
Advanced spectroscopy, microscopy, diffraction and thermal analysis of polyamide adhesives and prediction of their functional properties with solid-state NMR spectroscopy†
Kash A.
Bhullar
a,
Aaron
Meinel
a,
Kennedy
Maeder
 a,
Richard
Wuhrer
b,
Marianne
Gaborieau
*a and
Patrice
Castignolles
a
a,
Richard
Wuhrer
b,
Marianne
Gaborieau
*a and
Patrice
Castignolles
a
aWestern Sydney University, Australian Centre for Research on Separation Science (ACROSS), School of Science, Parramatta, 2150, Australia. E-mail: m.gaborieau@westernsydney.edu.au
bWestern Sydney University, Advanced Materials Characterisation Facility (AMCF), Parramatta, 2150, Australia
First published on 16th December 2020
Abstract
Adhesives are an essential class of industrial polymers with applications ranging from pressure-sensitive adhesives to hot-melt adhesives used for repairing conveyor belts in mines. The composition and homogeneity of a polyamide-based hot-melt adhesive (HMA) was revealed with attenuated total reflection (ATR)-FTIR and solid-state NMR spectroscopy. Analysis of the polyamide shows that it is obtained through sustainable manufacturing based on dimer acids. ATR-FTIR showed incorporation of an abrasion-resistant additive on the surface of the HMA but was unsuitable for other additives such as carbon black or an antistatic agent. Quantitative 13C NMR spectroscopy revealed heterogeneity in the distribution of an antistatic agent in the HMA, which was supported by observations with differential scanning calorimetry (DSC). 1H NMR relaxation and two-dimensional wideline separation (2D-WISE) NMR revealed differences in the molecular dynamics of functional groups in the polyamide resin and the additives dispersed in the resin matrix. 1H T2 relaxation revealed that the molecular mobility of the least mobile and moderately mobile components increased with increasing temperature and antistatic agent content. 2D-WISE NMR revealed a phase separation in the base resin matrix and plasticization of the whole sample at very high antistatic agent content. 1H T2 relaxation showed possible correlations with mechanical properties such as Young's modulus and Shore A hardness and weaker correlations with adhesive properties such as T-peel strength. This shows the suitability of NMR to assist product innovation through the design of better-performing HMAs or of HMAs for application in different climatic conditions.
Introduction
Hot-melt adhesives (HMAs) consist of a base resin (such as ethyl vinyl acetate, polyurethane, polyamide, or polyester) mixed with additives (such as coloring agent, antistatic agent, tackifier, or fillers) that complement the adhesive's properties to fulfill specific requirements.1 This type of adhesives offers many advantages such as joining dissimilar materials and hardening soon after application; however, one issue these multicomponent materials face is the heterogeneities in composition and molecular dynamics arising due to additives and base resin from the compounding process. The heterogeneities in HMAs can affect their mechanical and adhesive properties, which may lead to batch-to-batch variability in functional properties, thus requiring re-compounding and re-testing on numerous samples. Hence, it is of utmost importance for manufacturers to be able to characterize and understand the HMAs properties on a molecular level, in order to relate them with macroscopic properties (adhesive and mechanical) to design application-specific samples. Moreover, most studies rely on trial-and-error methods to develop HMAs with higher performance. Alternative methods are needed to design HMAs.Polyamide base resins utilized in HMA formulations are usually synthesized from bio-sourced diacids (such as dimer acids) with one or more different diamines (such as alkyl, aromatic, oligo(propylene oxide), piperidinyl-based diamine, and oligo(N-alkyl amine).2,3 Additives are added when specific properties are required for specific applications. They have diverse forms, i.e., particles, small molecules, oligomers, polymers. A few common examples include, carbon black (particles) for coloring, tackifying resins (oligomers or polymers) to improve stickiness, antistatic agents (polymers) to lower the electrical resistivity,4 abrasion-resistant (polymers) to decrease abrasion at the surface, plasticizers (small molecules or oligomers) to dilute polymer entanglement networks and increase flowability,1 stabilizers or antioxidants (small molecules, oligomers or polymers) to provide resistance to heat and oxidation.5 The base resin and the additives are blended or compounded in the extrusion or molding process into a granular, disc, rod, or brick form.
HMAs must strictly comply with industry standards (e.g., AS 4606-2012,6 and ASTM D257-937) for specific applications such as underground mining, where electrical discharges can ignite firedamp and cause explosions. Antistatic agents are added into insulating polymers to reduce electrical discharge by lowering the surface resistivity to be static dissipative (1012 to 105 Ω cm) or conductive (<105 Ω cm).4 Compliance with standard is usually assessed through the measurement of resistivity (surface or volume) and charge decay.8 However, this measures the antistatic agent's performance but does not yield information on its interaction with the resin matrix and its mechanism of action. If they are immiscible, it is unlikely to form (on the microscale) the percolation network required for best performance. Overcoming this poor performance then requires large quantities of antistatic agent, thereby increasing the production costs and possibly affecting adhesive or mechanical properties.
In dimer acid-based polyamide resins, due to the varying triacid and monoacid content, branching is present, and depending on the diamine content nanostructuration is likely to occur. In poly(n-alkyl acrylates) and polyethylene, the short- and long-chain branching affects molecular relaxation processes and causes changes in rheological properties.9,10 The research on dimer acid-based polyamides has focused on understanding their synthesis (or formulation) and their relation to functional properties (such as thermal, tensile and adhesive),2,11,12 while studies correlating chemical structure and functional properties were found to be scarce.
Spectroscopic techniques are valuable to characterize organic materials at the molecular level. Attenuated total reflectance Fourier-transform infrared (ATR-FTIR) spectroscopy is a straightforward and quick characterization technique that yields information on the average chemical composition of samples; it requires no tedious sample preparation.13 Numerous studies have shown the usefulness of ATR-FTIR spectroscopy to characterize the composition of polymeric-based materials, such as a HMAs,14 paints,13 or fibers.15
Solid-state nuclear magnetic resonance (NMR) spectroscopy is one of the most versatile and informative analytical tools available that has long been utilized to characterize the average molecular, and supramolecular structure of samples.161H line width analysis and Carr–Purcell–Meiboom–Gill (CPMG) analysis are potent tools to probe nanodomains and structural heterogeneity in polymer blends17 (such as percolation networks). CPMG allows the determination of transverse relaxation time (T2).18 The T2 for rubber-like materials plays an essential role in functional (adhesive and mechanical) properties as both are governed by the chemical and physical crosslinks of the polymer chains to form networks, the molecular weight between crosslinks, and the heterogeneity of the formed networks.19,20 Since chain mobility is linked to elastomer structure, chemical information can be compared with the functional properties.
Multidimensional NMR spectroscopy offers a diverse range of techniques for the elucidation of slow dynamic processes that are responsible for polymers softening above their glass transition temperature (Tg).21,22 Two-dimensional wideline separation (2D-WISE) is an elegant method to assess the chain dynamics of solid polymers and to correlate the chemical structure to segmental mobility by yielding a 1H spectrum for each resolved carbon resonance.23 The distinction between segmental mobility of specific functional groups gives information on favorable polymer–polymer interactions that affect miscibility; it is also indicative of phase behavior in phase-separated polymer blends.
In this work, the chemical structure and homogeneity of additive incorporation in polyamide adhesives were investigated using ATR-FTIR spectroscopy, solid-state 1H, and 13C NMR spectroscopy. Complementary methods such as X-ray diffraction (XRD), differential scanning calorimetry (DSC), and dynamic-thermal mechanical analysis (DTMA) were used to explore the thermal behavior, long order crystallinity and functional properties. The differences in structure and mobility of the HMA, the additives and the base resin were probed through changes in 1H T2 transverse relaxation time and with 2D-WISE. Furthermore, the changes in molecular dynamics were compared with functional properties to correlate with the viscoelastic and mechanical properties of the HMA.
Experimental
Samples
Ethanol (99%) and adamantane (99%) were purchased from Sigma-Aldrich. Hot-melt adhesives (HMAs) compounded by extrusion were provided as research samples by Imatech (Castle Hill, Australia). Compositions are shown in Table 1. The constituents of the different blends: the base resin (BaseResin1; a polyamide), antistatic agent (Antistatic1; a polyamide-b-poly(ethylene glycol)), abrasion-resistant additive (Antiabrasion1; a para-aramid), and three carbon blacks (CarbonBlack1, CarbonBlack2, and CarbonBlack3) were provided as pure samples. A sample of the purging agent (PurgingAgent1) used to clean the extruder was provided.| Sample | BaseResin1 (%) | Antiabrasion1 (%) | Antistatic1 (%) | CarbonBlack1 (%) | CarbonBlack2 (%) | CarbonBlack3 (%) |
|---|---|---|---|---|---|---|
| HMA1 | 84.99 | 0 | 15 | 0.01 | 0 | 0 |
| HMA2 | 94.99 | 5 | 0 | 0.01 | 0 | 0 |
| HMA3 | 94.9 | 5 | 0 | 0.1 | 0 | 0 |
| HMA4 | 94.9 | 0 | 5 | 0.1 | 0 | 0 |
| HMA5 | 90 | 0 | 0 | 0 | 10 | 0 |
| HMA6 | 95 | 0 | 0 | 0 | 0 | 5 |
| HMA7 | 99.99 | 0 | 0 | 0.01 | 0 | 0 |
Scanning electron microscopy
A JEOL 7001F FEGSEM scanning electron microscopy was used, with a Moran Scientific Microanalysis System (energy dispersive spectroscopy, EDS) and Amptek SDD detector with C2 window. Images were taken at an accelerating voltage of 15 kV and a working distance of 10 mm. The samples were coated with 30 nm of carbon. A backscattered electron detector was used for imaging.X-ray diffraction
HMA1 was prepared by cutting a flat piece of 1 × 1 × 0.3 cm from a HMA pellet. BaseResin1 was prepared by melting the pellets and cutting a flat piece of 1 × 1 × 0.3 cm. Antistatic1 was prepared by crushing a pellet between two circular aluminum plates (2 cm diameter and 1 cm thickness) using a bench vice into a flattened pellet of 0.5 × 0.5 × 0.1 cm. Each sample was placed on a XRD sample holder on top of clay support (Fig. S1, in ESI†). XRD data was acquired on a Bruker D8 Advance diffractometer over the 2θ range from 4° to 80° at a step size of 0.02° with a 12 mm variable slit. Scan time was 4 seconds, with a total scan time of 4 h 22 min. Incident radiation was Cu Kα II (λ = 0.154056 nm) with detection by a Bruker LYNXEYE silicon drift detector. The data was treated with Bruker TOPAS 3 software. Raw data (i.e., 2θ and intensity) was exported, 2θ was transformed to d-spacing (nm) using Braggs equation (eqn (1)):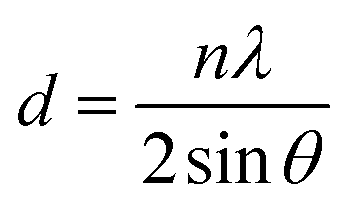 | (1) |
Differential scanning calorimetry
DSC measurements were undertaken on a Netzsch 204 F1 Phoenix under air mixture (80% nitrogen, 20% oxygen). Samples were cooled down to −150 °C, then heated at 10 °C min−1 to 10 °C lower than the onset of degradation temperature, thrice. The onset of degradation temperature of samples was measured with thermogravimetric analysis as the temperature at which 1% mass loss occurs (312 °C for BaseResin1, 274 °C for Antistatic1, 299 °C for HMA2, and 305 °C for HMA7, see Table S12†). The first heating and cooling steps were used to erase the sample's thermal history. Phase transitions temperatures, including Tg, were measured on both the second and third heating cycle with the average reported.Attenuated total reflection Fourier transform infrared spectroscopy
ATR-FTIR spectra were recorded with a Bruker Vertex 70 Spectrometer with a 2 cm−1 resolution and 64 scans. Good coverage of the ATR window crystal (diamond) was ensured by adjusting the sample position, without prior sample preparation, until a real spectrum showed distinct signals. A background was measured before each measurement. The data was recorded, normalized, and baseline corrected using the Bruker OPUS software suite.Solid-state NMR spectroscopy
Samples were packed in zirconia rotors with a 4 mm outer diameter and a 3 mm inner diameter. HMA samples were softened with a heat gun, then cylindrical pieces with the same diameter as the inner rotor diameter were punched out of the pellets (Fig. S2†) and transferred into the rotor. From HMA1, three samples were drawn in the same fashion from different positions of a pellet: HMA1-W from a position close to the outside of the pellet, HMA1-C1, and HMA1-C2 from two positions towards the center of the pellet.Solid-state NMR spectra were recorded on a Bruker DPX200 spectrometer operating at Larmor frequencies of 200 MHz and 50 MHz for 1H and 13C, respectively. A commercial double-resonance, magic-angle spinning (MAS) probe was used. For 1H and 13C single-pulse excitation (SPE-MAS) experiments, the 90° pulse was optimized using adamantane. For 2D-CPMG, the 1H 180° pulse was optimized using adamantane. For 13C cross-polarization (CP-MAS) experiments, power levels were optimized using a mixture of three 13C singly labeled alanines. The 1H and 13C chemical shifts scales were externally referenced using adamantane by setting the CH resonance to 1.64 and 38.48 ppm, respectively.24 Variable temperature experiments were calibrated using lead nitrate at various MAS rates (Table S2†).
1H SPE-MAS NMR spectra of HMA1-C1, HMA1-C2, HMA1-W, the BaseResin1, the Antistatic1, and the CarbonBlack1 were recorded at 10 kHz MAS with a 5 μs 90° pulse, a 10 s repetition delay, and 24 scans at 25, 50, 75 and 100 °C. 13C SPE-MAS NMR spectra of the BaseResin1, the Antistatic1, and the CarbonBlack1 were recorded at 10 kHz MAS, at room temperature and 50 °C, with a 5 μs 90° pulse, a 3 s repetition delay and 31![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 884, 17040 and 7740 scans respectively. 13C SPE-MAS NMR spectra of HMA1-W were acquired at 25, 50, 75, and 100 °C using a 5 μs 90° pulse, a 3 s repetition delay, and 7180, 20877, 33485 and 44574 scans, respectively.
884, 17040 and 7740 scans respectively. 13C SPE-MAS NMR spectra of HMA1-W were acquired at 25, 50, 75, and 100 °C using a 5 μs 90° pulse, a 3 s repetition delay, and 7180, 20877, 33485 and 44574 scans, respectively.
13C longitudinal relaxation times (T1) of HMA1-C2 were estimated at 50 °C and 10 kHz MAS with one-dimensional inverse recovery experiments with a 5 μs 90° pulse, for tested T1 values of 0.2, 0.4, 0.6, 0.7, 0.8, 1, and 2 s, with 109, 454, 272, 256, 152, 77, and 48 scans, respectively. Quantitative 13C SPE-MAS NMR spectra of HMA1-C1, HMA1-C2, and HMA1-W were measured at 50 °C and 10 kHz MAS with a 5 μs 90° pulse, a 9 s repetition delay, and 2665, 7740 and 2665 scans, respectively. The repetition delays were set at least five times longer than the T1 of the signals of interest between 0 and 100 ppm to ensure that the spectra were quantitative (Fig. S16†). The signal-to-noise ratio (SNR) was determined with an in-built function of Bruker Topspin 3.6 (SINO CAL).
1H transverse relaxation times (T2) were determined with a 2D-CPMG experiment25,26 at various temperatures from 23 to 85 °C. The spectra were recorded at 6.5 kHz MAS with a 3.0–3.2 μs 90° pulse, a 6.0–6.4 μs 180° pulse, a 300–320 μs delay between pulses, a time-domain size of 64000, a 0.64 s acquisition time, a 4.36 s repetition delay, and 32 scans. The number of loops (of inter-pulse delay +180° pulse) was incremented in the indirect dimension as follows: 1, 2, 3, 4, 5, 6, 10, 20, 30, 40, 60, 80, 120, 160, 180, 200, 240, 280, 320, 360 and 400, and the signal was fully decayed by the end of it. For each sample, a repeat T2 measurement was made 15 min after the original measurement to check for repeatability and to ensure that the temperature was equilibrated. The data set was then phase-corrected, and baseline-corrected in TopSpin 3.6 software. The data (signal integral vs. delay time) was then linearized and fitted using a multiple-step Levenberg-Marquardt linear fit to a multicomponent, single-exponential decay in Origin 2016 software (see ESI† for more details).
2D-WISE spectra were recorded at 10 kHz MAS and room temperature for HMA1-C1, HMA1-C2, HMA1-W, HMA1, HMA7, and the BaseResin1. A 3–4 μs 90° pulse was used, followed by a 180° pulse in the middle of the evolution time to refocus chemical shifts, and a 0.5 ms contact time for cross-polarization. The acquisition time in the indirect dimension was 0.2 ms with 40 increments unless otherwise specified. The acquisition time on the 13C channel was 25.6 ms with 4776–5144 scans and a 2 s relaxation delay.
Dynamic-thermal mechanical analysis
The DTMA data for HMA7 was acquired using a Netzsch TMA 402 F1/F3 Hyperion instrument with a fused-silica three-point flexural test module. The analysis was carried out at a constant frequency of 16 Hz, a strain of 0.1 N, and a temperature range from −30 to 75 °C at a heating rate of 2 °C min−1. The raw data obtained was Fourier-transformed using Netzsch Proteus Thermal analysis software. Young's modulus (E′) and the loss tangent (tanδ) were determined from the Fourier-transformed data as a function of temperature for the sample.
Shore A hardness
Hardness of HMA7 was measured with a handheld Precision Instrument Shore A digital durometer, that allows fast measurement. The durometer conforms with ISO 868-1986 & ISO 7619 international standards for rubber measurements with samples of a minimum thickness of 0.6 cm. Before each series of measurements, the validity and repeatability of the durometer was checked using known samples in known conditions. Once removed from the fridge, freezer, or oven, samples were measured within approximately 30 seconds to minimize the change in sample temperature. Ten measurements were done across the surface of the same sample for temperatures up to room temperature (−22, 3, and 23 °C), and 20 measurements for higher temperatures (37, 45, 55, 65, 75, 85, 95, 105 and 115 °C).T-peel strength
The samples were prepared according to ASTM D903-9827 with neoprene rubber strips (25.4 × 300 mm) as substrate. The adhesive was poured in between the two neoprene rubber strips sitting in an aluminum mold. The assembly was equilibrated to a specific temperature in an oven. Then the T-peel adhesion was measured at a peel rate of 254 mm min−1 with a tensile testing machine. When testing was carried out above room temperature, the sample assembly started to cool down during measurement, hence, a range of temperatures is given for T-peel strength values to account for that.Results and discussion
Assessment of additives incorporation into the HMA matrix
The spectroscopic analysis of HMA is focused on the quantification of additives in the HMA, compositional heterogeneity, and molecular dynamics. Structure elucidation was carried out to identify functional groups for quantification as well as to probe molecular dynamics. In base resin and HMAs, both are essential to identify whether and how the HMA can be made safer, better performing and cheaper to produce.Investigation of composition by ATR-FTIR spectroscopy
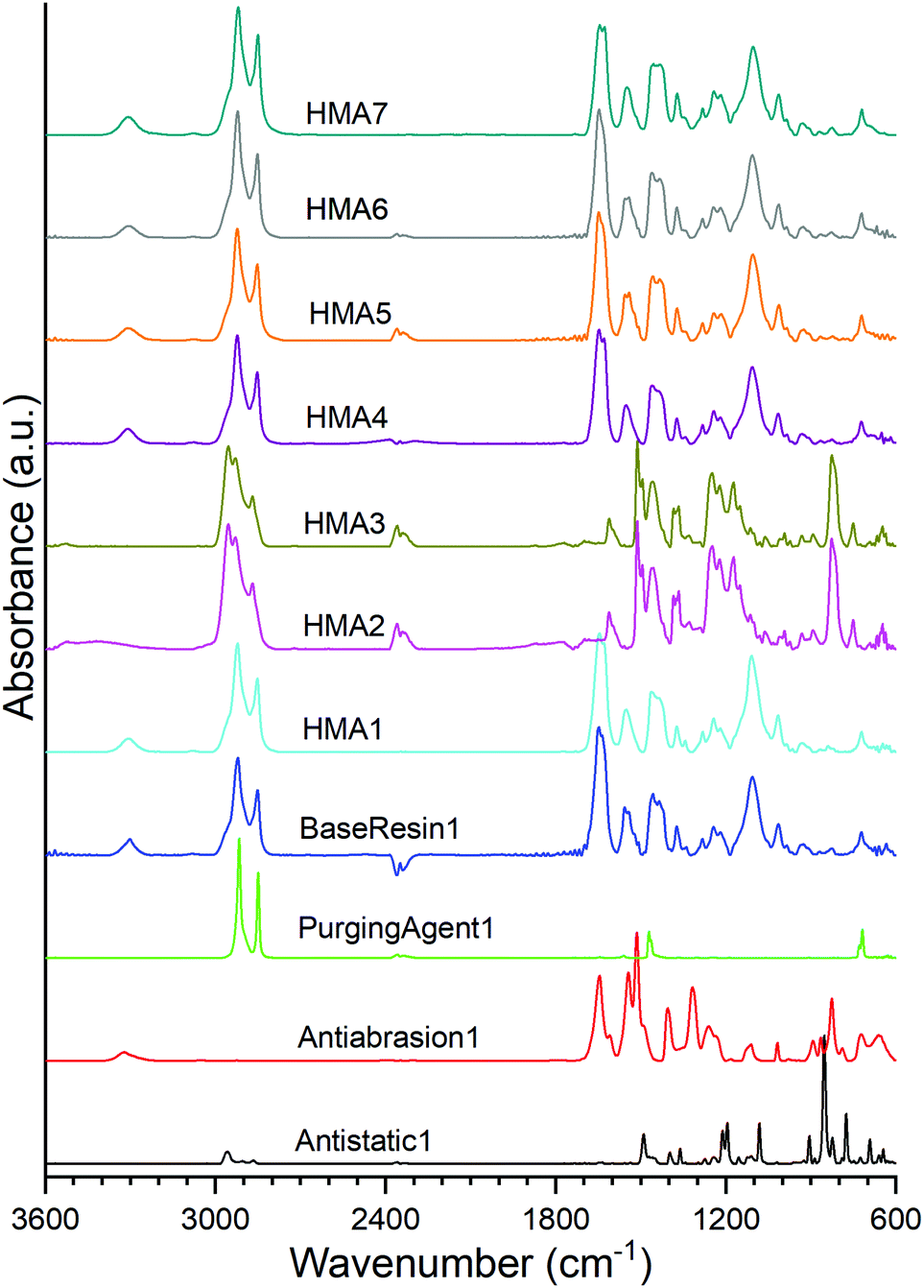
The polyamide nature of BaseResin1 was confirmed (Fig. 1) through the characteristic bands: 3307 cm−1 (N–H in the amide group stretching vibration), 2921 and 2850 cm−1 (CH2 asymmetric and symmetric stretching vibrations), 1644 and 1542 cm−1 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibration in the amide I and II bond), 1464 cm−1 (N–H deformation vibration), and 1243 cm−1 (C–N vibration).28 The band at 722 cm−1 confirmed the presence of vinylic components.29 The band at 1105 cm−1 confirmed the existence of a C–O–C bond within BaseResin1.30 The identified bands in BaseResin1 were indicative of a polyamide mainly composed of dimer acid and oligo(propylene oxide) diamine.
O stretching vibration in the amide I and II bond), 1464 cm−1 (N–H deformation vibration), and 1243 cm−1 (C–N vibration).28 The band at 722 cm−1 confirmed the presence of vinylic components.29 The band at 1105 cm−1 confirmed the existence of a C–O–C bond within BaseResin1.30 The identified bands in BaseResin1 were indicative of a polyamide mainly composed of dimer acid and oligo(propylene oxide) diamine.
 | ||
| Fig. 1 ATR-FTIR spectra of HMA samples, BaseResin1, PurgingAgent1, Antiabrasion1, and Antistatic1. The full band assignment is given in Table S1.† | ||
ATR-FTIR confirmed Antistatic1 contained poly(ethylene oxide) and polyamide through the presence of the bands at 2959 and 2868 cm−1 (CH2 asymmetric and symmetric stretching vibrations), 1489 cm−1 (C–H bending in CH2), 1435 cm−1 (–C–H(cis-) bending (rocking)), 1105 cm−1 (C–O (aliphatic saturated ether)), 1282 cm−1 (C–N (secondary amide, III) + CO (carbonyl) asymmetric deformation), 1243 and 1219 cm−1 (C–N stretching) and 826 cm−1 (CC stretching vibration of aromatic ring).28,31 In the case of the Antiabrasion1 additive, the para-aramid structure was confirmed with bands in the regions of 3307 cm−1 (stretching vibration for N–H in the amide group), 1644 cm−1 (CO (secondary amide, I)), 1611 and 1512 cm−1 (CC stretching vibration of aromatic ring), 1219 and 1243 cm−1–(C–N stretching), 930 cm−1 (CO-NH in-plane) and 826 cm−1 (CC stretching vibration of aromatic ring). Similar to previous studies,32 ATR FTIR was found to be unsuitable for analysis of carbon black additive (see Fig. S3†). In addition, all HMA samples were obtained by extrusion; however, due to band overlap it could not be concluded whether the HMA samples were contaminated with PurgingAgent1 (see ESI section S2†).
The ATR-FTIR spectra of HMA1, HMA4, HMA5, HMA6, HMA7 and BaseResin1 were congruent even though all the HMA samples had different additives. HMA2 and HMA3 ATR-FTIR spectra were congruent, which is consistent with these samples having the same composition; they showed significant differences with that of other HMA samples due to the strong absorption of the para-aramid polymer. There seems to be lower proportions of Antistatic1 present on the surface of HMA1 and HMA4 compared to the proportion of Antiabrasion1 present on the surface of HMA2 and HMA3. The strong absorption of the para-aramid polymer additive on the surface of the HMA sample indicated that the additive was present in its intended location, which was mainly on the surface of the HMA. The fact that the antistatic additive cannot be detected on the surface of the HMA samples indicates that the additive did not get compounded adequately with BaseResin1, meaning that it was not dispersed on the molecular level.
The antistatic agent needs to percolate through the resin matrix to provide the antistatic effect. If it does not, then it means the antistatic agent forms macroscopic aggregates rather than strings that form a network throughout the HMA pellet (Fig. S4†). To further investigate which of both possibilities is the case, the distribution and molecular dynamics of the additives (such as antistatic agent and abrasion resistant) were examined with solid-state NMR spectroscopy.
Determination of average composition and compositional heterogeneity by NMR spectroscopy
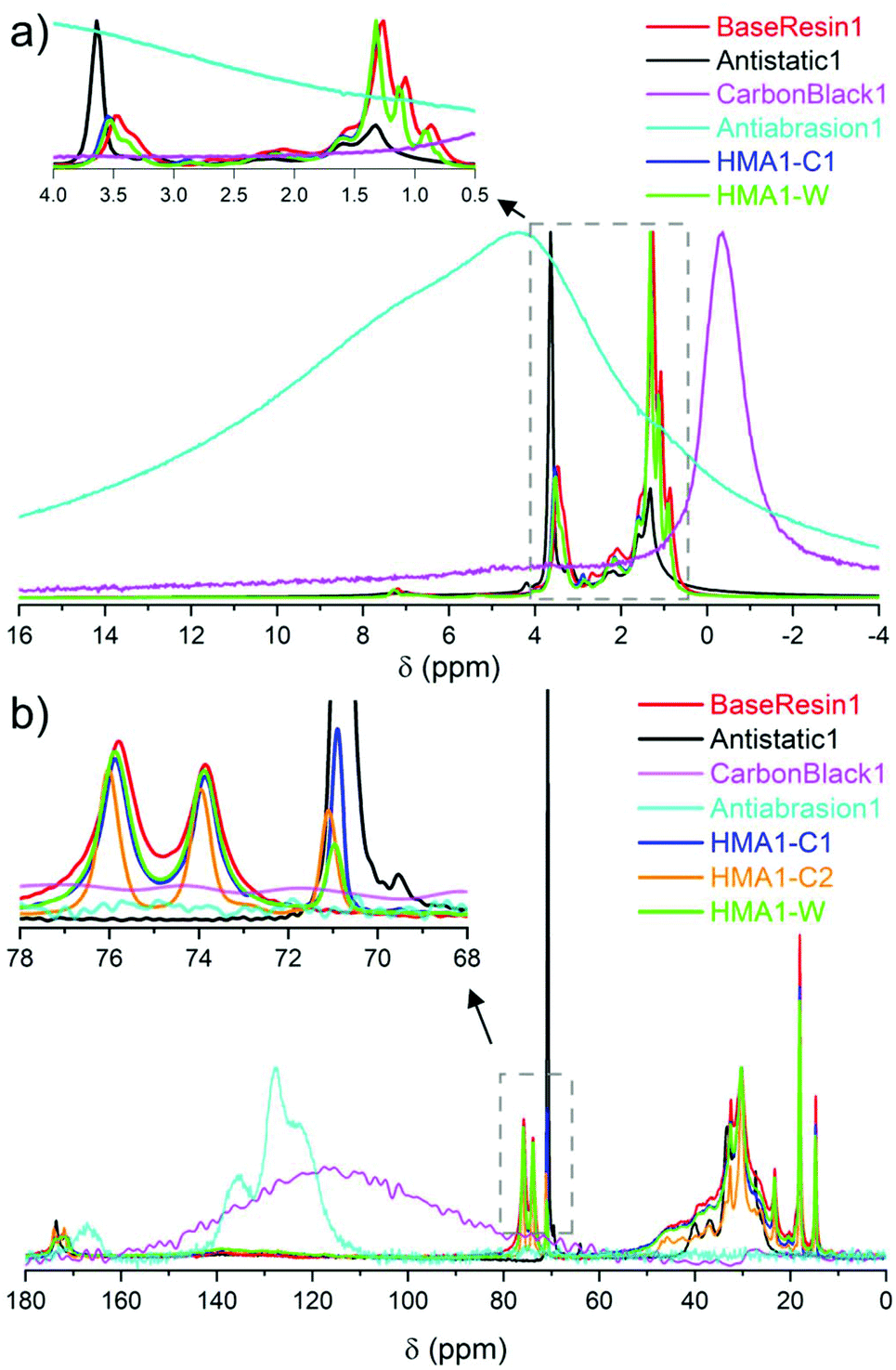 | ||
| Fig. 2 Partial (a) 1H NMR spectra of BaseResin1, Antistatic1, CarbonBlack1 HMA1-C1 and HMA1-W at 100 °C along with Antiabrasion1 at room temperature (normalized to the same maximal signal intensity), and (b) 13C SPE-MAS NMR spectra of BaseResin1, Antistatic1, CarbonBlack1 and Antiabrasion1 at room temperature, and HMA-C1, HMA-C2, and HMA-W at 50 °C (normalized to the same intensity for signals at 30 or 120 or 130 ppm signals, see Table S3† for 13C NMR signal assignment). | ||
The sparse CarbonBlack1 was not detected in HMA samples by 1H NMR, thus its quantification was not possible. Methylene in poly(ethylene oxide)34 from Antistatic1 was detected around 3.5–3.7 ppm but could not be used to quantify Antistatic1 due to signal overlap. Therefore, 13C solid-state NMR was sought to overcome the limited resolution faced in 1H solid-state NMR to quantify additives in HMA.
![[C with combining low line]](https://www.rsc.org/images/entities/b_char_0043_0332.gif) O at 170–175 ppm, –O–HR– at 70–80, –NH–H2–R at 39–50 ppm, –H2– at 21–39, and –H3 at 14–21 ppm were observed (Fig. 2b and Fig. S12–S14, see ESI† for individual spectra and Table S3† for signal assignment). The observed signals confirmed the dimer acid and oligo(propylene oxide) diamine composition of BaseResin1.35–38 The Antiabrasion1 spectrum showed broad aromatic signals around 115–140 ppm, and carbonyl-amide signals 160–175 ppm confirming the para-aramid structure. The CarbonBlack1 spectrum exhibited only a weak signal around 120 ppm (Fig. S15†),39 which made it difficult to distinguish it from the background signal observed on this probe.
O at 170–175 ppm, –O–HR– at 70–80, –NH–H2–R at 39–50 ppm, –H2– at 21–39, and –H3 at 14–21 ppm were observed (Fig. 2b and Fig. S12–S14, see ESI† for individual spectra and Table S3† for signal assignment). The observed signals confirmed the dimer acid and oligo(propylene oxide) diamine composition of BaseResin1.35–38 The Antiabrasion1 spectrum showed broad aromatic signals around 115–140 ppm, and carbonyl-amide signals 160–175 ppm confirming the para-aramid structure. The CarbonBlack1 spectrum exhibited only a weak signal around 120 ppm (Fig. S15†),39 which made it difficult to distinguish it from the background signal observed on this probe.
A distinct signal was observed for the Antistatic1 at 71 ppm in the HMA1-W, HMA1-C1, and HMA1-C2 spectra corresponding to the –O–H2–H2–O– of poly(ethylene oxide) (see the full signal assignment in Table S3†). This signal can be compared to the neighboring signals of BaseResin1 at 74 and 76 ppm to quantify Antistatic1 in HMA1. The Antistatic1 content (FAS) was defined as the percentage of antistatic agent in the sample expressed as the ratio of the concentrations of O–CH2 groups in the antistatic agent and in the base resin. It was quantified in HMA1 samples with eqn (2) to allow the assessment of the spatial distribution in HMA samples:
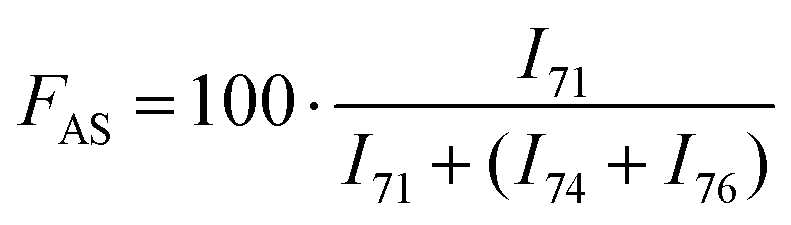 | (2) |
An accurate quantification requires a full resolution of the signals of interest. This was not the case at 25 °C for the signals at 74 and 76 ppm vs. the signal at 71 ppm (Fig. S11†), and performing a deconvolution would affect the accuracy of the quantification. At 50 °C, the resolution (Rs) between the adjacent signals of interest at 71 and 74 ppm was assessed through eqn (3):
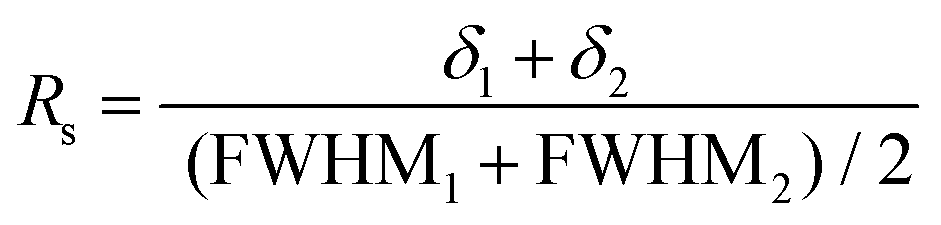 | (3) |
Obtaining quantitative information from 13C SPE-MAS NMR spectroscopy requires adequately long relaxation delays between scans to ensure full sample relaxation.40T1 values were estimated from the signals of interest with one-dimensional T1 inversion recovery on sample HMA-C1 (Fig. S16†). T1 for the signals of interest was shorter than 0.6 s. Hence, 13C SPE-MAS spectra are quantitative for these signals when recorded with a repetition delay longer than 3 s. A longer repetition delay of 9 s was chosen to ensure other signals in the spectra are quantitative as well (Fig. 2b).
The meaningful comparison of Antistatic1 contents in HMA samples required the knowledge of their precision. An empirical relationship was published between the relative standard deviation (RSD) of the degree of branching for polyacrylates and polyolefins and the signal-to-noise ratio (SNR) of the smallest signal used in the quantification (carbon at the branching point) as eqn (4).41
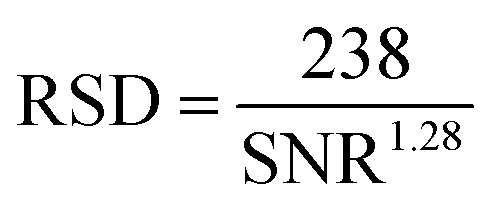 | (4) |
Eqn (4) was applied here using the SNR of the small signal at 71 ppm. The branching quantifications and the present antistatic agent quantification are similar in that their precision is limited by the signal-to-noise ratio of the smaller signal, used as the numerator of the fraction in both cases.41 SNR increased with the measurement time as expected (Fig. S12–S14 and S17 and S18†). Hence spectra with a large number of scans were recorded for the quantification (typically SNR greater than 20 which corresponds to about 100 min measurement, to reduce the RSD to around 5%).
F AS was found to be in decreasing order: HMA1-C2 (21.6 ± 0.5%), HMA1 (15.7 ± 0.4%), HMA1-C1 (11.2 ± 0.3%), and HMA1-W (5.6 ± 0.4%) (for SNR, RSD and standard error values see Table S4†). The percentage of Antistatic1 in an HMA pellet is higher in the center than in regions closer to the edge of the pellet.
Morphology characterized by scanning electron microscopy, X-ray diffraction and differential scanning calorimetry
SEM images of HMA1 and HMA7 were used to visualize the morphological changes in HMA with and without the formulation of Antistatic1 (Fig. 3a and c, for Antistatic1 and BaseResin1 see Fig. S48†). The HMA1 sample revealed two different morphological phases present. X-ray mapping revealed the aggregates dispersed in an HMA matrix to be richer in chlorine (red in Fig. 3d). This would be expected of Antistatic1. HMA7 showed one consistent phase with irregular filler materials. | ||
| Fig. 3 SEM images of (a) HMA7 and (c) HMA1 and X-ray mapping of (b) HMA7 and (d) HMA1. | ||
The organizational structure of HMA1, BaseResin1, and Antistatic1 were characterized by XRD (Fig. 4a). The three diffractograms exhibited broad peaks indicating semi-crystallinity. The influence of the Antistatic1 in the BaseResin1 matrix was not distinguishable in the diffractogram of HMA1. The presence of broad peaks suggests HMA1, BaseResin1, and Antistatic1 contain some nanostructuration (small ordered domains).
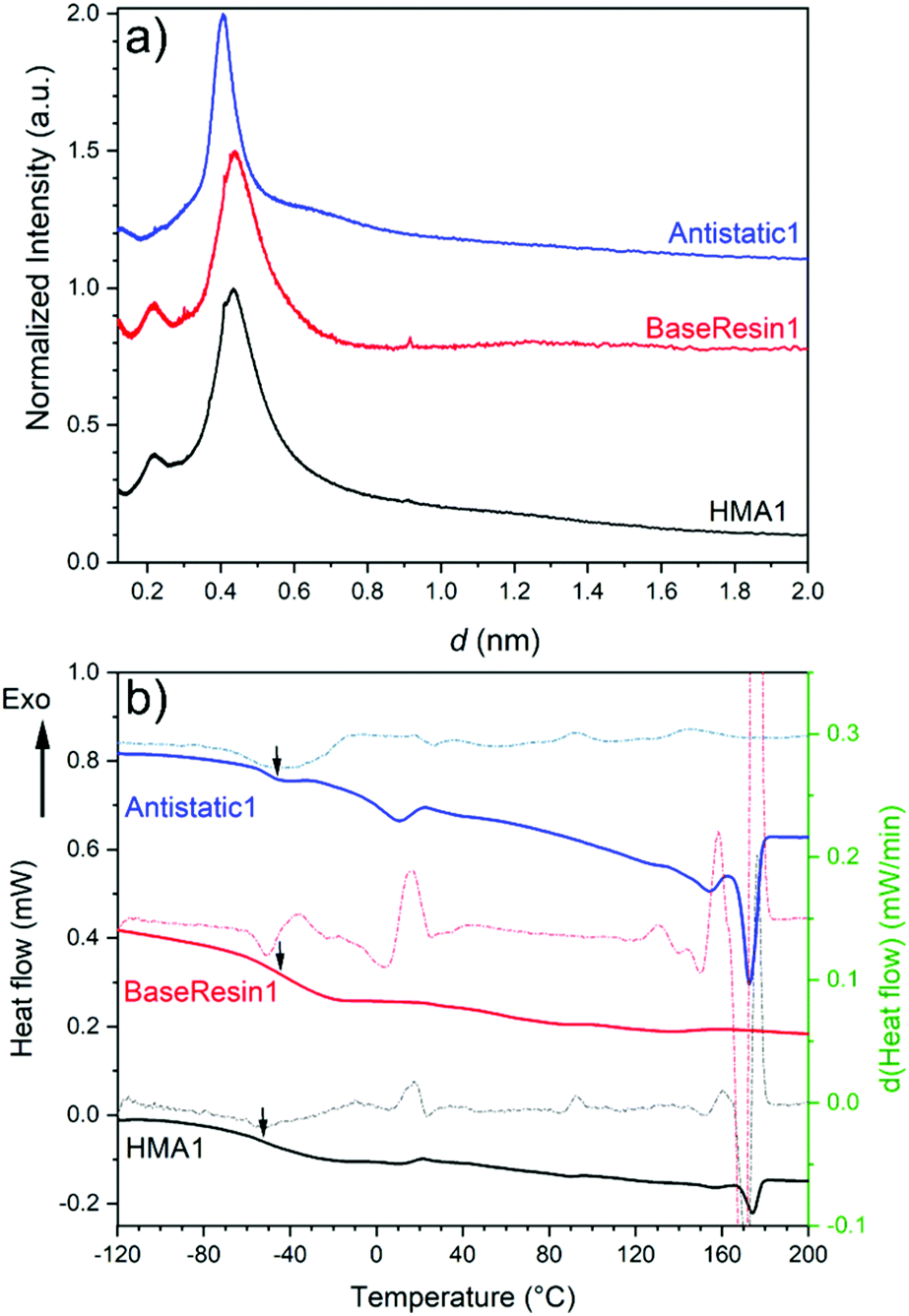 | ||
| Fig. 4 (a) X-ray diffractograms (see Table S11† for peak position, d-spacing, and intensity) and (b) DSC (solid line) and first derivative (dotted line) of Antistatic1 (blue), BaseResin1 (red), and HMA1 (black). Graphs were offset for readability. | ||
Miscibility in polymer blends (such as HMAs formed from BaseResin1 and Antistatic1) can be characterized by DSC. A single Tg generally indicates of full miscibility, while two Tgs indicate incomplete miscibility, therefore, heterogeneity in the material on a scale of tens of nanometers or smaller.42 No significant differences were seen in the Tg calculated from the second heating (Fig. 4b) and third heating (Table S13 and Fig. S52–S55†) in the DSC analysis of Antistatic1 (Tg = −50 °C), BaseResin1 (Tg = −37 °C), the HMA1 (Tg = −55 °C) and HMA7 (Tg = −49 °C). A single Tg does not always mean that the system is homogeneous and consists of a single phase (i.e., fully miscible) as there are several systems which exhibit phase separation but still have a single Tg. A melting transition was observed in HMA1 and Antistatic1 around 175 °C and not observed for BaseResin1. The transition revealed that Antistatic1 is incorporated in the HMA but not mixed at the molecular level. A molecularly dispersed antistatic agent would indeed not have retained crystalline domains large enough for their melting to be detected with DSC. This confirmed the existence of the microphase separation observed by SEM-EDS. The molecular dynamics was investigated to further probe heterogeneity on the nanoscale using various solid-state NMR spectroscopic methods.
Molecular dynamics of the HMA, antistatic agent and the base resin by NMR spectroscopy
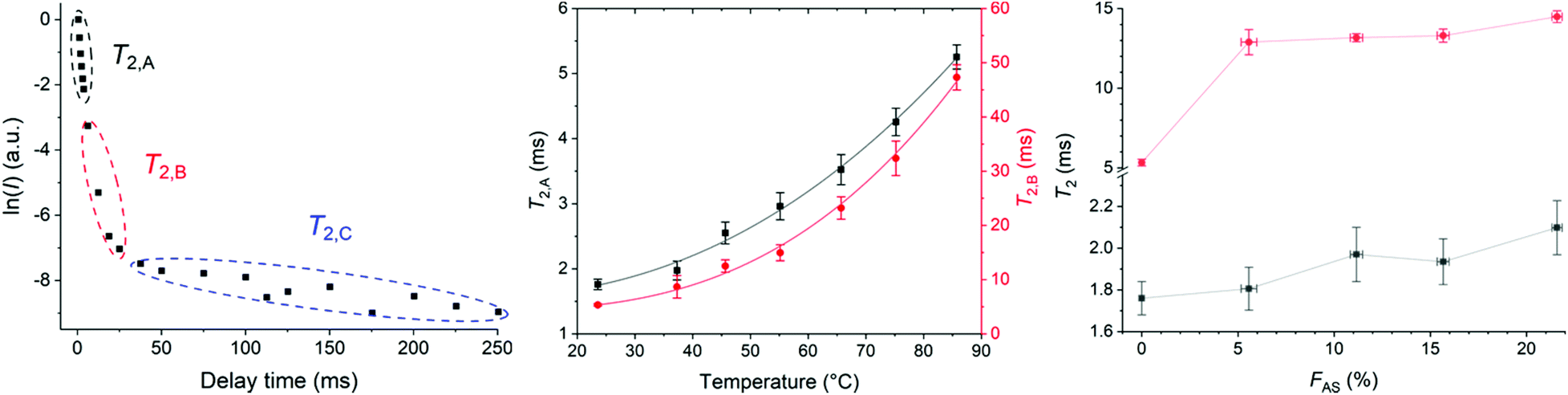 | ||
| Fig. 5 T 2 determined with CPMG. (a) Natural logarithm of the signal integral against delay time for HMA7 at 23 °C, (b) evolution of T2,A (squares) and T2,B (circles) with temperature in HMA7, with power fit (error bars represent the error obtained from the linear fit, black line, y = 1.55 + 1.47 × 10−4·x2.28 with R2 = 0.994 and red line, y = 4.33 + 1.14 × 10−4·x2.88 with R2 = 0.993, respectively). (c) Evolution of T2,A (squares) and T2,B (circles) with FAS in HMA7 and HMA1 samples. | ||
The pre-exponential factors showed the major contribution of component B (∼75–85%) compared to component A (∼10–30%) with a negligible contribution from component C at different temperatures (Table S9†). T2,A and T2,B both increased with increasing temperature indicating higher molecular mobility and decreased sample rigidity (Fig. 5b and Table S5†).43
T 2 relaxation times of HMA1 samples with different FAS were compared (Fig. 5c, Table S7†). Three components were determined with CMPG: the most mobile component with T2,A, the moderately mobile one with T2,B, and the most mobile one with T2,C. The pre-exponential factors showed the major contribution of component A (∼80%) compared to component B (∼20–30%) with a negligible contribution from component C (Table S10†). The increase in T2,A and T2,B with increasing FAS indicated that parts of the sample became more mobile with increasing Antistatic1 which would make the HMA less rigid and hence, could adversely affect the mechanical performance. It was unclear whether nanodomains of Antistatic1 formed within the BaseResin1 matrix as the individual relaxation times could not be directly attributed to Antistatic1 or BaseResin1. Hence, a 2D NMR method, 2D-WISE, was employed to investigate the microphase separations within HMA.
1D 13C NMR spectra extracted from 2D-WISE spectra of BaseResin1, HMA1 samples, and HMA7 (Fig. 6a) showed well-resolved signals. This includes the –O–H2–H2–O– signal of Antistatic1 around 71 ppm (except for BaseResin1 and HMA7), the –O–H(CH3)–H2–O– signals of BaseResin1 around 74–76 ppm, the –H2– broad signal from both Antistatic1 and BaseResin1 centered around about 30 ppm, the –O–CH(H3)–CH2–O– signal of the BaseResin1 around 18 ppm, and the –CH2–H3 signal of the BaseResin1 around 15 ppm. The carbonyl signal was not observed at 170–180 ppm due to the dependence on weak interactions from remote hydrogens, hence, lowered cross-polarization.44
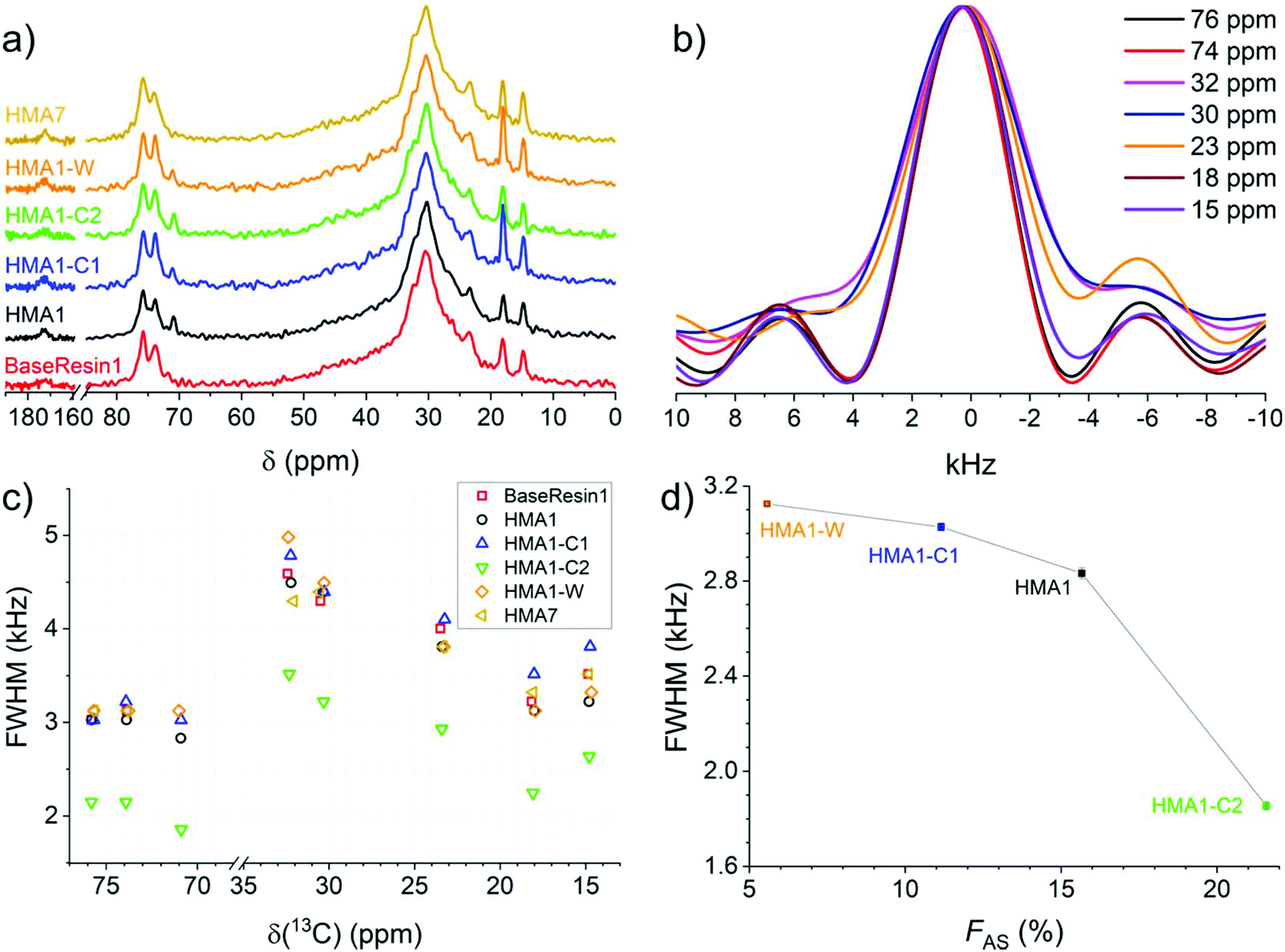 | ||
| Fig. 6 1D NMR spectra extracted from 2D-WISE spectra (normalized to the same maximal signal intensity): (a) 13C NMR spectra for BaseResin1, HMA1 samples, and HMA7, and (b) 1H NMR spectra for HMA7. FWHM of 1D 1H NMR spectra extracted from 2D-WISE spectra of BaseResin1, HMA1 samples, and HMA7 plotted against (c) the corresponding 13C chemical shifts, and (d) FAS for the signal at 71 ppm (see Fig. S37–S41† for 2D-WISE spectra, Fig. S42–S46† for their extracted 1D 1H NMR spectra, and Table S3† for 13C NMR signal assignment). | ||
1D 1H NMR spectra were extracted for the different 13C NMR signals to investigate the local molecular mobility (see Fig. 6b for HMA7, and Fig. S42–S46† for the other samples). For HMA7, the functional groups at 74 and 76 ppm (FWHM of ∼3 kHz) were more mobile than the functional groups at 15 and 18 ppm (FWHM of ∼3.5 kHz), which in turn were more mobile than functional groups at 30 and 32 ppm (FHWM of ∼4.5 kHz). The lower FWHM of the signals at 15 and 18 ppm was attributed to the fast rotation of the methyl groups. The significantly lower FWHM of functional groups at 74–76 ppm compared with that of methylene groups at 30 ppm indicates a higher mobility, and thus a phase separation.
The 1D 1H NMR spectra of HMA1 samples at 71 ppm show a decreasing FWHM with increasing Antistatic1 content (Fig. 6d). When FAS is well below 10%, the line width for the Antistatic1 signal at 71 pm and that of the BaseResin1 matrix at 74–76 ppm are similar indicating that AntiStatic1 either forms separate domains or is incorporated in the domains formed by the –O–CH(CH3)–CH2–O– segments of BaseResin1. The line width of the AntiStatic1 signal at 71 ppm becomes more and more different from that of the BaseResin1 signals at 74–76 ppm, indicating phase separation (Fig. 6c). All line widths in the sample with FAS above 20% are significantly lower than for other samples below a FAS of 20%, indicating plasticization. This increase in molecular mobility throughout the sample means that the antistatic agent present in large quantities acts as a plasticizer and thus, negatively affects the mechanical properties of the material, which is not desirable. It would be interesting to confirm the plasticization through other techniques (assessment of thermal or mechanical properties); however, the heterogeneity of HMA1 (from which HMA1-C1, HMA1-C2 and HMA1-W were sampled) makes this impractical.
Relating the structure of the HMA to its adhesive and mechanical properties
Adhesive and mechanical properties of elastomers are influenced by the molecular weight of the chains, the chemical and physical crosslinks of the chains to form networks, and the heterogeneity of the formed networks.19,20,45 No differences in Shore A hardness were observed on varying the CarbonBlack1 content (Tables S14 and S15†). HMA7's functional properties such as Young's modulus, Shore A hardness, and T-peel strength values decreased with increasing temperature (Fig. S56–S58 and Table S14†). Since chain mobility depends on the elastomers chemical nature and on higher levels of structure (e.g., molecular weight, branching, and crosslinking), the T2 relaxation times of HMA7 were plotted against Young's modulus, Shore A hardness, and T-peel strength at different temperatures (Fig. 7; for values see Table S14†).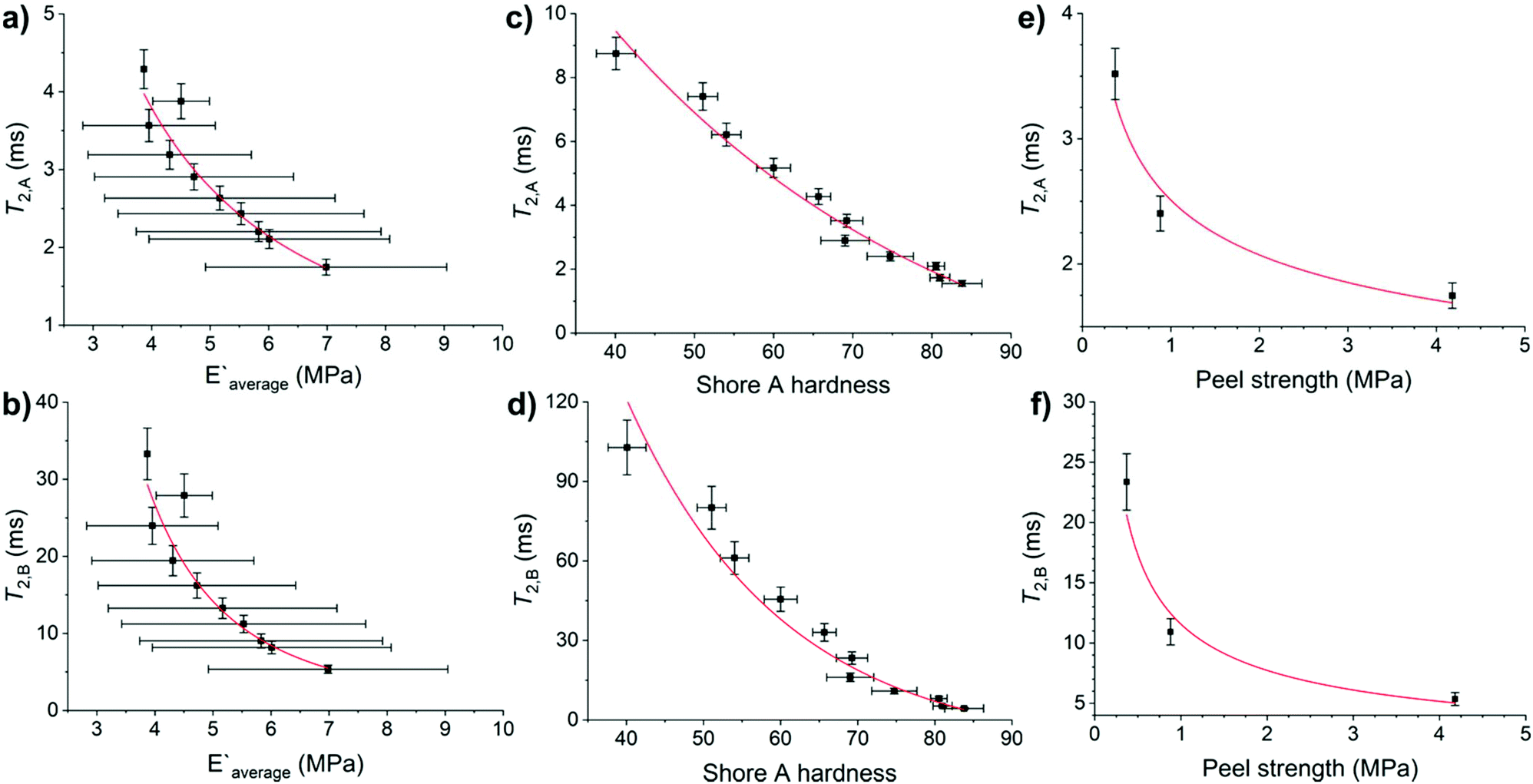 | ||
| Fig. 7 Relation of T2 relaxation times of HMA7 with its functional properties: (a) T2,A and (b) T2,B against Young's modulus, with allometric function fits (red lines), (c) T2,A and (d) T2,B against Shore A hardness, with exponential decay fits (red lines), and (e) T2,A and (f) T2,B against peel strength, with allometric function fits (red lines). Error bars are from linear fits for T2, from standard deviation for Young's modulus and Shore A hardness. Fit details are (a) y = 26.6 × x−1.41, R2 = 0.92, (b) y = 1361.6 × x−2.84, R2 = 0.93, (c) y = 31.3 × 10−x/44.1, R2 = 0.96, (d) y = 951.9 × e−x/20.3, R2 = 0.92, (e) y = 2.51 × x−0.28, R2 = 0.89 and (f) y = 11.6 × x−0.58, R2 = 0.89. | ||
T 2,A and T2,B relaxation times correlate well with Young's modulus and Shore A hardness (R2 = 0.92–0.96 with allometric or exponential fits, Fig. 7a–d). Their relationship with T-peel strength was weaker (R2 = 0.89 with allometric fits, Fig. 7e and f). Both relaxation components correlate well with Young's modulus and Shore A hardness as these functional properties are affected by increasing temperature with an increase in chain mobility leading to reduced rigidity and resistance to stress. T-peel strength is dependent on the chain mobility not only in the HMA but also in the adherend and hence, relations with T2,A and T2,B relaxation times were weaker as for other functional properties.
Conclusions
ATR-FTIR spectra of different HMA adhesive formulations with the same base resin allowed the qualitative identification of the chemical nature of the components and revealed the distribution of additives on the surface of the samples. However, quantifying additives using ATR-FTIR spectroscopy was not possible as the relevant individual signals could not be resolved. 13C SPE-MAS NMR spectroscopy complemented the qualitative identification of the chemical nature of the components from ATR-FTIR, and allowed the quantification of an antistatic agent in HMA samples in relatively short measurement time (typically a few hours). The differences in antistatic agent content in different parts of a sample showed inhomogeneous mixing of the additive during extrusion. The formulation homogeneity was also probed with SEM-EDS, DSC and XRD. SEM-EDS showed the existence of the microphase separation of antistatic agent within the HMA matrix. DSC allowed the observation of the antistatic agent melting transition in a HMA sample indicating its incorporation without being fully dispersed at the molecular level. XRD measurements of a HMA and its antistatic agent showed that both samples exhibited a semi-crystalline structure and the influence of the antistatic agent in the base resin matrix was not distinguishable.The chain dynamics in the HMA formulations was probed with solid-state NMR. 1H NMR relaxation revealed three T2 relaxation times. The relaxation times of the least mobile and moderately mobile components increased with increasing temperature, indicating an increase in molecular mobility as expected for elastomers. The relaxation times of the least mobile components increased with increasing antistatic agent content. 2D-WISE NMR allowed the correlation of chemical structure and molecular mobility and revealed a phase separation in the base resin matrix and the formation of nanodomains with increasing antistatic agent content. 2D-WISE NMR also revealed the plasticization of the whole sample at very high antistatic agent contents which could adversely affect the functional properties.
The T2 relaxation times of the least mobile and moderately mobile components showed good correlations with functional properties such as Young's modulus and Shore A hardness (mechanical properties) and weaker relations with T-peel strength (adhesive properties). The functional and adhesive properties are critical functional properties of hot-melt adhesives. The correlations between molecular dynamics and functional properties showed the suitability of NMR as a non-destructive method with minimal sample preparation to carry out batch analysis on HMA formulations.
In this work, for the first time solid-state NMR was utilized for polyamide resins and adhesives. High-resolution NMR spectroscopy experiments performed are expensive in terms of time and money and also require expert knowledge. Their implementation as standardized methods for quality control is possible via low-field NMR such as benchtop or portable NMR, especially relaxometry.46 This study shows the structural and thermal properties of dimer acid- and oligo(propylene oxide)-based polyamide resins for the first time and opens the way to other polymeric materials to be developed from these resins. The structural characterization of HMA formulations demonstrated here will enable the improvement of HMA formulations regarding the spatial distribution of additives. There are mines all over the world which require different HMAs for different environments. This work aids in the development of better-performing HMAs or of HMAs for application in different climatic conditions.
Conflicts of interest
The first author, Kash Bhullar, is the recipient of the Warwick J. Rule PhD scholarship funded by the company Imatech (see also Acknowledgment section in the manuscript). Richard Wuhrer, Patrice Castignolles, and Marianne Gaborieau are recipients of a partnership grant co-funded by Imatech and of the grant related to the Warwick J. Rule scholarship fully funded by Imatech.The authors have no other known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgements
The authors thank International Material and Technology Pty Ltd (Imatech) for the Warwick J. Rule Scholarship, Western Sydney University for Research Training Program funding, Scott Cheevers, Russell Eggers, Joel Thevarajah, and other (former or current) Imatech staff for providing samples and useful discussions on conditions on the mining site. The authors thank Imatech and Western Sydney University School of Science and Health for funding through a partnership grant in the early stages of the project. MG thanks Western Sydney University for an Academic Development Plan. The authors thank the Advanced Materials Characterisation Facility at Western Sydney University for support and discussions.References
- W. Li, L. Bouzidi and S. S. Narine, Ind. Eng. Chem. Res., 2008, 47, 7524–7532 CrossRef CAS.
- E. Hablot, B. Donnio, M. Bouquey and L. Averous, Polymer, 2010, 51, 5895–5902 CrossRef CAS.
- C. Rossitto, in Handbook of adhesives, ed. I. Skeist, Chapman & Hall, New York, NY, USA, Boston, Massachusetts, USA, 3 edn, 1990, ch. 28, pp. 478–498 Search PubMed.
- M. C. Grob and E. Minder, Plast. Addit. Compd., 1999, 1, 20–26 CrossRef CAS.
- S. T. Oeltjen and D. B. Malcolm, USA Patent5989385, 1999, p. 6 Search PubMed.
- AS 4606-2012: Grade S fire resistant and antistatic requirements for conveyor belting and conveyor accessories, Standards Australia, Sydney, NSW, Australia, 2017 Search PubMed.
- ASTM D257-14: Standard Test Methods for DC Resistance or Conductance of Insulating Materials, ASTM International, West Conshohocken, PA, USA, 2014, p. 18 Search PubMed.
- V. Dudler, M. C. Grob and D. Merian, Polym. Degrad. Stabil., 2000, 68, 373–379 CrossRef CAS.
- M. Gaborieau, R. Graf, S. Kahle, T. Pakula and H. W. Spiess, Macromolecules, 2007, 40, 6249–6256 CrossRef CAS.
- F. J. Stadler, C. Piel, K. Klimke, J. Kaschta, M. Parkinson, M. Wilhelm, W. Kaminsky and H. Munstedt, Macromolecules, 2006, 39, 1474–1482 CrossRef CAS.
- M. Feldmann and A. K. Bledzki, Compos. Sci. Technol., 2014, 100, 113–120 CrossRef CAS.
- X. M. Chen, H. Zhong, L. Q. Jia, J. C. Ning, R. G. Tang, J. L. Qiao and Z. Y. Zhang, Int. J. Adhes. Adhes., 2002, 22, 75–79 CrossRef CAS.
- S. Prati, E. Joseph, G. Sciutto and R. Mazzeo, Acc. Chem. Res., 2010, 43, 792–801 CrossRef CAS.
- E. Orgiles-Calpena, F. Aran-Ais, A. M. Torro-Palau and C. Orgiles-Barcelo, Int. J. Adhes. Adhes., 2016, 70, 218–224 CrossRef CAS.
- N. Vasanthan and D. R. Salem, J. Polym. Sci., Part B: Polym. Phys., 2001, 39, 536–547 CrossRef CAS.
- H. W. Spiess, Macromolecules, 2017, 50, 1761–1777 CrossRef CAS.
- F. Mellinger, M. Wilhelm and H. W. Spiess, Macromolecules, 1999, 32, 4686–4691 CrossRef CAS.
- C. Hedesiu, D. E. Demco, R. Kleppinger, A. A. Buda, B. Blumich, K. Remerie and V. M. Litvinov, Polymer, 2007, 48, 763–777 CrossRef CAS.
- Y. Nakamura, M. Adachi, Y. Kato, S. Fujii, M. Sasaki, Y. Urahama and S. Sakurai, J. Adhes. Sci. Technol., 2011, 25, 869–881 CrossRef CAS.
- V. M. Litvinov and A. A. Dias, Macromolecules, 2001, 34, 4051–4060 CrossRef CAS.
- P. Caravatti, P. Neuenschwander and R. R. Ernst, Macromolecules, 1986, 19, 1889–1895 CrossRef CAS.
- H. W. Spiess, Macromolecules, 2010, 43, 5479–5491 CrossRef CAS.
- K. Schmidt-Rohr, J. Clauss and H. W. Spiess, Macromolecules, 1992, 25, 3273–3277 CrossRef CAS.
- C. R. Morcombe and K. W. Zilm, J. Magn. Reson., 2003, 162, 479–486 CrossRef CAS.
- H. Y. Carr and E. M. Purcell, Phys. Rev., 1954, 94, 630–638 CrossRef CAS.
- S. Meiboom and D. Gill, Rev. Sci. Instrum., 1958, 29, 688–691 CrossRef CAS.
- ASTM D903-98: Standard Test Method for Peel or Stripping Strength of Adhesive Bonds, ASTM International, West Conshohocken, PA, USA, 2017, p. 3 Search PubMed.
- P. G. Kadam and S. T. Mhaske, J. Polym. Mater., 2013, 30, 65–78 CAS.
- E. Hablot, A. Tisserand, M. Bouquey and L. Avérous, Polym. Degrad. Stab., 2011, 96, 1097–1103 CrossRef CAS.
- B. Mailhot, S. Morlat-Thérias, M. Ouahioune and J.-L. Gardette, Macromol. Chem. Phys., 2005, 206, 575–584 CrossRef CAS.
- Z. Varga, J. Mihaly, S. Berenyi and A. Bota, Eur. Polym. J., 2013, 49, 2415–2421 CrossRef CAS.
- W. Zhang, R. S. Blackburn and A. Dehghani-Sanij, Scr. Mater., 2007, 57, 949–952 CrossRef CAS.
- R. M. Silverstein, F. X. Webster and D. Kiemle, in Spectrometric Identification of Organic Compounds, John Wiley & Sons, Inc., Hoboken, New Jersey, USA, 7th edn, 2005, ch. 3, pp. 127–203 Search PubMed.
- Y. C. Yu and W. H. Jo, J. Appl. Polym. Sci., 1994, 54, 585–591 CrossRef CAS.
- T. Ding, Q. Y. Liu, R. Shi, M. Tian, H. Yang and L. Q. Zhang, Polym. Degrad. Stab., 2006, 91, 733–739 CrossRef CAS.
- B. S. Holmes, W. B. Moniz and R. C. Ferguson, Macromolecules, 1982, 15, 129–132 CrossRef CAS.
- M. V. Mokeev and V. V. Zuev, Eur. Polym. J., 2015, 71, 372–379 CrossRef CAS.
- M. R. Krejsa, K. Udipi and J. C. Middleton, Macromolecules, 1997, 30, 4695–4703 CrossRef CAS.
- H. Darmstadt, C. Roy, S. Kaliaguine, G. Xu, M. Auger, A. Tuel and V. Ramaswamy, Carbon, 2000, 38, 1279–1287 CrossRef CAS.
- M. Gaborieau, L. Nebhani, R. Graf, L. Barner and C. Barner-Kowollik, Macromolecules, 2010, 43, 3868–3875 CrossRef CAS.
- M. Gaborieau, S. P. S. Koo, P. Castignolles, T. Junkers and C. Barner-Kowollik, Macromolecules, 2010, 43, 5492–5495 CrossRef CAS.
- D. S. Kaplan, J. Appl. Polym. Sci., 1976, 20, 2615–2629 CrossRef CAS.
- V. M. Litvinov and J. P. Penning, Macromol. Chem. Phys., 2004, 205, 1721–1734 CrossRef CAS.
- J. L. Koenig, in Spectroscopy of Polymers, Elsevier Science, New York, 2nd edn, 1999, ch. 8, pp. 383–384 Search PubMed.
- V. M. Litvinov, W. Barendswaard and M. van Duin, Rubber Chem. Technol., 1998, 71, 105–118 CrossRef CAS.
- D. Besghini, M. Mauri and R. Simonutti, Appl. Sci., 2019, 9, 1–33 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Sample preparation, ATR-FTIR, and 1H and 13C NMR spectra with detailed signal assignment, NMR probe temperature calibration, 1H NMR relaxation detailed data treatment, 2D-WISE spectra, scanning electron microscope images, thermogravimetric analysis, X-ray diffraction, differential scanning calorimetry, mechanical and adhesive tests. See DOI: 10.1039/d0py01348j |
| This journal is © The Royal Society of Chemistry 2021 |