
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTuning slow magnetic relaxation behaviour in a {Dy2}-based one-dimensional chain via crystal field perturbation†
Shui Yua,
Qinhua Zhangc,
Huancheng Hu*a,
Zilu Chen *a,
Dongcheng Liua,
Yuning Lianga and
Fupei Liang*ab
*a,
Dongcheng Liua,
Yuning Lianga and
Fupei Liang*ab
aState Key Laboratory for Chemistry and Molecular Engineering of Medicinal Resources, School of Chemistry and Pharmaceutical Sciences, Guangxi Normal University, Guilin 541004, P. R. China. E-mail: zlchen@mailbox.gxnu.edu.cn; siniantongnian@126.com; fliangoffice@yahoo.com
bGuangxi Key Laboratory of Electrochemical and Magnetochemical Functional Materials, College of Chemistry and Bioengineering, Guilin University of Technology, Guilin, 541004, P. R. China
cState Key Laboratory of Heavy Oil Processing, Institute of New Energy, College of Chemical Engineering, China University of Petroleum (East China), Qingdao 266580, P. R. China
First published on 24th March 2020
Abstract
Two novel {Dy2}-based one dimensional chain compounds {[Dy2(H3L)4(OAc)6]·2MeOH}n (1) and {[Dy2(H3L)4(OAc)4(NCS)2]·2MeOH}n (2) (H3L = 1,3-bis(2-hydroxynaphthalenemethyleneamino)-propan-2-ol) have been prepared under solvothermal conditions. Crystal structure analyses indicate that 1 and 2 feature similar 1D chain structures bearing dinuclear secondary building units. The difference between these two structures is that one chelated acetate ligand of Dy(III) ion in 1 is replaced by one monodentate coordinated NCS− ion in 2, leading to their different coordination numbers and geometry configurations to Dy(III) ion. Magnetic properties indicate that 1 and 2 display slow magnetic relaxation behavior with an effective energy barrier of 16.44(2) K in 1 and 8.02(2) K in 2, respectively, which is maybe attributed to the subtle crystal field perturbation of Dy(III) ions.
Introduction
Recently single-molecule magnets (SMMs) have attracted much attention because of their promising applications in ultrahigh-density information storage, molecular spintronic devices and quantum computing, etc.1–3 Comparing with transition metal ions, lanthanide (Ln) ions often possess large orbital angular momentum and inherent magnetic anisotropy, which contribute to improving the effective energy barrier (Ueff) and blocking temperature (TB) of SMM materials.4–7 As a result, Ln ions particularly Dy(III) ion have become ideal candidates to construct SMM materials since Ishikawa et al. reported the first Ln-based SMM of [Pc2Ln]−·TBA+ (Ln = Tb or Dy, Pc = dianion of phthalocyanine; TBA+ = N(C4H9)4+) in 2003.8 After that, numerous Ln-based SMM materials including discrete Lnx (x ≥ 1) coordination clusters, chainlike, two dimensional and three dimensional coordination compounds have been prepared and investigated in detail.9–11Aiming at enhancing the performance of Ln-based SMM materials, many effective efforts have been dedicated by researchers in the past decades.12–15 It is noteworthy that fine-tuning the symmetry of ligand field and/or the orientations of single-ion easy axes of lanthanide ions have been viewed as general and satisfactory approaches to increase the single-ion anisotropy of Ln(III) ions and further improve the values of Ueff and TB of SMM materials.16,17 For example, two essentially isomorphous planar {Dy4} coordination clusters of [Dy4(μ3-OH)2(hmmpH)2(hmmp)2Cl4]·3MeCN·MeOH and [Dy4(μ3-OH)2(hmmpH)2(hmmp)2(N3)4]·4MeOH {hmmpH2 = 2-[(2-hydroxy-ethylimino)methyl]-6-methoxyphenol} exhibit an anion-dependent magnetic slow relaxation behavior.18 When coordination water molecules in compound [Dy(acac)3(H2O)2] are replaced by 1,10-phenanthroline or its derivatives with large aromatic groups, the energy barrier can also be enhanced.19 Comparing with compound [Dy(H2L)(NO3)(H2O)(EtOH)](NO3)2·H2O (H2L = 2,6-diylbis(ethan-1-yl-1-ylidene)di(isonicotinohydrazide)), the slow magnetic behavior of [Dy(L)(NO3)(MeOH)2] is changed obviously by deprotonating H2L ligand and using different solvents.20 Therefore, it is necessary to design and prepare compounds with similar or isomorphous structure by tuning ligands to control the anisotropy of Dy(III) ions and further explore the structure–property relationships of SMM materials. But up to now, the investigations of this field are still limited.
In this context, we report two polyhydroxylamine-derived Schiff-base {Dy2}-based one dimensional chain compounds {[Dy2(H3L)4(OAc)6]·2MeOH}n (1) and {[Dy2(H3L)4(OAc)4(NCS)2]·2MeOH}n (2) featuring with similar structures. The different coordination groups of Dy(III) in 1 and 2 lead to different geometrical configurations and symmetries and coordination numbers of Dy(III) ions. The slow magnetic relaxation behaviors of 1 and 2 are obviously perturbed by their difference of crystal field for Dy(III) ion. The energy barrier is 16.44(2) K and 8.02(2) K with pre-exponential factor of 3.25 × 10−6 s and 5.6 × 10−5 s in 1 and 2, respectively.
Experimental
Materials and physical measurements
All chemicals were purchased from commercial companies and used directly without further purification. The Fourier transform infrared (FT-IR) data were collected on PerkinElmer Spectrum One FT-IR spectrometer using the corresponding KBr pellets in the wavenumber range of 4000–400 cm−1. The powder X-ray diffraction (PXRD) measurements were carried out on a Rigaku D/max 2500v/pc diffractometer equipped with Cu-Kα radiation (λ = 1.5418 Å) at 40 kV and 40 mA, with a step size of 0.02° in 2θ and a scan speed of 5° min−1. Elemental analyses for C, H and N elements were performed on an Elementar Micro cube C, H, N elemental analyzer. The TG analyses were conducted on a PerkinElmer Diamond TG/DTA thermal analyzer in a flowing nitrogen atmosphere with a heating rate of 5 °C min−1. All magnetic data were measured on a Quantum Design MPMS SQUID-XL-7 SQUID magnetometer. The magnetic data were corrected with a consideration of diamagnetic contribution from the sample and the sample holder.X-ray crystal structure analysis
The single crystal X-ray diffraction experiments were collected on SuperNova diffractometer with graphite monochromated Mo-Kα radiation (λ = 0.71073 Å). The single crystal structures were solved by the direct method using SHELXT21 and refined by means of full-matrix least-squares procedures on F2 by the SHELXL program.22 All non-hydrogen atoms (C, N, O, S and Dy) were located by different Fourier maps and subsequently refined with anisotropic displacement parameters. The H atoms attached on C and N atoms were refined at the geometrical sites. The details of crystallographic data of 1 and 2 are presented in Table S1.† Selected bond lengths and bond angles in 1 and 2 are listed in Tables S2 and S3,† respectively.Synthesis
Results and discussion
Structural characterization
![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) space group. The asymmetric unit of 1 contains one crystallographically independent Dy(III) ion, one H3L ligand, three OAc− ions and one lattice methanol molecule. Dy(III) ion is coordinated by nine O atoms from two H3L ligands and four carboxylate groups, giving a distorted muffin coordination geometry with Cs symmetry by SHAPE analysis (Table S4†). Two adjacent Dy(III) ions with the distance of 4.120 Å are bridged by two bidentate carboxylate oxygen atoms to form a dinuclear [Dy2O2] unit, and these [Dy2O2] units are further connected by H3L ligands to form 1D chain structure (Fig. 1d). The Dy(III)–O bond distances range from 2.285(2) to 2.564(3) Å and Dy–O–Dy bond angles are in the range of 49.70(12)° to 148.94(12)°.23,24
space group. The asymmetric unit of 1 contains one crystallographically independent Dy(III) ion, one H3L ligand, three OAc− ions and one lattice methanol molecule. Dy(III) ion is coordinated by nine O atoms from two H3L ligands and four carboxylate groups, giving a distorted muffin coordination geometry with Cs symmetry by SHAPE analysis (Table S4†). Two adjacent Dy(III) ions with the distance of 4.120 Å are bridged by two bidentate carboxylate oxygen atoms to form a dinuclear [Dy2O2] unit, and these [Dy2O2] units are further connected by H3L ligands to form 1D chain structure (Fig. 1d). The Dy(III)–O bond distances range from 2.285(2) to 2.564(3) Å and Dy–O–Dy bond angles are in the range of 49.70(12)° to 148.94(12)°.23,24
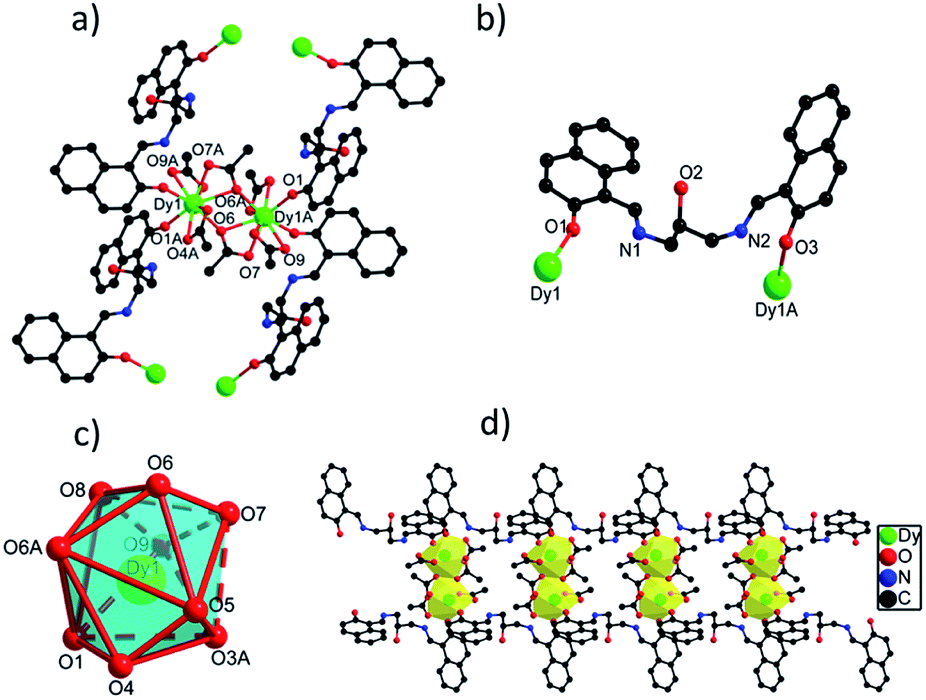 | ||
| Fig. 1 (a) The structure of {Dy2} core in 1 with selected atoms labelled. (b) The coordination mode of H3L ligand in 1. (c) The coordination polyhedron of Dy(III) ion in 1. (d) The 1D chain structure of 1. | ||
space group. The asymmetric unit of 2 contains one crystallographically independent Dy(III) ion, one H3L ligand, two carboxylate groups, one SCN− ion and one lattice methanol molecule. As depicted in Fig. S2,† the Dy(III) ion in 2 is coordinated by seven O atoms from two H3L ligands and three carboxylate groups and one N atom from one SCN− ion, showing a triangular dodecahedron with D2d symmetry by SHAPE analysis (Table S5†). Two neighboring Dy(III) ions with the distance of 4.027 Å are bridged by two O atoms from two carboxylate groups to form a [Dy2O2] unit, which are further linked to build 1D chain structure via bridging H3L ligands (Fig. 2d). The bond lengths of Dy(III)–O and Dy(III)–N are in the range of 2.270(4)–2.501(5) Å, and the angles of O–Dy(III)–O and O–Dy(III)–N range from 51.63(14) to 165.86(17)°.20,25 Interestingly, although different {Dy2} units exist in 1 and 2, they still exhibit similar 1D chain structures owing to the same bridging H3L ligands. The flexibility of H3L ligand leads to its slightly different coordination modes in 1 and 2, thus they show different crystal packing structures which are constructed from the one-dimensional chains through π⋯π interaction rooting from aromatic rings (Fig. S1†).
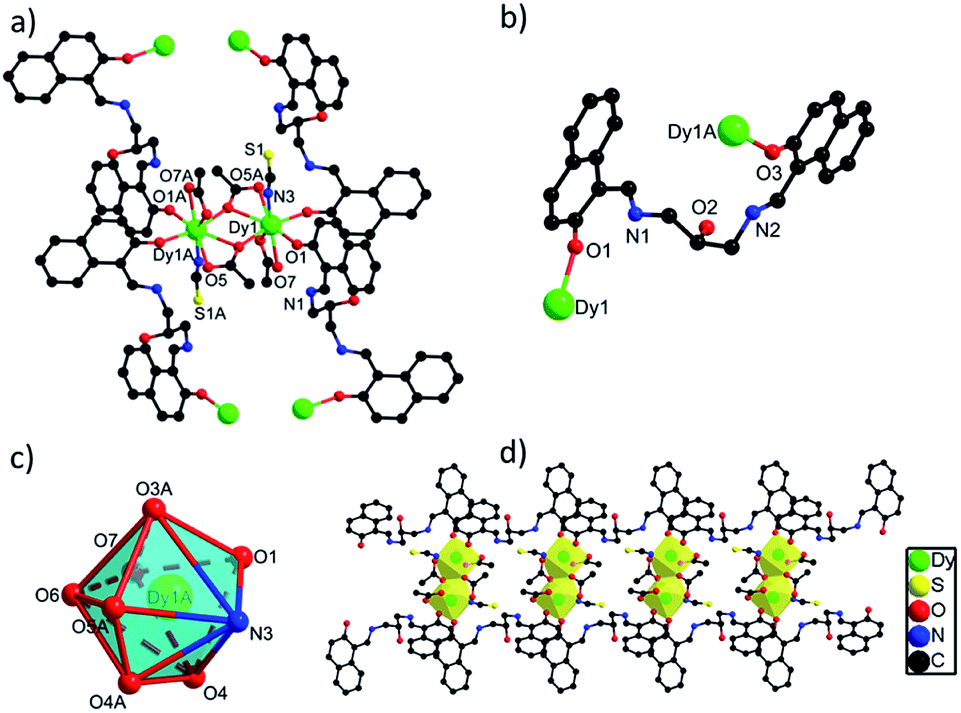 | ||
| Fig. 2 (a) The structure of {Dy2} unit in 2 with selected atoms labelled. (b) The coordination mode of H3L ligand in 2. (c) The coordination polyhedron of Dy(III) ion in 2. (d) The 1D chain structure of 2. | ||
Powder X-ray diffraction and thermogravimetric analysis
The experimental powder X-ray diffraction (PXRD) patterns of 1 and 2 match well with the corresponding simulated ones obtained from the single crystal X-ray diffraction data, respectively, confirming the high purity of the bulk samples of 1 and 2 (Fig. S3†). As shown in Fig. S4,† the thermogravimetric analysis (TGA) curve of 1 shows an obvious weight loss of 5.90% before 131 °C, due to the loss of MeOH molecules (calcd: 4.20%). With the temperature increasing, a platform is occurred before the collapse of the structure of 1 above 214 °C. The TGA curve of 2 exhibits a gradual weight loss of 4.75% below 248 °C, which is as well attributed to the loss of MeOH molecules (calcd: 4.20%). As the temperature is further increased, the structure of 2 starts to decompose.Magnetic properties
Direct current magnetic susceptibility measurements on polycrystalline samples of compounds 1 and 2 were performed in the temperature range of 1.8–300 K under an external field of 1000 Oe. As shown in Fig. 3, the χMT values at room temperature of 1 and 2 are 26.73 and 29.47 cm3 K mol−1, which are very close to the theoretical value of 28.34 cm3 K mol−1 for two uncoupled Dy(III) ion (6H15/2, L = 5, g = 4/3).26,27 For compound 1, the χMT value gradually decreases in the temperature range of 300 K to 178 K. Upon cooling, the value of χMT in 1 decreases rapidly and reaches a minimum of 22.36 cm3 K mol−1 at 1.8 K. For compound 2, the χMT value almost keeps constant up to 161 K, and then it reduces rapidly to 21.92 cm3 K mol−1 at 1.8 K. The behavior of 1 and 2 at low temperature can be attributed to thermal depopulation of excited Stark sub-levels, antiferromagnetic interactions between Dy(III) ions and/or large magnetic anisotropy of Dy(III) ions (Fig. 3 and 4).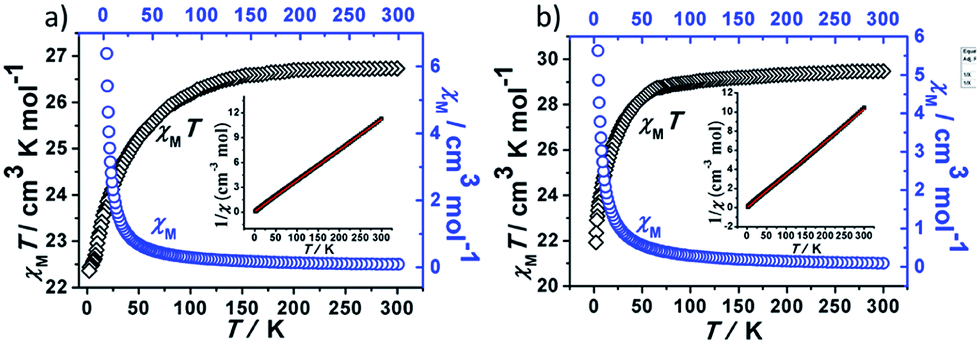 | ||
| Fig. 3 Plots of χM and χMT vs. T for 1 (a) and 2 (b). | ||
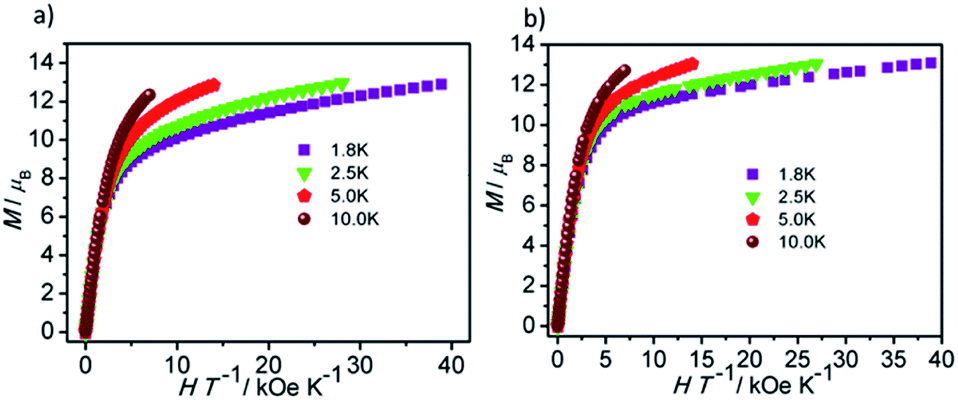 | ||
| Fig. 4 Plots of M vs. HT−1 for 1 (a) and 2 (b) measured at 1.8, 2.5, 5.0 and 10 K. | ||
The field-dependent magnetization for 1 and 2 at 1.8, 2.5, 5.0 and 10 K exhibit that the magnetization increases rapidly in the low field, and then slowly increases with the increasing applied field. The maximum values of M for 1 and 2 at 7 T are obviously lower than the theoretical saturated value of 20 Nβ for dinuclear Dy(III) compounds (Fig. S5†).28,29 Meanwhile, the non-superposition of the M vs. H/T plots for 1 and 2 at different temperatures in the high field further implies that the above mentioned behavior is probably due to the crystal-field-induced splitting of the Stark level, significant magnetic anisotropy and/or low-lying energy states.30–32
In order to investigate the dynamic magnetic properties of compounds 1 and 2, variable frequency alternating current (ac) magnetic susceptibilities were firstly performed with 3 Oe alternating field and 0 Oe direct current (dc) field at 2 K. No peaks are observed in the in-phase (χ′) vs. v and out-of-phase (χ′′) vs. v curves for 1 and 2, which may be attributed to the presence of quantum tunneling of magnetization (QTM). To suppress the QTM effect, different dc fields ranging from 200 to 4000 Oe were applied on 1 and 2, unfortunately, no obvious peaks were observed in χ′ vs. v and χ′′ vs. v plots for 1 and 2 (Fig. S7 and S8†). Therefore, for 1 and 2, the alternating current magnetic susceptibilities measurements were measured in the frequency range of 1–1000 Hz under zero dc field. As shown in Fig. 5, clear frequency dependence and temperature dependence of χ′′ signals can be observed although no obvious peaks were detected in 1 and 2, illuminating the existence of typical slow magnetic relaxation processes in 1 and 2. The obvious difference of χ′ signals vs. temperature plots and frequency dependencies plots for 1 and 2 (Fig. S9 and S10†) maybe derive from their different coordination numbers and geometry configurations and symmetries of the central Dy(III) ion.
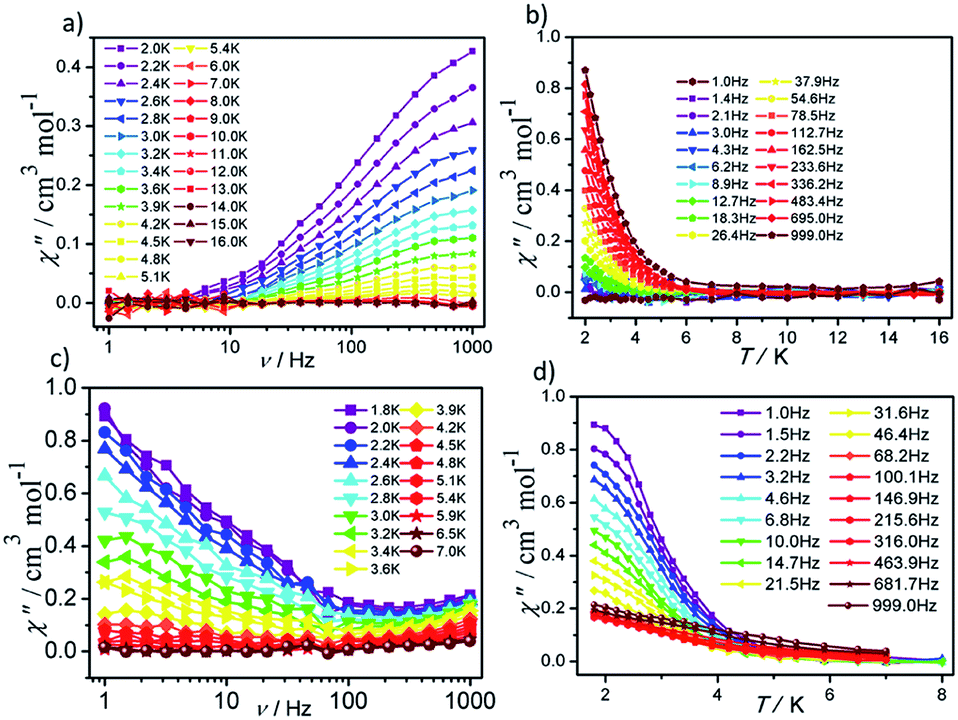 | ||
| Fig. 5 Frequency- and temperature-dependent out-of phase (χ′′) ac susceptibilities for 1 (a and b) and 2 (c and d) under zero dc field. | ||
The effective energy barrier (Ueff) and relaxation time (τ0) of 1 and 2 can be obtained by fitting ln(χ′′/χ′) vs. 1/T data with the following equation: ln(χ′′/χ′) = ln(ωτ0) + Ea/(kBT),33–35 and the excellent linear fitting between ln(χ′′/χ′) and 1/T can be depicted at different frequencies, yield Ueff ≈ 16.44 K and 8.02 K, τ0 ≈ 3.25 × 10−6 s and 5.6 × 10−5 s for 1 and 2, respectively. The different effective energy barrier and relaxation time for 1 and 2 are due to the different anisotropy of their Dy(III) ions from subtle crystal field perturbation caused by the change of one chelated acetate in 1 by one NCS− ligand (Fig. 6).
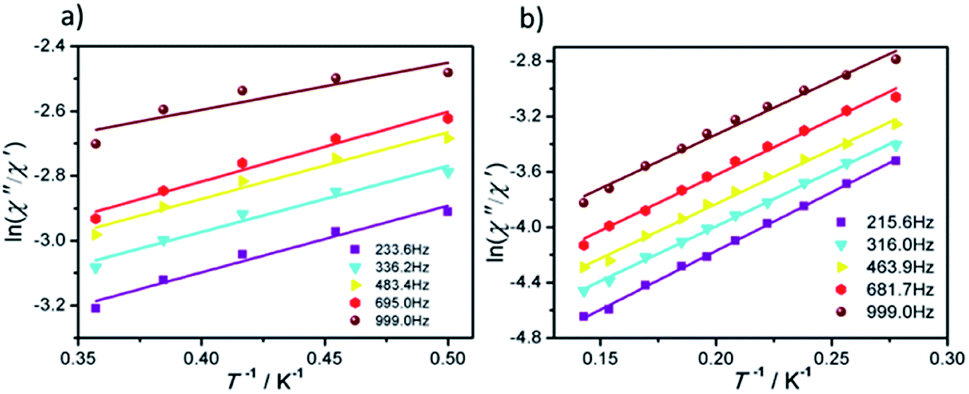 | ||
| Fig. 6 Plots of ln(χ′′/χ′) vs. 1/T for 1 (a) and 2 (b) under 0 Oe dc field, the solid lines represent the best fits. | ||
Conclusions
In conclusion, two new {Dy2}-based 1D chain structures bearing a Schiff-base ligand were constructed and characterized in detail. Magnetic properties indicate that they exhibit similar slow magnetic relaxation behavior under zero dc field. Interestingly, one chelated acetate ligand in 1 is replaced by one NCS− ion in 2, which leads to the different coordination geometries of their Dy(III) ions, and thus their different Dy(III) anisotropies. All of these factors results in their different effective energy barriers. This phenomena indicates that the slight crystal field perturbation of Dy(III) ions will affect the slow magnetic behavior for the compounds with similar structures. This work will enrich the methods of tuning the performance of SMM materials via crystal field perturbation effect.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by National Natural Science Foundation of China (grant no. 21661009 and 21901050), Guangxi Natural Science Foundation of China (grant no. 2018GXNSFBA050031), the project for improving the basic scientific research ability of young and middle-aged teachers in Guangxi Universities (grant no. 2019KY0054), the Key Project of Guangxi Normal University (grant no. 2018ZD003) and Innovation Project of Guangxi Graduate Education (title: Study on the synthesis and magnetic properties of dysprosium complexes).References
- L. Ungur, S.-Y. Lin, J. Tang and L. F. Chibotaru, Chem. Soc. Rev., 2014, 43, 6894–6905 RSC.
- D. N. Woodruff, R. E. Winpenny and R. A. Layfield, Chem. Rev., 2013, 113, 5110–5148 CrossRef CAS PubMed.
- J. Liu, Y. C. Chen, J. L. Liu, V. Vieru, L. Ungur, J. H. Jia, L. F. Chibotaru, Y. Lan, W. Wernsdorfer, S. Gao, X. M. Chen and M. L. Tong, J. Am. Chem. Soc., 2016, 138, 5441–5450 CrossRef CAS PubMed.
- Y.-X. Wang, Y. Ma, Y. Chai, W. Shi, Y. Sun and P. Cheng, J. Am. Chem. Soc., 2018, 140, 7795–7798 CrossRef CAS PubMed.
- B. S. Dolinar, D. I. Alexandropoulos, K. R. Vignesh, T. James and K. R. Dunbar, J. Am. Chem. Soc., 2018, 140, 908–911 CrossRef CAS PubMed.
- M. K. Singh and G. Rajaraman, Inorg. Chem., 2019, 58, 3175–3188 CrossRef CAS PubMed.
- A. Bhanja, R. Herchel, Z. Travnicek and D. Ray, Inorg. Chem., 2019, 58, 12184–12198 CrossRef CAS PubMed.
- N. Ishikawa, M. Sugita, T. Ishikawa, S.-y. Koshihara and Y. Kaizu, J. Am. Chem. Soc., 2003, 125, 8694–8695 CrossRef CAS PubMed.
- Z.-H. Zhu, X.-F. Ma, H.-L. Wang, H.-H. Zou, K.-Q. Mo, Y.-Q. Zhang, Q.-Z. Yang, B. Li and F.-P. Liang, Inorg. Chem. Front., 2018, 5, 3155–3162 RSC.
- A. B. Canaj, M. Siczek, M. Otreba, T. Lis, G. Lorusso, M. Evangelisti and C. J. Milios, Dalton Trans., 2016, 45, 18591–18602 RSC.
- J. Kobylarczyk, M. Liberka, P. Konieczny, S. Baran, M. Kubicki, T. Korzeniak and R. Podgajny, Dalton Trans., 2020, 49, 300–311 RSC.
- L. F. Chibotaru, L. Ungur and A. Soncini, Angew. Chem., Int. Ed., 2008, 47, 4126–4129 CrossRef CAS PubMed.
- J. Mayans, Q. Saez, M. Font-Bardia and A. Escuer, Dalton Trans., 2019, 48, 641–652 RSC.
- Y. N. Guo, G. F. Xu, W. Wernsdorfer, L. Ungur, Y. Guo, J. Tang, H. J. Zhang, L. F. Chibotaru and A. K. Powell, J. Am. Chem. Soc., 2011, 133, 11948–11951 CrossRef CAS PubMed.
- A. A. Patrascu, M. Briganti, S. Soriano, S. Calancea, R. A. Allao Cassaro, F. Totti, M. G. F. Vaz and M. Andruh, Inorg. Chem., 2019, 58, 13090–13101 CrossRef CAS PubMed.
- R. J. Blagg, C. A. Muryn, E. J. McInnes, F. Tuna and R. E. Winpenny, Angew. Chem., Int. Ed., 2011, 50, 6530–6533 CrossRef CAS PubMed.
- F. Habib, P. H. Lin, J. Long, I. Korobkov, W. Wernsdorfer and M. Murugesu, J. Am. Chem. Soc., 2011, 133, 8830–8833 CrossRef CAS PubMed.
- Y.-Z. Zheng, Y. Lan, C. E. Anson and A. K. Powell, Inorg. Chem., 2008, 47, 10813–10815 CrossRef CAS PubMed.
- G.-J. Chen, Y.-N. Guo, J.-L. Tian, J. Tang, W. Gu, X. Liu, S.-P. Yan, P. Cheng and D.-Z. Liao, Chem.–Eur. J., 2012, 18, 2484–2487 CrossRef CAS PubMed.
- J. Wang, H. Wang, Y. Ma, J. Tang, L. Li, Q. Wang, B. Zhao, P. Cheng and J. Ma, Cryst. Growth Des., 2019, 19, 3365–3371 CrossRef CAS.
- G. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- G. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- S.-D. Jiang and S.-X. Qin, Inorg. Chem. Front., 2015, 2, 613–619 RSC.
- K. Griffiths, P. Kumar, G. R. Akien, N. F. Chilton, A. Abdul-Sada, G. J. Tizzard, S. J. Coles and G. E. Kostakis, Chem. Commun., 2016, 52, 7866–7869 RSC.
- H. L. Wang, X. F. Ma, J. M. Peng, Z. H. Zhu, B. Li, H. H. Zou and F. P. Liang, Inorg. Chem., 2019, 58, 9169–9174 CrossRef CAS PubMed.
- S. Biswas, L. Mandal, Y. Shen and M. Yamashita, Dalton Trans., 2019, 48, 14096–14102 RSC.
- H. Ke, W. Wei, Y. Yang, J. Zhang, Y. Q. Zhang, G. Xie and S. Chen, Dalton Trans., 2019, 48, 7844–7852 RSC.
- K. H. Zangana, E. Moreno Pineda and R. E. Winpenny, Dalton Trans., 2015, 44, 12522–12525 RSC.
- W. Cao, C. Gao, Y. Q. Zhang, D. Qi, T. Liu, K. Wang, C. Duan, S. Gao and J. Jiang, Chem. Sci., 2015, 6, 5947–5954 RSC.
- J. Lu, X.-L. Li, Z. Zhu, S. Liu, Q. Yang and J. Tang, Dalton Trans., 2019, 48, 14062–14068 RSC.
- J. Vallejo, J. Cano, I. Castro, M. Julve, F. Lloret, O. Fabelo, L. Canadillas-Delgado and E. Pardo, Chem. Commun., 2012, 48, 7726–7728 RSC.
- Q. Zhou, F. Yang, D. Liu, Y. Peng, G. Li, Z. Shi and S. Feng, Inorg. Chem., 2012, 51, 7529–7536 CrossRef CAS PubMed.
- M. J. Liu, J. Yuan, Y. Q. Zhang, H. L. Sun, C. M. Liu and H. Z. Kou, Dalton Trans., 2017, 46, 13035–13042 RSC.
- H. Li, J. Sun, M. Yang, Z. Sun, J. Tang, Y. Ma and L. Li, Inorg. Chem., 2018, 57, 9757–9765 CrossRef CAS PubMed.
- I. A. Kühne, K. Griffiths, A.-J. Hutchings, O. P. E. Townrow, A. Eichhöfer, C. E. Anson, G. E. Kostakis and A. K. Powell, Cryst. Growth Des., 2017, 17, 5178–5190 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: IR curves, TGA curves, PXRD patterns, additional structural and magnetic figures, selected bond lengths and bond angles for the three titled compounds. CCDC 1984194 and 1984195. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0ra01604g |
| This journal is © The Royal Society of Chemistry 2020 |