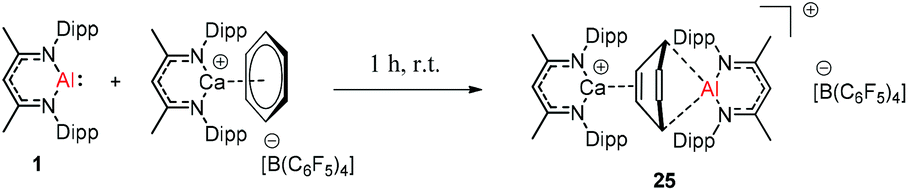DOI:
10.1039/C9DT04763H
(Perspective)
Dalton Trans., 2020,
49, 1351-1364
The unique β-diketiminate ligand in aluminum(I) and gallium(I) chemistry
Received
16th November 2019
, Accepted 17th December 2019
First published on 23rd December 2019
Abstract
Over the past few decades, β-diketiminate ligands have been widely used in coordination chemistry and are capable of stabilizing various metal complexes in multiple oxidation states. Recently, the chemistry of aluminum and gallium in their +1 oxidation state has rapidly emerged. NacNacM(I) (M = Al, Ga; NacNac = β-diketiminate ligand) shows a two coordinate metal center comparable with singlet carbene-like species. The metal center also possesses a formally vacant p-orbital. In this article we present an overview of the last 10 years for aluminum(I) and gallium(I) stabilized by β-diketiminate ligands that have been widely explored in bond breaking and forming species.
1. Introduction
Complexes containing a low-oxidation metal center is a key topic in modern organometallic chemistry as it leads to the development of new systems. A number of transition metal complexes were synthesized in a broad range of oxidation states to activate small molecules or used as catalysts in organic reactions.1 It was not until 1991 that the first structurally characterized molecular aluminum(I) compound was reported.2 Since then, the chemistry of Group 13 metals in the +1 oxidation state have played a great part in the development of p-block chemistry. The synthesis of compounds with aluminum and gallium in the +1 oxidation state is experimentally challenging and consistently hampered by their high reactivity and pronounced tendency towards disproportionation.3 The lone pair of electrons in MR (R = β-diketiminate) (M = Al, Ga) compounds can act as a basic site towards a range of Lewis acids, forming a σ donating bond.4 Donor–acceptor bonds have been observed in a range of main group elements, transition metals, and lanthanide and actinide metal complexes. The general MIR unit (M = Al, Ga) can be considered isolobal with singlet carbenes, CO, and CNR.
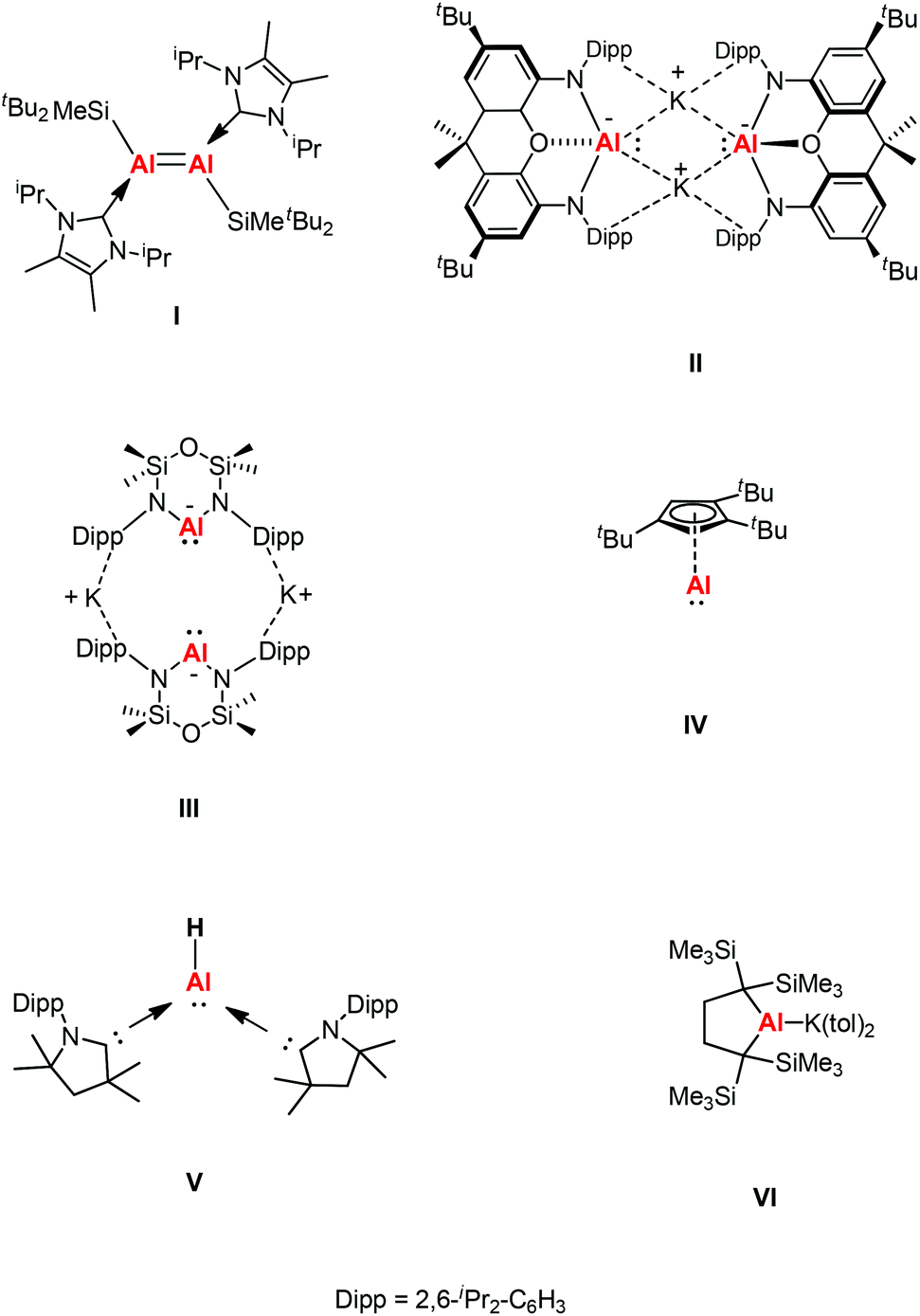
β-Diketiminate ligands have found widespread application as supporting ligands in metal-mediated catalysis.5 The stoichiometric transformations of NacNacAl(I) and NacNacGa(I) have also been explored widely owing to the lone pair of electrons and a formally vacant p-orbital on aluminum affording high electrophilic and nucleophilic reactivity.6 A comparison between main group elements and transition metals was drawn when main group species were found to have reactivity towards small molecules under ambient conditions. This was rationalized by main group species possessing donor/acceptor frontier orbitals which are separated by modest energy gaps, thus drawing comparisons with open-shell transition metal species.7 In 2000, Roesky et al. chose the β-diketiminate ligand to synthesize a more kinetically stable monomeric aluminum(I) compound Al{HC[C(Me)NDipp]2} (Dipp = 2,6-iPr2C6H3) (1).8a This was the first stable dicoordinate aluminum(I) compound to be prepared and structurally characterized in the solid-state. Later, Cui et al. also reported a β-diketiminate ligand stabilized aluminum(I) compound Al{HC[C(tBu)NDipp]2}.8b Computational studies of β-diketiminate stabilized heavier group metal complexes have shown that their metal lone pairs are associated with the HOMO−2.9 As a result, they are good σ donor ligands and poor π acceptors like N-heterocyclic carbenes (NHCs), and thus have the potential to display carbene-like chemistry. Inoue et al. reported the first neutral Al(I) compound containing an Al![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) Al double bond, which was achieved through the reductive dimerization of the corresponding N-heterocyclic carbene (NHC)-stabilized silyl substituted aluminum(III) dihalide (I, Fig. 1), and its reactivity toward the fixation and selective reduction of CO2, both of which can be accessed in a stoichiometric and catalytic fashion.10 In 2018, Aldridge and Goicoechea et al. reported the first isolation of a nucleophilic aluminyl anion [(NON)Al]− by employing a chelating ligand (NON = 4,5-bis(2,6-diisopropylanilido)-2,7-di-tert-butyl-9,9-dimethylxanthene) (II, Fig. 1), which acts as an unprecedented aluminum(I) nucleophile (e.g., in reactions with tBu3PAuI), and which effects the formal oxidative addition of the C–C bond in benzene.11 After that, Coles and co-workers also synthesized a two-coordinate N-heterocyclic aluminyl anion K[Al(NONDipp)] (NONDipp = [O(SiMe2NDipp)2]2−, Dipp = 2,6-iPr2C6H3, III, Fig. 1), which is able to undergo further reactions such as activation of elemental selenium to form an aluminum complex containing an aluminum–selenium multiple bond and with 1,3,5,7-cyclooctatetraene (COT) to give the first aluminum complex containing a reduced COT-ligand with a strong aromatic character, respectively.12 Both aluminyl anion complexes reacted with two abundant greenhouse gases (CO2 and N2O) via cycloaddition to generate a monoalumoxane anion.13 The first isolable example of a room temperature stable monomeric cyclopentadienylaluminum(I) derivative was reported by Braunschweig and co-workers, which was supported by a bulky 1,2,4-tri-tert-butylcyclopentadienyl (Cp3t) ligand (IV, Fig. 1).14a The same group also reported the first example of a monomeric Lewis base stabilized Al(I) hydride that can be isolated and handled under ambient conditions (V, Fig. 1).14b Very recently, Yamashita's group reported an alkyl-substituted aluminum anion that exhibits very strong basicity and nucleophilicity (VI, Fig. 1).14c These species have been observed to form both covalent and donor–acceptor bonds, revealing both the reducing and nucleophilic properties of these novel complexes.
Al double bond, which was achieved through the reductive dimerization of the corresponding N-heterocyclic carbene (NHC)-stabilized silyl substituted aluminum(III) dihalide (I, Fig. 1), and its reactivity toward the fixation and selective reduction of CO2, both of which can be accessed in a stoichiometric and catalytic fashion.10 In 2018, Aldridge and Goicoechea et al. reported the first isolation of a nucleophilic aluminyl anion [(NON)Al]− by employing a chelating ligand (NON = 4,5-bis(2,6-diisopropylanilido)-2,7-di-tert-butyl-9,9-dimethylxanthene) (II, Fig. 1), which acts as an unprecedented aluminum(I) nucleophile (e.g., in reactions with tBu3PAuI), and which effects the formal oxidative addition of the C–C bond in benzene.11 After that, Coles and co-workers also synthesized a two-coordinate N-heterocyclic aluminyl anion K[Al(NONDipp)] (NONDipp = [O(SiMe2NDipp)2]2−, Dipp = 2,6-iPr2C6H3, III, Fig. 1), which is able to undergo further reactions such as activation of elemental selenium to form an aluminum complex containing an aluminum–selenium multiple bond and with 1,3,5,7-cyclooctatetraene (COT) to give the first aluminum complex containing a reduced COT-ligand with a strong aromatic character, respectively.12 Both aluminyl anion complexes reacted with two abundant greenhouse gases (CO2 and N2O) via cycloaddition to generate a monoalumoxane anion.13 The first isolable example of a room temperature stable monomeric cyclopentadienylaluminum(I) derivative was reported by Braunschweig and co-workers, which was supported by a bulky 1,2,4-tri-tert-butylcyclopentadienyl (Cp3t) ligand (IV, Fig. 1).14a The same group also reported the first example of a monomeric Lewis base stabilized Al(I) hydride that can be isolated and handled under ambient conditions (V, Fig. 1).14b Very recently, Yamashita's group reported an alkyl-substituted aluminum anion that exhibits very strong basicity and nucleophilicity (VI, Fig. 1).14c These species have been observed to form both covalent and donor–acceptor bonds, revealing both the reducing and nucleophilic properties of these novel complexes.
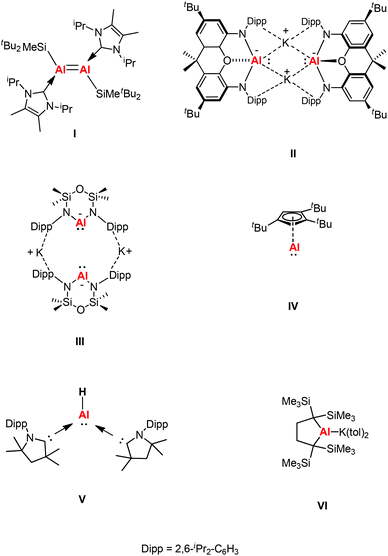 |
| | Fig. 1 Di-tert-butyl(methyl)silyl-substituted dialumene (I), anionic group 13 analogues of N-heterocyclic carbenes (II–III), monomeric 1,2,4-tri-tert-butylcyclopentadienylaluminum(I) (IV), Lewis base stabilized aluminum(I) hydride (V) and an alkyl-substituted aluminum anion (VI). | |
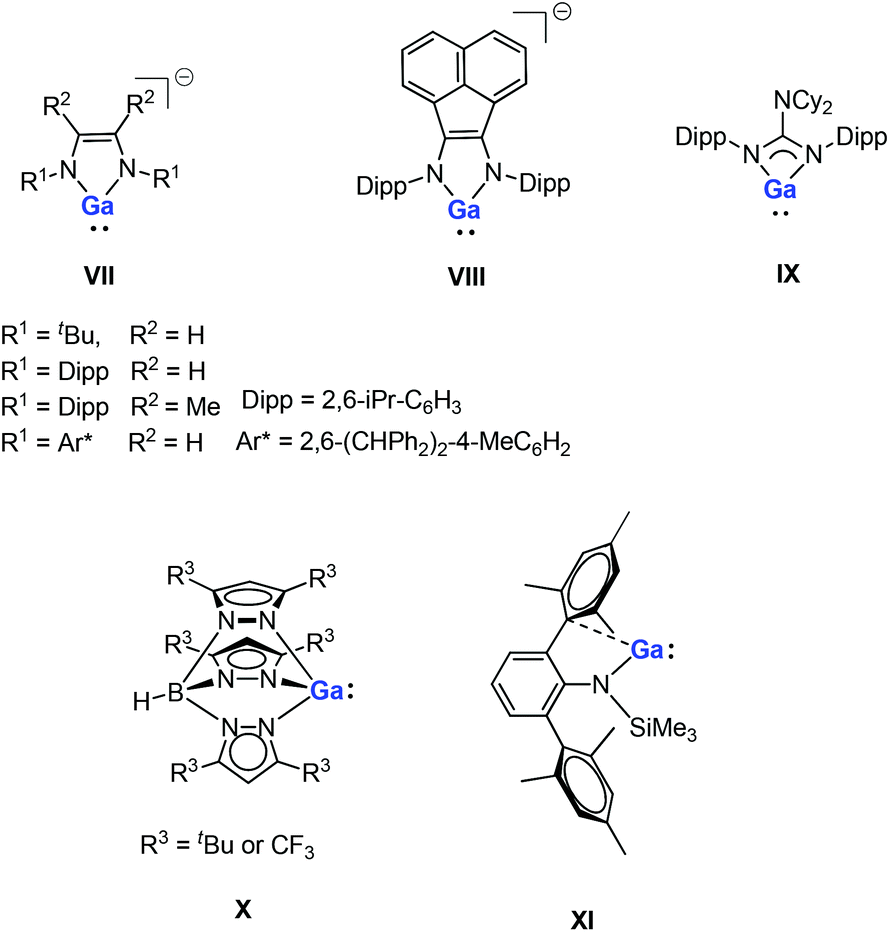
Gallium(I) compounds are often driven by the thermodynamic preference for the metal center to exist in the +3 oxidation state. Having said this, much of the reported chemistry of monomeric gallium(I) compounds is derived from the significant basicity of the metal through its lone pair of electrons. Gallium(I) compounds are generally more stable towards disproportionation than the corresponding aluminum(I) compounds. So far, several gallium(I) N-heterocycles have been reported.15,16 Bi- and tridentate ligand systems have been used in the preparation of a variety of neutral and anionic gallium(I) heterocycles (e.g., five-membered anionic complexes VII–VIII, a guanidinate complex IX and a monomeric tris(pyrazolylborate) complex X, see Fig. 2).16 These compounds have been prepared either by salt-metathesis reactions between alkali metal salts of the ligands and “GaI” or by alkali metal reduction of GaII or GaIII precursors. The monomeric example of GaI amide (XI, Fig. 2) can be considered to be having a quasi one-coordinate metal center, which also exhibits weak intramolecular arene interactions in the solid state.16m Recently, a pincer-type gallylene ligand has been successfully synthesized utilizing bis(phosphino)-terpyridine as an efficient scaffold for the Ir–GaI bond, which enabled various reactions at the Ir center by keeping the gallylene ligand intact.17
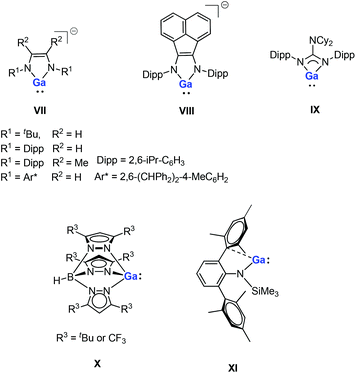 |
| | Fig. 2 Reported examples of gallium(I) N-heterocycles. | |
The β-diketiminate ligands typically provide monoanionic, bidentate support for metal complexes and offer a much higher degree of steric control through the choice of N-substituents. By tuning the steric and electronic properties of the supporting β-diketiminate ligands, the reactivity of the compounds can be significantly improved.18 Thus, complexes with a low-valent metal could be stabilized by the employment of sterically encumbering β-diketiminate ligands. It is interesting to note that with a redox-inactive metal bound and appropriate substituents, β-diketiminate ligands become redox-active ligands.19 Herein we present an overview that is of relevance to the corresponding bond activation by aluminum(I) and gallium(I) with β-diketiminate ligands.
2. Chemistry of DippNacNacAl(I) and DippNacNacGa(I)
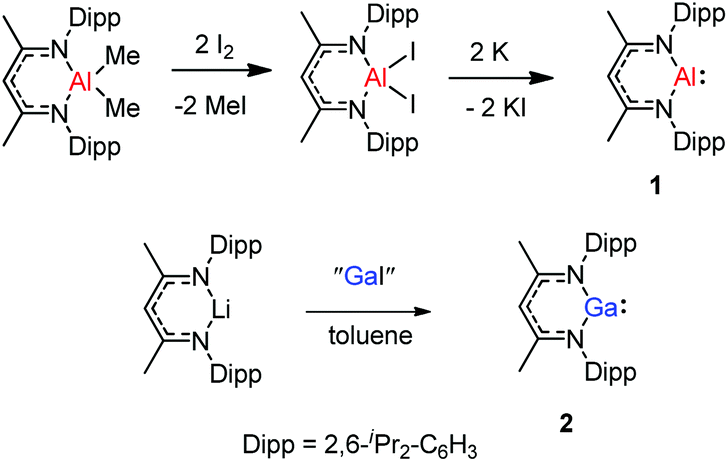
The monomeric aluminum carbenoid DippNacNacAl(I) (1) was prepared through reduction of the corresponding DippNacNacAlI2 with potassium (Scheme 1).8a At the same time, Power and co-workers reported a β-diketiminate stabilized Ga(I) monomer DippNacNacGa(I) (2) (Scheme 1).20 It was obtained by the reaction of [Li{HC(CMeNDipp)2}] with “GaI”. The remarkable thermal stability of the compounds toward disproportionation reaction (decomp. >150 °C) can be attributed to the steric bulk of the β-diketiminate ligand, which provides kinetic protection to the metal center. X-ray crystal structure analysis showed that compound 2 is monomeric and isostructural with its aluminum counterpart. With a singlet lone pair and formally empty p-orbital on the metal, the neutral heterocycles DippNacNacAl(I) and DippNacNacGa(I) have the potential to exhibit both nucleophilic and electrophilic characteristics.
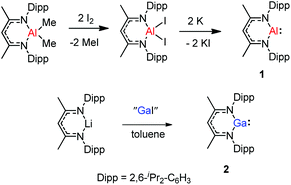 |
| | Scheme 1 Syntheses of DippNacNacAl(I) and DippNacNacGa(I). | |
2.1. Small-molecule activation
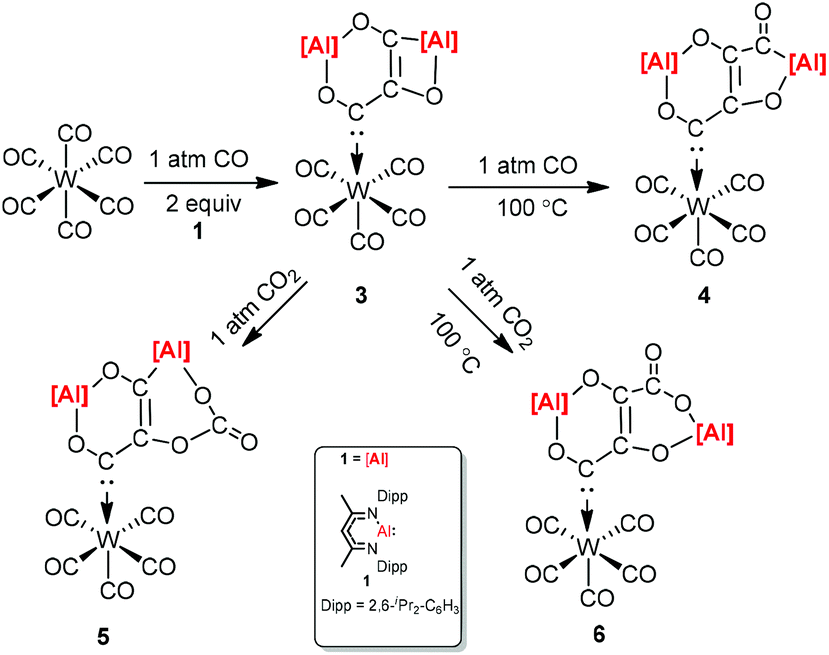
CO and CO2 activation by transition-metal complexes has been studied extensively for many years. However, the activation of CO and CO2 with Group 13 metal elements and their compounds has been explored scarcely.21 In 2018, Crimmin et al. reported carbon chain growth from C1 to C3 and to C4 by sequential reactions of CO and CO2 with a transition metal carbonyl complex in the presence of an aluminum(I) complex (Scheme 2).22 Warming a frozen suspension of [W(CO)6] with 2 equiv. of 1 under 1 atm of CO from −78 °C to r.t. in a benzene-d6 solvent results in the formation of the C3 homologated product 3. Heating the isolated and purified sample of 3 under one atmosphere of CO leads to the formation of the chain growth product 4. Further chain growth of C3 to a C4 fragment could be achieved upon the reaction of 3 with one atmosphere of CO2. Although the reaction of CO2 with 3 at 25 °C initially produces 5, when the sample 3 is heated at 100 °C for 48 h, it completely converts to 6. No reaction occurs between 1 and CO in the absence of [W(CO)6]. The gallium products of these reactions are not reported.
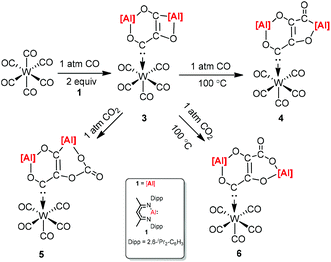 |
| | Scheme 2 Carbon chain growth by sequential reactions of CO and CO2 with [W(CO)6] and 1. | |
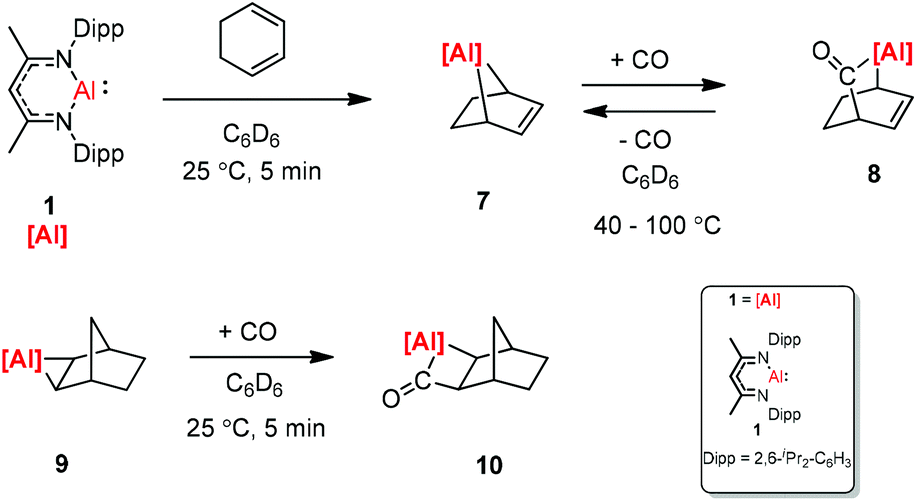
The [2.2.1] metallobicyclic compound 7 was synthesized by the cycloaddition of complex (1) with low-valent aluminum and 1,3-cyclohexadiene. The exposure of a C6D6 solution of 7 to one atmosphere of CO generated the insertion product 8. As shown in Scheme 3 the reaction mixture is reversible, when 8 is heated for longer time. Compound 9 was also studied in the reaction with CO, and the insertion of CO into the Al–C bond was observed. However, compound 10 decomposes at 25 °C within 12 h into an intractable mixture of products.23
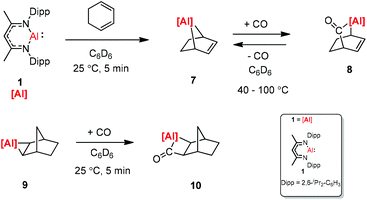 |
| | Scheme 3 Syntheses of [2.2.1] metallobicycle 7 and reversible CO insertion. | |
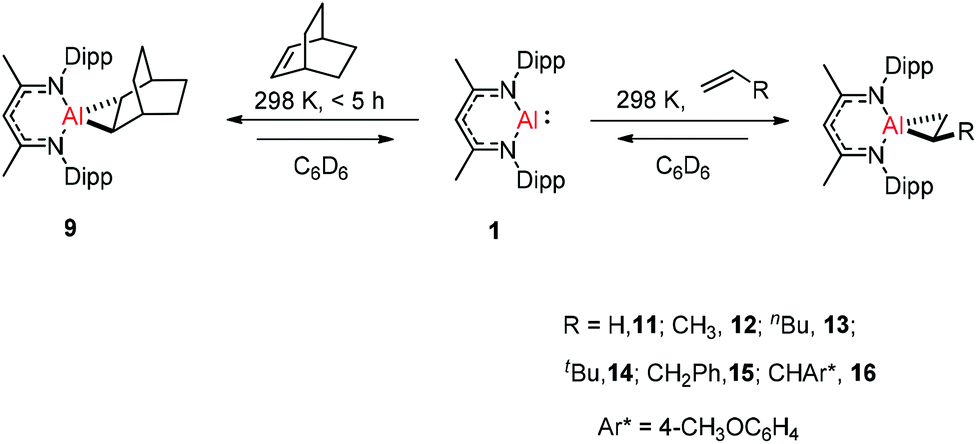
Crimmin and co-workers documented the first reversible addition of ethylene to aluminum(I) 1. The monomeric molecular aluminum(I) complex reacted with a series of terminal and strained alkenes including norbornene,24a ethylene, propylene, hex-1-ene, 3,3-dimethyl-1-butene, allylbenzene and 4-allylanisole. Remarkably all these reactions are reversible under mild conditions (Scheme 4).24b
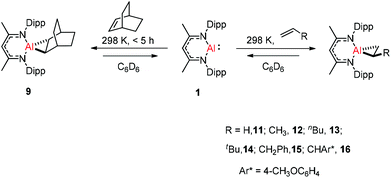 |
| | Scheme 4 Reversible alkene binding to 1. | |
2.2. Cleavage of the M–X single bond
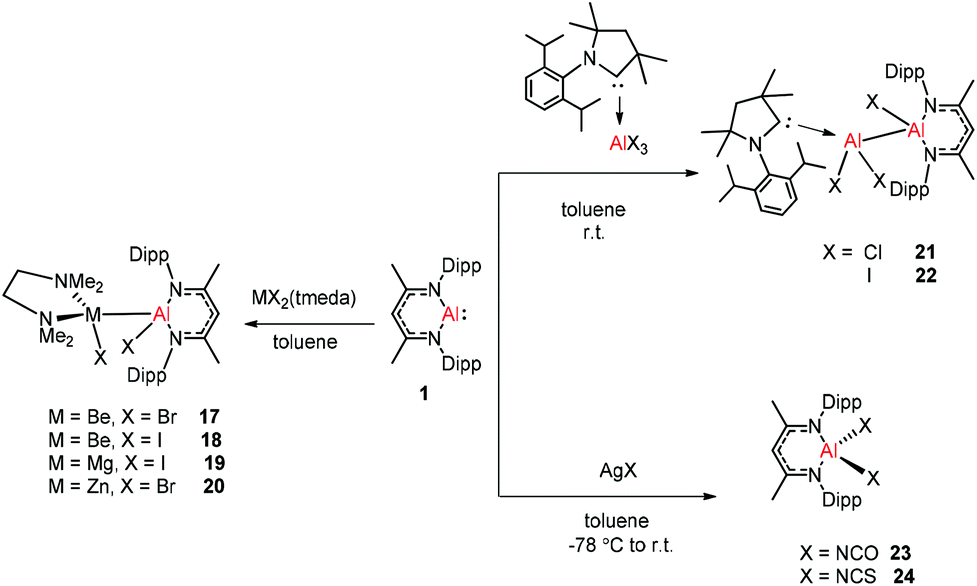
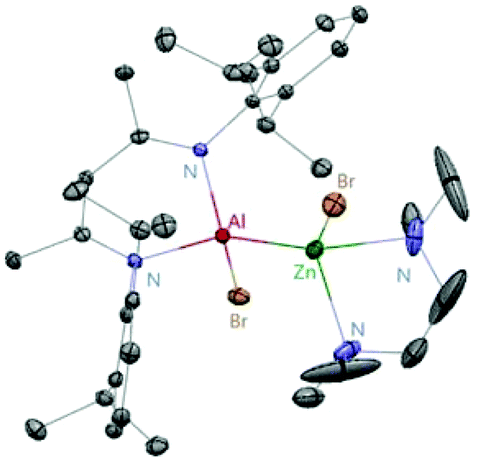
Aluminum(I) 1 has been developed to act as a synthon for the preparation of aluminum–metal bonded compounds via oxidative insertion of the Al center into metal-halogen linkages. Jones et al. reported the first example of molecular complexes containing an unsupported Be–Al bond. The Be–Al bonded complexes 17 and 18 were obtained as yellow crystalline solids from the reaction of DippNacNacAl(I) (1) with [BeX2(tmeda)] (X = Br or I, tmeda = tetramethylethylenediamine) in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 stoichiometry (Scheme 5). The Be–Al bond distances in 17 and 18 are 2.474(1) Å and 2.432(6) Å, respectively. They are significantly longer than the sum of single bond covalent radii of the elements (2.28 Å). DFT calculations reveal that the compounds with metal–metal bonds have a high s-character. This is consistent with similar Pauling electronegativities between Al and Be. The isostructural Mg–Al (19) and Zn–Al (20) analogues of these complexes have been isolated in the 1:1 reaction of DippNacNacAl(I) (1) with [MgI2(tmeda)] and [ZnBr2(tmeda)], respectively (Scheme 5).25 The composition of compound 20 was confirmed by means of single-crystal X-ray structural analysis (Fig. 3). Compound 20 has distorted tetrahedral Al and Zn centers. The Zn–Al bond distance is 2.471(1) Å, which is longer than the sum of covalent radii of the elements (2.44 Å). DippNacNacGa(I) (2) shows no reactivity towards [BeX2(tmeda)], even at elevated temperature. Roesky et al. reported unsymmetrical dialumanes by the disproportionation of DippNacNacAl(I) (1) with (Me2cAAC)AlX3 (X = Cl, I) adducts (Scheme 5). The Al–Al bond lengths in compound 21 (2.6327(11) Å) and compound 22 (2.5953(16) Å) are slightly shorter than those of symmetric Al–Al bond lengths, owing to the relaxation of the electrostatic repulsion between the Al atoms.26 The reactions of DippNacNacAl(I) (1) with AgX (X = OCN, SCN) resulted in compounds 23 and 24 containing two pseudohalide groups coordinated to the aluminum(III) center.27 The reactions proceed via oxidative addition of the pseudohalides and elimination of the silver metal.
:1 stoichiometry (Scheme 5). The Be–Al bond distances in 17 and 18 are 2.474(1) Å and 2.432(6) Å, respectively. They are significantly longer than the sum of single bond covalent radii of the elements (2.28 Å). DFT calculations reveal that the compounds with metal–metal bonds have a high s-character. This is consistent with similar Pauling electronegativities between Al and Be. The isostructural Mg–Al (19) and Zn–Al (20) analogues of these complexes have been isolated in the 1:1 reaction of DippNacNacAl(I) (1) with [MgI2(tmeda)] and [ZnBr2(tmeda)], respectively (Scheme 5).25 The composition of compound 20 was confirmed by means of single-crystal X-ray structural analysis (Fig. 3). Compound 20 has distorted tetrahedral Al and Zn centers. The Zn–Al bond distance is 2.471(1) Å, which is longer than the sum of covalent radii of the elements (2.44 Å). DippNacNacGa(I) (2) shows no reactivity towards [BeX2(tmeda)], even at elevated temperature. Roesky et al. reported unsymmetrical dialumanes by the disproportionation of DippNacNacAl(I) (1) with (Me2cAAC)AlX3 (X = Cl, I) adducts (Scheme 5). The Al–Al bond lengths in compound 21 (2.6327(11) Å) and compound 22 (2.5953(16) Å) are slightly shorter than those of symmetric Al–Al bond lengths, owing to the relaxation of the electrostatic repulsion between the Al atoms.26 The reactions of DippNacNacAl(I) (1) with AgX (X = OCN, SCN) resulted in compounds 23 and 24 containing two pseudohalide groups coordinated to the aluminum(III) center.27 The reactions proceed via oxidative addition of the pseudohalides and elimination of the silver metal.
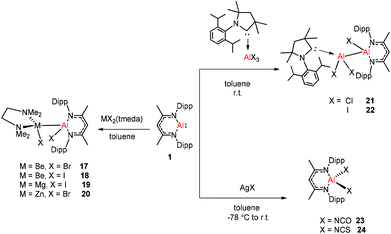 |
| | Scheme 5 Synthesis of compounds 17–24. | |
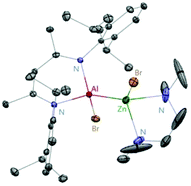 |
| | Fig. 3 X-ray single crystal structure of compound 20. | |
Harder et al. reported a combined attack of [(DippNacNac)Ca+·(C6H6)][B(C6F5)4−] and DippNacNacAl(I) (1), which led to the complete dearomatization of benzene to give C6H62− that chelates to the Al(III) center (Scheme 6).
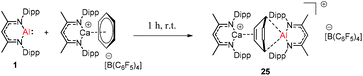 |
| | Scheme 6 Activation of benzene using Al(I) and Ca2+. | |
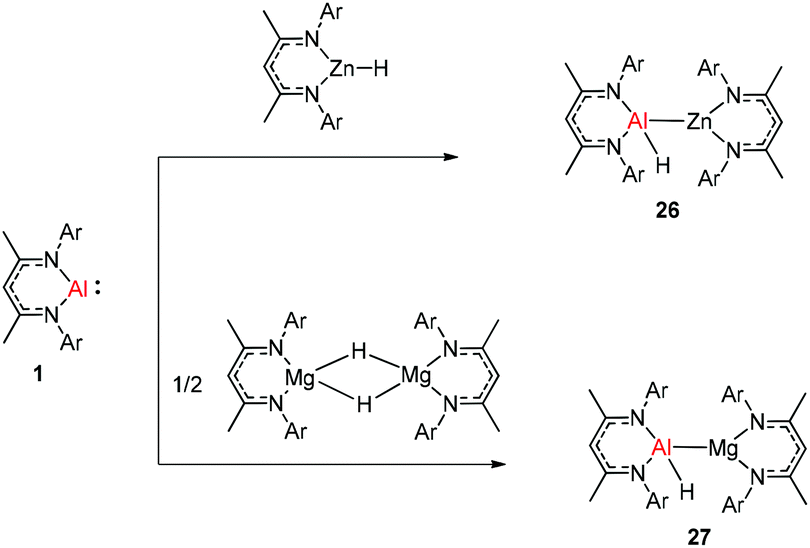
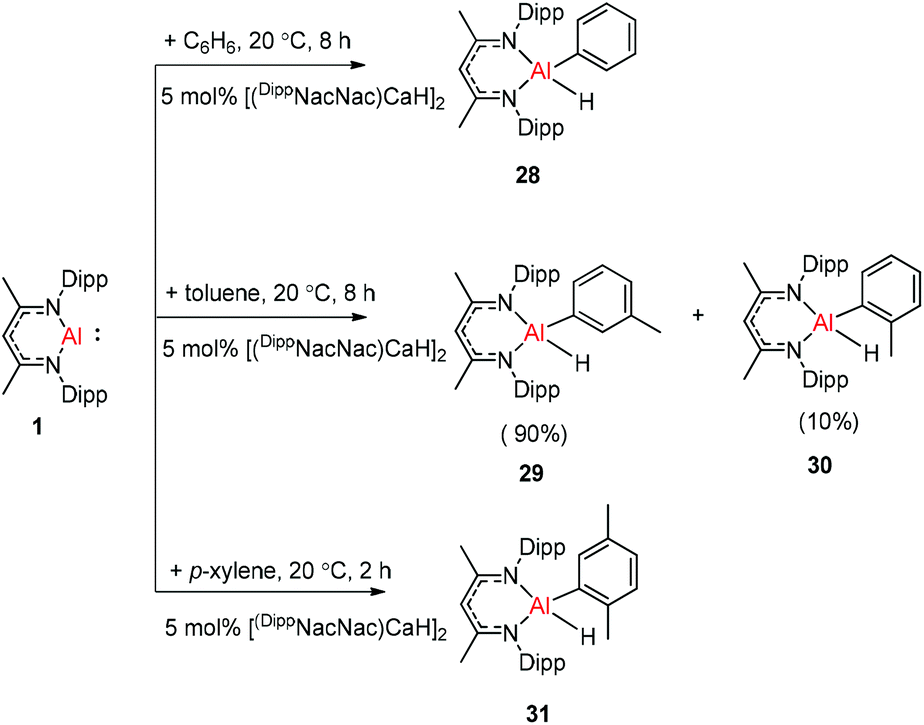
The molecular structure of 25, however, showed a heterobimetallic complex in which the C6H62− fragment is bridging to Ca and to the Al center.28a Crimmin et al. reported the first catalytic method for the transformation of C–H bonds in unactivated arenes (benzene, toluene and xylene) into C–Al bonds and proposed a mechanism by which the C–H bond is activated by an unusual Al–Pd intermetallic complex.28b Very recently, Harder et al. have described the stoichiometric reactions of DippNacNacAl(I) (1) with [(DippNacNac)MgH]2 and (DippNacNac)ZnH, respectively in benzene, where the Al center inserts into the metal hydride bond, which results in (DippNacNac)Al(H)M(DippNacNac) complexes (M = Zn 26, Mg 27) (Scheme 7).28c However, the reaction of the calcium hydride complex with 1 in benzene followed a different course leading to benzene C–H alumination. The cleavage of the sp2 C–H bond in unactivated arenes (benzene, toluene and xylene) by the low valent Al(I) complex 1 at room temperature has been achieved by the catalytic presence of [(DippNacNac)CaH]2 (Scheme 8). The possible pathway is the combined action of nucleophilic Al and arene activation by π-coordination to a Lewis acidic Ca center, which is parallel to the first aluminyl anions.11c
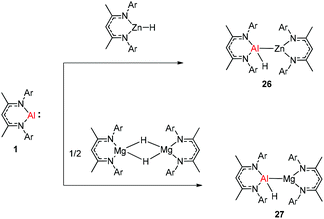 |
| | Scheme 7 Synthesis of (DippNacNac)Al(H)M(DippNacNac) complexes (M = Zn 26 and Mg 27). | |
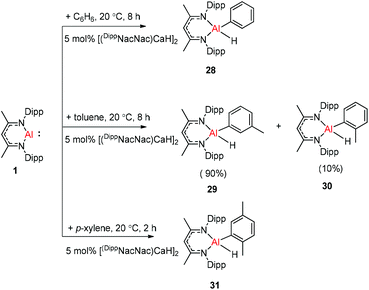 |
| | Scheme 8 Calcium hydride catalyzed C–H bond activation of unactivated arenes by oxidative addition of DippNacNacAl(I) (1). | |
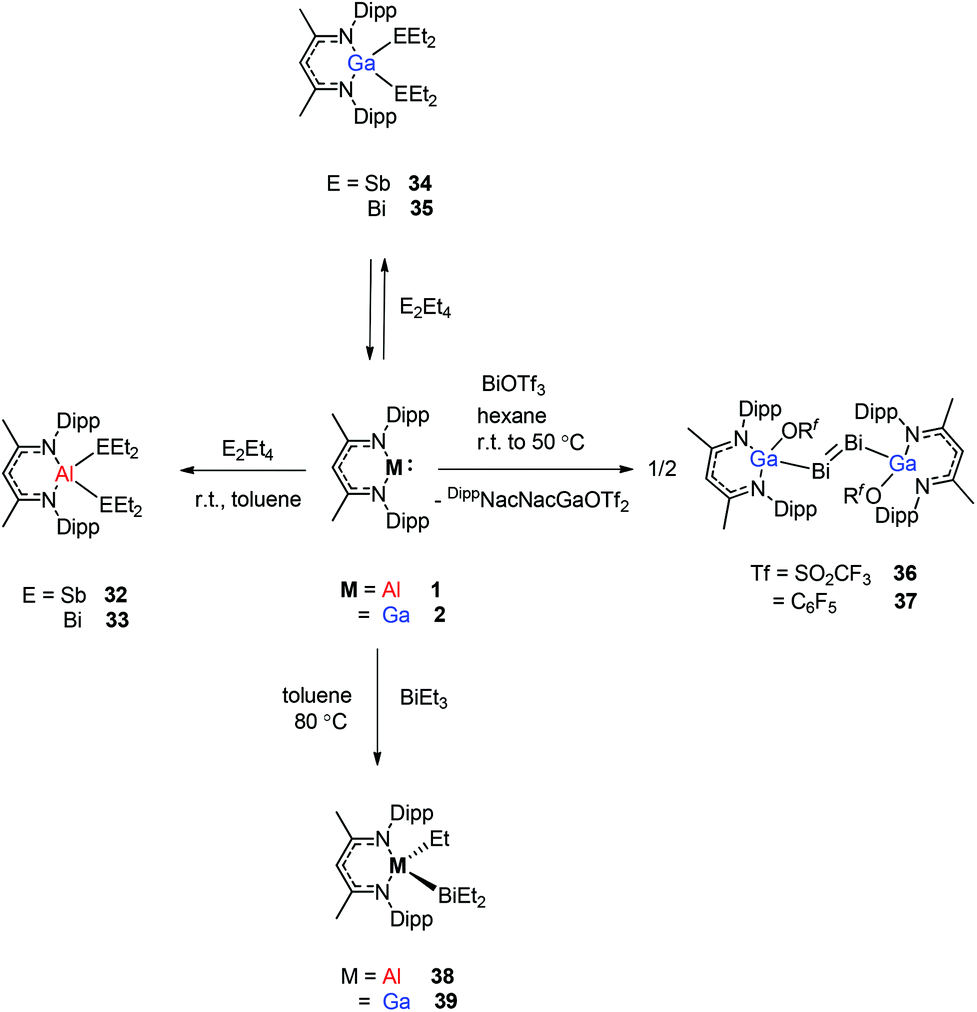
Schulz et al. reported that a solution of DippNacNacAl(I) (1) reacts with E2Et4 (E = Sb, Bi) in toluene to insert DippNacNacAl(I) (1) into the E–E bond with the formation of DippNacNacAl(EEt2)2 (E = Sb 32, Bi 33) (Scheme 9). Orange crystals of compound 33 were isolated from hexane at −30 °C, and slowly decompose in solution with the formation of BiEt3 and elemental Bi. The analogous reaction of DippNacNacGa(I) (2) with E2Et4 is fully reversible and temperature dependent. Analytically pure compounds 34 and 35 were isolated from the 1:1 mixture of DippNacNacGa(I) (2) and E2Et4, respectively (Scheme 9). By changing the molar ratio of DippNacNacGa(I) (2) and Bi2Et4 into a 1:2 ratio, 35 can be isolated in good yield.29 Fischer et al. have achieved the reaction of DippNacNacGa(I) (2) with Bi(OSO2CF3)3 and [{Bi-(OC6F5)3(toluene)}2] to yield [(RfO)(DippNacNac)GaBiBiGa((DippNacNac)(OTf)] (Tf = SO2CF336, C6F537). The dibismuthenes show short Bi–Bi bond lengths of 2.8111(2) and 2.8182(4) Å, respectively (Fig. 4). The reaction proceeds via oxidative addition of the Bi–O bond to DippNacNacGa(I) (2) and concomitant elimination of DippNacNacGa(OTf)2. Finally, Bi(OTf)3 adds to another equiv. of 2 with subsequent dimerization to form a new type of dibismuthene stabilized by NHC-related ligands.30 Schulz et al. described a similar insertion reaction of DippNacNacM with BiEt3 that leads to DippNacNacMEt(BiEt2) (M = Al 38 and Ga 39) (Scheme 9). The consecutive second activation proceeds at a higher temperature through the reductive elimination of DippNacNacMEt2, elemental Bi and BiEt3.31
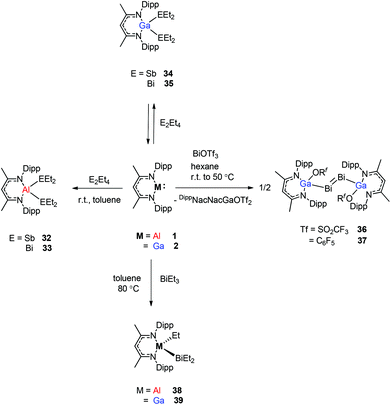 |
| | Scheme 9 Cleavage of Sb–Sb and Bi–Bi bonds by DippNacNacAl(I) and DippNacNacGa(I), respectively. | |
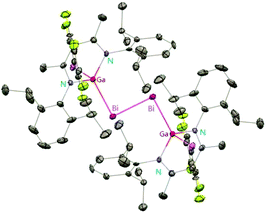 |
| | Fig. 4 X-ray single crystal structure of compound 36. | |
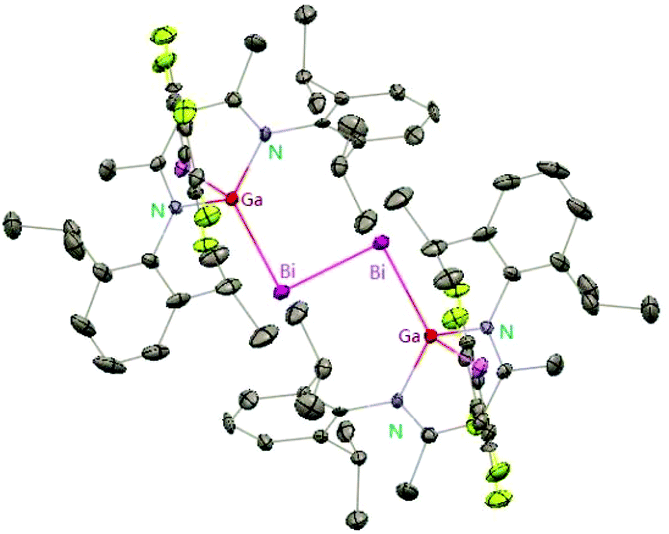
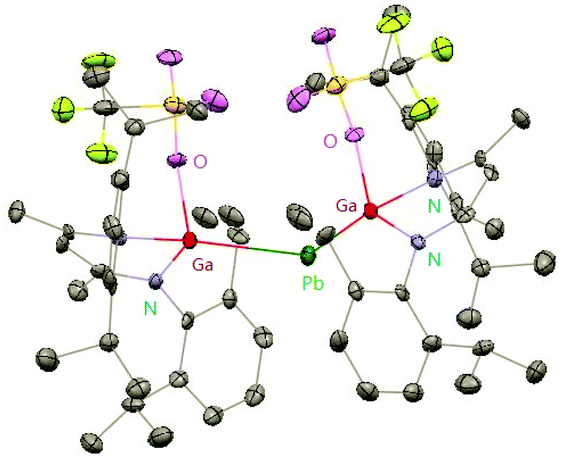
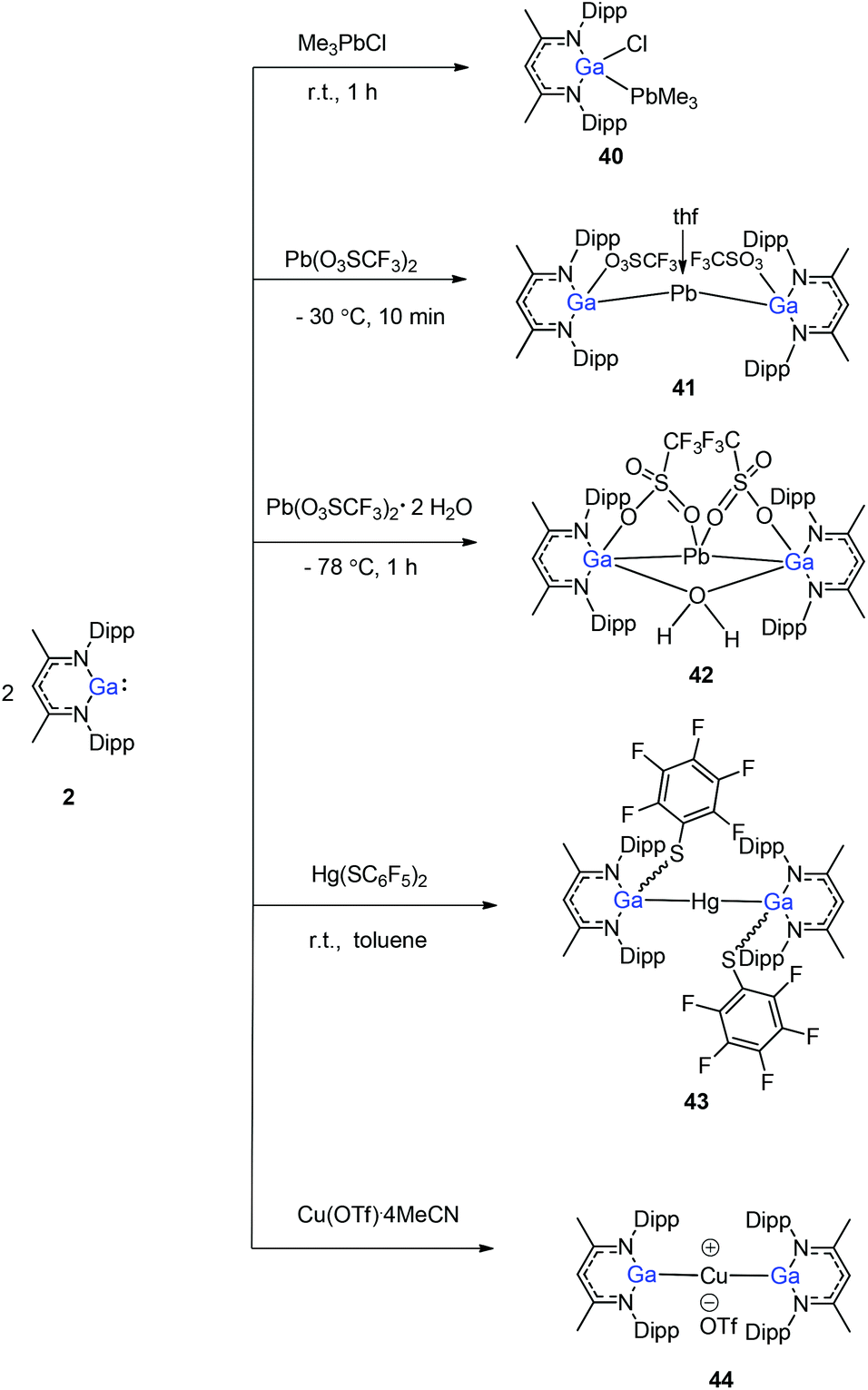
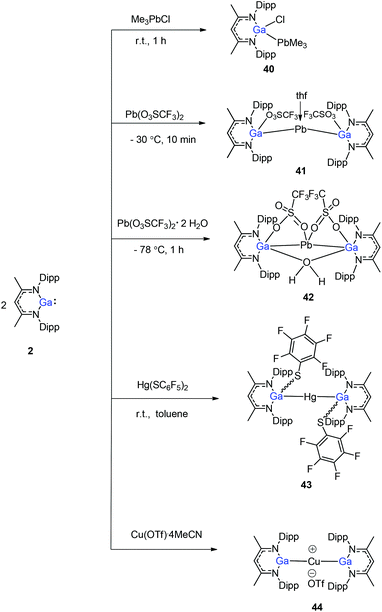
Heavy-metal complexes containing gallium-lead and gallium-mercury bonds were derived from the oxidative addition of DippNacNacGa(I) (2) with the corresponding metal precursors. The reaction of Me3PbCl with DippNacNacGa(I) (2) in THF at ambient temperatures afforded compound [{(DippNacNac)Ga(Cl)}PbMe3] (40) in high yield. In addition, the reaction between [Pb(OSO2CF3)2] and DippNacNacGa(I) (2) (two equiv.) leads to the complex 41 containing a Ga–PbII–Ga linkage (Fig. 5). When two equiv. of DippNacNacGa(I) (2) were treated with [Pb(OSO2CF3)2·2H2O] in THF, deep red crystals of 42 were formed in very poor yield (Scheme 10). The structure of the compound consists of a bent Ga–Pb–Ga backbone with a bridging triflate group between the Ga–Pb bond and a weakly interacting water molecule at the gallium center. Similarly, the reaction of mercury thiolate Hg(SC6F5)2 with DippNacNacGa(I) (2) (two equiv.) produced the bimetallic homoleptic compound 43 (Scheme 10).32 The linear cationic complex [{(DippNacNac)Ga}2Cu][OTf]·2C6H5F (44, Tf = SO2CF3) was obtained from the reaction of DippNacNacGa(I) (2) (two equiv.) with Cu(OTf)·4CH3CN.33
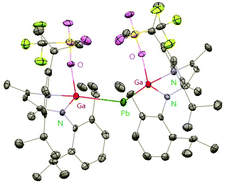 |
| | Fig. 5 X-ray single crystal structure of compound 41; the THF molecule attached to lead is omitted for clarity. | |
 |
| | Scheme 10 The oxidative addition of Pb/Hg/Cu salts to DippNacNacGa(I) (2). | |
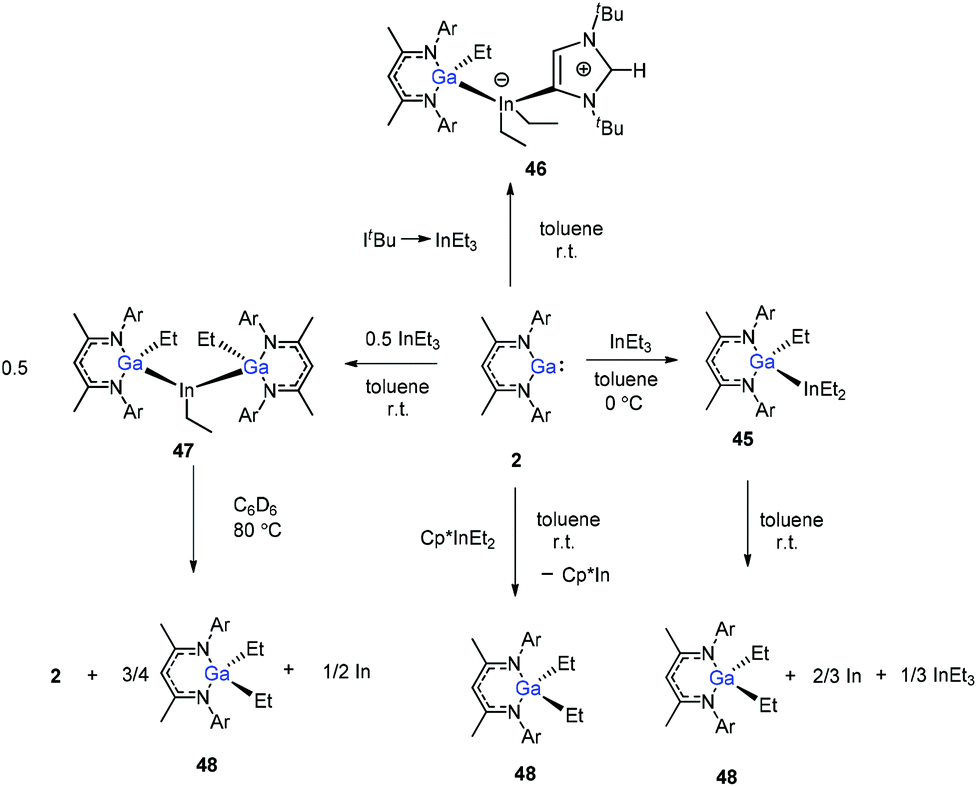
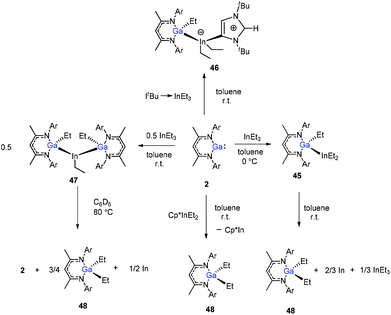
In addition, Schulz et al. showed that DippNacNacGa(I) (2) inserted into the In–C bond of InEt3 resulted in the formation of DippNacNacGaEt(InEt2) (45). The solution of 45 in benzene and toluene tends to decompose slowly at ambient temperature. But the reaction of DippNacNacGa(I) (2) with ItBu → InEt3 {ItBu = [C(Nt-Bu2CH)2]} resulted in a stable compound 46, where the carbene coordinates to the indium atom. The reaction of InEt3 with 2 equiv. of DippNacNacGa(I) (2) resulted in a double insertion product [DippNacNacGa(Et)]2InEt (47) (Scheme 11), while further insertion of DippNacNacGa(I) (2) into the remaining In–Et bond does not occur. Complex 57 gradually decomposes at 80 °C in C6D6, resulting in DippNacNacGa (2) and DippNacNacGaEt2 (48). A similar reaction between DippNacNacGa(I) (2) and Cp*InEt2 (Cp* = C5Me5) leads to the production of DippNacNacGaEt2 (48). These reactions proceeded via the oxidation of DippNacNacGa(I) (2) with the In–Et bond and subsequent reductive elimination of DippNacNacGaEt2 from the indium(III) center.35
 |
| | Scheme 11 Syntheses and thermal stability of compounds 45–48. | |
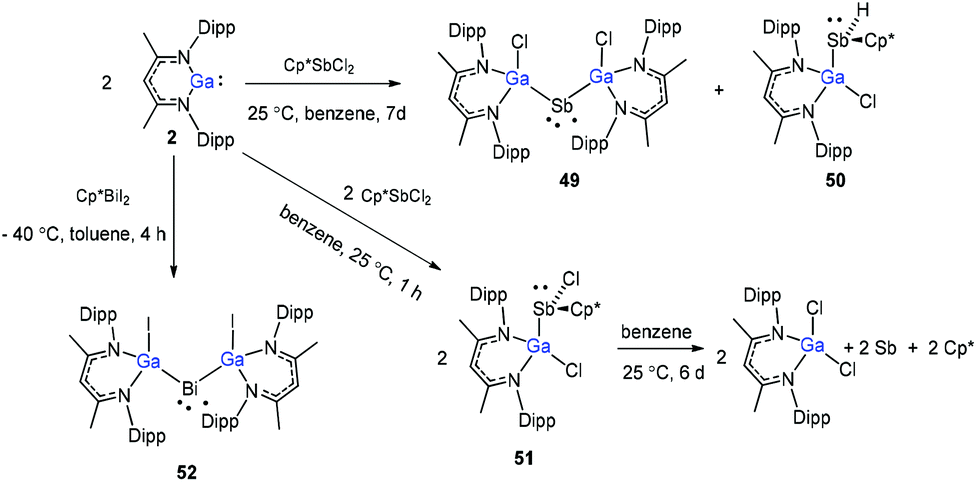
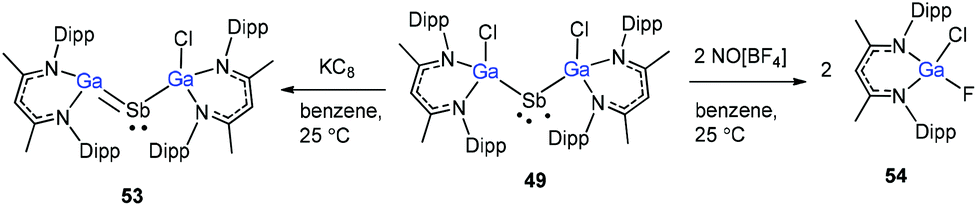
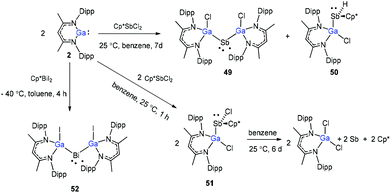
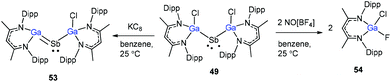
Schulz and co-workers have also reported a stibinyl radical [DippNacNac(Cl)Ga]2Sb˙ (49) by the reaction of two equiv. of DippNacNacGa(I) (2) with Cp*SbCl2 (Scheme 12), and the traces of DippNacNac(Cl)GaSb(H)Cp* (50) were formed as a byproduct. Similarly, the equimolar reaction yielded DippNacnac(Cl)GaSb(Cl)Cp* (51), which slowly decomposed to DippNacNacGaCl2, decamethylfulvalen (Cp*2) and the antimony metal. The analogous reaction of DippNacNacGa(I) (2) with Cp*BiI2 yielded a bismuthinyl radical [DippNacNac(I)Ga]2Bi˙ (52) (Scheme 12). Their formation illustrates the stepwise insertion of DippNacNacGa(I) (2) into the E–X bond of Cp*EX2 followed by the homolytic bond cleavage of the E–Cp* bond and elimination of Cp*2. Theoretical calculations showed the significant electron delocalization of the Sb and Bi unpaired radicals onto the Ga ligands. Compounds 49 and 52 adopt V-shaped geometries with Ga–E–Ga bond angles of 104.89(1)° (49) and 106.68(3)° (52), respectively. Compound 53 was isolated by the direct reaction of 49 with KC8 in benzene, which gives the first structurally characterized compound containing a GaSb double bond (Scheme 13). In contrast, the equimolar redox reaction of 2 with the single-electron oxidant [NO][BF4] occurred with the formation of DippNacNacGaClF (54).36
 |
| | Scheme 12 Synthesis of compounds 49–52. | |
 |
| | Scheme 13 Single-electron oxidation and reduction reactions of compound 49. | |
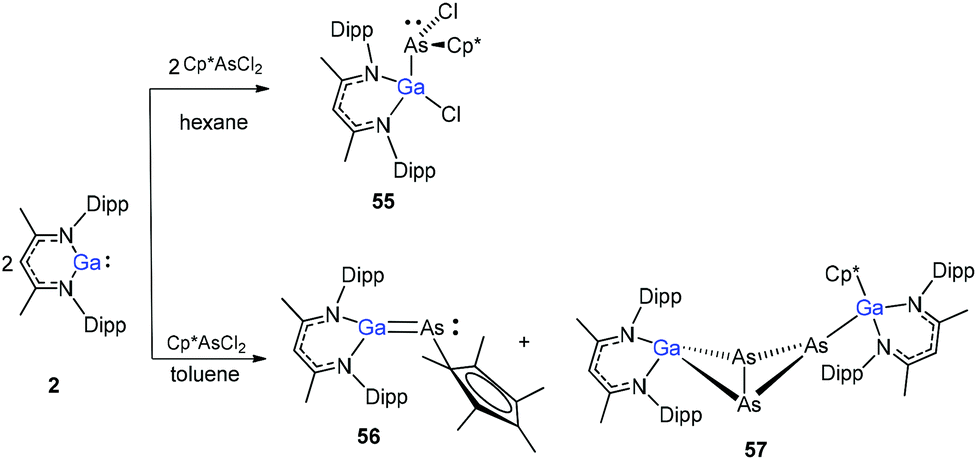
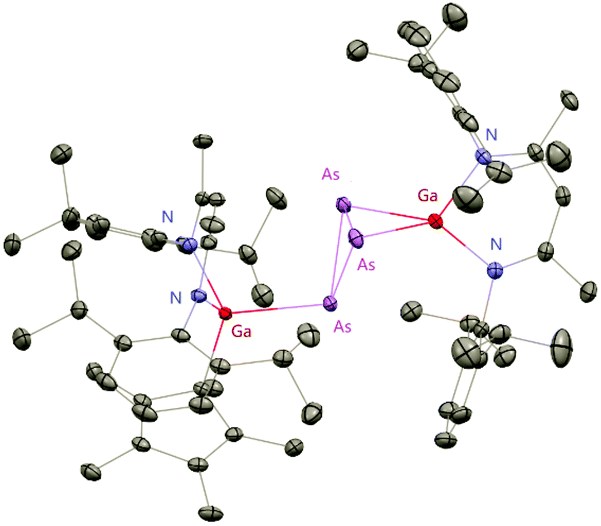
The Cp*AsCl2 (Cp* = C5Me5) reacted with one equiv. of DippNacNacGa(I) (2) with the insertion of DippNacNacGa(I) (2) into one As–Cl bond and the subsequent formation of DippNacNac(Cl)GaAs(Cl)Cp* (55) (Scheme 14). In contrast, the reaction of two equiv. of DippNacNacGa(I) (2) with Cp*AsCl2 proceeds with the formation of gallaarsene DippNacNacGaAsCp* (56) with a GaAs double bond and (DippNacNacGaCp*)(DippNacNacGa)(η1,η2,μ-As3) (57), containing a central GaAs3 butterfly type core. The central structural motif of 57 is the bridging As3 three-membered ring, which coordinates in an η1 and η2 fashion to two Ga atoms, respectively (Fig. 6).37
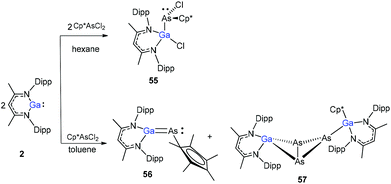 |
| | Scheme 14 The reaction of DippNacNacGa(I) (2) with Cp*AsCl2. | |
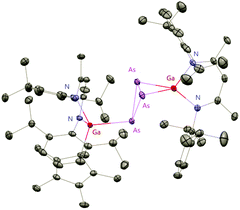 |
| | Fig. 6 X-ray single crystal structure of compound 57. | |
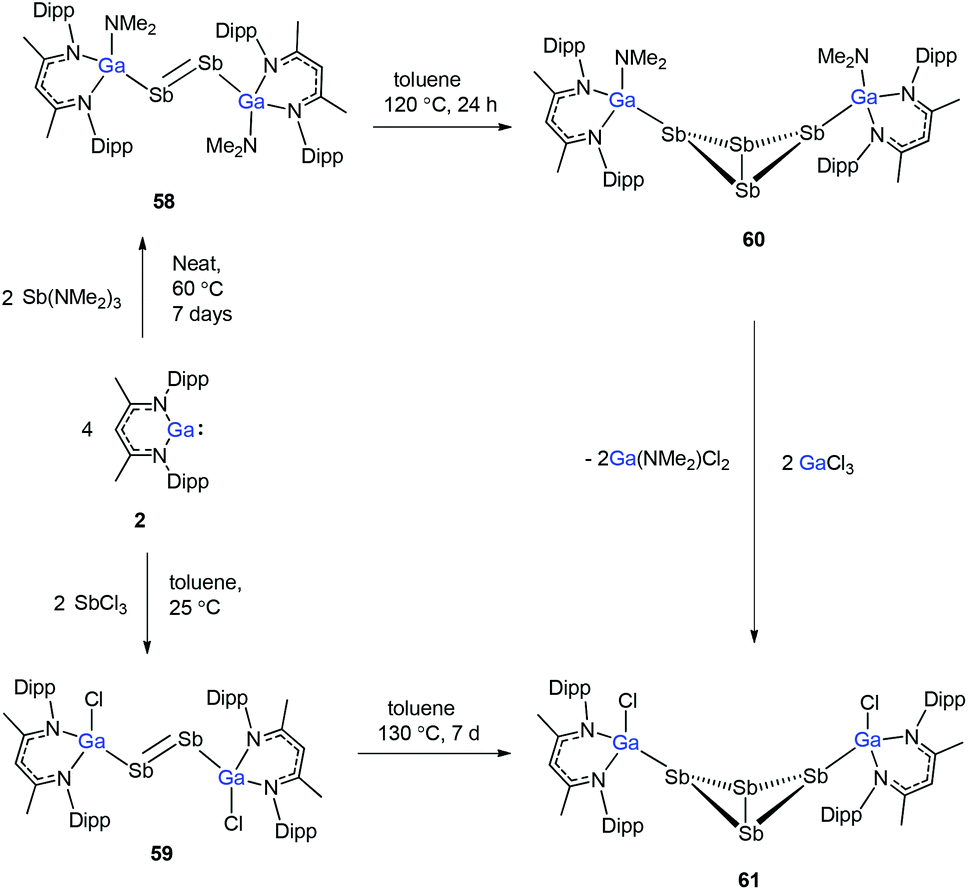
To evaluate the reduction potential of DippNacNacGa(I) (2) in detail, Schulz et al. studied the reactions of DippNacNacGa(I) (2) with other SbIII reagents. DippNacNacGa(I) (2) reacted with SbX3 (X = NMe2, Cl) in a 2:1 molar ratio to form Ga-substituted distibenes [(DippNacNacGaX)2Sb2] (X = NMe258, Cl 59) (Scheme 15). Heating a toluene solution of 58 at 120 °C for 24 h yielded [(DippNacNacGaNMe2)2(μ,η1:1-Sb4)] (60). But complex 59 required 7 days of heating at 130 °C to give [(DippNacNacGaCl)2( μ,η1:1-Sb4)] (61). Compound 58 can react with GaCl3 to form 59; in addition, 59 reacts with Li amide to form 58. The central Sb4 unit in 60 and 61 adopts a butterfly type conformation (Fig. 7). Interestingly, compound 60 reacted with GaCl3 in an amide/Cl exchange reaction with the subsequent formation of 61.38
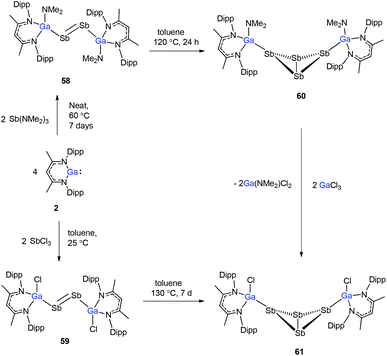 |
| | Scheme 15 Syntheses of compounds 58–61. | |
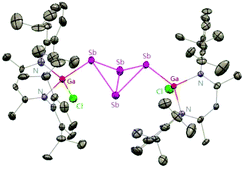 |
| | Fig. 7 X-ray single crystal structure of compound 61. | |
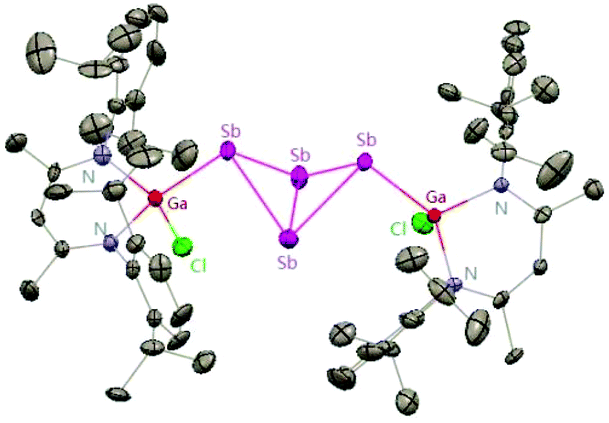
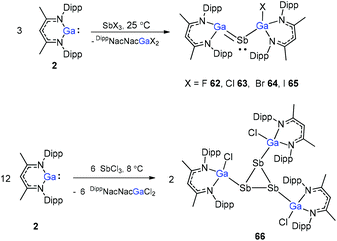
To better understand the reaction mechanism leading to compounds containing SbSb and GaSb double bonds, the reactions of SbCl3 with different equiv. of DippNacNacGa(I) (2) at different temperatures were investigated. The DippNacNacGaSbGa(X)DippNacNac (X = F 62, Cl 63, Br 64, and I 65) compounds were formed by a twofold insertion of DippNacNacGa(I) (2) into two Sb–X bonds, followed by an intramolecular elimination of DippNacNacGaX2 (Scheme 16). The reactions of two equiv. of DippNacNacGa(I) (2) with SbCl3 at 8 °C yielded cyclotristibine 66. The Sb–Sb bond (2.8205(3)–2.8437(3) Å) in cyclotristibine 66 is typical of the Ga–Sb single bond.39
 |
| | Scheme 16 Syntheses of gallastibenes containing GaSb double bonds and cyclotristibine. | |
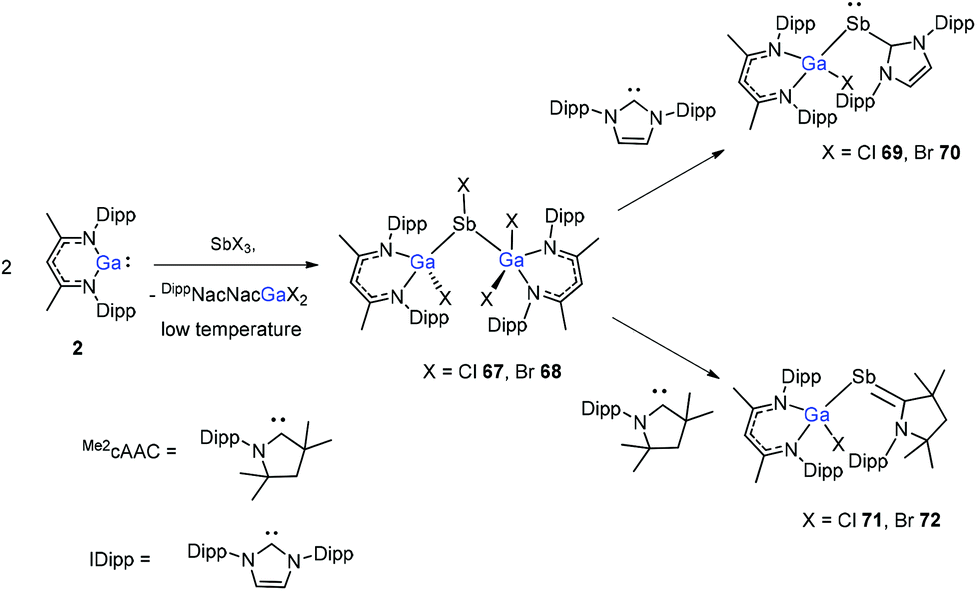
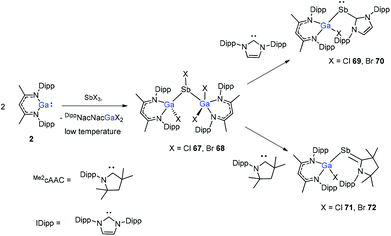
The insertion reactions of two equiv. of DippNacNacGa(I) (2) with SbX3 (X = Cl, Br) at low temperature produced a double-inserted product (DippNacNacGaX)2SbX (X = Cl 67, Br 68). In addition, the reaction of isolated compounds 67 and 68 with strong σ-donating carbenes (IDipp(1,3-bis-(2,6-diisopropylphenyl)imidazol-2-ylidene), Me2cAAC) yielded four carbene-stabilized stibinidenes (69–72) (Scheme 17). The studies reveal that IDipp-stabilized stibinidenes 69 and 70 show Sb–Ccarbene single bonds, whereas the Me2cAAC-stabilized derivatives 71 and 72 exhibit Sb–Ccarbene π-backbonding character.40
 |
| | Scheme 17 Syntheses of IDipp- (69 and 70) and Me2cAAC-stabilized stibinidenes (71 and 72). | |
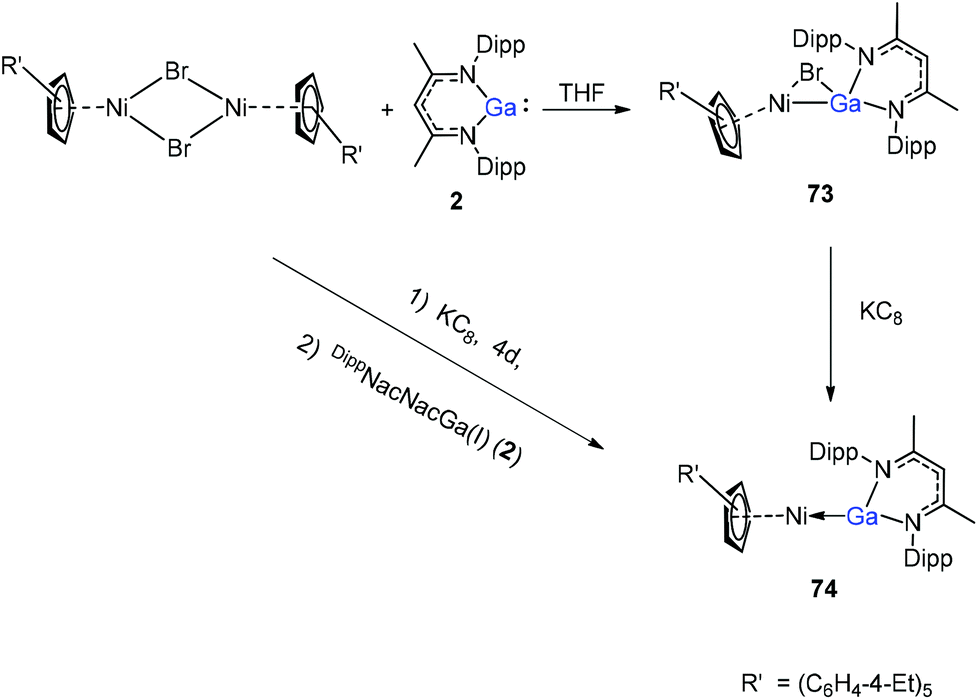
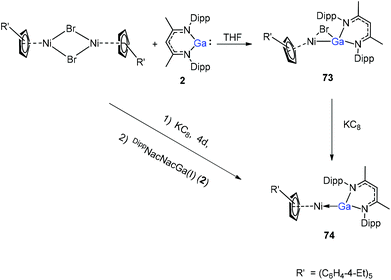
A red-brown complex 73 was obtained by the reaction of the half-sandwich complex [CpR′Ni(μ-Br)]2 (CpR′ = C5(C6H4-4-Et)5) with DippNacNacGa(I) (2) in THF (Scheme 18).41 The CpR′(centroid)–Ni–Ga linkage is bent, and the nickel atom is surrounded by an η5-coordinated CpR′ ligand and a σ-coordinated Ga(DippNacNac) ligand, while the bromide bridges the Ni–Ga bond. The reduction of 73 with KC8 afforded compound 74 where the “CpR′Ni(I)” fragment is trapped by DippNacNacGa. The reduction of [CpR′Ni(μ-Br)]2 with KC8 and the subsequent addition of DippNacNacGa(I) (2) also result in compound 74 albeit in smaller yield (Scheme 18).
 |
| | Scheme 18 Syntheses of complexes 73 and 74. | |
2.3. Cleavage of E′–E single and E′![[double bond, length as m-dash]](https://www.rsc.org/images/entities/h3_char_e001.gif) E double bonds
E double bonds
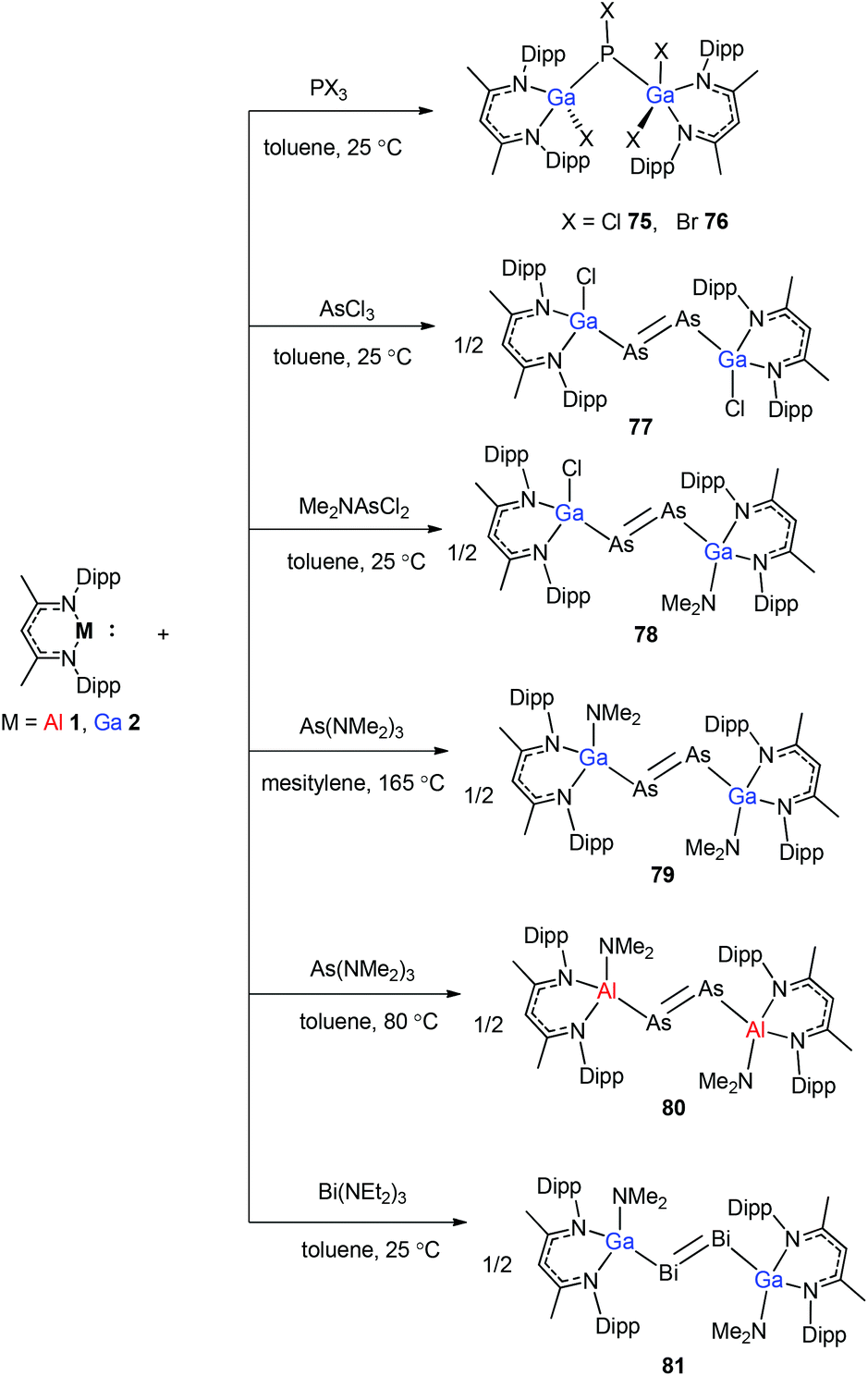
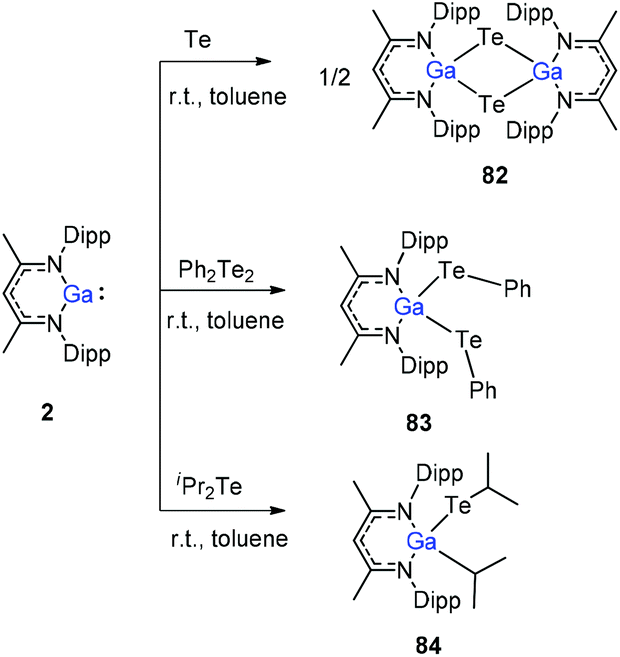
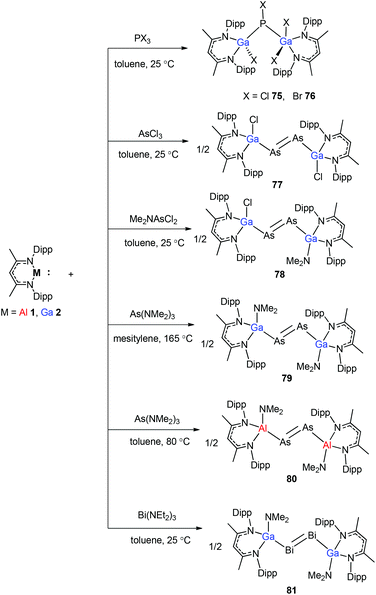
To stabilize Ga-coordinated dipnictenes of the type [DippNacNac(X)Ga]2E2 (E = P–Bi), the reactions of DippNacNacGa(I) (2) with phosphorus, arsenic, and bismuth halides and amides were studied.42 Two equiv. of DippNacNacGa(I) (2) reacted with PX3 (X = Cl, Br) in toluene at ambient temperature with the insertion of DippNacNacGa(I) (2) into two P–X bonds, which resulted in [DippNacNac(X)Ga]2PX (X = Cl 75, Br 76) (Scheme 19). Similar twofold insertion reactions into AsCl3 and the subsequent elimination of DippNacNacGaCl2 resulted in the formation of the stable Ga-coordinated diarsene species, which was isolated as a green crystalline solid 77 (Scheme 20). The analogous reaction with Me2NAsCl2 yielded unsymmetrically-substituted diarsene [DippNacNac(Cl)Ga]AsAs[Ga(NMe2)DippNacNac] (78) (Scheme 20). In contrast, the reaction of DippNacNacGa(I) (2) with As(NMe2)3 required much harsher reaction conditions. A mixture of DippNacNacGa(I) (2) and As(NMe2)3 heated at 165 °C for 5 days resulted in compound 79 (Scheme 19). Its analogous reaction with DippNacNacAl(I) (1) yielded [DippNacNac(Me2N)Al]2As2 (80) after heating at 80 °C for one day (Scheme 20). Finally, the reaction of DippNacNacGa(I) (2) with Bi(NEt2)3 also occurred with the insertion and elimination of DippNacNacGa(NEt2)2 and resulted in the corresponding Ga-substituted dibismuthene [DippNacNac(Et2N)Ga]2Bi2 (81) (Scheme 19). The reaction of 2 with elemental tellurium yielded the Te-bridged compound [DippNacNacGa-μ-Te]2 (82). Moreover, the cleavage of the Te–Te and Te–C bonds upon reactions of 2 with Ph2Te2 and iPr2Te resulted in the formation of DippNacNacGa(TePh)2 (83) and DippNacNacGa(iPr)Te(iPr) (84), respectively (Scheme 20).34
 |
| | Scheme 19 Syntheses of complexes 75–81. | |
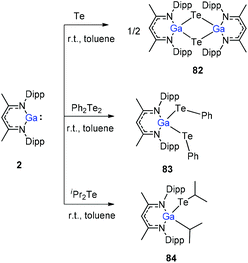 |
| | Scheme 20 Te–Te and Te–C bond cleavage reactions using DippNacNacGa(I) (2). | |
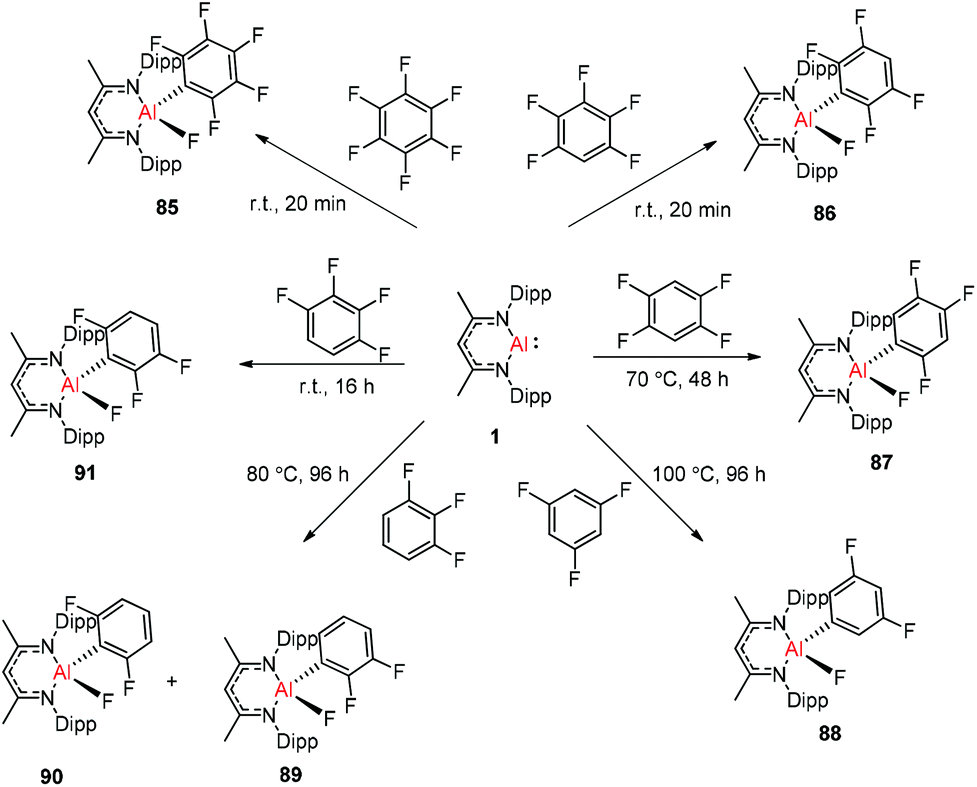
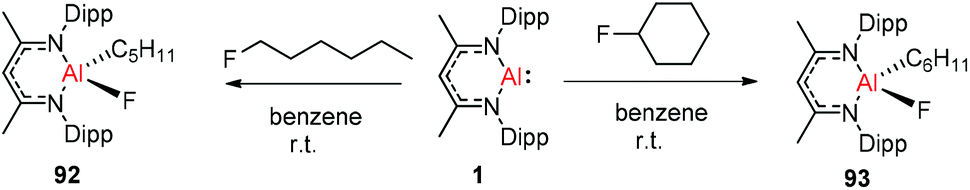
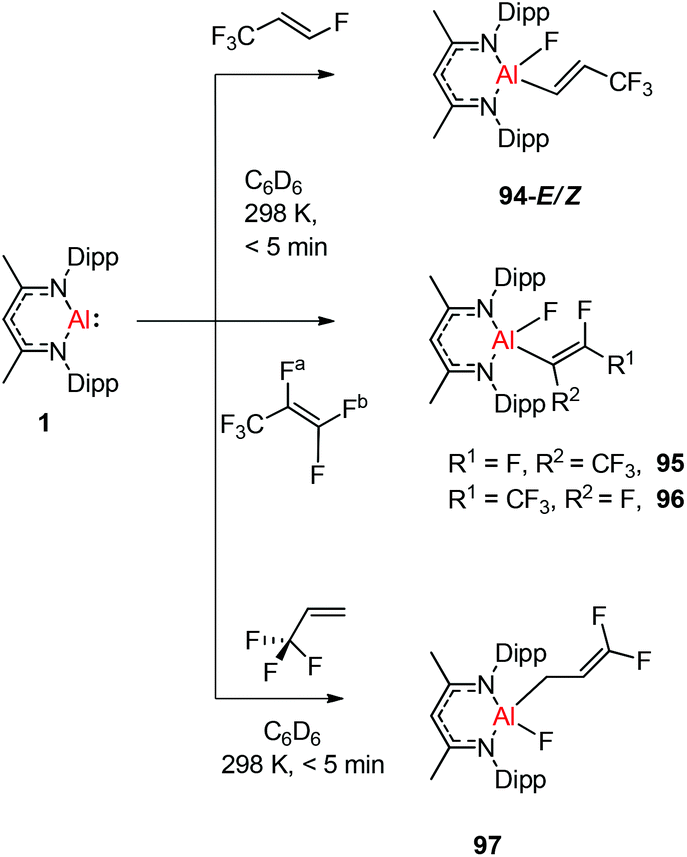
Nikonov and Crimmin groups separately reported the reactions of the monomeric Al(I) complex with various fluoroalkenes and fluoroarenes, resulting in the breaking of strong sp2 and sp3 C–F bonds. Aluminum(I) compound 1 undergoes a facile oxidative addition with aryl C–F bonds.43 The reaction of 1 with an excess of hexafluorobenzene or pentafluorobenzene resulted in compounds 85 and 86, respectively (Scheme 21). A further decrease in the number of fluorine atoms in the starting arene necessitates an increase in the reaction temperature to cleave the C–F bond. The cleaving ability decreases in the order o– > p– > m–. The addition of 1-fluorohexane or fluorocyclohexane to 1 at room temperature yielded the corresponding aluminum alkyl 92 and 93, respectively (Scheme 22).43a The reaction with (E)-1,3,3,3-tetrafluoro-propene(HFO-1234ze) resulted in the immediate formation of a 4:1 mixture of 94-E and 94-Z (Scheme 23). The addition of hexafluoropropene to 1 gave two products which were separated by fractional crystallization from hexane. 95 is formed from the internal sp2 C–F bond cleavage, while 96 is the result of breaking the terminal sp2 C–F bond trans to the CF3 group. The reaction of 1 with 3,3,3-trifluoropropene yielded 97 by the formation of a metallocyclopropane intermediate followed by β-fluoride elimination (Scheme 23).24a
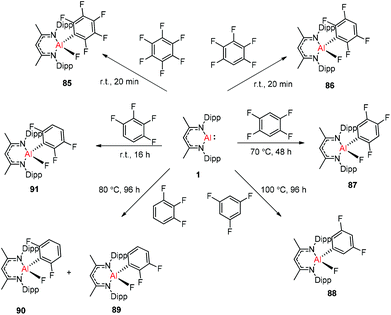 |
| | Scheme 21 Oxidation addition of C(sp2)–F bonds by 1. | |
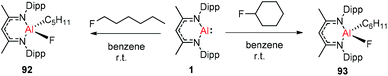 |
| | Scheme 22 Oxidation addition of C(sp3)–F bonds to 1. | |
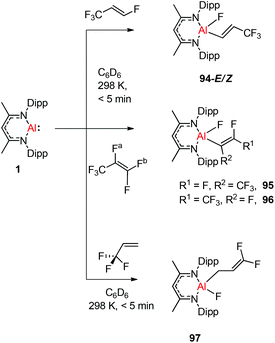 |
| | Scheme 23 Reactions of 1 with (E)-1,3,3,3-tetrafluoro-propene, hexafluoropropene and trifluoropropene, respectively. | |
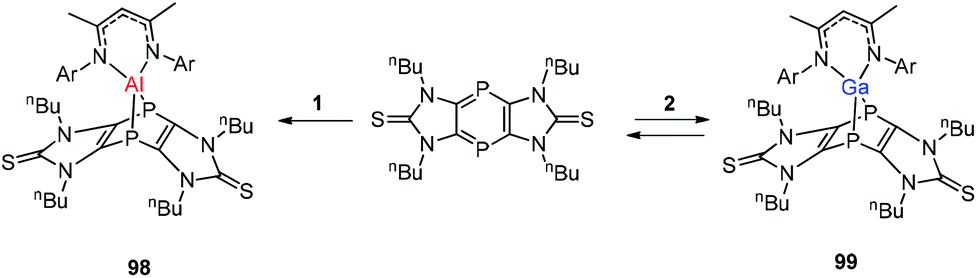
Streubel and co-workers described the reaction of monovalent compounds DippNacNacM (M = Al, Ga) with imidazole-2-thione based tricyclic 1,4-diphospinine, which produced the corresponding 7-metalla-1,4-diphosphanorbornadiene (98, 99) (Scheme 24).44a Previously Nikonov et al. described the oxidative cleavage of the CS bond at the metal center,44b while 98 and 99 undergo the [4 + 1] cycloaddition reaction, which is both kinetically and thermodynamically favorable.
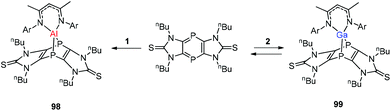 |
| | Scheme 24 Reaction of tricyclic 1,4-diphosphinine with DippNacNacAl(I) (1) and DippNacNacGa(I) (2) to get 7-metalla-1,4-diphosphanorbornadienes 98 and 99. | |
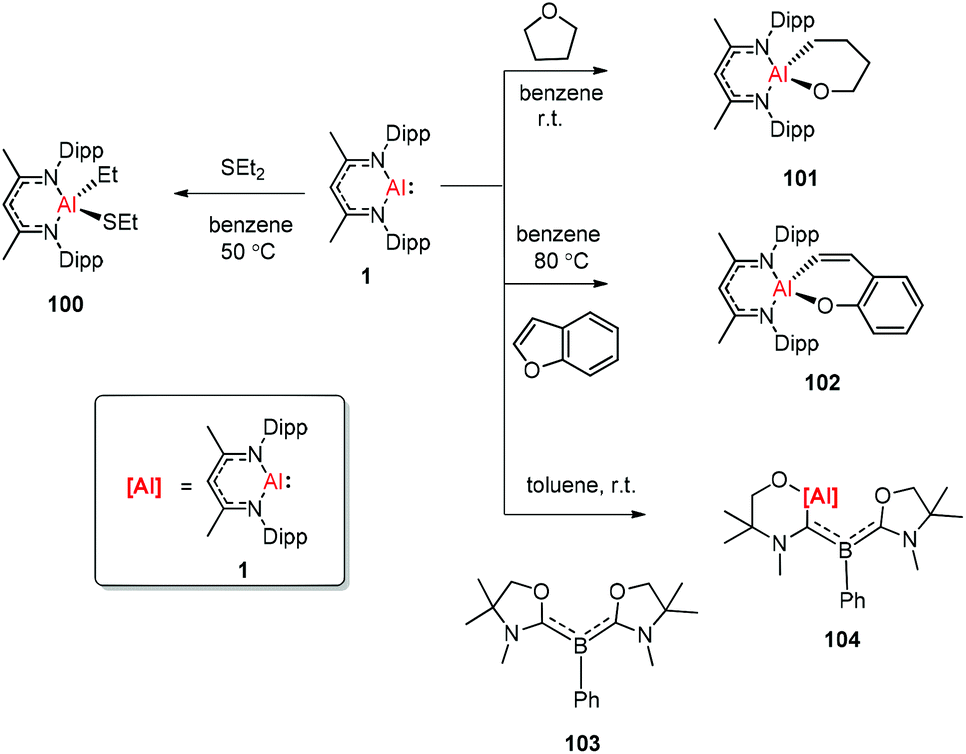
The aluminum(I) compound DippNacNacAl(I) (1) reacted with diethyl sulfide at 50 °C, which resulted in the oxidative addition of the C(sp3)–S bond. This is the first example of C(sp3)–S bond activation by a main-group element.45 The groups of Nikonov, Crimmin, and Kinjo independently reported the reactions of monomeric Al(I) compound 1 towards C–O bonds. The oxidative addition reaction of tetrahydrofuran with DippNacNacAl(I) (1) smoothly occurred at room temperature to give complex 101,43b while the reaction between 1 and benzofuran upon heating at 80 °C slowly converted it to product 102.43a The reaction of 1 with an equiv. amount of L′2PhB (103) (L′ = oxazol-2-ylidene) in toluene instantly occurred with the insertion of DippNacNacAl(I) (1) into the C–O bond, affording complex 104 involving an Al, N, and O mixed heterocyclic carbene or anionic (amino)(boryl)carbene derivative (Scheme 25).46
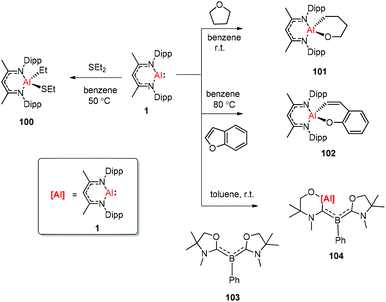 |
| | Scheme 25 C–O and C–S bond activation, respectively, by the aluminum(I) compound. | |
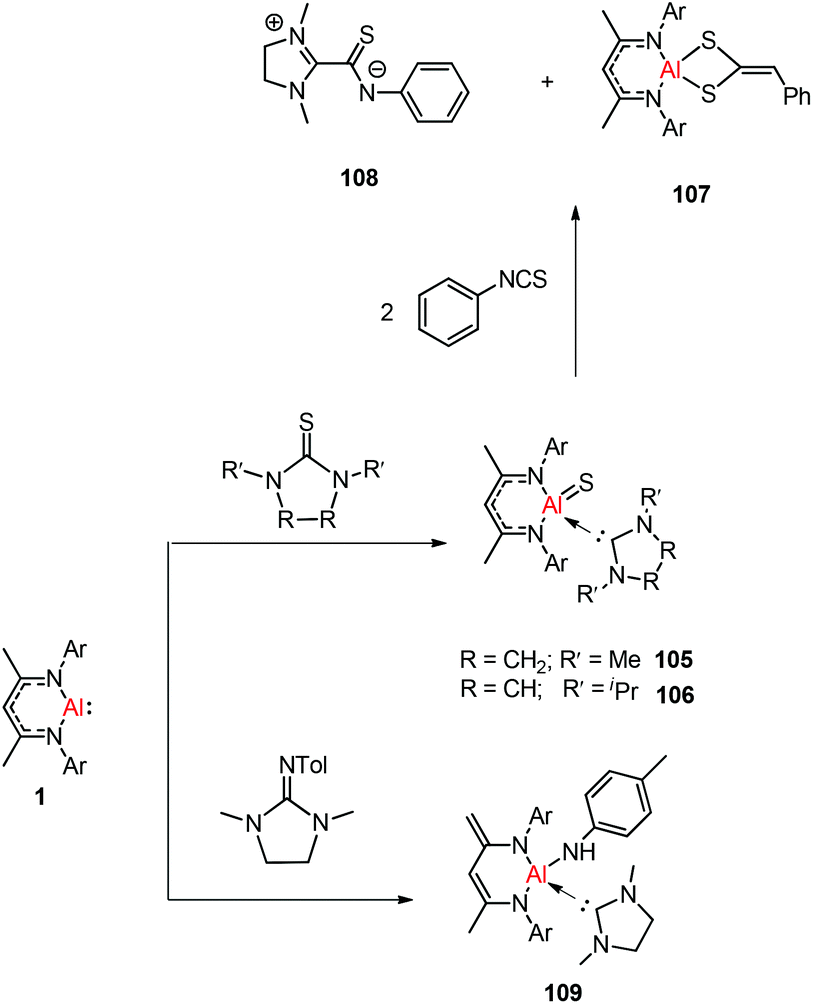
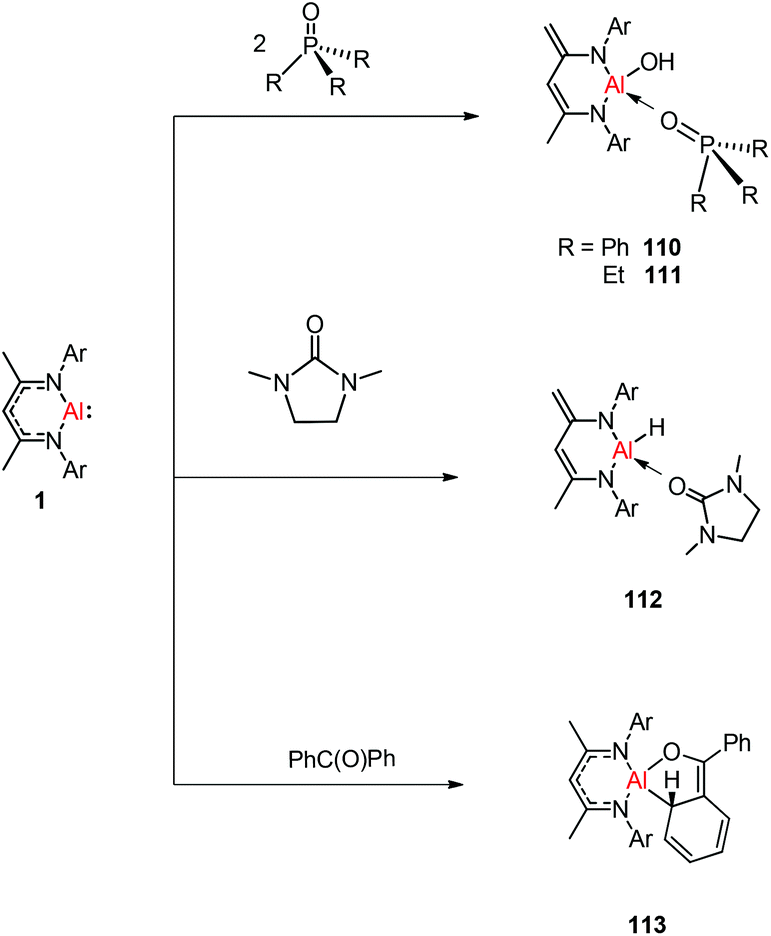
Treatment of DippNacNacAl(I) (1) with thiourea resulted in the first carbene-stabilized terminal aluminum sulfide complexes 105 and 106 by the oxidative cleavage of the CS bond. In contrast, the mixing of compound 1 and triphenylphosphine sulfide in a 1:1 ratio afforded a mixture of terminal sulfide DippNacNacAlS(SPPh3), unreacted 1, and free triphenylphosphine. The existence of the Al–S double bond in 105 and 106 was supported by DFT calculations. Complex 105 undergoes facile cycloaddition with phenyl isothiocyanate to form compound 107 along with zwitterion 108 obtained from the coupling between the liberated carbene and PhNCS (Scheme 26).44b To investigate the oxidative cleavage of the unsaturated bond of the CN unit, the reaction of DippNacNacAl(I) (1) with cyclic guanidine was accomplished and showed the unprecedented cleavage of the C–N multiple bond to give the carbene-ligated amido complex DippNacNacAl(NHTol)(SIMe) (SIMe = C{N(Me)CH2}2) (109). The splitting of the CN bond in 109 is the first example of the oxidative addition of the CN double bond to any metal center.47 The DFT study supported that the production of 109 occurs via an intermediate of aluminum imide as a result of the oxidative cleavage of TolNSIMe (SIMe = C{N(Me)CH2}2) by 1. The reactions of phosphine oxides with 1 occurred readily with the formation of hydroxyl derivatives DippNacNacAl(OH)(OPR3) (R = Ph 110, Et 111). The CO bond (179 kcal mol−1) is much stronger when compared with the PO bond (110 kcal mol−1). Therefore, the reaction of cyclic urea 1,3-dimethyl-2-imidazolidinone with 1 resulted in an unexpected aluminum hydride DippNacNacAlH(OSIMe) (SIMe = C{N(Me)CH2}2) 112, with the deprotonation of the weakly acidic methyl group in the backbone of the DippNacNac ligand.48a In contrast, the reactivity of 1 towards benzophenone afforded a ketylate species NacNacAl(η2(C,O)–OCPh2) (113) (Scheme 27). The latter compound undergoes easy cyclization reaction with an unsaturated substrate.48b
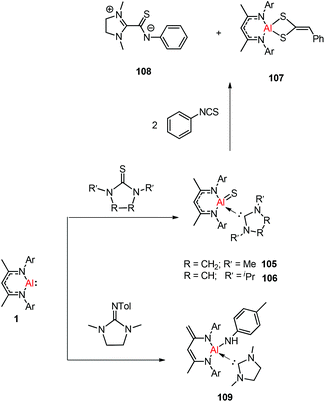 |
| | Scheme 26 Reactivity of 1 toward compounds with CS and CN unsaturated bonds. | |
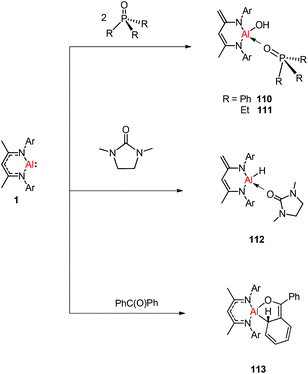 |
| | Scheme 27 Reactivity of 1 toward compounds with PO and CO unsaturated bonds. | |
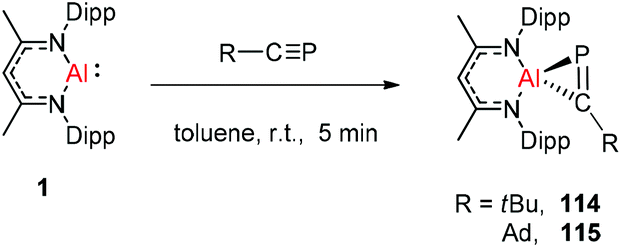
Recently Stephan et al. have reported the first heteroaluminirenes DippNacNacAl[C(R)P] (R = tBu or adamantyl) 114 and 115via the [1 + 2] cycloaddition reaction of the aluminum(I) complex DippNacNacAl(I) (1) with phosphaalkynes, which feature moderate three-centered 2π-electron aromaticity (Scheme 28). The compounds containing the AlCP ring can be used as synthons to prepare a series of unprecedented Al- and P-containing heterocyclic frameworks.49
 |
| | Scheme 28 Synthesis of phosphaaluminirenes 114 and 115. | |
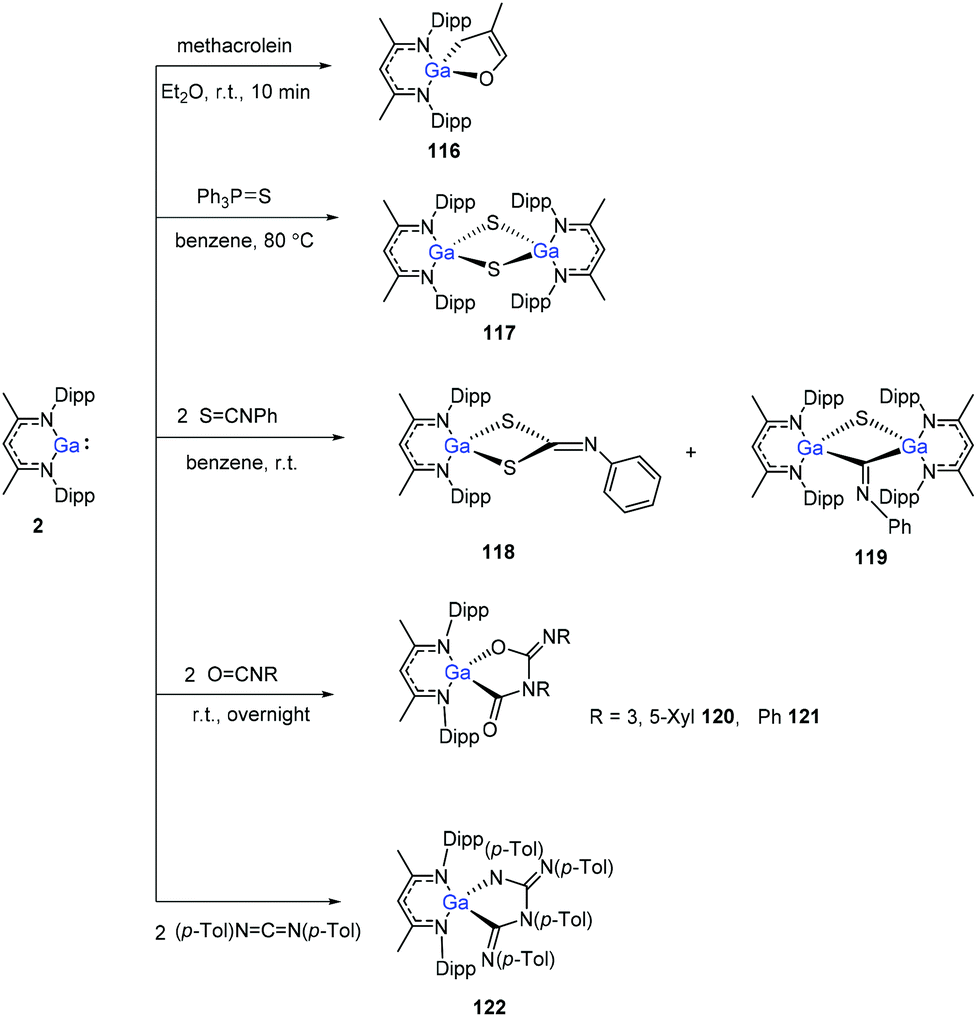
The Ga(I) compound easily undergoes cyclization with methacrolein at room temperature within 10 min to give gallium enolate 116. Unlike the aluminum congener 1, the gallium compound 2 does not cleave the PS bond of Et3PS, even upon heating to 80 °C. With Ph3PS, however, a slow reaction occurs upon heating to 80 °C to obtain the sulfide (DippNacNacGa–S)2 (117). Nevertheless, compound 2 readily reacts with two equiv. of PhNCS to give product 118via CS bond cleavage and cyclization, and also the dimer (DippNacNacGa)2(μ-S)(μ-CNPh) (119) at a ratio of 5:1 (3,5-Me2C6H3)NCO and PhNCO, respectively, reacts with 2, readily to form the coupling products 120 and 121. 1,3-Di-p-tolylcarbodiimide and DippNacNacGa(I) formed the coupling product 122 (Scheme 29).50
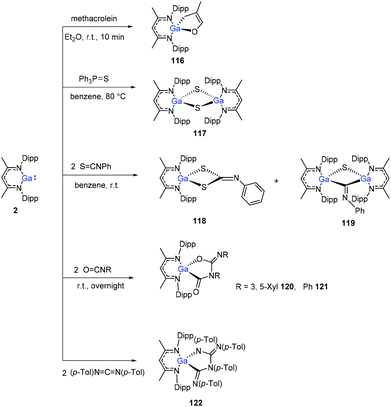 |
| | Scheme 29 Reactivity of 2 toward compounds with unsaturated bonds. | |
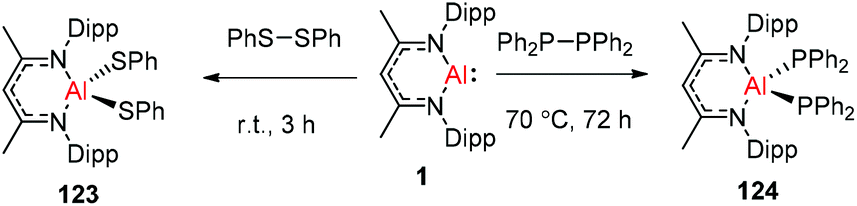
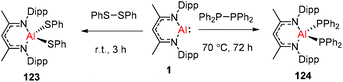
DippNacNacAl(I) (1) reacted with diphenyl disulfide to afford the symmetrically substituted bis(phenyl sulfide) aluminum complex 123via the cleavage of the S–S bond in diphenyl disulfide. In a similar fashion, the reaction of 1 with bulky tetraphenyl diphosphine resulted in the cleavage of the P–P bond over the course of 3 d at 70 °C to produce the novel aluminum bis(diphenyl phosphido) complex (124) (Scheme 30).45
 |
| | Scheme 30 P–P and S–S bond cleavage by 1. | |
2.4 H–X bond activation
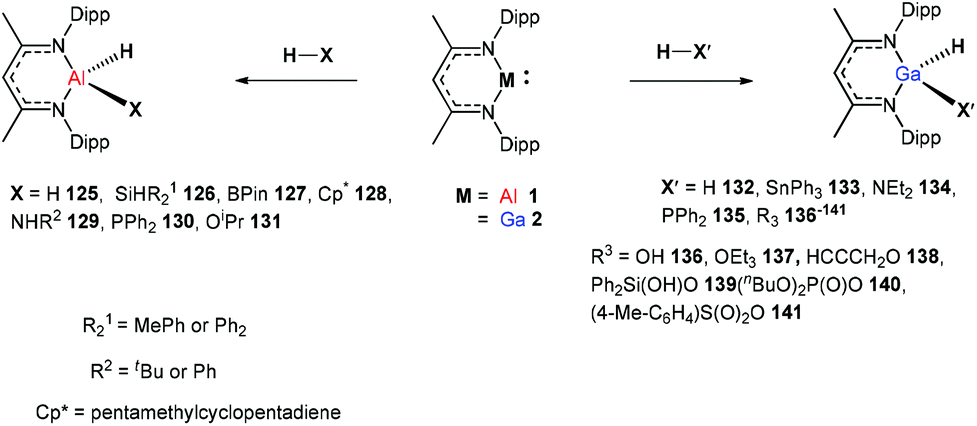
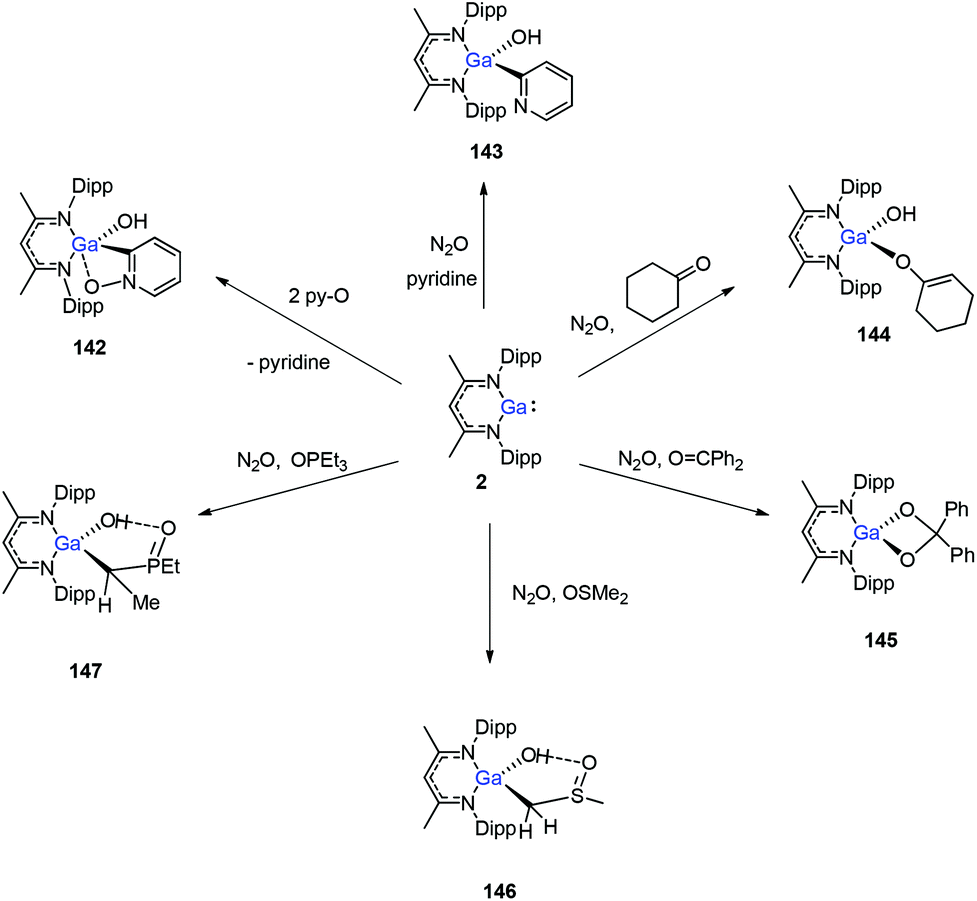
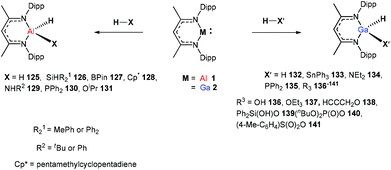
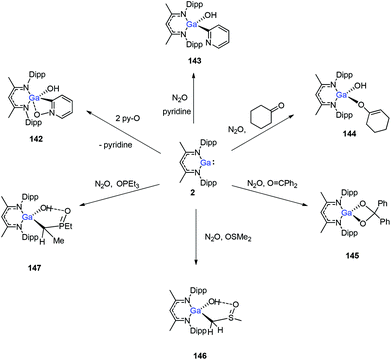
In 2014, Nikonov et al. reported the first examples of oxidative addition of a series of robust H–X bonds (X = H, B, C, Si, N, P, O) to a single Al(I) center. The addition of H3SiPh, HBPin, HPPh2, HOiPr, H2NtBu and H2NPh with 1 proceeded smoothly at ambient temperature, while the reaction of bulkier silane (H2SiMePh), H2, Cp*H with compound 1 required heating at 70 °C. The Al(I) compound reactivity toward the oxidative addition of DippNacNacAlH2 is reversible, proving the possibility of reductive elimination from the species DippNacNacAlH(X) (X = H, B, C, Si, N, P, O).51 Linti et al. described that DippNacNacGa(I) (2) undergoes facile oxidative addition reactions towards H2, HSnPh3, HNEt2, HPPh2, HOEt and H2O, leading to a series of gallium hydrides, DippNacNacGaH(X) (X = H, Sn, O, N, P), substituted by hydride, tin, alkoxy, amido and phosphido groups.52 The oxidative addition of DippNacNacGa(I) (2) with HCCCH2OH, Ph2Si(OH)2, (nBuO)2P(O)(OH) and (4-Me-C6H4)S(O)2(OH) resulted in the formation of compounds DippNacNacGa(H)(μ-O)CH2CCH (138), DippNacNacGa(H)(μ-O)SiPh2(OH) (139), DippNacNacGa(H)(μ-O)P(O)(OnBu)2 (140) and DippNacNacGa(H)(μ-O)S(O)2(C6H4-4-Me) (141), respectively at very low temperature (Scheme 31).53 Very recently, Nikonov et al. have reported the in situ oxidation of DippNacNacGa(I) (2) by N2O or pyridine oxide which results in the generation of NacNacGa(O) as a monomeric oxide intermediate leading to the C–H bond activation (Scheme 32).54 The oxidation of DippNacNacGa(I) (2) by using pyridine oxide led to the C–H bond activation of pyridine oxide, yielding DippNacNacGa(OH)(η1(C),κ1(O)-o-C5H4N–O) (142), and pyridine. A similar reaction between DippNacNacGa(I) (2) and N2O in the presence of pyridine and cyclohexanone leads to DippNacNacGa(OH)(o-C5H4N) (143) and DippNacNacGa(OH)(OC6H9) (144), respectively. The oxidation of DippNacNacGa(I) (2) in the presence of Ph2CO resulted in the isolation of compound DippNacNacGa(κ2-O2CPh2) (145), formed from the sequential oxidation and cyclization. The in situ oxidation of the mixtures of DippNacNacGa(I) (2) with OSMe2 and OPEt3 resulted in the sp3 C–H bond cleavage yielding compounds DippNacNacGa(CH2S(O)Me)OH (146) and DippNacNacGa(CH(Me)P(O)Et2)OH (147), respectively.
 |
| | Scheme 31 Oxidative addition of σ bonds to Al(I) and Ga(I) compounds, respectively. | |
 |
| | Scheme 32 Sequential oxidation/C–H activation of Ga(I). | |
3. Conclusions and perspectives
In conclusion, we report monomeric aluminum and gallium carbenoid complexes supported by β-diketiminate ligands which possess a lone pair of electrons and a formally vacant p orbital. These features afford high electrophilic and nucleophilic reactivity that could be used in bond activation and cleavage upon reactions with small molecules. The aluminum and gallium carbenoid complexes undergo a series of oxidative addition reactions with σ H–X and E′–E bonds where E is an element from groups 13 to 16. These compounds also demonstrate the oxidative cleavage of multiple bonds and enthalpically strong bonds (M–X, E′E). An extension of the synthetic approach is presented in this review for the synthesis of Group 13 metalloid complexes that consist of an unsupported M′–M (M′ = Al, Ga) bond. The oxidative cleavage of multiple bonds shows new and unusual reactivities. These reactivities have opened up a new realm in aluminum and gallium chemistry that could lead to many new products. Nevertheless, the compounds with low valent aluminum and gallium are still showing some limitations in catalytic applications.
However, dialumene (I) promoted both the catalytic and stoichiometric reduction of CO2 to value added C1 products, which is the first example of catalysis using a homonuclear main-group multiple bond. It is important to explore reductive elimination of C–C bonds, which leads to reversible bond activation paving the way towards catalytic applications. Recent developments in the isolation of aluminyl complexes (II) will likely extend the low oxidation state Al chemistry that is used to activate the C–C bonds. We look forward to conducting further studies of the Al(I) and Ga(I) complexes which will result in more unprecedented bond cleavage reactions and important future applications in catalysis.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
H. W. R. thanks the DFG for support of this work (RO 224/71-1). We also thank Prof. Z. Yang from the Beijing Institute of Technology for helpful discussions. This work is dedicated to Professor Reinhold Tacke on the occasion of his 70th birthday.
Notes and references
-
(a) R. H. Holm, Chem. Rev., 1987, 87, 1401–1449 CrossRef CAS
 ;
(b) N. Miyaura and A. Suzuki, Chem. Rev., 1995, 95, 2457–2483 CrossRef CAS ;
(c) E. M. McGarrigle and D. G. Gilheany, Chem. Rev., 2005, 105, 1563–1602 CrossRef CAS PubMed ;
(d) I. A. I. Mkhalid, J. H. Barnard, T. B. Marder, J. M. Murphy and J. F. Hartwig, Chem. Rev., 2010, 110, 890–931 CrossRef CAS PubMed ;
(e) B. Sarkar, D. Schweinfurth, N. Deibel and F. Weisser, Coord. Chem. Rev., 2015, 293–294, 250–262 CrossRef CAS .
;
(b) N. Miyaura and A. Suzuki, Chem. Rev., 1995, 95, 2457–2483 CrossRef CAS ;
(c) E. M. McGarrigle and D. G. Gilheany, Chem. Rev., 2005, 105, 1563–1602 CrossRef CAS PubMed ;
(d) I. A. I. Mkhalid, J. H. Barnard, T. B. Marder, J. M. Murphy and J. F. Hartwig, Chem. Rev., 2010, 110, 890–931 CrossRef CAS PubMed ;
(e) B. Sarkar, D. Schweinfurth, N. Deibel and F. Weisser, Coord. Chem. Rev., 2015, 293–294, 250–262 CrossRef CAS .
-
(a) C. Dohmeier, C. Robl, M. Tacke and H. Schnöckel, Angew. Chem., Int. Ed. Engl., 1991, 30, 564–565 (
Angew. Chem.
, 1991
, 103
, 594–595
) CrossRef ;
(b) S. Schulz, H. W. Roesky, H.-J. Koch, G. M. Sheldrick, D. Stalke and A. Kuhn, Angew. Chem., Int. Ed. Engl., 1993, 32, 1729–1731 CrossRef .
-
(a) W. Uhl, Angew. Chem., Int. Ed. Engl., 1993, 32, 1386–1397 (
Angew. Chem.
, 1993
, 105
, 1449–1461
) CrossRef ;
(b) C. Dohmeier, D. Loos and H. Schnöckel, Angew. Chem., Int. Ed. Engl., 1996, 35, 129–149 (
Angew. Chem.
, 1996
, 108
, 141–161
) CrossRef CAS .
- A. H. Cowley, Chem. Commun., 2004, 2369–2375 RSC .
-
(a) L. Bourget-Merle, M. F. Lappert and J. R. Severn, Chem. Rev., 2002, 102, 3031–3066 CrossRef CAS PubMed ;
(b) Y. Tsai, Coord. Chem. Rev., 2012, 256, 722–758 CrossRef CAS ;
(c) C. Chen, S. M. Bellows and P. L. Holland, Dalton Trans., 2015, 44, 16654–16670 RSC .
-
(a) H. W. Roesky and S. S. Kumar, Chem. Commun., 2005, 4027–4038 RSC ;
(b) S. Nagendran and H. W. Roesky, Organometallics, 2008, 27, 457–492 CrossRef CAS ;
(c) M. Asay, C. Jones and M. Driess, Chem. Rev., 2011, 111, 354–396 CrossRef CAS PubMed ;
(d) W. Li, X. Ma, M. G. Walawalkar, Z. Yang and H. W. Roesky, Coord. Chem. Rev., 2017, 350, 14–29 CrossRef CAS ;
(e) Y. Liu, J. Li, X. Ma, Z. Yang and H. W. Roesky, Coord. Chem. Rev., 2018, 374, 387–415 CrossRef CAS ;
(f) T. Chu and G. I. Nikonov, Chem. Rev., 2018, 118, 3608–3680 CrossRef CAS PubMed ;
(g) V. Nesterov, D. Reiter, P. Bag, P. Frisch, R. Holzner, A. Porzelt and S. Inoue, Chem. Rev., 2018, 118, 9678–9872 CrossRef CAS PubMed ;
(h) P. Bag, C. Weetman and S. Inoue, Angew. Chem., Int. Ed., 2018, 57, 14394–14413 CrossRef CAS PubMed .
-
(a) P. P. Power, Nature, 2010, 463, 171–177 CrossRef CAS PubMed ;
(b) C. Weetman and S. Inoue, ChemCatChem, 2018, 10, 4213–4228 CrossRef CAS ;
(c) R. L. Melen, Science, 2019, 363, 479–484 CrossRef CAS PubMed .
-
(a) C. Cui, H. W. Roesky, H.-G. Schmidt, M. Noltemeyer, H. Hao and F. Cimpoesu, Angew. Chem., Int. Ed., 2000, 39, 4274–4276 CrossRef CAS ;
(b) X. Li, X. Cheng, H. Song and C. Cui, Organometallics, 2007, 26, 1039–1043 CrossRef CAS ;
(c) H. Zhu, R. B. Oswald, H. Fan, H. W. Roesky, Q. Ma, Z. Yang, H.-G. Schmidt, M. Noltemeyer, K. Starke and N. S. Hosmane, J. Am. Chem. Soc., 2006, 128, 5100–5108 CrossRef CAS PubMed .
- Y. Garcĺa-Rodeja, F. M. Bickelhaupt and I. Fernández, Chem. – Eur. J., 2016, 22, 13669–13676 CrossRef PubMed .
-
(a) P. Bag, A. Porzelt, P. J. Altmann and S. Inoue, J. Am. Chem. Soc., 2017, 139, 14384–14387 CrossRef CAS PubMed ;
(b) C. Weetman, P. Bag, T. Szilvási, C. Jandl and S. Inoue, Angew. Chem., Int. Ed., 2019, 58, 10961–10965 CrossRef CAS PubMed .
-
(a) J. Hicks, P. Vasko, J. M. Goicoechea and S. Aldridge, Nature, 2018, 557, 92–95 CrossRef CAS PubMed ;
(b) J. Hicks, A. Mansikkamäki, P. Vasko, J. M. Goicoechea and S. Aldridge, Nat. Chem., 2019, 11, 237–241 CrossRef CAS PubMed ;
(c) J. Hicks, P. Vasko, J. M. Goicoechea and S. Aldridge, J. Am. Chem. Soc., 2019, 141, 11000–11003 CrossRef CAS PubMed .
-
(a) R. J. Schwamm, M. D. Anker, M. Lein and M. P. Coles, Angew. Chem., Int. Ed., 2019, 58, 1489–1493 CrossRef CAS PubMed ;
(b) M. D. Anker and M. P. Coles, Angew. Chem., Int. Ed., 2019, 58, 13452–13455 CrossRef CAS PubMed .
-
(a) J. Hicks, A. Heilmann, P. Vasko, J. M. Goicoechea and S. M. Aldridge, Angew. Chem., Int. Ed., 2019, 58, 17265–17268 CrossRef CAS PubMed ;
(b) D. Anker and M. P. Coles, Angew. Chem., Int. Ed., 2019, 58, 18429–18433 CrossRef .
-
(a) A. Hofmann, C. Pranckevicius, T. Tröster and H. Braunschweig, Angew. Chem., Int. Ed., 2019, 58, 3625–3628 CrossRef CAS PubMed ;
(b) S. K. Mellerup, Y. Cui, F. Fantuzzi, P. Schmid, J. T. Goettel, G. Bélanger-Chabot, M. Arrowsmith, M. Krummenacher, Q. Ye, V. Engel, V. Engels and H. Braunschweig, J. Am. Chem. Soc., 2019, 141, 16954–16960 CrossRef CAS PubMed ;
(c) S. Kurumada, S. Takamori and M. Yamashita, Nat. Chem., 2020, 12, 36–39 CrossRef CAS PubMed .
-
(a) S. L. Choong, W. D. Woodul, A. Stasch, C. Schenk and C. Jones, Aust. J. Chem., 2011, 64, 1173–1176 CrossRef CAS ;
(b) C. P. Sindlinger, S. R. Lawrence, S. Acharya, C. A. Ohlin and A. Stasch, Dalton Trans., 2017, 46, 16872–16877 RSC ;
(c) A. L. Hawley, C. A. Ohlin, L. Fohlmeister and A. Stasch, Chem. – Eur. J., 2017, 23, 447–455 CrossRef CAS PubMed .
-
(a) E. S. Schmidt, A. Jockisch and H. Schmidbaur, J. Am. Chem. Soc., 1999, 121, 9758–9759 CrossRef CAS ;
(b) E. S. Schmidt, A. Schier and H. Schmidbaur, J. Chem. Soc., Dalton Trans., 2001, 505–507 RSC ;
(c) R. J. Baker, R. D. Farley, C. Jones, M. Kloth and D. M. Murphy, J. Chem. Soc., Dalton Trans., 2002, 3844–3850 RSC ;
(d) I. L. Fedushkin, A. N. Lukoyanov, G. K. Fukin, S. Y. Ketkov, M. Hummert and H. Schumann, Chem. – Eur. J., 2008, 14, 8465–8468 CrossRef CAS PubMed ;
(e) I. L. Fedushkin, A. N. Lukoyanov, A. N. Tishkina, G. K. Fukin, K. A. Lyssenko and M. Hummert, Chem. – Eur. J., 2010, 16, 7563–7571 CrossRef CAS PubMed ;
(f) I. L. Fedushkin, A. N. Lukoyanov, G. K. Fukin, S. Y. Ketkov, M. Hummert and H. Schumann, Chem. – Eur. J., 2008, 14, 8465–8468 CrossRef CAS PubMed ;
(g) I. L. Fedushkin, A. N. Lukoyanov, A. N. Tishkina, G. K. Fukin, K. A. Lyssenko and M. Hummert, Chem. – Eur. J., 2010, 16, 7563–7571 CrossRef CAS PubMed ;
(h) D. Dange, S. L. Choong, C. Schenk, A. Stasch and C. Jones, Dalton Trans., 2012, 41, 9304–9315 RSC ;
(i) C. Jones, P. C. Junk, J. A. Platts and A. Stasch, J. Am. Chem. Soc., 2006, 128, 2206–2207 CrossRef CAS PubMed ;
(j) G. Jin, C. Jones, P. C. Junk, A. Stasch and W. D. Woodul, New J. Chem., 2008, 32, 835–842 RSC ;
(k) J. Overgaard, C. Jones, D. Dange and J. A. Platts, Inorg. Chem., 2011, 50, 8418–8426 CrossRef CAS PubMed ;
(l) M. C. Kuchta, J. B. Bonanno and G. Parkin, J. Am. Chem. Soc., 1996, 118, 10914–10915 CrossRef CAS ;
(m) R. J. Wright, M. Brynda, J. C. Fettinger, A. R. Betzer and P. P. Power, J. Am. Chem. Soc., 2006, 128, 12498–12509 CrossRef CAS PubMed .
- N. Saito, J. Takaya and N. Iwasawa, Angew. Chem., Int. Ed., 2019, 58, 9998–10002 CrossRef CAS PubMed .
-
(a) C. Jones, Nat. Rev. Chem., 2017, 1, 59 CrossRef CAS ;
(b) S. F. McWilliams and P. L. Holland, Acc. Chem. Res., 2015, 48, 2059–2065 CrossRef CAS PubMed .
- P. J. Chirik, Inorg. Chem., 2011, 50, 9737–9740 CrossRef CAS PubMed .
- N. J. Hardman, B. E. Eichler and P. P. Power, Chem. Commun., 2000, 1991–1992 RSC .
- J. A. B. Abdalla, I. M. Riddlestone, R. Tirfoin and S. Aldridge, Angew. Chem., Int. Ed., 2015, 54, 5098–5102 CrossRef CAS PubMed .
- R. Y. Kong and M. R. Crimmin, J. Am. Chem. Soc., 2018, 140, 13614–13617 CrossRef CAS PubMed .
- R. Y. Kong and M. R. Crimmin, Chem. Commun., 2019, 55, 6181–6184 RSC .
-
(a) C. Bakewell, A. J. P. White and M. R. Crimmin, Angew. Chem., Int. Ed., 2018, 57, 6638–6642 CrossRef CAS PubMed ;
(b) C. Bakewell, A. J. P. White and M. R. Crimmin, Chem. Sci., 2019, 10, 2452–2458 RSC .
- A. Paparo, C. D. Smith and C. Jones, Angew. Chem., Int. Ed., 2019, 58, 11459–11463 CrossRef CAS PubMed .
- B. Li, S. Kundu, H. Zhu, H. Keil, R. Herbst-Irmer, D. Stalke, G. Frenking, D. M. Andradac and H. W. Roesky, Chem. Commun., 2017, 53, 2543–2546 RSC .
- S. Sinhababu, S. Kundu, A. N. Paesch, R. Herbst-Irmer, D. Stalke and H. W. Roesky, Eur. J. Inorg. Chem., 2018, 2237–2240 CrossRef CAS .
-
(a) S. Brand, H. Elsen, J. Langer, W. A. Donaubauer, F. Hampel and S. Harder, Angew. Chem., Int. Ed., 2018, 57, 14169–14173 CrossRef CAS PubMed ;
(b) T. N. Hooper, M. Garçon, A. J. P. White and M. R. Crimmin, Chem. Sci., 2018, 9, 5435–5440 RSC ;
(c) S. Brand, H. Elsen, J. Langer, S. Grams and S. Harder, Angew. Chem., Int. Ed., 2019, 58, 15496–15503 CrossRef CAS PubMed .
- C. Ganesamoorthy, D. Bläser, C. Wölper and S. Schulz, Angew. Chem., Int. Ed., 2014, 53, 11587–11591 CrossRef CAS PubMed .
- G. Prabusankar, C. Gemel, P. Parameswaran, C. Flener, G. Frenking and R. A. Fischer, Angew. Chem., Int. Ed., 2009, 48, 5526–5529 CrossRef CAS PubMed .
- C. Ganesamoorthy, D. Bläser, C. Wölper and S. Schulz, Chem. Commun., 2014, 50, 12382–12384 RSC .
- G. Prabusankar, C. Gemel, M. Winter, R. W. Seidel and R. A. Fischer, Chem. – Eur. J., 2010, 16, 6041–6047 CrossRef CAS PubMed .
- G. Prabusankar, S. Gonzalez-Gallardo, A. Doddi, C. Gemel, M. Winter and R. A. Fischer, Eur. J. Inorg. Chem., 2010, 28, 4415–4418 CrossRef .
- C. Ganesamoorthy, G. Bendt, D. Bläser, C. Wölper and S. Schulz, Dalton Trans., 2015, 44, 5153–5159 RSC .
- C. Ganesamoorthy, D. Bläser, C. Wölper and S. Schulz, Organometallics, 2015, 34, 2991–2996 CrossRef CAS .
- C. Ganesamoorthy, C. Helling, C. Wölper, W. Frank, E. Bill, G. E. Cutsail III and S. Schulz, Nat. Commun., 2018, 9, 87–93 CrossRef PubMed .
- C. Helling, C. Wölper and S. Schulz, J. Am. Chem. Soc., 2018, 140, 5053–5056 CrossRef CAS PubMed .
-
(a) L. Tuscher, C. Ganesamoorthy, D. Bläser, C. Wölper and S. Schulz, Angew. Chem., Int. Ed., 2015, 54, 10657–10661 CrossRef CAS PubMed ;
(b) L. Tuscher, C. Helling, C. Ganesamoorthy, J. Kreger, C. Wölper, W. Frank, A. S. Nizovtsev and S. Schulz, Chem. – Eur. J., 2017, 23, 12297–12304 CrossRef CAS PubMed .
- J. Kreger, C. Ganesamoorthy, L. John, C. Wölper and S. Schulz, Chem. – Eur. J., 2018, 24, 9157–9164 CrossRef PubMed .
- J. Krüger, C. Wölper, L. John, L. Song, P. R. Schreiner and S. Schulz, Eur. J. Inorg. Chem., 2019, 11–12, 1669–1678 CrossRef .
- U. Chakraborty, B. Mühldorf, N. J. C. van Velzen, B. de Bruin, S. Harder and R. Wolf, Inorg. Chem., 2016, 55, 3075–3078 CrossRef CAS PubMed .
-
(a) L. Tuscher, C. Helling, C. Wölper, W. Frank, A. S. Nizovtsev and S. Schulz, Chem. – Eur. J., 2018, 24, 3241–3250 CrossRef CAS PubMed ;
(b) L. Song, J. Schoening, C. Wölper, S. Schulz and P. R. Schreiner, Organometallics, 2019, 38, 1640–1647 CrossRef CAS .
-
(a) M. R. Crimmin, M. J. Butler and A. J. P. White, Chem. Commun., 2015, 51, 15994–15996 RSC ;
(b) T. Chu, Y. Boyko, I. Korobkov and G. I. Nikonov, Organometallics, 2015, 34, 5363–5365 CrossRef CAS .
-
(a) A. Koner, B. M. Gabidullin, Z. Kelemen, L. Nyulászi, G. I. Nikonov and R. Streubel, Dalton Trans., 2019, 48, 8248–8253 RSC ;
(b) T. Chu, S. F. Vyboishchikov, B. Gabidullin and G. I. Nikonov, Angew. Chem., Int. Ed., 2016, 55, 13306–13311 CrossRef CAS PubMed .
- T. Chu, Y. Boyko, I. Korobkov, L. G. Kuzmina, J. A. K. Howard and G. I. Nikonov, Inorg. Chem., 2016, 55, 9099–9104 CrossRef CAS PubMed .
- L. Kong, R. Ganguly, Y. Li and R. Kinjo, Chem. – Eur. J., 2016, 22, 1922–1925 CrossRef CAS PubMed .
- T. Chu, S. F. Vyboishchikov, B. M. Gabidullin and G. I. Nikonov, J. Am. Chem. Soc., 2017, 139, 8804–8807 CrossRef CAS PubMed .
-
(a) T. Chu, S. F. Vyboishchikov, B. M. Gabidullin and G. I. Nikonov, Inorg. Chem., 2017, 56, 5993–5997 CrossRef CAS PubMed ;
(b) A. Dmitrienko, J. F. Britten, D. Spasyuk and G. I. Nikonov, Chem. – Eur. J., 2020, 26, 206–211 CrossRef CAS PubMed .
- L. L. Liu, J. Zhou, L. L. Cao and D. W. Stephan, J. Am. Chem. Soc., 2019, 141, 16971–16982 CrossRef CAS PubMed .
- A. Kassymbek, J. F. Britten, D. Spasyuk, B. Gabidullin and G. I. Nikonov, Inorg. Chem., 2019, 58, 8665–8672 CrossRef CAS PubMed .
- T. Chu, I. Korobkov and G. I. Nikonov, J. Am. Chem. Soc., 2014, 136, 9195–9202 CrossRef CAS PubMed .
- A. Seifert, D. Scheid, G. Linti and T. Zessin, Chem. – Eur. J., 2009, 15, 12114–12120 CrossRef CAS PubMed .
- E. Herappe-Mejía, K. Trujillo-Hernández, J. C. Garduño-Jiménez, F. Cortés-Guzmán, D. Martínez-Otero and V. Jancik, Dalton Trans., 2015, 44, 16894–16902 RSC .
- A. Kassymbek, S. F. Vyboishchikov, B. M. Gabidullin, D. Spasyuk, M. Pilkington and G. I. Nikonov, Angew. Chem., Int. Ed., 2019, 58, 18102–18107 CrossRef CAS PubMed .
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  ,
Soumen
Sinhababu
,
Soumen
Sinhababu