
Theoretical investigation on lithium polysulfide adsorption and conversion for high-performance Li–S batteries
Jianbo
Li†
,
Yanru
Qu†
,
Chunyuan
Chen
,
Xin
Zhang
* and
Mingfei
Shao
 *
*
State Key Laboratory of Chemical Resource Engineering, Beijing University of Chemical Technology, Beijing 100029, P. R. China. E-mail: shaomf@mail.buct.edu.cn; zhangx@mail.buct.edu.cn
First published on 25th November 2020
Abstract
Lithium–sulfur (Li–S) batteries have shown great application prospects as next-generation energy storage systems due to their high theoretical capacity and high energy density. However, the practical application of Li–S batteries is still hindered by several challenges, such as their sluggish sulfur redox kinetics and shuttle effect of lithium polysulfides (LiPSs). To date, significant research has been focused on the confinement adsorption and catalytic conversion of LiPSs using theoretical or/and experimental methods. Among them, theoretical calculations are highly attractive to observe complex LiPS conversion reactions, which facilitate the rational design of S mediators for high-performance Li–S batteries. In this review, we summarize and discuss the recent advances in the adsorption and conversion of LiPSs from the viewpoint of theoretical calculations. Moreover, a set of theoretical principles to guide the screening of suitable host materials for Li–S batteries is presented and discussed. Finally, some personal insights about the future challenges and the focus of research in this field are presented, which will push a milestone step toward high-efficiency and long-life Li–S batteries.
 Jianbo Li | Jianbo Li received his Master's Degree in 2018 from Beijing University of Chemical Technology (BUCT). Currently, he is a Ph.D. candidate under the supervision of Prof. Mingfei Shao in BUCT. His research interests currently focus on the fabrication of integrated electrodes for advanced batteries, such as Li–S batteries, Zn-ion batteries and Na-ion batteries. |
 Xin Zhang | Xin Zhang received her PhD Degree from Xiamen University in 2009. She worked as a Postdoctoral Researcher in Beijing Normal University from 2009 to 2011, after which she joined the staff of BUCT. She was a Visiting Scholar at the Western Michigan University (in 2007) and the University of Minnesota, Twin Cities (in 2015). Her current interests are theoretical and computational chemistry, focusing on the study of the solid surface properties, catalytic reaction mechanisms and electronic structures of functional materials. |
 Mingfei Shao | Mingfei Shao received his PhD Degree from Beijing University of Chemical Technology (BUCT) in 2014, after which he joined the staff of BUCT, and was promoted to Full Professor in 2018. He was also a visiting student at the University of Oxford in 2013. His research interests focus on host–guest materials and their applications in electrochemical and photoelectrochemical energy storage and conversions. |
1. Introduction
Lithium–sulfur (Li–S) batteries are considered as one of the most promising next-generation alternative batteries due to their high theoretical capacity of 1675 mA h g−1 and energy density of 2600 W h kg−1. Meanwhile, sulfur cathode with the merits of natural abundance, low cost and low toxicity has potential for large-scale commercial applications.1–5 Unfortunately, the practical performance of Li–S batteries is still far below the theoretical expectation because of their low sulfur utilization and shuttle effect of intermediate lithium polysulfides (LiPSs). Meanwhile, the insulation of S and discharge products (Li2S2/Li2S) hinders the electron transfer, which limits the reaction kinetics of S transformation. Therefore, it is very urgent to propose reasonable strategies to ameliorate the adsorption and catalytic conversion of LiPSs in Li–S batteries.6–12To achieve highly efficient LiPS transformation, the key strategy is to select an appropriate matrix with the properties of excellent conductivity and appropriate adsorption and catalytic conversion capacity for LiPSs. Conductive carbonaceous materials (such as porous carbon, carbon nanotube (CNT), and graphene)13–15 were firstly considered as an S host to overcome the electric insulation of S and its reduced products, which can greatly improve the utilization of S. However, the shuttle effect of intermediates LiPSs is hard to overcome by pure carbon materials, causing fast decay of capacity and low coulombic efficiency. Thus, numerous works focusing on polarizing carbon materials to anchor LiPSs by strong chemical absorption and formation of Li bonds16,17 have been reported, such as heteroatom (e.g., N, O, S, and P)18–22 and functional group doping (e.g., phosphate, sulfonic, and amino groups).23–27 Recently, chemical entrapment was investigated in Li–S batteries due to the relatively strong interactions between LiPSs and polar substrates (e.g., metal and metal compounds).28–30 Nevertheless, the incomplete conversion of LiPSs to the final product (Li2S2/Li2S) still occurred due to the sluggish redox kinetics.31,32 To overcome this issue, numerous studies have been carried out to investigate materials with high catalytic activities, which can accelerate the conversion kinetics and alter the activation energy.33,34 Unfortunately, although extensive related research on Li–S batteries have been reported, the multi-electron transfer and multi-step solid–liquid–solid phase conversion in the reaction process make the study of the LiPS adsorption and catalytic processes particularly complicated.
Theoretical calculations, such as density functional theory (DFT) calculation, are expected to become an alternative approach for revealing the adsorption and conversion process of LiPSs. Specially, the theoretical calculations involved in Li–S batteries are mainly divided into two aspects, i.e. the influence of the structure and components of the S host and the thermodynamics and kinetics involved in the chemistry of Li–S batteries.35 Based on the optimized geometrical and electronic structures of the host materials, the corresponding molecular orbital energy level, orbital interaction, charge distribution, electrochemical potentials, and bonding information can be obtained. The thermodynamic parameters involved in the conversion between LiPSs, including energetics, enthalpy, entropy, and Gibbs free energy, can be also obtained by theoretical calculations and statistical mechanics, which are very beneficial to reveal the electrochemical reaction of Li–S batteries. Based on the above information, the energy, enthalpy and Gibbs free energy changes in the conversion process of various S species can be determined, which facilitates the confirmation of possible conversion pathways to confirm the reaction mechanism in Li–S batteries. To date, various density functional calculations have been extensively conducted to provide effective guidance in designing an advanced Li–S battery system. For example, Zhang et al.36 showed the complete mechanistic understanding of the interaction between non-metallic monolayer materials and LiPSs with the assistance of DFT. Wu et al.37 used DFT to research the thermodynamic behavior of LiPS intermediates, showing that the stable molecular structure for short chain LiPSs is linear sawtooth, while the ring shape is stable for long chains. Notably, they demonstrated the linear relationship between the chemisorption of LiPSs and specific capacity, directly bridging the relevance between adsorption ability and battery performance. Therefore, theoretical calculations play a vital role in exploring the transformation of LiPSs, which can provide a new approach for selecting materials with suitable adsorption and catalytic conversion capacity of LiPSs.
Recently, some impactful and insightful reviews have been well summarized on Li–S batteries considering theory and experiment.11,35 For example, Zhang et al.35 summarized the recent progress in computational approaches applied to Li–S batteries and emphasized the significance of combining theory and experiment. However, there are no reviews providing a comprehensive introduction to quantify the adsorption and conversion capabilities of LiPSs on various host materials based on theoretical calculations. Thus, in this review, we highlight the irreplaceable role of theoretical calculations in understanding the complex electrode reaction of Li–S batteries and the mechanism of the adsorption and conversion of LiPSs. Subsequently, we summarize the research progress of theoretical mechanisms in the strategies of designing mediator (e.g., modified carbon materials, MOF/COF, polymer, and metal-based materials, Fig. 1) to enhance the adsorption capacity of LiPSs and regulate the catalytic ability of intermediate LiPS products. According to the theoretical investigation results, we further present rational theoretical guidance for screening suitable carrier materials, which will help reduce useless attempts and achieve a substantial improvement in the performance of Li–S batteries. Finally, based on the state-of-art progress, some personal insights about the future challenges and the focus of research work in this hot research field are concluded, which will accelerate the commercialization of advanced Li–S batteries.
 | ||
| Fig. 1 Illustration of mechanism research for the confinement adsorption and catalytic conversion in Li–S batteries on various host materials. | ||
2. Theoretical investigation of LiPS confinement adsorption
The shuttle effect of soluble LiPSs results in a low utilization of S and poor cycle stability, making it difficult to realize their large-scale application Li–S batteries. Therefore, the rational design of S hosts to restrain LiPSs has become the key to Li–S battery research. Benefitting from their excellent electrical conductivity, large specific surface area and light weight, carbonaceous materials have been widely used as the S host in the Li–S battery system.13,14,22,24 However, it is difficult for non-polar carbon materials to completely suppress the shuttle effect due to the weak interaction between LiPSs and carbon atoms, which usually cause fast capacity decay during the cycling process.Based on this, extensive approaches have been proposed to influence the polarity of pure carbon by introducing functional heteroatoms or functional groups.25,26 Modified carbonaceous materials can greatly improve the adsorption capacity for LiPSs efficiently through both physical and chemical trapping. When modified carbonaceous materials are used as the host of S, the performance of Li–S batteries greatly improves. Nevertheless, it is hard to judge the detailed working mechanism only from experimental aspects. Therefore, DFT calculations have been used to analyze the effects of absorption energy, geometrical and electronic structure variations of various heteroatom doping on modified carbonaceous materials.
2.1. LiPS confinement adsorption on heteroatom-doped carbon
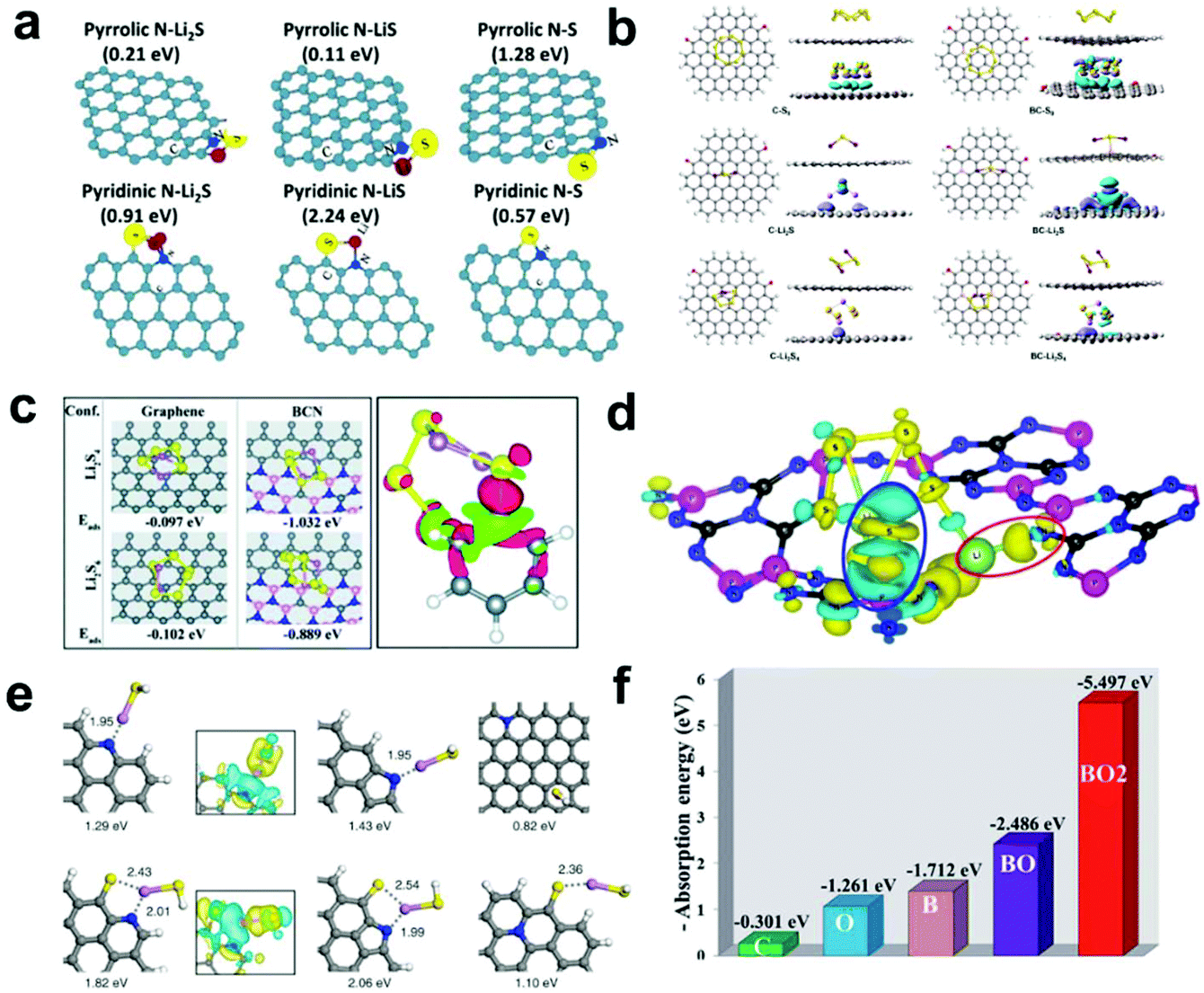 | ||
| Fig. 2 (a) Optimized geometric configurations for graphene sheet containing: pyrrolic N and pyridinic N to absorb S species. Reprinted with permission from ref. 44. Copyright 2014, Wiley-VCH. (b) Optimized geometric configurations of S species absorbed on pristine carbon and B-doped carbon surfaces. Reprinted with permission from ref. 49. Copyright 2018, Springer Nature. (c) Optimized geometric configurations of S species on pristine graphene and borocarbonitride nanotubes (left) and the charge density difference map of a B atom with an Li2S4 molecule (right). Reprinted with permission from ref. 58. Copyright 2020, The Royal Society of Chemistry. (d) Schematic diagram of the charge density difference after the adsorption of Li2S6 on carbon nitride phosphorus-22. Reprinted with permission from ref. 59. Copyright 2019, American Chemical Society. (e) Optimized configurations for the binding of LiSH to N-doped graphene and N,S-codoped graphene. Charge density difference isosurfaces are shown in the insets. Reprinted with permission from ref. 54. Copyright 2016, the American Chemical Society. (f) Corresponding adsorption energies on different surfaces of multi-walled CNTs. Reprinted with permission from ref. 46. Copyright 2017, The Royal Society of Chemistry. | ||
As the neighbor element to N, boron (B)-doping is also widely employed in Li–S batteries. B-doping can reduce the electron density of delocalized π electrons on carbon atoms and make the carbon surface positively polarized, which helps to adsorb negatively charged LiPSs.45–48 Yu et al.49 reported a hybrid structure of carbon spheres/graphene with a high B-doping content of 6.51 wt%. The corresponding DFT calculations showed that the B-doped carbon framework possessed a larger interaction energy with S species (e.g., −1.18, −2.79, and −1.15 eV for S8, Li2S, and Li2S4, respectively) than that of pristine carbon (−0.67, −0.82 eV for Li2S and Li2S4, respectively) (Fig. 2b). Furthermore, the obvious charge density difference map deformations further prove the much stronger chemical interactions between the B-doped carbon framework and S species, which effectively suppress the shuttle effect and improve long-term cyclability. Besides, S-doped carbon materials also show a positive influence on attracting LiPSs and inhibiting dissolution.50 DFT calculations illustrated that the interaction between doped S atoms and LiPSs is stronger than that with C atoms, which will efficiently provide high binding energy with LiPSs and inhibit the shuttle effect.
To guide the screening and design of scaffolding carbonaceous materials with chemical dopants, a series of heteroatom-doped (B, N, O, F, P, S and Cl) graphene nanoribbons was modeled and their binding behaviors with sulfur species were evaluated by Zhang's group.51 Based on the configuration, binding energy, bond length, and charge transfer of the carbon skeleton with different doped heteroatoms, they concluded the rational design principles of heteroatom-doped carbon materials. On one hand, the doped atoms need to possess lone pairs of electrons, higher electronegativity and smaller atomic radius that matches Li. On the other hand, the introduced heteroatom can form delocalized π bonds and stable bond structures. With these conditions fulfilled, suitable chemical doping of N or O heteroatoms with an extra pair of electrons or even co-doping is more favorable to facilitate a better anchoring effect than that of other heteroatoms (B, F, S, P, and Cl), which delivers an improvement in the reversible capacity and coulombic efficiency.
Moreover, Manthiram et al.54 studied the absorption mechanisms of different modified types of graphene materials, including N, S and co-doped materials, and their adsorption capacity for LiPSs was further investigate by theoretical calculations (Fig. 2e). Through detailed structural analysis, they proposed three adsorption mechanisms between different interfaces and LiPSs (the dissociative mechanism, the destructive mechanism, and the intact adsorption mechanism). Besides, the DFT results also showed that N, S co-doped graphene can suppress the shuttle effect most effectively, which is consistent with previous experimental results. Similarly, Tao et al.46 conducted DFT calculations to expound the absorption mechanism of B and O dual-doped multi-walled CNTs. The corresponding adsorption energies on different surfaces of the multi-walled CNTs are shown in Fig. 2f. Evidently, the adsorption ability is enhanced as the amount of B and O doping increases, and doping species containing both B and O show the best adsorption ability.
According to the above results, carbon material doped with heteroatoms as an S host can deliver better electrochemical properties in Li–S batteries. However, it is difficult to analyze the influence of the structure and size of carbon material on the performance of Li–S batteries by theoretical calculations.
2.2. LiPS confinement adsorption on functional group-modified carbon
Introducing functional groups in the carbon matrix is also an effective method to improve both the binding property to LiPSs and conductivity of carbon. Oxygen-containing functional groups can form a strong attraction between Li (with low electronegativity) and O (with high electronegativity), which is beneficial to adsorb LiPSs and inhibit the shuttle effect.60 Sawangphruk et al.61 investigated the influence of the interaction between LiPSs and functionalized carbon fiber paper on the electrochemical performance of an Li–S battery by DFT. Fig. 3a shows the graphene model modified with different functional groups (–OH, –COOH, and –C–O–C), and the corresponding binding energy. According to the calculation results, it can be observed that the Li atoms in LiPSs form a strong Li bond with the treated carbon fiber paper and ethylenediamine-modified carbon fiber paper, which effectively inhibits the shuttle effect. Besides, they also found that the “initial discharge capacity and capacity retention rate” are approximately proportional to the “adsorption energy”. Thus, based on this discovery, the adsorption energy can be considered an important factor in determining the performance of Li–S batteries. | ||
| Fig. 3 (a) Proposed models of graphene layer and calculated binding energies of different oxygen functional groups. Reprinted with permission from ref. 61. Copyright 2018, the American Chemical Society. (b) Structure and IR spectra of polyethylenimine/Li–S species deduced from DFT analysis. Reprinted with permission from ref. 63. Copyright 2016, the American Chemical Society. (c) Mechanism of the ion selectivity and anchoring to the LiPSs on the surface of S–rGO. Reprinted with permission from ref. 25. Copyright 2017, the American Chemical Society. (d) Theoretical calculation of molecular binding among the carbon surface, CH3–O–PO3H2 and S or Li2S. Reprinted with permission from ref. 26. Copyright 2018, Elsevier. (e) Nitrogen-doped multilayer graphene-coated Celgard PP separator and its mechanism to chemically bind LiPSs. Reprinted with permission from ref. 64. Copyright 2019, the American Chemical Society. | ||
Amine-functionalized carbon materials also show effective anchoring of sulfur species in the Li–S cathode.62 Lou's group24 reported that modifying amino functional groups on reduced graphene can effectively restrain the sulfur and discharge products, and thus the specific capacity of Li–S batteries was still maintained at 80% after 350 cycles. Besides, Archer et al.63 designed amine-functionalized CNTs in a cathode by tethering polyethylenimine polymers to hydroxyl/carboxyl-functionalized multiwall CNTs, which showed an effective anchoring effect for LiPSs in the Li–S battery cathode. In addition, DFT calculations verified the strong affinity between the LiPSs and polyethylenimine-functionalized CNTs (Fig. 3b). The binding energy of LiPSs on polyethylenimine (1.24 eV) is much higher than that of graphene (0.34 eV). Meanwhile, DFT calculation analyzed the comparison of the IR spectra of the pure polyethylenimine and LiPS/polyethylenimine mixture. A new peak at 640 cm−1 appears in the mixture of polyethylenimine and LiPSs, which is attributed to the formation of an Li–N bond in the mixture, indicting the strong bonding between the amine groups and LiPSs. When used in Li–S batteries, the S/polyethylenimine–CNT composite cathodes provided a strong LiPSs anchoring ability and stable battery cycle performance.
Moreover, the sulfonic groups on carbon materials have been proven to improve the utilization of soluble LiPSs and more efficiently convert Li2S2/Li2S. Wen et al.25 ingeniously proved that the sulfonic groups interact with the terminal Li ion in LiPSs via the Lewis acid–base principle (Fig. 3c). They found that the binding energy between the sulfonic group and LiPSs is more stable than that between LiTFSI and the sulfonic group, and thus it is more inclined to anchor LiPSs via an Li bond. Meanwhile, Zhang et al.26 reported a dihydrogen phosphate-modified carbon matrix prepared from luffa sponge via phosphoric treatment (Fig. 3d). Based on DFT calculations, the binding energy of S and LiPSs on CH3–O–PO3H2 was determined to be 1.24 eV and 1.81 eV, which are much higher than that of pristine carbon (0.83 eV and 0.30 eV), respectively, showing the significant enhancement of binding to S and LiPSs.
To reveal the synergistic impact of polar functional groups on the cycling performance of Li–S batteries, Knibbe et al.64 calculated the adsorption energies between multi-architectural graphene with functional groups (N, –OH, –COOH, and –SO3−) and LiPSs using DFT (Fig. 3e). The DFT results showed that the longer the S chain, the weaker the adsorption capacity of the functional groups. Meanwhile, LiPSs are more likely to form an Li–O bond with oxygen in the –OH group, which can contribute to achieving LiPS binding and accelerated charge transfer. In addition, due to the coordinated effect of pyridinic N and –SO3−, modified graphene exhibits a stronger binding energy with LiPSs, leading to a higher areal capacity in Li–S batteries.
The above results indicate that the introduction of electron-rich functional groups can effectively inhibit the shuttle of LiPSs, which improves the cycle stability of the battery. However, the stability of functional groups may be affected by the complex organic electrolytes.
2.3. LiPS confinement adsorption on MOF/COF/polymers
As a new class of crystalline organic–inorganic hybrid materials, metal organic frameworks (MOF) have many unique physical and chemical properties, such as high surface area, unique pore structure, diverse chemical composites and facile synthesis, and thus they have attracted increasing attention in the field of energy storage and conversion.65–70 Especially in Li–S batteries, MOF materials can be employed as a host material to load S and confine LiPSs, or as a separator to suppress the shuttle effect.71–73 Xiao et al.74 reported a novel nickel-based MOF (Ni–MOF), which could remarkably immobilize LiPSs and achieve high capacity retention up to 89% after 100 cycles at 0.1 C. To better understand the functioning mechanism, DFT calculations were performed to investigate the interactions between Ni–MOF and LiPSs. The results of the calculated binding energies indicated that LiPSs with different chain lengths are quite stable within the pores of Ni-MOF. In addition, the interaction becomes stronger when the chain length of LiPSs increases. Thus, to further prove the strong interaction between transition metal ions and LiPSs, they synthesized a Co-MOF and used it as an S host for Li–S batteries. The electrochemical performance results showed that Co-MOF had better conductivity, but it delivered poor cycle stability. Subsequently, theoretical calculations confirmed that the poor stability of Co-MOF is due to the poor coordination ability between Co and LiPSs and the lower binding energy with LiPSs than that of Ni-MOF (Fig. 4a). Thus, the above result proved the Lewis acid–base interaction between MOF and LiPSs, which may provide important insights and opportunities to develop Li–S battery technology.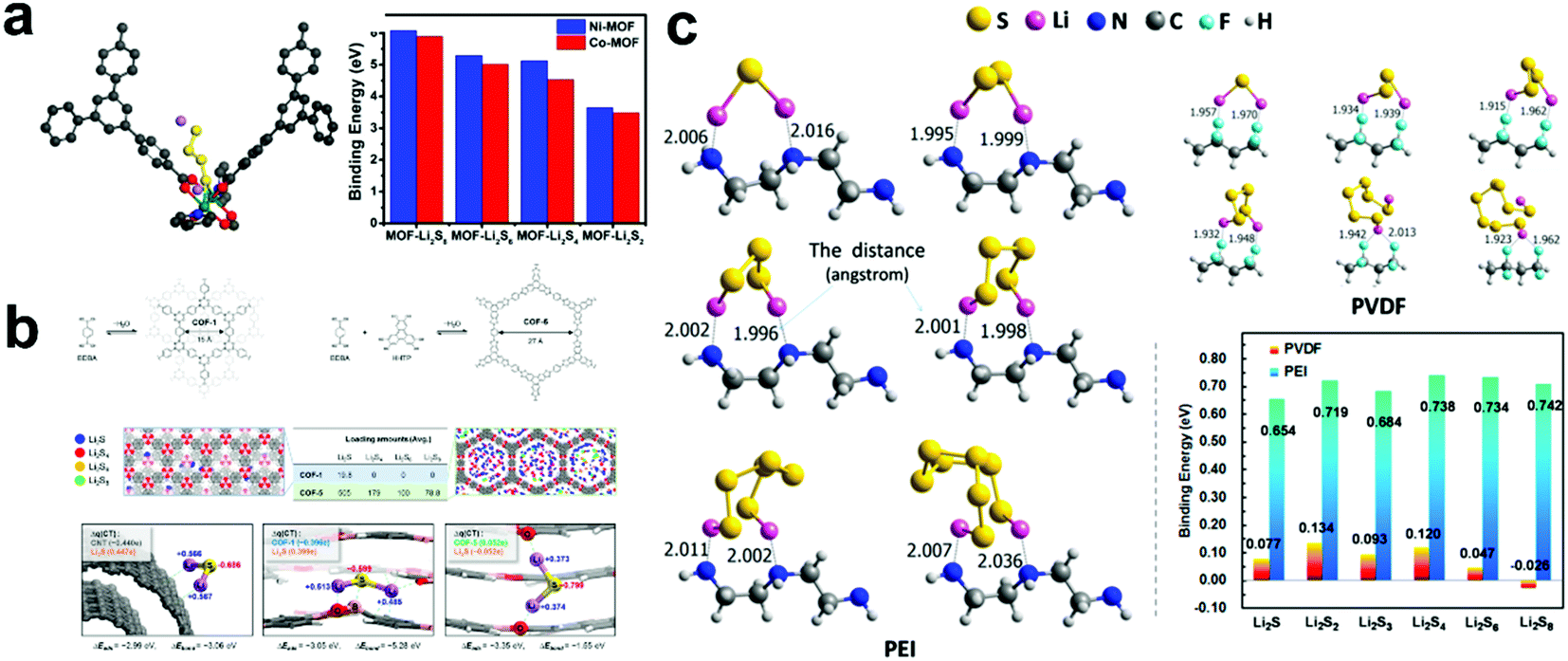 | ||
| Fig. 4 (a) Illustration of the interaction between LiPSs and Ni–MOF and the binding energies of LiPSs to Ni–MOF or Co–MOF. Reprinted with permission from ref. 74. Copyright 2018, The Royal Society of Chemistry. (b) Structures of COF-1 and COF-5. Initial binding configurations of LiPSs species in the COF-1 (blue box, left) and COF-5 (green box, right) and binding configurations, total adsorption energies, and chemical bond formation energies of Li2S on the CNT surface (left) and in the bulk of COF-1 (center) and COF-5 (right). Reprinted with permission from ref. 77. Copyright 2016, the American Chemical Society. (c) Possible interactions and theoretically calculated adsorption binding energies between LiPSs and PVDF and the linear PEI, respectively. Reprinted with permission from ref. 88. Copyright 2018, the American Chemical Society. | ||
Covalent organic frameworks (COFs) possess the advantages of structural diversity, low density, high thermal stability and permanent porosity, and thus have attracted considerable attention in catalysis and especially in Li–S batteries.75,76 Lee et al.77 proposed the concept of molecularly designed, hierarchical porous chemical traps for LiPSs in Li–S batteries. Based on this concept, they fabricated a “microporous COF-net on mesoporous CNT-net” hybrid architecture as a barrier layer, which significantly improved the performance of Li–S batteries. Subsequently, the influence of pore size on the LiPS adsorption capacity was further explored through DFT calculation. COF-1 (micropore size = 0.7 nm) and COF-5 (2.7 nm) were selected as models for the analysis (Fig. 4b) and the total absorption energy calculations (including bond formation, reconstruction of the host structure and structural change of LiPSs) and local density of states of the LiPS–absorbed host was analyzed. The results showed that COF-1 with a well-designed micropore size had better selective binding to LiPSs, which can significantly improve the performance of Li–S batteries.
Recently, polymers also have received a great deal of attention as promising materials in Li–S batteries due to their good conductive and electrochemical stability with various roles.78–87 Ye et al.88 proved that the commercially available water–soluble linear polyethyleneimine, [–(CH2)2–NH–]n, is a bifunctional polymer binder (Fig. 4c). Subsequently, to further quantify this conclusion, they studied the bonding strength between LiPSs and linear polyethylenimine via DFT calculations, and compared it with the most commonly used commercial binder, poly(vinylidene difluoride). The results showed that the binding energy between polyethylenimine and LiPS (0.654–0.742 eV) is much higher than that between LiPSs and poly(vinylidene difluoride) (<0.134 eV). In addition, electrochemical tests also proved that linear polyethylenimine can significantly improve the cathode performance, thereby increasing the capacity and slowing down the capacity decay.
Although MOF/COF/polymers show better performances than nonpolar carbon materials in trapping soluble LiPSs via strong chemical bonding, the conductivity of MOF/COF/polymers is still not satisfactory to obtain a good rate capability when used as S host materials. Moreover, due to the complex molecular structure and multi-level pores of MOF/COF/polymers, the theoretical calculation and experimental results usually show a certain deviation.
3. Mechanism research of LiPS catalytic conversion on metal-based materials
Besides the effective adsorption of LiPSs, the reversible conversion of insoluble Li2S to soluble LiPSs during cycling is still a huge challenge. However, due to the potential difference between the different components, and since the oxidation of insoluble short-chain Li2S requires a higher activation energy, it is an effective strategy to perform a reasonable host design to degrade the reaction activation energy and promote mutual conversion.Recently, metal-based materials, such as metal, metal oxides, metal sulfides, metal nitrides, and metal carbide, have been regarded as the most promising catalysts to suppress the shuttle effect and improve the kinetics of the LiPS redox reaction.89–95 Based on various known or unknown materials, theoretical calculation can quickly screen suitable catalysts, which can significantly reduce the number of experiments.
3.1. LiPS catalytic conversion on metals
Arava et al.96 firstly demonstrated that the LiPS shuttle process in Li–S batteries can be controlled by electrocatalysis. They applied graphene to load Pt and Ni nanoparticles (Pt/graphene and Ni/graphene, respectively) to ensure a high surface area, and their Tafel plots and corresponding exchange current density values were derived to verify their catalytic effect during the charge/discharge process. The experimental results showed that both Pt/graphene and Ni/graphene could reduce the overpotential and improve the specific capacity. Furthermore, He et al.97 proved the coordinated adsorption of LiPSs by Pt and Ni as a metal catalyst via theoretical calculations. The calculation results showed that the adsorption capacity of the Pt@Ni core–shell catalyst for LiPSs is higher than that of its single components (Pt and Ni) (Fig. 5a). Meanwhile, the Pt@Ni core–shell catalyst (0.101 eV) had a much lower decomposition barrier for insoluble Li2S than its single components (Pt: 0.486 eV and Ni: 0.388 eV), indicating that it has a positive effect on the oxidation process of Li2S. Therefore, the bimetallic catalyst exhibited a coordinated catalytic effect, which not only realized the effective adsorption of LiPSs, but also realized the catalytic oxidation of Li2S, resulting in an improvement in the specific capacity and cycle stability of Li–S batteries.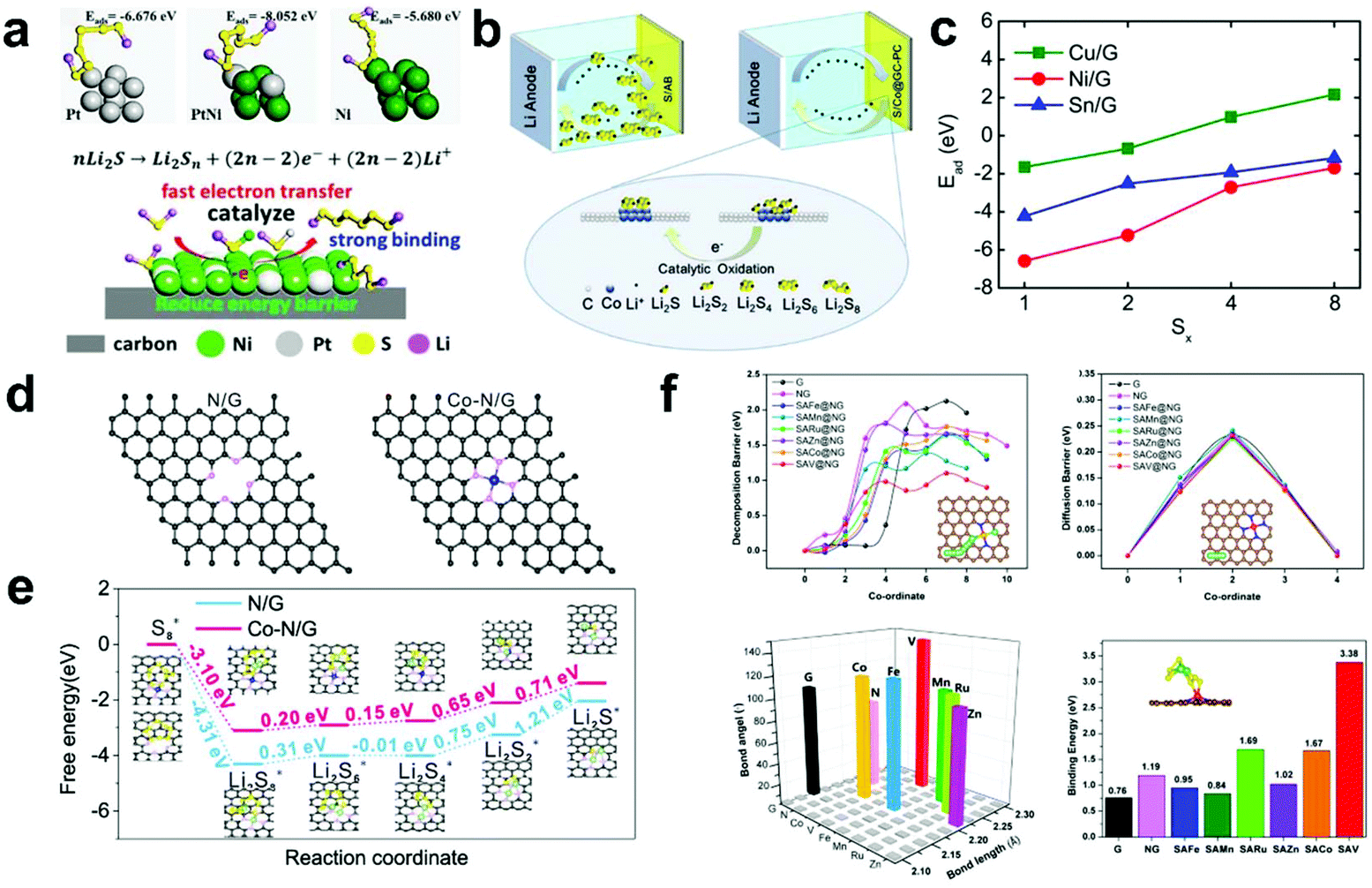 | ||
| Fig. 5 (a) Simulation of the interaction between LiPSs and catalyst and schematic illustration of the catalytic oxidation mechanism. Reprinted with permission from ref. 97. Copyright 2019, Wiley-VCH. (b) Schematic diagram for the effect of Co on the catalytic decomposition of LiPSs. Reprinted with permission from ref. 98. Copyright 2018, the American Chemical Society. (c) Calculated adsorption energies of LiPSs on the surface of metal/graphene systems. Reprinted with permission from ref. 103. Copyright 2018, the American Chemical Society. (d) Structures of nitrogen-doped graphene and Co–nitrogen-doped graphene and (e) energy profiles for the reduction of LiPSs on nitrogen-doped graphene and Co–nitrogen-doped graphene substrates. Reprinted with permission from ref. 110. Copyright 2019, the American Chemical Society. (f) Theoretical understanding of Li2S decomposition, Li-ion diffusion, bond angle and length of Li2S and binding energy on different substrates. Reprinted with permission from ref. 111. Copyright 2020, the American Chemical Society. | ||
Although noble metal catalysts show excellent catalytic conversion capabilities, it is difficult from them to meet large-scale applications due to their high cost and scarce reserves. Owning to the fact that S species easily form chemical bonds on the surface of transition metals, transition metals also show effective ability to bind LiPSs species. In addition, the ability of transition metals to bind LiPSs is related to their d-band center and the P-band centers of S species. Therefore, it is of great significance to select suitable transition metals to adsorb LiPSs. Besides, introducing substrates, such as graphene, can optimize the position of the d-band and improve its adsorption capacity.
Metal cobalt (Co) is the most widely researched metal due to its superior catalysis and anchoring ability. Huang et al.98 reported a Co@graphitic carbon–porous composite derived from ZnCo–MOF-5 microtubes and conducted DFT calculations to reveal the effect of Co on the catalytic decomposition of Li2S (Fig. 5b). On the 111-crystal plane of Co, the adsorption energy for Li2S is as high as 4.39 eV, indicating a strong interaction between Li2S and Co. In addition, the decomposition barrier of Li2S on Co (0.13 eV) is much lower than that of carbon materials (1.8 eV), indicating that the Co catalyst can reduce the decomposition activation energy. Thus, the introduction of a Co catalyst not only improves the adsorption capacity of LiPSs, but also effectively reduces the decomposition barrier, thereby improving the cycle stability and specific capacity in Li–S batteries.
To explore the influence of different metals on the adsorption capacity of LiPSs, Wang et al.99 further investigated the interaction of Cu, Ni, and Sn with graphene or defective graphene with LiPSs via DFT studies (Fig. 5c). The calculation results showed that composites of metal and graphene can effectively improve the overall ion/electronic conductivity. Besides, they can form a strong chemical bond to anchor LiPSs. Meanwhile, they found that the localized d states of Cu and Ni can undergo electron transfer, thereby improving the adsorption energy of LiPSs, while for the main group metal Sn, it can hardly change the adsorption energy of LiPSs.
Compared with the original graphene, the doped modified graphene can show the effect of synergistic adsorption and catalytic conversion with the metal catalyst.100,101 Wu et al.102 designed super hierarchical Co-embedded N-doped porous carbon nanosheets as a host for S. According to the results of their theoretical calculations, the adsorption energy of Li2S4 on the Co substrate (0.83 eV) is higher than that of graphene (0.54 eV) and N-doped graphene (0.55 eV). Meanwhile, the charge analysis showed that electrons are transferred from Co to N-doped graphene, which enables the N atoms to obtain additional pairs of electrons and helps to form Li–N bonds with LiPSs. Due to the electron transfer effect and the strong chemical interaction, the decomposition of Li2S is accelerated with the low energy barrier of 1.27 eV and 0.85 eV for Co-embedded graphitic N-doped graphene and Co-embedded pyridinic N-doped graphene, respectively. Thus, the above theoretical calculation results indicate that Co-embedded pyridinic N-doped porous carbon nanosheets play a vital role in synergistically binding LiPSs and accelerating the decomposition of Li2S.
To gain a thorough understand of the metal-embedded N-doped carbon matrix, a series of metal–nitrogen/graphene (including Cr, Mn, Fe, Co, Ni, and Cu in metal–N4 graphene) anchoring and catalytic conversion effects on LiPSs were designed by Ding’ group.103 The theoretical calculation results showed that Cr–, Mn–, Fe–, Co–, and Cu–nitrogen/graphene deliver the strongest interaction with LiPSs (Fig. 5c). This proves once again that the synergy between the metal and N atoms not only effectively binds the soluble LiPSs to inhibits the shuttle effect, but also guarantees the conversion effect of LiPSs in Li–S batteries.
Single-atom catalysts (SACs) are dispersed catalysts in monodisperse atoms on a substrate, which show the maximum increase in catalytic activity and provide a model system for in-depth exploration of catalytic reaction pathways.104–109 To reveal the catalytic mechanism of single atoms in Li–S batteries, Wan et al.110 explored the reaction process of LiPSs on different substrate materials (N-doped graphene with and without Co atoms) via theoretical calculations (Fig. 5d and e). From the calculation results, the formation and decomposition of solid Li2S/Li2S2 need to overcome a high Gibbs free energy, which is the rate-limiting step in the whole conversion process. In addition, the DFT results also proved that the Co atom N-doped graphene interface has a lower the formation and dissociation energy of Li2S (1.37 eV and 1.43 eV, respectively) than that of N-doped graphene (2.06 and 2.29 eV, respectively), which further proves that the Co–N–C centers in Co atom N-doped graphene serve as active centers and can accelerate the phase transition to improve the utilization of S.
Furthermore, to quickly screen suitable SAC catalysts candidates, Cui et al.111 designed 10 composite materials (including graphene, N-doped graphene, single-atom Fe, Mn, Ru, Zn, Co, Cu, V, and Ag on N-doped graphene) for the decomposition of Li2S via theoretical simulations. Subsequently, they succeeded in loading various SAC catalysts on graphene by controlling the experimental conditions. Among the 10 composite materials, they found that the single V active catalysis sites exhibit the smallest barrier for decomposition of Li2S (1.10 eV, Fig. 5f), indicating that it can effectively promote the decomposition of Li2S. When the single V active catalysis was used in Li–S batteries, it showed a higher specific capacity and good stability, which is consistent with the theoretical calculation results. This work offers theoretical guidance for the precise synthesis of single-atom catalysts and greatly reduces the workload of screening, which provides opportunities for the development of advanced Li–S batteries.
According to the results of experiments and calculations, the introduction of metals can effectively improve the redox reaction kinetics and S utilization. However, the reported theoretical calculation models are all from the perspective of single atoms or clusters, which are not consistent with actual materials. Thus, in future, it is necessary to reduce the size of the catalyst and increase the degree of its dispersion to further improve the utilization efficiency of the catalyst.
3.2. LiPS catalytic conversion on single metal compounds
Among the various metal oxides, TiO2 shows the strongest ability to adsorb LiPSs due to the strong chemical interaction between Ti4+ and LiPSs. In addition, TiO2 also exhibits the ability to catalytically convert LiPSs. However, the conductivity of TiO2 and its ability to bind LiPSs need to be further improved for the real application of Li–S batteries. Zhang et al.117 designed oxygen-deficient TiO2 to be used as a novel functional host in Li–S batteries (Fig. 6a). In their work, Li2S4 and the TiO2 (100) surface was selected as the research model (Fig. 6b). The theoretical calculation results showed that the introduction of oxygen vacancies effectively inhibits the shuttle of LiPSs and improves the catalytic conversion kinetics of S and LiPSs.
 | ||
Fig. 6 (a) Crystal and electronic structures of bulk TiO2 after geometry optimization. (b) Tilted and top view of Li2S4 adsorbed on TiO2 (100) with oxygen deficiency. Reprinted with permission from ref. 117. Copyright 2019, the Royal Society of Chemistry. (c) Adsorption test, adsorption binding energies and relative models of LiPSs on the surface of metal oxides. Reprinted with permission from ref. 118. Copyright 2016, Nature. (d) Optimized geometries of Li2S8 on RuO2 (110) and graphene surfaces; adsorption binding energies of LiPSs on RuO2 (110) and neat graphene; and adsorption test for neat graphene and graphene/RuO2 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. Reprinted with permission from ref. 119. Copyright 2019, Elsevier. :1. Reprinted with permission from ref. 119. Copyright 2019, Elsevier. | ||
Furthermore, Cui et al.118 explored the valid option supported by the proposed appropriate criteria for oxide selection. They synthesized a series of non-conductive metal oxide nanoparticle-decorated carbon flakes. DFT calculations were conducted to reveal the binding energies and sites of these compounds. The most favorable binding site on the metal oxides of Li2S is two Li atoms bonding with the oxygen atom, and the binding energy between Li2S and CeO2, Al2O3, La2O3, MgO, and CaO is 6.33, 7.12, 5.85, 5.71, and 5.49 eV, respectively (Fig. 6c). Fast diffusion can effectively hinder the shuttle effect and accelerate the redox kinetics. Therefore, DFT calculations were also conducted to investigate the diffusivity of the S species on the surface of various metal oxides. The calculated results and electrochemical performances show that MgO, La2O3, and CeO2 with a carbon matrix as the cathode materials can achieve high capacity and excellent cycling performance due to the optimized balance between absorption and surface diffusion.
The above polar metal oxides show strong adsorption capacity for LiPSs, but their own poor conductivity limits their application prospects. Considering this problem, Kim et al.119 prepared a graphene/RuO2/S composite and used it as a host for S cathodes, which showed a good conductivity and catalytic performance. Subsequently, the strong interaction between RuO2 and LiPSs was further studied through theoretical calculations. On the entire surface of RuO2, Li in the LiPSs is preferentially combined with the oxygen atom of RuO2, and S forms a bond with the Ru atoms on the RuO2 (110) plane (Fig. 6d). This result indicates that the polar RuO2 nanoparticles have a strong interaction with the LiPSs, which can effectively suppress the shuttle effect. Therefore, the composite electrode exhibited an excellent electrochemical performance with high residual capacities (389 mA h g−1 after 800 cycles at 1 C).
Although the unique advantages of oxides cause them to be widely studied, they still have the potential risk of the decomposition of organic electrolytes and most of them suffer from poor conductivity, which can lower the rate capability. Therefore, conductive bases are always required to couple with metal oxides with rational structures when applied in Li–S batteries.
Due to their good electrical conductivity and strong ability to adsorb LiPSs, cobalt sulfides (CoxSy, Co3S4, CoS2, and Co9S8) are considered the most promising catalysts in Li–S batteries.122–124 Mao et al.125 prepared a novel hybrid catalyst structure via the in situ growth of nano-CoS2 in 3D honeycomb-like hierarchical porous graphitic carbon for Li–S batteries. The systematic experiment proved that effectively adsorbed and catalytically transformed LiPSs were achieved on the hierarchical porous graphitic carbon. In addition, theoretical calculations further showed that the composite material exhibited a strong anchoring effect on LiPSs (−2.65 eV) and enabled a reduction in the conversion overpotential for the conversion soluble Li2S4 to insoluble Li2S2 (Fig. 7a).
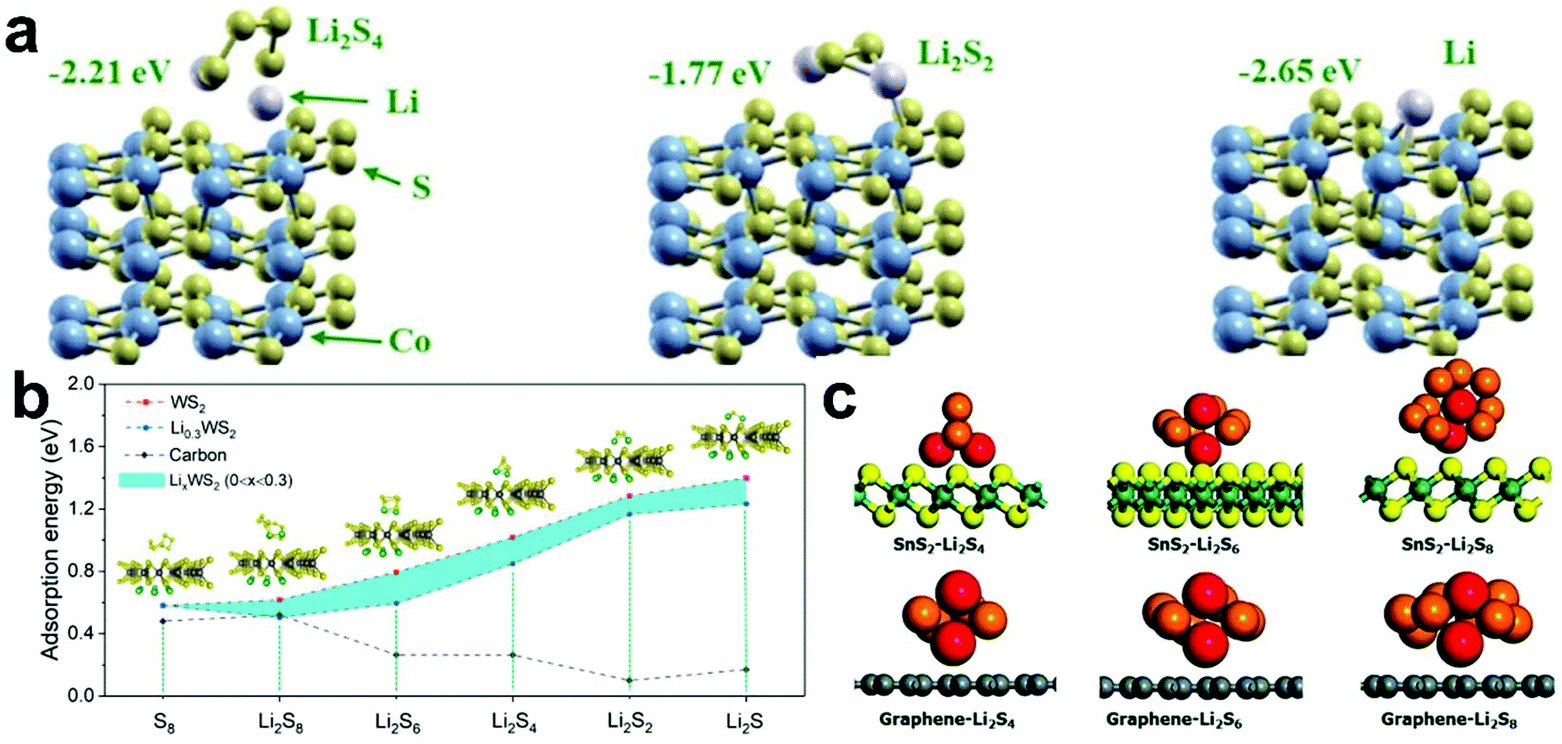 | ||
| Fig. 7 (a) Optimized geometries of LiPSs on CoS2. Reprinted with permission from ref. 125. Copyright 2019, the American Chemical Society. (b) Adsorption binding energies for LiPSs on LixWS2 (0 ≤ x ≤ 0.3) and carbon. Reprinted with permission from ref. 126. Copyright 2018, the American Chemical Society. (c) Optimized geometries of LiPSs on SnS2 and graphene. Reprinted with permission from ref. 127. Copyright 2019, The Royal Society of Chemistry. | ||
Additionally, other metal sulfides have also been researched in Li–S batteries by DFT calculations. For example, WS2 shows an absorption energy of 0.51–1.4 eV even after it is combined with Li+ extracted from LiPSs during the discharge process, and the WS2–rGO–CNT cathode exhibited a stable cycling performance (90.7% capacity retention after 100 cycles) and good rate capability (614 mA h g−1 at 2 C) (Fig. 7b).126 Kim et al.127 also proved that a strong chemical interaction and physical interaction exists between LiPSs and SnS2 (Fig. 7c). When SnS2 was modified on a separator, this specially coated SnS2-modified separator not only captured LiPSs efficiently by a strong chemical and physical interaction, but also ensured the rapid diffusion of Li ions.
Although most metal sulfides exhibit improved conductivity compared with metal oxides, they still exhibit internal resistance in the electrode and highly depend on the effective use of carbon materials. Therefore, the novel design and practical application of Li–S batteries still require deeper investigation.
Meanwhile, Chen et al.133 used the 3D structure of ZIF-67 to obtain a hierarchical porous polyhedron nanocatalyst via a carbonization and selenization process (Fig. 8a). Besides, Fig. 8b shows that the prepared composite electrode exhibits a satisfactory catalytic conversion performance for accelerating the redox reaction of LiPSs. Subsequently, theoretical calculations further revealed that the CoSe (110) surface exhibited a strong interface interaction with LiPSs on the atomic scale (Fig. 8c), indicating that it has a stronger ability to adsorb LiPSs. Furthermore, the prepared hierarchical porous polyhedron nanocatalyst not only provides abundant adsorption sites for LiPSs, but also accelerates the conversion of LiPSs and the formation and decomposition of Li2S. Therefore, the hierarchical porous polyhedron nanocatalyst/S cathode exhibited a satisfactory electrochemical performance and stability. Moreover, comprehensive consideration of the morphology, structure and adsorption-catalysis ability for the S host material design in this work provides a reference for the development of advanced Li–S batteries.
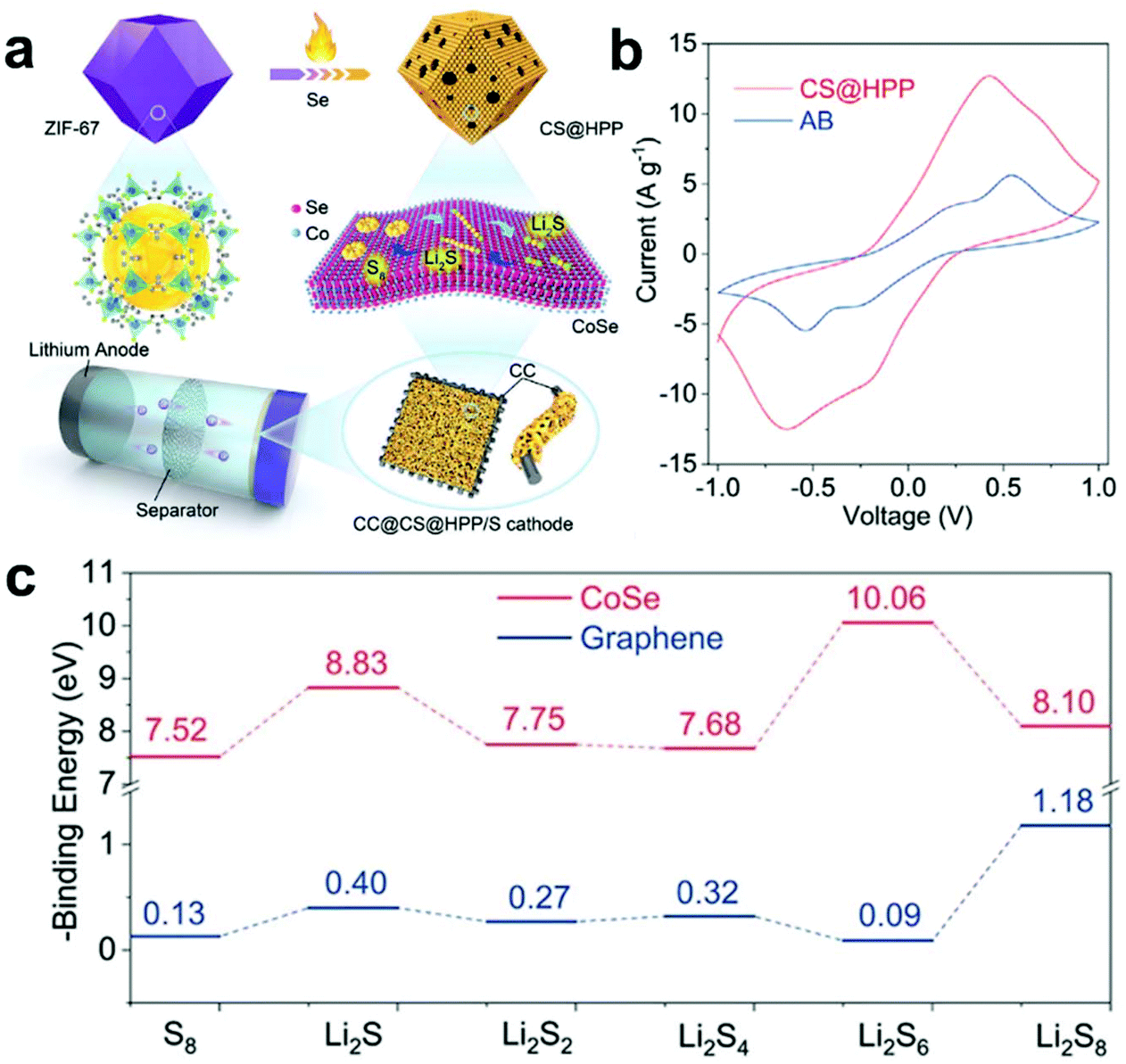 | ||
| Fig. 8 (a) Schematic illustration for the synthesis of CoSe@hierarchical porous polyhedron. (b) CV curves of Li2S6 symmetrical cell with CoSe@hierarchical porous polyhedron and acetylene black. (c) Binding energies of LiPSs with the (110) plane of CoSe and (001) plane of graphene. Reprinted with permission from ref. 133. Copyright 2020, Wiley-VCH. | ||
Although they show much more applied potential as an additive in Li–S batteries, the use of metal selenides as electrocatalysts to confine and catalyze LiPSs is still in its infancy.
Among the various metal nitrides, TiN has attracted much attention due to its excellent electrical conductivity and catalytic activity. Ding et al.138 reported that carbon@TiN dual shell nanospheres were used as a host of S, which simultaneously achieved the physical confinement, chemical adsorption and catalytic conversion of LiPSs. The DFT calculations showed that LiPSs can form Li–N bonds and Ti–S bonds with TiN. Therefore, carbon@TiN exhibited a strong ability to adsorb LiPSs, making it exhibit a higher S utilization. Fig. 9a–c show the adsorption structures, adsorption energies, and charge density difference plots for Li2S4, Li2S6, and Li2S8 on TiN (200), respectively. It is worth noting that the long-chain Li2S8 exhibits different reaction paths on the surface of TiN and TiO2, where the adsorption energy of TiN on Li2S8 is as high as 7.83 eV, and Li2S8 tends to degrade into two shorter chains on their surfaces, and the corresponding TiO2 also shows a higher binding energy (3.71 eV), but has no tendency to fracture. Therefore, the carbon@TiN dual-shell nanosphere-S composites showed a faster conversion rate and stronger ability to adsorb LiPSs in Li–S batteries.
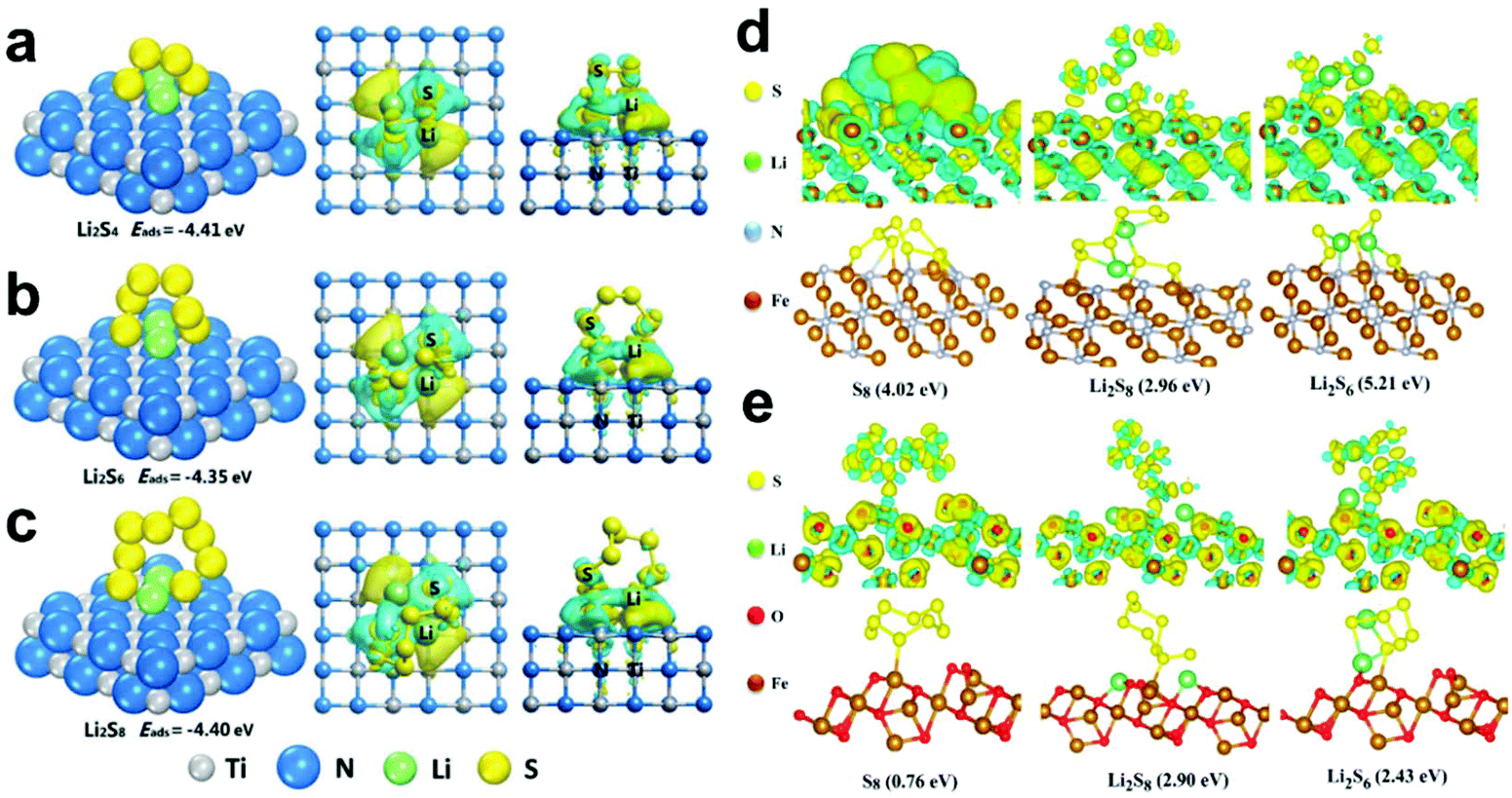 | ||
| Fig. 9 Adsorption structures, adsorption energies, and charge density difference plots for (a) Li2S4, (b) Li2S6, and (c) Li2S8 on TiN (200). Reprinted with permission from ref. 138. Copyright 2019, Elsevier. Optimized configurations for the binding of S8 and LiPSs to (d) Fe2N (011) and (e) Fe3O4 (311). Reprinted with permission from ref. 139. Copyright 2019, the American Chemical Society. | ||
Besides, Zheng et al.139 also conducted DFT calculations to confirm the absorption ability of yolk–shelled Fe2N@carbon nanoboxes. As shown in Fig. 9d, Fe2N delivered a much higher binding energy with different S species (S8: −4.02 eV, Li2S8: −2.96 eV and Li2S6: −5.21 eV) than that of Fe3O4 (Fig. 9e) and N-doped carbon. Consequently, the strong ability of faster conversion and chemical adsorption of LiPSs significantly improved the cycle performance of the Li–S batteries.
The above metal nitrides show improved performance and can be used as a guide for their application in energy storage. However, the strict conditions for the preparation of metal nitrides with catalytic efficiency needs to be considered.
Kuang et al.144 added an iron (Fe) source to biomass to form a 2D Fe3C/mesoporous carbon composite via annealing. Due to the addition of Fe, the conductivity, adsorption capacity and catalytic activity of the material were improved, thereby improving the performance of Li–S batteries.
Subsequently, theoretical calculations were performed to further explore the ability of Fe3C to adsorb and convert LiPSs (Fig. 10a). Two S atoms of Li2S6 formed a strong chemical interaction with three Fe atoms of Fe3C (3.85 eV), indicating its strong ability to adsorb LiPSs. Zhang et al.141 also used theoretical calculations to verify the adsorption and catalytic activity of Fe3C for LiPSs. The results showed that the bond length of LiPSs increased significantly and LiPSs were prone to redox reactions when adsorbed on the surface of Fe3C, which further illustrated the catalytic ability of Fe3C.
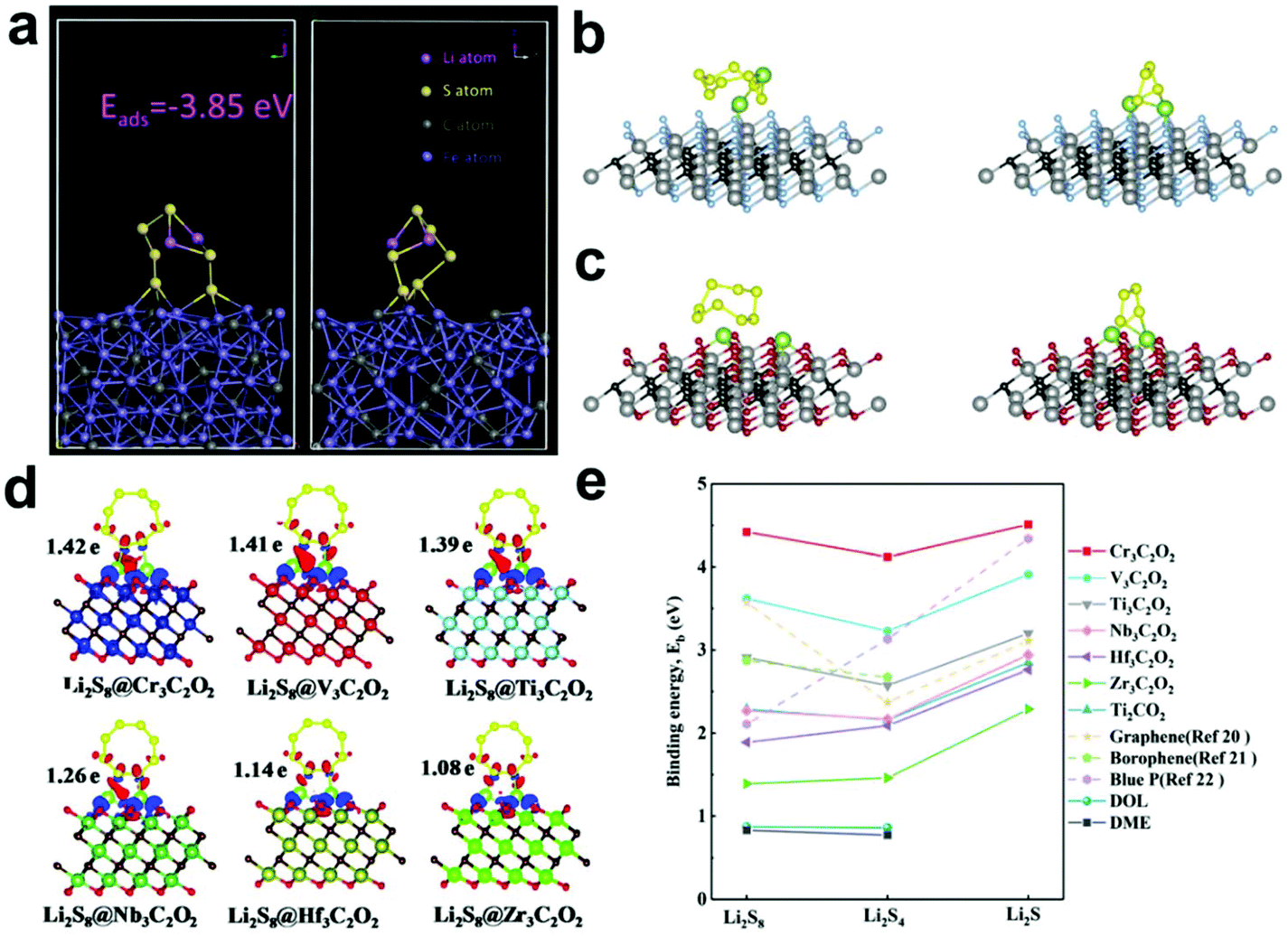 | ||
| Fig. 10 (a) Optimized geometries of Li2S6 on Fe3C. Reprinted with permission from ref. 144. Copyright 2019, Elsevier. Optimized configurations of Li2S8 and Li2S4 anchored on (b) Ti2CF2 and (c) Ti2CO2, respectively. Reprinted with permission from ref. 146. Copyright 2019, Elsevier. (d) Differential charge density between Li2S8 and M3C2O2 surfaces. (e) Binding energies of LiPSs on M3C2O2 MXenes. Reprinted with permission from ref. 147 Copyright 2019, The Royal Society of Chemistry. | ||
MXenes are a fascinating large family of 2D early transition metal carbides/carbonitrides for anchoring materials due to their interactive surface and structural stability. Nazar's group145 as a pioneer firstly reported Mxene TiC as a sulfur host material in Li–S batteries with high conductivity and highly active 2D surfaces to chemically bond LiPSs. The 70 wt% S/TiC composite still exhibited a stable performance owning to its strong interaction with the LiPS species by S–Ti bonding.
To get a further understanding of the anchoring effect, Chung et al.146 performed a DFT study on F and O-functionalized Ti2C (Fig. 10b and c), respectively. The calculated results indicated that Ti2CF2 and Ti2CO2 present two different mechanisms. By calculating the binding energy and average charge of S of LiPSs, Ti2CF2 is considered to trap LiPSs through strong interactions with the substrates, while Ti2CO2 converts soluble LiPSs into insoluble S. To analyze the catalytic activity of Ti2CO2, the density of states (DOS) of Ti2CO2 and Ti2CF2 were calculated. After the absorption LiPSs, the changes in DOS indicate more electrons are donated by the LiPSs to Ti2CO2 than to Ti2CF2, and these large amounts of electrons fill the empty 3d states on Ti of Ti2CO2. The 3d states of Ti are dominant at the bottom of the conduction band; consequently, charge transfer will lead to a change in Ti2CO2 to metal from semiconductor, and thus Ti2CO2 can support redox reactions by supplying its own free electrons.
Besides the above MXenes, other transition metals-based MXenes have also been studied. Fan et al.147 systematically investigated a series of transition metal (Cr, V, Ti, Nb, HF and Zr)-based MXenes, M3C2O2, via DFT calculations (Fig. 10d and e). By analyzing the binding energy and lattice constant of the selected MXene model, Cr3C2O2 showed the strongest adsorption of LiPSs. It is worth mentioning that a smaller lattice constant can result in a stronger adsorption effect. In particular, O-terminated MXenes with a lattice constant in the range of 2.88–3.31 Å are effective adsorption sites for LiPSs. This discovery provides a new strategy for screening suitable electrodes.
Thus, the above results show that metal carbides exhibit a relatively stronger LiPS adsorption capacity than that of other metal compounds. However, an excessive adsorption capacity will lead to LiPS chain scission and desorption difficulties.
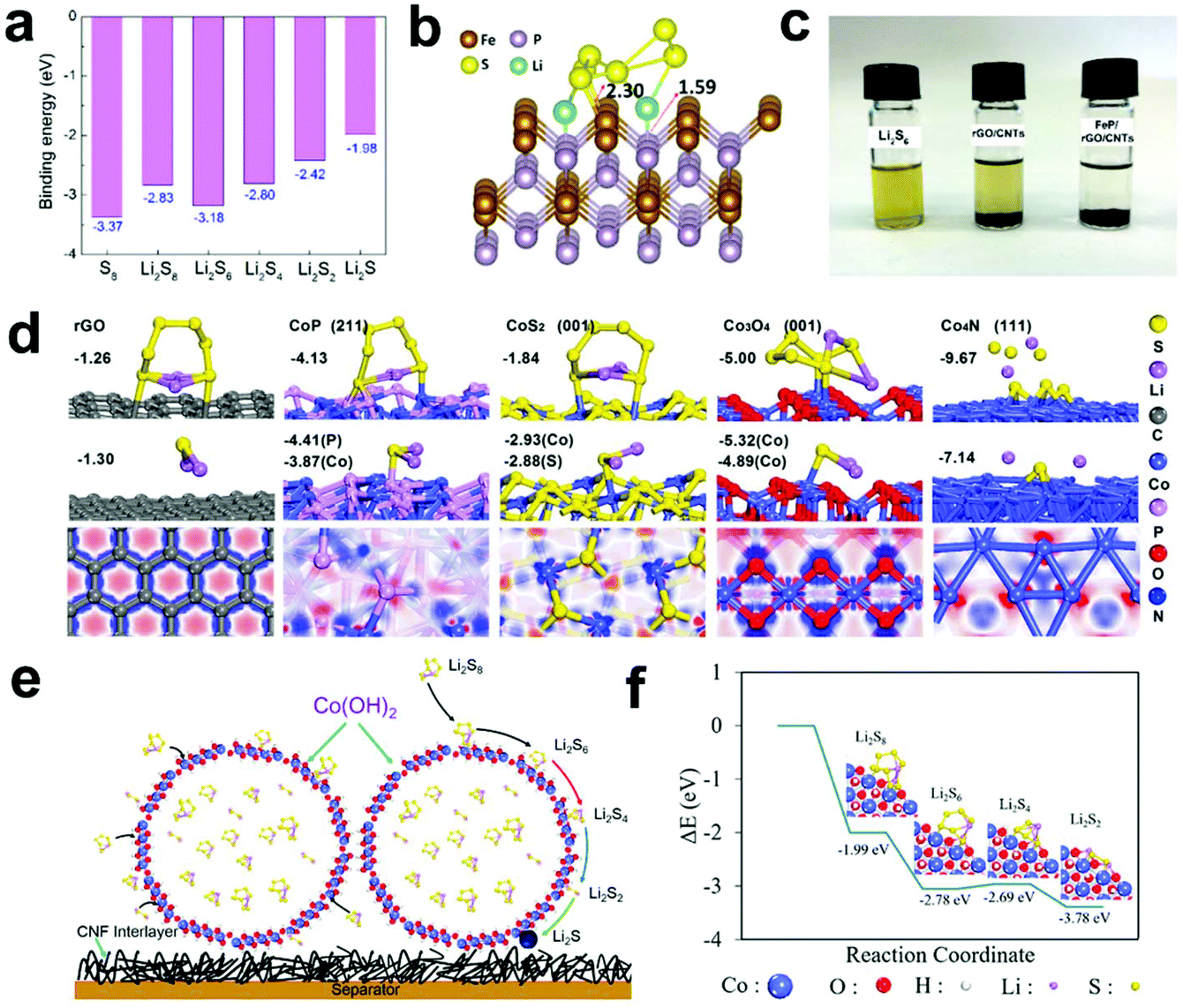 | ||
| Fig. 11 (a) Binding energies of S species and (b) geometry of Li2S6 binding on FeP (111). (c) Adsorption tests of rGO/CNTs and FeP/rGO/CNTs with Li2S6. Reprinted with permission from ref. 152. Copyright 2020, Elsevier. (d) Adsorption configurations and energies of LiPSs on rGO and Co-based compounds and surface electron density difference of rGO and Co-based compounds. Reprinted with permission from ref. 153. Copyright 2018, Elsevier. (e) Schematic depiction of Co(OH)2 for the chemical adsorption and catalytic decomposition of LiPSs and (f) binding energies of LiPSs at the edge of Co(OH)2. Reprinted with permission from ref. 154. Copyright 2019, the American Chemical Society. | ||
To explore the influence of the p-band of metal compounds on Li–S batteries, Qian et al.153 designed a series of cobalt-based compounds with different anions (CoP, CoS2, Co3O4, and Co4N). Subsequently, they used theoretical calculations to understand the interface of cobalt-based compounds at the atomic level. Fig. 11d shows the optimized adsorption configurations with the corresponding energetic information of Li2S6 on different compounds. It can be seen that the adsorption capacity of Co4N is the strongest, while the adsorption capacity of CoP is in the middle range, and the adsorption capacity of carbon is the weakest. In addition, the adsorption capacity of Li2S also shows a similar adsorption trend. In subsequent diffusion studies, it was found that the surface of CoP exhibits the lowest energy barrier, indicating that CoP has the fastest diffusion behavior. In Li–S batteries, S@rGO/CoP also showed the best rate performance and cycle stability. This result further proved that the ability to adsorb LiPSs at an appropriate level and a low migration barrier is beneficial to maximize the performance of Li–S batteries.
Sun et al.154 prepared a S@Co(OH)2 core–shell composite material with a high sulfur content (80%), which effectively improved the electrode conductivity and inhibited the shuttle effect under the coordination of the carbon–nanofiber-based interlayer (Fig. 11e). Subsequently, they analyzed the adsorption and decomposition of LiPSs on the Co(OH)2 interface via DFT calculations (Fig. 11f). The calculation results show that Co(OH)2 is thermodynamically favorable for the adsorption and catalytic conversion of LiPSs.
Besides the above materials, an increasing number of metal compounds are selected to improve the performance of Li–S batteries. Unfortunately, a single-component metal compound is difficult to solve the complex problems involved in Li–S batteries.
3.3. LiPS catalytic conversion on composites
Composite metal compounds are constructed by coupling two or more nanocrystals with different band-gaps, which have attracted extensive attention recently owning to their superior properties and wide applications in photocatalysis, sensors and energy storage. Benefiting from the internal electric field at heterointerfaces, they can facilitate charge transport and enhance the reaction kinetics.155–163Typically, TiO2 has strong adsorption for LiPSs but low electrical conductivity. In contrast, TiN exhibits higher conductivity and catalytic activity, but its ability to adsorb LiPSs is worse than that of TiO2. Based on this, Yang et al.164 designed a TiO2–TiN heterostructure/graphene complex and applied it as the blocking layer of Li–S batteries. To explore the coordination mechanism of the composite heterojunction, they used DFT calculations to analyze the adsorption and migration processes of LiPSs on the surfaces of TiO2 and TiN. Fig. 12a shows the optimized geometries of the Li2S4 adsorption on the TiO2 (110) and TiN (200) surfaces. The calculation results show that the TiO2 exhibits strong adsorption capacity, but it is difficult to convert LiPSs into Li2S due to its poor conductivity. Excitingly, the migration barrier for Li2S4 on TiO2 (110) is only about 1.04 eV, which can make the LiPSs diffuse rapidly from TiO2 to TiN. Meanwhile, TiN with better electrical conductivity is beneficial for the rapid and reversible redox conversion of LiPSs. Therefore, the coordinated adsorption and conversion of LiPSs on the interface of the prepared composite material are achieved.
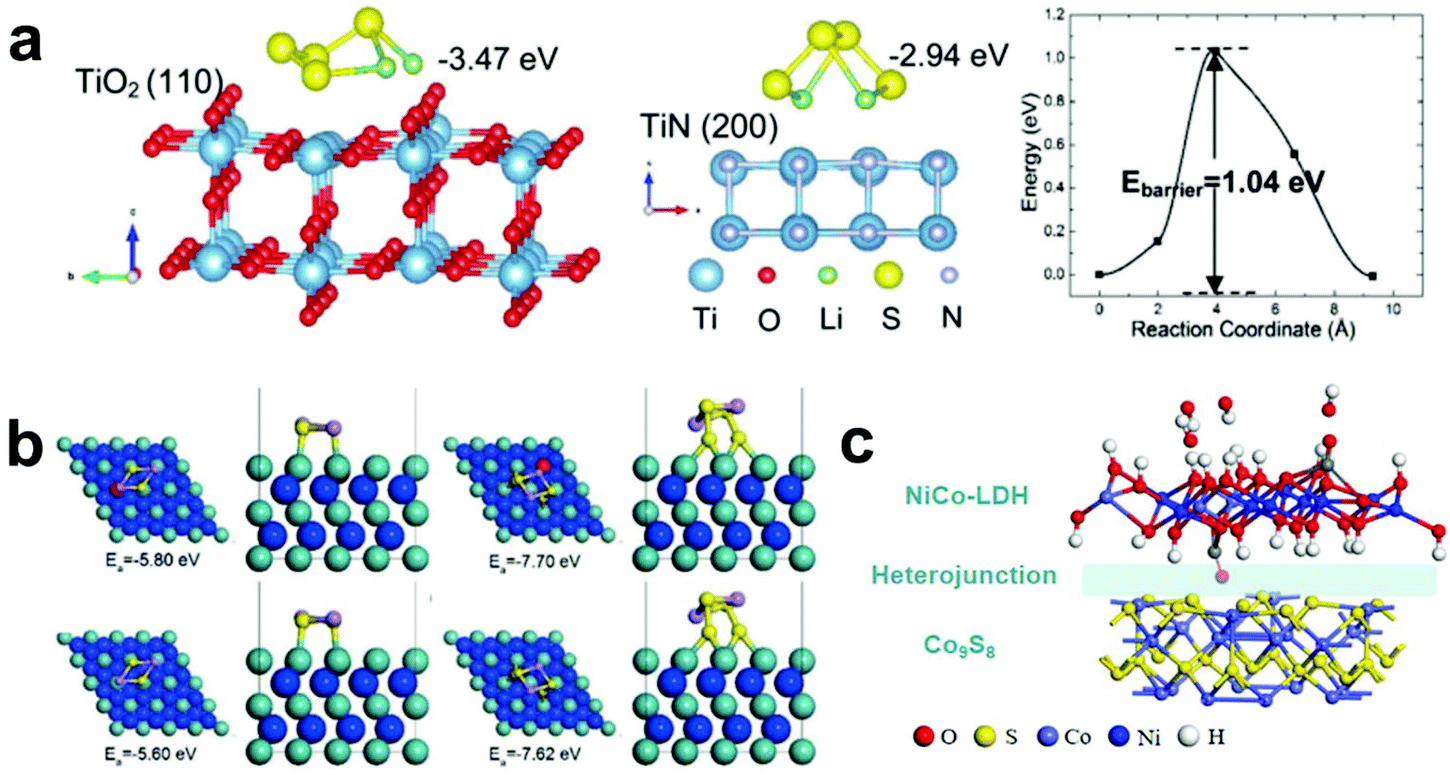 | ||
| Fig. 12 (a) Calculation and experimental demonstration of the trapping–diffusion–conversion of LiPSs on TiO2, TiN and the TiO2–TiN heterostructure. Reprinted with permission from ref. 164. Copyright 2017, The Royal Society of Chemistry. (b) Optimized configurations of V–MoN–Li2S2, V–MoN–Li2S4, MoN–Li2S2 and MoN–Li2S4. Reprinted with permission from ref. 165. Copyright 2018, Wiley-VCH. (c) Optimized geometries of H–LDH/Co9S8. Reprinted with permission from ref. 166. Copyright 2020, Elsevier. | ||
Similarly, to explore the adsorption behavior of LiPSs on the interface of MoN–VN, Qiao et al.165 used theoretical calculations to obtain the adsorption energy of LiPSs on the interface of MoN (002) without and with V doping. The calculation results showed that V–MoN exhibits a much higher adsorption energy for Li2S2 and Li2S4 (5.60 and 7.62, eV, respectively, Fig. 12b) than that of MoN, which proved that the introduction of V improved its adsorption capacity of LiPSs. To further study the reasons why V can enhance the adsorption capacity of LiPSs, they analyzed the DOS of Mo atoms on the surface of different substrate materials (MoN and V–MoN) via theoretical calculations. The calculation results show that the V atoms change the electronic state of Mo on the surface, and thereby the ability to adsorb LiPSs is significantly improved.
Besides, Deng et al.166 obtained an NiCo–LDH/Co9S8 heterojunction structure with a hollow nanocage morphology through an ingenious design, which delivered ultralong cycle stability as the cathode for Li–S batteries. They used theoretical calculations to further reveal the working mechanism of the conversion of LiPSs with NiCo–LDH/Co9S8 (Fig. 12c). The energy band gap of Co9S8 (0.28 eV) is much lower than that of NiCo–LDH (1.74 eV) and their conduction band (CB) and valence band (VB) almost overlap, which makes Co9S8 show better electronic conductivity than NiCo–LDH. In addition, the charge analysis shows that NiCo–LDH transfers electrons to the surface of Co9S8, which makes the heterojunction interface form an interfacial internal electric field and effectively improve the overall electronic conductivity. Moreover, the hollow nanocage morphology can achieve both physical and chemical adsorption of LiPSs, thereby effectively improving the LiPS conversion kinetics to inhibit the shuttle effect. Furthermore, the hollow structure is conducive to achieving a high S loading and alleviates volume changes during charging and discharging. Therefore, the prepared S@H–LDH/Co9S8 exhibits an ultralong cycle life of 1500 cycles with a decay rate of 0.047% per cycle.
To meet the requirements of complex Li–S battery reactions, the designed catalyst should have all the characteristics of strong adsorption capacity, good conductivity and high efficiency conversion capacity, which are very difficult to be integrated into a single catalyst. Therefore, the construction of heterojunctions with multiple catalytic materials can effectively solve this problem, which will make it possible to improve the development of Li–S batteries.
4. Conclusions
In summary, a significant amount of research has focused on the fundamental understanding of the chemistry of Li–S batteries, and controlling the dissolution of LiPSs using both calculation and experiment. Particularly, ‘sulfiphilic’ cathode host surfaces are regarded as an effective strategy, which have attracted widespread attention. They include N- and S-doped carbons, MOFs/COFs and metal-based materials, which can achieve LiPS binding with surfaces via polar or Lewis acid–base interactions. Subsequently, we compared the adsorption capacity of different materials for LiPSs (taking Li2S4 as an example, Fig. 13). The results show that the adsorption capacity of modified carbon materials has been significantly improved compared with pure carbon materials, but it is still lower than that of polar compounds. Among the many polar compounds, metal nitrides and metal carbides a show relatively high adsorption capacity. According to the literature, excessive adsorption capacity may make the desorption of LiPSs difficult. Among them, metal phosphides and composite metal compounds with suitable adsorption capacity are the most promising to achieve the effective adsorption of LiPSs. | ||
| Fig. 13 Adsorption energies of Li2S4 on various substrates. | ||
Meanwhile, the sluggish redox reaction between long chain and short chain LiPSs is another main reason for the low utilization of S and notorious shuttle effect, which seriously hinders the improvement of the Li–S battery performance. Therefore, for the construction of advanced Li–S batteries, a suitable adsorption capacity and catalytic activity of LiPSs are indispensable for the S host material. To date, numerous catalysts have been reported, achieving the efficient adsorption and fast conversion of LiPSs in the Li–S battery. For example, transition metal compounds can effectively confine LiPSs, lower the reaction energy barrier and accelerate the reaction rate, which are the most promising to push a big step forward in Li–S batteries. Based on previous research results, the mechanisms of different materials to improve the performance of Li–S batteries are summarized in Table 1.
| Performance improvement mechanism | Improvement principle | Representative materials |
|---|---|---|
| Physical absorption | 1. Multi-level structure design | Porous carbon, hollow CNTs, and graphene |
| 2. Multi-level pore design | ||
| 3. Improve the electron conductivity | ||
| 4. Larger specific surface area | ||
| Chemical interaction | 1. Polarization treatment of carbon matrix | Nitrogen doped graphene, PPy, PANI, g-C3N4, and TiO2 |
| 2. Introduction polar compound | ||
| Catalytic mechanism | 1. Reduce reaction energy barrier | Metal particles (e.g., Pt, Ni, and Co) and metal compounds (e.g., MnO2, CoxSy, CoP, TiN, and FeC) |
| 2. Improve diffusion rate of LiPS | ||
| 3. Accelerate the nucleation and decomposition of Li2S | ||
| Trapping–diffusion conversion | Synergistic effects: LiPS strongly adsorbed, the adsorbed LiPSs rapidly diffuse and LiPS is easily converted to Li2S by fast charge transfer | Heterostructures (e.g., TiO2–TiN, MoC–MoOx, TiO2–MXene, TiN–sulfur-doped oxide layer, and TiC–graphene graphene–TiC) |
Although significant progress has been made in the research on Li–S batteries, there is still an urgent requirement for in-depth understanding to provide guidance and help tackle the high future energy demands. There is no doubt that the introduction of theoretical calculations can guide the rational design research and accelerate the development of advanced Li–S batteries. In the geometrical structure, numerous works focused on the optimization of the adsorption state of special sites to obtain structural (bond length) and energy (adsorption energy) data, and clarify the ability of special sites to bind S or Li in specific species (moderate/strong anchoring). In the electronic structure, the common analysis is DOS, charge analysis (Bader and Mulliken), CDD, and band structure, which can give a qualitative description of the electron transfer. Unfortunately, the current theoretical calculation work is still at the level of qualitative research. Accordingly, the following suggestions for the rational design of advanced Li–S batteries using theoretical calculation methods are proposed for reference.
4.1. Fundamental mechanism for S transformation
Accelerating the redox conversion between LiPSs and Li2S/Li2S2 is an important strategy to inhibit the shuttle of LiPSs. Therefore, a highly efficient catalytically active S host material is the key to improving the performance of Li–S batteries. Although many catalysts have been reported to achieve significant effects in the conversion of LiPSs, the internal mechanism of scission and formation of S–S bonds in LiPS chains is still not clear. Thus, it is necessary to focus on the exploration of more detailed paths for LiPSs based on theoretical calculations. Meanwhile, the phase transition of Li2S is recognized as a decisive step, where complex solid–liquid phase transitions need further analysis and disclosure.To gain a deeper understanding of the reaction mechanism, more calculation models need to be employed in the Li–S battery system. CHE (computational hydrogen electrode) and CEP (constant electrode potential) models can effectively calculate the Gibbs free energy of intermediate species at a specific potential, and thus reveal the potential-determining step (PDS). Transition states can be investigated via methods such as nudged elastic band (NEB) and DIMER, giving rise to the details of LiPS diffusion. In addition, most DFT calculations focus on a single LiPS molecule, rather than the actual system containing a large number of LiPSs and electrolyte molecules. One way to address this problem is to simulate the actual solvation system and the reaction behaviors of multiple LiPS species in a limited space by molecular dynamics simulations. Ab initio molecular dynamics (AIMD) can give more accurate information such as the diffusion coefficient and diffusion barrier of Li. Another way is to introduce an explicit model containing electrolyte molecules, which makes it possible to study the effect of solvation and cations.
4.2. Material optimization and design
To achieve large-scale applications in the future, catalyst materials with low cost and large-scale production (e.g., metal oxides and sulfides) should become the focus of Li–S battery research. However, considering the poor conductivity of this type of catalyst itself, it is important to combine it with materials with excellent conductivity (e.g., graphene and CNT). In addition, it is necessary to ensure that the host materials have a large surface area to achieve a high S loading. According to the research results on the reaction mechanism and electrochemical process, high-throughput screening and even machine learning can be used to predict potential high-performance Li–S battery materials. This scheme needs to select parameters as the screening space, such as electronegativity, coordination number and adsorption energy of key intermediate species. At this stage, due to the distinction of synthetic methods and reaction conditions, to date, there is no systematic deep understanding of the structure–activity relationship of Li–S battery materials. Also, how to refine the descriptors and use them in the screening of Li–S battery remain a challenge.Although the road to the commercialization of Li–S batteries faces many challenges, it will usher in greater development to solve the above problems through calculation and characterization in the future.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (21922501 and 21521005) and the Fundamental Research Funds for the Central Universities (XK1802-6 and XK1803-05).Notes and references
- A. Manthiram, Y. Fu, S. Chung, C. Zu and Y. Su, Chem. Rev., 2014, 114, 11751 CrossRef CAS PubMed.
- Z. W. Seh, Y. Sun, Q. Zhang and Y. Cui, Chem. Soc. Rev., 2016, 45, 5605 RSC.
- A. Bhargav, J. R. He, A. Gupta and A. Manthiram, Joule, 2020, 4, 285 CrossRef.
- M. Zhao, B.-Q. Li, H.-J. Peng, H. Yuan, J.-Y. Wei and J.-Q. Huang, Angew. Chem., Int. Ed., 2020, 59, 12636 CrossRef CAS PubMed.
- H. Yuan, H. J. Peng, B. Q. Li, J. Xie, L. Kong, M. Zhao, X. Chen, J. Q. Huang and Q. Zhang, Adv. Energy Mater., 2019, 9, 1802768 CrossRef.
- W.-P. Wang, J. Zhang, J. Chou, Y.-X. Yin, Y. You, S. Xin and Y.-G. Guo, Adv. Energy Mater., 2020, 2000791 Search PubMed.
- L. Kong, X. Chen, B. Q. Li, H. J. Peng, J. Q. Huang, J. Xie and Q. Zhang, Adv. Mater., 2018, 30, 1705219 CrossRef PubMed.
- L. Zhou, D. L. Danilov, R.-A. Eichel and P. H. L. Notten, Adv. Energy Mater., 2020, 2001304 Search PubMed.
- Y. S. Fu, Z. Wu, Y. F. Yuan, P. Chen, L. Yu, L. Yuan, Q. R. Han, Y. J. Lan, W. X. Bai, E. J. Kan, C. X. Huang, X. P. Ouyang, X. Wang, J. W. Zhu and J. Lu, Nat. Commun., 2020, 11, 845 CrossRef CAS PubMed.
- S. Y. Zhou, S. Yang, X. W. Ding, Y. C. Lai, H. G. Nie, Y. G. Zhang, D. Chan, H. Duan, S. M. Huang and Z. Yang, ACS Nano, 2020, 14, 7538 CrossRef CAS PubMed.
- D. Liu, C. Zhang, G. Zhou, W. Lv, G. Ling, L. Zhi and Q. H. Yang, Adv. Sci., 2018, 5, 1700270 CrossRef PubMed.
- M. Zhao, B.-Q. Li, H.-J. Peng, H. Yuan, J.-Y. Wei and J.-Q. Huang, Angew. Chem., Int. Ed., 2020, 59, 12636 CrossRef CAS PubMed.
- T. Dhawa, S. Chattopadhyay, M. Sreemany, G. De and S. Mahanty, J. Am. Chem. Soc., 2018, 130, 109 Search PubMed.
- G. Hu, C. Xu, Z. Sun, S. Wang, H. M. Cheng, F. Li and W. Ren, Adv. Mater., 2016, 28, 1603 CrossRef CAS PubMed.
- T. H. Zhou, Y. Zhao, G. M. Zhou, W. Lv, P. J. Sun, F. Y. Kang, B. H. Li and Q.-H. Yang, Nano Energy, 2017, 39, 291 CrossRef CAS.
- T.-Z. Hou, W.-T. Xu, X. Chen, H.-J. Peng, J.-Q. Huang and Q. Zhang, Angew. Chem., Int. Ed., 2017, 56, 8178 CrossRef CAS PubMed.
- X. Chen, Y.-K. Bai, C.-Z. Zhao, X. Shen and Q. Zhang, Angew. Chem., Int. Ed., 2020, 59, 11192 CrossRef CAS PubMed.
- A. Manthiram, X. Yu and S. Wang, Nat. Rev. Mater., 2017, 2, 16103 CrossRef CAS.
- Y.-S. Su and A. Manthiram, Nat. Commun., 2012, 3, 1166 CrossRef PubMed.
- T. Z. Hou, X. Chen, H. J. Peng, J.-Q. Huang, B.-Q. Li, Q. Zhang and B. Li, Small, 2016, 12, 3283 CrossRef CAS PubMed.
- M. Rana, B. Luo, M. R. Kaiser, I. Gentle and R. Knibbe, J. Energy Chem., 2020, 42, 195 CrossRef.
- W. Lv, Z. Li, Y. Deng, Q.-H. Yang and F. Kang, Energy Storage Mater., 2016, 2, 107 CrossRef.
- M. Zhang, W. Chen, L. X. Xue, Y. Jiao, T. Y. Lei, J. W. Chu, J. W. Huang, C. H. Gong, C. Y. Yan, Y. C. Yan, Y. Hu, X. F. Wang and J. Xiong, Adv. Energy Mater., 2020, 10, 1903008 CrossRef CAS.
- Z. Y. Wang, Y. F. Dong, H. J. Li, Z. B. Zhao, H. B. Wu, C. Hao, S. H. Liu, J. S. Qiu and X. W. Lou, Nat. Commun., 2014, 5, 5002 CrossRef CAS PubMed.
- Y. Lu, S. Gu, J. Guo, K. Rui, C. H. Chen, S. P. Zhang, J. Jin, J. H. Yang and Z. Y. Wen, ACS Appl. Mater. Interfaces, 2017, 9, 14878 CrossRef CAS PubMed.
- Y. H. Cui, Q. Zhang, J. W. Wu, X. Liang, A. P. Baker, D. Y. Qu, H. Zhang, H. Y. Zhang and X. H. Zhang, J. Power Sources, 2018, 378, 40 CrossRef CAS.
- X. W. Ding, S. Yang, S. Y. Zhou, Y. X. Zhan, Y. C. Lai, X. M. Zhou, X. J. Xu, H. G. Nie, S. M. Huang and Z. Yang, Adv. Funct. Mater., 2020, 2003354 CrossRef CAS.
- Y. Liang, C. Z. Zhao, H. Yuan, Y. Chen, W. Zhang, J.-Q. Huang, D. Yu, Y. Liu, M. M. Titirici, Y. L. Chueh, H. Yu and Q. Zhang, InfoMat, 2019, 1, 6 CrossRef CAS.
- D. Larcher and J. M. Tarascon, Nat. Chem., 2015, 7, 19 CrossRef CAS PubMed.
- Y. Z. Song, W. L. Cai, L. Kong, J. S. Cai, Q. Zhang and J. Y. Sun, Adv. Energy Mater., 2019, 1901075 Search PubMed.
- Q. P. Wu, Z. G. Yao, X. J. Zhou, J. Xu, F. H. Cao and C. L. Li, ACS Nano, 2020, 14, 3365 CrossRef CAS.
- J. Qian, F. J. Wang, Y. Li, S. Wang, Y. Y. Zhao, W. L. Li, Y. Xing, L. Deng, Q. Sun, L. Li, F. Wu and R. J. Chen, Adv. Funct. Mater., 2020, 30, 2000742 CrossRef CAS.
- D. Luo, Z. Zhang, G. R. Li, S. B. Cheng, S. Li, J. D. Li, R. Gao, M. Li, S. Sy, Y.-P. Deng, Y. Jiang, Y. F. Zhu, H. Z. Dou, Y. F. Hu, A. P. Yu and Z. W. Chen, ACS Nano, 2020, 14, 4849 CrossRef CAS PubMed.
- M. Z. Sun, Z. Wang, X. Li, H. B. Li, H. S. Jia, X. X. Xue, M. Jin, J. Q. Li, Y. Xie and M. Feng, J. Mater. Chem. A, 2020, 8, 11818 RSC.
- X. Chen, T. Z. Hou, K. A. Persson and Q. Zhang, Mater. Today, 2019, 22, 142 CrossRef CAS.
- Y. P. Zheng, H. H. Li, H. Y. Yuan, H. H. Fan, W. L. Li and J. P. Zhang, Appl. Surf. Sci., 2018, 434, 596 CrossRef CAS.
- Q. Liu, D. B. Mu, B. R. Wu, L. Wang, L. Gai and F. Wu, RSC Adv., 2017, 7, 33373 RSC.
- T. Z. Hou, X. Chen, H. J. Peng, J. Q. Huang, B. Q. Li, Q. Zhang and B. Li, Small, 2016, 12, 3283 CrossRef CAS PubMed.
- Y. Z. Shu, X. L. Li, J. J. Ye, W. Gao, S. Cheng, X. C. Zhang, L. Ma and Y. S. Ding, Energy Technol., 2019, 1901057 Search PubMed.
- L. Y. Zhao, R. He, K. T. Rim, T. Schiros, K. S. Kim, H. Zhou, C. Gutierrez, S. P. Chockalingam, C. J. Arguello, L. Palova, D. Nordlund, M. S. Hybertsen, D. R. Reichman, T. F. Heinz, P. Kim, A. Pinczuk, G. W. Flynn and A. N. Pasupathy, Science, 2011, 333, 999 CrossRef CAS.
- G. M. Zhou, Y. B. Zhao and A. Manthiram, Adv. Energy Mater., 2015, 5, 1402263 CrossRef.
- Y. Wang, J. Y. Huang, X. B. Chen, L. Wang and Z. Z. Ye, Carbon, 2018, 137, 368 CrossRef CAS.
- W. L. Li, Y. S. Ye, J. Qian, Y. Xing, W. Qu, N. X. Zhang, L. Li, F. Wu and R. J. Chen, ChemElectroChem, 2019, 6, 1094 CrossRef CAS.
- K. Han, J. M. Shen, S. Q. Hao, H. Q. Ye, C. Wolverton, M. C. Kung and H. H. Kung, ChemSusChem, 2014, 7, 2545 CrossRef CAS PubMed.
- C. P. Yang, Y. X. Yin, H. Ye, K. C. Jiang, J. Zhang and Y.-G. Guo, ACS Appl. Mater. Interfaces, 2014, 6, 8789 CrossRef CAS PubMed.
- C. B. Jin, W. K. Zhang, Z. Z. Zhuang, J. G. Wang, H. Huang, Y. P. Gan, Y. Xia, C. Liang, J. Zhang and X. Y. Tao, J. Mater. Chem. A, 2017, 5, 632 RSC.
- F. Wu, J. Qian, W. P. Wu, Y. S. Ye, Z. G. Sun, B. Xu, X. G. Yang, Y. H. Xu, J. T. Zhang and R. J. Chen, Nano Res., 2017, 10, 426 CrossRef CAS.
- J. P. Paraknowitsch and A. Thomas, Energy Environ. Sci., 2013, 6, 2839 RSC.
- W. Ai, J. W. Li, Z. Z. Du, C. J. Zou, H. F. Du, X. Xu, Y. Chen, H. B. Zhang, J. F. Zhao, C. M. Li, W. Huang and T. Yu, Nano Res., 2018, 11, 4562 CrossRef CAS.
- X. L. Ma, G. Q. Ning, Y. Wang, X. Y. Song, Z. H. Xiao, L. Q. Hou, W. Yang, J. S. Gao and Y. F. Li, Electrochim. Acta, 2018, 269, 83 CrossRef CAS.
- T.-Z. Hou, X. Chen, H.-J. Peng, J.-Q. Huang, B.-Q. Li, Q. Zhang and B. Li, Small, 2016, 12, 3283 CrossRef CAS.
- W. Ai, Z. M. Luo, J. Jiang, J. H. Zhu, Z. Z. Du, Z. X. Fan, L. H. Xie, H. Zhang, W. Huang and T. Yu, Adv. Mater., 2014, 26, 6186 CrossRef CAS PubMed.
- Q. Pang, J. Tang, H. Huang, X. Liang, C. Hart, K. C. Tam and L. F. Nazar, Adv. Mater., 2015, 27, 6021 CrossRef CAS PubMed.
- G. Zhou, E. Paek, G. S. Hwang and A. Manthiram, Nat. Commun., 2015, 6, 7760 CrossRef CAS.
- X. X. Gu, C.-J. Tong, C. Lai, J. X. Qiu, X. X. Huang, W. L. Yang, B. Wen, L.-M. Liu, Y. L. Hou and S. Q. Zhang, J. Mater. Chem. A, 2015, 3, 16670 RSC.
- F. He, K. Li, C. Yin, Y. C. Ding, H. Tang, Y. Wang and Z. J. Wu, J. Power Sources, 2018, 373, 31 CrossRef CAS.
- C.-Y. Liu and E. Y. Li, ACS Appl. Energy Mater., 2018, 1, 455 CrossRef CAS.
- M. Z. Yang, D. Shi, X. C. Sun, Y. L. Li, Z. Y. Liang, L. Zhang, Y. L. Shao, Y. Z. Wu and X. P. Hao, J. Mater. Chem. A, 2020, 8, 296 RSC.
- V. Do, Deepika, M. S. Kim, M. S. Kim, K. R. Lee and W. I. Cho, ACS Appl. Mater. Interfaces, 2019, 11, 11431 CrossRef CAS PubMed.
- J. Balach, H. K. Singh, S. Gomoll, T. Jaumann, M. Klose, S. Oswald, M. Richter, J. Eckert and L. Giebeler, ACS Appl. Mater. Interfaces, 2016, 8, 14586 CrossRef CAS PubMed.
- T. Maihom, S. Kaewruang, N. Phattharasupakun, P. Chiochan, J. Limtrakul and M. Sawangphruk, J. Phys. Chem. C, 2018, 122, 7033 CrossRef CAS.
- S. Kosasang, N. Ma, S. Duangdangchote, P. Chiochan, N. Chanlek and M. Sawangphruk, Carbon, 2019, 155, 553 CrossRef CAS.
- L. Ma, H. L. L. Zhuang, S. Y. Wei, K. E. Hendrickson, M. S. Kim, G. Cohn, R. G. Hennig and L. A. Archer, ACS Nano, 2016, 10, 1050 CrossRef CAS.
- M. Rana, Q. He, B. Luo, T. E. Lin, L. B. Ran, M. Li, I. Gentle and R. Knibbe, ACS Cent. Sci., 2019, 5, 1946 CrossRef CAS PubMed.
- Z. Q. Wang, R. Tan, H. B. Wang, L. Y. Yang, J. T. Hu, H. B. Chen and F. Pan, Adv. Mater., 2018, 30, 1704436 CrossRef.
- Y. J. Li, S. Y. Lin, D. D. Wang, T. T. Gao, J. W. Song, P. Zhou, Z. K. Xu, Z. H. Yang, N. Xiao and S. J. Guo, Adv. Mater., 2020, 32, 1906722 CrossRef CAS PubMed.
- J. W. Zhou, R. Li, X. X. Fan, Y. F. Chen, R. Han, W. Li, J. Zheng, B. Wang and X. G. Li, Energy Environ. Sci., 2014, 7, 2715 RSC.
- H. Q. Jiang, X.-C. Liu, Y. S. Wu, Y. F. Shu, X. Gong, F.-S. Ke and H. X. Deng, Angew. Chem., Int. Ed., 2018, 57, 3916 CrossRef CAS PubMed.
- Q. Q. Wang, D. J. Liu and X. Q. He, Acta Phys.-Chim. Sin., 2019, 35, 740 Search PubMed.
- G. M. Qu, X. X. Zhang, G. T. Xiang, Y. R. Wei, J. M. Yin, Z. H. Wang, X. L. Zhang and X. J. Xu, Chin. Chem. Lett., 2020, 31, 2007 CrossRef CAS.
- Z. Q. Wang, B. X. Wang, Y. Yang, Y. J. Cui, Z. Y. Wang, B. L. Chen and G. D. Qian, ACS Appl. Mater. Interfaces, 2015, 7, 20999 CrossRef CAS PubMed.
- S. Y. Bai, X. Z. Liu, K. Zhu, S. C. Wu and H. S. Zhou, Nat. Energy, 2016, 1, 16094 CrossRef CAS.
- Y. X. Yang, Z. H. Wang, T. Z. Jiang, C. Dong, Z. Mao, C. Y. Lu, W. Sun and K. N. Sun, J. Mater. Chem. A, 2018, 6, 13593 RSC.
- J. M. Zheng, J. Tian, D. X. Wu, M. Gu, W. Xu, C. M. Wang, F. Gao, M. H. Engelhard, J.-G. Zhang, J. Liu and J. Xiao, Nano Lett., 2014, 14, 2345 CrossRef CAS PubMed.
- H. P. Liao, H. M. Ding, B. J. Li, X. P. Ai and C. Wang, J. Mater. Chem. A, 2014, 2, 8854 RSC.
- H. P. Liao, H. M. Wang, H. M. Ding, X. S. Meng, H. Xu, B. S. Wang, X. P. Ai and C. Wang, J. Mater. Chem. A, 2016, 4, 7416 RSC.
- J. T. Yoo, S.-J. Cho, G. Y. Jung, S. H. Kim, K.-H. Choi, J.-H. Kim, C. K. Lee, S. K. Kwak and S.-Y. Lee, Nano Lett., 2016, 16, 3292 CrossRef CAS PubMed.
- Z. M. Liu, X. He, C. Fang, L. E. Camacho-Forero, Y. Z. Zhao, Y. B. Fu, J. Feng, R. Kostecki, P. B. Balbuena, J. H. Zhang, J. X. Lei and G. Liu, Adv. Funct. Mater., 2020, 2003605 CrossRef CAS.
- J. Y. Liu, P. Zhou, Z. H. Shen, H. G. Zhang, M. X. Jiang, Y. Q. Han, J. J. Li and J. W. Li, Energy Technol., 2019, 7, 1801158 CrossRef.
- W. Chen, T. Y. Lei, T. Qian, W. Q. Lv, W. D. He, C. Y. Wu, X. J. Liu, J. Liu, B. Chen, C. L. Yan and J. Xiong, Adv. Energy Mater., 2018, 8, 1702889 CrossRef.
- S. Y. Wei, L. Ma, K. E. Hendrickson, Z. Y. Tu, L. A. Archer and J. Am, Chem. Soc., 2015, 137, 12143 CrossRef CAS PubMed.
- J. B. Liao, J. L. Wang, Z. Liu and Z. B. Ye, ACS Appl. Energy Mater., 2019, 2, 6732 CrossRef CAS.
- S. Y. Wei, L. Ma, K. E. Hendrickson, Z. Y. Tu and L. A. Archer, J. Am. Chem. Soc., 2015, 137, 12143 CrossRef CAS PubMed.
- S. Karuppiah, B. Kalimuthu, M. A. Nazrulla, S. Krishnamurty and K. Nallathamby, J. Mater. Chem. A, 2019, 7, 10067 RSC.
- Y. Yao, W. F. Feng, M. L. Chen, X. W. Zhong, X. J. Wu, H. B. Zhang and Y. Yu, Small, 2018, 14, 1802516 CrossRef PubMed.
- W. Q. Wang, D. Y. Wang, G. Y. Wang, M. Y. Zheng and G. C. Wang, Adv. Energy Mater., 2020, 10, 1904026 CrossRef CAS.
- X. Luo, X. B. Lu, X. D. Chen, Y. Chen, C. Y. Yu, D. W. Su, G. X. Wang and L. F. Cui, J. Energy Chem., 2020, 50, 63–72 CrossRef.
- J. B. Liao, Z. Liu, X. D. Liu and Z. B. Ye, J. Phys. Chem. C, 2018, 122, 25917 CrossRef CAS.
- Z. H. Shen, Z. L. Zhang, M. Li, Y. F. Yuan, Y. Zhao, S. Zhang, C. L. Zhong, J. Zhu, J. Lu and H. G. Zhang, ACS Nano, 2020, 14, 6673 CrossRef CAS PubMed.
- Z. W. Seh, W.-Y. Li, J. J. Cha, G.-Y. Zheng, Y. Yang, M. T. McDowell, P.-C. Hsu and Y. Cui, Nat. Commun., 2013, 4, 1331 CrossRef.
- X. Liang, C. Hart, Q. Pang, A. Garsuch, T. Weiss and L. F. Nazar, Nat. Commun., 2015, 6, 5682 CrossRef PubMed.
- Z. Li, J.-T. Zhang and X.-W. Lou, Angew. Chem., Int. Ed., 2015, 54, 12886 CrossRef CAS PubMed.
- K. K. Xiao, Z. Chen, Z. Liu, L. L. Zhang, X. Y. Cai, C. S. Song, Z. F. Fan, X. H. Chen, J. L. Liu and Z. X. Shen, J. Power Sources, 2020, 455, 227959 CrossRef CAS.
- J. Yu, J. W. Xiao, A. Li, Z. Yang, L. Zeng, Q. F. Zhang, Y. J. Zhu and L. Guo, Angew. Chem., Int. Ed., 2020, 59, 13071 CrossRef CAS PubMed.
- M. Z. Sun, Z. Wang, X. Li, H. B. Li, H. S. Jia, X. X. Xue, M. Jin, J. Q. Li, Y. Xie and M. Feng, J. Mater. Chem. A, 2020, 8, 11818 RSC.
- H. A. Salem, G. Babu, C. V. Rao and L. M. R. Arava, J. Am. Chem. Soc., 2015, 137, 11542 CrossRef PubMed.
- Y. Liu, W. Kou, X. C. Li, C. Q. Huang, R. B. Shui and G. H. He, Small, 2019, 15, 1902431 CrossRef PubMed.
- Y.-Q. Lu, Y.-J. Wu, T. Sheng, X.-X. Peng, Z.-G. Gao, S.-J. Zhang, L. Deng, R. Nie, J. Swiatowska, J.-T. Li, Y. Zhou, L. Huang, X.-D. Zhou and S.-G. Sun, ACS Appl. Mater. Interfaces, 2018, 10, 13499 CrossRef CAS PubMed.
- X. L. Yao, J. J. Xu, Z. L. Hong, G. R. Li, X. W. Wang, F. Lu, W. H. Wang, H. Liu, C. D. Liang, Z. Lin and W. C. Wang, J. Phys. Chem. C, 2018, 122, 3263 CrossRef CAS.
- Y. Z. Wang, D. Adekoya, J. Q. Sun, T. Y. Tang, H. L. Qiu, L. Xu, S. Q. Zhang and Y. L. Hou, Adv. Funct. Mater., 2019, 29, 1807485 CrossRef.
- J. B. Li, C. Y. Chen, Y. W. Chen, Z. H. Li, W. F. Xie, X. Zhang, M. F. Shao and M. Wei, Adv. Energy Mater., 2019, 1901935 CrossRef CAS.
- S. H. Liu, J. Li, X. Yan, Q. F. Su, Y. H. Lu, J. S. Qiu, Z. Y. Wang, X. D. Lin, J. L. Huang, R. L. Liu, B. N. Zheng, L. Y. Chen, R. W. Fu and D. C. Wu, Adv. Mater., 2018, 30, 1706895 CrossRef PubMed.
- T. Q. Zhang, Z. Chen, J. X. Zhao and Y. H. Ding, Diamond Relat. Mater., 2018, 90, 72 CrossRef CAS.
- B. Qiao, A. Wang, X. Yang, L. F. Allard, Z. Jiang, Y. Cui, J. Liu, J. Li and T. Zhang, Nat. Chem., 2011, 3, 634 CrossRef CAS PubMed.
- D. Zhang, S. Wang, R. M. Hu, J. N. Gu, Y. L. S. Cui, B. Li, W. H. Chen, C. T. Liu, J. X. Shang and S. B. Yang, Adv. Funct. Mater., 2020, 30, 2002471 CrossRef CAS.
- L. Z. Fang, Z. G. Feng, L. Cheng, R. E. Winans and T. Li, Small Methods, 2020, 2000315 CrossRef CAS.
- Y. J. Li, J. B. Wu, B. Zhang, W. Y. Wang, G. Q. Zhang, Z. W. Seh, N. Zhang, J. Sun, L. Huang, J. J. Jiang, J. Zhou and Y. M. Sun, Energy Storage Mater., 2020, 30, 250 CrossRef.
- Y. J. Li, G. L. Chen, J. R. Mou, Y. Z. Liu, S. F. Xue, T. Tan, W. T. Zhong, Q. Deng, T. Li, J. H. Hu, C. H. Yang, K. Huang and M. L. Liu, Energy Storage Mater., 2020, 28, 196 CrossRef.
- Z. F. Zhong, Acta Phys.-Chim. Sin., 2019, 35, 1047 Search PubMed.
- Z. Z. Du, X. J. Chen, W. Hu, C. H. Chuang, S. Xie, A. Hu, W. S. Yan, X. H. Kong, X. J. Wu, H. X. Ji and L.-J. Wan, J. Am. Chem. Soc., 2019, 141, 3977 CrossRef CAS PubMed.
- G. M. Zhou, S. Y. Zhao, T. S. Wang, S.-Z. Yang, B. Johannessen, H. Chen, C. W. Liu, Y. S. Ye, Y. C. Wu, Y. C. Peng, C. Liu, S. P. Jiang, Q. F. Zhang and Y. Cui, Nano Lett., 2020, 20, 1252 CrossRef CAS PubMed.
- W. J. Dong, D. Wang, X. Y. Li, Y. Yao, X. Zhao, Z. Wang, H.-E. Wang, Y. Li, L. H. Chen, D. Qian and B.-L. Su, J. Energy Chem., 2020, 48, 259 CrossRef.
- Z. Li, J. Zhang, B. Guan, D. Wang, L. M. Liu and X. W. Lou, Nat. Commun., 2016, 7, 13065 CrossRef CAS PubMed.
- X. Liu, J.-Q. Huang, Q. Zhang and L. Q. Mai, Adv. Mater., 2017, 29, 1601759 CrossRef.
- N. N. Hu, X. S. Lv, Y. Dai, L. L. Fan, D. B. Xiong and X. F. Li, ACS Appl. Mater. Interfaces, 2018, 10, 18665 CrossRef CAS PubMed.
- M. Ding, S. Z. Huang, Y. Wang, J. P. Hu, M. E. Pam, S. Fan, Y. M. Shi, Q. Ge and H. Y. Yang, J. Mater. Chem. A, 2019, 7, 25078 RSC.
- H.-E. Wang, K. L. Yin, N. Qin, X. Zhao, F.-J. Xia, Z.-Y. Hu, G. L. Guo, G. Z. Cao and W. J. Zhang, J. Mater. Chem. A, 2019, 7, 10346 RSC.
- X. Y. Tao, J. G. Wang, C. Liu, H. T. Wang, H. B. Yao, G. Y. Zheng, Z. W. Seh, Q. X. Cai, W. Y. Li, G. M. Zhou, C. X. Zu and Y. Cui, Nat. Commun., 2016, 7, 11203 CrossRef CAS PubMed.
- J.-Q. Huang, J.-Q. Huang, W. G. Chong, J. Cui, S. S. Yao, B. L. Huang and J.-K. Kim, J. Energy Chem., 2019, 35, 204 CrossRef.
- X. Chen, H.-J. Peng, R. Zhang, T.-Z. Hou, J.-Q. Huang, B. Li and Q. Zhang, ACS Energy Lett., 2017, 2, 795 CrossRef CAS.
- J. T. Liu, S. H. Xiao, L. Chang, L. Lai, R. Wu, Y. Xiang, X. Q. Liu and J. S. Chen, J. Energy Chem., 2021, 56, 343 CrossRef.
- J. Pu, Z. Shen, J. Zheng, W. Wu, C. Zhu, Q. Zhou, H. Zhang and F. Pan, Nano Energy, 2017, 37, 7 CrossRef CAS.
- X. B. Liao, Z. H. Li, Q. He, L. X. Xia, Y. Li, S. H. Zhu, M. M. Wang, H. Wang, X. Xu, L. Q. Mai and Y. Zhao, ACS Appl. Mater. Interfaces, 2020, 12, 9181 CrossRef CAS PubMed.
- C. George, A. J. Morris, M. H. Modarres and M. D. Volder, Chem. Mater., 2016, 28, 7304 CrossRef CAS PubMed.
- G. Ai, Q. Q. Hu, L. Zhang, K. H. Dai, J. Wang, Z. J. Xu, Y. Huang, B. Zhang, D. J. Li, T. Zhang, G. Liu and W. F. Mao, ACS Appl. Mater. Interfaces, 2019, 11, 33987 CrossRef CAS PubMed.
- S. Z. Huang, Y. Wang, J. P. Hu, Y. V. Lim, D. Z. Kong, Y. Zheng, M. Ding, M. E. Pam and H. Y. Yang, ACS Nano, 2018, 12, 9504 CrossRef CAS PubMed.
- B. Moorthy, S. Kwon, J.-H. Kim, P. Ragupathy, H. M. Lee and D. K. Kim, Nanoscale Horiz., 2019, 4, 214 RSC.
- J. Yang, H. Gao, S. Men, Z. Shi, Z. Lin, X. Kang and S. Chen, Adv. Sci., 2018, 5, 1800763 CrossRef.
- C.-Y. Fan, Y.-P. Zheng, X.-H. Zhang, Y.-H. Shi, S.-Y. Liu, H.-C. Wang, X.-L. Wu, H.-Z. Sun and J.-P. Zhang, Adv. Energy Mater., 2018, 8, 1703638 CrossRef.
- D. Cai, B. K. Liu, D. H. Zhu, D. Chen, M. J. Lu, J. M. Cao, Y. H. Wang, W. H. Huang, Y. Shao, H. R. Tu and W. Han, Adv. Energy Mater., 2020, 10, 1904273 CrossRef CAS.
- L. Najafi, S. Bellani, R. Oropesa-Nunez, M. Prato, B. Martin-Garcia, R. Brescia and F. Bonaccorso, ACS Nano, 2019, 13, 3162 CrossRef CAS PubMed.
- W. Tian, B. Xi, Z. Feng, H. Li, J. Feng and S. Xiong, Adv. Energy Mater., 2019, 9, 1901896 CrossRef.
- Z. Q. Ye, Y. Jiang, L. Li, F. Wu and R. J. Chen, Adv. Mater., 2020, 2002168 CrossRef CAS.
- Z. J. Chen, W. Lv, F. Y. Kang and J. Li, J. Phys. Chem. C, 2019, 123, 25025 CrossRef CAS.
- Z. H. Li, Q. He, X. Xu, Y. Zhao, X. W. Liu, C. Zhou, D. Ai, L. X. Xia and L. Q. Mai, Adv. Mater., 2018, 30, 1804089 CrossRef.
- C. C. Li, J. J. Shi, L. Zhu, Y. Y. Zhao, J. Lu and L. Q. Xu, Nano Res., 2018, 11, 4302 CrossRef CAS.
- Z. X. Hao, J. Chen, L. X. Yuan, Q. M. Bing, J. Y. Liu, W. L. Chen, Z. Li, F. R. Wang and Y. H. Huang, Small, 2019, 15, 1902377 CrossRef CAS PubMed.
- Y. K. Wang, R. F. Zhang, Y.-C. Pang, X. Chen, J. X. Lang, J. J. Xu, C. H. Xiao, H. L. Li, K. Xi and S. J. Ding, Energy Storage Mater., 2019, 16, 228 CrossRef.
- W. W. Sun, C. Liu, Y. J. Li, S. Q. Luo, S. K. Liu, X. B. Hong, K. Xie, Y. M. Liu, X. J. Tan and C. M. Zheng, ACS Nano, 2019, 13, 12137 CrossRef CAS PubMed.
- W. Kou, G. H. Chen, Y. Liu, W. X. Guan, X. C. Li, N. Zhang and G. H. He, J. Mater. Chem. A, 2019, 7, 20614 RSC.
- Y. Z. Wang, M. Li, L. C. Xu, T. Y. Tang, Z. Ali, X. X. Huang, Y. L. Hou and S. Q. Zhang, Chem. Eng. J., 2019, 358, 962 CrossRef CAS.
- G. Y. Jiang, N. Zheng, X. Chen, G. Y. Ding, Y. H. Li, F. G. Sun and Y. S. Li, Chem. Eng. J., 2019, 373, 1309 CrossRef CAS.
- Y. Z. Song, Z. T. Sun, Z. D. Fan, W. L. Cai, Y. L. Shao, G. Sheng, M. L. Wang, L. X. Song, Z. F. Liu, Q. Zhang and J. Y. Sun, Nano Energy, 2020, 70, 104555 CrossRef CAS.
- H. X. Li, S. Ma, H. Q. Cai, H. H. Zhou, Z. Y. Huang, Z. H. Hou, J. J. Wu, W. J. Yang, H. B. Yi, C. P. Fu and Y. F. Kuang, Energy Storage Mater., 2019, 18, 338 CrossRef.
- X. Liang, A. Garsuch and L. F. Nazar, Angew. Chem., Int. Ed., 2015, 54, 3907 CrossRef CAS PubMed.
- E. S. Sim, G. S. Yi, M. Je, Y. B. Lee and Y.-C. Chung, J. Power Sources, 2017, 342, 64 CrossRef CAS.
- N. Li, Q. Q. Meng, X. H. Zhu, Z. Li, J. L. Ma, C. X. Huang, J. Song and J. Fan, Nanoscale, 2019, 11, 8485 RSC.
- S. Z. Huang, Y. V. Lim, X. M. Zhang, Y. Wang, Y. Zheng, D. Z. Kong, M. Ding, S. Y. A. Yanga and H. Y. Yang, Nano Energy, 2018, 51, 340 CrossRef CAS.
- H.-J. Peng, Z.-W. Zhang, J.-Q. Huang, G. Zhang, J. Xie, W.-T. Xu, J.-L. Shi, X. Chen, X. B. Cheng and Q. Zhang, Adv. Mater., 2016, 28, 9551 CrossRef CAS PubMed.
- Z. Q. Ye, Y. Jiang, T. Feng, Z. H. Wang, L. Li, F. Wu and R. J. Chen, Nano Energy, 2020, 70, 104532 CrossRef CAS.
- K. K. Xiao, Z. Chen, Z. Liu, L. L. Zhang, X. Y. Cai, C. S. Song, Z. F. Fan, X. H. Chen, J. L. Liu and Z. X. Shen, J. Power Sources, 2020, 455, 227959 CrossRef CAS.
- S. Z. Huang, Y. V. Lim, X. M. Zhang, Y. Wang, Y. Zheng, D. Z. Kong, M. Ding, S. Y. A. Yanga and H. Y. Yang, Nano Energy, 2018, 51, 340 CrossRef CAS.
- J. B. Zhou, X. J. Liu, L. Q. Zhu, J. Zhou, Y. Guan, L. Chen, S. W. Niu, J. Y. Cai, D. Sun, Y. C. Zhu, J. Du, G. M. Wang and Y. T. Qian, Joule, 2018, 2, 2681 CrossRef CAS.
- Y.-X. Mo, J.-X. Lin, Y.-J. Wu, Z.-W. Yin, Y.-Q. Lu, J.-T. Li, Y. Zhou, T. Sheng, L. Huang and S.-G. Sun, ACS Appl. Mater. Interfaces, 2019, 11, 4065 CrossRef CAS PubMed.
- Y. J. Lia, P. Xu, G. L. Chen, J. R. Mou, S. F. Xue, K. Li, F. H. Zheng, Q. F. Dong, J. H. Hu, C. H. Yang and M. L. Liu, Chem. Eng. J., 2020, 380, 122595 CrossRef.
- Y. Y. Xiao, Y. T. Liu, G. H. Qin, P. Y. Han, X. Y. Guo, S. X. Cao and F. S. Liu, Composites, Part B, 2020, 193, 108004 CrossRef CAS.
- R. C. Wang, C. Luo, T. S. Wang, G. M. Zhou, Y. Q. Deng, Y. B. He, Q. F. Zhang, F. Y. Kang, W. Lv and Q.-H. Yang, Adv. Mater., 2020, 200031 Search PubMed.
- R. P. Fang, S. Y. Zhao, Z. Sun, D.-W. Wang, R. Amal, S. G. Wang, H.-M. Cheng and F. Li, Energy Storage Mater., 2018, 10, 56 CrossRef.
- Y. Z. Song, W. Zhao, L. Kong, L. Zhang, X. Y. Zhu, Y. L. Shao, F. Ding, Q. Zhang, J. Y. Sun and Z. F. Liu, Energy Environ. Sci., 2018, 11, 2620 RSC.
- R. C. Wang, C. Luo, T. S. Wang, G. M. Zhou, Y. Q. Deng, Y. B. He, Q. F. Zhang, F. Y. Kang, W. Lv and Q.-H. Yang, Adv. Mater., 2020, 2000315 CrossRef CAS PubMed.
- B. Zhang, C. Luo, Y. Q. Deng, Z. J. Huang, G. M. Zhou, W. Lv, Y.-B. He, Y. Wan, F. Y. Kang and Q.-H. Yang, Adv. Energy Mater., 2020, 2000091 CrossRef CAS.
- M. Waqas, Y. P. Han, D. J. Chen, S. Ali, C. Zhen, C. Feng, B. T. Yuan, J. C. Han and W. D. He, Energy Storage Mater., 2020, 27, 333 CrossRef.
- Q. Liu, X. S. Wang, J. L. Wang and X. Huang, Acta Phys.-Chim. Sin., 2019, 35, 1099 Search PubMed.
- T. H. Zhou, W. Lv, J. Li, G. M. Zhou, Y. Zhao, S. X. Fan, B. L. Liu, B. H. Li, F. Y. Kang and Q.-H. Yang, Energy Environ. Sci., 2017, 10, 1694 RSC.
- C. Ye, Y. Jiao, H. Y. Jin, A. D. Slattery, K. Davey, H. H. Wang and S.-Z. Qiao, Angew. Chem., Int. Ed., 2018, 57, 1 CrossRef.
- S. X. Chen, J. H. Luo, N. Y. Li, X. X. Han, J. Wang, Q. Deng, Z. L. Zeng and S. G. Deng, Energy Storage Mater., 2020, 30, 187 CrossRef.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |