
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBiomass valorisation over metal-based solid catalysts from nanoparticles to single atoms
Cecilia
Mondelli
 *a,
Gökalp
Gözaydın
b,
Ning
Yan
*b and
Javier
Pérez-Ramírez
*a
*a,
Gökalp
Gözaydın
b,
Ning
Yan
*b and
Javier
Pérez-Ramírez
*a
aInstitute for Chemical and Bioengineering, Department of Chemistry and Applied Biosciences, ETH Zurich, Vladimir-Prelog-Weg 1, 8093 Zürich, Switzerland. E-mail: cecilia.mondelli@chem.ethz.ch; jpr@chem.ethz.ch
bDepartment of Chemical and Biomolecular Engineering, National University of Singapore, 4 Engineering Drive 4, 117585, Singapore. E-mail: ning.yan@nus.edu.sg
First published on 27th May 2020
Abstract
Heterogeneous catalysts are vital to unlock superior efficiency, atom economy, and environmental friendliness in chemical conversions, with the size and speciation of the contained metals often playing a decisive role in the activity, selectivity and stability. This tutorial review analyses the impact of these catalyst parameters on the valorisation of biomass through hydrogenation and hydrodeoxygenation, oxidation, reforming and acid-catalysed reactions, spanning a broad spectrum of substrates including sugars and platform compounds obtained from (hemi)cellulose and lignin derivatives. It outlines multiple examples of classical structure sensitivity on nanoparticle-based materials with significant implications for the product distribution. It also shows how the recently emphasised application of metals in the form of ultrasmall nanoparticles (<2 nm), clusters and single atoms, while fulfilling superior metal utilisation and robustness, opens the door to unprecedented electronic and geometric properties. The latter can lead to facilitated activation of reactants as well as boosted selectivity control and synergy between distinct active sites in multifunctional catalysts. Based on the analysis conducted, guidelines for the selection of metals for diverse applications are put forward in terms of chemical identity and structure, and aspects that should be explored in greater depth for further improving the exploitation of metals in this research field and beyond are highlighted.
 Cecilia Mondelli | Cecilia Mondelli conducted her PhD studies on infrared studies of heterogeneously catalysed reactions under the supervision of Dr Rinaldo Psaro at the University of Milan, Italy, earning her degree in 2007. She then moved to the ETH Zurich to continue activities in heterogeneous catalysis as a postdoctoral fellow in the group of Prof. Alfons Baiker. In 2010, she joined the group of Prof. Javier Pérez-Ramírez at the same institution, where she took a position as a scientist in 2011 and lecturer in 2017. Her research pursues the valorisation of renewable and abundant feedstocks such as biomass and CO2 to green chemicals and fuels over tailored catalytic systems. |
 Gökalp Gözaydın | Gökalp Gözaydın received BSc and MSc degrees in Chemical Engineering from Izmir Institute of Technology in 2014 and 2016, respectively. He conducted his Master thesis on the valorisation of biomass waste in hot compressed water under the supervision of Prof. Aslı Yüksel. Since 2017, he has been pursuing his PhD under the supervision of Prof. Ning Yan at the National University of Singapore, focusing on the valorisation of chitin into N-containing chemicals. |
 Ning Yan | Ning Yan received BSc and PhD degrees in Chemistry from Peking University working with Prof. Yuan Kou. After a Marie Curie Fellowship at the École Polytechnique Fédérale de Lausanne in Switzerland with Prof. Paul Dyson, he joined the Department of Chemical and Biomolecular Engineering at the National University of Singapore as an assistant professor in 2012 and was promoted to a tenured associate professor in 2018. Ning Yan works in advanced catalysis, renewable energy, and sustainable chemistry, for which he was recognised by recent awards from the Royal Society of Chemistry, American Chemical Society and National University of Singapore, among others. |
 Javier Pérez-Ramírez | Javier Pérez-Ramírez holds the Chair of Catalysis Engineering at ETH Zurich. His research pursues the design of heterogeneous catalysts and reactor concepts tackling current and future energy, resource, and environmental challenges of society. His work has been recognised by several awards, most recently the Paul H. Emmett Award in Fundamental Catalysis of the North American Catalysis Society. He directs a National Competence Center of Research in Catalysis in Switzerland and has a visiting appointment at the National University of Singapore within the Flagship Green Energy Program. |
Key learning points(1) Size tuning of metallic nanoparticles is effective to alter the product distribution in many hydrogenation, oxidation and reforming reactions due to distinct structure sensitivity of competitive reactions.(2) Isolated sites in the lattice of porous materials are the only active speciation for titanium and tin in acid-catalysed reactions. (3) Moving from conventional nanoparticles to ultrasmall nanoparticles (<2 nm), clusters and single atoms appears beneficial to improve the activity, selectivity, stability and synergy between catalytic sites in multifunctional systems. (4) Single-atom catalysts should be more rigorously compared to nanoparticle-based catalysts and the electronic properties and metal–host interactions should be better assessed to unlock their full potential. (5) Information about the behaviour of low-nuclearity metal species in biomass conversions should be gained. |
Introduction
Metals are central components of heterogeneous catalysts, which have been widely applied in the form of well-defined nanoparticles (NPs). Scientist have for a long time realised that the size of such structures matters. Since terraces, edges and corners often exhibit distinct intrinsic capabilities in adsorbing and transforming reactants, particle size tuning has been broadly exploited to enhance catalyst activity and selectivity. Recently, the introduction of single metal atom catalysts has brought a paradigm shift in heterogeneous catalysis. Similarly to homogeneous complexes, the metal is atomically dispersed, all catalytic centres are equivalent and their electronic properties can be modulated through tailored metal–host interactions, leading to unprecedented performances. While excellent reviews have described the effects of NP size and the implications associated with the transition from NPs to single atoms in traditional catalysis applications,1,2 here we discuss these aspects specifically in relation to the efforts made in the last decade to valorise biobased feedstocks into a spectrum of chemicals, fuels and fuel additives.Metals in reduced and oxidised forms have taken centre stage as active sites in a realm of transformations applied to upgrade biomass, including hydrogenation and hydrodeoxygenation (HDO), assisted by molecular hydrogen or by hydrogen donors, oxidation, reforming and acid-catalysed conversions, comprising hydrolysis, dehydration, epi-/isomerisation, aldol-condensation, the retro-aldol reaction and transfer-hydrogenation. Initial studies focused on their chemical identity, with different metals being deposited onto carriers with loadings typically in the range of 5–10 wt% except for acid-mediated reactions, where oxidised metals have been used in the form of bulk or supported metal oxides or isolated centres in porous materials. Owing to the relatively high metal content, non-noble metals have been investigated in addition to noble species.
The presence of multiple functional groups in bioderived substrates has posed great challenges to catalyst selectivity and stability. Indeed, such moieties, as well as the polar solvents required for their conversion, can act as ligands, causing blocking and leaching of the metal centres, and may lead to fouling. These issues have triggered widespread attention towards catalyst nanoengineering. Early studies placed emphasis on tuning the electronic and geometric properties of the metal through the addition of a second metal or via confinement in carriers with tailored porosity, with the downside of generating more complex catalyst architectures and compositions.3 In line with a general trend in catalysis research, the last few years have witnessed increasing efforts toward a more effective use of the active metal phase by precise control of its size.
Following the classical concept of structure sensitivity of reactions, the particle dimensions have been adjusted to enrich the surface with sites able to boost the yield of desired products. In the majority of cases, the dimensions of the metal aggregates have been reduced, producing ultrasmall NPs, typically having a size of <2 nm, small clusters and single atoms. These smaller entities would improve the metal utilisation, boosting the activity and reducing the ecologic footprint of the catalyst, and strengthen the interaction with the carrier, modulating the electronic properties as well as hindering sintering phenomena and leaching. Moreover, they could foster proximity to additional catalytic centres of different nature to conduct one-pot multi-step transformations, driving emerging technologies toward environmentally and economically advantageous process intensification.
This tutorial review overviews the effects of the size and speciation of metals used in the chemocatalytic valorisation of biomass in four sections, each dedicated to a family of reactions (Fig. 1). Within each section, relevant substrate classes are considered, and for each class, heterogeneous catalysts based on noble and non-noble metals are presented. The discussions transition from NPs to clusters and/or single atoms, highlighting general trends and peculiarities in their behaviour and stressing relevant synthetic strategies and carrier properties applied to achieve control of the metal nuclearity. A conclusion section organises the learnings and outlines open challenges in the exploitation of metals in sustainable applications, especially at the level of isolated and low-nuclearity species.
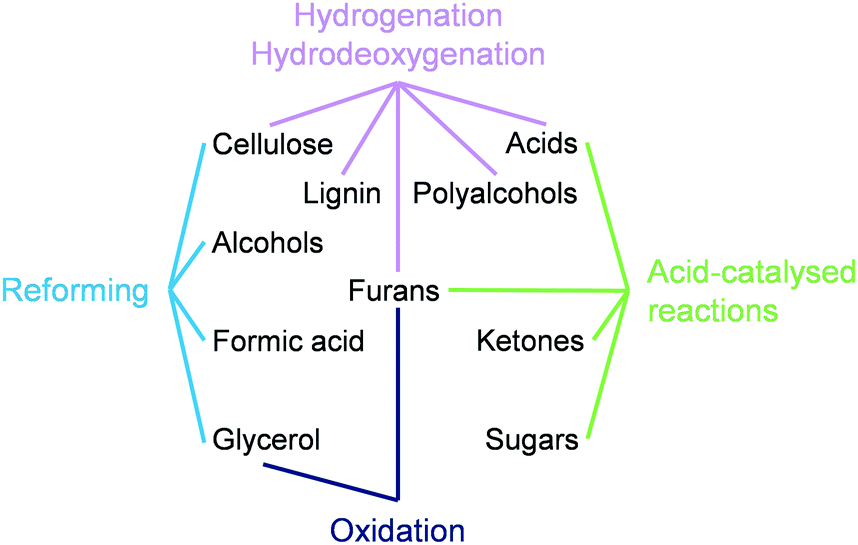 | ||
| Fig. 1 Reaction classes and substrate types for which the role of the particle size and speciation of the active metal has been investigated in biomass valorisation. | ||
Hydrogenation and hydrodeoxygenation reactions
H2-mediated reactions, hydrogenation and HDO, are momentous strategies for the upgrading of biomass-derived feedstocks. A wide array of catalysts containing noble (i.e., Pt, Ru, Pd, Cu and Au) and non-noble (i.e., Ni and Co) metals carried on various supports have been proposed for the hydrogenation of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C and CO bonds of the carbohydrate-derived compounds 5-hydroxymethylfurfural (5-HMF), furfural and levulinic acid (LA), and for the HDO of carboxylic acids (succinic acid) and lignin-derived compounds (phenol, m-creosol and vanillin). In the past couple of years, direct correlations between the metal particle size and catalytic activity and selectivity have been sought after in the context of establishing an economically viable biorefinery. To bridge catalysis and sustainable chemistry technology, the introduction of innovative and facile methodologies for NP synthesis and fine-tuning of isolated metallic sites has been widely addressed to control the number of adsorption sites and their intrinsic catalytic properties.
C and CO bonds of the carbohydrate-derived compounds 5-hydroxymethylfurfural (5-HMF), furfural and levulinic acid (LA), and for the HDO of carboxylic acids (succinic acid) and lignin-derived compounds (phenol, m-creosol and vanillin). In the past couple of years, direct correlations between the metal particle size and catalytic activity and selectivity have been sought after in the context of establishing an economically viable biorefinery. To bridge catalysis and sustainable chemistry technology, the introduction of innovative and facile methodologies for NP synthesis and fine-tuning of isolated metallic sites has been widely addressed to control the number of adsorption sites and their intrinsic catalytic properties.
Furans
Among the multifarious platform chemicals derived from biomass components, furans have paramount importance because of the abundance of carbohydrates in lignocellulosic materials. In this context, the structure sensitivity of two competing vapour-phase reactions, i.e., decarbonylation and hydrogenation of the carbonyl group of furfural, over Pt NPs dispersed on mesoporous silica (Pt/MCF-17) was systematically studied.4 Pt/MCF-17 was synthesised by the immobilisation of polyvinylpyrrolidone (PVP)-capped Pt NPs on the support. Polyhedral NPs (>3 nm) substantially produced furfuryl alcohol (FOL), while Pt NPs of less than 2 nm favoured the generation of furan. Specifically, the selectivity of FOL and the turnover rate (TOR) were enhanced up to 66% and 70 times, respectively, at 200 °C and 0.93 bar H2 within a Pt NP size range of 1.5–7.1 nm, corresponding to changes in the activation energy up to 7-fold. On the contrary, the activation energy of furfural decarbonylation was insensitive to the Pt NP size. It should be noted that the shape of the metal NPs was another factor that affected the product distribution. For Pt NPs of comparable size, an octahedral morphology (6.2 nm) was more favourable to synthesise FOL, while cube-shaped structures (6.8 nm) almost formed FOL and furan in comparable amounts. Along this line, the same group introduced 3-dimensional Pt NP-based catalysts with various mesoporous oxide supports (TiO2, Nb2O5, Al2O3 and Ta2O5), which were synthesised by a soft-templating method using induced capillary inclusion.5 Similar prevailing decarbonylation product formation was observed regardless of the carrier with 1.9 nm Pt NPs. In contrast, acidic oxide supports (TiO2 and Nb2O5) favoured the hydrogenation of the carbonyl group, yielding more FOL and 2-methylfuran. These findings highlight that the selectivity can be driven by the nature of the carrier and of the catalyst surface sites, i.e., corner, terrace and edge sites, controlled by the particle size and shape.Cai et al. gathered mechanistic insight via density functional theory (DFT) calculations and microkinetic modelling to ascertain the underlying structure-dependence of platinum catalysts in furfural transformation.6 Three different surfaces, including flat Pt(111), stepped Pt(211) and the Pt55 cluster, were selected to represent the reactive adsorption sites (corner, terrace and step) on Pt NPs. It should be stressed that, while suffering from material and pressure gaps, which are not straightforward to fill, DFT simulations could provide, here and in the studies outlined later on, precious information about the adsorption of reactants and the reaction mechanism, consolidating experimentally-driven insights into the catalytic systems investigated. Thermodynamically, the corner sites on the stepped Pt(211) and Pt55 cluster were more suitable for chemoselective decarbonylation than those on the flat Pt(111) surface, which were favourable for hydrogenation. Consequently, larger Pt NPs, with their surface being dominated by flat and stepped structures, led to the synthesis of FOL, whereas smaller Pt NPs, containing more corner sites, mainly promoted the generation of furan via decarbonylation due to the low activation barrier. Combining results from DFT calculations and microkinetic modelling, it was concluded that a higher abundance of corner sites on small Pt NPs (<1.4 nm) was key to favour furan formation at 200 °C and 0.93 bar.
Very recently, Durndell et al. disclosed the influence of the size of Ru NPs supported on SiO2 and SBA-15 to derive structure–activity relations for liquid-phase furfural conversion.7 The variation of the Ru NP size also governed the two competing reactions, i.e., hydrogenation and decarbonylation, distinctly from the aforementioned Pt NPs. Although FOL was selectively produced over Ru NPs of any size (2–25 nm), there is a clear dependence of the decarbonylation activity on the particle dimensions. The turnover frequency (TOF) for furan formation was 98 h−1 over 2.3 nm Ru NPs, and sharply decreased to 1 h−1 over 24 nm Ru NPs at 10 bar H2. In strong contrast, the TOF of carbonyl hydrogenation only changed from 237 to 307 h−1, indicating its moderate structure dependency. This opposite product distribution pattern was due to the distinct fraction of low-coordinated sites on the differently sized NPs.
Ultrasmall Ru NPs deposited onto TiO2 were produced through a method that exploited the ligand effect of tannic acid, a bio-derived polyphenol, on Ru3+ and Ti4+ cations contained in the metal precursors (Fig. 2a).8 Ru NPs with a size of 1.6 nm were obtained after calcination at 500 °C, which exhibited the best catalytic performance for selective FOL production (96%). Catalysts treated at higher temperatures (600–700 °C) possessed a comparatively larger particle size, in line with the detection of a diffraction line of metallic ruthenium in the X-ray powder diffraction (XRD) pattern, and yielded ca. 4–5-fold higher amounts of tetrahydrofurfuryl alcohol (THFA) at 40 °C and 40 bar after 7 h (Fig. 2b).
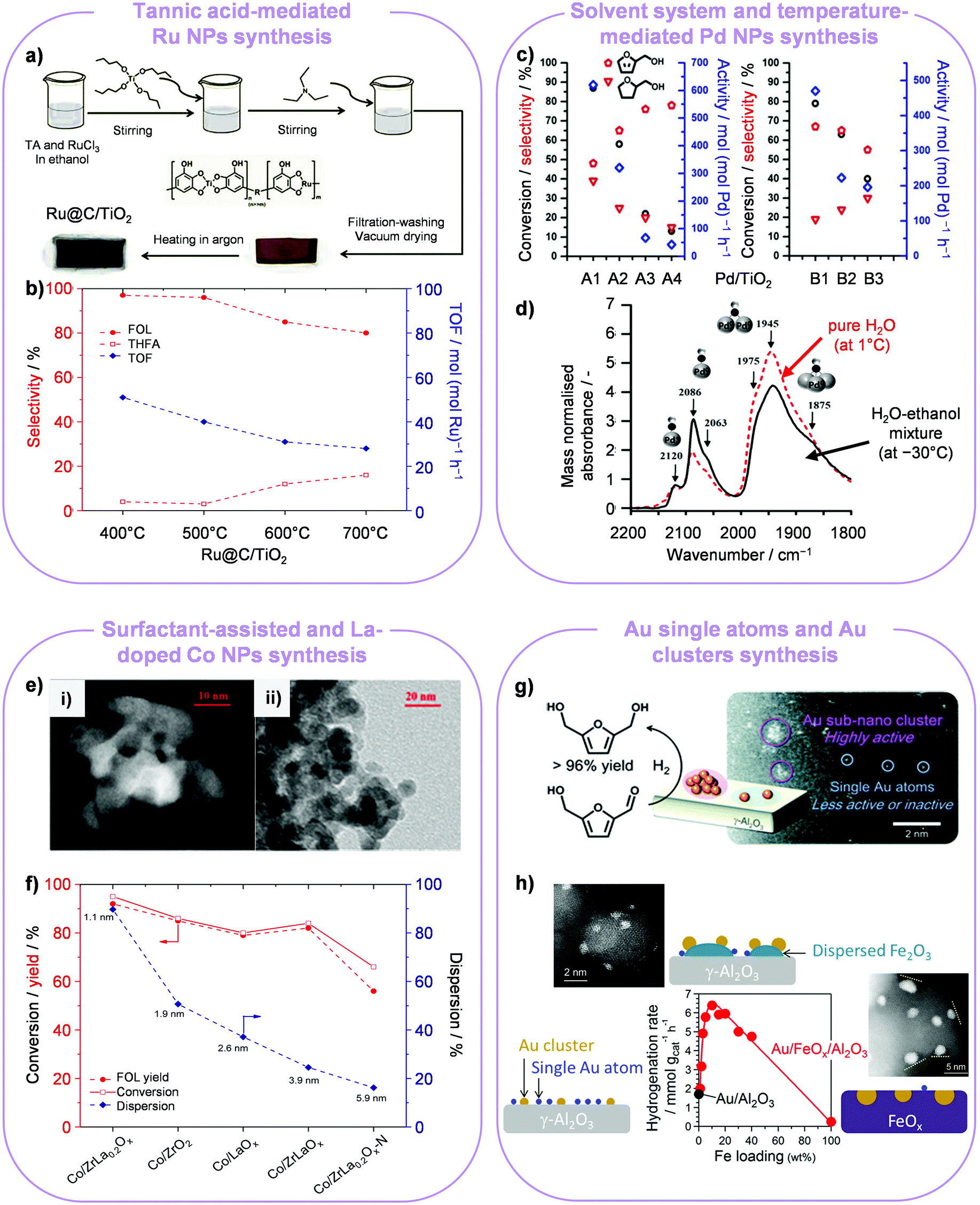 | ||
| Fig. 2 Metal NP syntheses for furan hydrogenation. (a and b) Ru NPs of distinct size are attained using tannic acid as a chelating agent and tuning the heat treatment temperature. FOL production is favoured on smaller NPs, while larger NPs also form some THFA. Reproduced with permission from ref. 8. Copyright 2017 Wiley-VCH. (c and d) A water–ethanol mixture and a lower temperature lead to smaller Pd NPs on titania with respect to pure water and higher temperature, as highlighted by the relative amount of linearly and two/three-fold bound CO on their surface. The two catalyst sets (water, A1: 1 °C (2.5 nm), A2: 25 °C (2.7 nm), A3: 50 °C (2.9 nm), A4: 75 °C (5.2 nm); water–ethanol, B1: −30 °C (1.4 nm), B2: 1 °C (2.1 nm), B3: 25 °C (3.4 nm)) exhibit opposite selectivity trends to FOL and THFA depending on the particle size. Reproduced with permission from ref. 9. Copyright 2017 American Chemical Society. (e) Microscopy images of Co/ZrLa0.2Ox obtained without (i) and with (ii) the use of a surfactant and (f) catalytic performance of these and reference Co-based catalysts, indicating how a smaller cobalt particle size correlates to higher FOL yields. Reproduced with permission from ref. 10. Copyright 2018 American Chemical Society. (g) Au single atoms supported on Al2O3 are inferior to Au clusters in the hydrogenation of 5-HMF. Reprinted with permission from ref. 14. Copyright 2013 Royal Society of Chemistry. (h) Promotional effect of FeOx decoration on the formation of smaller Au clusters showing appreciable hydrogenation activity under milder conditions. Reprinted with permission from ref. 15. Copyright 2016 American Chemical Society. | ||
The influence of the temperature of a sol-immobilisation route using polyvinyl alcohol (PVA) on tailoring the Pd particle size was explored.9 It was elucidated that a lower temperature facilitated the generation of small-sized particles, i.e., 2.5 nm at 1 °C in pure water and 1.4 nm at −30 °C in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ethanol–water mixture. In furfural hydrogenation, the selectivity to FOL increased whereas the THFA selectivity dropped with increasing particle size in pure water, but the opposite trend was observed in the alcohol–water mixture (Fig. 2c). These opposing results could be rationalised considering the fraction of available active corner and edge sites on the surface, which depends on the size of the NPs as well as on their interaction with PVA. According to infrared spectroscopy using CO as a probe molecule, carbon monoxide mostly bonded linearly over available corner and edge sites of the Pd NPs attained in the binary medium, which promoted the FOL selectivity by binding furfural in a perpendicular mode (Fig. 2d). On the contrary, bridged and 3-fold bonded CO species were most abundant over the Pd NPs prepared in water, indicating that furfural binds with either the aldehydic group or the furan ring, accelerating THFA generation.
:1 ethanol–water mixture. In furfural hydrogenation, the selectivity to FOL increased whereas the THFA selectivity dropped with increasing particle size in pure water, but the opposite trend was observed in the alcohol–water mixture (Fig. 2c). These opposing results could be rationalised considering the fraction of available active corner and edge sites on the surface, which depends on the size of the NPs as well as on their interaction with PVA. According to infrared spectroscopy using CO as a probe molecule, carbon monoxide mostly bonded linearly over available corner and edge sites of the Pd NPs attained in the binary medium, which promoted the FOL selectivity by binding furfural in a perpendicular mode (Fig. 2d). On the contrary, bridged and 3-fold bonded CO species were most abundant over the Pd NPs prepared in water, indicating that furfural binds with either the aldehydic group or the furan ring, accelerating THFA generation.
Focusing on the alternative use of non-noble metals, Ma et al. demonstrated the synthesis of a Co-loaded and rare-earth metal-doped ZrO2 catalyst for the selective hydrogenation of furfural under mild conditions.10 Doping with even a very small amount of La (Co/ZrLa0.2Ox) more than doubled the surface area of the support, leading to the formation of ultrasmall Co NPs (1.1 nm). The addition of a surfactant in the synthesis increased the support surface area and decreased the cobalt average particle size without influencing the crystalline phase of ZrO2, which remained tetragonal (Fig. 2e). Moreover, it led to strong metal–support interactions (SMSI). Well-dispersed tiny cobalt nanoclusters on ZrLa0.2Ox exhibited superior catalytic activity than larger Co NPs on the same carrier, yielding 95% FOL in water at 40 °C and 20 bar H2 after 10 h (Fig. 2f).
Similar observations regarding the correlation of SMSI and the particle size distribution with the catalytic performance were made by Goyal et al.11 The proposed organic matrix deposition method induced strong interactions between the metal and a nitrogen-doped mesoporous carbon material, resulting in higher nickel dispersion on the support compared with conventional synthesis methods such as impregnation, sol–gel, adsorption and co-precipitation. In the catalytic reaction of 5-HMF to DMF, superior activity was attained over small-sized Ni NPs (<5 nm) due to SMSI effects.
Other valuable chemicals, such as 2-methyltetrahydrofuran (2-MTHF) and tetrahydrofuran (THF), were targeted in the hydrogenation of furfural via single-step ring hydrogenation and decarboxylation over Pd/C catalysts. The use of NaBH4 as a reducing agent decreased the average particle size of palladium owing to the incorporation of boron in the metal lattice. Thus, the smallest Pd NPs (4.8 nm) were formed by NaBH4 reduction, which selectively produced 2-MTHF (40%) and THF (27%) with complete hydrogenation of FOL, while larger Pd NPs (22.4 nm) reduced using formaldehyde and 100% H2 (18.0 nm) led to lower selectivities of 2-MTHF and THF. Additionally, the generation of FOL was favoured over smaller particles, suggesting that ring hydrogenation and decarbonylation can be manipulated by the type of reducing agent. Another support featuring an inherent large pore diameter for the conversion of 5-HMF is mesoporous silica.12 Highly dispersed and thermally stable Ru clusters anchored within nanosized mesoporous zirconium silica (Ru/MSN-Zr-20) displayed higher hydrogenation activity in aqueous media at room temperature and 5 bar H2. The average Ru NP size was affected by either the channel length of mesoporous silica or the presence of Zr, following the trend Ru/MCM-41 (3.8 nm) > Ru/MSN (1.6 nm) > Ru/MSN-Zr-20 (1.1 nm). Based on X-ray photoelectron spectroscopy (XPS), the Ru/MSN-Zr catalyst contained Ruδ+ species, suggesting the creation of electron deficiency for tiny Ru NPs. Therefore, both the small size of the Ru NPs and electron deficiency were proposed as the origin of the superior hydrogenation ability. The overall TOF of 5-HMF hydrogenation was clearly size-dependent for sub-2 nm particles, whereas it varied only slightly for NPs between 2.0 and 14.4 nm. Moreover, the selectivity shifted from 2,5-dihydroxymethylfuran to 2,5-dihydroxymethyl-tetrahydrofuran over smaller Ru NPs (1.1–1.6 nm).
Small-sized Au NPs were preferred in the hydrogenation of aldehydes because they are more active compared to large Au NPs. This could be attributed to fact that the more abundant under-coordinated sites on smaller Au NPs remarkably promoted the dissociative adsorption of H2 and the adsorption of the CO group.13 Accordingly, Au-based catalysts comprising metal NPs of variable size, tuned by altering the calcination temperature and atmosphere (H2 or air), were investigated for the hydrogenation of 5-HMF to 2,5-bis(hydroxymethyl)-furan (BHF) (Fig. 2g).14 The catalytic activity was influenced by both the acid–base properties of the supports, with basic supports (i.e., γ-Al2O3, CeO2 and La2O3) favouring BHF formation and acidic supports (i.e., TiO2, ZrO2 and Ta2O5) predominantly leading to ring opening products, and the size of Au NPs. Activity tests revealed that Au/Al2O3 containing a mixture of Au clusters (0.88 nm) and single atoms was far more efficient in 5-HMF hydrogenation than 0.1 wt%-Au/Al2O3 and 0.5 wt%-Au/Al2O3 catalysts, which mostly contained single atoms as well as larger Au NPs.
Modification with iron oxide (FeOx) was proposed to control the formation of Au clusters and decrease the frequency of Au single atoms to promote the hydrogenation activity and, hence, be able to carry out the reaction under milder conditions (80 °C and 10 bar H2).15 High-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) revealed that Au supported on Al2O3 in the absence of Fe comprises a high fraction of single atoms, while the 10 wt% Fe loaded Au/Al2O3 catalyst contains ultrasmall Au NPs (0.6–1.2 nm), confirming that the modification with FeOx favours Au agglomeration. Thus, the 5-HMF hydrogenation rate was ca. 3.8-fold higher over the Fe-containing catalysts compared with the Fe-free material (6.4 vs. 1.7 mmol gcat−1 h−1), whereby the number of corner atoms in Au NPs could be regarded as a rate determining factor for hydrogenation (Fig. 2h). It is worth stressing that the incorporation of Fe at high loading (20 wt%) led to a substantially reduced activity (6.0 mmol gcat−1 h−1), due to the burying of a considerable amount of gold beneath FeOx upon reduction.
For conversions involving consecutive hydrogenation steps, the particle size was found to impact the performance to different extents. For instance, Nakagawa et al. studied the two-step total hydrogenation of furfural to THFA, comprising the hydrogenation of the CO bond to yield the FOL intermediate and further hydrogenation of the cyclic CC bonds.16 In the first step, the TOF value did not show any dependence on the size of Ni NPs supported on SiO2 in the range probed (2.6–12.8 nm). Yet, the second step proved to be structure-dependent, i.e., smaller Ni NPs with higher metal dispersion seemed to enhance the hydrogenation of FOL and the selectivity towards THFA. This could be caused by the weaker adsorption of FOL on the catalyst surface, which is likely dependent on the size of the Ni NPs.
Acids
Acid-catalysed carbohydrate transformations enable the production of organic acids and their esters, serving as platforms for a large spectrum of value-added chemicals. Among the latter, γ-valerolactone (GVL) has been particularly targeted owing to its numerous applications. This compound is obtained from LA and its methyl ester through hydrogenation and ring closure, typically conducted over Ru supported on an acidic carrier. In this context, it has been reported that the use of a Zr-functionalised spherical mesoporous silica support (SMS) not only fostered the dispersion of ruthenium but also minimised the agglomeration of the ultrasmall Ru NPs (1.24–1.32 nm).17 The best catalyst, containing 8.2 wt% Zr in silica and 4.5 wt% Ru (Ru/Zr5SMS), offered higher activity and selectivity under mild conditions than Ru/MCM-41 (1.72 nm) with an identical Ru loading. Moreover, Ru/Zr5SMS and Ru/SMS (1.49 nm) exhibited enhanced reusability compared to Ru/MCM-41 since the SMS support hinders ruthenium sintering.The HDO of an aliphatic carboxylic acid, propanoic acid (PAc), was studied over Pd/SiO2 in the size range of 1.9–12.4 nm. The catalytic activity was weakly structure-sensitive since the TOF was similar for NPs between 3.1 and 12.4 nm. The decarbonylation of PAc to yield ethane dominated in the low size range and hydrogenation to propionaldehyde became more pronounced for increasing the Pd particle size.18 Evaluation of the crystal structure through Van Hardeveld and Hartog statistics hinted that low-coordinated sites (corners and edges) were catalytically inactive, since a 7-fold decrease in their fraction did not have any significant effect on the reactions. In contrast, the variation in catalytic performance correlated well with the change in the amount of Pd(111) and Pd(100) facets exposed by the NPs, which increased by 30%.
Very recently, the selective hydrogenolysis of 2-furan carboxylic acid (FCA) to a high value aliphatic compound bearing two terminal oxygen-containing functional groups, 5-hydroxyvaleric acid (5-HVA), was demonstrated on Pt/SiO2 and Pt/Al2O3.19,20 The Pt/SiO2 catalyst was able to suppress the cleavage of δ-C–O bonds and the hydrogenation of the furan ring at low temperature (40 °C) and 30 bar H2 in water.19 The TOR of FCA conversion and the cleavage of α-C–O bonds towards selective 5-HVA synthesis were tuned by controlling the platinum particle size. Specifically, the TOR of FCA conversion reduced from 108.0 to 99.6 molFCA molPt-surface−1 h−1 while the 5-HVA selectivity rose from 73.3 to 80% when the particle size increased from 2.6 to 7.5 nm. The selectivity of the product containing a saturated furan ring (tetrahydrofuran-2-carboxylic acid, THFCA) followed a contrary trend. These results suggest that the higher portion of terrace sites on the larger particles facilitated 5-HVA formation, which was explained through diffuse-reflectance Fourier transform infrared spectroscopy of adsorbed CO (CO-DRIFTS) and DFT calculations by a lowering of the activation barrier and the electron-withdrawing effect of the carboxylic group of FCA. Pt/Al2O3 also selectively transformed FCA into 5-HVA derivatives at 100 °C and 40 bar H2 in methanol.20 5-HVA derivatives were associated with a total selectivity of 62%, with methyl 5-hydroxyvalerate making up 55% of them. By-products formed through ring hydrogenation (i.e., THFCA and its methyl ester) and C–O hydrogenolysis (i.e., valeric acid, VA). The noble metal played a greater role than the support in the performance. Although the solvent type had a limited influence on the reaction, the undesired ring hydrogenation products were favoured moving from shorter to longer and from primary to tertiary alcohols, and using low polarity solvents. The activity of Pt/Al2O3 substantially decreased after the fourth cycle and could be only partially recovered through calcination. Based on transmission electron microscopy (TEM), the particle size grew from 2.9 to 3.1 nm. Hence, sintering accompanied fouling. Using Pt NPs of similar size (2–3 nm) alternatively supported on titania and in the presence of molybdenum oxide as a modifier (Pt–MoOx–TiO2), FCA was selectively hydrodeoxygenated to VA rather than 5-HVA with a yield of 51% in water at 140 °C under 15 bar H2.21
Although Au single-atom catalysts (SACs) showed negligible activity in the hydrogenation of 5-HMF,14,15 maximising the dispersion and the metal utilization granted unrivalled performances when using other noble metals as well as non-noble metals. For instance, Pd SACs were superior catalysts for the selective hydrogenation of succinic acid (SA) to γ-butyrolactone.22 Pd1/γ-AlOOH showed 30- and 1100-fold higher metal-based activity than Pd13/γ-AlOOH and Pd55/γ-AlOOH, respectively, owing to the higher adsorption energies of Pd single atoms on the (010) support surface. On the basis of the correlation between HAADF-STEM and kinetic analyses, catalysts with a higher Pd loading possessing few single Pd atoms (28.2% of the total metal) and larger Pd NPs (1.27 nm) were associated with a higher activation energy than low-loading catalysts, for which the contribution of single atoms reached more than 65% (132.5 vs. 83.5 kJ mol−1). The prominent role of the Pd single atoms in the catalyst performance was corroborated by correlating the activity to the fraction of palladium featuring this speciation.
A single atom alloy (SAA) Pt–Cu catalyst, i.e., containing Pt atoms atomically dispersed on Cu NPs, was introduced for the efficient production of ethanol from cellulose-derived methyl glycolate (MG).23 The best ethanol selectivity (76.7%) was achieved with 0.1 wt% Pt (0.1Pt–Cu/SiO2), which contained Pt–Cu NPs of 1.63 nm in size, based on TEM. Lower or higher Pt loading in the catalyst led to drastically reduced selectivity. The superior catalytic response of 0.1Pt–Cu/SiO2 stemmed from the higher Cu dispersion and ratio between Cu+ and Cu0, investigated by N2O chemisorption, CO-DRIFTS and X-ray absorption near edge structure (XANES). Mechanistically, dissociation of MG and H2 took place on Cu+ and Pt sites, respectively, while the hydrogenation of the C2-moiety occurred on adjacent Cu0 after spill over to these centres.
Lignin derivatives
The valorisation of aromatic biopolymers contained in lignin into biofuels and higher-value chemicals remains particularly challenging due to the recalcitrance and heterogeneity of this biomass component, explaining the use of model compounds and phenols in the majority of studies. Pursuing the application of more abundant transition metals, HDO of these substrates has been carried out favourably on acid/base supported Ni catalysts. Schutyser et al. screened a series of Ni-based catalysts (Ni/CeO2, Ni/ZrO2, Ni/TiO2, Ni/SiO2, Ni/γ-Al2O3 and Ni/MgO) and proposed a sequential transformation of lignin into polymer building blocks, caprolactones.24 Softwood lignin was firstly converted into 4-alkylguaiacols by hydrotreating, which underwent ring hydrogenation and demethylation, producing 4-alkylcyclohexanols (CHols). These were further dehydrogenated into 4-alkylcyclohexanones (CHones), which were oxidised to caprolactones. Amphoteric supports like ZrO2 and CeO2 favoured the formation of the alcohols and ketones. The TOFs of guaiacol, 4-n-propylguaiacol, 2-methoxycyclohexanol (2-MeOCHol) and other alcohols and ketones over Ni/CeO2 were promoted with a higher metal loading (Fig. 3a), which induced the Ni NPs to grow from 2.5 (1 wt% Ni) to 20 nm (12 wt% Ni). This pattern clearly indicated that the conversion of guaiacol was structure sensitive and larger NPs were more active.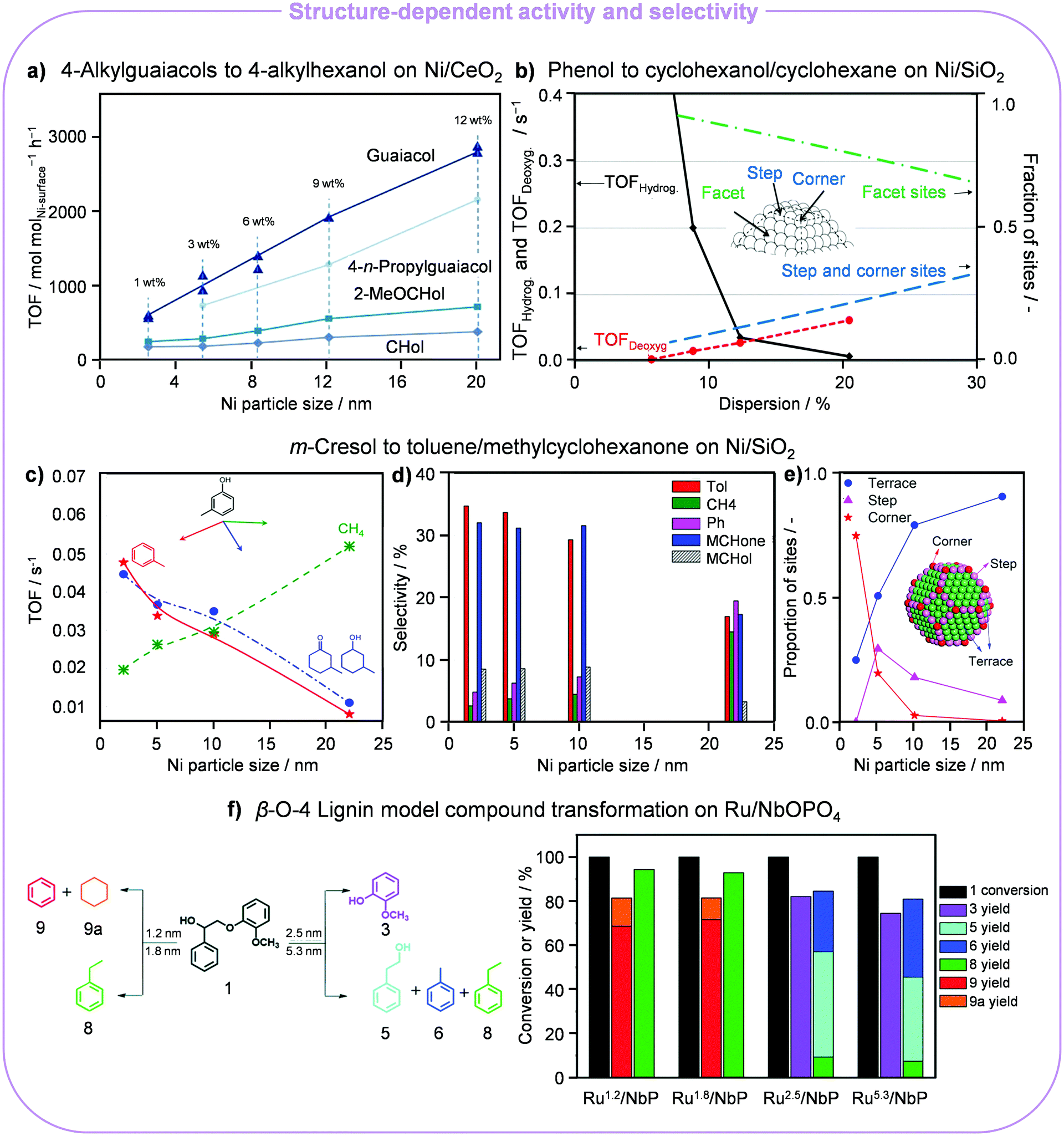 | ||
| Fig. 3 Particle size effects in the H2-assisted transformation of lignin derivatives. (a) Dependence of the TOF for guaiacol, 4-n-propylguaiacol, 2-MeOCHol and CHol hydrogenation on the Ni particle size for 12 wt% Ni/CeO2. Reproduced with permission from ref. 24. Copyright 2015 Wiley-VCH. (b) Relative amount of facet, step and corner sites depending on the size of Ni NPs in Ni/SiO2 and their impact on the hydrodeoxygenation of phenol. Reproduced with permission from ref. 25. Copyright 2015 Elsevier B.V. Role of the Ni NP size in the (c) TOF, (d) major hydrodeoxygenated product selectivity and (e) distribution of active sites in the NPs in the upgrading of cresol. Reproduced with permission from ref. 26. Copyright 2018 American Chemical Society. (f) Size-dependent product distribution in the hydrogenolysis of a β-O-4 lignin model compound over Ru/NbOPO4. Reproduced with permission from ref. 28. Copyright 2018 Royal Society of Chemistry. | ||
HDO of phenol over Ni/SiO2 (5–22 nm) in the liquid phase at 275 °C and 100 bar H2 also showed structure sensitivity.25 Both consecutive hydrogenation and deoxygenation steps depended on the size of the metal NPs. Larger particles proved more active to hydrogenate phenol to cyclohexanol with a monotonically increasing yield, while the yield of cyclohexane decreased. Accordingly, NPs of 10 nm were optimal. Elucidating the effect of the particle size was also key for the identification of the rate-limiting step. Due to the higher fraction of step and corner sites, the adsorption of oxygenated compounds and the cleavage of the C–O bond were favoured on smaller Ni NPs according to the surface site population theory (Fig. 3b). In contrast, abundant facets on larger Ni NPs (>10 nm) fostered the hydrogenation of the benzene ring, which can adsorb in a flat geometry over the contiguous metallic sites. Conversely, an opposite size dependency trend was illustrated for the HDO of m-cresol over Ni/SiO2 (2–22 nm) at 300 °C and 1 bar H2.26 Here, increasing the Ni NP size from 2 to 22 nm decreased the TOF of m-cresol conversion and facilitated the TOF of toluene (Tol), 3-methylcyclohexanone (MCHone), 3-methylcyclohexanol (MCHol), phenol (Ph) and CH4 formation (Fig. 3c and d). This was due to the fact that defect sites (steps and corners), abundant on small Ni NPs, foster deoxygenation and hydrogenation to yield Tol, MCHone and MCHol, while terrace sites, plentiful on larger NPs, are more active for C–C hydrogenolysis to CH4 and Ph (Fig. 3e). DFT calculations revealed that the position of defect sites is key to define how phenol is adsorbed and regulates the activation barrier of direct dehydroxylation, whereby a lower barrier was determined for defect-containing Ni(211) (120.5 kJ mol−1) than for Ni(111) and Ni(211). For the HDO of eugenol, small Co NPs supported on SiO2 and Al2O3 (<3 nm) showed a good performance to cleave Caryl–OCH3 and conduct aromatic ring hydrogenation, attaining propylcyclohexanol with a yield of >90% at 200 °C and 10 bar for 2 h.27 Conversely, the yield of propylcyclohexanol was lower over Co/CeO2, Co/ZrO2 and Co/HZSM-5 with respect to Co/SiO2 and Co/Al2O3, in which the Co NPs were in the range of 13–20 nm. This discrepancy indicated that higher cobalt dispersion was the main reason for the superior catalytic activity. However, Co/TiO2 surpassed Co/SiO2 and Co/Al2O3, yielding >99.9% of propylcyclohexanol even though the size of Co NPs on titania was larger. This result was explained based on the SMSI created by TiOy layers on the metal, modifying its electronic properties.
Considering the synthesis of aromatic compounds from lignin, ruthenium supported on niobium-based carriers (Ru/NbOPO4) exhibited a strong particle size effect.28 While larger Ru NPs (2.5–5.3 nm) favoured the production of oxygen-bearing chemicals like 2-methoxyphenol (75–82%) and 2-phenylethanol (38–47%), smaller aggregates (1.2–1.8 nm) could mediate the demanding C–O bond cleavage, leading to aromatic hydrocarbons with benzene and ethylbenzene yields of up to 72 and 94%, respectively (Fig. 3f). Furthermore, larger particles also favoured the generation of Tol by further hydrogenation or decarbonylation of 2-phenylethanol. DFT calculations indicated that surface models including step and edge sites, as in small Ru NPs, are associated with lower activation energies for C–O bond scission.
Other Ru catalysts carried on Nb-based solids were developed by the same research group.29 The catalyst with the smaller Ru particle size (Ru/Nb2O5-layer, 0.9 nm and 66.5% dispersion) outperformed (up to 88% selectivity) the commercial Ru/NbPO-CBMM catalyst (1.8 nm and 34.0% dispersion) and Ru/HY-340 (1.9 nm and 33.3% dispersion) in the formation of arenes from enzymatic lignin. When the Ru particles were too small for the aromatic ring to adsorb, niobium species activated the Caromatic–O bond to yield arenes. On the contrary, the benzene ring of phenolic monomers adsorbed directly on larger Ru NPs to form cycloalkanes.28,29 The superior arene productivity of Ru/Nb2O5-layer seems to be linked not only to the dispersion but also to the presence of Ruδ+ species, favouring arene selectivity, which are detected for this catalyst as well as Ru/HY-340, i.e., due to support effects.
Single ruthenium atoms supported on mesoporous graphitic carbon nitride (Ru1/mpg-C3N4) were reported to facilitate either hydrogenation or HDO of a typical lignin model compound, i.e., vanillin, in water.30 The atomic distribution of the metal was corroborated by HAADF-STEM, XRD, TEM and X-ray absorption fine structure (XAFS). Such Ru SACs mediated the selective hydrogenation or HDO of vanillin at distinct temperatures. Specifically, vanillyl alcohol was produced at lower temperature (60 °C), while the deoxygenated product, 2-methoxy-p-cresol, was formed together with vanillyl alcohol at 120 °C and fully converted into 2-methoxy-p-cresol at 140 °C. The selectivity switch was rationalised based on DFT calculations, indicating that although 2-methoxy-p-cresol is thermodynamically more stable, higher energy barriers have to be overcome to produce this chemical, which cannot happen at the lower reaction temperature.
Bimetallic catalysts were demonstrated to circumvent the handicaps of noble metals and nickel-based catalysts, i.e., the availability and cost of the former and low activity at low temperatures (<120 °C) and low dispersion of the latter.31 A Ni85Ru15 catalyst (2 nm) showed enhanced activity towards aromatic monomer formation by the cleavage of β-O-4 linkages under mild conditions (100 °C and 1 bar). This finding is explained based on the facilitated reduction of nickel in the presence of the second metal, which makes it possible to apply lower temperatures upon catalyst activation, hence fostering metal dispersion.
To investigate the validity of the systems developed for the transformation of lignin model compounds when using real feedstocks, some studies included tests with native lignin.24,28,31 For instance, Schutyser et al. carried out the conversion of pine sawdust-derived lignin oil in methanol over Ru/C.24 Alternatively, lignin oil mainly comprising 4-n-propylguaiacol was converted to n-propylCHol with a 73% yield over 3 wt% Ni/CeO2 (ca. 5–6 nm) at 300 °C and 20 bar H2. The impact of the size of Ru NPs on the product distribution reported above was mirrored in the transformation of enzymatically-derived lignin from corncob.28 Smaller particles supported on NbOPO4 (1.2 nm) were still substantially active for the cleavage of C–O bonds, yielding 90.6% monomers and 68.7% C7–C9 arenes, while larger particles on the same carrier (5.3 nm) mainly led to oxygen-containing compounds (26.6%). Furthermore, the performance of the bimetallic catalyst Ni85Ru15 for the depolymerization of birch wood sawdust-extracted organosolv lignin into monomeric aromatics was also consistent with that observed with model compounds, being more active than pure metal catalysts.31
Polyalcohols
Concerning sugar alcohols, Liu et al. reported an effective Cu–SiO2 catalyst (4.0–38.0 nm) for the selective hydrogenolysis of xylitol into C2 and C3 polyols without generating methane.32 With the increment of the Cu particle size, determined by dissociative N2O chemisorption, the TOF value and the selectivity of combined alcohols (ethylene glycol, propylene glycol and glycerol) monotonically improved, reaching an optimum value (140.8 h−1 TOF and 66.2% combined alcohol selectivity) for a size of 10.8 nm (90% Cu–SiO2, calcined at 550 °C) at 200 °C and 60 bar for 2 h. On the other hand, the selectivities of lactic acid and glycolic acid had an opposite pattern until 10.8 nm. Recalcination and re-reduction after use enabled the original conversion and C2 and C3 polyol selectivities to be retained for four consecutive cycles, effectively removing the deposited coke. Shifting focus onto sorbitol hydrogenolysis, Wang et al. emphasised the correlation between the particle size of Cu carried on carbon and the TOF and product distribution.33 Larger size and lower dispersion of copper were observed at a higher metal loading. The variation of these properties was reflected utterly in the TOF, which improved up to a Cu NP size of 14 nm (from 10.4 to 54.8 h−1) due to the enlargement of the active Cu(111) surface and remained stable upon further particle growth. Besides, the combined selectivity to 1,2-propandiol and lactic acid decreased constantly from 62.7 (5 wt% Cu/C, 5.8 nm) to 50.7% (25 wt% Cu/C, 14 nm), while the selectivity to glycerol rose from 10.2% to 25.0% over the catalyst containing Cu NPs of 14.0 nm. For the 25 wt% Cu/C catalyst, progressive sintering upon cycling under hydrothermal conditions led to a decrease of the sorbitol conversion and an increase of the selectivity to lactic acid.When considering the valorisation of glycerol, a burgeoning by-product of biodiesel production from fatty acids, the ReOx–Au/CeO2 catalyst offered unique features for allyl alcohol synthesis with molecular hydrogen.34 Indeed, the hydrogenation of the CC bond contained in the desired product was hindered by using a combination of Au, ReOx and CeO2, which is less active in hydrogenation as compared to noble metals (Pd, Pt, Ru, etc.), and by tuning the particle size of the Au cocatalyst. ReOx–Au0.6/CeO2 (25 nm) and ReOx–Au0.3/CeO2 (12 nm) prepared by impregnation led to an allyl alcohol selectivity of 83 and 95%, respectively, with the 1-propanol selectivity limited to 11 and 3%, at 140 °C for 24 h. In stark contrast, when the size of the Au NPs was reduced to 3 nm, by use of a deposition–precipitation method, the selectivity to the allyl alcohols dropped to 62% and that of 1-propanol rose to 30%. Based on H2-TPR, the authors suggested that the Au cocatalyst boosted the reducibility of rhenium oxide.
Other substrates
The fabrication of (ultrasmall) Pt NPs (0.7–5.3 nm) through stabilisation with a resorcinol–formaldehyde resin opened up a new route to the regioselective hydrogenation of quinolone with unrivalled activity under ambient conditions.35 Specifically, Pt NPs of 1.2 nm were associated with a 6-times higher TOF value than Pt NPs of 5.3 nm. The former exhibited almost complete conversion and selectivity towards 1,2,3,4-tetrahydroquinoline (99% for both parameters) within 80 min and appreciable recyclability (5 runs). Based on XANES and high-resolution valence-bond XPS and proton nuclear magnetic resonance spectroscopy (1H NMR), the particle size directly related to the d-band vacancy and the adsorption strength of quinolone and hydrogen, respectively.Another unconventional method enabled the production of high-loading Ni–N–C SACs for the valorisation of cellulose, outperforming conventional Ni NPs supported on activated carbon (Ni/AC) in terms of stability.36 Although the TOF of glycolaldehyde hydrogenation on fresh Ni/AC was ca. 11-times higher than on fresh Ni–N–C, the latter catalyst could be recycled 7 times after use in one-pot cellulose conversion under harsh conditions, while the activity over used Ni/AC (after the second cycle) halved. This evidence was attributed to the strong covalent bonding between nickel and the nitrogen atoms in the host, preventing leaching and aggregation.
Oxidation reactions
In view of the oxygenated nature of biomass components, oxidation reactions are considerably less widely applied in their upgrading than H2-assisted transformations. Still, they play a relevant role in the valorisation of two of the most important sugar-derived platforms, 5-HMF and furfural, into 2,5-diformylfuran (DFF) and 2,5-furandicarboxylic acid (FDCA), potential precursors for the polymer industry, as well as in the conversion of glycerol, a side stream in the conversion of fatty acids into biodiesel, into value-added products. The impact of the size of classical noble metal NPs applied in the oxidation of these substrates on the activity and selectivity has been explored in a few studies. When non-noble metals are considered, isolated sites embedded in zeolite matrices or carbon supports and low-nuclearity clusters stabilised on silicalite stand as significant speciations for effective upgrading of both furanic compounds and the triol.Furans
Siankevich et al. studied the oxidation of 5-HMF into FDCA with molecular oxygen using Pt NPs stabilised by PVP in water37 having five different average sizes between ca. 2 and 5 nm. They showed that the rate of the catalytic oxidation decreases as the size of the NPs increases, as expected in view of the reduced metal surface available to activate and convert the substrate. Moreover, they evidenced distinct product distributions after a catalytic run of 6 h for catalysts containing NPs of distinct sizes, uncovering the structure sensitivity of the consecutive oxidation steps (Fig. 4a). Specifically, the oxidation of the CH2OH moiety in 5-HMF to the CHO group in DFF is favoured over larger NPs, whereas the oxidation of the latter into the COOH group in FDCA is favoured over smaller NPs. These findings are in line with the reaction rate constants estimated based on kinetic modelling, indicating that the oxidation of the alcohol function in 5-HMF to the aldehydic group in DFF is the most facile step over the catalyst with bigger metal NPs. However, the smallest Pt NPs are the most active in relation to the full oxidation of 5-HMF to FDCA. Overall, the latter match the performance of benchmark systems but advantageously operate under milder conditions without the need for additives.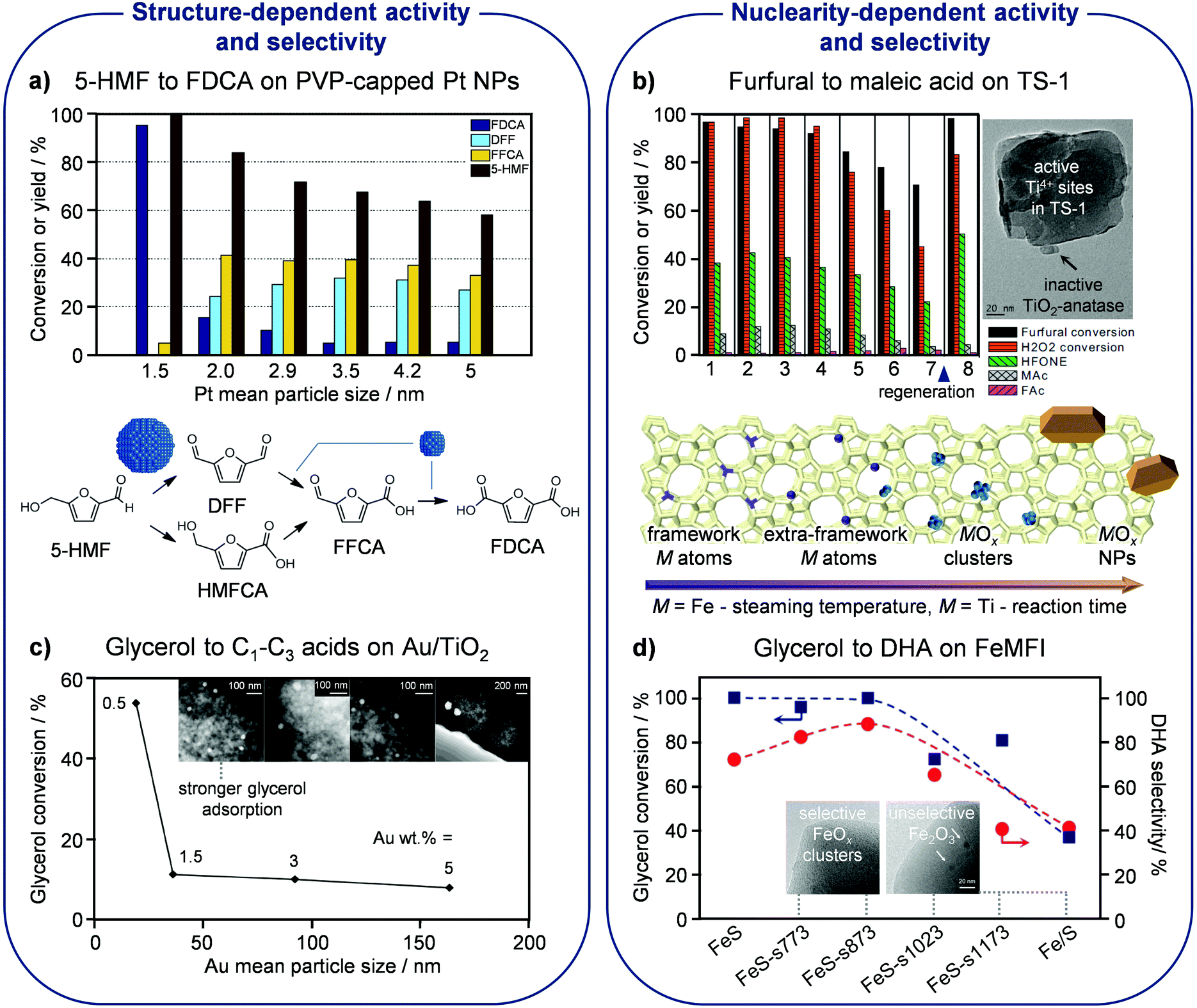 | ||
| Fig. 4 Particle size and nuclearity effects in oxidation reactions. (a) Smaller Pt NPs encapsulated in polyvinylpyrrolidone (PVP) exhibit higher activity and selectivity in the oxidation of 5-HMF to FDCA due to higher dispersion and favored DFF oxidation, respectively. Reproduced with permission from ref. 37. Copyright 2014 Elsevier B.V. (b) The maleic acid yield is depleted in subsequent catalytic cycles over titanium-silicalite (TS-1) due to the progressive extraction of Ti4+ cations from the MFI lattice and their aggregation into inactive TiO2. Reproduced with permission from ref. 40. Copyright 2017 Elsevier B.V. (c) Smaller NPs of Au supported on titania show higher activity in glycerol oxidation due to stronger adsorption of the reactant. Reproduced with permission from ref. 41. Copyright 2017 American Chemical Society. (d) High selectivity to DHA in glycerol oxidation is associated with low-nuclearity extraframework clusters of oxidic iron on silicalite (S), which can be obtained by mild steaming of an iron-containing hydrothermally-prepared solid. Reproduced with permission from ref. 42. Copyright 2015 American Chemical Society. | ||
Artz et al. have reported the preparation of a Ru-based catalyst showing 7-times higher productivity than the conventional Ru/C catalyst in the aerobic oxidation of 5-HMF to DFF, by using a covalent triazine framework as a carrier.38 The nitrogen atoms contained therein served as anchoring sites for the homogeneous Ru salt used as a precursor, minimising the sintering of the noble metal upon reduction in H2. The authors claimed that no nanoparticles formed upon this procedure based on the absence of defined aggregates in the TEM images collected. Hence, they indicated that the activated catalyst contains clusters, with the high metal dispersion being the reason for its superiority. The mesoporous volume and surface area of the support were stressed as relevant parameters to reach this result. The catalyst suffered from deactivation due to strong substrate and product adsorption, but reactivation under a H2 flow enabled the recovery of a large fraction of the original activity, permitting the recycling of the solid in four further catalytic runs. Despite the fact that the study suggests strong particle size effects, a more accurate determination of the dimensions of the metal aggregates would be required to derive a more quantitative relation between size and performance.
Zhou et al. introduced a Co-based SAC supported on N-doped carbon (named Co SAs/N@C) obtained from a cobalt/zinc-lignin (Co/Zn-L) complex for the oxidation of alcohols and aldehydes, including furfural and 5-HMF, to the corresponding carboxylic acids in the presence of molecular oxygen and Na2CO3.39 The catalyst performance is generally inferior to that of materials based on noble metals, but surpasses that of an analogue, in which Co NPs were supported on the same carrier, by a factor of 2, upon 5-HMF oxidation in relation to both selectivity and activity. The latter observation was explained not only in terms of dispersion, but also of the lower energy barrier for the activation of furfural on the single atom catalyst, in line with the behaviour of undercoordinated single metal sites in a variety of reactions. While no insights were offered into the evolution of adsorbates on the catalyst surface, the main role of oxygen in removing abstracted hydrogen in the form of water was proven by experiments using 18O2. It should be remarked that such a direct comparison of catalysts featuring the metal in NP or single atom form is rare in the studies reporting SACs, and the exact reason why single-atom species lower the reaction barrier remains unclear.
Another example of a SAC applied in the oxidation of furfural, to maleic acid in this case, comprises titanium silicalite applied by Alba-Rubio et al. in an aqueous medium containing hydrogen peroxide as the oxidant (Fig. 4b).40 Titanium silicalite, also called TS-1, is a pure-silica framework with MFI topology in which titanium isomorphously substitutes silicon, introduced by Enichem almost four decades ago. The isolated lattice Ti4+ species showed remarkable activity and selectivity in this application as well as in a number of other oxidation reactions, and could also mediate other transformations due to their intrinsic Lewis-acid character (vide infra). The mentioned study highlighted that the catalyst performance depends on the quality of the active sites, with extraframework titanium atoms forming inactive TiOx clusters blocking the access to the active isolated sites. Upon reaction, the catalyst suffered from fouling and moderate titanium leaching. Whereas the carbonaceous deposits could be successfully removed by calcination in order to reuse the catalyst in subsequent catalytic cycles, the loss of titanium is irreversible and, since it affected both speciations of the metals, it brought beneficial and detrimental consequences. While the oxidic deposits were cleared, improving diffusion in the catalyst micropores, some of the desired catalytic centres were also washed out in the long term.
Glycerol
The effect of the metal particle size in the aerobic oxidation of glycerol in the aqueous phase has been investigated by D’Agostino et al. on Au catalysts supported on titania.41 The dimensions of the Au NPs were varied in the range of ca. 20–165 nm by altering the loading of the metal on the carrier between 0.5 and 5 wt%. In line with previous studies, the particle size distribution was narrower for the catalyst with lower loading and was augmented significantly for materials containing more of the noble metal. The catalytic data indicated a decrease of the catalyst activity with an increase of the gold content (Fig. 4c). The superiority of smaller Au NPs was explained through 1H NMR using proton relaxation methods. These analyses uncovered that small gold particles can adsorb glycerol more strongly than the molecules of the water solvent, which has clear consequences for the performance, thus providing the first experimental support of earlier indications from computational and theoretical studies. Accurate control of the particle size (distribution) is thus recommended for this and other applications of gold catalysts in glycerol upgrading.Glycerol can be oxidised at different carbon atoms and to distinct extents, leading, in association with acid-catalysed reactions often simultaneously promoted under the reaction conditions applied, to an array of products with diverse relevance. The partially oxidised derivative dihydroxyacetone (DHA) has received great attention because it serves as a substrate for the production of lactic acid, a monomer used for the manufacture of biodegradable plastics, through Lewis-acid mediated isomerisation (vide infra). High selectivity to DHA (88%) was attained in a gas-phase process at 350 °C by Lari et al. through the precise tuning of the agglomeration degree of iron accommodated in a silicalite matrix.42 Highly dispersed iron species, including isolated cations or tiny FeOx clusters in extraframework positions of silicalite, were active and exclusively produced DHA, except for a small amount of CO2 (Fig. 4d). In stark contrast, more clustered iron species, like Fe2O3 particles supported on the same pure-silica zeolite, were shown to promote competitive oxidation and dehydration reactions, mainly leading to acetol, pyruvaldehyde and pyruvic acid, thus reaching a selectivity to DHA of only 40%. Suitable iron speciation was attained by hydrothermal synthesis of silicalite with 0.75 wt% of the transition metal followed by controlled steaming at a mild temperature of 500 °C. Accordingly, iron introduced in the silicalite lattice was extracted to extraframework positions, while its aggregation into large clusters or NPs was hindered, as occurred upon applying higher steaming temperatures. Only a slight decline of the catalytic performance was detected with time on stream due to minor sintering of the iron species.
Reforming reactions
Formic acid and alcohols, obtained as target or side products in biomass valorisation processes, are preferred substrates for reforming reactions owing to their comparably high hydrogen content. Platinum and platinum-group metals have been mainly applied to these transformations in the form of NPs of distinct sizes as well as single atoms, often cooperating with sites carried by the support to reach higher efficiency.Formic acid
Regarding the use of formic acid as a hydrogen vector, Koh et al. supported ultrasmall palladium NPs (ca. 2 nm) on amine-functionalised SBA-15 (SBA-15-N/Pd, Fig. 5a, top).43 They showed that such a catalyst could generate hydrogen through formic acid dehydrogenation at ambient temperature with a remarkable TOF per mole of metal in the catalyst (293 h−1) and with close to 100% selectivity to CO2, i.e., without producing any CO and water according to a competitive path. They also demonstrated that it could be reused without appreciable performance loss in repeated cycles, which was in line with an increase of the Pd particle size from 1.9 to only 2.4 nm after the fifth use. Since the particle size distribution was moderately broader after the last cycle, including particles of ca. 3.5 nm, these results imply the lack of pronounced structure-sensitivity of the reaction. The amine functional groups inside the mesoporous silica channels and their ability to stabilise the Pd NPs were claimed to play important roles in limiting the agglomeration of the metal upon reduction. Besides, such functional groups were supposed to donate electron density to Pd, thereby enhancing its activity in the reaction. Moreover, they would partake in the reaction mechanism too. Specifically, they could serve as Brønsted-basic sites, facilitating the deprotonation of formic acid into a formate intermediate, as corroborated by the control experiments with added n-propylamine (PA, Fig. 5a), while the ultrasmall Pd NPs would catalytically activate the C–H bond of the resulting formate species. Accordingly, the small size of the Pd NPs would be beneficial to increase the number of proximal basic and redox sites.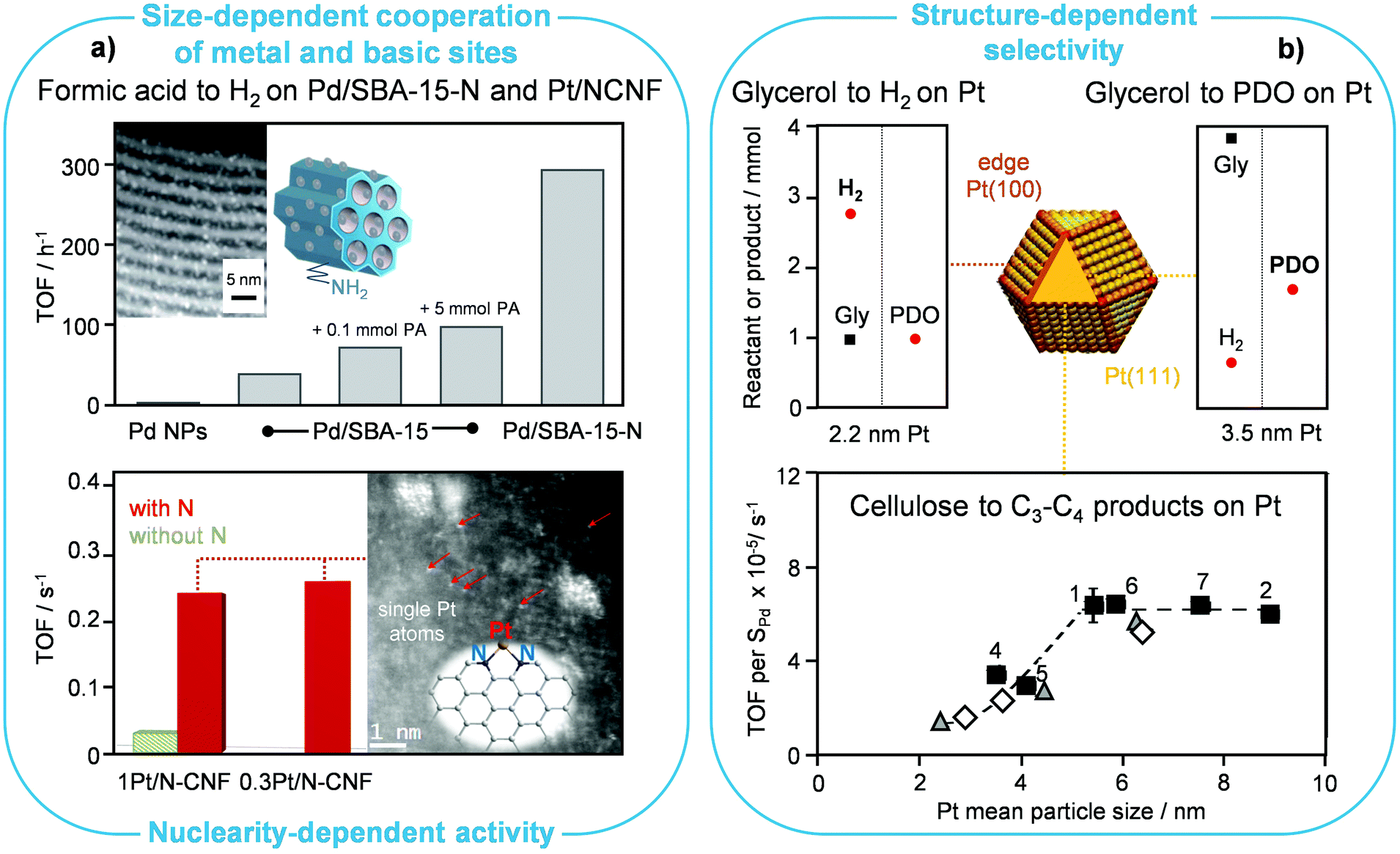 | ||
| Fig. 5 Particle size and speciation effects in reforming reactions. (a) Formic acid is more effectively reformed to H2 on small Pd NPs, which are in closer contact with the amine groups anchored on the SBA support (reproduced with permission from ref. 43. Copyright 2014 Royal Society of Chemistry), and on single Pt atoms stabilised on nitrogen-containing carbon nanofibers (reproduced with permission from ref. 44. Copyright 2016 American Chemical Society). (b) The formation of H2 from glycerol (Gly) is favoured on (100) edges of Pt NPs, while the production of 1,2-propanediol (PDO) from the same substrate (reproduced with permission from ref. 46. Copyright 2018 Elsevier B.V.) and of C3–C4 compounds from cellulose (reproduced with permission from ref. 47. Copyright 2017 Wiley-VCH) is better mediated by (111) surfaces, leading to distinct preferred Pt NP sizes in the different cases. | ||
Another type of N-containing carrier, i.e., mesoporous N-doped carbon nanofibers (N-CNF), was used by Bulushev et al. to anchor Ru, Pd and Pt in the form of single atoms, reaching with the latter metal up to 1-order-of-magnitude higher rates of formic acid decomposition than over corresponding NP-based catalysts (Fig. 5a, bottom).44 The TOF associated with this Pt-based SAC is apparently calculated based on the exposed metal and is ca. 3 times higher than for the catalyst of Koh et al. described above.43 The identification of isolated metal sites by electron microscopy, their reduced ability to chemisorb CO and the oxidised state determined by XPS hinted that a major fraction of platinum is present in the form of single atoms. The bonding of the latter to the support and formic acid adsorption were investigated by quantum-chemical calculations comparing Pt atoms attached to the divacancy with two pyridinic N atoms, near to graphitic type N atoms, to two pyridinic N atoms on the armchair edge, or to two pyridinic N atoms on the zigzag edge. The simulations indicate that the strongest interaction of formic acid leading to dissociation is exclusive to the third type of Pt atoms, which assume a ionic/electron-deficient state and coordinate the substrate in a square planar geometry, while the other sites can be considered as inactive. Such single atoms would increase the selectivity of the reaction, since an inferior value was determined over analogous catalysts supported on unfunctionalised carbon nanofibers featuring metallic NPs. They would also prevent aggregation, thus explaining the only minor drop in activity of the catalyst under 45 h on stream. It should be remarked that such an insightful face-to-face comparison of the performance of single atoms and nanostructures is one of the very few presented so far in the literature.
Alcohols
Steam reforming of ethanol has been extensively studied.45 Nonetheless, the role of the particle size in the catalyst performance remains not fully understood. Mechanistic studies uncovered that the desired dehydrogenation/hydrogenation of CHx species, activation of water and oxidation of C* species are favoured on low-coordination metal sites, while undesired reactions, like CHx hydrogenation to methane and the growth of carbon filaments, preferably occur on higher-coordination metal sites. In agreement with these findings, testing of Co NPs with an average size of 3–8 nm obtained through a colloidal route and supported on SiO2 indicated that smaller particles were more active in ethanol steam reforming. Similarly, the TOF increased with decreasing particle size for Co NPs deposited on carbon nanofibers. Moreover, the smaller Co NPs underwent deactivation via fouling and sintering to a lower extent. Specifically, the average carbon deposition rate diminished from 0.010 to 0.002 gC h−1 upon decreasing the metal size in the Co/SiO2 catalysts. For Ni, Rh and Pt, theoretical simulations put forward the type of surface termination that would enhance C–H cleavage, CO oxidation and carbon growth, providing guidelines to identify the most suited size for NPs of these metals. In the case of Pt NPs, it was calculated that graphene would start forming fibres moving to the support rather than layers covering the metal NPs when the latter are smaller than ca. 2 nm. For Ni NPs, particles up to 10 nm are resistant to fouling. The metal particle size was also indicated to play a role in the facility with which the metal would form detrimental oxidised species during the reaction. The latter information is especially relevant for tests in which oxygen is added to the feed stream to facilitate hydrogen removal from the surface. It was suggested that oxidation might be more relevant than fouling at small particle sizes for Co NPs, but no experimental correlations corroborated such a statement.When considering the aqueous-phase reforming of glycerol on Pt/Al2O3 catalysts, the selectivity to valuable liquid and gas products could be tailored using Pt NPs of distinct sizes.46 All catalysts contained 2 wt% of the noble metal, which was aggregated to NPs between 2.25 and 3.61 nm, based on X-ray diffraction analyses, by applying different reduction temperatures in the range of 25 to 90 °C. Smaller Pt NPs, rich in edge sites such as those of (100) terminations, which are responsible for the dehydrogenation of glycerol to glyceraldehyde in the reforming process, formed H2 as the main product with a yield of 5.5% (Fig. 5b, top). In contrast, larger Pt NPs, mainly exhibiting (111) surfaces able to dehydrate glycerol to hydroxyacetone, led to 1,2-propanediol with a yield of 12.5%.
Cellulose
Ogo et al. investigated the hydrothermal conversion of biomass cellulose to light hydrocarbons over Pt/zeolite catalysts, finding a Pt/NH4-USY catalyst as the material maximising the production of C3 + C4 hydrocarbons (14.5 C% after 72 h reaction time) even at 170 °C without hydrogen or other expensive reagents.47 The conversion of cellulose to liquid products was mainly promoted over the acidic sites of the zeolite carrier, while their further upgrading took place on the metallic sites. The size of the Pt NPs was varied by altering the loading (1–3 wt%), the metal incorporation method and the calcination temperature, and was stable under the reaction conditions. The TOF value increased with an increase of the Pt NP size up to 5 nm, while levelling off for larger metal structures (Fig. 5b, bottom). This evidence was explained based on the catalytic site requirement for the dissociation of the carbonyl group of glucose to enable hydrocarbon formation, including several metal atoms and step-edge sites. Such sites can be provided by (311) and (331) surfaces, which are only exposed if the NP size exceeds 5 nm.Acid-catalysed reactions
Acid-catalysed reactions take centre stage in the conversion of biomass into chemicals and fuels. They are required for the decompositon of (hemi)cellulose into its building units (i.e., hydrolysis) as well as in a number of transformations (i.e., dehydration, epi-/isomerisation, retro-aldolisation, aldol-condensation, and transfer-hydrogenation) valorising these monosaccharides into platform molecules and further upgraded products. Metals in oxidised forms best mediate these reactions, whereby a major role is played by non-noble metals stabilised as single atoms in porous crystalline frameworks. In this context, a rich body of literature investigated zeolites and zeotype materials, but a few significant alternatives based on metal organic frameworks (MOFs) have also been reported.Sugars, ketones and furans
Lewis-acid metals such as Al, Ga, Sn, Ti and Zr were incorporated into amorphous porous materials like MCM-41 and SBA-15 and zeolites.48 Among the latter, the MFI and BEA frameworks were most extensively explored as hosts for such well-defined single atoms. Zeolite-based materials showed higher efficiency in the conversion of hexoses and trioses, especially in aqueous media, with those containing tin exhibiting the best performances. This is due to the intrinsically stronger Lewis acidity of this metal as well as the lack of its significant Brønsted-acid character, typically mediating competitive pathways, in both isolated and aggregated forms. As for the latter, SnOx clusters would not take part in the reactions. Hence, the detrimental effect of clustering simply is inefficient metal utilisation.Sn-containing beta zeolites prepared using hydrothermal synthesis displayed unrivalled activity and selectivity in the isomerisation of glucose to fructose and mannose, xylose to xylulose, lactose to lactulose and DHA to lactic acid and alkyl lactates, in the one-pot conversion of glucose to lactic acid (through consecutive isomerisation, retroaldol reaction and isomerisation), in the conversion of 5-HMF into furan derivatives and of furan into p-xylene, and in the C–C coupling between DHA and formaldehyde (Fig. 6a). This is due to the high quality of the tin sites, i.e., virtually all tin atoms present in such materials isomorphously substitute silicon atoms in the lattice. Still, since the crystallisation of these materials requires fluoride ions and a long time and produces large crystals, in which bulk tin atoms are typically underutilised due to diffusional constraints, a multiplicity of methods were devised to incorporate the catalytic centres in the lattice in a more facile manner and limit the size of the zeolite particles. Seed-assisted hydrothermal syntheses or dry-gel conversion protocols belong to alternative bottom-up approaches, while direct metallation and demetallation–metallation comprise relevant top-down strategies. The latter methods reach inferior site quality, i.e., a portion of tin does not enter the lattice but deposits on the external surface and in the pores in the form of inactive SnOx species. Still, the Lewis-acid metal is mainly introduced at the surface of the zeolite crystals, which helps in counterbalancing that drawback. Through soft or hard templating in hydrothermal syntheses and with alkaline-assisted post-synthetic metallation it was also shown to be possible to concomitantly tune the porosity of zeolites, thus granting improved access to large bio-derived substrates.
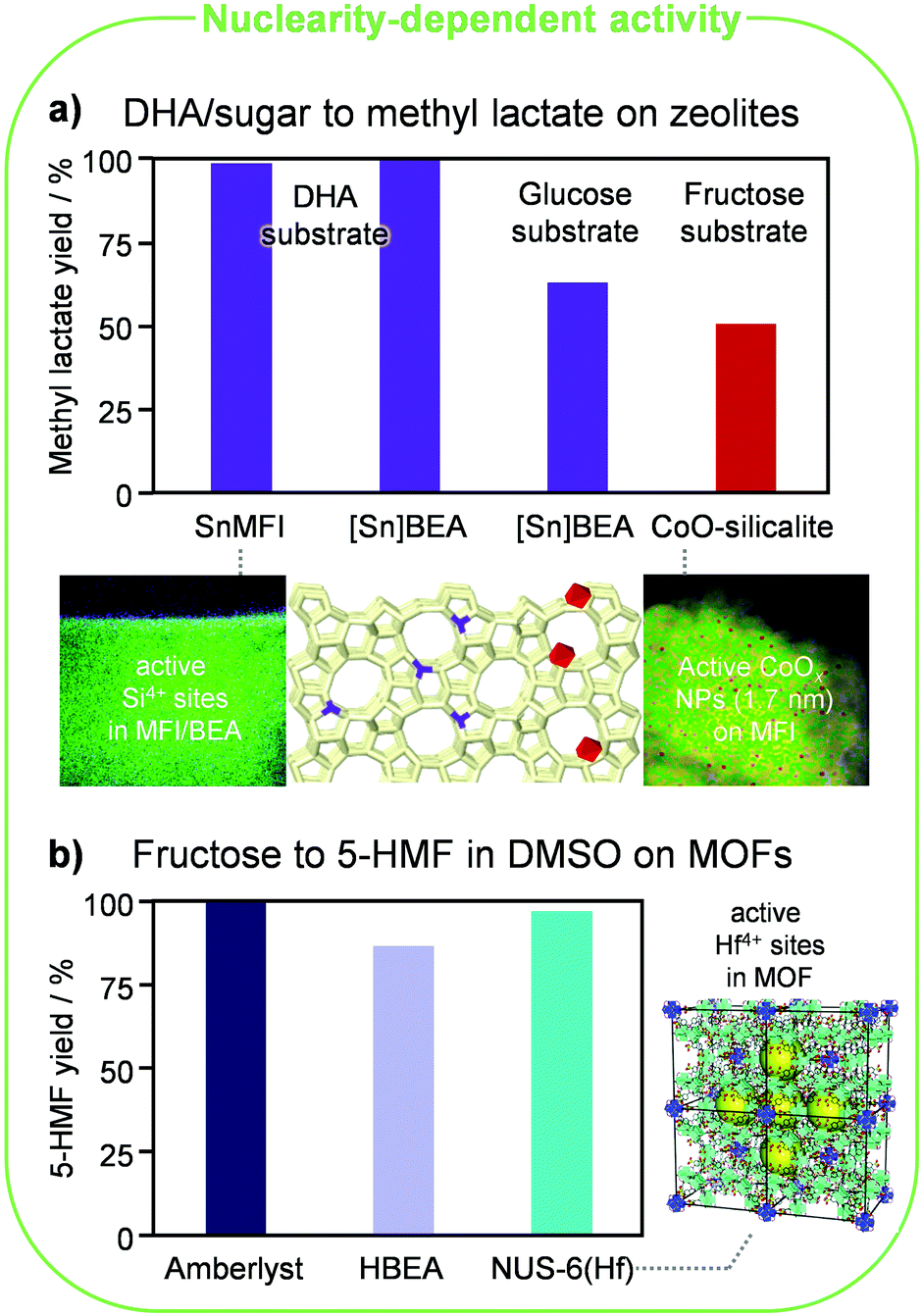 | ||
| Fig. 6 Nuclearity and coordination effects in acid-catalysed reactions. (a) Framework Sn4+ cations (purple) in silicalite (reproduced with permission from ref. 49. Copyright 2016 Royal Society of Chemistry) and Al-free BEA zeolites and small extraframework NPs of CoOx (red) supported on silicalite (reproduced with permission from ref. 50. Copyright 2019 American Chemical Society) effectively catalyse the conversion of monosaccharides and DHA to methyl lactate. The square brackets in the catalyst name underline that the guest atom is embedded in the lattice. (b) Hf4+ cations in the NUS-6 MOF reach superior performance compared to the best conventional heterogeneous catalysts in the dehydration of fructose to 5-HMF in DMSO. The yields reported were determined after 1 and 2 h at 100 and 120 °C for the MOF and the other two catalysts, respectively. Reproduced with permission from ref. 52. Copyright 2017 Royal Society of Chemistry. | ||
The active site in Sn-beta catalysts was shown to comprise lattice tin atoms in an open form, implying that one of the four Sn–O bonds is hydrolysed, forming a stannanol, coordinating two water molecules. In spite of their hydrophobic surface, tin zeolites with BEA and MFI frameworks displayed rapid deactivation upon isomerisation of DHA and xylose in water.49 Thorough investigations in the less frequently applied but more industrially relevant continuous mode related this phenomenon to an interplay of distinct mechanisms, with zeolite amorphisation and tin site restructuring dominating over tin leaching and catalyst fouling, and being more pronounced for BEA solids and for materials obtained though post-synthetic procedures. A better scenario was evidenced when using methanol as a solvent. Due to its inferior basic character, irreversible structural alterations of zeolite and tin sites were minimised and the activity was depleted to a lower extent and mostly due to the deposition of carbonaceous products. Calcination was effective in restoring the initial activity almost completely. It is worth noting that life-cycle analysis proved methanol to be a preferable medium for the manufacture of lactic acid from glycerol compared to water from both an economic and environmental viewpoint.
Silicalite has been alternatively used to encapsulate sintering-resistant clusters of CoO for the conversion of fructose into methyl lactate.50 Ultrasmall CoO NPs (∼1.7 nm) were attained through a bottom-up steam-assisted method with Co loadings smaller than 2 wt% and led to nearly 100-fold higher Co-mass-based activity (mgmethyl lactate mgCo−1) compared with CoO or Co3O4 particles located outside the silicalite framework. However, the catalyst remains moderately inferior to the best Sn-containing beta zeolite applied to the more complex glucose-to-DHA conversion, i.e., an additional initial isomerisation step is required when starting from the aldose (Fig. 6a). Since the CoO phase formed strong covalent bonds with the zeolite framework, this catalyst exhibited good reusability after removal of coke through facile calcination. This contrasts with the fate of other metal oxide catalysts, which irreversibly deactivated in the regeneration step due to particle coalescence.
Several MOFs containing distinct metal centres and linkers were studied as catalysts for the valorisation of biobased compounds, typically attaining inferior performances to classic inorganic catalysts, except for a few materials that deserve a mention. Hf-based MOFs introduced by Rojas-Buzo et al. showed high selectivity and efficiency in the cross-aldol condensation of furanic carbonyls with acetone at 100 °C, attaining an almost full yield of the desired products.51 The metal centres in such materials are hexacoordinated, i.e., coordinatively unsaturated, and highly accessible due to the size of the pores (4.8 and 18.4 Å). NMR studies with isotopically-labelled acetone corroborated that acid–base pairs in the MOF comprising the metal centre and the oxygen bound to it, respectively, mediate the soft enolisation of acetone via abstraction of the α-proton. The catalyst Hf-MOF-808 could be reused 5 times with only a minor depletion in catalytic activity, which could be fully recovered by Soxhlet extraction. Moreover, although the presence of added water (10 wt% with a molar water/furfural ratio of 60) led to a 25% activity loss compared to the dry reaction, the condensation product was attained with a yield of 76% upon a 4-fold longer reaction. This diverges from findings for traditional base catalysts, which strongly deactivate under similar conditions.
Other MOFs displaying remarkable performance are MIL-101Cr-SO3H-15% and NUS-6(Hf), which are among the best heterogeneous catalysts reported for the conversion of fructose into 5-HMF in dimethylsulphamide (DMSO).52 While sulphonic acid groups anchored onto the first material rather than the chromium cations are the actual active sites in the first, hafnium sites are responsible for 5-HMF formation over the second. This solid was prepared through a hydrothermal route with 2-sulfoterephthalate linkers. The structure features some partially missing linkers, giving rise to additional mesopores with a size of ca. 4 nm (indicated by yellow spheres in Fig. 6b). This catalyst yielded 98% 5-HMF after 1 h at 100 °C, being thus superior to top traditional heterogeneous catalysts such as an Amberlyst resin and a zeolite beta in protonic form, which required 2 h and 120 °C to reach comparable and still moderately inferior yields, respectively (Fig. 6b). While DMSO is an appealing solvent because it hinders the generation of humins and hence catalyst deactivation by fouling, product separation from this medium is challenging due to the formation of toxic sulphur compounds at the high temperatures needed for its distillation. Therefore, despite the relevant results, it is difficult to foresee a technical process for this reaction based on this MOF-based catalytic system.
Acids
For the esterification of LA to ethyl levulinate, UiO-66Zr MOF catalysts, displaying octahedral cavities, tetrahedral cavities and narrow triangular windows with diameters of 11, 8 and 6 Å, respectively, are superior to effective heterogeneous catalysts such as Amberlyst-15, a zeolite HZSM-5, a mesoporous silica HMCM-22 and sulphated TiO2 and ZrO2.52 For instance, at an ethanol/LA ratio of 5, UiO-66Zr attained a conversion of 49% after 4 h at 78 °C, while Amberlyst-15 and sulphated TiO2 reached conversion levels of 55 and 40%, respectively, after 5 h at 70 °C. In contrast to these results, the MOF is inferior to a material containing dodecatungstophosphoric acid deposited onto a desilicated zeolite and sulphated TiO2 nanorods. Indeed, it reached full conversion of the acid and a yield of 95% of the ester at the same temperature in 8 h of reaction only at higher ethanol/levulinic acid ratios, while the above-mentioned systems attained conversions of 82–83% under comparable or higher temperatures with a lower alcohol excess. Clearly, the catalyst activity and stability are here the main issues, while the reaction does not pose significant selectivity challenges. Interestingly, the importance of open metal sites for the MOF activity was highlighted through a study of the effect of the linker deficiency on the performance. Specifically, materials exhibiting higher linker deficiencies, determined on the basis of the mass loss of organic ligands in thermogravimetric analyses, correlated with up to 1-order-of-magnitude higher activity and the corresponding rate constants were higher. In all cases, the coordination number of the metal centre was lower than 12, i.e., the theoretical maximum expected for the UiO-66 structure. Hence, particle size control, i.e., metal site isolation, does not guarantee per se the highest performance of the MOF.Conclusions and outlook
This review has outlined the effects of the metal particle size in a wide range of biomass upgrading strategies, spanning redox reactions, i.e., hydro(deoxy)genation, oxidation and reforming, to a number of acid-catalysed transformations, involving most relevant substrates like furans, sugars, alcohols and polyols, ketones, acids and lignin derivatives. The findings are graphically summarised in Table 1.| Metal | Size | Decarbonylation | CO hydrogenation |
Ring CC hydrogenation |
Ring opening (C–O hydrogenolysis) | Deoxygenation | Ester/acid reduction | Oxidation | Reforming | Acid-catalysed reactions |
|---|---|---|---|---|---|---|---|---|---|---|
| Pt |

|

|

|

|

|

|
||||
| Pd |

|

|

|

|

|

|
||||
| Ni |

|

|

|

|
||||||
| Ru |

|

|

|

|
||||||
| Co |

|

|

|

|

|

|

|
|||
| Au |

|

|

|
|||||||
| Cu |

|

|

|
|||||||
| Ti, Sn |

|

|

|
|||||||
| Fe |

|

|
Concerning H2-mediated reactions, most studies suggest that smaller NPs are preferable for decarbonylation of furans, with the benefit from decreasing particle size being stronger for Pd and Ru than for Pt. They also hint that smaller Pd and Co NPs are more suited for their hydrogenation and larger Cu NPs for their hydrogenolysis. These findings relate to the relative amount of furan adsorbing through the carbonyl or through the ring. Pd SACs generally emerge as superior catalysts for furan hydrogenation. The response of gold to the degree of agglomeration contrasts with the general trend, with single atoms being inactive and clusters or small NPs representing the most adequate speciation for H2-assisted furan valorisation. In the upgrading of lignin derivatives (i.e., guaiacols and phenol), larger Ni NPs and smaller Pt NPs are more active in ring hydrogenation and larger NPs of both metals better mediate C–O bond scission. Smaller Ru NPs in Nb-containing catalysts foster the generation of arenes owing to their greater capability to cleave aromatic and aliphatic C–O bonds. A seemingly underlying trend is that ring hydrogenation becomes favoured over C–O scission on larger metal particles due to the requirement of a flat adsorption geometry.
Also in oxidations, the particle size has a strong influence on the activity and selectivity patterns. For furan upgrading on noble metals, enhanced conversion levels of the substrate are achieved upon increasing the active site dispersion, e.g., using single atoms of cobalt or clusters of ruthenium. When non-noble metals such as titanium in silicalite are used, a sharp transition is observed, i.e., isolated cations in the lattice of the host comprise the only active speciation. In terms of selectivity, the formation of partially rather than fully oxidised products is favoured on larger Pt NPs. In glycerol valorisation, Au NPs of ca. 20 nm size show higher activity than larger particles due to higher metal availability as well as to improved adsorption capability. This contrasts the findings in the field of hydrogenation reactions, where such Au NPs would already exceed the optimal clustering degree. Still, this observation reinforces the conclusion that gold works better in agglomerated form. Iron zeolites displayed high chemoselectivity under the strong prerequisite that the transition metal remains anchored at extraframework positions of the silicate in the form of isolated sites or low-nuclearity clusters, with more agglomerated species activating paths to an array of secondary products.
In the case of biomass reforming, two main impacts of the particle size have been described. On one hand, decreasing the dimensions of the metal phase to small NPs and single atoms, mainly comprised of platinum and palladium, increases the proximity of redox centres to basic nitrogen-containing functions of the host material, fostering their synergetic roles in the valorisation of formic acid, and improves the sintering-resistance of the metal. With regard to single Pt atoms, an electron deficient state was claimed as the origin of the superior performance. On the other hand, tuning the particle size is effective in controlling the consequences of fouling and directing the selectivity to either gaseous or liquid products in the reforming of ethanol and glycerol, respectively.
Concerning Lewis-acid catalysed conversions, non-noble metals, mostly tin, hosted as single cations in the lattice of all-silica zeolites or embedded as tiny oxidic clusters into the same materials stand as the only speciation effective in mediating the valorisation of sugars, furans, acids and ketones. In stark contrast, even slightly more agglomerated oxide phases of this metal have displayed no activity in the same conversion routes, similarly to titanium catalysts applied in oxidations. The strong bonds between the metal and the matrix hinder irreversible deactivation phenomena, such as leaching, to an appreciable extent. However, the stability of tin centres in aqueous biomass upgrading appears inferior to that of (noble) metals anchored in isolated form onto host materials used in H2-assisted reactions. Single metal atoms, especially hafnium and zirconium, included in MOFs also show appreciable activity in this class of transformations when under-coordinated.
While metal isolation has led to very significant improvements in various biomass-to-value routes, only a very few studies have rigorously compared the performance of SACs, in term of activity, selectivity and stability, with NP-based counterparts featuring the same composition under identical conditions, as stressed above. It is clear that more comparative studies are required to assess the actual standing and potential of single metal atoms in a sensible and comprehensive manner, with a rigorous expression of the rate per exposed metal active site. This will help the more promising applications in this lively arena of heterogeneous catalysis further develop towards practical realisation. To this end, a deeper characterisation of the electronic properties of single atoms and of the nature of the interactions with the host should also be conducted in the future. Moreover, there is now a sharp transition from NPs to single atoms, with low-nuclearity clusters remaining mostly unexplored. Future studies should fill this gap, investigating their behaviour, which cannot be easily extrapolated due to the non-linearity of effects at small scales. This shall also be instrumental to unlock superior performances in conversions already investigated or mediate unprecedented reactions, as well as to gather further awareness of the contribution of secondary species possibly generated in the synthesis of isolated species in diverse applications of this research field. All these tasks shall require the development of improved or novel synthesis strategies to control the metal nuclearity and ensure high site quality.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the ETH Zurich and the National University of Singapore Flagship Green Energy Program (R-279-000-553-646 and R-279-000-553-731) for financial support. G. G. thanks the SINGA scholarship for supporting his PhD study at NUS.Notes and references
- R. J. White, R. Luque, V. L. Budarin, J. H. Clark and D. J. Macquarrie, Chem. Soc. Rev., 2009, 38, 481–494 RSC.
- X.-F. Yang, A. Wang, B. Qiao, J. Li, J. Liu and T. Zhang, Acc. Chem. Res., 2013, 46, 1740–1748 CrossRef CAS PubMed.
- P. Mäki-Arvela, B. Holmbom, T. Salmi and D. Y. Murzin, Catal. Rev., 2007, 49, 197–340 CrossRef.
- V. V. Pushkarev, N. Musselwhite, K. An, S. Alayoglu and G. A. Somorjai, Nano Lett., 2012, 12, 5196–5201 CrossRef CAS PubMed.
- K. An, N. Musselwhite, G. Kennedy, V. V. Pushkarev, L. Robert Baker and G. A. Somorjai, J. Colloid Interface Sci., 2013, 392, 122–128 CrossRef CAS PubMed.
- Q.-X. Cai, J.-G. Wang, Y.-G. Wang and D. Mei, AIChE J., 2015, 61, 3812–3824 CrossRef CAS.
- L. J. Durndell, G. Zou, W. Shangguan, A. F. Lee and K. Wilson, ChemCatChem, 2019, 11, 3927–3932 CrossRef CAS.
- Z. Zhang, J. Song, Z. Jiang, Q. Meng, P. Zhang and B. Han, ChemCatChem, 2017, 9, 2448–2452 CrossRef CAS.
- S. M. Rogers, C. R. A. Catlow, C. E. Chan-Thaw, A. Chutia, N. Jian, R. E. Palmer, M. Perdjon, A. Thetford, N. Dimitratos, A. Villa and P. P. Wells, ACS Catal., 2017, 7, 2266–2274 CrossRef CAS.
- Y. Ma, G. Xu, H. Wang, Y. Wang, Y. Zhang and Y. Fu, ACS Catal., 2018, 8, 1268–1277 CrossRef CAS.
- R. Goyal, B. Sarkar, A. Bag, N. Siddiqui, D. Dumbre, N. Lucas, S. K. Bhargava and A. Bordoloi, J. Catal., 2016, 340, 248–260 CrossRef CAS.
- J. Chen, F. Lu, J. Zhang, W. Yu, F. Wang, J. Gao and J. Xu, ChemCatChem, 2013, 5, 2822–2826 CrossRef CAS.
- S. Chen, R. Wojcieszak, F. Dumeignil, E. Marceau and S. Royer, Chem. Rev., 2018, 118, 11023–11117 CrossRef CAS PubMed and reference therein.
- J. Ohyama, A. Esaki, Y. Yamamoto, S. Arai and A. Satsuma, RSC Adv., 2013, 3, 1033–1036 RSC.
- J. Ohyama, Y. Hayashi, K. Ueda, Y. Yamamoto, S. Arai and A. Satsuma, J. Phys. Chem. C, 2016, 120, 15129–15136 CrossRef CAS.
- Y. Nakagawa, H. Nakazawa, H. Watanabe and K. Tomishige, ChemCatChem, 2012, 4, 1791–1797 CrossRef CAS.
- Y. Kuwahara, Y. Magatani and H. Yamashita, Catal. Today, 2015, 258, 262–269 CrossRef CAS.
- Y. K. Lugo-José, J. R. Monnier, A. Heyden and C. T. Williams, Catal. Sci. Technol., 2014, 4, 3909–3916 RSC.
- Q. Sun, S. Wang and H. Liu, ACS Catal., 2019, 11413–11425 CrossRef CAS.
- T. Asano, H. Takagi, Y. Nakagawa, M. Tamura and K. Tomishige, Green Chem., 2019, 21, 6133–6145 RSC.
- T. Asano, M. Tamura, Y. Nakagawa and K. Tomishige, ACS Sustainable Chem. Eng., 2016, 4, 6253–6257 CrossRef CAS.
- C. Zhang, L. Chen, H. Cheng, X. Zhu and Z. Qi, Catal. Today, 2016, 276, 55–61 CrossRef CAS.
- C. Yang, Z. Miao, F. Zhang, L. Li, Y. Liu, A. Wang and T. Zhang, Green Chem., 2018, 20, 2142–2150 RSC.
- W. Schutyser, S. Van den Bosch, J. Dijkmans, S. Turner, M. Meledina, G. Van Tendeloo, D. P. Debecker and B. F. Sels, ChemSusChem, 2015, 8, 1805–1818 CrossRef CAS PubMed.
- P. M. Mortensen, J.-D. Grunwaldt, P. A. Jensen and A. D. Jensen, Catal. Today, 2016, 259, 277–284 CrossRef CAS.
- F. Yang, D. Liu, Y. Zhao, H. Wang, J. Han, Q. Ge and X. Zhu, ACS Catal., 2018, 8, 1672–1682 CrossRef CAS.
- X. Liu, W. Jia, G. Xu, Y. Zhang and Y. Fu, ACS Sustainable Chem. Eng., 2017, 5, 8594–8601 CrossRef CAS.
- L. Dong, L.-L. Yin, Q. Xia, X. Liu, X.-Q. Gong and Y. Wang, Catal. Sci. Technol., 2018, 8, 735–745 RSC.
- D. Ma, S. Lu, X. Liu, Y. Guo and Y. Wang, Chin. J. Catal., 2019, 40, 609–617 CrossRef CAS.
- S. Tian, Z. Wang, W. Gong, W. Chen, Q. Feng, Q. Xu, C. Chen, C. Chen, Q. Peng, L. Gu, H. Zhao, P. Hu, D. Wang and Y. Li, J. Am. Chem. Soc., 2018, 140, 11161–11164 CrossRef CAS PubMed.
- J. Zhang, J. Teo, X. Chen, H. Asakura, T. Tanaka, K. Teramura and N. Yan, ACS Catal., 2014, 4, 1574–1583 CrossRef CAS.
- H. Liu, Z. Huang, C. Xia, Y. Jia, J. Chen and H. Liu, ChemCatChem, 2014, 6, 2918–2928 CrossRef CAS.
- X. Wang, A. K. Beine, P. J. C. Hausoul and R. Palkovits, ChemCatChem, 2019, 11, 4123–4129 CrossRef CAS.
- S. Tazawa, N. Ota, M. Tamura, Y. Nakagawa, K. Okumura and K. Tomishige, ACS Catal., 2016, 6, 6393–6397 CrossRef CAS.
- L. Bai, X. Wang, Q. Chen, Y. Ye, H. Zheng, J. Guo, Y. Yin and C. Gao, Angew. Chem., Int. Ed., 2016, 55, 15656–15661 CrossRef CAS PubMed.
- W. Liu, Y. Chen, H. Qi, L. Zhang, W. Yan, X. Liu, X. Yang, S. Miao, W. Wang, C. Liu, A. Wang, J. Li and T. Zhang, Angew. Chem., Int. Ed., 2018, 57, 7071–7075 CrossRef CAS PubMed.
- S. Siankevich, G. Savoglidis, Z. Fei, G. Laurenczy, D. T. L. Alexander, N. Yan and P. J. Dyson, J. Catal., 2014, 315, 67–74 CrossRef CAS.
- J. Artz, S. Mallmann and R. Palkovits, ChemSusChem, 2015, 8, 672–679 CrossRef CAS PubMed.
- H. Zhou, H. Xu, X. Wang and Y. Liu, Green Chem., 2019, 21, 2923–2927 RSC.
- A. C. Alba-Rubio, J. L. G. Fierro, L. León-Reina, R. Mariscal, J. A. Dumesic and M. López Granados, Appl. Catal., B, 2017, 202, 269–280 CrossRef CAS.
- C. D’Agostino, G. Brett, G. Divitini, C. Ducati, G. J. Hutchings, M. D. Mantle and L. F. Gladden, ACS Catal., 2017, 7, 4235–4241 CrossRef.
- G. M. Lari, C. Mondelli and J. Pérez-Ramírez, ACS Catal., 2015, 5, 1453–1461 CrossRef CAS.
- K. Koh, J. E. Seo, J. H. Lee, A. Goswami, C. W. Yoon and T. Asefa, J. Mater. Chem. A, 2014, 2, 20444–20449 RSC.
- D. A. Bulushev, M. Zacharska, A. S. Lisitsyn, O. Y. Podyacheva, F. S. Hage, Q. M. Ramasse, U. Bangert and L. G. Bulusheva, ACS Catal., 2016, 6, 3442–3451 CrossRef CAS.
- D. Zanchet, J. B. O. Santos, S. Damyanova, J. M. R. Gallo and J. M. C. Bueno, ACS Catal., 2015, 5, 3841–3863 CrossRef CAS.
- J. Callison, N. D. Subramanian, S. M. Rogers, A. Chutia, D. Gianolio, C. R. A. Catlow, P. P. Wells and N. Dimitratos, Appl. Catal., B, 2018, 238, 618–628 CrossRef CAS.
- S. Ogo, Y. Okuno, H. Sekine, S. Manabe, T. Yabe, A. Onda and Y. Sekine, ChemistrySelect, 2017, 2, 6201–6205 CrossRef CAS.
- P. Y. Dapsens, C. Mondelli and J. Pérez-Ramírez, Chem. Soc. Rev., 2015, 44, 7025–7043 RSC.
- G. M. Lari, P. Y. Dapsens, D. Scholz, S. Mitchell, C. Mondelli and J. Pérez-Ramírez, Green Chem., 2016, 18, 1249–1260 RSC.
- Y. Yan, Z. Zhang, S.-M. Bak, S. Yao, X. Hu, Z. Shadike, C.-L. Do-Thanh, F. Zhang, H. Chen, X. Lyu, K. Chen, Y. Zhu, X. Lu, P. Ouyang, J. Fu and S. Dai, ACS Catal., 2019, 9, 1923–1930 CrossRef CAS.
- S. Rojas-Buzo, P. García-García and A. Corma, Green Chem., 2018, 20, 3081–3091 RSC.
- A. Herbst and C. Janiak, CrystEngComm, 2017, 19, 4092–4117 RSC.
| This journal is © The Royal Society of Chemistry 2020 |