
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePd-Catalysed carbonylative Suzuki–Miyaura cross-couplings using Fe(CO)5 under mild conditions: generation of a highly active, recyclable and scalable ‘Pd–Fe’ nanocatalyst†
Zhuangli
Zhu
ab,
Zhenhua
Wang
a,
Yajun
Jian
 a,
Huaming
Sun
a,
Guofang
Zhang
a,
Jason M.
Lynam
c,
C. Robert
McElroy
c,
Thomas J.
Burden
c,
Rebecca L.
Inight
c,
Ian J. S.
Fairlamb
*c,
Weiqiang
Zhang
*a and
Ziwei
Gao
*a
a,
Huaming
Sun
a,
Guofang
Zhang
a,
Jason M.
Lynam
c,
C. Robert
McElroy
c,
Thomas J.
Burden
c,
Rebecca L.
Inight
c,
Ian J. S.
Fairlamb
*c,
Weiqiang
Zhang
*a and
Ziwei
Gao
*a
aKey Laboratory of Applied Surface and Colloid Chemistry, Ministry of Education, School of Chemistry & Chemical Engineering, Shaanxi Normal University, 199 South Chang'an Road, Xi'an, China. E-mail: zwq@snnu.edu.cn; zwgao@snnu.edu.cn
bTianjin Normal University, 393 West Binshui Road, Tianjin, China
cDepartment of Chemistry, University of York, Heslington, York, North Yorkshire YO10 5DD, UK. E-mail: ian.fairlamb@york.ac.uk
First published on 7th January 2021
Abstract
The dual function and role of iron(0) pentacarbonyl [Fe(CO)5] has been identified in gaseous CO-free carbonylative Suzuki–Miyaura cross-couplings, in which Fe(CO)5 supplied CO in situ, leading to the propagation of catalytically active Pd–Fe nanoparticles. Compared with typical carbonylative reaction conditions, CO gas (at high pressures), specialised exogenous ligands and inert reaction conditions were avoided. Our developed reaction conditions are mild, do not require specialised CO high pressure equipment, and exhibit wide functional group tolerance, giving a library of biaryl ketones in good yields.
Introduction
Pd-Catalysed three-component coupling reactions of aryl boronic acids, carbon monoxide (CO) and aryl halides/pseudo-halides, generally known as carbonylative Suzuki–Miyaura cross-coupling (C-SMCC) reactions,1 display numerous synthetic advantages, particularly the ready availability of commercially available starting materials, which are typically thermally-, air- and water-stable. The C-SMCC reactions exhibit wide functional group compatibility, accompanied by broad substrate scope.2 Typically, the reaction represents a straightforward and convenient process allowing access to a myriad of symmetrical and unsymmetrical biaryl ketones,3 which are omnipresent in natural products and other synthetic target compounds, with some exhibiting favourable bioactivity and material properties.4Over the last few decades we have seen practical advances in C-SMCCs, especially enabled through the design of new Pd (pre)catalysts.5 However, the toxic and flammable CO gas potentially hinders wider adoption of C-SMCCs, especially so in academic laboratories, for which expertise and specialised equipment might be limited, with health and safety concerns being at the forefront of any decision-making process. It is also known that the CO-insertion efficiency is reduced when employing electron-deficient aryl halides,6 which can typically be overcome by the adoption of an expensive exogenous ligand or high pressure in CO (Fig. 1a).7
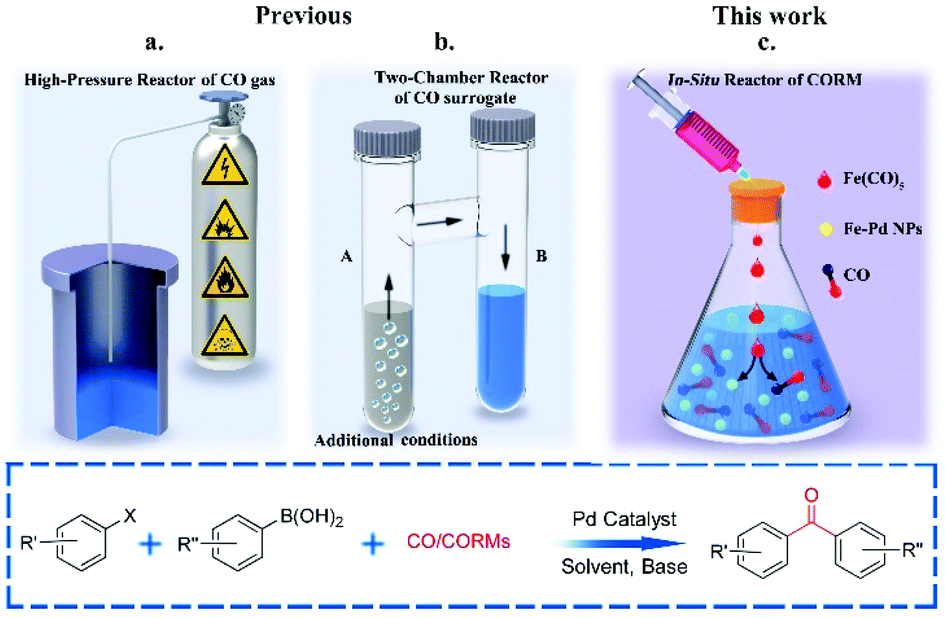 | ||
| Fig. 1 Pd-Catalysed carbonylative-Suzuki–Miyaura cross-couplings (C-SMCC)@ (a) historical and industrially relevant carbonylative high pressure system; (b) two-chamber, with independent generation of CO; and (c) in situ generation of CO from a metal-based CO–RM, the metal fragment capable of modifying catalytic efficacy. | ||
Moving the field forward, the development of a carbonylation procedure employing the in situ generation of CO, as a substitute for gaseous CO, is appealing and arguably necessary for general synthetic chemistry laboratories. With this point in mind several research teams have replaced CO gas with safer sources of molecular CO, i.e. in the form of CO-surrogates.8,9 Indeed, leaders in the field are Skrydstrup10et al., who have developed Pd-catalysed carbonylation reactions using ex situ CO generated from CO-surrogates and a specially designed two-chamber reactor (Fig. 1b). Several CO surrogates have been employed, e.g. 9-methyl-9H-fluorene-9-carbonyl chloride (COgen), silacarboxylic acids (SilaCOgen), alcohols, carbon dioxide, metal carbonyls and formic acid. These CO-surrogates degrade in a controllable manner, by different mechanisms, supplying CO gas for Pd-catalysed carbonylation reactions.10,11
With synthetic verstaility in mind, the well-controlled in situ generation of CO is essential for the development of a safe and convenient one-pot C-SMCC methodology. Transition metal carbonyls (TMCs) have been utilised as CO releasing molecules (CO-RMs) for therapeutic delivery of CO in biological systems,12 a field which we have contributed to independently, and for which much is now known about CO-release rates in different media. Many readily available homoleptic TMCs are attractive CO-RMs, since many carbonyls per CO-RM can in principle be delivered in solution. More importantly, the cleavage of the M–CO bond of TMCs can be controlled by the variation of temperature. Ni(CO)4 has been employed in the aminocarbonylation of vinyl halides13a and carbonylative olefination of aryl halides.13b Larhed et al.14 pioneered Pd-catalysed carbonylation of O, N nucleophiles using Cr(CO)6 and Mo(CO)6. The solid CO-RMs were activated by microwave heating, and CO was generated in situ for carbonylation. Later, other solid TMCs were evaluated as CO-RMs for transition metal-catalysed organic transformations.10 In all cases, the metal is thought to be a waste element within the reaction. Indeed, a limitation identified was that the low valence transition metal residue could poison the active Pd catalyst species, with reactions being sluggish, leading to poor yields of the carbonylative products,15a a potential issue for more challenging substrates.15b,c Maes et al. have developed methodologies employing isocyanides as CO-surrogates in Pd-catalysed cross-couplings, offering an alternative approach.16
In light of highly effective and catalytically competent Pd/Fe nanoparticles being discovered for C–C cross-coupling reactions,17 we recognised that iron carbonyls could potentially play the role of CO–RMs, while modifying the properties and catalytic activity of the Pd catalyst through beneficial interactions with Fe, i.e. where the CO ligands are utilised for the carbonylation process and the waste Fe (oxidised) creating a more efficient Pd–Fe catalytic system in situ. Due to the low-price and earth abundance of iron, we envisaged that an economical, safer and environmentally friendly Pd-catalysed C-SMCC methodology using Fe(CO)5 could be developed.
Herein, we report a benign C-SMCC reaction methodology, which does not require the use of CO gas or exogenous specialised ligands, where Fe(CO)5 provides CO for the reaction, generating well-defined, stable and catalytically competent Fe/Pd nanoparticles in situ, which can be further recycled in multiple reaction runs (Fig. 1c).
Results and discussion
The C-SMCC of p-iodo-acetophenone 1a with phenylboronic acid 2a to give 3a was chosen as the benchmark reaction to evaluate the viability of Fe(CO)5 as a CO source and Pd catalyst modifier. A reaction pathway to the non-carbonylative Suzuki–Miyaura cross-coupled product 3a′ is competitive; thus it is critical to identify reaction conditions enabling high selectivity for 3a. The reaction parameters, including (pre)catalysts, solvents and bases, were screened to establish the best system for the gaseous CO-free C-SMCC methodology (Table 1). Interestingly, without a phosphine ligand, all Pd precursors, Pd(OAc)2 {note: Pd3(OAc)6}, Pd2(dba)3 (dba = 1,4-dibenzylidene acetone), Pd(acac)2 (acac = acetylacetonate) and polymeric PdCl2, exhibited high conversion and selectivity (entries 1–5), using as little as 0.5 mol% Pd (pre)catalyst loading (entry 2). The presence of phosphine ligands suppressed the carbonylation product 3a yield (entries 6–12). Poor product selectivity shows that the phosphine ligands are able to facilitate the oxidative addition of 1a to the in situ generated “L–Pd0” species, but slow down the subsequent CO insertion into the Pd–C bond. We also recognise that the phosphine ligands could influence Pd catalyst speciation, negatively in terms of the carbonylation selectivity in this system.|
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst | Ligand | 3a | 3a’ | |
| Solvent/base | %b | ||||
| a Reaction conditions: 1a (0.434 mmol), 2a (1.75 eq.), Fe(CO)5 (0.15 mmol), and [Pd] (1 mol%, unless otherwise stated), 80 °C, 12 h. Entries 1–12: anisole and K2CO3 were used. b Detected by 1H NMR and confirmed by purification by silica gel chromatography. c Pd3(OAc)6, referred to as Pd(OAc)2, (0.5 mol% in Pd). | |||||
Effect of catalyst and ligand (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :1) :1)
|
|||||
| 1 | Pd(OAc)2 | 90 | 7 | ||
| 2 | Pd(OAc)2c | 86 | 8 | ||
| 3 | Pd2(dba)3 | 80 | 7 | ||
| 4 | Pd(acac)2 | 89 | 7 | ||
| 5 | PdCl2 | 80 | 18 | ||
| 6 | PdCl2(PPh3)2 | 5 | 94 | ||
| 7 | Pd(OAc)2 | PPh3 | 64 | 31 | |
| 8 | Pd(OAc)2 | CyJohnPhos | 31 | 22 | |
| 9 | Pd(OAc)2 | PCy3 | 71 | 21 | |
| 10 | Pd(OAc)2 | Dppf | 70 | 25 | |
| 11 | Pd(OAc)2 | Xphos | 40 | 12 | |
| 12 | Pd(OAc)2 | Xantphos | 48 | 14 | |
| Effect of solvent (3 mL) and base (2 eq.) | |||||
| 13 | Pd(OAc)2 | Toluene/K2CO3 | 87 | 11 | |
| 14 | Pd(OAc)2 | Dioxane/K2CO3 | 60 | 7 | |
| 15 | Pd(OAc)2 | Anisole/K3PO4 | 83 | 10 | |
| 16 | Pd(OAc)2 | Anisole/DIPEA | 51 | 1 | |
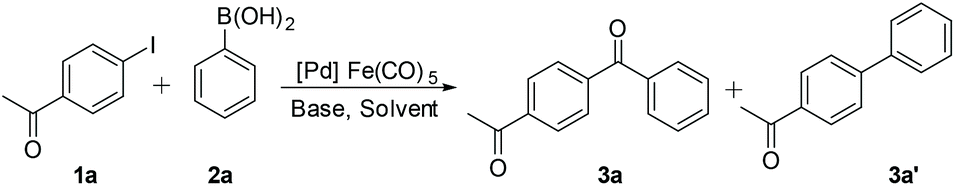
The effects of the solvent and base were further examined. It was found that the C-SMCC reaction proceeded well in relatively less polar solvents, such as anisole and toluene. The inorganic bases, K2CO3 and K3PO4, exhibited high substrate conversion and superior selectivity when compared against the organic base, DIPEA (entries 13–16). Other TMCs were also assessed as CORMs. Indeed, the ferrous-system performed considerably better than Mo(CO)6 and Cr(CO)6 (Table S2, see the ESI†). It is of particular note that using gaseous CO (balloon pressure) in place of Fe(CO)5 failed to effect a successful C-SMCC reaction, with only the SMCC biaryl product 3a′ being formed. The outcome demonstrates that Fe(CO)5 not only acts as a CO source, but also functions positively vide infra to enhance this specific Pd-catalysed C-SMCC reaction in some way.
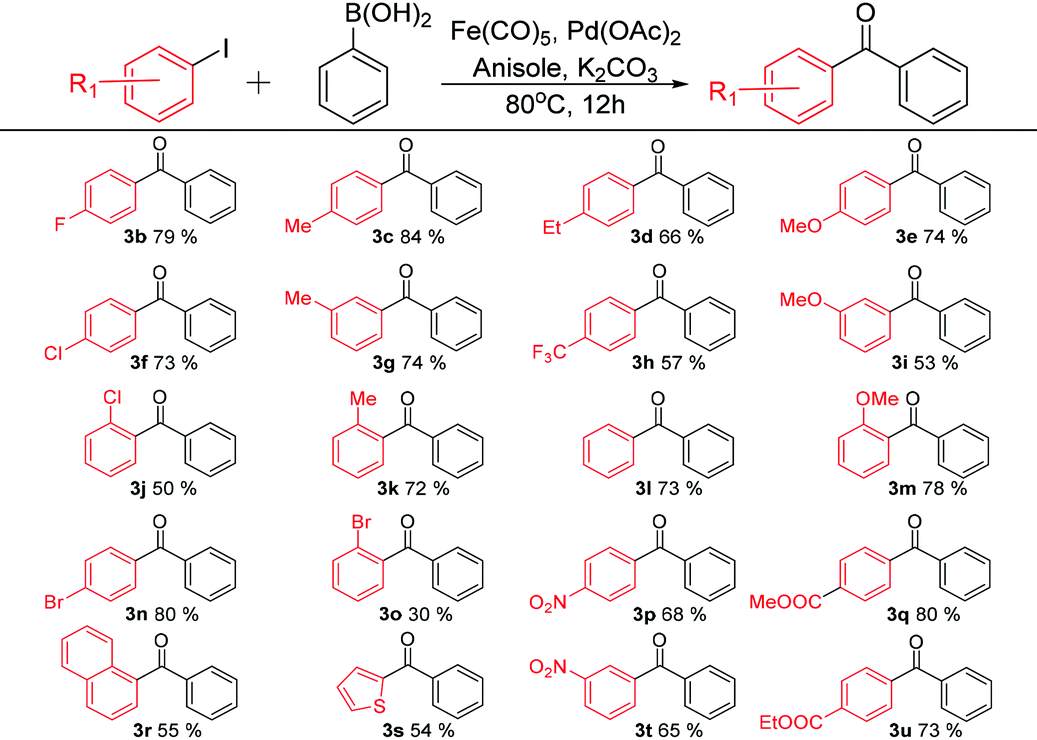
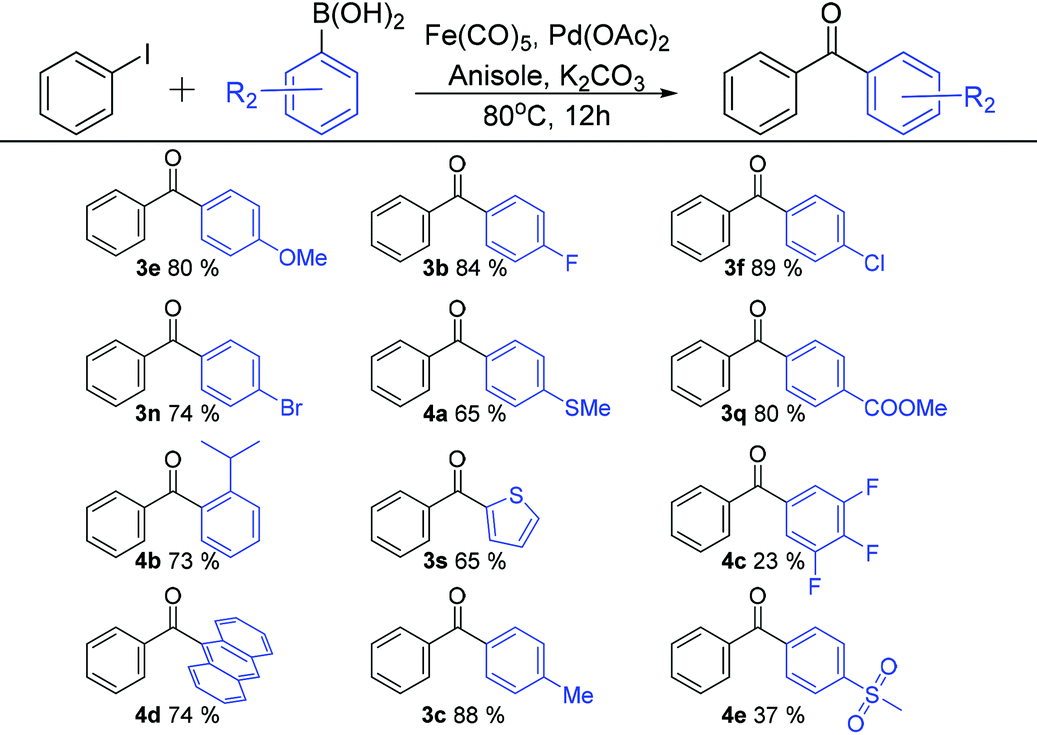
With the optimised reaction conditions in hand {0.15 mmol Fe(CO)5, 1 mol% Pd(OAc)2, anisole and K2CO3}, various functional biaryl ketones were synthesized using the new C-SMCC reaction differentiated by changes in aryl substituents, either electron-donating or electron-withdrawing, of aryl iodides (Table 2) and those of phenylboronic acids (Table 3), to evaluate the scope and limitations of the new C-SMCC methodology.
|
|
Table 2 shows the effects of substituents in the reactions of aryl iodides. Under the best identified reaction conditions, the para-halogenated (Cl, Br, and F) aryl iodides gave diaryl ketones in good yields (p-F, 3b, 79%, p-Cl, 3f, 73%, and p-Br, 3n, 74%). However, ortho-Cl and Br substituents led to only modest yields of cross-coupled products with 50% (3j) and 30% (3o) biaryl ketone products being generated respectively. We tentatively attribute the lowering of the yield to steric effects, which retard the oxidation addition step.18 The presence of a methyl group at para-, meta- and ortho-positions led to good product yields (p-Me, 3c, 84%, m-Me, 3g, 74%, and o-Me, 3k, 72%). The corresponding biaryl ketone products possessing methoxy groups at para-, meta- and ortho-positions were isolated in good yields, with 74% for 3e, 53% for 3i and 78% for 3m, respectively. Meanwhile, as the length of the alkyl chain was increased to ethyl a 66% yield of 3d was recorded. As for electron-withdrawing substituents, similar trends were noted. For example, the weakly electron-withdrawing ester group, p-CO2Me, gave a good yield of 80% (3q), with the p-CO2Et group giving a 73% yield of 3u. In addition, the stronger electron-withdrawing substituents only lower the yields marginally (p-CF3, 3h, 57%, p-NO2, 3p, 68%, and m-NO2, 3t, 65%). We further succeeded in synthesizing the biaryl ketones containing naphthalene (3r, 55%) and thiophene (3s, 54%), which are key intermediates to interesting luminescent materials.19
As shown in Table 3, different organoboronic acids were utilised in our new C-SMCC methodology. Most of the organoboronic acids underwent carbonylative coupling with good yields. The halogenated aryl iodides were converted into diaryl ketones in very good yields (e.g. p-F, 3b, 84%, p-Cl, 3f, 89%, and p-Br, 3n, 74%). Both electron-donating (p-Me, 3c, 88% and p-OMe, 3e, 80%) and electron-withdrawing substituents (p-CO2Me, 3q, 84%) also gave good yields. The 9-fluorenyl arylboronic acid (4d, 74%) was pleasingly formed with a good yield. Notably, thio-moieties were well tolerated. Thus, the biarylketone products bearing p-MeS and 2-thiophene were both isolated in 65% yield (4a and 3s). A sterically encumbered substituent, o-iPr, gave rise to a 73% yield of 4b, whilst the organoboronic acids bearing several fluorine atoms or a p-SO2Me group gave lower yields of 23% (4c) and 37% (4e), respectively.
Encouraged by the results presented above, we subsequently carried out a C-SMCC reaction on a gram scale to demonstrate its practical utility. With 5 mmol of 1a, product 3a was formed in 86% yield under our optimised conditions (Scheme 1). Furthermore, the phenylenebis–phenylmethanones were synthesized via the C-SMCC reactions of various diiodobenzenes with phenylboronic acid, in up to 86% yield (Scheme 1, 5a–c). The results highlight the significant potential of this C-SMCC for practical applications.
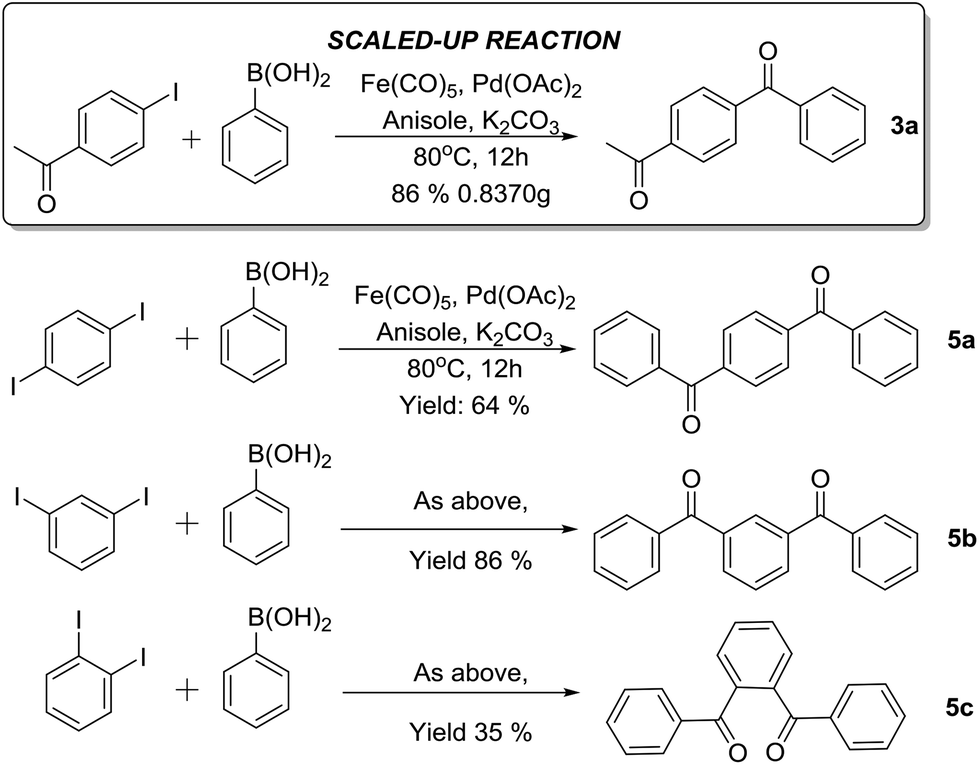 | ||
| Scheme 1 Examples and gram-scale reaction in practical applications. | ||
Finally, we have tested the C-SMCC methodology using 4-bromo-anisole and 4-bromo-6-methyl-2-pyrone as exemplar brominated substrates (employing standard conditions as described in Table 2, in the presence and absence of activating DPPF and Xantphos ligands which we hypothesised might assist with C–Br bond activation and CO-migratory insertion steps). However, in the case of 4-bromo-anisole it was left unreacted. For 4-bromo-6-methyl-2-pyrone, a small amount of 4-phenyl-6-methyl-2-pyrone was formed (7%) when the exogenous Xantphos ligand was added (by traditional SMCC reaction).
Green metrics using the CHEM21 toolkit
Clark et al. developed a unified metrics toolkit to evaluate the global sustainability of chemical reactions. It was a successful toolkit developed by the CHEM21 consortium consisting of academic and industrial members.20a The toolkit is built upon a comprehensive and holistic range of criteria enabling the measurement of how green a reaction is. The toolkit has been applied in the assessment of the synthesis of multicomponent products,20b opioid scaffolds,20c dialkyl chloramines,20d ionic liquids,20e solvents,20f peptides20g and tryptophan C–H bond functionalization chemistry.20i,h We recognized that the toolkit would be useful for initial appraisal of the newly developed C-SMCC methodology described above. The synthesis of 3a from 4-iodoacetophenone 1a and phenylboronic acid 2a (Scheme 1) was compared with a relevant synthesis involving an activated Pd precatalyst possessing a bis-chelating N-heterocyclic carbene ligand.21 The literature procedure employed 13.8 bar (200 psi) of CO and 0.01 mol% of Pd precatalyst. The methodology from the Chem21 Metrics Toolkit at the first path (key reaction) level was adopted to compare the results, which are collated in Table 4, including the reagent and product stoichiometries.|
|
||||
|---|---|---|---|---|
| Metric | Method A | Flag | Method B | Flag |
| Yield | 86% |

|
99.0% |

|
| Conversion | 94.3% |

|
100% |
|
| Selectivity | 91.2% |

|
99% |

|
| Reaction mass efficiency (RME) | 23.2% | 16.7% | ||
| Atom economy (AE) | 39.2% | 42.0% | ||
| Solvents | Anisole |
|
Toluene |

|
| Health and safety | H300, H330 |

|
H360, H372 |

|
| Process mass intensity (PMI) | 40.0 | 25.5 | ||
| PMI (reaction chemicals) | 4.3 | 6.0 | ||
| PMI (reaction solvents) | 35.7 | 19.5 | ||
| Catalyst | Yes |

|
Yes |
|
| Reactor | Batch |

|
Batch |

|
| Elements | B, Pd, I |

|
B, Pd, I |

|
| Energy | 80 °C |

|
120 °C |

|
The data in Table 4 show that the green metrics for the two synthetic methodologies are broadly similar. Surprisingly, this includes atom economy (AE), with our work only being slightly poorer due to one fragment of “Fe(CO)2” not making it into the final product (note: the final destination for Fe is in the form of Fe2O3vide infra which is required for Pd catalyst enhancement). However, the AE is dominated by the iodine and boronic acid leaving groups, a necessary and mandatory requirement for the arylation process. More significant however is the Reaction Mass Efficiency (RME) being significantly better for our work. This is because the CO–RM, Fe(CO)5, is an efficient and mild CO source, while the reported reaction conditions employ a 23 molar excess under significant pressure. The same effect can be seen in the Process Mass Intensity (PMI) reaction chemicals. These results are likely more significant, as 3× refills and purges of CO are necessary for the reported21 pressurised reaction vessel. The inclusion of this practical and necessary requirement gives an RME of 6.7% and a PMI of 14.9. Neither reaction scores well on health and safety, due to the need for liberation of CO (which is formally a necessary requirement for a carbonylation reaction). Lastly, the lower temperature and absence of the need to pressurize give an improved energy demand for our C-SMCC methodology.
Mechanistic studies
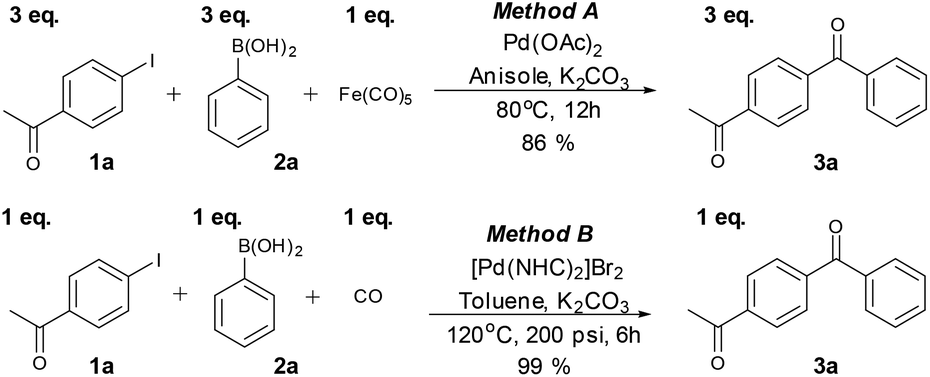
To assess further the properties of the metal species formed in the catalytic C-SMCC reactions, a series of control experiments were conducted (Scheme 2). Initially, addition of Hg (known to generally suppress surface catalysis) diminished the catalytic activity of the Pd–Fe catalytic system, with neither the carbonylative (3a) nor non-carbonylative cross-coupling products (3a′) detected (Scheme 2a). The Hg poison test supports a heterogeneous/pseudo-heterogeneous Pd-catalysed C-SMCC reaction using Fe(CO)5.22 We succeeded in separating the catalytic Pd–Fe nanoparticle material, which also catalysed a subsequent carbonylative coupling reaction using either additional CORM or CO gas, proceeding with 67% and 70% yields, respectively. Surprisingly, the catalytic Pd–Fe nanoparticles produced the carbonylative product independent of an added CO-source (3a, in 15% yield), indicating that CO resides within the Pd–Fe particles, i.e. as a CO-reservoir. FT-IR spectroscopic analysis of the fresh isolated metal catalyst material revealed three characteristic CO bands of the particles at 2106, 2029 and 1866 cm−1 (Fig. S6, see ESI†), suggesting that the particle surface consists of several carbonyl ligands, e.g. η1-Pd-CO and μ2-Fe-CO, which derive from the transfer process of CO from CO–RM to ketone products during the Pd-catalysed C-SMCC reaction. It is important to note that the C-SMCC reaction does not occur without added Pd(OAc)2 (Scheme 2b). Starting from Pd(OAc)2, the Pd-catalysed C-SMCC reaction with CO gas was found to afford only a 13% yield of biaryl ketone 3a (Scheme 2c), indicating again that Fe(CO)5 not only acted as a CO–RM, but also enhanced Pd-catalyst performance.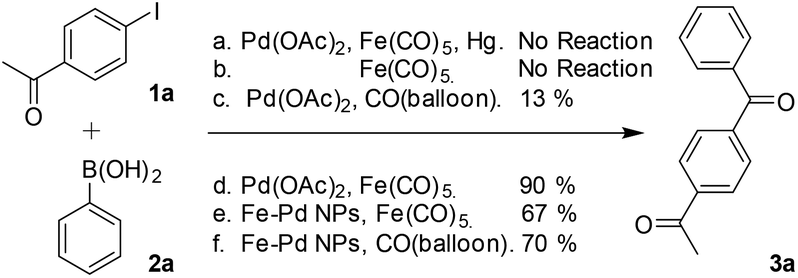 | ||
| Scheme 2 Control experiments to probe catalyst behaviour. | ||
To further characterize the catalytically active Pd–Fe particles, Transmission Electron Microscopy (TEM) measurements were conducted. The experiments showed the presence of 2 nm sized Pd–Fe nanoparticles over a large area (histogram inset in Fig. 2a). Lattice fringes were observed in the high-resolution TEM images (Fig. 2b), showing that the inter-planar distances approximate to 0.226 nm and 0.198 nm, corresponding to the (200) and (111) planes of the Pd metal. The assignment was confirmed using Fast Fourier Transform (FFT) and inverse FFT images (inset in Fig. 2b).
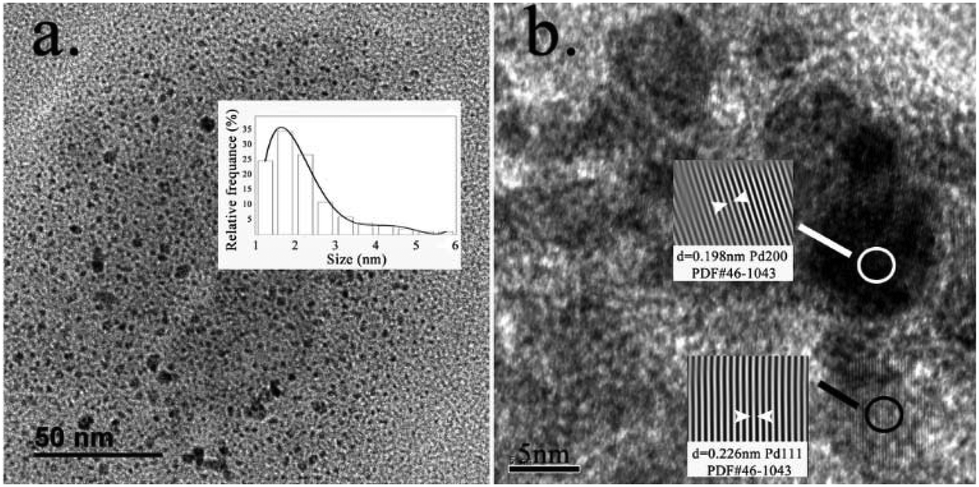 | ||
| Fig. 2 (a) HRTEM images of size distribution and (b) lattice fringes. | ||
The chemical state of the Pd–Fe nanoparticle surface was analysed by XPS (Fig. 3 and ESI S3†). Fig. 3 shows the XPS core level spectra for Pd 3d and Fe 2p, characterized by spin–orbit splitting (Pd 3d5/2, 3d3/2 and Fe 2p3/2, 2p1/2 components), where the peaks at 335.6 and 340.8 eV result from Pd 3d5/2 and Pd 3d3/2, respectively. The Pd species are characterised as being in an oxidation state of zero. FeIII was confirmed based on the peaks at 710.6 and 724.1 eV, corresponding to Fe 2p3/2 and Fe 2p1/2, respectively. The side peaks were attributed to the oxide; thus Fe exists in the form of Fe2O3. The EDAX mapping analysis confirmed the composition of the Pd–Fe nanoparticles deriving from Fe, Pd and O, which is fully consistent with our deduction (ESI S4†). SEM images show the morphology of the Pd–Fe nanoparticles (Fig. S5, see the ESI†).
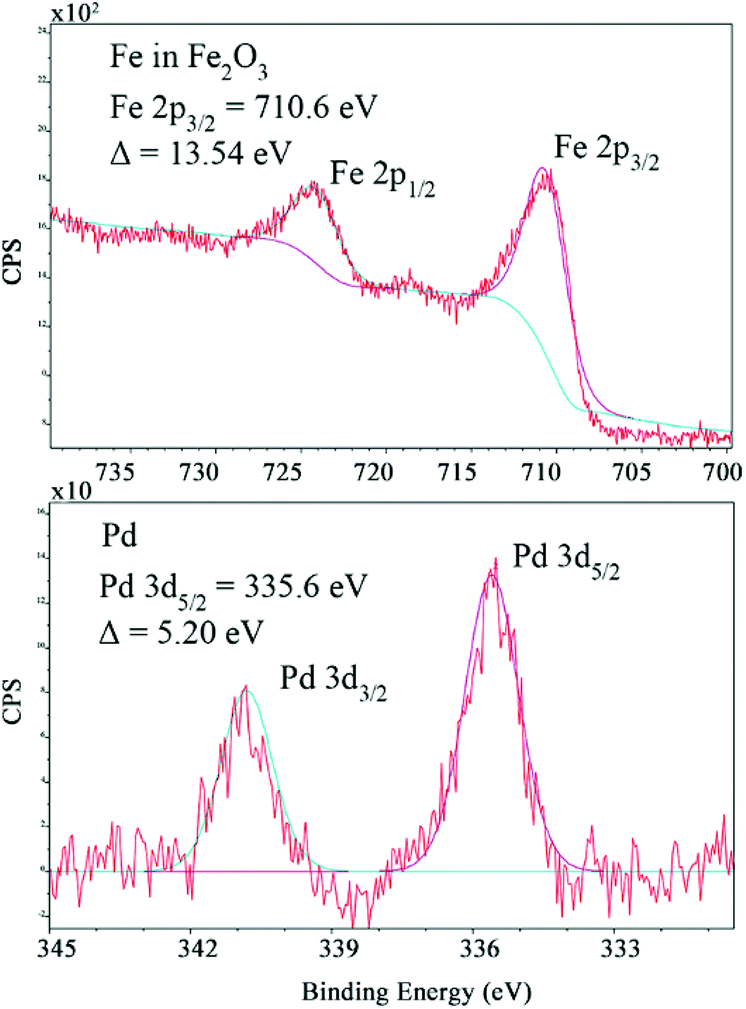 | ||
| Fig. 3 XPS images of the Pd–Fe catalyst nanoparticle material, with Fe2O3 and Pd0 prominent species. | ||
Finally, the recyclability of the Pd–Fe nanoparticles was assessed. The Pd–Fe nanoparticles were first isolated from a reaction of 1a + 2a → 3a, by filtration, and then sequentially washed with water and ethyl acetate. The C-SMCC reaction 1a + 2a → 3a was then recharged with fresh Fe(CO)5 and the isolated catalyst. At each cycle we found that the isolated catalyst can be recycled giving 3a, with the yields of 3a going from 90 to 50% over the 4 cycles. The deactivation of the heterogeneous Pd–Fe nanocatalyst species can be attributed to an excess Fe residue being deposited onto the heterometallic surface, either blocking the catalytically active sites or causing a restructuring of the catalyst surface (Fig. 4A).15,21 Interestingly, in the absence of Fe(CO)5, the isolated Pd–Fe nanoparticles also independently catalyzed the efficient SMCC reaction of 1a + 2a → 3a′ (Fig. 4B). A small amount of carbonylative product 3a is formed in the first cycle, formed from residual CO from the Pd–Fe nanoparticle catalyst. Crucially, the yield of 3a′ was over 80% for the subsequent cycles (2–5). These results are in accordance with the unique cooperative activity of a Pd–Fe bimetallic catalytic system reported by Lipshutz et al. for standard SMCC reactions (Fig. 4B).17
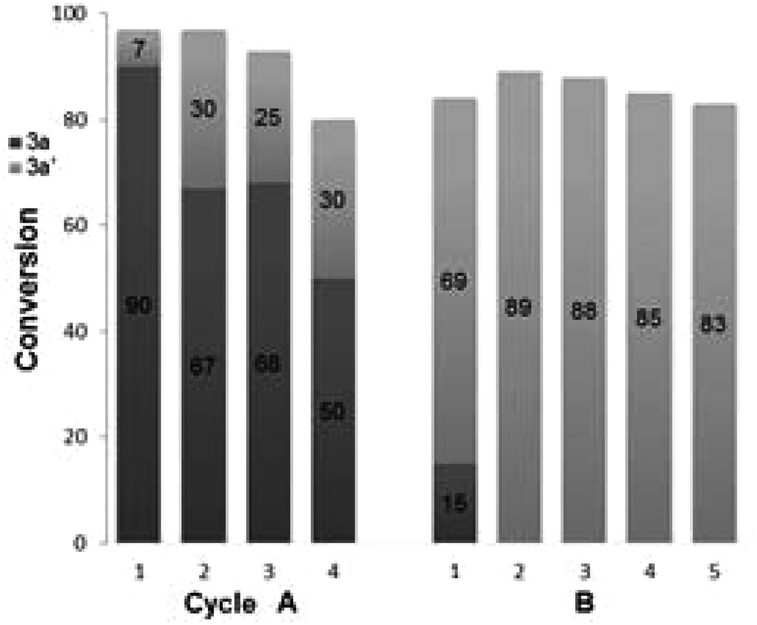 | ||
| Fig. 4 Recyclability of the Suzuki–Miyaura carbonylative cross-coupling (cycle A) and feasibility of the Pd–Fe nanoparticle catalyst in the SMCC reaction of iodobenzene and phenylboronic acid to give benzophenone (cycle B). | ||
Conclusions
In conclusion, we have developed a highly efficient synthetic strategy for accessing biaryl ketones via CO-gas free carbonylative Suzuki–Miyaura cross-couplings, using anisole as a green solvent.23 The green credentials of the methodology were compared with those of a recently reported reaction employing a novel and highly active Pd precatalyst possessing a bis-chelating carbene ligand. The Fe–Pd nanoparticles generated in situ from mixing ‘Pd(OAc)2’ and Fe(CO)5 are the key factor in the high catalytic efficiency observed in our study. The structural features of the in situ Fe–Pd nanoparticles were evaluated by TEM, EDAX, XPS, SEM and FT-IR spectroscopy, demonstrating the narrow size distribution, averaged ∼2 nm, which is the so-called ‘Goldilocks size’ for effective cross-coupling catalysis under reducing conditions.24 We believe that the particles consist primarily of Fe2O3 and the Pd metal, possessing a relaxed porous surface. Based on the above structural information, the efficacy of the C-SMCCs can be reasonably attributed to the synergistic effect of bimetallic Pd–Fe NPs, demonstrating the superiority over monometallic catalysis, which ought to facilitate further applications in catalysis, materials science, and organic synthesis. Studies directed toward understanding metal–metal interactions and cooperativity, particularly in bimetallic NPs,25 and their applications, are currently underway.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the grant from the National Natural Science Foundation of China (21371112 and 21446014), the 111 Project (B14041), the Fundamental Funds Research for the Central Universities (No. GK201501005, GK201503029, and 2016CSY002), the grant from the Fundamental Doctoral Fund of Ministry of Education of China (20120202120005), and the Program for Changjiang Scholars and Innovative Research Team in University (IRT_14R33). I. J. S. F. is grateful to the Royal Society for University Research Fellowship (2004-12). We thank Syngenta and the University of York for the iCASE funding and studentship to T. J. B. We also acknowledge the Leverhulme Trust for funding, enabling us to initiate this research (W. Z., I. J. S. F., and J. M. L.).Notes and references
- T. Ishiyama, H. Kizaki, N. Miyaura and A. Suzuki, Tetrahedron Lett., 1993, 34, 7595 CrossRef CAS.
- Y. Zhong and W. Han, Chem. Commun., 2014, 50, 3874 RSC.
- D. G. Hall, Boronic Acids-Preparation and Applications in Organic Synthesis, Medicine and Materials, Wiley-VCH, Weinheim, 2011, vol. 2 Search PubMed.
- (a) K. Maeyama, K. Yamashita, H. Saito, S. Aikawa and Y. Yoshida, Polym. J., 2012, 44, 315 CrossRef CAS; (b) W. Sharmoukh, K. C. Ko, C. Noh, J. Y. Lee and S. U. Son, J. Org. Chem., 2010, 75, 6708 CrossRef CAS; (c) F. Boscá and A. Miranda, J. Photochem. Photobiol., B, 1998, 43, 1 CrossRef; (d) G. Dormán and G. D. Prestwich, Biochemisty, 1994, 33, 5661 CrossRef; (e) S. Budavari, The Merck Index, Merck, Rahway, USA, 11th edn, 1989 Search PubMed.
- For some recent reviews on Pd-catalyzed carbonylation, see: (a) X.-F. Wu, H. Neumann and M. Beller, Chem. Rev., 2013, 113, 1 CrossRef CAS; (b) X.-F. Wu, H. Neumann and M. Beller, Chem. Soc. Rev., 2011, 40, 4986 RSC; (c) R. Grigg and S. P. Mutton, Tetrahedron, 2010, 66, 5515 CrossRef CAS; (d) A. Brennführer, H. Neumann and M. Beller, Angew. Chem., Int. Ed., 2009, 48, 4114 CrossRef; (e) M. Beller and X.-F. Wu, Transition Metal Catalyzed Carbonylation Reactions, Springer-Verlag, Berlin Heidelberg, 2013 CrossRef; (f) Z.-L. Zhu, W.-Q. Zhang and Z.-W. Gao, Prog. Chem., 2016, 28(11), 1616 Search PubMed.
- A. Petz, G. Péczely, Z. Pintér and L. Kollár, J. Mol. Catal. A: Chem., 2006, 255, 97 CrossRef CAS.
- F. Jin and W. Han, Chem. Commun., 2015, 51, 9133 RSC.
- I. J. S. Fairlamb, S. Grant, P. McCormack and J. Whittall, Dalton Trans., 2007, 8, 859 RSC.
- T. Morimoto and K. Kakiuchi, Angew. Chem., Int. Ed., 2004, 43, 5580 CrossRef CAS.
- (a) S. D. Friis, A. T. Lindhardt and T. Skrydstrup, Acc. Chem. Res., 2016, 49(4), 594 CrossRef CAS; (b) P. Hermange, A. T. Lindhardt, R. H. Taaning, K. Bjerglund, D. Lupp and T. Skrydstrup, J. Am. Chem. Soc., 2011, 133, 6061 CrossRef CAS; (c) A. Ahlburg, A. T. Lindhardt, R. H. Taaning, A. E. Modvig and T. Skrydstrup, J. Org. Chem., 2013, 78, 10310 CrossRef CAS.
- P. Gautam and B. M. Bhanage, Catal. Sci. Technol., 2015, 5, 4663 RSC.
- For the recent progress in CORMs, see: (a) S. García-Gallego and G. J. L. Bernardes, Angew. Chem., Int. Ed., 2014, 53, 9712 CrossRef; (b) U. Schatzschneider, Br. J. Pharmacol., 2014, 172, 1638 CrossRef.
- (a) E. J. Corey and L. S. Hegedus, J. Am. Chem. Soc., 1969, 91, 1233 CrossRef CAS; (b) E. Yoshisato, M. Ryang and S. Tsutsumi, J. Org. Chem., 1968, 34, 1500 CrossRef.
- (a) N.-F. K. Kaiser, A. Hallberg and M. Larhed, J. Comb. Chem., 2002, 4, 109 CrossRef CAS; (b) J. Wannbeg and M. Larhed, J. Org. Chem., 2003, 68, 5750 CrossRef.
- (a) S. R. Borhade, A. Sandström and P. I. Arvidsson, Org. Lett., 2013, 15, 1056 CrossRef CAS; (b) F. Jafarpour, P. Rashidi-Ranjbar and A. O. Kashani, Eur. J. Org. Chem., 2011, 2128 CrossRef CAS; (c) W. J. Ang, L.-C. Lo and Y. Lam, Tetrahedron, 2014, 70, 8545 CrossRef CAS.
- (a) T. Vlaar, E. Ruijter, B. U. W. Maes and R. V. A. Orru, Angew. Chem., Int. Ed., 2013, 52, 7084 CrossRef CAS; (b) J. W. Collet, T. R. Roose, E. Ruijter, B. U. W. Maes and R. V. A. Orru, Angew. Chem., Int. Ed., 2020, 59, 540 CrossRef CAS.
- S. Handa, Y. Wang, F. Gallou and B. H. Lipshutz, Science, 2015, 349, 1087 CrossRef CAS.
- M. Alami, C. Amatore, S. Bensalem, A. Choukchou-Brahim and A. Jutand, Eur. J. Inorg. Chem., 2001, 2001, 2675 CrossRef.
- (a) L. Kumar, S. K. Dhawan, M. N. Kamalasanan and S. Chandra, Thin Solid Films, 2003, 441, 243 CrossRef CAS; (b) A. Banerjee, A. Sahana, S. Das, S. Lohar, B. Sarkar, S. K. Mukhopadhyay, J. S. Matalobos and D. Das, Dalton Trans., 2013, 42, 16387 RSC; (c) Y. Niko, Y. Hiroshige, S. Kawauchi and G. Konishi, J. Org. Chem., 2012, 77, 3986 CrossRef CAS; (d) Z. Zhu, E. Shao, S. Xu, H. Sun, G. Zhang, Z. Xie, W. Zhang and Z. Gao, RSC Adv., 2016, 6, 76883 RSC.
- (a) C. R. McElroy, A. Constantinou, L. C. Jones, L. Summerton and J. H. Clark, Green Chem., 2015, 17, 3111 CAS; (b) S. Abou-Shehada, P. Mampuys, B. U. W. Maes, J. H. Clark and L. Summerton, Green Chem., 2017, 19, 249 RSC; (c) N. McStay, Z. Molphy, A. Coughlan, A. Cafolla, V. McKee, N. Gathergood and A. Kellett, Nucleic Acids Res., 2017, 45, 527 CrossRef CAS; (d) A. J. Blacker and K. E. Jolley, Beilstein J. Org. Chem., 2015, 11, 2408 CrossRef CAS; (e) A. Jordan, A. Haiß, M. Spulak, Y. Karpichev, K. Kümmerer and N. Gathergood, Green Chem., 2016, 18, 4374 RSC; (f) S. Jin, F. Byrne, C. R. McElroy, J. Sherwood, J. H. Clark and A. J. Hunt, Faraday Discuss., 2017, 202, 157 RSC; (g) K. E. Jolley, W. Nye, C. G. Niño, N. Kapur, A. Rabion, K. Rossen and A. J. Blacker, Org. Process Res. Dev., 2017, 21, 1557 CrossRef CAS; (h) A. J. Reay, L. A. Hammarback, J. T. W. Bray, T. Sheridan, D. Turnbull, A. C. Whitwood and I. J. S. Fairlamb, ACS Catal., 2017, 7, 5174 CrossRef CAS; (i) A. J. Reay, T. J. Williams and I. J. S. Fairlamb, Org. Biomol. Chem., 2015, 13, 8298 RSC.
- W. Mansour, M. Fettouhi and B. E. Ali, Appl. Organomet. Chem., 2020, 34, e5636 CAS.
- T. Storr, A. Firth, K. Wilson, K. Darley, C. Baumann and I. J. S. Fairlamb, Tetrahedron, 2008, 64, 6125 CrossRef CAS.
- We note that anisole, as a green solvent, has an excellent sustainability ranking, particularly in hydroformylation, see: F. G. Delolo, E. N. dos Santos and E. V. Gusevskaya, Green Chem., 2019, 21, 1091 RSC.
- A. J. Reay and I. J. S. Fairlamb, Chem. Commun., 2015, 51, 16289 RSC.
- For lead references, see: K. Mishra, N. Basavegowdaa and Y. R. Lee, Catal. Sci. Technol., 2015, 5, 2612 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details and characterization. See DOI: 10.1039/d0gc03036h |
| This journal is © The Royal Society of Chemistry 2021 |