H1 exclusively adopts the Z-configuration (>99%) at room temperature (Fig. S2†). Irradiation with 405 nm light yielded a photostationary state (PSS) consisting of 90% E-H1, while irradiation of the resulting solution with UV light (365 nm) yielded a mixture consisting of 70% Z-H1 at the PSS (Fig. S2A and S2B†). In addition, UV-Vis spectroscopy was conducted for photoisomerization studies (Fig. S2C†). The switching cycles of H1 in DMSO under alternating light irradiation were confirmed from the absorbance change at 373 nm (Fig. S2D†). The photoisomerization of the prepared BCP1 (PEG550-b-[OPLA8-H1-OPLA9]) was studied and monitored by 1H NMR spectroscopy in CDCl3 (Fig. 3, S13 and S14†). The irradiation of BCP containing Z-BCP1 (>99%) with blue light (λ = 405 nm) yielded a PSS consisting of 85% E-BCP1, while the irradiation of the resulting solution with UV light (365 nm) yielded a mixture containing 91% of the Z isomer at the PSS (Fig. 3B and C). The degree of conversion between E- and Z-isomers of H1 embedded in BCP is comparable to that of an individual H1 molecule, which proves the functionality of H1 in the BCP. The isomerization of H1 in the self-assembled structures of BCP1 was studied by UV-Vis absorption spectroscopy over time (Fig. S15†). Although the isomerization rate of H1 embedded in the BCP membrane was decreased, the photoisomerization was not compromised which resulted in shape transformation of the polymersomes.
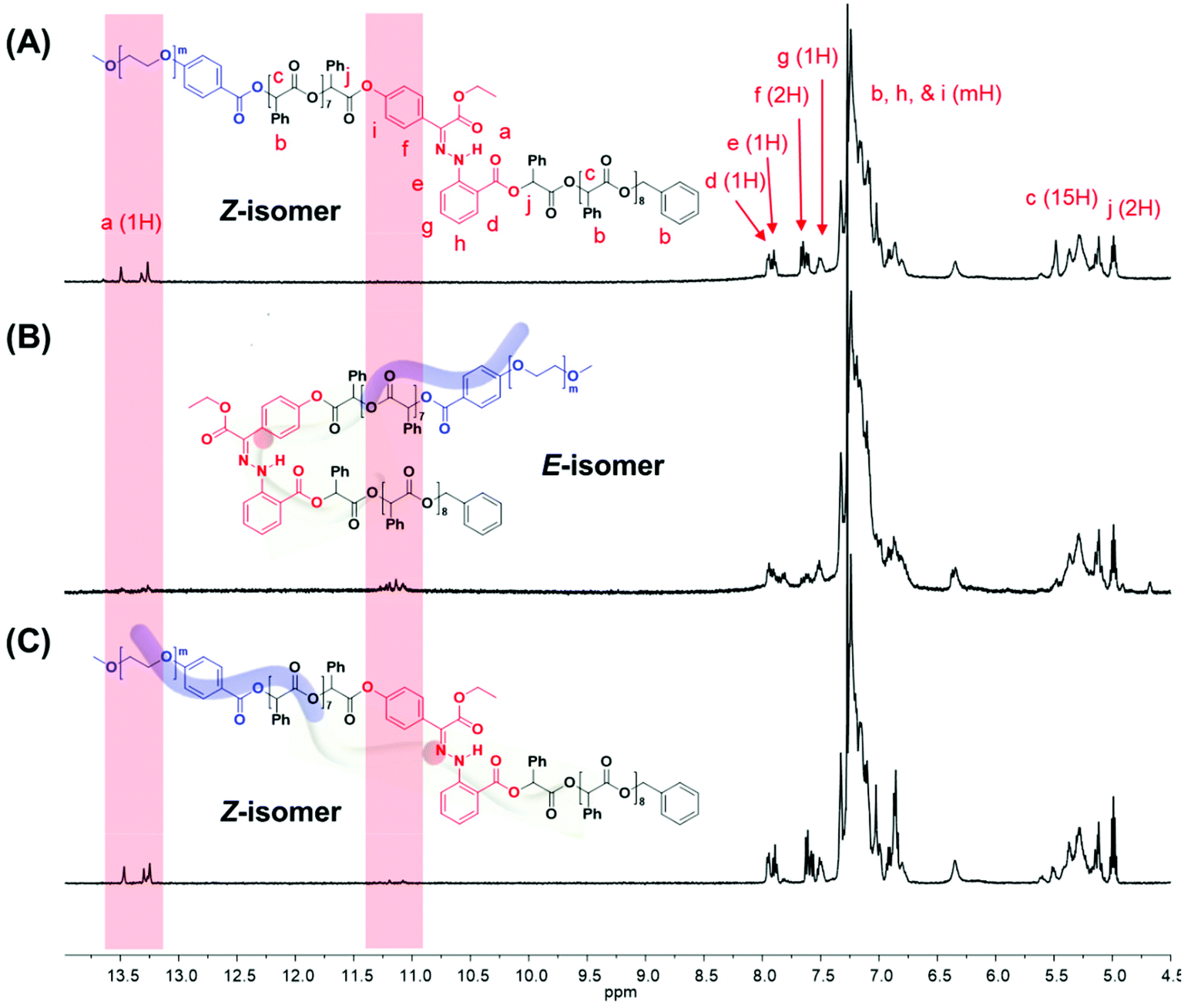 | ||
| Fig. 3 1H NMR spectra of amphiphilic block copolymer containing hydrazone-based photoswitches (A) before and (B) after irradiation with blue light (405 nm), followed by (C) photoirradiation with UV light (365 nm). | ||
This observation was confirmed by cryogenic transmission electron microscopy (cryo-TEM). The cryo-TEM images of the self-assembled structures show that BCP1 self-assembled into round vesicles (Fig. 4A). The polymersomes underwent shape transformation and formed flower-like structures upon irradiation with 405 nm light for 2 h (Fig. 4B), which subsequently were broken into smaller polymersomes upon irradiation with UV light (Fig. 4C). The size of the self-assembled nanoparticles was determined from the hydrodynamic diameter of the nanoparticles suspended in water, which was measured by dynamic light scattering (DLS) analysis. The average diameter of the BCP1 nanoparticles was 534 nm initially, decreased upon blue-light irradiation followed by an increase to 612 nm upon UV light irradiation. We reasoned that this change arose from the shape transformation of the polymersomes of BCP1 as observed by cryo-TEM (Fig. S18A†).
 | ||
| Fig. 4 Cryo-TEM images and 3D graphic representations of self-assembled structures of PEG550-b-[OPLA8-H1-OPLA9] under different light sources. The morphologies were observed (A) before and (B) after blue-light irradiation, followed by (C) UV light irradiation. | ||
 | ||
| Fig. 5 Cryo-TEM images of self-assembled structures of (A–C) PEG1000-b-[OPLA8-H1-OPLA9] and (D–F) PEG1000-b-[H1-OPLA17] under different light sources. The morphologies were observed (A and D) before and (B and E) after blue light irradiation, followed by (C and F) UV light irradiation. | ||
When the BCPs are irradiated with 405 nm light, the H1 units in both BCP3 and BCP4 undergo E–Z isomerization; this resulted in a distinct shape transformation in the case of BCP3, while no visible shape change was observed in the case of BCP4 although both BCP3 and BCP4 have the same molecular weights. This indicates that controlling the position of the photoswitch in the BCP is critical for shape transformation. When H1 is located at the middle of the hydrophobic chain, the self-assembled nanostructures underwent significant shape transformation due to the photoisomerization of the photoswitch. In contrast, no shape change was observed when H1 was located at the junction of the hydrophilic and hydrophobic blocks.
A plausible explanation could be that the E/Z isomerization of the photoswitch in the middle of the hydrophobic OPLA block would introduce a kink, which causes the conformational change of the hydrophobic chain (Fig. 3). This change might lead to the increase of the interfacial area of the compartmentalizing membrane of the polymersome. Assuming the constant volume of the internal water-filled compartment, the membrane would develop protrusion to compensate the increased interfacial area, which results in the shape transformation of polymersomes.
Effect of the number of hydrazone photoswitches in the BCP backbone
We found that the BCP3 polymersomes underwent shape transformation when H1 was located at the center of the hydrophobic chain in BCP3 (PEG1000-b-[OPLA8-H1-OPLA9]. The presence of H1 within the hydrophobic core of the self-assembled BCP that underwent photo-induced configurational change resulted in a change in the bilayer membrane of the polymersomes. Therefore, we investigated the effect of the number of H1 in the hydrophobic chain on the extent of shape transformation. For this, we synthesized of BCP5 composed of a hydrophilic PEG block and hydrophobic blocks (Đ = 1) with two H1 units (Scheme S7† and Fig. 6). BCP5 comprising discrete hydrophobic chains with multiple H1 units and PEG hydrophilic blocks with a molecular weight (Mn) of 1000 was characterized by 1H NMR spectroscopy and MALDI-TOF spectrometry (Fig. S16 and S17†); the results are summarized in Table 1.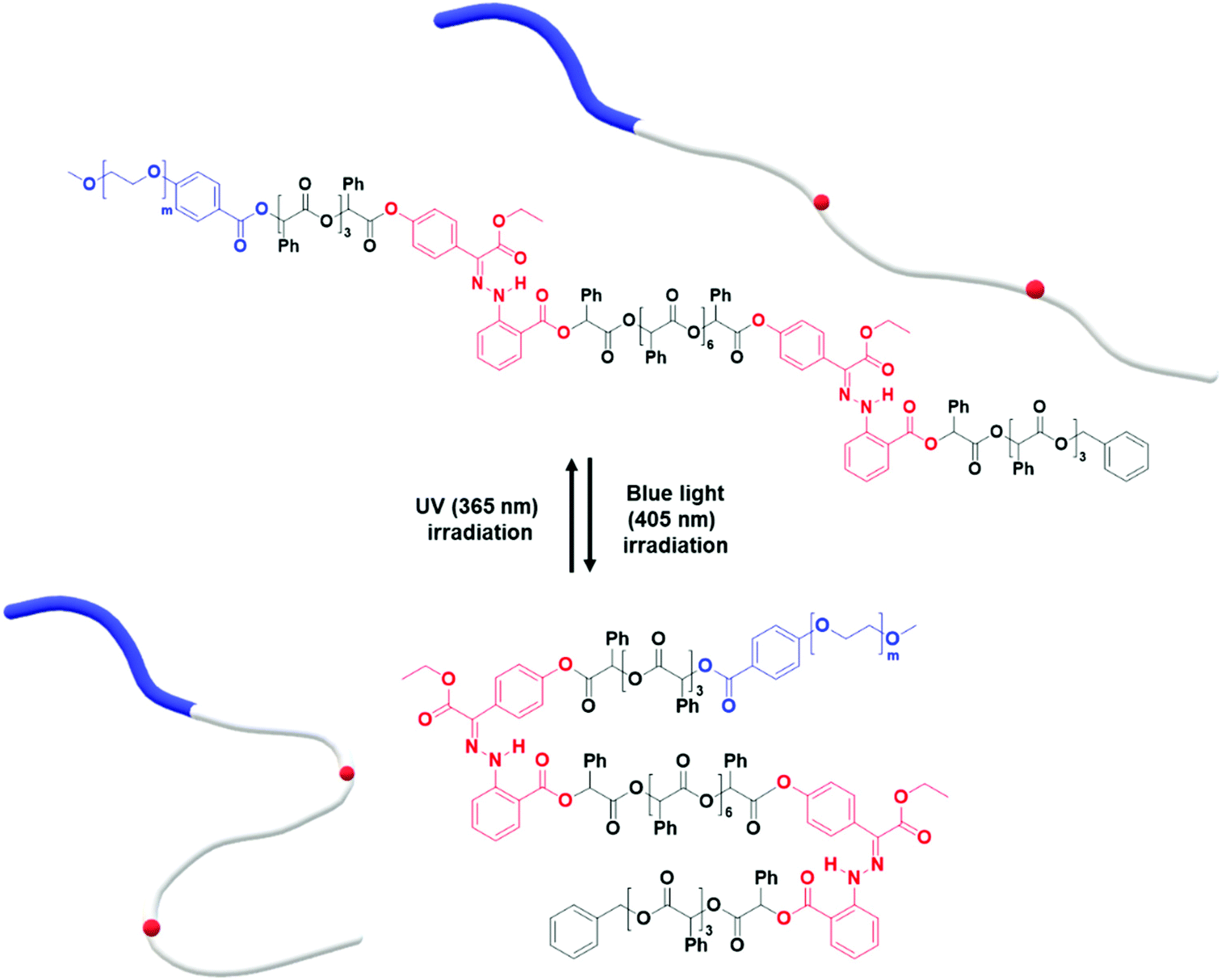 | ||
| Fig. 6 Photo-induced isomerization of two photoswitches in the hydrophobic chain of PEG1000-b-[OPLA4-H1-OPLA8-H1-OPLA4]. | ||
:acetone ratio reached 1:2. The high acetone content imparts fluidity to the sample with higher crystallinity due to an increase in the number of photoswitches. The induced configurational change in H1 was confirmed by the shape transformation of the polymersomes as observed by TEM. The prepared BCP5 (PEG1000-b-[OPLA4-H1-OPLA8-H1-OPLA4]) self-assembled into spherical vesicles (Fig. 7A). The average diameter of the polymeric vesicles was 890 nm, which decreased upon blue-light irradiation corresponding to the enhanced change in the shape of the nanostructures observed by TEM (Fig. S26†).
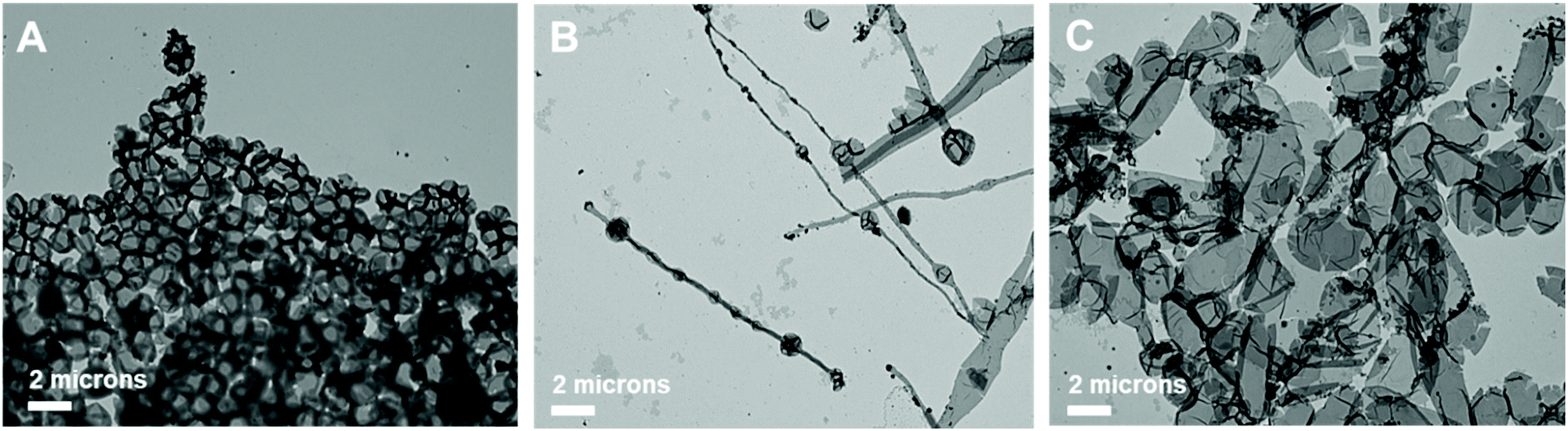 | ||
| Fig. 7 TEM images of self-assembled structures of PEG1000-b-[OPLA4-H1-OPLA8-H1-OPLA4] under different light sources. The morphologies were observed (A) before and (B) after blue-light irradiation, followed by (C) UV light irradiation. The PEG1000-b-[OPLA4-H1-OPLA8-H1-OPLA4] samples were treated with phosphotungstic acid solution for negative staining. | ||
The self-assembled nanoparticles were irradiated with 405 nm light for 2 h at room temperature. After blue-light irradiation, the polymeric vesicles formed elongated structures (Fig. 7B and Fig. S25†) with an average diameter of 520 nm, which indicates that the Z–E isomerization of the multiple H1 units enhanced the shape transformation effect as confirmed by the significant decrease in the hydrodynamic diameter of the nanoparticles. Upon UV light irradiation, the elongated structures transformed into a mixture of round and tubular vesicles (Fig. 7C). The remarkable reversibility of shape transformation because of which most of the particles regained their initial morphology, indicates that the reversible photoisomerization of the hydrazone-based photoswitches enhanced with an increase in the number of photoswitches in the BCP backbone. This in turn affected the extent of shape transformation of the nanostructures.
Further, the pristine samples were irradiated under blue light for different irradiation times (15, 30, and 120 min) at room temperature. Fig. 8 shows the TEM images of the irradiated self-assembled polymer vesicles. After 15 min of blue light irradiation, spherical vesicles formed with few elongated structures (Fig. 8B), which indicates the limited Z to E isomerization of H1 embedded in BCP5. Furthermore, the number of elongated structures increased with increasing irradiation time (Fig. 8C and D), which indicates an increase in the concentration of E-isomer. The ratio of the elongated structures to the spherical vesicles increases as the ratio of Z- isomers to E-isomers varies with irradiation time.
 | ||
| Fig. 8 TEM images of self-assembled structures of PEG1000-b-[OPLA4-H1-OPLA8-H1-OPLA4] under blue-light irradiation at different irradiation times. The morphologies were observed (A) before blue light irradiation and, after (B) 15 min, (C) 30 min, and (D) 120 min of blue light irradiation. The PEG1000-b-[OPLA4-H1-OPLA8-H1-OPLA4] samples were treated with phosphotungstic acid solution for negative staining. | ||
Conclusion
In summary, we synthesized BCPs composed of hydrophilic PEG blocks and OPLA blocks containing hydrazone-based photoswitches in specific positions. The BCPs self-assembled into nanostructures via a cosolvent self-assembly method. We found that the configurational change of the hydrazone-based photoswitches can be translated to macroscopic actuation such as the shape transformation of the self-assembled nanoparticles. The nanoparticles constructed with BCPs with the hydrazone-based photoswitch embedded at the middle of the hydrophobic OPLA chains exhibited dramatic membrane deformation, with reversible shape transformation from polymeric vesicles to urchin-like structures. In contrast, the self-assembled nanostructures of BCPs with the hydrazone-based photoswitches embedded at the junction of the hydrophilic and hydrophobic blocks did not undergo shape transformation upon irradiation with different light sources. This indicates that the position of the switch in the hydrophobic OPLA chain is a decisive factor that determines the shape transformation of the nanoparticles driven by the light-induced configurational change of the hydrazone-based photoswitches. Furthermore, with an increase in the number of photoswitches embedded in the OPLA chains, the extent of shape transformation is significantly enhanced. The on-demand actuation of our system can contribute significantly to the design and development of BCPs for the fabrication of polymersomes for a wide range of potential applications including delivery systems for cargo molecules.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work was supported by the National Research Foundation (NRF) of Korea (2019R1A2C3007541), Creative Materials Discovery Program through the NRF funded by Ministry of Science and ICT (2020M3D1A1070053) and Seoul National University (SNU) for the support by the Creative-Pioneering Re-searchers Programs (30520190050).References
- F. Meng, Z. Zhong and J. Feijen, Biomacromolecules, 2009, 10, 197–209 CrossRef CAS PubMed.
- O. Onaca, R. Enea, D. W. Hughes and W. Meier, Macromol. Biosci., 2009, 9, 129–139 CrossRef CAS PubMed.
- M.-H. Li and P. Keller, Soft Matter, 2009, 5, 927–937 RSC.
- X. Hu, Y. Zhang, Z. Xie, X. Jing, A. Bellotti and Z. Gu, Biomacromolecules, 2017, 18, 649–673 CrossRef CAS.
- Y. Wang and D. S. Kohane, Nat. Rev. Mater., 2017, 2, 17020 CrossRef CAS.
- U. Kauscher, M. N. Holme, M. Björnmalm and M. M. Steven, Adv. Drug Delivery Rev., 2019, 138, 259–275 CrossRef CAS.
- S. Hocine, A. Brulet, L. Jia, J. Yang, A. Di Cicco, L. Bouteillerce and M.-H. Li, Soft Matter, 2011, 7, 2613–2623 RSC.
- B. Yan, J. He, P. Ayotte and Y. Zhao, Macromol. Rapid Commun., 2011, 32, 972–976 CrossRef CAS PubMed.
- A. Peyret, E. Ibarboure, A. Tron, L. Beauté, R. Rust, O. Sandre, N. D. McClenaghan and S. Lecommandoux, Angew. Chem., Int. Ed., 2017, 56, 1566–1570 CrossRef CAS.
- S. C. Larnaudie, A. Peyret, L. Beauté, P. Nassoy and S. Lecommandoux, Langmuir, 2019, 35, 8398–8403 CAS.
- E. Mabrouk, D. Cuvelier, F. Brochard-Wyart, P. Nassoy and M.-H. Li, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 7294 CrossRef CAS.
- E. Amstad, S.-H. Kim and D. A. Weitz, Angew. Chem., 2012, 124, 12667–12671 CrossRef.
- X. Wang, J. Hu, G. Liu, J. Tian, H. Wang, M. Gong and S. Liu, J. Am. Chem. Soc., 2015, 137, 15262–15275 CrossRef CAS PubMed.
- Y. Sun, F. Gao, Y. Yao, H. Jin, X. Li and S. Lin, ACS Macro Lett., 2021, 10, 525–530 CrossRef CAS.
- B. Yan, X. Tong, P. Ayotte and Y. Zhao, Soft Matter, 2011, 7, 10001–10009 RSC.
- A. H. Gelebart, D. J. Mulder, M. Varga, A. Konya, G. Vantomme, E. W. Meijer, R. L. B. Selinger and D. J. Broer, Nature, 2017, 546, 632–636 CrossRef CAS PubMed.
- M. R. Molla, P. Rangadurai, L. Antony, S. Swaminathan, J. J. de Pablo and S. Thayumanavan, Nat. Chem., 2018, 10, 659–666 CrossRef CAS PubMed.
- G. Wang, X. Tong and Y. Zhao, Macromolecules, 2004, 37, 8911–8917 CrossRef CAS.
- X. Tong, G. Wang, A. Soldera and Y. Zhao, J. Phys. Chem. B, 2005, 109, 20281–20287 CrossRef CAS PubMed.
- X. Liu and M. Jiang, Angew. Chem., Int. Ed., 2006, 45, 3846–3850 CrossRef CAS PubMed.
- W. Su, Y. Luo, Q. Yan, S. Wu, K. Han, Q. Zhang, Y. Gu and Y. Li, Macromol. Rapid Commun., 2007, 28, 1251–1256 CrossRef CAS.
- W. Su, H. Zhao, Z. Wang, Y. Li and Q. Zhang, Eur. Polym. J., 2007, 43, 657–662 CrossRef CAS.
- D. Wang, H. Ren, X. Wang and X. Wang, Macromolecules, 2008, 41, 9382–9388 CrossRef CAS.
- L. Lin, Z. Yan, J. Gu, Y. Zhang, Z. Feng and Y. Yu, Macromol. Rapid Commun., 2009, 30, 1089–1093 CrossRef CAS PubMed.
- Q. Jin, G. Liu, X. Liu and J. Ji, Soft Matter, 2010, 6, 5589–5595 RSC.
- K. Chen, G. Xue, G. Shen, J. Cai, G. Zou, Y. Li and Q. Zhang, RSC Adv., 2013, 3, 8208–8210 RSC.
- R. Dong, B. Zhu, Y. Zhou, D. Yan and X. Zhu, Polym. Chem., 2013, 4, 912–915 RSC.
- Q. Ye, M. Huo, M. Zeng, L. Liu, L. Peng, X. Wang and J. Yuan, Macromolecules, 2018, 51, 3308–3314 CrossRef CAS.
- L. Li, S. Cui, A. Hu, W. Zhang, Y. Li, N. Zhou, Z. Zhang and X. Zhu, Chem. Commun., 2020, 56, 6237–6240 RSC.
- L. Li, Y. Li, S. Wang, L. Ye, W. Zhang, N. Zhou, Z. Zhang and X. Zhu, Polym. Chem., 2021, 12, 3052–3059 RSC.
- Y. Sun, F. Gao, Y. Yao, H. Jin, X. Li and S. Lin, ACS Macro Lett., 2021, 10, 525–530 CrossRef CAS.
- W. Wen and A. Chen, Polym. Chem., 2021, 12, 2447–2456 RSC.
- A. Altomare, F. Ciardelli, B. Gallot, M. Mader, R. Solaro and N. Tirelli, J. Polym. Sci., Part A: Polym. Chem., 2001, 39, 2957–2977 CrossRef CAS.
- J. del Barrio, L. Oriol, C. Sanchez, J. L. Serrano, A. Di Cicco, P. Keller and M.-H. Li, J. Am. Chem. Soc., 2010, 132, 3762–3769 CrossRef CAS PubMed.
- E. Blasco, J. L. Serrano, M. Piñol and L. Oriol, Macromolecules, 2013, 46, 5951–5960 CrossRef CAS.
- G. Cheng and J. Perez-Mercader, Chem. Mater., 2019, 31, 5691–5698 CrossRef CAS.
- H. Qian, S. Pramanik and I. Aprahamian, J. Am. Chem. Soc., 2017, 139, 9140–9143 CrossRef CAS PubMed.
- B. Shao and I. Aprahamian, Chem, 2020, 6, 2162–2173 CAS.
- X. Guo, B. Shao, S. Zhou, I. Aprahamian and Z. Chen, Chem. Sci., 2020, 11, 3016–3021 RSC.
- A. Ryabchun, Q. Li, F. Lancia, I. Aprahamian and N. Katsonis, J. Am. Chem. Soc., 2019, 141, 1196–1200 CrossRef CAS PubMed.
- J. M. Lee, M. B. Koo, S. W. Lee, H. Lee, J. Kwon, Y. H. Shim, S. Y. Kim and K. T. Kim, Nat. Commun., 2020, 11, 56 CrossRef CAS PubMed.
- M. B. Koo, S. W. Lee, J. M. Lee and K. T. Kim, J. Am. Chem. Soc., 2020, 142, 14028–14032 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1py00981h |
| This journal is © The Royal Society of Chemistry 2021 |