
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNovel light-emitting clays with structural Tb3+ and Eu3+ for chromate anion detection†
Stefano Marchesi a,
Chiara Bisio*ab and
Fabio Carniato*a
a,
Chiara Bisio*ab and
Fabio Carniato*a
aDipartimento di Scienze e Innovazione Tecnologica, Università degli Studi del Piemonte Orientale “Amedeo Avogadro”, Viale Teresa Michel 11, 15121-Alessandria, Italy. E-mail: fabio.carniato@uniupo.it
bCNR-SCITEC, Istituto di Scienze e Tecnologie Chimiche “G. Natta”, Via C. Golgi 19, 20133-Milano, Italy
First published on 19th August 2020
Abstract
Tb3+ and Eu3+ ions were encapsulated for the first time in the inorganic layers of a synthetic saponite clay following a one-pot synthetic approach. The co-presence of the two metal ions led to tuneable light-emitting properties, promoted by an efficient Tb3+ → Eu3+ energy transfer and enhanced Stokes shift character. To our knowledge, the so-prepared luminescent material was tested for the first time as an optical sensor for the detection of chromate anions in water.
Over the past years, synthetic smectite clays, and in particular saponite, have attracted great interest in the scientific community, due to their peculiar physico-chemical properties, such as controlled and tuneable chemical composition and specific surface area, excellent chemical versatility, high thermal stability and robustness, adjustable surface acidity and relatively simple and low cost synthesis.1–4
Saponite materials have been used in several applications, spanning from polymer science to heterogeneous catalysis.5–9 Examples of their applications in the agricultural and construction fields, for environmental purposes (i.e. recovering of heavy metals from soils and waters) and in optical/optoelectronic devices are also reported.10–15
Recently, a flourishing research branch on interesting layered and porous materials (including silicas, silicates, clays, MOFs…)16,17 has been related to their combination with rare-earth ions. Lanthanides represent key elements for many technologies used in our society (i.e. in energy devices, for biomedical uses…) owing to their unique electronic, magnetic, optical and catalytic properties.18–20 Preliminary studies on the preparation of lanthanide-containing layered and/or porous materials with specific functionalities have been made, with newfound applications as sensors for targets of interests (i.e. biothiols, surfactants, fingerprint detection…), as luminescent thermometers,21 or in bio-imaging fields (i.e. as oral MRI contrast agents).22–30 In clay materials, for example, the lanthanides (i.e. Eu3+, Tb3+, Gd3+…) were normally incorporated as complexes or free ions into the clay interlayer space by using post-synthesis intercalation procedures.24,30 However, these synthetic protocols are quite expensive and require long preparation times. Moreover, depending on the complexes' stability, metal leaching processes can occur, thus leading to a potential environmental contamination and to toxic effects in different organisms. These disadvantages could be overcome by design synthetic strategies based on the direct insertion of the lanthanide ions in the clay synthesis gel, as previously performed with vanadium and niobium metals.5,31,32
Based on these considerations, in this study, two luminescent lanthanides, Tb3+ and Eu3+, were simultaneously incorporated in the structure of a synthetic saponite clay (hereafter named Na-TbEuSAP), using a single-step hydrothermal approach. A solid with only Tb3+ ions was also prepared as a reference (hereafter named Na-TbSAP). A multi-technique characterization approach was employed to investigate the physico-chemical properties of the solids. These novel light-emitting saponites were also tested as potential optical sensors for inorganic anions in water. In this respect, it was proved that the lanthanide complexes, when incorporated in different hybrid solids or gels (i.e. metal–organic frameworks, polymers…), can be employed as efficient sensors for different kinds of substrates (ions and organic molecules) by exploiting the so-called “antenna effect” phenomena.33,34 To the best of our knowledge, the lanthanide-based clays studied in the literature has never been investigated as sensors for the detection of anionic species.
Among the anions, the chromate (CrO42−), a common source of the hexavalent chromium (Cr(VI)) which is regarded as the most toxic and dangerous form of this element, was selected for the sensor tests.35–38 The CrO42− is an environmentally-hazardous inorganic pollutant commonly present in soils and waters, and it has been found in several public drinking waters worldwide as well near particular industrial plants.35–38 The amount of chromium in water and soil is strictly regulated by international and national policies: i.e. the limit in drinking waters for EPA (United Sates Environmental Protection Agency) is of 100 ppb, while in Italy is even lower (50 ppb).39,40 Thus, the development of efficient sensors for Cr(VI) compounds, especially in water media, is therefore of great importance in terms of public health and safety.
In detail, Na-TbEuSAP and Na-TbSAP samples were prepared by a modified one-pot hydrothermal synthesis protocol, followed by sodium-exchange process (Scheme S1 in the ESI†).5,31,32 Eu3+ and Tb3+ were introduced in the form of chloride salts during the synthesis gel along with the silicon, aluminium and magnesium precursors. The final materials were then submitted to ionic exchange procedure to replace the different cations located in the saponite gallery (i.e. Al3+, Mg2+, H3O+) with Na+ ions.
The lanthanides loading in the final materials was determined by inductively coupled plasma mass spectrometry (ICP-MS). The Tb3+ amount was found to be 0.021 and 0.031 mmol g−1 for TbSAP and TbEuSAP samples, respectively. For both solids, the Na+-exchange procedure did not alter the amount of Tb3+ species. For TbEuSAP sample, the Eu3+ content resulted to be 0.023 mmol g−1, without modification after the ion-exchange procedure. The differences in the Tb3+ concentrations observed in the two materials could be ascribed to a certain variability in the chemical composition of the samples, as testified by the different cationic exchange capacity of TbSAP and TbEuSAP (see below). In fact, numerous parameters (i.e. the acidity of the gel during the condensation phase, the amount of water, etc…) including the co-presence of two different metal ions in competition with similar structural sites can positively or negatively affect the final loading of the metal ions. In general, however, the results suggest that both metals are mainly located in the framework of the saponite clay, as schematically proposed in Fig. 1B.
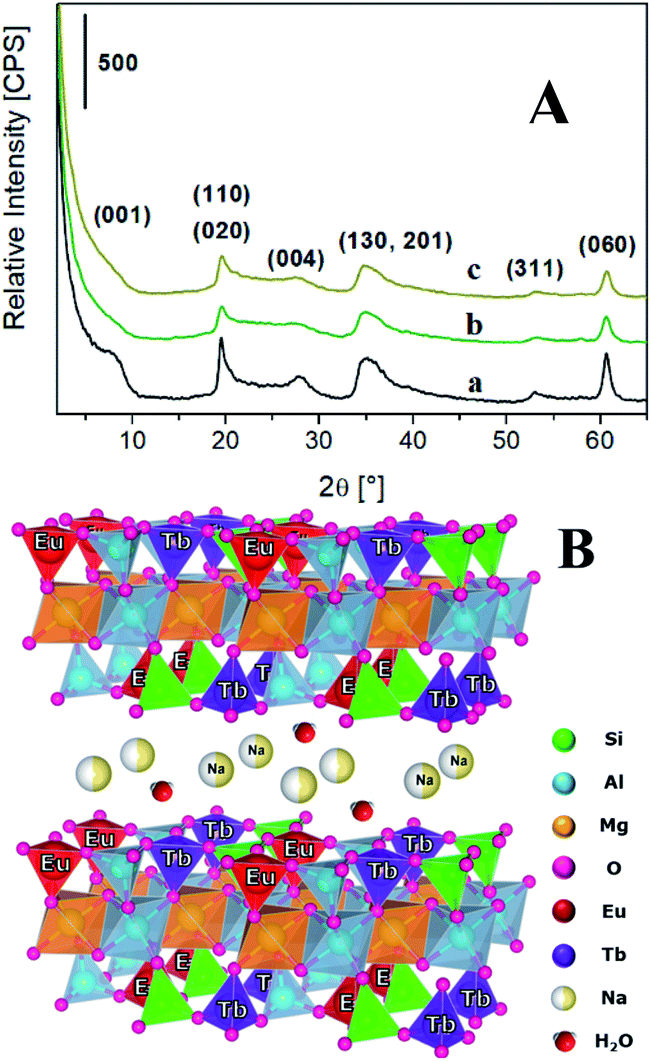 | ||
| Fig. 1 (A) X-ray diffraction pattern of Na-SAP (a), Na-TbSAP (b) and Na-TbEuSAP (c) samples. (B) Schematic representation of the Na-TbEuSAP clay. | ||
The inclusion of the lanthanides ions during the synthesis did not alter the layered structure of the saponite,1–7,12,22,23 as indicated by the X-ray powder diffraction (XRPD) analyses (Fig. 1A). In particular, the X-ray patterns of Na-TbSAP and Na-TbEuSAP showed the typical reflections of the saponite clay. Nevertheless, the signal associated to the basal plane (001) in both samples, with d-spacing value in the 7.4–8.0° 2θ range, is less defined respect to the parent Na-SAP sample, thus indicating a greater structural disorder of the lamellae packing.1
The cation-exchange capacity (CEC) of the clays, evaluated by UV-Visible spectroscopy analysis30,31 (Fig. S1 in the ESI†), was estimated to be 41.6 ± 4.8 meq./100 g and 61.2 ± 5.2 meq./100 g for Na-TbSAP and Na-TbEuSAP, respectively. These values are in line with the results obtained for other metal-containing saponites.5,31,32
The solids showed high thermal stability, as indicated by the thermogravimetric analyses (TGA). The TGA (Fig. S2A in the ESI†) and the derivative curves (DTG) (Fig. S2B in the ESI†) of Na-TbSAP (curve a) and Na-TbEuSAP (curve b) show a first weight loss around 100 °C related to the evaporation of physisorbed water, followed by a gradual release of the interlamellar water (with an initial dehydroxylation of the layered structure) in the 150–750 °C range. A third weight loss around 825 °C is correlated to the collapse of the saponite framework, with formation of the relative metal oxides.1–4
The photophysical properties of the luminescent saponites were thoroughly investigated by photoluminescence (PL) spectroscopy. The excitation spectra at solid state of Na-TbSAP and Na-TbEuSAP (Fig. 2A), collected at the most intense emission line of Tb3+ at 545 nm, showed a common broad band with a maximum at 270 nm associated to the 7F6-5d electronic transition of Tb3+.41–43 Both spectra are also composed by another characteristic peak of Tb3+, associated to its intra-4f8 electronic transitions (7F6–5DJ, J = 2,3).41–43 When monitored at the most intense emission line of Eu3+ at 615 nm (Fig. 2Ac), the spectrum of Na-TbEuSAP presented the same bands, previously observed, along with additional peaks ascribed to the intra-4f6 transitions of Eu3+ (7F0,1–5HJ, 5DJ, 5LJ and 5GJ), with a λmax at 395 nm (7F0–5L6).44–47 The presence of the Tb3+ transitions in the spectrum of Na-TbEuSAP analysed at the emission line of Eu3+ is a clear indication of the occurrence of a Tb3+ → Eu3+ metal-to-metal energy transfer (MMET) process.48–51 This hypothesis was confirmed by analysing the emission spectra at the solid state of Na-TbEuSAP under irradiation at λmax of Eu3+ (395 nm) and Tb3+ (270 nm) (Fig. 2B).
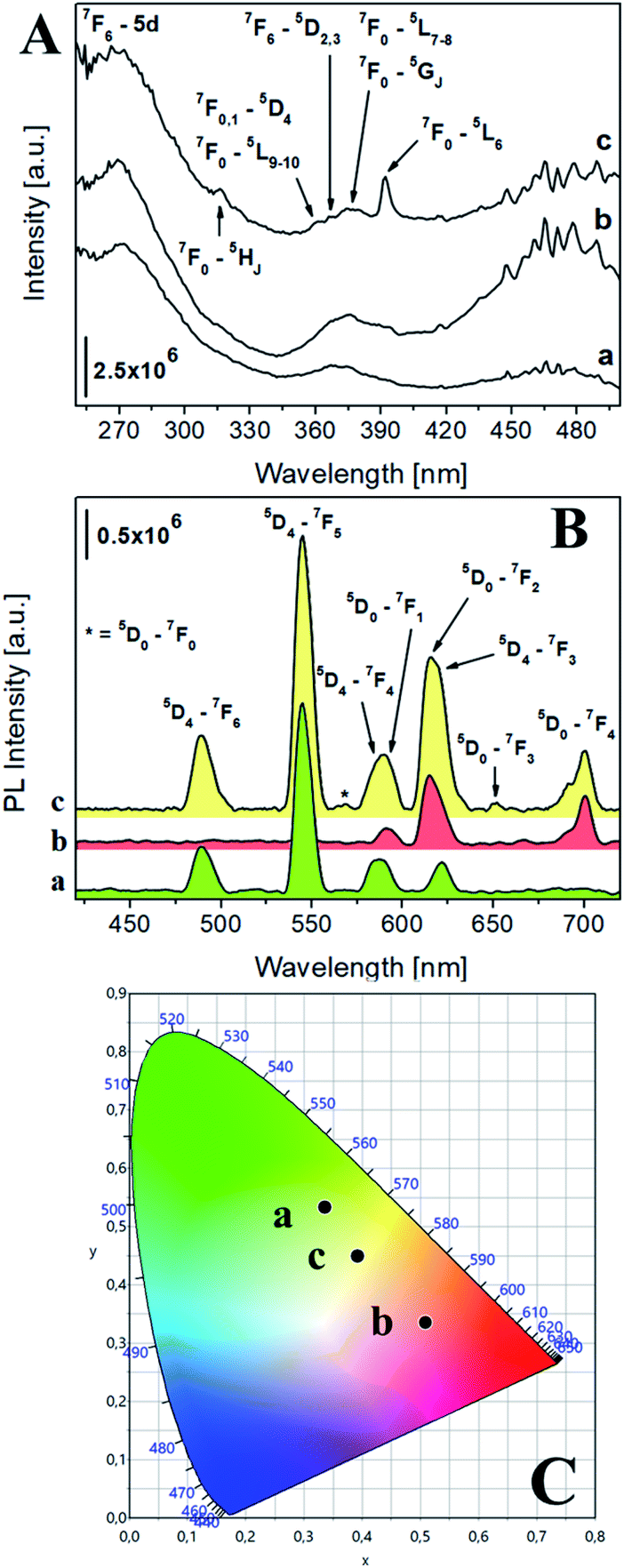 | ||
| Fig. 2 (A) Excitation spectra at solid state of Na-TbSAP (a, λem = 545 nm) and Na-TbEuSAP (b, λem = 545 nm, c, λem = 615 nm). (B) Emission spectra at solid state of Na-TbSAP (a, λexc = 270 nm) and Na-TbEuSAP (b, λexc = 395 nm, c, λexc = 270 nm); the intensity of Na-TbSAP spectrum was normalized (divided by five) for a better comparison. (C) CIE 1931 xy chromaticity diagrams derived from emission spectra in frame B. The colour of each emission spectrum in frame B is associated to its corresponding xy coordinates (frame C and Table S2†). | ||
Indeed, the emission spectrum collected at 270 nm showed emission peaks of both Tb3+ (5D4–7FJ, J = 6–3)41–43 and Eu3+ (5D0–7FJ, J = 0–4).45–49 The Tb3+ → Eu3+ energy transfer photo-enhanced the luminescence of europium, as indicated by the increased intensity of the 5D0–7F2 emission band of Eu3+ at 615 nm (compared to the same band obtained after direct excitation at 395 nm) of ca. 90%. This also suggests that Tb3+ and Eu3+ are probably located in close proximity, thus ensuring an efficient optical communication between them.48–51
Additional information on the local chemical environment surrounding the lanthanides were extrapolated from the collected emission spectra. An heterogeneous distribution of the Eu3+ sites in the Na-TbEuSAP is derived by the presence of two weak peaks for the 5D0–7F0 transition at ca. 560 nm (Fig. 2Bc).52 The multiplicity of this band is commonly associated with the number of distinct chemical domains around the europium, since both its initial and final states are non-degenerated.52 Moreover, the Eu3+ and Tb3+ centres in both clays are placed in a low symmetry environment, as suggested by the high values of their asymmetry factor (R > 2) (Table S1 in the ESI†). The R factor was calculated from the intensity ratio of electric-dipole/ED 5D0 → 7F2 on magnetic-dipole/MD 5D0 → 7F1 for Eu3+,44–47,53,54 and of ED 5D4 → 7F5/MD 5D4 → 7F6 for Tb3+.55
PL spectra allowed to extrapolate the colorimetric features of the samples, calculating their chromaticity coordinates (xy) and related parameters (RGB and Hex) according to CIE 1931 colour spaces (Fig. 2C and Table S2 in the ESI†).56 A modulation of the colour emission was observed for the bi-functional saponite. Indeed, while the direct excitation of Eu3+ at 395 nm in Na-TbEuSAP led to an emission in the visible red region, the sample after excitation at 270 nm showed a blue shift towards a final yellow emission. As a comparison, the emission colour of Na-TbSAP is green.
The parameters that describe the Tb3+–Eu3+ energy transfer process were derived by considering the experimental lifetimes of donor (D, Na-TbSAP) and donor–acceptor (DA, Na-TbEuSAP) systems, measured by time-resolved fluorescence spectroscopy.48–51,57–59 The intensity decay curves of 5D4 excited state of Tb3+ at λem = 545 nm (5D4–7F6) were collected at solid state, under excitation at 370 nm (Fig. S3 in the ESI†). The curves were fitted with a bi-exponential function, from which the average τ values were calculated. The energy transfer rate (kEnT) and efficiency (EEnT) parameters were calculated from eqn (1) and (2), using the lifetimes of the donor (Tb3+) in the presence (τDA) or absence (τD) of the acceptor (Eu3+), measured at 545 nm:57–59
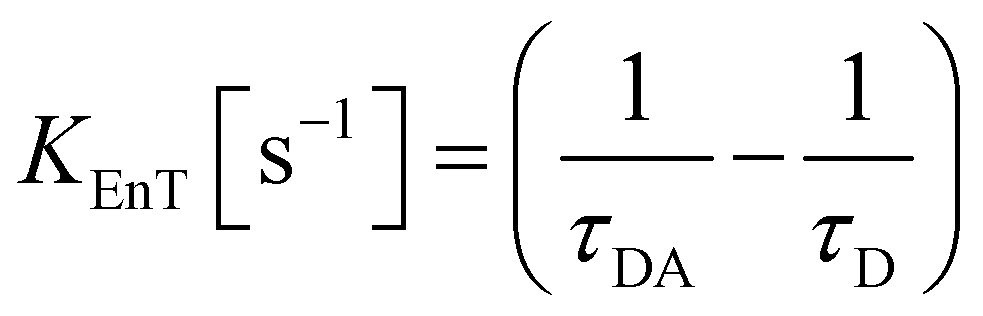 | (1) |
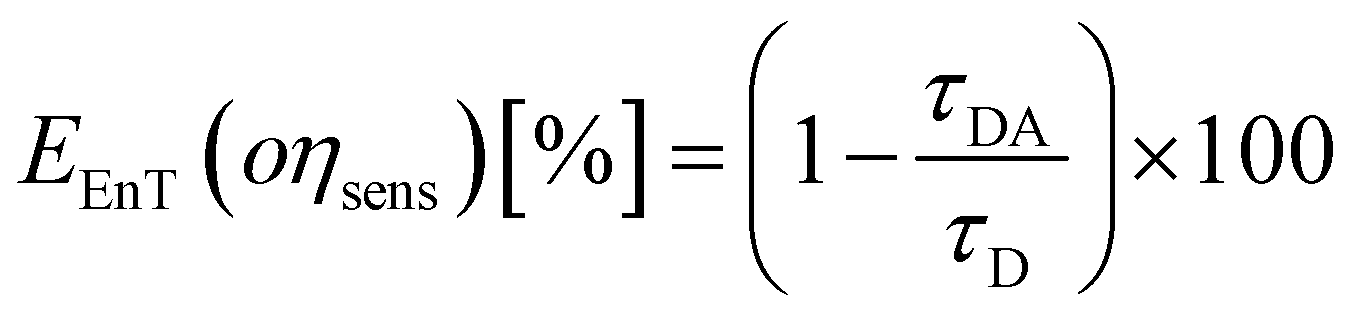 | (2) |
The results, reported in Table 1, showed a high energy transfer efficiency of ca. 81%, with a rate constant of 74![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 100 s−1. Surprisingly, the lifetime decay for Na-TbSAP is very low (5.66 × 10−5 s). A possible explanation is related to the fact that the first coordination sphere of the metal contains coordinated water molecules, adsorbed or intercalated in the solid material, which essentially determine the site lifetimes.
100 s−1. Surprisingly, the lifetime decay for Na-TbSAP is very low (5.66 × 10−5 s). A possible explanation is related to the fact that the first coordination sphere of the metal contains coordinated water molecules, adsorbed or intercalated in the solid material, which essentially determine the site lifetimes.
| τD [s] | τDA [s] | kEnT [s−1] | EEnT [%] |
|---|---|---|---|
| 5.66 × 10−5 | 1.09 × 10−5 | 7.41 × 104 | 80.74 |
Finally, the luminescence at solid state of Na-TbSAP and Na-TbEuSAP proved to be stable even after 1 h of irradiation at 270 nm, as demonstrated in photobleaching tests reported in Fig. S4.†
Considering their interesting photophysical properties, the luminescent saponite samples were preliminarily tested for the optical detection of the chromate anion (CrO42−) in water. For the sensing test, the samples were dispersed in water and then put in contact with different concentrations of CrO42−, from 0.0001 to 1 mM. Subsequently, for each experimental point, an emission spectrum was collected. The PL spectra were obtained at the two main λexc of Tb3+ and Eu3+ (270 and 395 nm, respectively), monitoring their emission bands at 545 and 615 nm (see in the ESI† for an accurate description of the procedure). The analyses were performed on the aqueous suspensions of Na-TbSAP and Na-TbEuSAP samples under continuous stirring, at room temperature. Prior the sensing tests, the suspensions stability of the saponites dispersed in water were carefully assessed by dynamic light scattering (DLS) analysis, performed at 25 °C (Fig. S5 in the ESI†). The suspensions remained qualitatively homogeneous and stable, with a hydrodynamic diameter of the particles of 16 and 35 nm (polydispersity index (PDI) of 0.71 and 0.65) for Na-TbSAP and Na-TbEuSAP, respectively. Nevertheless, the PDI index calculated for these samples must be carefully considered for two reasons: (i) the light emission properties of these samples can interfere with the laser source used for the analysis; (ii) the samples are composed by lamellae with different aspect ratio. Such particles morphology can influence the quality of the data analysis.
The emission spectra of the aqueous suspensions of Na-TbSAP and Na-TbEuSAP treated with progressive CrO42− concentrations are reported in Fig. S6 in the ESI.† The suspensions of the samples analysed were stirred continuously at room temperature during the sensing tests, to maintain a good stability over time. First, the spectra collected before the addition of CrO42− anions (black curves (a) in Fig. S6†) showed the typical emission peaks of Tb3+ and Eu3+,41–47 as observed in the solid-state analyses reported in Fig. 2B. The spectra of pristine samples in water showed no intensity reduction over time, thus confirming the high suspensions stability in the experimental conditions adopted, without any effect of particles aggregation or deposition on the final optical properties.
Afterwards, the samples were put in contact with the CrO42− anion. In the presence of increasing amount of CrO42− (from red (b) to blue (c) curves in Fig. S6†), the intensity of the peaks at 545 and 615 nm decreased for both solids, thus suggesting that both metals are probably involved in the interaction with the anion.
The evolution of the normalized PL intensity of the band at 545 nm (λexc = 270 nm) as a function of the CrO42− concentration is reported in Fig. 3A. For both Na-TbSAP and Na-TbEuSAP samples, an exponential decay of the intensity of the band at 545 nm is observed. In particular, a marked photoluminescence decrease after contacting the samples with a CrO42− concentration of 1 μM (= 52 ppb of Cr(VI), lower than the EPA value of 100 ppb) is visible.35–38 Indeed, the fluorescence is almost quenched for both samples with a CrO42− concentration of 500 μM. In these experimental conditions, the Na-TbEuSAP displayed a PL intensity attenuation at 1 μM of 63% compared to the 15% of Na-TbSAP, implying a more rapid CrO42− detection response which could be ascribed to differences in the chemical composition of the samples (i.e. loadings and localization of the metal sites,…).
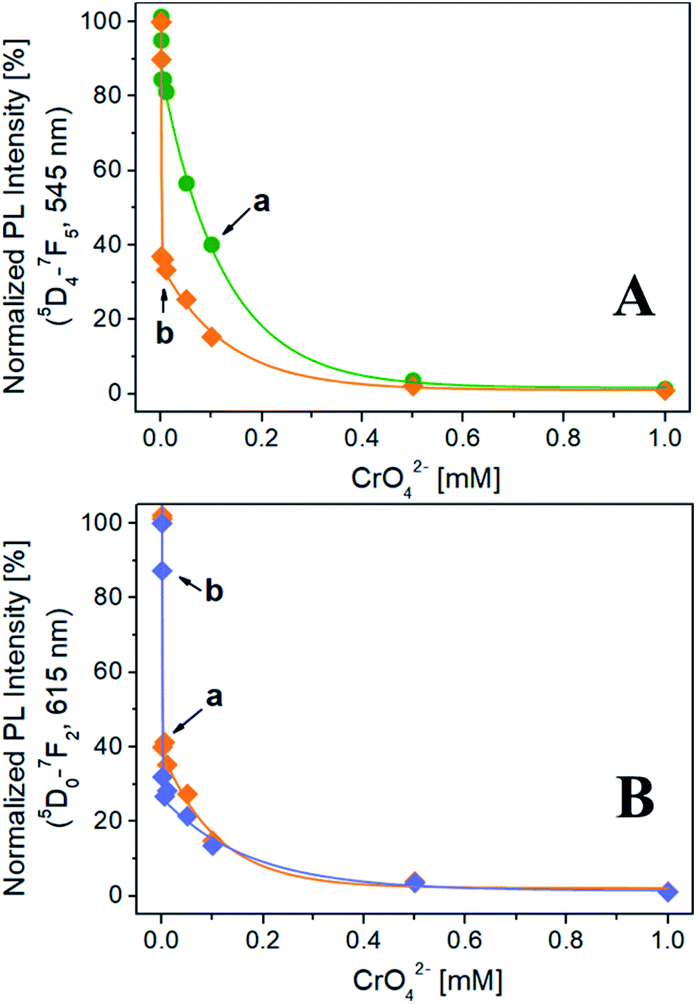 | ||
| Fig. 3 (A) Normalized PL intensity of the band at 545 nm in the presence of different CrO42− concentrations for Na-TbSAP (a) and Na-TbEuSAP (b). (B) Normalized PL intensity of the 615 nm band as a function of the different CrO42− concentrations for Na-TbEuSAP at λexc = 270 (a) and 395 nm (b). | ||
Similar results have been obtained by monitoring the normalized PL intensity of the band at 615 nm of Na-TbEuSAP, under excitation at 270 nm and 395 nm (Fig. 3B). The peak intensity quickly decreases with a similar trend under both excitations. Under direct excitation at 395 nm, however, a slightly more rapid decline of the PL intensity was detected.
These preliminary tests demonstrated high sensing capabilities of Tb3+/Eu3+-saponite clays, with a rapid recognition of CrO42− in water at low concentrations due to a marked quenching process, especially for Na-TbEuSAP sample. The sensing performance also proved to be highly modular: indeed, it is possible to easily monitor two distinct emission bands (545 and 615 nm) under two separate irradiations (270 and 395 nm).
The quenching mechanism is probably governed by diffusional phenomena of the CrO42− anions between the clay particles. As commonly known, the saponite clays are very efficient cationic exchanger solids.1 The diffusion of anions inside the interlayer space of clay is unlikely to happen. For these reasons, our hypothesis is that the interaction between the CrO42− and the Tb3+/Eu3+ sites may occur on the galleries entrance or on the particle's surfaces, between the clay tactoids, in accessible framework tetrahedral sites.60 The interaction metal/substrate could also be favoured by the swelling of the saponite lamellae in water, which results in more exposed lanthanides centres to surrounding CrO42− anions.61
The quenching efficiency was clarified in more detail with the aid of the Stern–Volmer relationship, which describes the kinetics of photophysical deactivation processes between a quencher molecule (Q) and an excited fluorophore (A*).62–65 In our case, Q is the CrO42− and A* is Eu3+ and Tb3+ ions. In general, two quenching processes are usually encountered: (1) static or SQE (formation of a Q–A ‘complex’ in the ground-state) and (2) dynamic or DQE (collisional). SQE and DQE are governed by Stern–Volmer equations (eqn (S1) and (S2)), reported in the ESI.†62–65 By plotting the evolution of F0/F vs. [Q], where F0 and F are the fluorescence intensities of the 545 or 615 nm band without and in the presence of the quencher at different concentrations, it was possible to extrapolate the type of quenching process and calculate the relative quenching constants.62–65 The Stern–Volmer plots reported in Fig. S7 in the ESI† exhibited an upward curvature in all the cases analysed, which is indicative of a quadratic relationship between fluorescence intensity and the anion concentrations, as reported in the literature.63 This non-linear dependence, even at low concentrations, suggest that both static and dynamic quenching are occurring with the same fluorophore, in both clays. Similar behaviour was previously documented in the literature by Lin et al. to explain complex quenching mechanisms occurred for MnO2 nanosheets towards Au nanocluster fluorescence.63 The phenomenon is governed by a modified Stern–Volmer equation (eqn (S3) in the ESI†), from which (KD × KS) and (KD + KS) parameters can be derived.63 The combined quenching dynamic (KD) and static (KS) constants, obtained by fitting the data in Fig. S7† (Table S3 in the ESI†), indicate that the quenching efficiency is dependent on the excitation wavelength, the monitored emission signal and the presence of both lanthanides. In particular, the quenching seems to be more pronounced for Na-TbEuSAP.
The performance demonstrated for both samples in these preliminary tests are extremely encouraging, and are comparable to those of other luminescent probes based on MOFs mostly containing transitions metals and Eu3+,66 Tb3+-complexes,67 coordinated polymers with Zn2+ and Tb3+,68 and carbon quantum dots.69
Conclusions
In this study, for the first time two luminescent lanthanides ions (Tb3+ and Eu3+) were successfully introduced in the framework of synthetic saponite clays through a simple one-pot hydrothermal method. The obtained samples, containing in the structure Tb3+ or a simultaneous combination of Tb3+ and Eu3+ ions, possessed good cation-exchange capacity, layered structure and high thermal stability. The luminescent clay showed interesting photoluminescence features. In particular, the co-presence of both metal centres in the framework gives rise to an efficient Tb3+ → Eu3+ energy transfer, that greatly enhances the europium luminescence. Moreover, the clay demonstrated a high stoke shift and tuneable emission colours. These excellent photophysical properties were exploited for the optical recognition of chromate anion in water, which is a common environmental pollutant and a source of hexavalent chromium. In the tests, all the luminescent solids demonstrated high photoluminescence sensing capabilities, directly exploiting the intrinsic optical properties of the two lanthanides, with a rapid detection response at very low concentrations (at μM level) due to a complex mixed static/dynamic quenching mechanism involved in the process.The simple one-pot synthetic method and the attractive physico-chemical properties of these luminescent clays are extremely encouraging for the future development of new metal-containing clay materials with one or more functionalities. The materials prepared in this study could find application in the rational design of effective fluorescent sensors for targets of interest.
The use of photoluminescence spectroscopy for these purposes is expanding and has some important advantages respect to traditional elemental analyses techniques: (i) the sample does not require specific treatments before analysis, (ii) the measurement is fast and easy to perform, (iii) can be applied to different types of matrices. One of the main disadvantages is related to the poor selectivity for the identification of multiple species. This specific topic along with a more comprehensive overview of the sensing properties of the lanthanide-clays (i.e. detection limits and reusability) are currently under investigation.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are fully grateful to Mr Marco Barbetta for his help in the experiments and to Dr Elena Perin (DiSIT, Università del Piemonte Orientale, Alessandria, Italy) for ICP-MS analyses. Financial support from the Università del Piemonte Orientale (FAR-2019) is also acknowledged.Notes and references
- (a) F. Carniato, G. Gatti and C. Bisio, New J. Chem., 2020, 44, 9969–9980 RSC; (b) G. Paul, C. Bisio, I. Braschi, M. Cossi, G. Gatti, E. Gianotti and L. Marchese, Chem. Soc. Rev., 2018, 47, 5684–5739 RSC.
- D. Costenaro, G. Gatti, F. Carniato, G. Paul, C. Bisio and L. Marchese, Microporous Mesoporous Mater., 2012, 162, 159–167 CrossRef CAS.
- C. Bisio, G. Gatti, E. Boccaleri, G. Superti, H. Pastore and M. Thommes, Microporous Mesoporous Mater., 2008, 107, 90–101 CrossRef CAS.
- (a) C. Bisio, G. Gatti, E. Boccaleri, L. Marchese, L. Bertinetti and S. Coluccia, Langmuir, 2008, 24, 2808–2819 CrossRef CAS PubMed; (b) C. Bisio, F. Carniato, G. Paul, G. Gatti, E. Boccaleri and L. Marchese, Langmuir, 2011, 27, 7250–7257 CrossRef CAS PubMed.
- F. Carniato, C. Bisio, R. Psaro, L. Marchese and M. Guidotti, Angew. Chem., Int. Ed., 2014, 53, 10095–10098 CrossRef CAS PubMed.
- F. Carniato, C. Bisio, G. Gatti, E. Boccaleri, L. Bertinetti, S. Coluccia, O. Monticelli and L. Marchese, Angew. Chem., Int. Ed., 2009, 48, 6059–6061 CrossRef CAS PubMed.
- M. Guidotti, R. Psaro, N. Ravasio, M. Sgobba, F. Carniato, C. Bisio, G. Gatti and L. Marchese, Green Chem., 2009, 11, 1173–1178 RSC.
- G. Mata, R. Trujillano, M. A. Vicente, C. Belver, M. Fernández-García, S. A. Korili and A. Gil, Appl. Catal., A, 2007, 327, 1–12 CrossRef CAS.
- L. A. Utracki, M. Sepehr and E. Boccaleri, Polym. Adv. Technol., 2007, 18, 1–37 CrossRef CAS.
- M. Mokhtar, Materials, 2017, 10, 760–772 CrossRef PubMed.
- M. I. Boyanov, D. E. Latta, M. M. Scherer, E. J. O'Loughlin and K. M. Kemner, Chem. Geol., 2017, 464, 110–117 CrossRef CAS.
- S. Marchesi, F. Carniato, M. Guidotti, M. Botta, L. Marchese and C. Bisio, New J. Chem., 2020, 44, 10033–10041 RSC.
- Y.-C. Lee, T.-H. Lee, H.-K. Han, W. J. Go, J.-W. Yang and H.-J. Shin, Photochem. Photobiol., 2010, 86, 520–527 CrossRef CAS PubMed.
- F. Olivero, F. Carniato, C. Bisio and L. Marchese, J. Mater. Chem., 2012, 22, 25254–25261 RSC.
- X. Chen, Y. Xu, H. Li and B. Liu, Sens. Actuators, B, 2017, 246, 344–351 CrossRef CAS.
- (a) J. Rocha, L. D. Carlos, F. A. A. Paz and D. Ananias, Chem. Soc. Rev., 2011, 40, 926–940 RSC; (b) J. Rocha and L. D. Carlos, Curr. Opin. Solid State Mater. Sci., 2003, 7, 199–205 CrossRef CAS.
- (a) J. Rocha, L. D. Carlos, A. Ferreira, J. P. Rainho, D. Ananias and Z. Lin, Mater. Sci. Forum, 2004, 455–456, 527–531 CAS; (b) D. Ananias, A. Ferreira, J. Rocha, P. Ferreira, J. P. Rainho, C. Morais and L. D. Carlos, J. Am. Chem. Soc., 2011, 123, 5735–5742 CrossRef PubMed.
- (a) V. Balzani, P. Ceroni and A. Juris, Photochemistry and Photophysics: Concepts, Research, Applications, 2014, Wiley-VCH Verlag GmbH & Co, Weinheim, ISBN: 978-3-527-33479-7 Search PubMed; (b) J.-C. G. Bünzli, Coord. Chem. Rev., 2015, 293–294, 19–47 CrossRef.
- (a) A. F. Mingo, S. C. Serra, S. Baroni, C. Cabella, R. Napolitano, I. Hawala, I. M. Carnovale, L. Lattuada, F. Tedoldi and S. Aime, Magn. Reson. Med., 2016, 78, 1523–1532 CrossRef PubMed; (b) D. Parker, Handbook on the Physics and Chemistry of Rare Earths, ed. J.-C. G. Bünzli and V. K. Pecharsky, Elsevier, Amsterdam, 2016, vol. 50, pp. 269–299, ISBN: 978-0-444-63851-9 Search PubMed.
- F. Carniato, L. Tei and M. Botta, Eur. J. Inorg. Chem., 2018, 4936–4954 CrossRef CAS.
- (a) D. Ananias, F. A. A. Paz, L. D. Carlos and J. Rocha, Chem.–Eur. J., 2018, 24, 11926–11935 CrossRef CAS PubMed; (b) J. Rocha, C. D. S. Brites and L. D. Carlos, Chem.–Eur. J., 2016, 22, 14782–14795 CrossRef CAS PubMed.
- S. L. C. Pinho, H. Faneca, C. F. G. C. Geraldes, J. Rocha, L. D. Carlos and M.-H. Delville, Eur. J. Inorg. Chem., 2012, 2828–2837 CrossRef CAS.
- M. Jin, D. E. M. Spillane, C. F. G. C. Geraldes, G. R. Williams and S. W. A. Bligh, Dalton Trans., 2015, 44, 20728–20734 RSC.
- (a) S. Marchesi, F. Carniato, C. Bisio, L. Tei, L. Marchese and M. Botta, Dalton Trans., 2018, 47, 7896–7904 RSC; (b) D. Lalli, S. Marchesi, F. Carniato, C. Bisio, L. Tei, L. Marchese and M. Botta, Dalton Trans., 2020, 49, 6566–6571 RSC.
- (a) D. Talarico de Araujo, K. J. Ciuffi, E. J. Nassar, M. A. Vicente, R. Trujillano, P. S. Calefi, V. Rives and E. H. de Faria, J. Phys. Chem. C, 2017, 121, 5081–5088 CrossRef; (b) H. Li, M. Li, Y. Wang and W. Zhang, Chem.–Eur. J., 2014, 20, 10392–10396 CrossRef CAS PubMed.
- (a) Y. Wang, P. Li, S. Wang and H. Li, J. Rare Earths, 2019, 37, 451–467 CrossRef CAS; (b) S.-J. Ruy, A. Kim, M. D. Kim, S. W. Hong, S. S. Min, J.-H. Lee, J.-K. Lee and H. Jung, Appl. Clay Sci., 2014, 101, 52–59 CrossRef.
- X. Chen, Y. Wang, R. Chai, Y. Xu, H. Li and B. Liu, ACS Appl. Mater. Interfaces, 2017, 9, 13554–13563 CrossRef CAS PubMed.
- D. Yang, Y. Wang, Y. Wang and H. Li, Sens. Actuators, B, 2016, 235, 206–212 CrossRef CAS.
- J. A. Peters and K. Djanashvili, Eur. J. Inorg. Chem., 2012, 1961–1974 CrossRef CAS.
- E. M. Kovacs, E. E. Baradacs, P. Konya, P. Kovacs-Palffy, S. Harangy, E. Kuzmann, J. Konya and N. M. Nagy, Colloids Surf., A, 2017, 522, 287–294 CrossRef CAS.
- L. Ostinelli, S. Recchia, C. Bisio, F. Carniato, M. Guidotti, L. Marchese and R. Psaro, Chem.–Asian J., 2012, 7, 2394–2402 CrossRef CAS PubMed.
- D. Costenaro, C. Bisio, F. Carniato, S. L. Safronyuk, T. V. Kramar, M. V. Taran, M. F. Starodub, A. M. Katsev and M. Guidotti, ChemistrySelect, 2017, 2, 1812–1819 CrossRef CAS.
- (a) F. Liu, W. Gao, P. Li, X.-M. Zhang and J.-P. Liu, J. Solid State Chem., 2017, 253, 202–210 CrossRef CAS; (b) Z. Zhou, Q. Wang and C. Tan, Soft Mater., 2014, 12, 98–102 CrossRef CAS.
- (a) P. D. Beer and P. A. Gale, Angew. Chem., Int. Ed., 2001, 40, 486 CrossRef CAS; (b) C. Suksai and T. Tuntulani, Chem. Soc. Rev., 2003, 32, 192 RSC.
- (a) H. Sun, J. Brocato and M. Costa, Curr. Environ. Health Rep., 2015, 2, 295–303 CrossRef CAS PubMed; (b) H. Oliveira, J. Bot., 2012, 1–8 Search PubMed.
- (a) NASA Principal Center for Regulatory Risk Analyses and Communication, Regulatory Considerations for Chromium, http://www.rracps.org, retrieved on May 2020 Search PubMed; (b) IARC (International Agency for Research on Cancer), Volume 100C: Arsenic, Metals, Fibres, and Dusts, 2012, Lyon, ISBN: 978-92-832-0135-9 Search PubMed.
- S. Mishra and R. N. Bharagava, J. Environ. Sci. Health, Part C: Environ. Carcinog. Ecotoxicol. Rev., 2015, 34, 1–32 CrossRef PubMed.
- A. K. Shanker, C. Cervantes, H. Loza-Tavera and S. Avudainayagam, Environ. Int., 2005, 31, 739–753 CrossRef CAS PubMed.
- (a) https://www.culligan.it/cromo-nelle-acque/, retrieved on May 2020; (b) Ministero della Salute (Italia), Direzione Generale della Prevenzione Sanitaria, Scheda “Acque Potabili – Parametri, Cromo”, 2016, http://www.salute.gov.it/portale/temi/documenti/acquepotabili/parametri/scheda_CROMO.pdf, retrieved on May 2020 Search PubMed; (c) https://www.epa.gov/sdwa/chromium-drinking-water, retrieved on May 2020.
- R. Bevan, Word Health Organization (WHO) Guidelines for Drinking-water Quality, Chromium in Drinking-water, 2019, retrieved on May 2020 Search PubMed.
- S. Cotton, Lanthanide and Actinide Chemistry, 2016, John Wiley & Sons, Inc., ISBN: 978-0-470-01005-1 Search PubMed.
- Q. Li, T. Li and J. Wu, J. Phys. Chem. B, 2001, 105, 12293–12296 CrossRef CAS.
- A. Podhorodecki, N. V. Gaponenkop, M. Banski, T. Kim and J. Misiewicz, ECS Trans., 2010, 28, 81–88 CrossRef CAS.
- Y. Wang and N. Lin, Photochem. Photobiol. Sci., 2011, 10, 42–47 RSC.
- S. Marchesi, F. Carniato and E. Boccaleri, New J. Chem., 2014, 38, 2480–2485 RSC.
- S. Marchesi, F. Carniato and E. Boccaleri, ChemPlusChem, 2015, 80, 915–918 CrossRef CAS PubMed.
- S. Marchesi, C. Bisio, E. Boccaleri and F. Carniato, ChemPlusChem, 2020, 85, 176–182 CrossRef CAS.
- (a) D. T. de Lill, A. de Bettencourt-Dias and C. L. Cahill, Inorg. Chem., 2007, 46, 3960–3965 CrossRef CAS PubMed; (b) A. M. Kaczmarek and P. Van Der Voort, Materials, 2020, 13, 566–593 CrossRef CAS PubMed.
- M. O. Rodrigues, J. D. L. Dutra, L. A. O. Nunes, G. F. de Sá, W. M. de Azevedo, P. Silva, F. A. A. Paz, R. O. Freire and S. A. Junior, J. Phys. Chem. C, 2012, 116, 19951–19957 CrossRef CAS.
- X. Zhou, L. Chen, Z. Feng, S. Jiang, Y. Pang, L. Li and G. Xiang, Inorg. Chim. Acta, 2018, 469, 576–5820 CrossRef CAS.
- X.-Y. Li, W.-J. Shi, X.-Q. Wang, L.-N. Ma, L. Hou and Y.-Y. Wang, Cryst. Growth Des., 2017, 17, 4217–4224 CrossRef CAS.
- K. Binnemans and C. Gorller-Walrand, J. Rare Earths, 1996, 14, 173–180 Search PubMed.
- S. F. Tang, A. Babai and A. V. Mudring, Angew. Chem., Int. Ed., 2008, 47, 7631–7638 CrossRef CAS PubMed.
- P. Zhang, Y. Wang, H. Liu and Y. Chen, J. Mater. Chem., 2011, 21, 18462–18466 RSC.
- H. Yin, Y. Li, J. Bai, M. Ma and J. Liu, J. Materiomics, 2017, 3, 144–149 CrossRef.
- I. P. Sahu, D. P. Bisen, R. K. Tamrakar, K. V. R. Murthy and M. Mohapatra, Journal of Science: Advanced Materials and Devices, 2017, 2, 59–68 Search PubMed.
- (a) A. Katiyar, S. Yadav, P. G. Smirniotis and N. G. Pinto, J. Chromatogr. A, 2006, 1122, 13 CrossRef CAS PubMed; (b) M. Kruk, M. Jaroniec, Y. Sakamoto, O. Terasa-ki, C. H. Ko and R. Ryoo, J. Phys. Chem. B, 2000, 104, 292 CrossRef CAS.
- K. Binnemans, Coord. Chem. Rev., 2015, 295, 1–45 CrossRef CAS.
- (a) F. Olivero, F. Carniato, C. Bisio and L. Marchese, Chem.–Asian J., 2013, 9, 158–165 CrossRef PubMed; (b) F. Cucinotta, F. Carniato, A. Devaux, L. De Cola and L. Marchese, Chem.–Eur. J., 2012, 18, 15310–15315 CrossRef CAS PubMed.
- (a) R. A. Schoonheydt and C. T. Johnston, The surface properties of clay minerals, Layered Mineral Structures and their Application in Advanced Technologies, M. F. Brigatti and A. Mottana, 2011 Search PubMed; (b) A. Doi, M. Ejtemaei and A. V. Nguyen, Miner. Eng., 2019, 143, 105929 CrossRef CAS.
- (a) K. Norrish, Nature, 1954, 1, 120–134 Search PubMed; (b) M. L. Whittaker, L. N. Lammers, S. Carrero, B. Gilbert and J. F. Banfield, PNAS, 2019, 116, 22052–22057 CrossRef CAS PubMed; (c) M. A. Chapperl, D. A. Laird, M. L. Thompson, H. Li, B. J. Teppen, V. Aggarwal, C. T. Johnston and S. A. Boyd, Environ. Sci. Technol., 2005, 39, 3150–3156 CrossRef PubMed; (d) H. Suquet, C. de la Calle and H. Pezerat, Clays Clay Miner., 1975, 23, 1–9 CrossRef CAS.
- E. Blatt, R. C. Chatelier and W. H. Sawyer, Biophys. J., 1986, 50, 349–356 CrossRef CAS PubMed.
- S. Lin, H. Cheng, Q. Ouyang and H. Wei, Anal. Methods, 2016, 8, 3935–3940 RSC.
- M. H. Gehlen, J. Photochem. Photobiol., C, 2020, 42, 100338 CrossRef CAS.
- M. A. Omary and H. H. Patterson, Encyclopedia of Spectroscopy and Spectrometry, Luminescence Theory, 2017, Elsevier Ltd., pp. 636–653 Search PubMed.
- (a) X. Zhuang, N. Zhang, X. Zhang, Y. Wang, L. Zhao and Q. Yang, Microchem. J., 2019, 104498 Search PubMed; (b) S. Mukherjee, S. Ganguly, D. Samanta and D. Das, ACS Sustainable Chem. Eng., 2020, 8, 1195–1206 CrossRef CAS; (c) T. Kundu, K. Manna, A. K. Jana and A. Natarajan, New J. Chem., 2019, 43, 13263–13270 RSC; (d) W. Liu, Y. Wang, Z. Bai, Y. Li, Y. Wang, L. Chen, L. Xi, J. Diwu, Z. Chai and S. Wang, ACS Appl. Mater. Interfaces, 2017, 9, 16448–16457 CrossRef CAS PubMed; (e) A. K. Jana and S. Natarajan, ChemPlusChem, 2017, 82, 1153–1163 CrossRef PubMed.
- W. Yang, J. Xia, G. Zhou, T. Hu, D. Ye, D. Jiang and Q. Li, Bull. Korean Chem. Soc., 2019 DOI:10.1002/bkcs.11495.
- (a) X. Feng, R. Li, L. Wang, S. W. Ng, G. Qin and L. Ma, CrystEngComm, 2015, 17, 7878–7887 RSC; (b) T.-Y. Gu, M. Dai, D. J. Young, Z.-G. Ren and J.-P. Lang, Inorg. Chem., 2017, 56, 4668–4678 CrossRef PubMed.
- (a) Y. Liu, Z. Chen, W. Li, C. Ma, P. Wu, X. Wu, S. Li and S. Liu, Microchim. Acta, 2018, 185 Search PubMed; (b) R. Vaz, J. Bettini, J. G. F. Júnior, E. D. S. Lima, W. G. Botero, J. C. C. Santos and M. A. Schiavon, J. Photochem. Photobiol., A, 2017, 346, 502–511 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra05693f |
| This journal is © The Royal Society of Chemistry 2020 |