
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Steric hindrance dependence on the spin and morphology properties of highly oriented self-doped organic small molecule thin films†
Daniel
Powell
,
Eric V.
Campbell
 ,
Laura
Flannery
,
Jonathan
Ogle
,
Sarah E.
Soss
and
Luisa
Whittaker-Brooks
*
,
Laura
Flannery
,
Jonathan
Ogle
,
Sarah E.
Soss
and
Luisa
Whittaker-Brooks
*
Department of Chemistry, University of Utah, Salt Lake City, UT 84112, USA. E-mail: luisa.whittaker@utah.edu
First published on 27th November 2020
Abstract
Introducing charge carriers is of paramount importance for increasing the efficiency of organic semiconducting materials. Various methods of extrinsic doping, where molecules or atoms with large/small reduction potentials are blended with the semiconductor, can lead to dopant aggregation, migration, phase segregation, and morphology alteration. Self-doping overcomes these challenges by structurally linking the dopant directly to the organic semiconductor. However, for their practical incorporation into devices, self-doped organic materials must be cast into thin-films, yet processing methods to allow for the formation of continuous and uniform films have not been developed beyond simple drop-casting. Whilst self-doped organic molecules afford the remarkable ability to position dopants with molecular precision and control of attachment mode, their steric bulk inevitably disrupts the crystallization on surfaces. As such, there is great interest in the development of processing modalities that allow deposited molecules to converge to the thermodynamic minimum of a well-ordered and highly crystalline organic thin film instead of getting trapped in local disordered minima that represent metastable configurations. By contrasting drop casting, ultrasonic deposition, and physical vapor deposition, we investigate the free energy landscape of the crystallization of sterically hindered self-doped perylene diimide thin films. A clear relationship is established between processing conditions, the crystallinity and order within the deposited films, the dopant structures and the resulting spin density. We find physical vapor deposition to be a robust method capable of producing smooth, continuous, highly ordered self-doped organic small molecule thin-films with tailored spin concentrations and well-defined morphologies.
Introduction
Organic semiconductors (OSCs) can generally be subdivided into two major categories, i.e., polymers and small molecules. The latter has attracted considerable attention from the semiconductor community because they possess many unique advantages over their polymeric counterparts. Chain length distribution, polymer architecture, local and global morphological ordering, and polymer microstructure are confounded by even small variations in monomer selection and synthetic procedures.1–3 Organic small molecules are more structurally homogeneous and synthetically modular than polymers, making them attractive for organic semiconductor applications.4 The intended use of a given small molecule OSC will dictate the specific properties it must possess. Some applications require highly specific properties that may be noninterchangeable between platforms, such as particular orientation, electronic structure, or photophysical properties.3–13 There are, however, some general characteristics of functional small molecule OSCs that are generally commutable. Speaking in the broadest sense, a good OSC will have a substantial degree of preferential molecular organization and it will be effectively doped to account for their low charge carrier concentrations.14Doping is an essential step in achieving any functional OSC regardless of its categorization.15,16 While several doping strategies have been employed for polymeric systems (e.g., extrinsic doping, intrinsic doping, thermally activated in situ doping, etc.), small molecule OSCs are almost ubiquitously doped with other small molecules possessing desirable ionization potentials.15,17–26 Molecular n-type doping is achieved by mixing low ionization potential containing species with the OSC and subsequently optimizing solution-based processing conditions to obtain ordered thin-films. A number of debilitating issues typically arise when molecular doping is employed. For example, when a dopant is mixed with a host OSC, the solubilities of the respective species may be very different from one another.27,28 Solubility differences between the host and dopant results in inhomogeneous mixing. Phase segregation of the solvent orthogonal species and subsequent aggregation of the dopant during film fabrication have been the historically canonical hurdles in OSC development.22,29 Inhomogeneous mixing of dopants is detrimental because aggregation leads to local grain boundaries that decrease electron mobility values and stifle doping efficiency.30,31 Furthermore, phase segregation is known to occur at even modest doping concentrations (<0.1%).19 There is a wealth of literature solely dedicated to improving the miscibility between dopants and OSC hosts, owing to the great importance of achieving a homogeneous distribution. However, this process is time-consuming and may yield only limited results for a given pair. Furthermore, it has been demonstrated that entirely different morphological phases may arise when optimizing dopant concentration.32,33 In such scenarios, changes in the carrier concentration from doping are convoluted with changes to a material's altered electronic structure via morphological differences. This convolution makes it very difficult to precisely determine the source or nature of any material observable that is simultaneously sensitive to both carrier concentration and morphology (e.g., electrical conductivity, electron mobility, etc.). Elevation or suppression of the observable can only be adequately described in a system where the morphology is primarily unchanged while the carrier concentration is modified or vice versa.13,29,34 Beyond intrinsic material parameters, the deconvolution of these properties is essential for the optimization of several electronic devices, such as organic thermoelectrics, where the figure of merit, ZT, is maximized by fine tuning the carrier concentration for a particular material quality factor.34 Finally, the introduction of dopants modifies the density of states near the Fermi-energy such that populated states now exist at some energy relative to the highest occupied molecular orbital (HOMO) or lowest unoccupied molecular orbital (LUMO) of the OSC. Upon charge transfer, the energies of these states distort in ways that may be very difficult to predict, leading to a broadened density of states.15,35 The physical distribution of dopants within a host matrix directly affects the density of states, which is relevant for a number of applications.7,15,20 Self-doping intrinsically mitigates these issues (vide infra).
Furthermore, perylene diimides (PDIs) with covalently-bound amino functional groups, such as those depicted in Fig. 1A, have been shown to form stable radical anions at room temperature and have demonstrated great promise in many applications including photovoltaic transport layers, n-type thermoelectrics, battery cathodes, WORM memory devices, biological and chemical sensing, as well as chromic and thermotropic liquid crystals for thin-film polarizers.36–49 However, any future incorporation of these materials into practical next-generation functional devices will require a robust processability method for producing high-quality thin-films. Despite the rising interest, attempts to fabricate thin-films have been limited to drop-casting from chloroform solutions (or water after methylation of the amine), which often produces rough and discontinuous films with little to no control over thickness.37,46,50 As such, drop-casting is usually not amenable to large scale processing and is seen as an inferior method of thin-film production. Here, we report a robust physical vapor deposition fabrication process capable of yielding highly oriented, uniform, and continuous self-doped organic thin-films. We believe this method can be extended to the fabrication of other self-doped organic small molecule thin-films that are not amenable to solution processing.
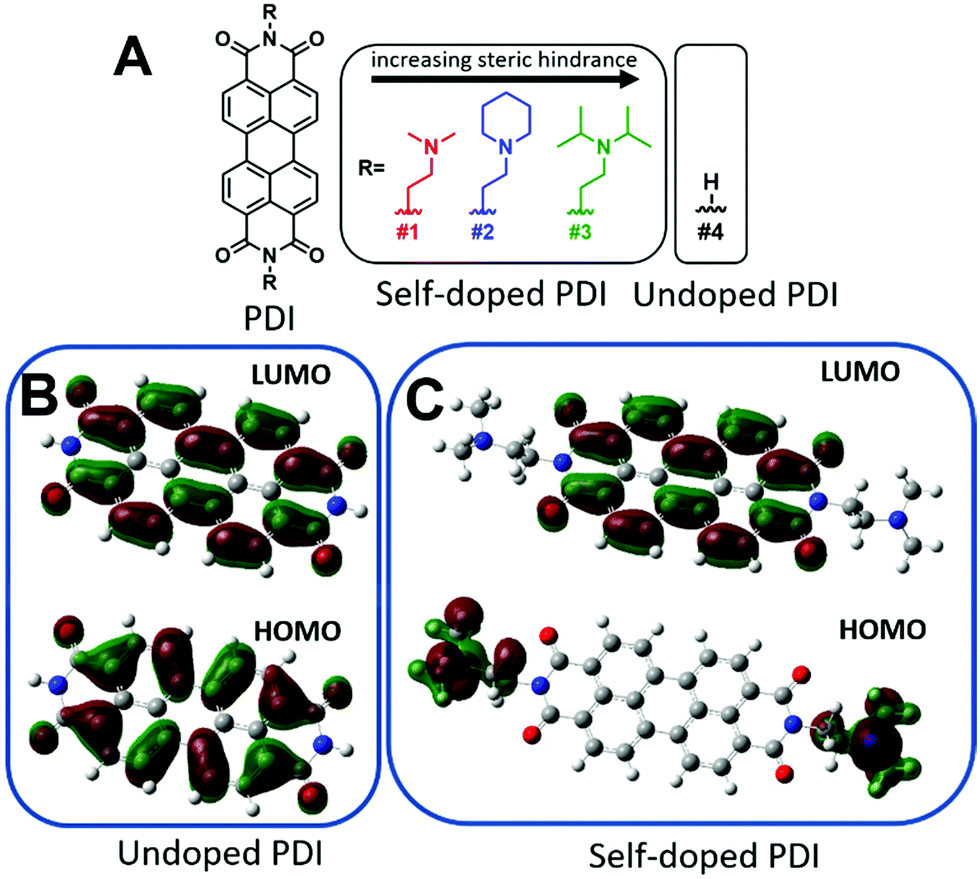 | ||
| Fig. 1 (A) Chemical structures for self-doped and undoped PDIs. The self-doped PDIs are ordered as a function of increasing steric hindrance. Gaussian simulated molecular orbitals for undoped and self-doped PDIs in their charge neutral forms. (B) HOMO and LUMO depictions for undoped PDI. (C) HOMO and LUMO depictions for self-doped PDI. | ||
Results and discussion
Self-doping, a process known by several names in the small molecule literature including “intrinsic doping,” or “push–pull,” can be generally understood to mean an n-type organic semiconductor that has been doped by a covalently bound electron source.36,51–54Fig. 1 depicts the series of self-doped organic molecules (perylene diimides, PDIs) that were synthesized and fully characterized (Fig. S1–S6, ESI†) using a simplified method developed in our laboratory (#4 was purchased directly from Sigma Aldrich without further purification). Experimental details are available in the supporting information. Very little is known about the impact of self-dopant architecture on the doping process. Regarding PDIs, previous reports have been limited to tertiary and quaternary methylamines, yet there are several reasons one may choose to modify this architecture.36,37,46,47 Dimethylamines attached to chromophores are known to undergo a photoinduced electron transfer process to generate the PDI radical anion PDI˙−.55–57 However, the precise variables affecting the reactivity or subsequent stability of the resultant radical amines R′-NR2˙+ are unclear. Some architectures are prone to degradation of the amine moiety while others exhibit pronounced stability.50,58,59 However, since there are no clearly established self-dopant design principles for PDIs, the optimal dopant architecture remains largely unknown. For example, carbon may act as an inductive group capable of donating electron density to stabilize organic radical formation, or conversely, the added steric hindrance may impede doping. Prior to this work, it was utterly unknown how the addition of aliphatic groups about the nitrogen source might affect radical formation. Moreover, the ideal thin-film processing method will be translatable to other architectures as the dopant structure is optimized. Thus, the aliphatic head groups added to our PDI molecules serve two essential functions: to observe changes in spin concentration through dopant structural modification and to challenge the robustness of our thin-film processing method.To investigate if the dopant structures provided in Fig. 1A are capable of doping PDIs, we performed Gaussian simulations to yield the electronic structure of the self-doped PDI molecules of interest. Fig. 1B show the Gaussian simulated frontier molecular orbitals for the neutral forms in the ground state of both undoped and self-doped PDIs. While the exact values of HOMO and LUMO orbitals in solid matrices are generally miscalculated by DFT, the relative ordering of orbital energies in ground state molecules is reliable. We do not report the calculated values for the frontier molecular orbital energies, but rather use them to depict the location of the HOMO and LUMO within the molecules. As depicted in Fig. 1B, the HOMO and LUMO of undoped PDIs are centralized on the PDI core. Contrastingly, the host and dopant in self-doped systems are comprised of a single set of molecular orbitals (Fig. 1C). As shown in the simulated frontier molecular orbitals for self-doped PDIs, prior to electron transfer, the HOMO and LUMO are localized on the amino electron source and the electron-accepting core, respectively. The presence of a PDI radical anion was experimentally confirmed via absorption spectroscopy studies (Fig. S7, ESI†). The donor and acceptor moieties are in close proximity to one another in this system, and their segregation from one another may be suppressed. This configuration eliminates any need to experimentally optimize host/dopant mixing, which imparts ease of use to these materials not present in molecularly doped systems. This arrangement thus allows for thin-film fabrication methods to be used that traditionally are not feasible for extrinsically doped systems, but that might otherwise be desirable.
Here, we investigated three different processing methods for the fabrication of self-doped PDI thin films, namely drop-casting, spray coating, and physical vapor deposition (Fig. 2). Traditionally, the overwhelming majority of PDI thin films have been solution processed via spin-coating, but to date no tertiary amine self-doped PDIs have been successfully spin-coated. We attempted to make spin-coated films but were unsuccessful due to poor substrate adhesion. This issue persisted despite any surface modification techniques we employed, including the use of self-assembled monolayers (SAMs) to modify the sample–substrate interactions and plasma cleaning. Consequently, we used a previously reported method of drop-casting for comparison where 1 mg mL−1 chloroform solutions of compounds 1–3 were drop-casted onto clean glass slides at room temperature.50 An SEM image of drop-cast compound #2 in Fig. 3A depicts large needle-like structures with sizes ranging from 5–100 μm in length that readily form on the substrate surface during solvent evaporation and tend to aggregate in sizable clusters near the substrate edges. These discontinuous aggregates vary widely in agglomerate size from hundreds of microns to several millimeters. Various substrates were tested, including glass, ITO, silicon, and silicon dioxide at various temperatures, both with and without plasma cleaning. However, we were unable to produce fully continuous drop-cast films for compounds #1–3 under any experimental conditions. Thin-film continuity is critical to material performance as discontinuities inhibit charge carrier flow.60,61 The as-prepared drop-cast thin films have poor substrate affinity; a common issue for many small molecule organic semiconductors. To produce more uniform films, we spray coated the compounds onto glass slides. Substrates were heated to 60 °C during the deposition process and 1 mg mL−1 chloroform solutions of each compound were passed through an ultrasonic spray nozzle at a controlled flow rate of 2 mL min−1. The formation of larger needles was mitigated with the aid of the ultrasonic nozzle, which atomizes the solution as it passes through the tip. Additionally, smaller crystallites on the order of 0.5–2 μm in length tend to be more evenly distributed across the substrate surface. However, aggregates still form at the edges of the spray droplets before solvent evaporation, as depicted in the SEM micrograph shown in Fig. 3B. While the films are more homogeneous across the substrate surface than those obtained by drop-casting, all sprayed films contain discontinuities regardless of the experimental conditions. Longer spraying results in better substrate coverage but does not mitigate crystallite formation and aggregation.
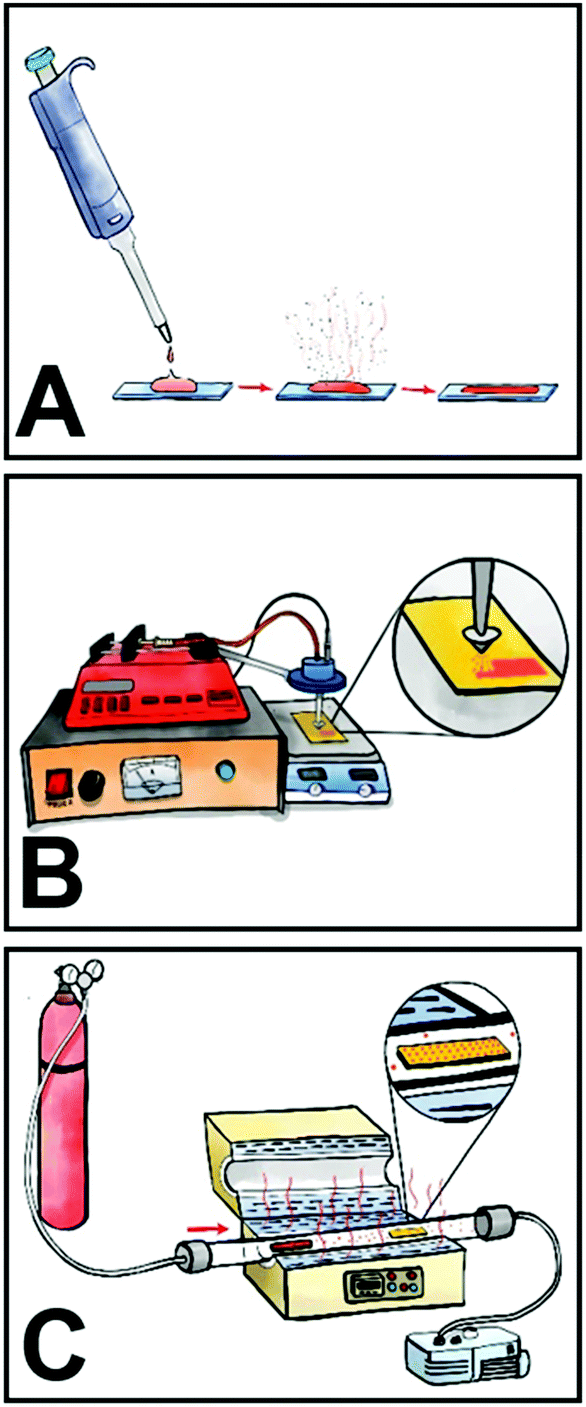 | ||
| Fig. 2 Depiction of the three thin film processing methods investigated. (A) Drop-casting and dried in air at room temperature. (B) Ultrasonic spray coating onto heated substrates. (C) Physical vapor deposition in a tube furnace under vacuum. | ||
 | ||
| Fig. 3 SEM images of self-doped PDI (compound #2) (A) drop-cast (B) ultrasonically sprayed and (C) vapor deposited into thin films. | ||
Physical vapor deposition (PVD) is a well-established method of producing thin-films.62 The technique requires users to heat materials under vacuum for film deposition, which may be a potential source of degradation. In contrast to thermal evaporation, PVD protocols take advantage of a temperature gradient across a tube furnace which help with the fabrication of thin films with more controlled morphologies. Thermogravimetric analysis (TGA) was performed on compounds 1–3 to determine their thermal stability and we observed that they begin to degrade at 320 °C, 300 °C, and 350 °C, respectively (Fig. S8–S10, ESI†). Therefore, we selected 305 °C, 295 °C, and 310 °C as the deposition temperatures for compounds #1, #2, and #3, respectively. Additionally, PVD is carried out under vacuum (<1 × 10−4 Torr) which further imparts stability to compounds 1–3. All of the films produced with this method are found to be fully continuous and evenly coat the substrate (Fig. 3C). The color of the thin-films depends on the sample thickness, which ranged from black to bright red. The film thickness is controlled by altering the deposition time and sample loading. Additionally, large crystallite aggregates are fully mitigated with this method, making it superior to the other methods tested.
In addition to continuity, surface roughness has been shown to disrupt the morphology of organic thin-film materials. For example, surface roughness is known to affect AC and DC transport properties, including electron mobility and conductivity.61,63 We measured the average surface roughness of PVD films using atomic force microscopy (AFM). Due to the extreme heterogeneity and diversity in crystallite size within drop-cast films, we were unable to measure the roughness of these samples with AFM. Additionally, the roughness of spray-coated films varies widely across the film surface from spot to spot by several hundred nanometers, likely due to spray droplet evaporation effects. Average surface roughness values are consistently found to be several hundred nanometers but are nevertheless measurable, indicating that they are considerably smoother than drop-cast films. The lowest surface roughness values measured for spray-coated films of compounds #1–3 are 228 nm, 153 nm, and 151 nm, respectively. For PVD films, the average roughness is between one and two orders of magnitude smoother than sprayed films, with average roughness values measured to be 30 nm, 3 nm, and 13 nm for compounds 1, 2, and 3, respectively (Fig. S11, ESI†).
To investigate the impact of thin-film processing on molecular orientation, we performed grazing incidence wide-angle X-ray scattering (GIWAXS) for all three processing methods of compounds 1–3, as shown in Fig. 4. Drop-cast and spray-coated films of 1–3 exhibit several reflections along the qz, corresponding to lamellar stacking structures that are preferentially oriented along φ = 0°, some of which exhibit a strong degree of mosaicity. Drop-cast films of 2 and 3 also have several diffraction peaks oriented near φ = 45°. From this information, we can surmise that compounds 1–3 tend to orient themselves edge-on with respect to the substrate during solvent evaporation. Compounds 1 and 3 also display more Bragg reflections along the qz than compound 2 for both spraying and drop-casting, indicative of greater long-range ordering in these films. PVD produces thin-films that appear to be highly crystalline with very little mosaicity in any of the exhibited Bragg reflections. When compared against one another, compound 3 appears to have the greatest degree of mosaicity in the qxy peaks. To have a more quantitative comparative description, we measured the azimuthal orientation of the (001) reflection observed in the GIWAXS patterns for all the films using the mosaicity factor (MF) as an evaluative tool for morphology.64 MF is given by the equation:
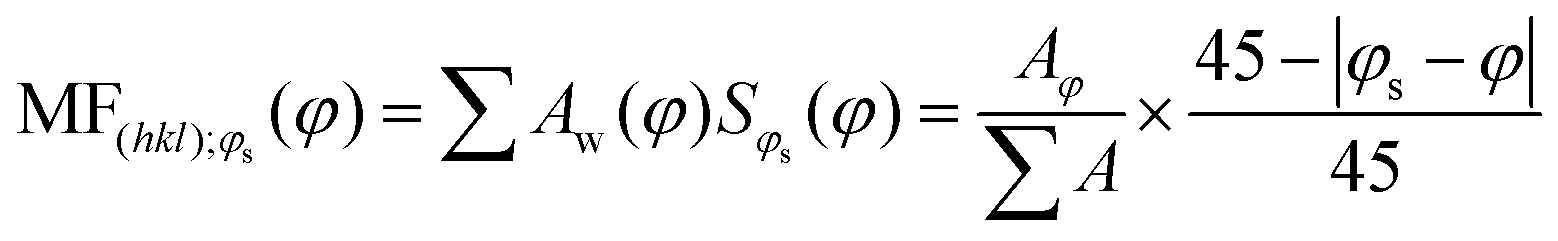 | (1) |
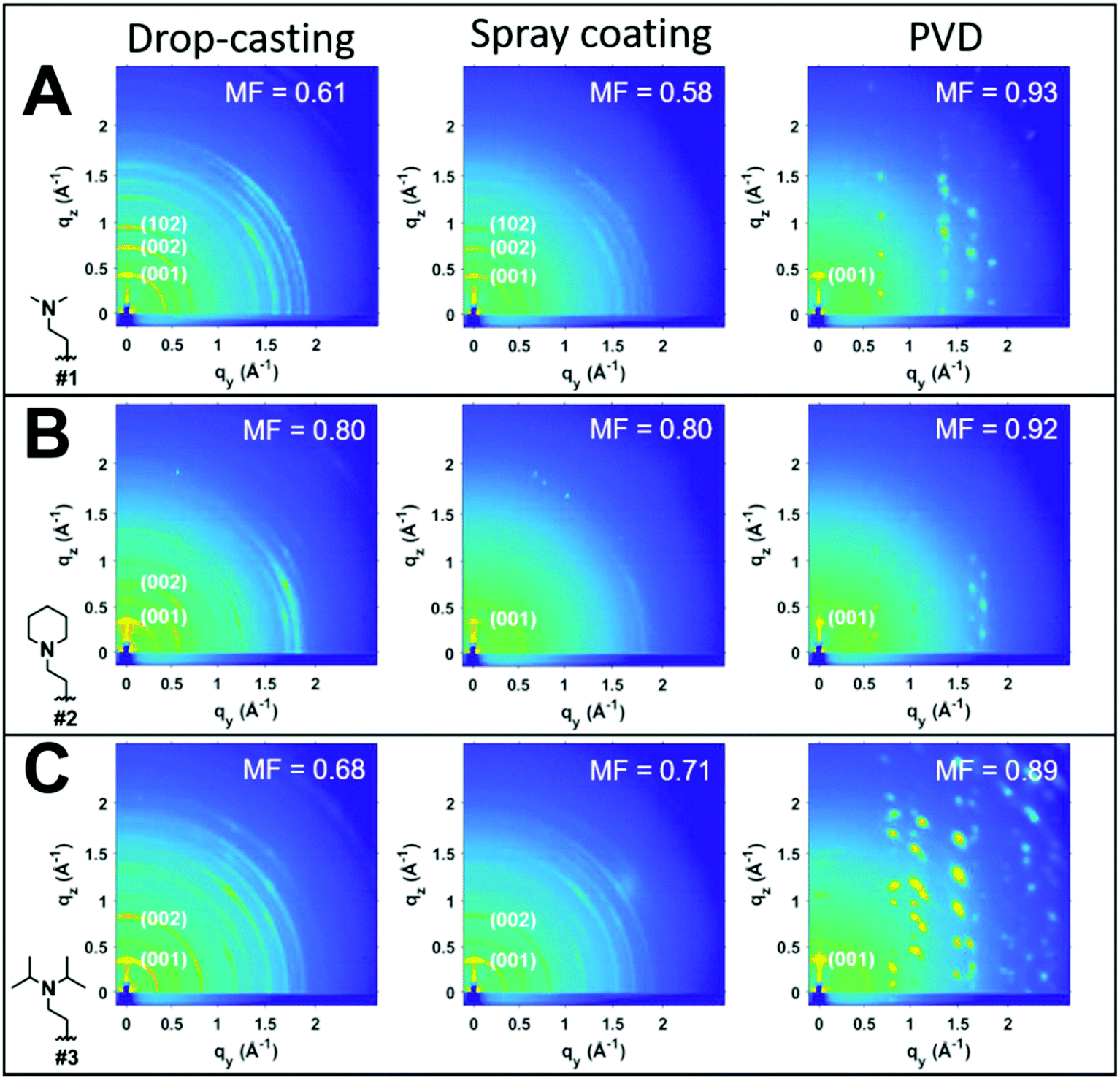 | ||
| Fig. 4 GIWAXS images of thin films. From left to right, the processing methods were drop-casting, spray coating, and PVD. Corresponding GIWAXS images of compound #1 (A), compound #2 (B), and compound #3 (C) as shown. | ||
The least oriented film in the series was the spray coated film of compound 1 with an MF = 0.58, while the PVD film of 1 was also the film with the smallest degree of mosaicity with an MF = 0.93. No difference in the orientation of the (001) plane was observed between drop-casting and spray coating of compound 2. Ultrasonic spraying improved the orientation of compound 3, with MF values of 0.68 and 0.71 for drop-casting and spraying, respectively. However, the modest changes in MF values for both drop-casting and spray coating indicates that these methods do not significantly alter the orientation of the crystallites. As stated previously, however, ultrasonic spray coating is a superior method to drop-casting due to its ability to minimize the formation of larger crystallites. We found PVD to be the most robust method as it created continuous, crystalline films with a strong degree of preferential orientation with MF values of 0.93, 0.92, and 0.89 for compounds #1, 2, and 3, respectively.
Since PVD requires sample heating, we tested the effect that annealing might have on the self-doping process. It is important that self-doping is not impaired by the film fabrication method for future applications. Via electron paramagnetic resonance (EPR) studies, we measured the spin concentration in powder samples, spray coated, and PVD film samples (Fig. S12, ESI†). Here we found that all processing methods yield the same spin concentrations within statistical error, demonstrating that PVD is a robust method for preparing self-doped thin-films with no observed degradation of the self-doped PDI molecule. To our knowledge, this is the first example of a self-doped PDI molecule processed via a vapor deposition process that yields very oriented, highly crystalline, and pinhole free thin films.
Assessing the stability of the radical amines is an important looming question in regard to self-doped systems given the thermodynamic instability often observed for amine radical cations. A few reports have shown that structural degradation of the radical amine can occur during sample annealing.50,55 However, others have observed pronounced stability of amine radicals attached to chromophores due to the formation of exciplexes.65–68 To test the stability of the radical amines, we performed solid state C13 NMR (SSNMR) on both heated and un-heated samples of compounds 1–3 in both air and under a nitrogen atmosphere (Fig. S13–S15, ESI†). No structural degradation was observed in samples equilibrated to air or nitrogen, nor was any sample degradation observed upon heating to 120 °C up to 7 days in nitrogen. However, in air, compound #2 showed clear signs of sample degradation in both the aromatic (120–140 ppm) and aliphatic (20–60 ppm) regions of the SSNMR spectrum (Fig. S14, ESI†). This demonstrates that subsequent reactivity of R′-NR2˙+ radical cation requires the presence of atmospheric oxidants, and involves a dealkylation mechanism that requires a specific construct of the dopant. Interestingly, this effect did not seem to be related to sterics of R′-NR2˙+, and instead may be related to the presence of longer hydrocarbon chains or an abundance of hydrogen in the alpha position.50,55 Moreover, this data demonstrates that R′-NR2˙+ radical cations are likely present in our systems rather than undergoing any further reaction under inert conditions.
Given that the PVD fabrication process allows for the systematic control of the morphology of our self-doped PDI thin films, we performed quantitative EPR measurements to investigate the impact that the addition of aliphatic head groups has on the generation of charge carriers. Fig. 5A shows the EPR spectra for undoped and self-doped PDIs. Compounds 1 and 3 have a first derivative Lorentzian line shape while compound 2 is a Gaussian–Lorentzian mixture due to dipolar broadening. The spin concentrations reported are defined as the number of radicals (i.e. EPR active states) present per 1 mole of sample. TEMPO, a widely known stable organic radical containing 1 spin equivalent per molecule, was used to construct a standard calibration curve. We then use this calibration curve to obtain the total number of spins, or unpaired electrons, in the samples. As depicted, the undoped PDI is not EPR spin active. However, upon the addition of aliphatic head groups which serve as dopants to the PDI molecules, we observe radical formation and an increase in spin concentrations. The average spin concentrations for self-doped PDIs are provided in Fig. 5B. We determined that even modest increases in the steric hindrance of the electron source results in a ten-fold decrease in doping efficiency, an observation which is consistent between both EPR and absorption studies (1021 to 1020 spins per mole when going from PDI self-doped with moiety #1 to #3, respectively). From EPR alone a ∼1–0.1% doping efficiency for 1–3 is obtained. However, the absorption spectra of the respective thin films (Fig. S4, ESI†) are indicative of a much higher concentration of the radical anion PDI˙−, pointing to a larger true doping efficiency (>5%). This discrepancy could plausibly be explained by the presence of spin-paired states which result in a magnetic moment of zero, and are thus EPR silent. We have already demonstrated that R′-NR2˙+ radical cations are present within our doped systems, leading us to propose that R′-NR2˙+ couples to PDI˙− following electron transfer, some of which are spin-paired. This coupled state is known as a heteroexcimer or exciplex, which imparts stability to what would otherwise be unstable radicals. Exciplexes have been demonstrated in similar constructs of tertiary amines covalently attached to chromophores.65–68 The fluorescence signal for compounds 1–3 thin films were also fully quenched, consistent with the formation of exciplexes, and leads us to propose that these and other self-doped PDIs may be ideally suited to organic light emitting diode (OLED) applications.69 We also note the possibility that spin pairing between PDI˙− and PDI˙− is also plausible if R′-NR2˙+ were to act as a reversible photoacid, or undergo some other reversible transformation.
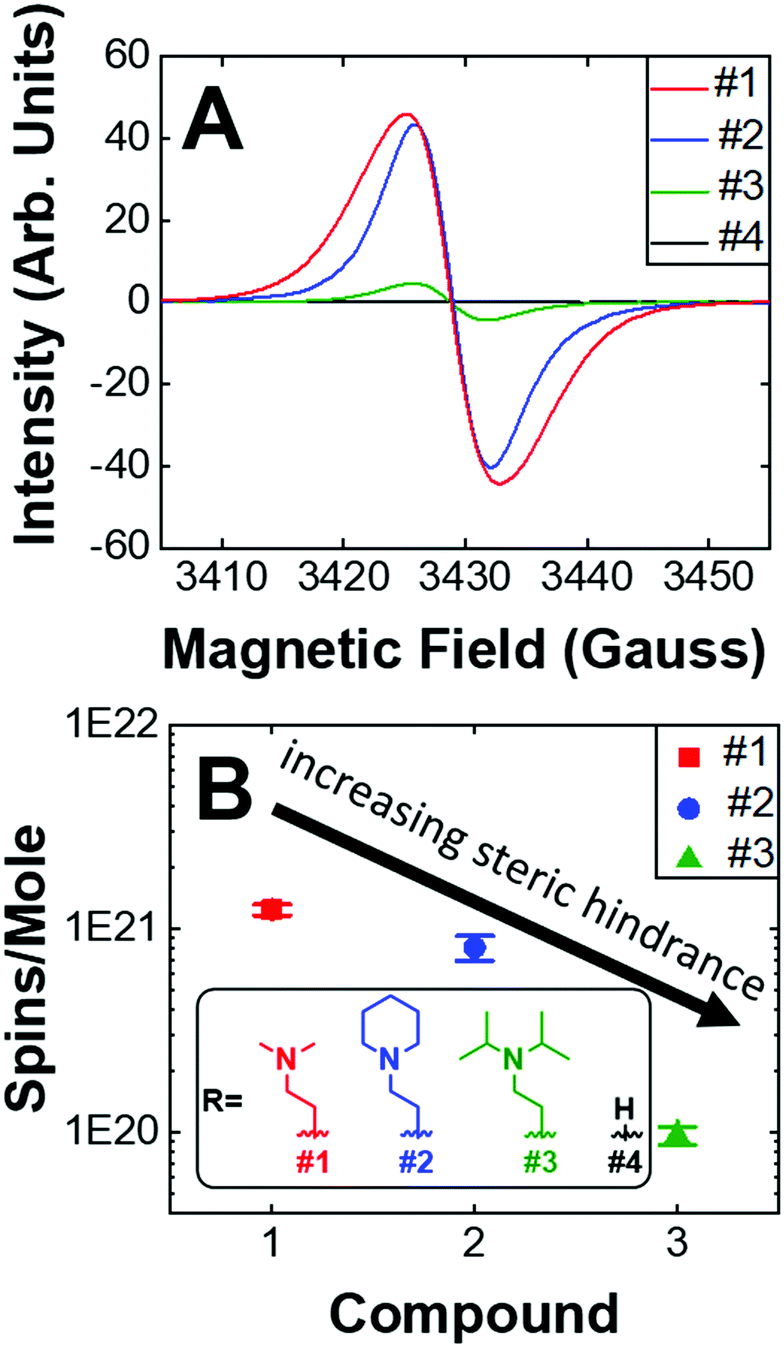 | ||
| Fig. 5 (A) Raw EPR spectra. Maximum intensities of compounds 1–4. (B) Average spin concentrations of compounds #1–3. Compound 4 is not spin active. (inset) Dopant structures and control compound #4. | ||
We also investigated the air stability self-doped PDI thin films. Fig. S16 (ESI†) shows the EPR spectra for compounds 1–3 which were measured under both a nitrogen atmosphere and after 3 days of air exposure. We observed that the spin concentration of #1 decreased by 60%, #2 decreased by 32%, and #3 decreased by 12% following air exposure. The more hindered amines are thus the most resistant to oxidation, which is a major challenge in n-type systems. The addition of carbonaceous substituents about the nitrogen source on the one hand increases the distance between donor and acceptor thus decreasing electron transfer efficiency, while on the other hand the same also imparts stability by shielding the radicals from oxidation. It remains to be seen how the incorporation of other heteroatoms, such as phosphorus, oxygen, or sulfur, may impact self-dopant performance in PDIs. It is anticipated that steric hindrance will play a key role in future work to establish dopant design principles and structural optimization.
Conclusions
To summarize, self-doping is a compelling doping strategy for PDIs and other small-molecule organic semiconductors that has recently emerged because it intrinsically mitigates phase segregation, dopant aggregation, and the need for host/dopant miscibility investigations. Self-doping also induces the increase in charge carrier concentrations in organic small molecules. For practical purposes, however, these materials must be easily processable into high-quality thin-films for them to be viable in functional devices. Self-doped PDIs have shown great promise in many applications, yet previous attempts at making thin-films have been limited to drop-casting from chloroform or water yielding poorly controlled morphologies. Here, we improve on earlier methods for thin-film production of self-doped materials. The elimination of host/dopant mixing enables us to utilize vapor deposition during film fabrication. We compare this method with two solution processing methods, namely drop-casting and ultrasonic spraying. We demonstrate that ultrasonic spray can be used to produce more homogeneous films than drop-casting without drastically altering the structures of the crystallites that form upon solvent evaporation. More importantly, we have demonstrated that physical vapor deposition is a robust method for fabricating smooth, continuous, and crystalline thin-films of self-doped perylene diimide derivatives with high spin carrier concentrations. We have also demonstrated that the elevated temperatures present during PVD film fabrication do not hinder the self-doping process in organic thin-films. This has allowed us to conclude that the introduction of bulky dopant structures deleteriously impacts the spin concentrations in self-doped organic thin films while simultaneously imparting stability to atmospheric oxidation of both the radical anion and cation. We propose that R′-NR2˙+ couples to PDI˙− to form transient exciplexes whose lowered energy imparts improved n-type stability, and is amenable to a variety of applications including OLEDs. We believe our work may enable the fabrication of other self-doped organic thin-films with controllable morphology and high spin carrier concentrations.Experimental section
All reagents were purchased directly from commercial sources and did not require further purification. 3,4,9,10-Perylene tetracarboxylic dianhydride was purchased from Acros Organics, 3,4,9,10-perylene tetracarboxylic diimide was purchased from Combi-Blocks, and 2-aminoethyl diisopropylamine was purchased from Alfa Aesar. Both N-(2-aminoethyl)piperidine and 2-dimethylaminoethylamine were purchased from Sigma Aldrich.Synthesis
Compounds 1, 2, and 3 were synthesized and purified according to a simplified method developed in our laboratory (Scheme 1). Briefly, 4 equivalents of 1,1-dimethylethylenediamine (compound 1, 2 mmol), 2-piperidinoethylamine (compound 2, 2 mmol), or N,N-diisopropylethylenediamine (compound 3, 2 mmol) were added to 1 equivalent of perylene-3,4,9,10-tetracarboxylic dianhydride (0.5 mmol) suspended in DMF (20 mM) in a round-bottom flask equipped with a condenser and stirred overnight at 120 °C. Upon cooling to room-temperature, 50 mL of acetone was added to the mixture and allowed to stand for 10 minutes. The slurry was then transferred to a centrifuge tube and centrifuged at 6000 rpm for 2 minutes and the supernatant was decanted. To remove unreacted starting material during the synthesis of compound 3, the product was treated with 30 mL of 0.1 M aqueous NaOH and washed with water. All solids were subsequently re-suspended and centrifuged 3 times with both acetone and diethyl ether (50 mL each wash). | ||
| Scheme 1 Reaction scheme for the synthesis of self-doped PDIs. | ||
Nuclear magnetic resonance
Proton NMR experiments were performed on a 500 MHz VXR-class spectrometer. All peaks are reported in units of δ and ppm relative to residual internal CHCl3 (7.26 ppm).Compound 11H NMR: (500 MHz, chloroform-d) δ 8.69 (d, J = 7.9 Hz, 4H), 8.62 (d, J = 8.0 Hz, 4H), 4.37 (t, J = 7.0 Hz, 4H), 2.70 (t, J = 7.0 Hz, 4H), 2.37 (s, 12H).
Compound 21H NMR: (500 MHz, chloroform-d) δ 8.64 (d, J = 7.9 Hz, 4H), 8.55 (d, J = 8.1 Hz, 4H), 4.40–4.36 (m, 4H), 2.73–2.67 (m, 4H), 2.58 (s, 8H), 1.63–1.58 (m, 8H), 1.48–1.43 (m, 4H).
Compound 31H NMR: (500 MHz, chloroform-d) δ 8.71 (d, J = 8.0 Hz, 4H), 8.66 (d, J = 8.1 Hz, 4H), 4.21 (t, J = 7.8 Hz, 4H), 3.12–3.06 (m, 4H), 2.75 (t, J = 7.8 Hz, 4H), 1.07 (d, J = 6.5 Hz, 24H).
Solid state NMR (SSNMR) spectra were collected on a chemagnetics spectrophotometer operating at 50.3 MHz 13C. The samples were loaded into a 5 mm rotor and data were collected at room temperature while spinning at 6 kHz and referenced to the methyl signal of an external sample of hexamethylbenzene. Room temperature samples were equilibrated in a nitrogen glovebox environment and in open air for several days, while heated samples were annealed at 120 °C for 7 days in air and nitrogen prior to analysis.
Mass spectrometry
MALDI mass spectrometry was performed on a Maldi micro MX ToF instrument with 1 mg mL−1 samples in chloroform.Absorption spectroscopy
Absorption studies were carried out with a 300 W xenon lamp and silicon photodetector attached to a monochromator in transmission geometry.Thermogravimetric analysis (TGA)
TGA was performed on a TA Instrument Q500 series equipped with an evolved gas furnace. Samples were placed in an aluminum pan and heated under atmospheric pressure at 10 °C min−1 from room temperature to 500 °C under continuous 10 mL min−1 nitrogen flow.Thin film preparation
All glass substrates were cleaned and washed in a sonicator bath for 15 minutes with Alconox detergent, deionized water, acetone, and isopropyl alcohol and dried with a nitrogen gun prior to film fabrication. Drop-cast and sprayed films were prepared from 1 mg mL−1 chloroform solutions. Spray coated films were prepared using an MTI MSK-SP-01A ultrasonic power supply (40 KHz, 0.5 amps) equipped with a cone-shaped spray nozzle. The distance between the substrate and nozzle was fixed to 10 cm. An EQ-300SP-LD digitally controlled infusion syringe pump was used to control the dispense rate at 2 mL min−1. Substrates were heated to 60 °C during the spray deposition process. Vapor deposited films were prepared in an MTI OTF-1200X tube furnace. 15 mg of fresh self-doped PDI was used for each sample batch. Sample boats were placed in the center of the tube furnace heating zone and substrates were placed 1 cm outside the heating zone. A vacuum level of 4.0 × 10−4 Torr and an argon flow rate of 60 sccm were set before the furnace heating cycle was initiated. The furnace temperature was ramped at 10 °C min−1 until final temperatures were reached and held constant for 30 minutes. Final temperatures of 305 °C, 295 °C, and 310 °C were set for compounds 1, 2, and 3 respectively. Under vacuum, these materials are expected to be considerably more stable. The film thickness could be controlled by the amount of material loaded into the sample boat as well as the deposition time.Gaussian simulations
All calculations were completed using the Gaussian 09 software package. Optimized geometries and frequencies of the neutral species were computed at the DFT level using the B3LYP functional and a 6-311G** basis set.Atomic force microscopy and scanning electron microscopy
AFM images were collected on a Bruker dimension icon atomic force microscope in tapping mode. SEM images were obtained on a FEI Nova NanoSEM 630 that was equipped with a field emission gun and operated at a 10 keV accelerating voltage.Grazing incidence wide-angle X-ray scattering
GIWAXS samples were all collected at the D1 station of the Cornell High Energy Synchrotron Source. The X-ray beam energy was 13.0 keV with synthetic multilayer optics (Mo/B4C, 30 Å d-spacing), and the sample to detector distance (SDD) was 190 mm. The incident angle for all GIWAXS patterns was 0.14°. Scattered intensity was collected on a two-dimensional CCD detector comprising 1024 × 1024 pixels with a size of 46.9 μm each. All images were background corrected and processed using the GIXSGUI graphical user interface. Mosaicity factors were calculated using the Shekiefactor.m matlab code.Electron paramagnetic resonance (EPR) spectroscopy
EPR experiments were performed on a Bruker EMX X-band rectangular cavity CW instrument. All samples were measured at room temperature. The instrument was calibrated for quantitative EPR measurements using TEMPO, a widely known stable organic radical, to construct a standard calibration curve. Samples were prepared in triplicate, weighed, and transferred to 5 mm quartz EPR tubes. Special care was taken to reduce static electricity in the weighing environment for error minimization with the aid of an anti-static wristband, fan ionizer, and an alpha ionizing cartridge. Thin film samples were prepared by cleaving quartz microscope slides into substrates sized to fit inside a 5 mm EPR tube. Substrates were cleaned (same procedure as glass slides) and weighed.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the NSF under award # DMR 1824263. EPR studies were supported by the NSF under award # CBET 2016191. LWB would also like to acknowledge the financial support from the Research Corporation for Science Advancement (USA) through a Cottrell Scholar Award. Use of the Stanford Synchrotron Radiation Lightsource, SLAC National Accelerator Laboratory, is supported by the US DOE, Office of Science, Office of Basic Energy Sciences under contract DE-AC02-76SF00515. GIWAXS studies were performed under NSF award # CBET 2018413. NMR results included in this report were recorded at the David M. Grant NMR Center, a University of Utah Core Facility. Funds for construction of the Center and the helium recovery system were obtained from the University of Utah and the National Institutes of Health under awards # 1C06RR017539-01A1 and 3R01GM063540-17W1, respectively. NMR instruments were purchased with support of the University of Utah and the National Institutes of Health under award # 1S10OD25241-01. The authors would like to thank Dr Jesse Powell, M. D. for helping with the graphic visualizations presented in this work.References
- A. D. Scaccabarozzi and N. J. Stingelin, J. Mater. Chem. A, 2014, 2, 10818–10824 RSC.
- M. K. Kiesewetter, E. J. Shin, J. L. Hedrick and R. M. Waymouth, Macromolecules, 2010, 43, 2093–2107 CrossRef CAS.
- W. Hou, Y. Xiao, G. Han and J.-Y. Lin, Polymers, 2019, 11, 143 CrossRef.
- A. Mishra and P. Bäuerle, Angew. Chem., Int. Ed., 2012, 51, 2020–2067 CrossRef CAS.
- A. Zevalkink, D. M. Smiadak, J. L. Blackburn, A. J. Ferguson, M. L. Chabinyc, O. Delaire, J. Wang, K. Kovnir, J. Martin, L. T. Schelhas, T. D. Spark, S. D. Kang, M. T. Dylla, J. Snyder, B. R. Ortiz and E. S. Toberer, Appl. Phys. Rev., 2018, 5, 021303 Search PubMed.
- H. Klauk, Chem. Soc. Rev., 2010, 39, 2643 RSC.
- B. Russ, A. Glaudell, J. J. Urban, M. L. Chabinyc and R. A. Segalman, Nat. Rev. Mater., 2016, 1, 16050 CrossRef CAS.
- R. Kroon, D. A. Mengistie, D. Kiefer, J. Hynynen, J. D. Ryan, L. Yu and C. Müller, Chem. Soc. Rev., 2016, 45, 6147–6164 RSC.
- N. Lu, L. Li and M. Liu, Phys. Chem. Chem. Phys., 2016, 18, 19503–19525 RSC.
- J. Rivnay, S. Inal, A. Salleo, R. M. Owens, M. Berggren and G. G. Malliaras, Nat. Rev. Mater., 2018, 3, 17086 CrossRef CAS.
- S. Muench, A. Wild, C. Friebe, B. Häupler, T. Janoschka and U. S. Schubert, Chem. Rev., 2016, 116, 9438–9484 CrossRef CAS.
- Y. Hinuma, Y. Kumagai, I. Tanaka and F. Oba, Phys. Rev. B, 2017, 95, 075302 CrossRef.
- S. N. Patel, A. M. Glaudell, K. A. Peterson, E. M. Thomas, K. A. O’Hara, E. Lim and M. L. Chabinyc, Sci. Adv., 2017, 3, e1700434 CrossRef.
- B. Lüssem, M. Riede and K. Leo, Phys. Status Solidi, 2013, 210, 9–43 CrossRef.
- I. Salzmann, G. Heimel, M. Oehzelt, S. Winkler and N. Koch, Acc. Chem. Res., 2016, 49, 370–378 CrossRef CAS.
- B. Lüssem, C. M. Keum, D. Kasemann, B. Naab, Z. Bao and K. Leo, Chem. Rev., 2016, 116, 13714–13751 CrossRef.
- Y. Ikenoue, J. Chiang, A. O. Patil, F. Wudl and A. J. Heeger, J. Am. Chem. Soc., 1988, 110, 2983–2985 CrossRef CAS.
- G.-H. Kim, L. Shao, K. Zhang and K. P. Pipe, Nat. Mater., 2013, 12, 719–723 CrossRef CAS.
- S. Rossbauer, C. Müller and T. D. Anthopoulos, Adv. Funct. Mater., 2014, 24, 7116–7124 CAS.
- I. Salzmann, G. Heimel, S. Duhm, M. Oehzelt, P. Pingel, B. M. George, A. Schnegg, K. Lips, R. P. Blum, A. Vollmer and N. Koch, Phys. Rev. Lett., 2012, 108, 1–5 CrossRef.
- V. A. Kolesov, C. Fuentes-Hernandez, W.-F. Chou, N. Aizawa, F. A. Larrain, M. Wang, A. Perrotta, S. Choi, S. Graham, G. C. Bazan, T.-Q. Nguyen, S. R. Marder and B. Kippelen, Nat. Mater., 2017, 16, 474–480 CrossRef CAS.
- I. Salzmann and G. Heimel, J. Electron Spectrosc. Relat. Phenom., 2015, 204, 208–222 CrossRef CAS.
- R. A. Schlitz, F. G. Brunetti, A. M. Glaudell, P. L. Miller, M. A. Brady, C. J. Takacs, C. J. Hawker and M. L. Chabinyc, Adv. Mater., 2014, 26, 2825–2830 CrossRef CAS.
- M. L. Tietze, J. Benduhn, P. Pahner, B. Nell, M. Schwarze, H. Kleemann, M. Krammer, K. Zojer, K. Vandewal and K. Leo, Nat. Commun., 2018, 9, 1182 CrossRef.
- C. K. Mai, R. A. Schlitz, G. M. Su, D. Spitzer, X. Wang, S. L. Fronk, D. G. Cahill, M. L. Chabinyc and G. C. Bazan, J. Am. Chem. Soc., 2014, 136, 13478–13481 CrossRef CAS.
- D. X. Long, M. Karakawa and Y.-Y. Noh, Phys. Chem. Chem. Phys., 2016, 18, 23904–23909 RSC.
- L. Qiu, J. Liu, R. Alessandri, X. Qiu, M. Koopmans, R. W. A. Havenith, S. J. Marrink, R. C. Chiechi, L. J. Anton Koster and J. C. Hummelen, J. Mater. Chem. A, 2017, 5, 21234–21241 RSC.
- J. Liu, L. Qiu, G. Portale, M. Koopmans, G. ten Brink, J. C. Hummelen and L. J. A. Koster, Adv. Mater., 2017, 29, 1701641 CrossRef.
- H. Un, S. A. Gregory, S. K. Mohapatra, M. Xiong, E. Longhi, Y. Lu, S. Rigin, S. Jhulki, C. Yang, T. V. Timofeeva, J.-Y. Wang, S. K. Yee, S. Barlow, S. R. Marder and J. Pei, Adv. Energy Mater., 2019, 9, 1900817 CrossRef.
- J. Euvrard, A. Revaux, P.-A. Bayle, M. Bardet, D. Vuillaume and A. Kahn, Org. Electron., 2018, 53, 135–140 CrossRef CAS.
- I. E. Jacobs, E. W. Aasen, J. L. Oliveira, T. N. Fonseca, J. D. Roehling, J. Li, G. Zhang, M. P. Augustine, M. Mascal and A. J. Moulé, J. Mater. Chem. C, 2016, 4, 3454–3466 RSC.
- D. T. Duong, C. Wang, E. Antono, M. F. Toney and A. Salleo, Org. Electron., 2013, 14, 1330–1336 CrossRef CAS.
- E. H. Suh, Y. J. Jeong, J. G. Oh, K. Lee, J. Jung, Y. S. Kang and J. Jang, Nano Energy, 2019, 58, 585–595 CrossRef CAS.
- S. D. Kang, G. J. Snyder, S. Dongmin Kang and J. G. Snyder, Nat. Mater., 2017, 16, 252–257 CrossRef.
- M. Oehzelt, N. Koch and G. Heimel, Nat. Commun., 2014, 5, 4174 CrossRef CAS.
- Z. Wang, N. Zheng, W. Zhang, H. Yan, Z. Xie, Y. Ma, F. Huang and Y. Cao, Adv. Energy Mater., 2017, 7, 1700232 CrossRef.
- T. H. Reilly, A. W. Hains, H.-Y. Chen and B. A. Gregg, Adv. Energy Mater., 2012, 2, 455–460 CrossRef CAS.
- S. Bettini, Z. Syrgiannis, R. Pagano, L. Dordević, L. Salvatore, M. Prato, G. Giancane and L. Valli, ACS Appl. Mater. Interfaces, 2019, 11, 17079–17089 CrossRef CAS.
- G. Echue, G. C. Lloyd-Jones and C. F. J. Faul, Chem. – Eur. J., 2015, 21, 5118–5128 CrossRef CAS.
- J. Min, Z.-G. Zhang, Y. Hou, C. O. Ramirez Quiroz, T. Przybilla, C. Bronnbauer, F. Guo, K. Forberich, H. Azimi, T. Ameri, E. Spiecker, Y. Li and C. J. Brabec, Chem. Mater., 2015, 27, 227–234 CrossRef CAS.
- H. Zhang, L. Xue, J. Han, Y. Q. Fu, Y. Shen, Z. Zhang, Y. Li and M. Wang, J. Mater. Chem. A, 2016, 4, 8724–8733 RSC.
- M. Milton, Q. Cheng, Y. Yang, C. Nuckolls, R. Hernández Sánchez and T. J. Sisto, Nano Lett., 2017, 17, 7859–7863 CrossRef CAS.
- H. J. Kim, W. B. Jung, H. S. Jeong and H. T. Jung, J. Mater. Chem. C, 2017, 5, 12241–12248 RSC.
- M. E. El-Khouly, A. El-Refaey, S. Y. Shaban and M. M. El-Hendawy, Photochem. Photobiol. Sci., 2018, 17, 1098–1107 RSC.
- W. Deng, J. Yu, Y. Qian, R. Wang, Z. Ullah, S. Zhu, M. Chen, W. Li, Y. Guo, Q. Li and L. Liu, Electrochim. Acta, 2018, 282, 24–29 CrossRef CAS.
- B. Russ, M. J. Robb, F. G. Brunetti, P. L. Miller, E. E. Perry, S. N. Patel, V. Ho, W. B. Chang, J. J. Urban, M. L. Chabinyc, C. J. Hawker and R. A. Segalman, Adv. Mater., 2014, 26, 3473–3477 CrossRef CAS.
- J. Li, C. Yang, Y. Chen and W.-Y. Lai, New J. Chem., 2016, 40, 8886–8891 RSC.
- W. Ji, X. Zhang, J. Zhao, Y. Gao, W. Song and Y. Ozaki, Analyst, 2018, 143, 1899–1905 RSC.
- Y. Guan, Y. Zakrevskyy, J. Stumpe, M. Antonietti and C. F. Faul, Chem. Commun., 2003, 894–895 RSC.
- B. Russ, M. J. Robb, B. C. Popere, E. E. Perry, C.-K. Mai, S. L. Fronk, S. N. Patel, T. E. Mates, G. C. Bazan, J. J. Urban, M. L. Chabinyc, C. J. Hawker and R. A. Segalman, Chem. Sci., 2016, 7, 1914–1919 RSC.
- H. Liu, L. Huang, X. Cheng, A. Hu, H. Xu, L. Chen and Y. Chen, ACS Appl. Mater. Interfaces, 2017, 9, 1145–1153 CrossRef CAS.
- W. Chen, W. Jiao, D. Li, X. Sun, X. Guo, M. Lei, Q. Wang and Y. Li, Chem. Mater., 2016, 28, 1227–1235 CrossRef CAS.
- N. Wu, C. Wang, B. R. Bunes, Y. Zhang, P. M. Slattum, X. Yang and L. Zang, ACS Appl. Mater. Interfaces, 2016, 8, 12360–12368 CrossRef CAS.
- F. Bureš, RSC Adv., 2014, 4, 58826–58851 RSC.
- H. Liu, G. Yin, Q. Li, G. Liu, S. Pu and H. Zhang, Dyes Pigm., 2019, 165, 319–326 CrossRef CAS.
- B. Rybtchinski, L. E. Sinks and M. R. Wasielewski, J. Phys. Chem. A, 2004, 108, 7497–7505 CrossRef CAS.
- H. Wu, H. Wang, L. Xue, Y. Shi and X. Li, J. Phys. Chem. B, 2010, 114, 14420–14425 CrossRef CAS.
- M. J. Ahrens, M. J. Tauber and M. R. Wasielewski, J. Org. Chem., 2006, 71, 2107–2114 CrossRef CAS.
- Y. Matsunaga, K. Goto, K. Kubono, K. Sako and T. Shinmyozu, Chem. – Eur. J., 2014, 20, 7309–7316 CrossRef CAS.
- A. Karki, G. A. H. Wetzelaer, G. N. M. Reddy, V. Nádaždy, M. Seifrid, F. Schauer, G. C. Bazan, B. F. Chmelka, P. W. M. Blom and T. Nguyen, Adv. Funct. Mater., 2019, 29, 1901109 CrossRef.
- V. Coropceanu, J. Cornil, D. A. da Silva Filho, Y. Olivier, R. Silbey and J.-L. Brédas, Chem. Rev., 2007, 107, 926–952 CrossRef CAS.
- L. Flannery, H. Gálvez, W. Nimens, A. A. Rahman and L. Whittaker-Brooks, Polyhedron, 2019, 170, 334–358 CrossRef CAS.
- N. Karl, Synth. Met., 2003, 133–134, 649–657 CrossRef CAS.
- J. Ogle, D. Powell, E. Amerling, D.-M. Smilgies and L. Whittaker-Brooks, CrystEngComm, 2019, 21, 5707–5720 RSC.
- T. Okada, M. Migita, N. Mataga, Y. Sakata and S. J. Misumi, J. Am. Chem. Soc., 1981, 103, 4715–4720 CrossRef CAS.
- P. Van Haver, N. Helsen, S. Depaemelaere, M. Van der Auweraer and F. C. De Schryver, J. Am. Chem. Soc., 1991, 113, 6849–6857 CrossRef CAS.
- A. M. Swinnen, M. Van der Auweraer, F. C. De Schryver, K. Nakatani, T. Okada and N. Malaga, J. Am. Chem. Soc., 1987, 109, 321–330 CrossRef CAS.
- D. R. G. Brimage and R. S. Davidson, J. Chem. Soc. D, 1971, 1385–1386 RSC.
- Q. Wang, Q. Tian, Y. Zhang, X. Tang and L. Liao, J. Mater. Chem. C, 2019, 7, 11329–11360 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ma00822b |
| This journal is © The Royal Society of Chemistry 2021 |