
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhoto-triggered hydrogen atom transfer from an iridium hydride complex to unactivated olefins†
Mirjam R.
Schreier
,
Björn
Pfund
,
Xingwei
Guo‡
 * and
Oliver S.
Wenger
*
* and
Oliver S.
Wenger
*
Department of Chemistry, University of Basel, St. Johanns-Ring 19, 4056 Basel, Switzerland. E-mail: xingwei_guo@mail.tsinghua.edu.cn; oliver.wenger@unibas.ch
First published on 6th August 2020
Abstract
Many photoactive metal complexes can act as electron donors or acceptors upon photoexcitation, but hydrogen atom transfer (HAT) reactivity is rare. We discovered that a typical representative of a widely used class of iridium hydride complexes acts as an H-atom donor to unactivated olefins upon irradiation at 470 nm in the presence of tertiary alkyl amines as sacrificial electron and proton sources. The catalytic hydrogenation of simple olefins served as a test ground to establish this new photo-reactivity of iridium hydrides. Substrates that are very difficult to activate by photoinduced electron transfer were readily hydrogenated, and structure–reactivity relationships established with 12 different olefins are in line with typical HAT reactivity, reflecting the relative stabilities of radical intermediates formed by HAT. Radical clock, H/D isotope labeling, and transient absorption experiments provide further mechanistic insight and corroborate the interpretation of the overall reactivity in terms of photo-triggered hydrogen atom transfer (photo-HAT). The catalytically active species is identified as an Ir(II) hydride with an IrII–H bond dissociation free energy around 44 kcal mol−1, which is formed after reductive 3MLCT excited-state quenching of the corresponding Ir(III) hydride, i.e. the actual HAT step occurs on the ground-state potential energy surface. The photo-HAT reactivity presented here represents a conceptually novel approach to photocatalysis with metal complexes, which is fundamentally different from the many prior studies relying on photoinduced electron transfer.
1 Introduction
Photoinduced electron transfer (PET) can readily be initiated with many photoactive metal complexes, but photo-triggered hydrogen atom transfer (photo-HAT) reactivity of coordination compounds is comparatively rare.1 Much progress has been made in recent years regarding the development of new photosensitizers, particularly including complexes of Earth-abundant metals2–6 and purely organic (metal-free) variants,7 but they all exhibit the well-established PET reactivity. Consequently, the vast majority of current photochemical studies, including most of the work by the photoredox community,8 still operate on the basis of PET. Even though increasingly thermodynamically challenging PET reactions have been accomplished lately,9–18 sometimes in combination with an applied electrochemical potential,19–21 such reactions are limited by the redox properties of the photosensitizer and the substrates. To overcome thermodynamic limitations imposed by PET, proton-coupled electron transfer (PCET)22 has been exploited successfully in photoredox catalysis for substrates with sufficiently polar functional groups that can form hydrogen bonds.23 Similarly, the interaction of Lewis acids with polar functional groups can help activate substrates that would be difficult to engage in pure PET chemistry.24–27We hypothesized that the photochemical hydrogenation of olefins would be an ideal test ground for exploring the photo-HAT reactivity of a metal complex. Whilst alkene hydrogenation is of course an extremely well-developed field, photochemical methods are very scarce and largely limited to substrates with strongly electron-withdrawing substituents to permit typical PET chemistry,28 or to the reduction with solvated electrons.29,30 For the present work, we chose olefins that do not contain electron-withdrawing groups and which therefore cannot be activated by PET due to their very negative reduction potentials (Ered < −2.7 V vs. SCE31). From the perspective of PET, these olefins are unactivated.
HAT is a key process in many photochemical reactions,32,33 but usually requires auxiliary reagents such as thiols or ascorbate which act as H-atom donors in a secondary, light-independent step that does not involve the photosensitizer.34–37 Photoreactions, in which a metal complex donates or accepts an H-atom in an electronically excited state, are rare.33,38–43 For the conceptually related proton-coupled electron transfer (PCET) reactions, there exist numerous compounds which exhibit PCET reactivity from electronically excited states, but these systems typically rely on polar O–H or N–H groups which can form hydrogen bonds.44–47
The HAT reactions considered herein do not proceed directly from an electronically excited state, but instead occur after an initial reductive excited-state quenching process with a tertiary amine. The sequence of reductive excited-state quenching followed by single-electron transfer to the substrate is rather common in organic photoredox catalysis as well as energy-related photochemical research, but here, the situation is conceptually different: reductive quenching leads to an H-atom donor, which can then undergo HAT with olefins. Thermal HAT from metal complexes in their electronic ground state has attracted significant attention48,49 and homolytic splitting of metal-hydride bonds upon excitation with visible light is known for a few selected examples,50,51 but we are unaware of prior reports of photo-triggered HAT from metal complexes to unactivated olefins. The reductive excited-state quenching step involved in our photo-triggered HAT gives access to a particularly reactive metal hydride with a very low M–H bond dissociation free energy (BDFE), and this enables uncommon photochemistry that goes beyond the well-established single electron transfer (SET) reactivity. From the synthetic viewpoint, our approach offers the possibility to form C–H bonds, complementing recent work on photo-triggered PCET that focused on the cleavage of C–H bonds.52,53
Metal hydride complexes play important roles for proton and CO2 reduction,54,55 and recently tungsten hydrides provided fundamental insight into multi-site PCET reactions.56–58 To some extent their reactivity can be regarded as HAT-like, but with coupled proton and electron transfer reactions occurring to separate base and oxidant molecules.22 Iridium cyclopentadienyl hydride complexes are a particularly well investigated class of compounds,59 which have been shown to function as photoacids60 (Fig. 1). Furthermore, they are able to form H2 upon irradiation with visible light61,65 or when used in photo-electrocatalytic settings in water or CH3CN.66 “Self-quenching” reactions, in which an electron is transferred from an excited [Cp*Ir(α-diimine)H]+ complex (Cp* = pentamethylcyclopentadienyl) to an iridium hydride in the ground state, are responsible for photochemical H2 formation in water and CH3CN.67 By contrast, in neat CH2Cl2 the “self-quenching” products (comprised of one oxidized iridium hydride complex and one reduced iridium hydride complex) undergo sequential hydride transfer and HAT, enabling the hydrodechlorination of two equivalents of CH2Cl2 to CH3Cl.64
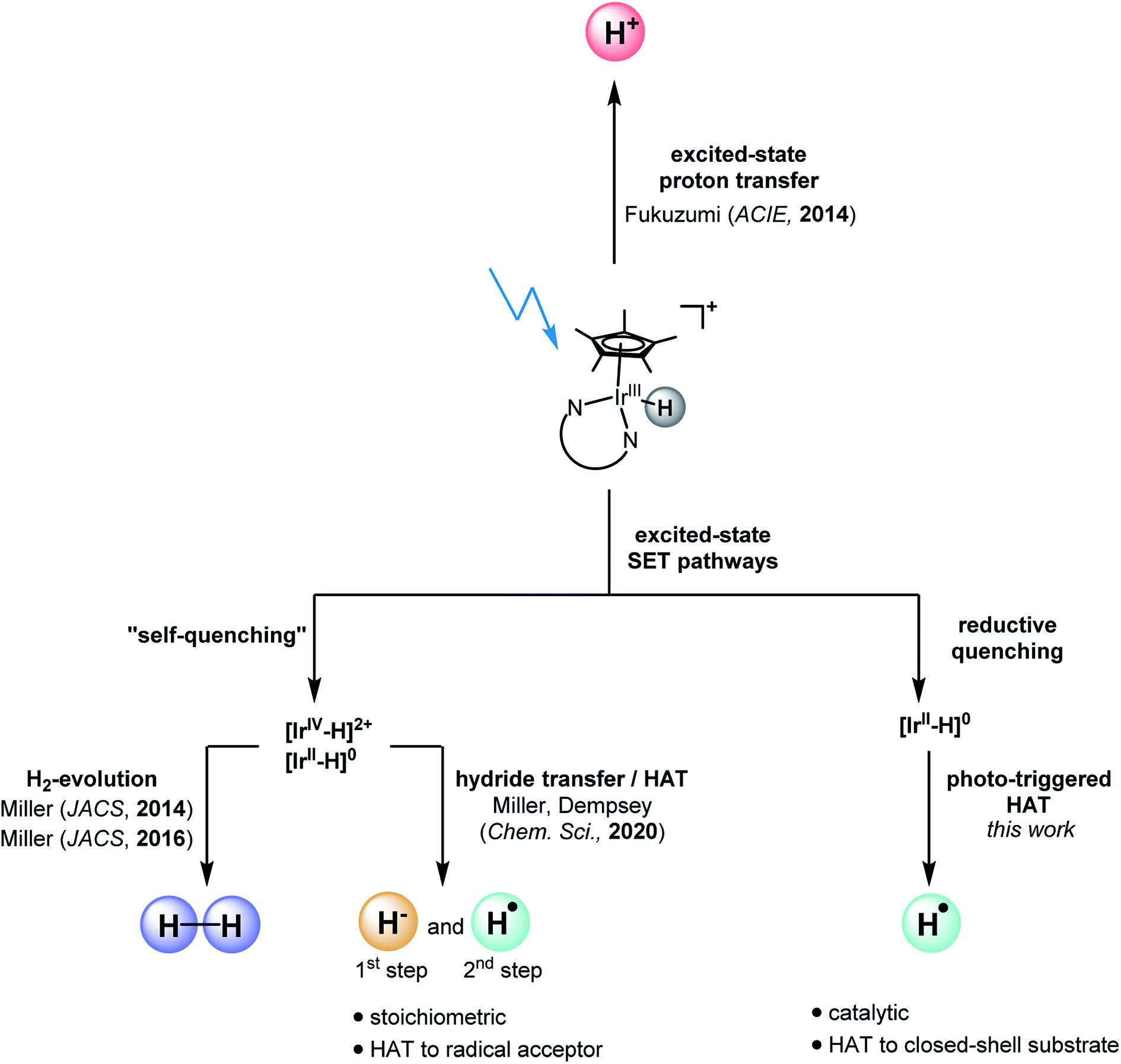 | ||
| Fig. 1 Different photo-reactivities: iridium hydrides can undergo excited-state proton transfer60 or photodriven dihydrogen formation.61,65,67 Herein we demonstrate that photo-HAT represents an additional reaction pathway for iridium hydride complexes, complementing recent work demonstrating sequential hydride and HAT transfer.64 | ||
In the present work, [Cp*Ir(phen)(H)]+ (phen = 1,10-phenanthroline) was formed in situ via photo-irradiation of [Cp*Ir(phen)Cl]+ in CH3CN in presence of excess triethylamine (TEA), which served as a combined electron and proton source. Continued irradiation then promoted [Cp*Ir(phen)(H)]+ to its (relatively long-lived) 3MLCT state. In presence of 250 mM TEA, reductive excited-state quenching to form [Cp*Ir(phen)(H)]0 rather than “self-quenching” is the dominant reaction pathway. For the closely related [Cp*Ir(bpy)(H)]0 complex (bpy = 2,2′-bipyridine), an IrII–H bond dissociation free energy (BDFE) of 43.9 kcal mol−1 was reported previously.67
We anticipated that such a low IrII–H BDFE would make [IrII–H] complexes suitable for olefin hydrogenation via photo-triggered HAT. Activation of olefins by thermal (i.e., not light-driven) metal-hydride catalyzed hydrogen atom transfer (MHAT)48,68 is typically performed with metal-hydrides that have BDFEs ranging from ≈50 to 60 kcal mol−1.70,71 Our results presented below indicate that the key-step in our photoinduced hydrogenation of olefins (Fig. 2) is indeed an initial HAT from the [IrII–H] species to the substrate. After successful activation of the olefin starting material (SM) via photo-triggered HAT, a radical intermediate (RI˙) is formed. The latter can undergo two different onward reactions: (i) either RI˙ can accept a second H-atom to form the hydrogenation product H, or (ii) an H-atom can be abstracted from a different position of radical intermediate RI˙ to give the rearranged products (E)-R or (Z)-R.69,72 In this work, we attempted to maximize the hydrogenation and to minimize the rearrangement yield, but the main point of the study was to establish a new type of photo-reactivity of metal complexes, as noted above.
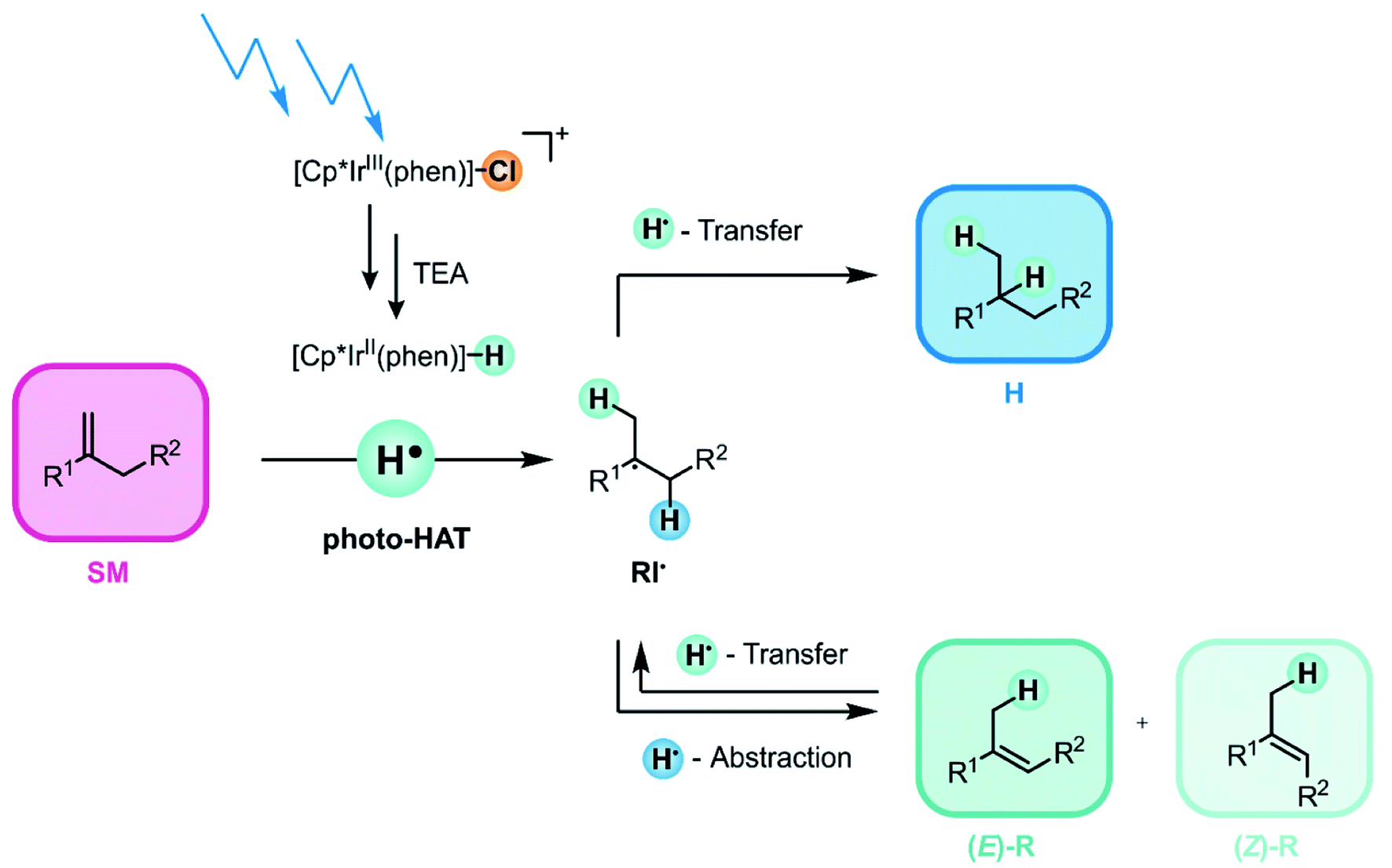 | ||
| Fig. 2 Visible-light driven catalytic hydrogenation of electron-neutral olefins based on photo-triggered hydrogen atom transfer (photo-HAT) from an iridium(II) hydride (Cp* = pentamethylcyclopentadienyl; phen = 1,10-phenanthroline), see text for details. | ||
A very recent study reported on the photoenzymatic hydrogenation of vinyl pyridines, using “ene” reductases in combination with Ru(bpy)3Cl2 as a catalytic system (with some modest product yields also observable in absence of the enzyme).73 Mechanistically, this formal hydrogenation proceeded via a sequence of reduction, protonation and HAT steps. Such combined enzyme-photoredox catalysis approaches hold much promise for enantioselective conversions, but they are not straightforward to implement.74–76
Our iridium complex serves both as photosensitizer and as catalyst of HAT. Due to this dual role, no fine tuning of different catalytic cycles is needed,77 as is sometimes necessary when merging photoredox chemistry with traditional transition metal catalysis or with enzyme catalysts. The hydride complex is formed in situ from a robust Ir(III) precursor complex, and HAT occurs to stable olefin substrates and does not require highly reactive radicals as reaction partners. Thus, alongside the abovementioned very recent study by Miller, Dempsey, and coworkers,64 our work opens a conceptually new avenue to photoredox chemistry, exploiting visible-light excitation of a simple metal complex for inducing H-atom rather than electron transfer.
2 Results and discussion
2.1 Reaction optimization
We started our reaction screening with (3-methoxyprop-1-en-2-yl)benzene (1-SM) as substrate, because geminal disubstituted olefins with aromatic substituents are particularly well-suited acceptors for HAT.78 In an initial experiment, [Cp*Ir(phen)Cl]Cl was used as iridium hydride precursor and diisopropylethylamine (DIPEA) was employed as sacrificial donor. 85% conversion of 1-SM was observed when irradiating the reaction mixture at 470 nm for 44 h (Table 1, entry 1) under inert atmosphere. However, the chemoselectivity of the reaction was limited, as expressed by a 1-H/1-R product ratio of 2.8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. When the sacrificial donor was changed to triethylamine (TEA), both the conversion (90%) and the chemoselectivity (1-H/1-R 3.2:1) of the reaction were slightly increased (entry 2). Upon addition of excess phenanthroline (30 mol%), the chemoselectivity of the reaction improved drastically in favor of the hydrogenation product 1-H (1-H/1-R 14.7:1, entry 3) and the conversion further increased (94%). When the reaction was performed in the dark (but otherwise identical conditions), no conversion of 1-SM was observed (entry 4). Moreover, no conversion of 1-SM was detected when either the iridium catalyst (entry 5) or the sacrificial donor (entry 6) was absent.
:1. When the sacrificial donor was changed to triethylamine (TEA), both the conversion (90%) and the chemoselectivity (1-H/1-R 3.2:1) of the reaction were slightly increased (entry 2). Upon addition of excess phenanthroline (30 mol%), the chemoselectivity of the reaction improved drastically in favor of the hydrogenation product 1-H (1-H/1-R 14.7:1, entry 3) and the conversion further increased (94%). When the reaction was performed in the dark (but otherwise identical conditions), no conversion of 1-SM was observed (entry 4). Moreover, no conversion of 1-SM was detected when either the iridium catalyst (entry 5) or the sacrificial donor (entry 6) was absent.
|
|
|||||
|---|---|---|---|---|---|
| Entrya | Catalyst | Sacrificial donor | Additive (mol%) | Conversionb [%] | 1-H/1-Rb |
| a Reaction conditions: 1-SM (50 mM), [Cp*Ir(phen)Cl]Cl (2.5 mM), sacrificial donor (250 mM) and 1,10-phenanthroline (0 or 15 mM) in 0.5 mL degassed CD3CN. Irradiation of the sample occurred at 470 nm (7.5 W) for 44 h, while the temperature was kept at 50 °C. See ESI Section 1 for details regarding the photoreactor and ESI Section 3.1 for experimental details. b Conversions and H/R product ratios were determined by 1H-NMR spectroscopy. See ESI Section 3.1.1 for details. c Product formation was confirmed by GC/MS spectrometry. See ESI Section 3.1.2 for details. d Reaction was performed in the dark. | |||||
| 1 | [Cp*Ir(phen)Cl]Cl | DIPEA | — | 85 | 2.8:1 |
| 2 | [Cp*Ir(phen)Cl]Cl | TEA | — | 90 | 3.2:1 |
| 3 | [Cp*Ir(phen)Cl]Cl | TEA | phen (30) | 94 |
14.7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :1 :1
|
| 4 | [Cp*Ir(phen)Cl]Cl | TEA | — | 0 | n.d. |
| 5 | — | TEA | — | 0 | n.d. |
| 6 | [Cp*Ir(phen)Cl]Cl | — | — | 0 | n.d. |
2.2 Substrate scope
As noted in the introduction, the olefin hydrogenation reaction mainly serves as a test ground to establish a new type of metal hydride photo-reactivity, but nevertheless it seemed useful to explore its substrate scope in the interest of gaining a reactivity picture that is as complete as possible. We therefore probed a range of substrates (Fig. 3) and determined hydrogenation and rearrangement product yields by a combination of quantitative NMR and GC/MS analyses in presence of an internal standard. We focused mostly on geminal disubstituted olefins, because the lack of substituents on their terminal olefinic carbon atom permits for faster initial HAT than what would be possible with other olefins.78 Furthermore, the tertiary radical intermediate formed after initial HAT to this substrate class can be rather stable, particularly when using aromatic geminal disubstituted olefins, which result in benzylic radical intermediates.78 Consequently, the new method can be applied to a range of substrates of this type (1-SM–5-SM), tolerating both electron-withdrawing and electron-donating substituents and affording the different hydrogenation products in high yields (88–99%) and with high chemoselectivities (H/R ratios exceeding 12.7:1). Both para- and meta-substituents on the aromatic ring are well tolerated, and no dehalogenation was observed for both chloro-substituted substrates (4-SM and 5-SM), as opposed to what would be expected for PET with strong photoreductants.10,79–81
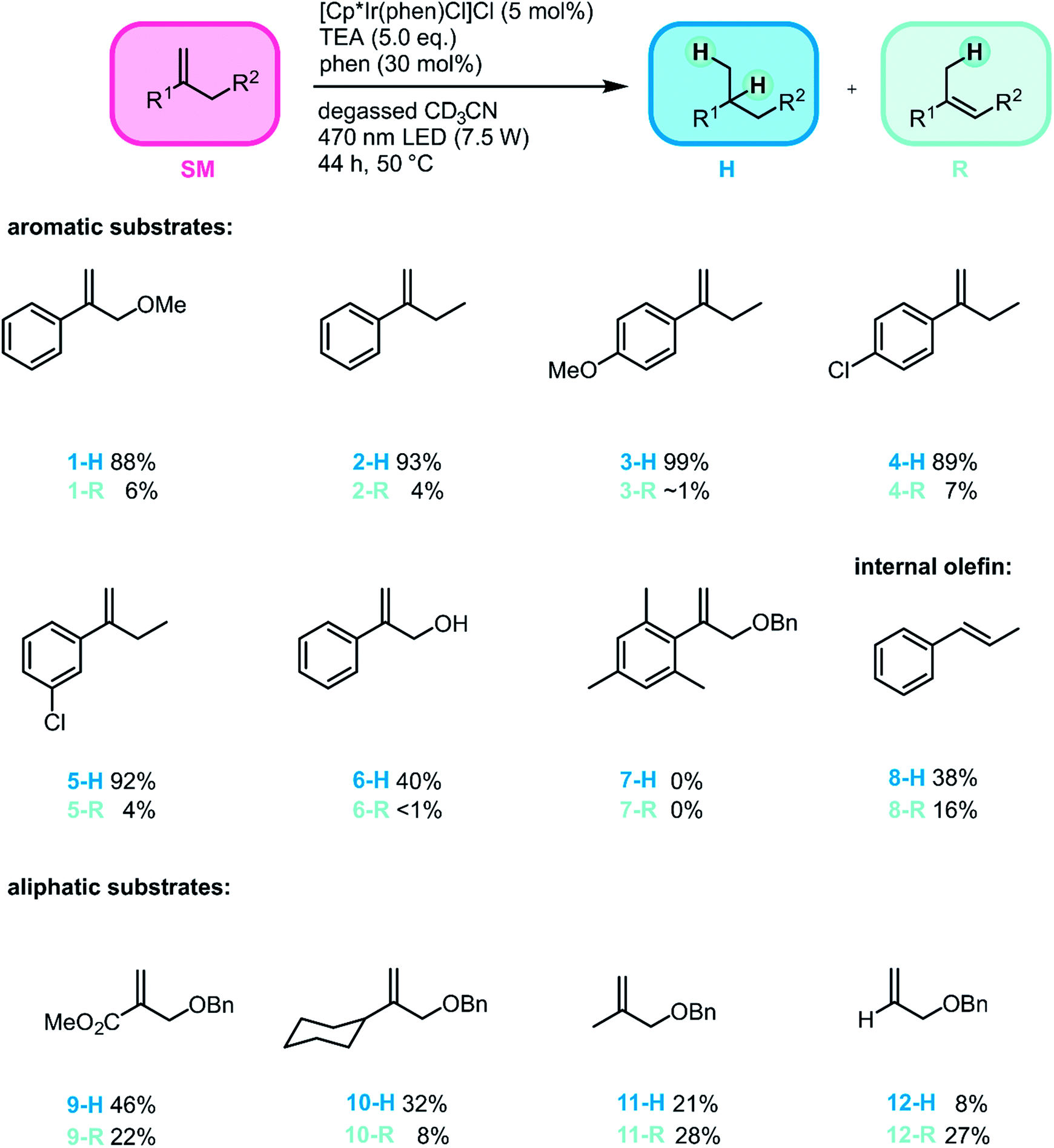 | ||
| Fig. 3 Substrate scope for the photo-triggered hydrogenation of olefins promoted by an iridium hydride. For simplicity, only one isomer of the rearranged products (R) is shown. Reaction conditions: substrate (50 mM), [Cp*Ir(phen)Cl]Cl (2.5 mM), TEA (250 mM) and 1,10-phenanthroline (15 mM) in 0.5 mL degassed CD3CN. Irradiation of the sample occurred at 470 nm (7.5 W) for 44 h, while the temperature was kept at 50 °C, see Section 1 of the ESI† for details regarding the photoreactor. Yields and conversions were determined by 1H-NMR spectroscopy and the yields of the hydrogenation products (1-H to 5-H, 8-H, 9-H and 11-H) were likewise confirmed by GC-MS spectrometry. See ESI Section 3.2† for details. In the case of product 8-R, R means isomerization to cis-1-phenyl-1-propene. In the case of products 6-H/6-R, 10-H/10-R, and 12-H/12-R the product conversion could not be determined by GC-MS spectrometry. Therefore, the yields for these substrates were determined based on 1H-NMR spectroscopy exclusively. | ||
The conversion of substrate 6-SM with its free hydroxyl-group was considerably lower, affording the hydrogenation product in only 40% yield. This observation suggests that the catalytic turnover is slowed down by coordinating substituents on the substrate, even if the substrate coordinates only weakly and reversibly, as expected for 6-SM. When using the mesityl-substituted substrate 7-SM the reaction was completely shut down. The combined observations made for substrates 6-SM and 7-SM suggest that coordination to the Ir center is undesirable, but sterically very demanding substituents can impede HAT completely.
To explore whether our method can also be applied to internal olefins, trans-1-phenyl-1-propene (8-SM) was tested as substrate, affording propylbenzene (8-H) in 38% yield. The lower conversion of this internal olefin in comparison to terminal olefins is in line with prior studies, which found that an additional methyl substituent at the terminal carbon of 1,1-diphenylethylene slows down the thermal HAT from CpCr(CO)3H to various olefins by about 800 times.78 Furthermore, with 8-SM the formation of the hydrogenation product was less favored than for 1-SM–6-SM. Significant isomerization to cis-1-phenyl-1-propene was observed, whereas no isomerization to the thermodynamically less favored terminal olefin (allylbenzene) was detected.
To gain insight into how the stability of the presumed radical intermediates influences the outcome of the overall reaction, a series of terminal olefins (9-SM–12-SM) with different non-aromatic substituents at the disubstituted olefinic carbon atom was investigated. The hydrogenation yield decreases along the series 1-SM > 9-SM > 11-SM > 12-SM, reflecting the stability of the different radicals formed after initial HAT to these substrates along the substituent series Ph > COOMe > CH3 > H (Fig. 4A).78 Furthermore, the chemoselectivity of the reaction is highly dependent on the stability of the formed radical intermediate. While the hydrogenation product is the major product for all investigated aromatic substrates (1-SM–6-SM), the formation of the rearranged product becomes increasingly important for aliphatic olefins. Specifically, H/R ratios decrease from ca. 15:1 for 1-SM to roughly 2:1 for 9-SM, and finally about 1:3 for 12-SM. This observation reflects the fact that for aliphatic substrates, the newly formed C–H bond in the radical intermediate (RI˙ in Fig. 2) is roughly 10 kcal mol−1 (ref. 82 and 83) weaker compared to aromatic substrates (see ESI Section 4.1† for details). Consequently, H-atom abstraction to yield rearranged products is faster for aliphatic (e.g., 11-SM or 12-SM) than for aromatic substrates (e.g., 1-SM).
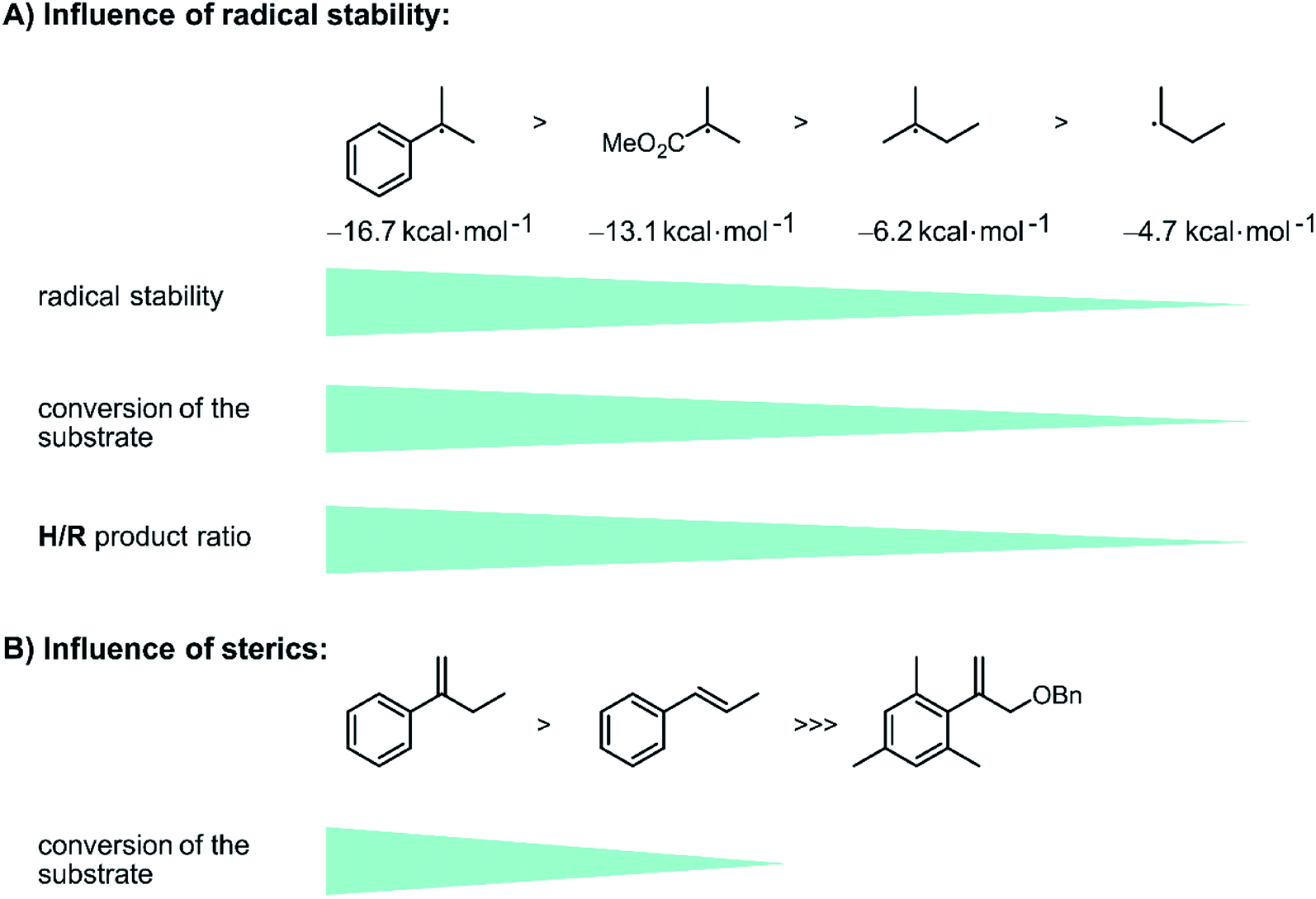 | ||
| Fig. 4 Influence of radical stability and sterics on reactivity of the substrates and product distribution: (A) the conversion of the substrate decreases with decreasing radical stability (reference value: methyl radical = 0 kcal mol−1).84 The chemo-selectivity changes towards the rearranged product when aliphatic substrates are used instead of aromatic substrates. (B) Introducing sterics on the substrates hinders the initial HAT and lowers the reactivity of the substrate, see text for details. | ||
The very good correlation between the reactivity (and chemoselectivity) of the different substrates and the stability of the formed radical intermediates RI˙ (Fig. 4A) is fully in line with hydrogenation via an initial photo-HAT. The investigated olefin substrates have very negative reduction potentials (below −2.7 V vs. SCE)31 and photoexcited [Cp*Ir(α-diimine)X] species (X = Cl−, H−) do not have this much reducing power (see ESI Section 4.1† for details),67 hence PET is not a viable reaction pathway.
2.3 Monitoring the reaction progress over time
Further mechanistic insight was gained by monitoring the reaction progress at different reaction times of photo-irradiation (Fig. 5A), using the same conditions as for the substrates in Fig. 3. Continuous hydrogenation of the olefin substrate (1-SM) was observed over the first 20 h, and a final conversion of 94% was reached after 44 h (Fig. 5B, blue trace). Interestingly, the ratio of the different reaction products (1-H/1-R) changed over the reaction course. At the beginning of the reaction the 1-H/1-R ratio was only 3:1, but then it increased to 14.7:1 after 44 h. Moreover, the total amount of rearranged product ((E)-R + (Z)-R) even slightly decreased over the reaction course (Fig. 5B, turquoise traces). This is because the rearranged products still contain an olefinic moiety and as such can be hydrogenated over time (leftward arrow in the bottom part of Fig. 2).
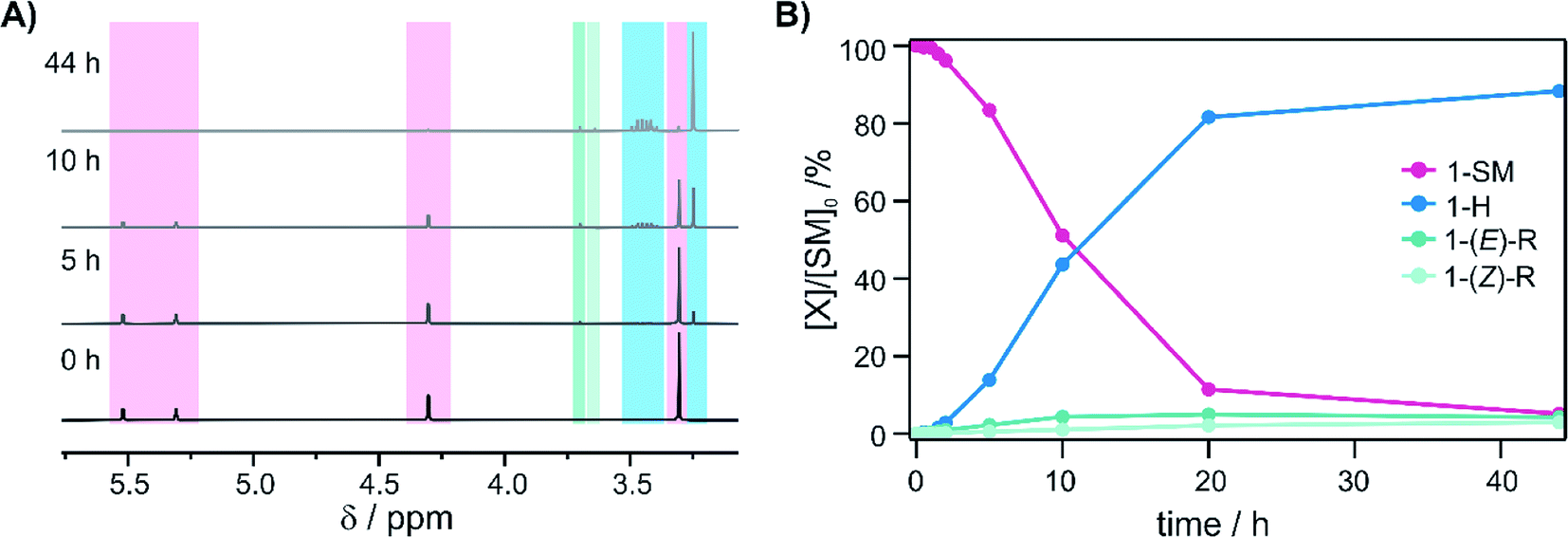 | ||
| Fig. 5 Reaction progress as a function of time: (A) 1H-NMR spectra of the reaction mixture after different time intervals following irradiation with a 7.5 W 470 nm LED (NMR acquisition occurred in the dark, using 8 aliquots of the same stock solution irradiated in parallel for different amounts of time). The characteristic signals of the starting material (1-SM, pink background) disappear, while formation of the hydrogenation product (1-H, blue background) and the rearranged products (1-(E)-R and 1-(Z)-R, turquoise backgrounds) are observable. (B) Conversion of the substrate (1-SM, pink trace) and 1H-NMR yields of the different products (1-H, blue trace; 1-(E)-R and 1-(Z)-R, turquoise traces) over the reaction course. | ||
2.4 Probing the role of iridium hydride
Next we monitored the reaction progress when using [Cp*Ir(phen)(H)](PF6) instead of [Cp*Ir(phen)Cl]Cl as iridium source. With [Cp*Ir(phen)Cl]Cl, conversion of 1-SM was only observed after an initial lag phase of approximately 1 hour, during which essentially no hydrogenation of the substrate was observed (yellow trace in Fig. 6A). This indicates that [Cp*Ir(phen)Cl]+ first must be converted to the catalytically active species. When instead using [Cp*Ir(phen)(H)](PF6) (see ESI Sections 2.1 and 4.2† for details), no lag phase is observed (green trace in Fig. 6A), and the hydrogenation product 1-H begins to form without any significant delay. This suggests that the initial lag phase when using [Cp*Ir(phen)Cl]+ is due to the light-driven conversion of the chloro-complex to [Cp*Ir(phen)(H)]+ in presence of TEA, and furthermore that [Cp*Ir(phen)(H)]+ is a catalytically relevant species. However, even with that iridium(III) hydride, no substrate conversion occurred when the reaction mixture was stirred in the dark (entry 2 in Fig. 6B), indicating that [Cp*Ir(phen)(H)]+ must be activated by visible light in order for the HAT to occur. Evidently, the [Cp*Ir(phen)(H)]+ complex in its electronic ground state is not a sufficiently good H-atom donor for the olefin substrates considered here (see ESI Section 4.1† for further details).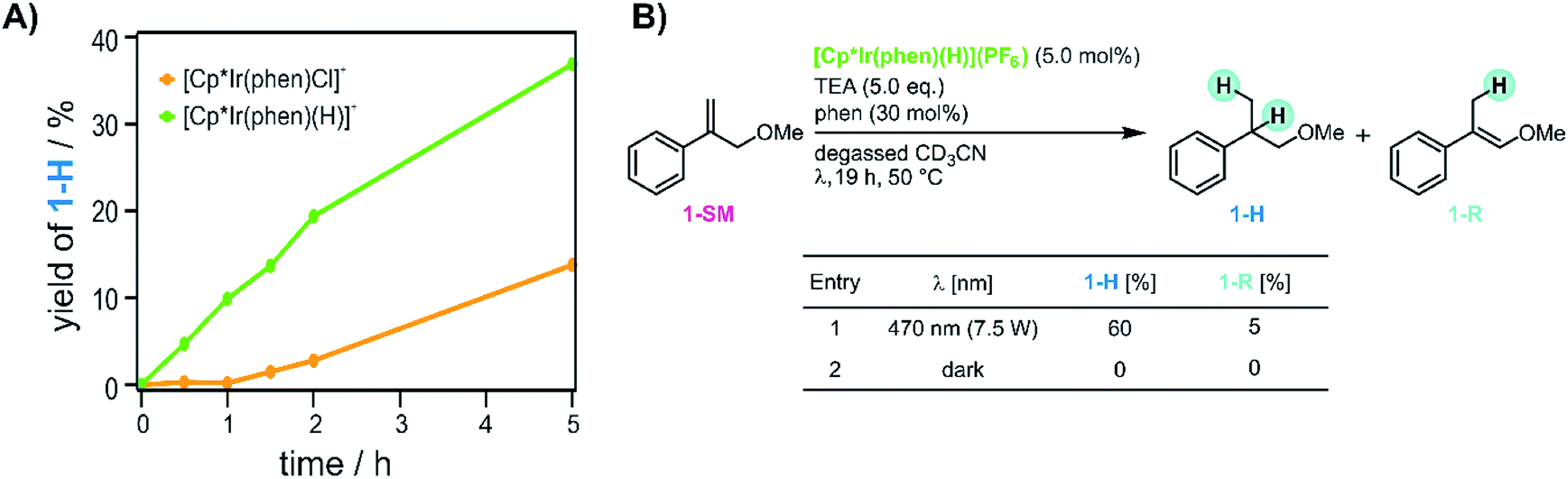 | ||
| Fig. 6 Role of iridium hydride in the reaction mixture. (A) Comparison of the different reaction time profiles when using [Cp*Ir(phen)Cl]+ (yellow trace) or [Cp*Ir(phen)(H)]+ (green trace). (B) Olefin hydrogenation with [Cp*Ir(phen)(H)](PF6), yields determined by 1H-NMR spectroscopy. The yellow trace in panel A is the same data set as the blue trace in Fig. 5B. | ||
The electrochemical and chemical conversion of [Cp*Ir(bpy)Cl]+ to [Cp*Ir(bpy)(H)]+ is well established,66,85 but so far does not seem to have been studied in photochemical settings. For related rhodium complexes, the photochemical conversion of chloro- to hydride complexes has been explored to some extent.86–88 When irradiating a 80 μM solution of [Cp*Ir(phen)Cl]+ in CH3CN in presence of 1.0 mM TEA with an LED at 455 nm, the UV-Vis absorption spectrum of [Cp*Ir(phen)Cl]+ converts slowly but steadily to the spectral signature of [Cp*Ir(phen)(H)]+. The series of spectra shown in Fig. S33† were recorded after different irradiation times and have an isosbestic point at 375 nm, in line with expectation based on the UV-Vis spectra of neat [Cp*Ir(phen)Cl]+ and [Cp*Ir(phen)(H)]+.
To probe this conversion under conditions resembling more closely those of the reactions in Section 2.2, 3.5 mM [Cp*Ir(phen)Cl]+ in CD3CN containing 350 mM TEA (but no olefin substrate) was irradiated at 470 nm. The 1H-NMR spectra recorded (in the dark) after photo-irradiation for 2 hours provide clear evidence for the formation of [Cp*Ir(phen)(H)]+ (ESI, Section 4.2†). This set of experiments furthermore demonstrates that prolonged photo-irradiation leads to dissociation of the phen ligand, which explains why the photochemical hydrogenation proceeds better in the presence of excess phen (Table 1).
Presumably, this photoreaction proceeds via an initial reductive dissociation of the chloro-ligand, thereby leading to [Cp*Ir(phen)]0. Protonation of that species (with TEA˙+ as proton source) can then in principle lead directly to the formation of [Cp*Ir(phen)(H)]+, but this is speculative and our data does not provide insight into the individual elementary steps involved in this conversion. Whilst TEA is principally an electron donor, its one-electron oxidized form TEA˙+ is competent to act either as acid or H-atom donor.89
Having established that [Cp*Ir(phen)(H)]+ forms from [Cp*Ir(phen)Cl]+ in the course of continuous photo-irradiation in presence of excess TEA, the next logical step is to elucidate possible onward photochemical reactions of that iridium(III) hydride species. [Cp*Ir(phen)(H)]+ exhibits 3MLCT photoluminescence with a lifetime (τ) of 148 ns in deaerated CH3CN at a [Cp*Ir(phen)(H)]+ concentration of 0.2 mM with 0.6 mM NBu4PF6 at room temperature (Fig. S36†). TEA induces reductive 3MLCT quenching with a rate constant of 4.5 × 108 M−1 s−1 according to Stern–Volmer luminescence quenching experiments (inset of Fig. S36†), thereby leading to the respective iridium(II) hydride, [Cp*Ir(phen)(H)]0.
That iridium(II) hydride species cannot be observed directly by transient absorption spectroscopy, because this is a highly reactive species that does not accumulate in detectable concentrations. However, UV-Vis transient absorption spectroscopy provides unambiguous evidence for a follow-up transient species, namely the iridium(I) complex [Cp*Ir(phen)]0, which forms from the iridium(II) hydride [Cp*Ir(phen)(H)]0 after H-atom release. This iridium(I) species exhibits diagnostic absorption bands near 600 and 640 nm (Fig. 7C, vertical black lines), which are observable after pulsed 445 nm excitation of [Cp*Ir(phen)(H)]+ in presence of excess TEA in CH3CN (Fig. 7A). An analogous experiment with a triarylamine electron donor (TAA-OMe) instead of TEA provides clear evidence for the triarylamine radical cation (absorption band at 720 nm in Fig. 7B/D) superimposed with the spectroscopic signature of the iridium(I) species. Thus, it is clear that both TEA and the triarylamine act as electron donors to the 3MLCT-excited [Cp*Ir(phen)(H)]+ complex.
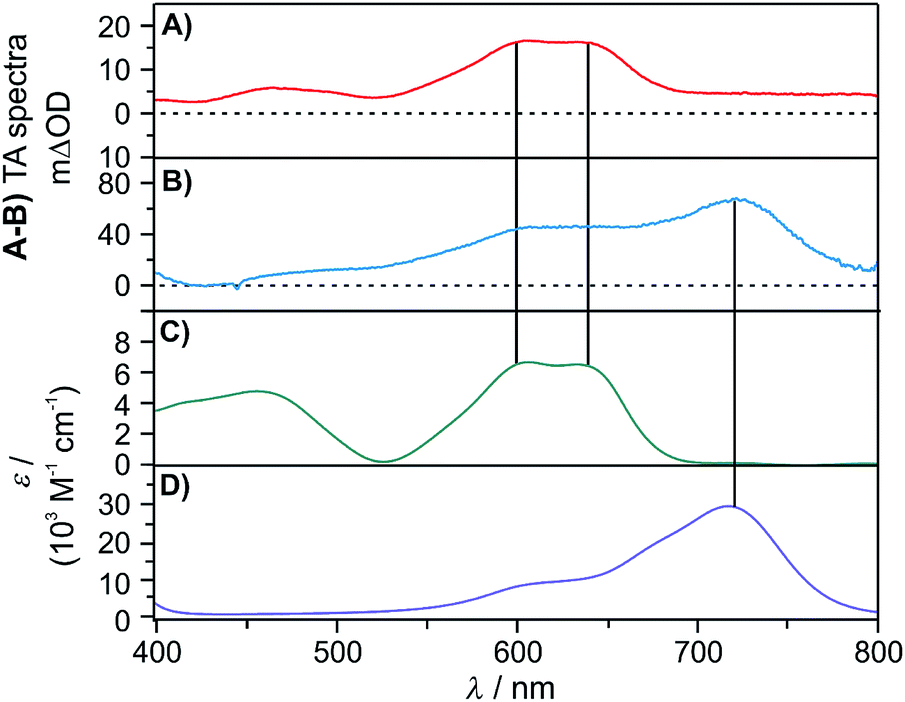 | ||
| Fig. 7 (A) Transient absorption spectrum of a solution containing [Cp*Ir(phen)(H)](PF6) (0.5 mM) and TEA (10 mM). The spectrum was measured at a time delay of 60 ns after excitation at 445 nm (9 mJ) and was time-integrated over 100 ns. (B) Transient absorption spectrum of a solution containing [Cp*Ir(phen)(H)](PF6) (0.5 mM) and TAA-OMe (10 mM) in deaerated CH3CN. The spectrum was measured at a time delay of 10 ns after excitation at 445 nm (9 mJ) and was time-integrated over 100 ns. See ESI Section 2.6† for the molecular structure of TAA-OMe. (C) Absorption spectrum of [Cp*Ir(phen)]0 in deaerated CH3CN at room temperature obtained after deprotonation of [Cp*Ir(phen)(H)]+ with KOtBu (1.3 eq.). (D) Absorption spectrum of TAA-OMe˙+ in deaerated CH3CN after chemical oxidation of a TAA-OMe solution (31 μM) with NOBF4 (3.0 eq.). | ||
In the presence of the olefin substrates, we expect that [Cp*Ir(phen)(H)]0 acts as H-atom donor, leading to the [Cp*Ir(phen)]0 complex and the radical intermediates (RI˙ in Fig. 2). In the transient absorption experiments of Fig. 7, where there is no olefin substrate present, [Cp*Ir(phen)]0 is expected to form via H2 evolution from two equivalents of [Cp*Ir(phen)(H)]0 as illustrated in Fig. S37.† This reaction is exergonic by ca. 16 kcal mol−1.67
2.5 Radical clock experiment
The reactivity patterns identified above (Fig. 4) are fully compatible with a HAT mechanism. To gain further mechanistic insight, an experiment with (1-(2-phenylcyclopropyl)vinyl)benzene (13-SM), a radical clock-type substrate, was performed (Fig. 8). The exact rate constant for the ring-opening of the radical formed after HAT to this particular substrate (13-RI1˙) is not known, but based on structurally closely related compounds (ESI Section 4.5†) it seems plausible that the rate constant (kRO) for the opening of the cyclopropane-ring in 13-RI1˙ (leading to 13-RI2˙) occurs with a rate on the order of 108 s−1.90,91 Under our standard reaction conditions (Fig. 8, top left) using [Cp*Ir(phen)Cl]Cl, the olefin substrate 13-SM converts exclusively to the ring-opened product 13-RO (71% yield after 44 h). No formation of the ring-retention hydrogenation product (13-RR) is observed. The observation of the ring-opened product 13-RO is consistent with a radical mechanism, and furthermore indicates that the intramolecular ring-opening reaction is faster than secondary HAT. Because 13-RO still contains an olefinic functional group, it reacts onwards to hydrogenation and rearranged products, analogously to substrates 1-SM–12-SM (ESI Section 4.5†).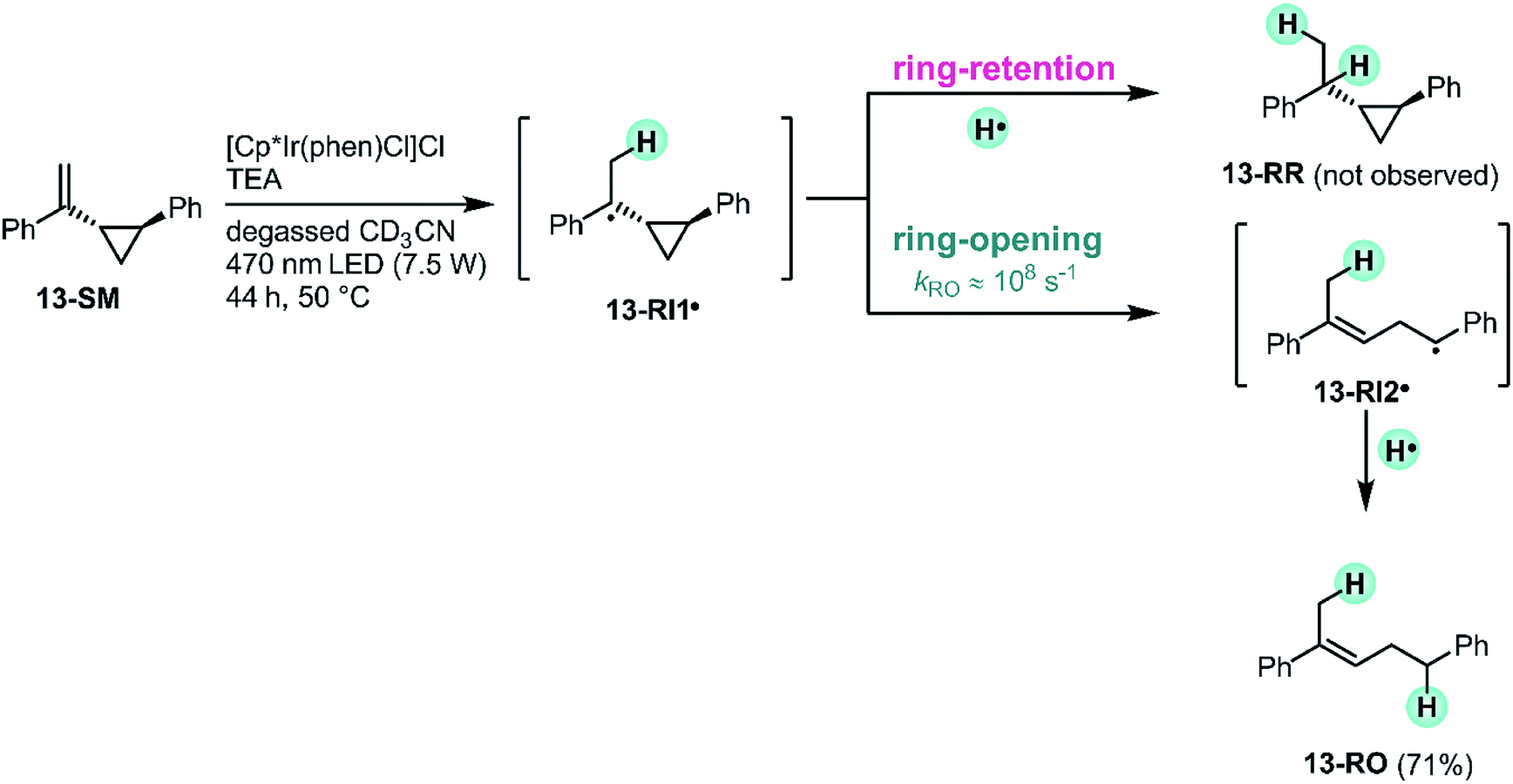 | ||
| Fig. 8 Radical clock experiment with a cyclopropane substituted olefin confirms that the reaction proceeds via a radical intermediate. For simplicity, only the (E)-isomer of the ring-opened product 13-RO is shown. | ||
2.6 Experiment with a deuterated substrate probing the reversibility of primary HAT
The prior section confirmed the radical nature of the reaction and provided already some insight into the kinetics of secondary HAT to the radical intermediate. By exploring the visible-light induced hydrogenation of deuterated substrate 1-SM-d4 we sought to probe the initial HAT step in more detail. After an initial HAT to 1-SM-d4 (kHAT1 in Fig. 9), the formed radical intermediate (1-RI˙-d4) can in principle directly react onwards to the hydrogenation (1-H-d4) or the rearranged (1-R-d3) products. On the other hand also a reverse HAT (H-atom abstraction from 1-RI˙-d4 to afford 1-SM-d3, k−HAT1 in Fig. 9) could occur prior to product formation. Thus, the deuterated substrate is useful to probe the reversibility of initial HAT: if reverse HAT from 1-RI˙-d4 were kinetically competitive with onward reaction to 1-H-d4 or 1-R-d3, there should be proton incorporation into the originally fully deuterated double bond (bond labeled “a” in Fig. 9) and some 1-SM-d3 should be formed.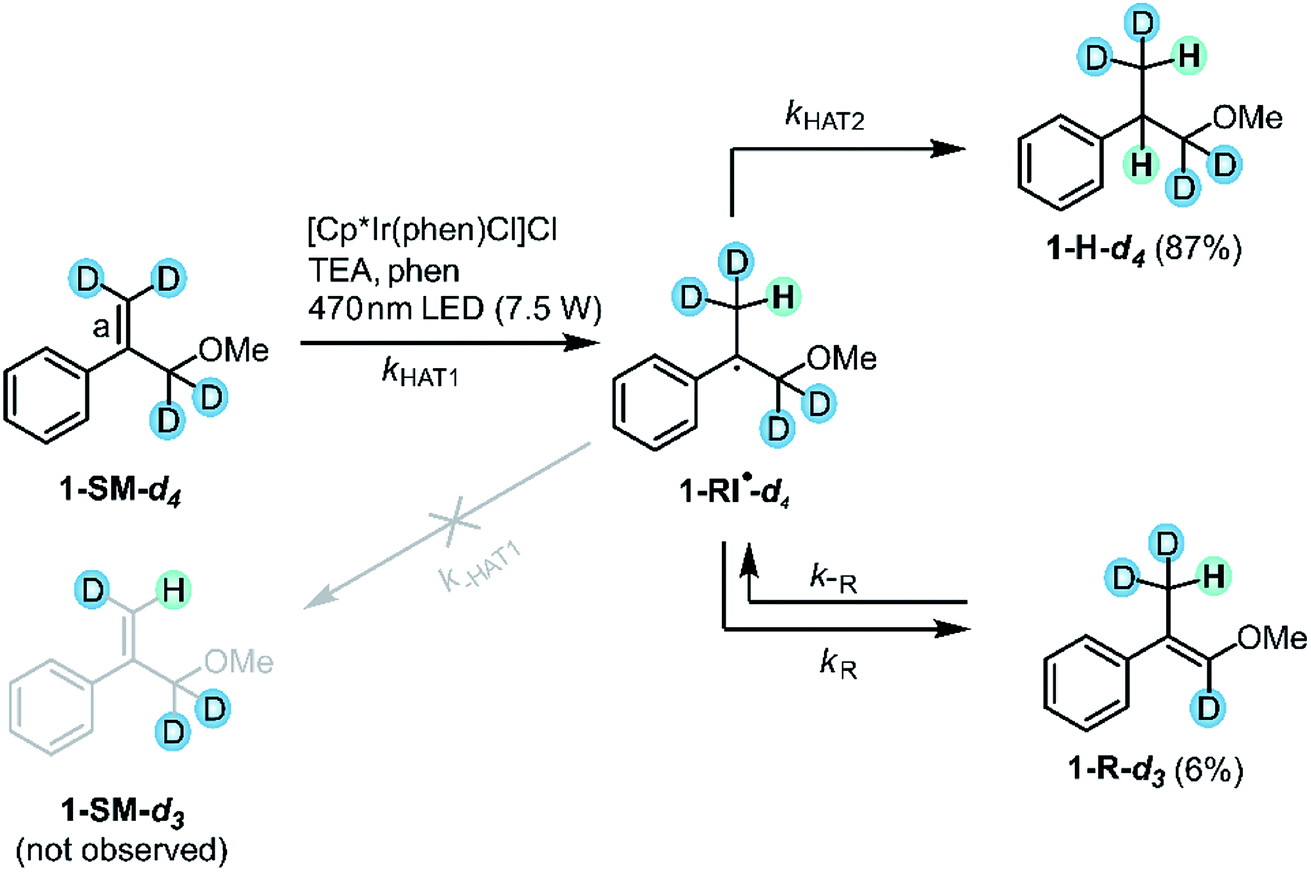 | ||
| Fig. 9 Experiment with deuterated substrate 1-SM-d4: no proton incorporation into the terminal double bond (bond labeled “a”) was observed, indicating that H-atom abstraction from 1-RI˙-d4 to afford 1-SM-d3 is negligible (crossed gray leftward arrow labeled k−HAT1). For simplicity, only the (E)-isomer of the rearranged product 1-R-d3 is shown. | ||
When the deuterated substrate (1-SM-d4) was subjected to the standard conditions (see Fig. 3) and the reaction mixture was irradiated for 44 h, both hydrogenation (1-H-d4, 87%) and isomerization (1-R-d3, 6%) products were observed. No formation of 1-SM-d3 was detectable throughout the irradiation process (ESI Section 4.6†). Thus, H-atom abstraction from the radical intermediate (1-RI˙-d4) to form 1-SM-d3 can be neglected. Evidently, secondary HAT (kHAT2) yielding the final hydrogenation product 1-H-d4 as well as isomerization (kR) leading to the rearranged product 1-R-d3 are both faster than reverse HAT (k−HAT1). The fast onward reaction to give preferably the hydrogenation product 1-H-d4 is likely due to the presence of TEA oxidation products such as TEA˙+ or TEA˙.89,92 These highly reactive species presumably serve as the H-atom sources for a secondary HAT to yield the final hydrogenation product.
As noted previously for thermal MHAT, the chemoselectivity of the overall reaction strongly depends on the concentration of the H-atom donor.72 When the concentration of the H-atom donor is relatively high, such as for a system where CpCr(CO)3H was readily regenerated under H2 pressure,78 hydrogenation products are formed preferably. With lower H˙ concentrations, however, which was the case in a previously investigated Co–H/H2 system,72 the rearranged products (1-R-d3) are formed preferably. The preferential formation of hydrogenation products in our study therefore indicates an elevated (local) H-atom donor concentration, presumably due to the presence of iridium hydrides and TEA oxidation products in sufficiently close proximity.
2.7 Proposed reaction mechanism
From the various studies in the prior sections, the mechanistic picture in Fig. 10 emerges. Before the actual catalytic cycle initiates, the catalyst precursor [Cp*Ir(phen)Cl]+ first needs to be converted into [Cp*Ir(phen)(H)]+ (Section 2.4). The [Cp*Ir(phen)(H)]+ complex with the metal in its +III oxidation state is not yet reactive enough to catalyze the HAT-step to the olefin substrate (SM), as demonstrated by the lack of conversion when the reaction mixture is stirred in the dark (Fig. 6B, entry 2). The IrIII–H BDFE of [Cp*Ir(phen)(H)]+ is not known, but for a related complex the IrIII–H BDFE is 69 kcal mol−1,93 which is too high for formation of the radical intermediates RI˙ (ESI Section 4.1†).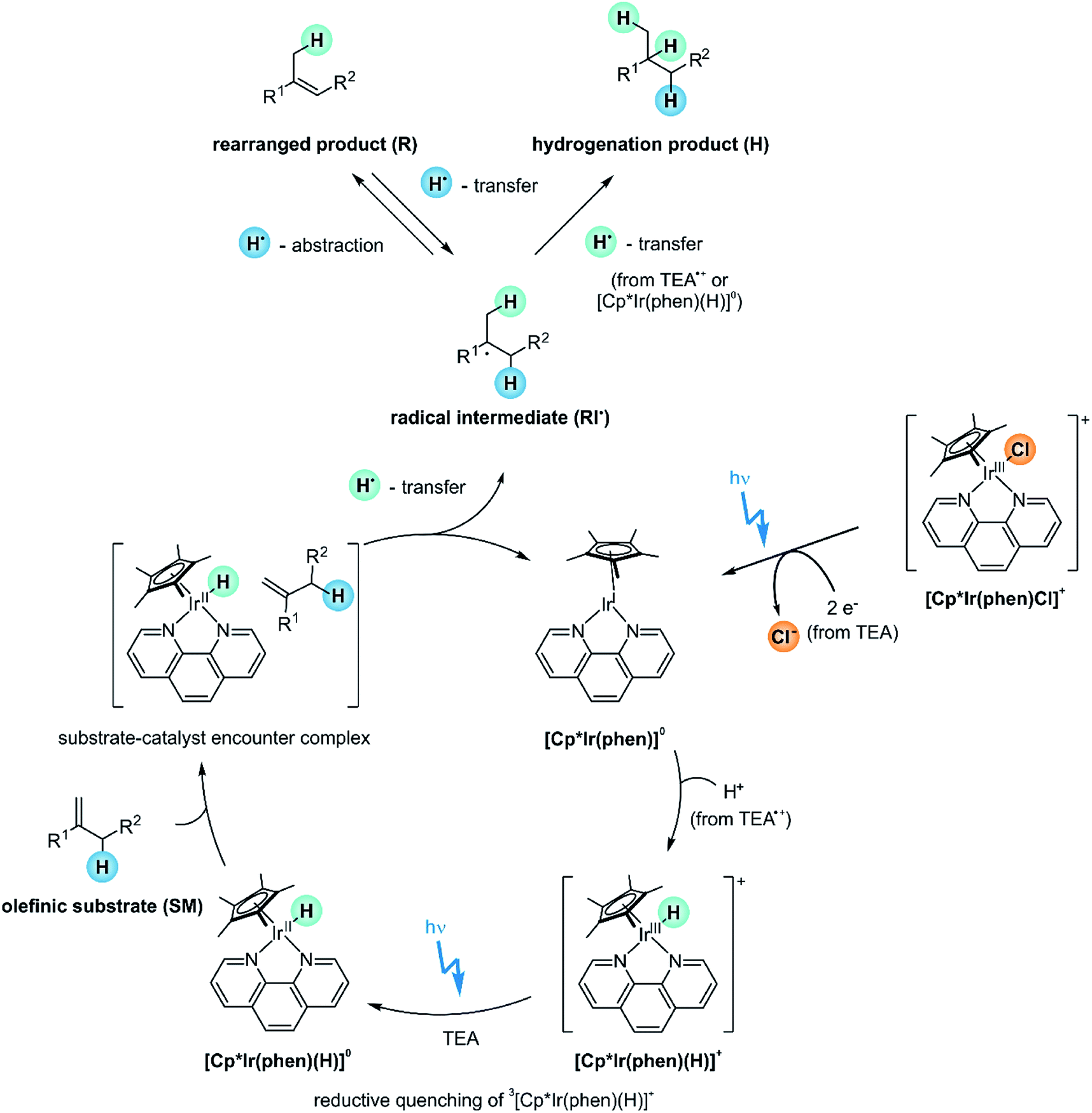 | ||
| Fig. 10 Proposed reaction mechanism. For simplicity, only the (E)-isomer of the rearranged product is shown. | ||
Photoexcitation of [Cp*Ir(phen)(H)]+ populates a long-lived 3MLCT excited state (τ of 148 ns at a [Cp*Ir(phen)(H)]+ concentration of 0.2 mM),59,67 which is quenched reductively by TEA with a rate constant of kq = 4.5 × 108 M−1 s−1 to form [Cp*Ir(phen)(H)]0 (ESI Section 4.3†). This iridium(II) hydride is likely the key catalytic intermediate, acting as H-atom donor vis-à-vis the olefin substrates. At the relevant TEA concentrations for our olefin hydrogenations (250 mM), the rate for reductive 3MLCT excited state quenching exceeds the rate for “self-quenching” by at least one order of magnitude.
Direct detection of [Cp*Ir(phen)(H)]0 by transient absorption spectroscopy is not possible, because this is a highly reactive and short-lived species, which does not accumulate in detectable concentrations. Instead, [Cp*Ir(phen)]0, which is formed from [Cp*Ir(phen)(H)]0 after H-atom release, is readily detected (Section 2.4).
Based on previously determined electrochemical potentials for analogous bpy-based (instead of phen-based) iridium complexes, neither the 3MLCT-excited [Cp*Ir(phen)(H)]+ (E0′(IV/III*) ≈ −1.3 vs. SCE,67 ESI Section 4.1†) nor the [Cp*Ir(phen)(H)]0 complex (E0′(III/II) ≈ −1.4 V vs. SCE,67 ESI Section 4.1†) have the reducing power to permit electron transfer to the olefin substrates (Ered < −2.7 V vs. SCE).31 Consequently, olefin hydrogenation cannot occur via a PET pathway (ESI Section 4.1†).
However, [Cp*Ir(phen)(H)]0 exhibits an IrII–H BDFE of approximately 44 kcal mol−1,67 considerably lower than the M–H BDFEs of common MHAT-catalysts, which are typically in the range of 50–60 kcal mol−1.70,71 This very weak IrII–H bond permits an initial HAT to the olefin substrates (ESI Section 4.1†). The achievable hydrogenation yields for the various substrates 1-SM–12-SM reflect the relative stabilities of the involved (charge-neutral) radical intermediates RI˙ (Fig. 4), as expected for HAT. Furthermore, the radical clock experiment (Fig. 8) underpins the radical pathway and speaks against a mechanism involving olefin insertion into the Ir–H bond.
No conversion at all is observed with the sterically demanding mesityl-substituted olefin 7-SM (Fig. 3), suggesting that the catalyst and 7-SM cannot approach sufficiently closely to undergo HAT. The formation of sufficiently intimate contacts, different however from actual substrate ligation to the metal center or olefin insertion into the M–H bond, seems to be a key requirement for successful reaction.
For substrates undergoing successful photo-HAT, the formed radical intermediate (RI˙) can either react via secondary HAT to yield the hydrogenation product (H, top right in Fig. 10) or rearrange to the isomerization product (R, top left in Fig. 10). TEA oxidation products such as TEA˙+ or TEA˙ likely serve as H-atom sources for secondary HAT. Whilst the hydrogenation products (H) are inert and do not react onwards, the rearranged products (R) can be activated via HAT from [Cp*Ir(phen)(H)]0 to re-form radical intermediate (RI˙). Thus, irradiation over extended time periods drives the overall reaction towards the hydrogenation product (Fig. 5B). Excess phen ligand (30 mol%) in the reaction mixture enhances that H/R chemoselectivity by decelerating catalyst degradation and enabling photo-reactions over longer irradiation periods (Section 2.4).
3 Conclusions
This work illustrates a new type of photo-reactivity for an archetypal metal hydride complex, complementing prior studies on photoacid and “self-quenching” behavior of this well-known class of compounds.59–67 From a photochemical perspective, the discovery of photo-HAT from the [Cp*Ir(phen)(H)]+ complex could represent an important breakthrough, because the vast majority of previously explored metal-based photosensitizers operate on the basis of photoinduced electron transfer. Consequently, photoredox catalysis until now largely relied on single electron transfer, but the possibility of photo-HAT could open completely new perspectives. Our new insights complement recent photophysical and -chemical studies of iridium hydride59–67 and related complexes.94,95 In particular, recent work established that the hydrodechlorination of CH2Cl2 proceeds via excited-state “self-quenching” producing a pair of [IrIV–H]2+ and [IrII–H]0 species, which then undergoes (sequential) hydride transfer and HAT to form two equivalents of CHCl3 from two molecules of CH2Cl2 solvent. Here, the [IrII–H]0 H-atom donor is formed via reductive 3MLCT-excited state quenching of the [IrIII–H]+ species, which itself is formed from the robust [IrIII–Cl]+ precursor in photochemical fashion. In our case, the overall HAT reaction is catalytic and occurs to olefin substrates rather than carbon-centered radicals derived from CH2Cl2 solvent. Contrary to previously explored polyoxometallate-based photo-HAT systems,38–43 the iridium hydride explored herein operates under visible-light irradiation.We focused on the photoinduced hydrogenation of relatively simple olefins as a test ground to explore the reactivity patterns of photo-HAT, but prior studies on thermal (i.e., light-independent) reactions established that metal-catalyzed HAT has numerous applications in organic synthetic chemistry.68 Compared to thermal HAT, the photo-HAT with [Cp*Ir(phen)(H)]+ has the advantage that even lower M–H BDFEs (ca. 44 kcal mol−1 for the reductive quenching product [Cp*Ir(phen)(H)]0) are reachable, which stands in contrast to the traditional thermal manner, where most M–H BDFEs are above 50 kcal mol−1.70 This gain in driving force of around ΔΔG ≈ 6 kcal mol−1 can in principle translate to an approximately 104 times faster reaction rate. Very low M–H BDFEs are typically unstable to bimolecular H2 evolution,65–67 but in the presence of 50 mM olefin substrate our hydrogenation reactions proceeded well and exhibited the typical reactivity patterns of HAT.
In our reactions, even aliphatic substrates yielding highly reactive radical intermediates were activated successfully. The investigated olefin substrates have strongly negative reduction potentials (<−2.7 V vs. SCE)31 and therefore cannot be activated easily by the traditional path of photoinduced electron transfer.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Swiss National Science Foundation through the NCCR Molecular Systems Engineering. Felix Glaser is thanked for skillful experimental support.References
- D. M. Arias-Rotondo and J. K. McCusker, Chem. Soc. Rev., 2016, 45, 5803–5820 RSC.
- K. S. Kjær, N. Kaul, O. Prakash, P. Chábera, N. W. Rosemann, A. Honarfar, O. Gordivska, L. A. Fredin, K. E. Bergquist, L. Häggström, T. Ericsson, L. Lindh, A. Yartsev, S. Styring, P. Huang, J. Uhlig, J. Bendix, D. Strand, V. Sundström, P. Persson, R. Lomoth and K. Wärnmark, Science, 2019, 363, 249–253 CrossRef.
- J. D. Braun, I. B. Lozada, C. Kolodziej, C. Burda, K. M. E. Newman, J. van Lierop, R. L. Davis and D. E. Herbert, Nat. Chem., 2019, 11, 1144–1150 CrossRef CAS.
- R. Hamze, J. L. Peltier, D. Sylvinson, M. Jung, J. Cardenas, R. Haiges, M. Soleilhavoup, R. Jazzar, P. I. Djurovich, G. Bertrand and M. E. Thompson, Science, 2019, 363, 601–606 CrossRef CAS.
- C. Förster and K. Heinze, Chem. Soc. Rev., 2020, 40, 1057–1070 RSC.
- O. S. Wenger, J. Am. Chem. Soc., 2018, 140, 13522–13533 CrossRef CAS.
- J. C. Theriot, C. H. Lim, H. Yang, M. D. Ryan, C. B. Musgrave and G. M. Miyake, Science, 2016, 352, 1082–1086 CrossRef CAS.
- L. Marzo, S. K. Pagire, O. Reiser and B. König, Angew. Chem., Int. Ed., 2018, 57, 10034–10072 CrossRef CAS.
- L. Zhang and L. Jiao, J. Am. Chem. Soc., 2019, 141, 9124–9128 CrossRef.
- H. Yin, Y. Jin, J. E. Hertzog, K. C. Mullane, P. J. Carroll, B. C. Manor, J. M. Anna and E. J. Schelter, J. Am. Chem. Soc., 2016, 138, 16266–16273 CrossRef CAS.
- J. J. Devery, J. D. Nguyen, C. Dai and C. R. J. Stephenson, ACS Catal., 2016, 6, 5962–5967 CrossRef CAS.
- P. Herr, F. Glaser, L. A. Büldt, C. B. Larsen and O. S. Wenger, J. Am. Chem. Soc., 2019, 141, 14394–14402 CrossRef CAS.
- J. H. Shon, S. Sittel and T. S. Teets, ACS Catal., 2019, 9, 8646–8658 CrossRef CAS.
- T. U. Connell, C. L. Fraser, M. L. Czyz, Z. M. Smith, D. J. Hayne, E. H. Doeven, J. Agugiaro, D. J. D. Wilson, J. L. Adcock, A. D. Scully, D. E. Gómez, N. W. Barnett, A. Polyzos and P. S. Francis, J. Am. Chem. Soc., 2019, 141, 17646–17658 CrossRef CAS.
- F. Glaser, C. Kerzig and O. S. Wenger, Angew. Chem., Int. Ed., 2020, 59, 10266–10284 CrossRef CAS.
- M. Giedyk, R. Narobe, S. Weiß, D. Touraud, W. Kunz and B. König, Nat. Catal., 2020, 3, 40–47 CrossRef CAS.
- M. Neumeier, D. Sampedro, M. Májek, V. a. de la Peña O'Shea, A. Jacobi von Wangelin and R. Pérez-Ruiz, Chem.–Eur. J., 2018, 24, 105–108 CrossRef CAS.
- J. P. Cole, D.-F. Chen, M. Kudisch, R. M. Pearson, C.-H. Lim and G. M. Miyake, J. Am. Chem. Soc., 2020 DOI:10.1021/jacs.0c05899.
- H. Kim, H. Kim, T. H. Lambert and S. Lin, J. Am. Chem. Soc., 2020, 2087–2092 CrossRef CAS.
- N. G. W. Cowper, C. P. Chernowsky, O. P. Williams and Z. K. Wickens, J. Am. Chem. Soc., 2020, 142, 2093–2099 CrossRef CAS.
- J. P. Barham and B. König, Angew. Chem., Int. Ed., 2019, 59, 2–18 Search PubMed.
- J. W. Darcy, B. Koronkiewicz, G. A. Parada and J. M. Mayer, Acc. Chem. Res., 2018, 51, 2391–2399 CrossRef CAS.
- E. C. Gentry and R. R. Knowles, Acc. Chem. Res., 2016, 49, 1546–1556 CrossRef CAS.
- M. Kuss-Petermann and O. S. Wenger, Chem.–Eur. J., 2017, 23, 10808–10814 CrossRef CAS.
- G. E. M. Crisenza, D. Mazzarella and P. Melchiorre, J. Am. Chem. Soc., 2020, 142, 5461–5476 CrossRef CAS.
- T. P. Yoon, Acc. Chem. Res., 2016, 49, 2307–2315 CrossRef CAS.
- R. Brimioulle and T. Bach, Science, 2013, 342, 840–843 CrossRef CAS.
- C. Pac, M. Ihama, M. Yasuda, Y. Miyauchi and H. Sakurai, J. Am. Chem. Soc., 1981, 103, 6495–6497 CrossRef CAS.
- R. Naumann, F. Lehmann and M. Goez, Chem.–Eur. J., 2018, 24, 13259–13269 CrossRef CAS.
- C. Kerzig, X. Guo and O. S. Wenger, J. Am. Chem. Soc., 2019, 141, 2122–2127 CrossRef CAS.
- H. Senboku, H. Komatsu, Y. Fujimura and M. Tokuda, Synlett, 2001, 418–420 CrossRef CAS.
- Y. Y. Loh, K. Nagao, A. J. Hoover, D. Hesk, N. R. Rivera, S. L. Colletti, I. W. Davies and D. W. C. MacMillan, Science, 2017, 1187, 1182–1187 CrossRef.
- L. Capaldo and D. Ravelli, Eur. J. Org. Chem., 2017, 2056–2071 CrossRef CAS.
- M. H. Shaw, V. W. Shurtleff, J. A. Terrett, J. D. Cuthbertson and D. W. C. MacMillan, Science, 2016, 352, 1304–1308 CrossRef CAS.
- J. Twilton, M. Christensen, D. A. DiRocco, R. T. Ruck, I. W. Davies and D. W. C. MacMillan, Angew. Chem., Int. Ed., 2018, 57, 5369–5373 CrossRef CAS.
- X. Q. Hu, J. R. Chen and W. J. Xiao, Angew. Chem., Int. Ed., 2017, 56, 1960–1962 CrossRef CAS.
- X. Guo and O. S. Wenger, Angew. Chem., Int. Ed., 2018, 57, 2469–2473 CrossRef CAS.
- S. Protti, M. Fagnoni and D. Ravelli, ChemCatChem, 2015, 7, 1516–1523 CrossRef CAS.
- V. De Waele, O. Poizat, M. Fagnoni, A. Bagno and D. Ravelli, ACS Catal., 2016, 6, 7174–7182 CrossRef.
- P. J. Sarver, V. Bacauanu, D. M. Schultz, D. A. Dirocco, Y. Lam, E. C. Sherer and D. W. C. MacMillan, Nat. Chem., 2020, 12, 459–467 CrossRef CAS.
- J. J. Zhong, W. P. To, Y. Liu, W. Lu and C. M. Che, Chem. Sci., 2019, 10, 4883–4889 RSC.
- D. M. Schultz, F. Lévesque, D. A. DiRocco, M. Reibarkh, Y. Ji, L. A. Joyce, J. F. Dropinski, H. Sheng, B. D. Sherry and I. W. Davies, Angew. Chem., Int. Ed., 2017, 56, 15274–15278 CrossRef CAS.
- G. Laudadio, Y. Deng, K. Van Der Wal, D. Ravelli, M. Nuño, M. Fagnoni, D. Guthrie, Y. Sun and T. Noël, Science, 2020, 96, 92–96 CrossRef.
- J. J. Concepcion, M. K. Brennaman, J. R. Deyton, N. V. Lebedeva, M. D. E. Forbes, J. M. Papanikolas and T. J. Meyer, J. Am. Chem. Soc., 2007, 129, 6968–6969 CrossRef CAS.
- J. C. Lennox, D. A. Kurtz, T. Huang and J. L. Dempsey, ACS Energy Lett., 2017, 2, 1246–1256 CrossRef CAS.
- O. S. Wenger, Chem.–Eur. J., 2011, 17, 11692–11702 CrossRef CAS.
- Y. Deng, J. A. Roberts, S.-M. Peng, C. K. Chang and D. G. Nocera, Angew. Chem., Int. Ed., 1997, 2124–2127 CrossRef CAS.
- S. W. M. Crossley, C. Obradors, R. M. Martinez and R. A. Shenvi, Chem. Rev., 2016, 116, 8912–9000 CrossRef CAS.
- J. Hartung, M. E. Pulling, D. M. Smith, D. X. Yang and J. R. Norton, Tetrahedron, 2008, 64, 11822–11830 CrossRef CAS.
- R. N. Perutz and B. Procacci, Chem. Rev., 2016, 116, 8506–8544 CrossRef CAS.
- J. A. Bandy, F. G. N. Cloke, G. Cooper, J. P. Day, R. B. Girling, R. G. Graham, J. C. Green, R. Grinter and R. N. Perutz, J. Am. Chem. Soc., 1988, 110, 5039–5050 CrossRef CAS.
- C. M. Morton, Q. Zhu, H. Ripberger, L. Troian-Gautier, Z. S. D. Toa, R. R. Knowles and E. J. Alexanian, J. Am. Chem. Soc., 2019, 141, 13253–13260 CrossRef CAS.
- M. E. Ener, J. W. Darcy, F. S. Menges and J. M. Mayer, J. Org. Chem., 2020, 85, 7175–7180 CrossRef CAS.
- F. Gloaguen and T. B. Rauchfuss, Chem. Soc. Rev., 2009, 38, 100–108 RSC.
- Y. A. Small, D. L. Dubois, E. Fujita and J. T. Muckerman, Energy Environ. Sci., 2011, 4, 3008–3020 RSC.
- M. Bourrez, R. Steinmetz, S. Ott, F. Gloaguen and L. Hammarström, Nat. Chem., 2015, 7, 140–145 CrossRef CAS.
- T. Liu, M. Guo, A. Orthaber, R. Lomoth, M. Lundberg, S. Ott and L. Hammarström, Nat. Chem., 2018, 10, 881–887 CrossRef CAS.
- T. Huang, E. S. Rountree, A. P. Traywick, M. Bayoumi and J. L. Dempsey, J. Am. Chem. Soc., 2018, 140, 14655–14669 CrossRef CAS.
- D. Sandrini, M. Maestri and R. Ziessel, Inorg. Chim. Acta, 1989, 163, 177–180 CrossRef CAS.
- T. Suenobu, D. M. Guldi, S. Ogo and S. Fukuzumi, Angew. Chem., Int. Ed., 2003, 42, 5492–5495 CrossRef CAS.
- S. M. Barrett, C. L. Pitman, A. G. Walden and A. J. M. Miller, J. Am. Chem. Soc., 2014, 136, 14718–14721 CrossRef CAS.
- K. R. Brereton, C. N. Jadrich, B. M. Stratakes and A. J. M. Miller, Organometallics, 2019, 38, 3104–3110 CrossRef CAS.
- C. L. Pitman, K. R. Brereton and A. J. M. Miller, J. Am. Chem. Soc., 2016, 138, 2252–2260 CrossRef CAS.
- S. M. Barrett, B. M. Stratakes, M. B. Chambers, D. A. Kurtz, C. L. Pitman, J. L. Dempsey and A. J. M. Miller, Chem. Sci., 2020, 11, 6442–6449 RSC.
- R. Ziessel, J. Am. Chem. Soc., 1993, 115, 118–127 CrossRef CAS.
- C. L. Pitman and A. J. M. Miller, ACS Catal., 2014, 4, 2727–2733 CrossRef CAS.
- M. B. Chambers, D. A. Kurtz, C. L. Pitman, M. K. Brennaman and A. J. M. Miller, J. Am. Chem. Soc., 2016, 138, 13509–13512 CrossRef CAS.
- S. A. Green, S. W. M. Crossley, J. L. M. Matos, S. Vásquez-Céspedes, S. L. Shevick and R. A. Shenvi, Acc. Chem. Res., 2018, 51, 2628–2640 CrossRef CAS.
- S. W. M. Crossley, F. Barabé and R. A. Shenvi, J. Am. Chem. Soc., 2014, 136, 16788–16791 CrossRef CAS.
- J. Choi, M. E. Pulling, D. M. Smith and J. R. Norton, J. Am. Chem. Soc., 2008, 130, 4250–4252 CrossRef CAS.
- R. G. Pearson, Chem. Rev., 1985, 85, 41–49 CrossRef CAS.
- G. Li, J. L. Kuo, A. Han, J. M. Abuyuan, L. C. Young, J. R. Norton and J. H. Palmer, J. Am. Chem. Soc., 2016, 138, 7698–7704 CrossRef CAS.
- Y. Nakano, M. J. Black, A. J. Meichan, B. A. Sandoval, M. M. Chung, K. F. Biegasiewicz, T. Zhu and T. K. Hyster, Angew. Chem., Int. Ed., 2020, 59, 10484–10488 CrossRef CAS.
- X. Guo, Y. Okamoto, M. R. Schreier, T. R. Ward and O. S. Wenger, Chem. Sci., 2018, 9, 5052–5056 RSC.
- Z. C. Litman, Y. Wang, H. Zhao and J. F. Hartwig, Nature, 2018, 560, 355–359 CrossRef CAS.
- K. Lauder, A. Toscani, Y. Qi, J. Lim, S. J. Charnock, K. Korah and D. Castagnolo, Angew. Chem., Int. Ed., 2018, 57, 5803–5807 CrossRef CAS.
- T. J. Whittemore, C. Xue, J. Huang, J. C. Gallucci and C. Turro, Nat. Chem., 2020, 12, 180–185 CrossRef CAS.
- J. Choi, L. Tang and J. R. Norton, J. Am. Chem. Soc., 2007, 129, 234–240 CrossRef CAS.
- C. Kerzig and O. S. Wenger, Chem. Sci., 2019, 2, 11023–11029 RSC.
- A. U. Meyer, T. Slanina, A. Heckel and B. König, Chem.–Eur. J., 2017, 23, 7900–7904 CrossRef CAS.
- R. Matsubara, T. Yabuta, U. Md Idros, M. Hayashi, F. Ema, Y. Kobori and K. Sakata, J. Org. Chem., 2018, 83, 9381–9390 CrossRef CAS.
- L. Yu-Ran, Comprehensive Handbook of Chemical Bond Energies, Taylor & Francis Group, Boca Raton, 2007 Search PubMed.
- D. J. Goebbert and P. G. Wenthold, Int. J. Mass Spectrom., 2006, 257, 1–11 CrossRef CAS.
- J. Hioe and H. Zipse, in Encyclopedia of Radicals in Chemistry, Biology and Materials, John Wiley & Sons, Ltd, 2012 Search PubMed.
- M. Ladwig and W. Kaim, J. Organomet. Chem., 1992, 439, 79–90 CrossRef CAS.
- T. Ghosh, T. Slanina and B. König, Chem. Sci., 2015, 6, 2027–2034 RSC.
- K. T. Oppelt, E. Wöβ, M. Stiftinger, W. Schöfberger, W. Buchberger and G. Knör, Inorg. Chem., 2013, 52, 11910–11922 CrossRef CAS.
- L. Zedler, A. K. Mengele, K. M. Ziems, Y. Zhang, M. Wächtler, S. Gräfe, T. Pascher, S. Rau, S. Kupfer and B. Dietzek, Angew. Chem., Int. Ed., 2019, 58, 13140–13148 CrossRef CAS.
- P. J. DeLaive, T. K. Foreman, D. G. Whitten and C. Giannotti, J. Am. Chem. Soc., 1980, 102, 5627–5631 CrossRef CAS.
- R. Hollis, L. Hughes, V. W. Bowry and K. U. Ingold, J. Org. Chem., 1992, 57, 4284–4287 CrossRef CAS.
- J. Masnovi, E. G. Samsel and R. M. Bullock, J. Chem. Soc., Chem. Commun., 1989, 1044–1045 RSC.
- Y. Pellegrin and F. Odobel, C. R. Chim., 2017, 20, 283–295 CrossRef CAS.
- J. A. M. Simões and J. L. Beauchamp, Chem. Rev., 1990, 90, 629–688 CrossRef.
- J. C. Deaton, C. M. Taliaferro, C. L. Pitman, R. Czerwieniec, E. Jakubikova, A. J. M. Miller and F. N. Castellano, Inorg. Chem., 2018, 57, 15445–15461 CrossRef CAS.
- C. L. Pitman and A. J. M. Miller, Organometallics, 2017, 36, 1906–1914 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc01820a |
| ‡ Current address: Center of Basic Molecular Science (CBMS), Department of Chemistry, Tsinghua University, Beijing 100084, China. |
| This journal is © The Royal Society of Chemistry 2020 |