
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOptimization and comparison of statistical tools for the prediction of multicomponent forms of a molecule: the antiretroviral nevirapine as a case study†
Rogeria
Nunes Costa
 *a,
Duane
Choquesillo-Lazarte
b,
Silvia Lucía
Cuffini
a,
Elna
Pidcock
c and
Lourdes
Infantes
*d
*a,
Duane
Choquesillo-Lazarte
b,
Silvia Lucía
Cuffini
a,
Elna
Pidcock
c and
Lourdes
Infantes
*d
aInstituto de Ciência e Tecnologia, Universidade Federal de São Paulo, 12331-280 São José dos Campos, Brazil. E-mail: rogeria.ncosta@gmail.com
bLaboratorio de Estudios Cristalográficos, IACT, CSIC-Universidad de Granada, Avda. de las Palmeras 4, 18100 Armilla, Granada, Spain
cCambridge Crystallographic Data Centre, 12 Union Road, Cambridge, UK
dInstituto de Química Fisica Rocasolano, Consejo Superior de Investigaciones Científicas, 28006, Madrid, Spain. E-mail: xlourdes@iqfr.csic.es
First published on 3rd September 2020
Abstract
In the pharmaceutical area, to obtain structures with desired properties, one can design and perform a screening of multicomponent forms of a drug. However, there is an infinite number of molecules that can be used as co-formers. Aiming to avoid spending time and money in failed experiments, scientists are always trying to optimize the selection of co-formers with high probability to co-crystallize with the drug. Here, the authors propose the use of statistical tools from the Cambridge Crystallographic Data Centre (CCDC) to select the co-formers to be used in a pharmaceutical screening of new crystal forms of the antiretroviral drug nevirapine (NVP). The H-bond propensity (HBP), coordination values (CV), and molecular complementarity (MC) tools were optimized for multicomponent analysis and a dataset of 450 molecules was ranked by a consensus ranking. The results were compared with CosmoQuick co-crystal prediction results and they were also compared to experimental data to validate the methodology. As a result of the experimental screening, three new co-crystals – NVP–benzoic acid, NVP–3-hydroxybenzoic acid, and NVP–gentisic acid – were achieved and the structures are reported. Since each tool assesses a different aspect of supramolecular chemistry, a consensus ranking can be considered a helpful strategy for selecting co-formers. At the same time, this type of work proves to be useful for understanding the target molecule and analyzing which tool may exhibit more significance in co-former selection.
Introduction
Crystal engineering is an important area of science since it allows one to study and to understand how the arrangement of the atoms and molecules, as well as the intermolecular and intramolecular interactions, will affect the crystalline structure and, consequently, its properties.1–4 Exploring many aspects of solid-state supramolecular chemistry, crystal engineering is a field of study that could be used to understand a wide variety of materials, such as semi-conductors, metalorganic or organic materials, including pharmaceutical compounds.1,4,5In the pharmaceutical area, crystal engineering has been intensely explored during the last few years, with the aim to design new forms of pharmaceutical compounds with desired properties, especially, multicomponent forms, such as co-crystals, salts, and solvates.6–8 However, for a complete screening of new multi-component forms of a molecule, e.g., an active pharmaceutical ingredient (API), there is an infinite number of other molecules and solvents that can potentially interact with the target molecule. These molecules and solvents can be called co-former molecules or simply co-formers.
In general, the process adopted for the screening of new forms is 1) identify the functional groups of the target molecule and identify which other groups could interact preferentially, 2) select co-formers containing those functional groups that could interact with the API to form co-crystals, salts and/or solvates of the target molecule, 3) perform the experimental process and identify through solid-state characterization if the new forms were obtained and 4) determine if the desired properties were obtained. As one can imagine, this process could be expensive in terms of time and money. Moreover, one should face the fact that new polymorphs can be found, which increases the number of possibilities.
In order to optimize this cocrystal screening, reducing the number of co-formers to be tested is desirable. A tool capable of reliably predicting which co-formers are most likely to form the multicomponent system with the target molecule is required.
Nowadays, there are some approaches based on different parameters to assess the propensity of a drug and a co-former to co-crystallize. To mention a few, there are computational methods that evaluate the energetic driving force for cocrystal formation;9 methods based on electrostatic potentials of molecular surfaces (MEPS) of both components to estimate the stability of the multicomponent compared to that of the individual molecules;10 thermodynamic methods as COSMOQuick software11,12 that uses the excess enthalpy of an undercooled melt of a drug and a co-former; and methods based on supramolecular knowledge that evaluate all possible interactions between the target molecule and the co-former and determine if they are robust enough to be preferred against the formation of the individual components.
Therefore, supramolecular knowledge-based tools use the probabilities of formation of intermolecular interactions or the interacting ability of the atoms. In this work, the authors aimed to evaluate the use of three13 statistical knowledge-based tools from CCDC for selecting co-formers with a high probability to form new multicomponent forms of the antiretroviral nevirapine (NVP). The named tools are molecular complementarity (MC),14 hydrogen bond propensity (HBP),15 and coordination values (CV),16–19 available in CSD_Materials. Only MC returns a pass/fail decision by default, HBP and CV provide likelihoods of hydrogen bond interactions to form, and functional groups to participate in hydrogen bonding, respectively. All three tools have been optimized for a multicomponent analysis. A function that scores the probability of a co-former–API multicomponent formation has been developed for each tool. Based on these scores, the co-formers were ranked from the best to the worst probabilities in each tool. For a combination of the three results, a consensus score was calculated for each of the co-formers as the sum of the position obtained in the three techniques (HBP, CV, and MC). Finally, if the co-formers are ordered by their ascending consensus score, this consensus ranking gives the co-formers ordered by their relative potential ability to form a cocrystal with NVP.
The COSMOQuick software11,12 was also used to evaluate the probability of the selected co-formers to form NVP co-crystals through thermodynamic aspects. The results were added to the consensus ranking. This consensus ranking is a result of the sum of MC, HBP, CV, and COSMOQuick classifications.
Aiming to validate the methodology adopted, an experimental procedure to obtain multicomponents was performed with 38 molecules. Since the experimental work was conducted concurrently with the development of the methodology, these co-formers were not selected based on the results of the theoretical screening but represent a selection of molecules readily available to us. Three new co-crystals were found and their structures are reported: NVP–benzoic acid (NVP–BZC), NVP–3-hydroxybenzoic acid (NVP–3HBZC), and NVP–gentisic acid (NVP–GTS) (Fig. 1). These three co-formers are part of a family of carboxyl and hydroxyl substituted benzene compounds, that includes another two compounds, the salicylic acid and the 4-hydroxybenzoic acid, whose co-crystals with NVP have been previously reported.20,21
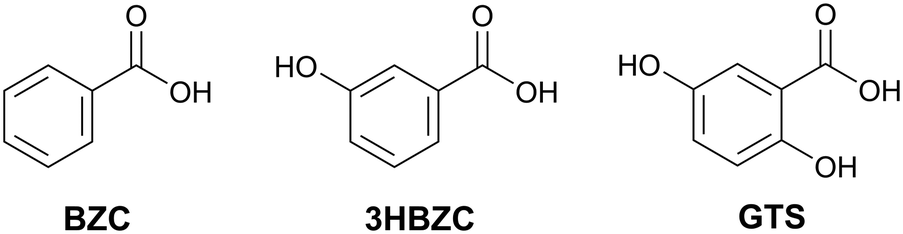 | ||
| Fig. 1 2D chemical diagram for benzoic acid (BZC), 3-hydroxybenzoic acid (3HBZC), and gentisic acid (GTS). | ||
Methodology
Selection of co-formers and the generation of appropriate coordinate files for statistical analysis
A dataset of 450 co-formers was selected for this study. All molecules are part of the generally recognised as safe (GRAS) list. Care was taken to include in the list of co-formers all those molecules that appear in the literature as previously tested with NVP and those 38 compounds selected for our experimental work.The multicomponent analyser tools are provided with molecular geometry and coordinates. Besides, values are dependent on the molecular conformation which is responsible for the chemical group accessibility and also for the molecular shape. The molecular geometry information for the major part of the molecules used in this study is available from the Cambridge Structure Database (CSD). ConQuest v.1.9.0 software was employed to obtain this information for each molecule and by using the Mercury v.3.9.0 software, mol2 files were obtained for each molecule. Mercury tools were used to guarantee that each file exhibits just a single molecule, with no charges, no disorder, and containing all H atoms with bond distances normalized.
In the cases where no information was available in the CSD, mol2 files containing the molecular three-dimensional information were constructed through the Chem3D software (PerkinElmer Informatics).
Molecular conformation can affect calculations for CV and MC. The conformation of a co-former modifies the accessible surface area of the atoms available for the intra- and intermolecular interactions and, consequently, the coordination values are affected. Furthermore, the MC tool considers the length of the axes of the virtual box in which the molecule is contained. So, the shape of the molecule could affect this box size and, therefore, the results of the MC. Thus, it was necessary to obtain the most representative conformations for every co-former. All molecules were analysed by the CSD Conformer Generator tool.22,23 For the rigid molecules, only one conformation was used. However, as the degree of conformational freedom for the co-formers increased, so did the number of molecular conformations used as input for the CV and MC tools. Only the best output value was kept for each tool and co-former molecule.
A Python API script was developed to write mol2 files containing the NVP and the co-former molecule. These files were used as input for HBP and CV methods to assess the interactions between the two molecules.
Input for the thermodynamic tool
The COSMOQuick11,12 v.1.7 was used to predict the tendency of cocrystal formation. This tool requires the simplified molecular input line entry specification or SMILES of a molecule as input data. So, the SMILES for the 450 selected molecules were generated using the ChemDraw software (PerkinElmer Informatics).Multicomponent analysed tools
The three CCDC tools (HBP, CV, and MC) are available in the CSD-materials module in the Mercury package and were modified to be able to evaluate or quantify the stability of a multicomponent versus its individual structures. This was achieved by writing bespoke python scripts, utilizing the CSD Python API. After the calculations, for each tool, the co-former molecules were sorted from the best to the worst values. Then, every co-former was assigned a ranking position for each tool. Initially, two consensus scores were calculated. The consensus score A is the sum of HBP, CV, and MC ranking positions; and the consensus score B was calculated by adding the COSMOQuick ranking position to the consensus score A. After the analysis of the results, we considered it necessary to calculate a third consensus classification where the CVs were removed, and it is the consensus score C.The new structures
A screening of 38 compounds to achieve new cocrystals of NVP was conducted using the liquid-assisted grinding (LAG) method. NVP raw material was manufactured by Nortec Química. Potential co-formers were selected from the GRAS list: acetylsalicylic acid, adipic acid, L-alanine, 4-aminobenzoic acid, L-arginine, L-ascorbic acid, L-aspartic acid, benzamide, benzoic acid, caffeine, catechol, citric acid, L-cystine, fumaric acid, gallic acid, gentisic acid, glutamic acid, L-glutamine, glutaric acid, glycolic acid, hippuric acid, hydroquinone, 3-hydroxybenzoic acid, 4-hydroxybenzoic acid, isonicotinamide, maleic acid, malonic acid, nicotinamide, nicotinic acid, orcinol, oxalic acid, phloroglucinol, resorcinol, succinic acid, theobromine, theophylline, urea, and vanillin. 150 mg of stoichiometric (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) amounts of NVP and co-former were ground in the presence of 4 or 5 drops of chloroform. Different milling conditions were applied and they are detailed in the ESI.† Specifically, in the case of NVP–BZC, NVP–3HBZC, and NVP–GTS, the sample mixtures and the solvent were placed in an agate jar with two steel balls (Φ = 5 mm) and milled using the mixer mill MM200 (Retsch), with a vibration frequency of 25 Hz, during 30 minutes. Chloroform was selected because it does not form NVP solvate crystals. All co-formers as well as the chloroform were at ACS grade. Powder samples were analysed through powder X-ray diffraction (PXRD) to identify the formation of new crystalline phases.
:1) amounts of NVP and co-former were ground in the presence of 4 or 5 drops of chloroform. Different milling conditions were applied and they are detailed in the ESI.† Specifically, in the case of NVP–BZC, NVP–3HBZC, and NVP–GTS, the sample mixtures and the solvent were placed in an agate jar with two steel balls (Φ = 5 mm) and milled using the mixer mill MM200 (Retsch), with a vibration frequency of 25 Hz, during 30 minutes. Chloroform was selected because it does not form NVP solvate crystals. All co-formers as well as the chloroform were at ACS grade. Powder samples were analysed through powder X-ray diffraction (PXRD) to identify the formation of new crystalline phases.
Crystallization and single-crystal X-ray diffraction (SCXRD)
From the screening of 38 molecules, three of them presented a PXRD pattern in agreement with new phases and possible multicomponent forms. Potential co-crystals, NVP–BZC, NVP–3HBZC, and NVP–GTS, were recrystallized in chloroform and ethyl acetate. Single crystals were obtained in both solvents with different quality and the best were selected for a single-crystal X-ray diffraction (SCXRD) analysis. SCXRD experiments for NVP–BZC were performed at room temperature in a Bruker D8 Venture diffractometer (Photon 100 CMOS detector and MoKα radiation from Incoatec micro source), whilst for NVP–3HBZC and NVP–GTS, experiments were conducted in a Bruker D8 Venture diffractometer equipped with a CMOS Photon 100 detector using CuKα radiation at room temperature for the former and at 120 K for the latter. The diffraction images were analysed (indexed, integrated, and scaled) using the Apex3 software.24 Crystal structures were solved through the direct methods and refined by full-matrix-block least-squares in the SHELXL software.25 All non-hydrogen atoms were anisotropically refined, and all hydrogen atoms were placed in idealized geometries according to the riding model. In the three crystal structures, the co-former molecules are situated in the vicinity of a centre of symmetry inducing disorder. Connectivity and rigid body restraints were necessary to describe and refine these fragments.Methodology validation and generation of ROC curves
Receiver operator characteristic (ROC) curves were obtained to evaluate the performance of the methodology proposed. A ROC curve plots the sensitivity against (1-specificity). The sensitivity is the number of true positive predictions over the total number of positive observations, whilst the specificity is given by the number of false-positive predictions over the total number of negative observations. A binary classifier system was used to correlate predictive and experimental results (Scheme 1) and its discriminative threshold (number of molecules cutoff) varied from low to higher values.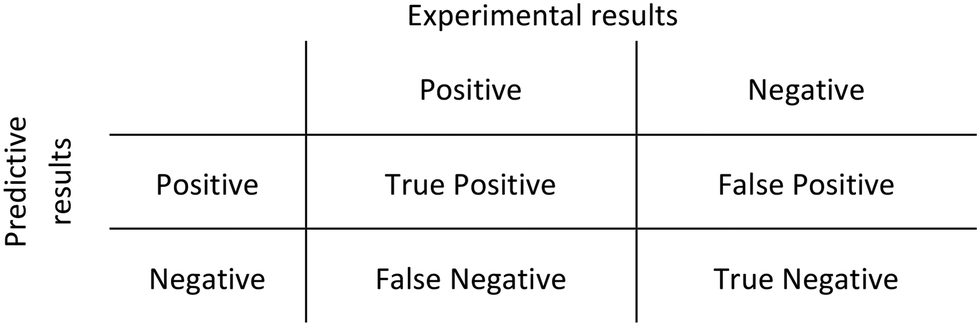 | ||
| Scheme 1 Binary classifier system used to construct ROC curves. | ||
A ROC curve has been obtained for each evaluated tool as well as for the consensus rankings. Besides, the area under curve (AUC) was calculated to evaluate the discriminative power of each tool. ROC curves and AUC values were obtained using OriginLab software.
Results and discussion
Nevirapine and its multicomponent forms
NVP (11-cyclopropyl-4-methyl-5,11-dihydro-6H-dipyrido[3,2-b:2′,3′-e][1,4]diazepin-6-one) (Fig. 2) is an antiretroviral drug used in the treatment of HIV-1 infection.26,27 This drug exhibits a low-aqueous solubility and there is interest in improving its solubility through the formation of multicomponent structures.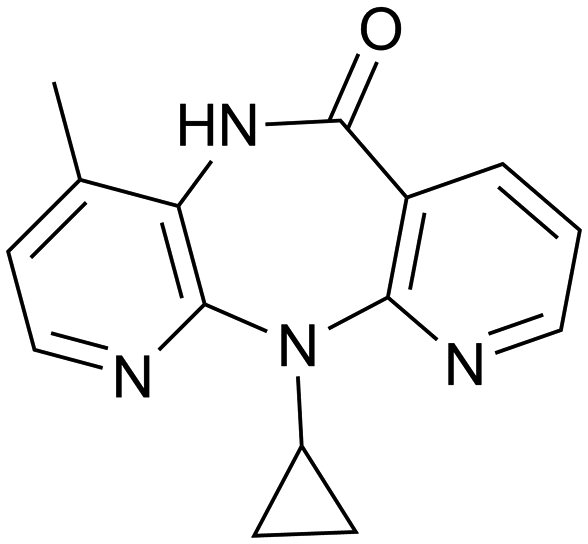 | ||
| Fig. 2 Molecular diagram of nevirapine (NVP). | ||
A total of 76 molecules have been tested to form multicomponent with NVP (Tables 1 and S1†), and depending on the results they were grouped into three clusters: G1 – molecules that form multicomponent structures with NVP and are reported in the CSD database or here (highlighted in green), G2 – molecules referred into the literature as forming co-crystals even though their structure is not yet reported (highlighted in yellow), and G3 – molecules that have been attempted, but no group has reported forming multicomponents yet (highlighted in red). The second group also includes the eutectic systems, since, even when the multicomponent structure has not been formed, there are weaker interactions between the two molecules that are responsible for the eutectic behaviour. There are 25 molecules in the first cluster, G1; 17 molecules in the second, G2; and 34 in the third, G3. For the molecules in the groups G2 and G3, a list of the techniques used in the trials are listed in Table S2.†
| Co-former | CV ranking | MC ranking | HBP ranking | Consensus ranking A | COSMO ranking | Consensus ranking B | Consensus ranking C | Tested by our team | Tested in literature | CSD refcode |
|---|---|---|---|---|---|---|---|---|---|---|
| a Toluene does not exhibit donor or acceptor groups. Thus, the HBP and CV ranking positions do not reflect an adequate position. b The co-former has suffered deprotonation, resulting in a salt multicomponent form. ★ this symbol indicates that two or more groups have tested the co-former, but just one of them has obtained the multicomponent form. | ||||||||||
| G1 | ||||||||||
| Maleic Acid | 73 | 158 | 33 | 29 | 42 | 5 | 10 | ✗ | ✓20,43 | LATQII20 |
| 4-Hydroxybenzoic acid | 56 | 85 | 117 | 24 | 32 | 4 | 11 | ✓21 | ✓43 | POZCOZ21 |
| Salicylic acid | 109 | 121 | 90 | 44 | 30 | 11 | 13 | ✓21 | ✓20 | LATQUU,20 LATQUU01, and LATQUU02 (ref. 21) |
| Ethanol | 222 | 234 | 65 | 126 | 134 | 97 | 61 | ✗ | ✓30 | OKETII30 |
| Glutaric acid | 84 | 93 | 67 | 21 | 279 | 51 | 66 | ✗ | ✓20,43 | LATQEE20 |
| Trichloroacetic acidb | 235 | 372 | 69 | 201 | 10 | 113 | 76 | ✓35 | QUDWET35 | |
| Naphthalene-1,5-disulfonic acidb | 317 | 201 | 225 | 252 | 26 | 155 | 77 | ✓35 | QUDWOD35 | |
| 3,5-Dinitrosalicylic acidb | 364 | 272 | 206 | 328 | 22 | 180 | 98 | ✓35 | QUDWIX35 | |
| n-Butanol | 170 | 83 | 153 | 78 | 291 | 120 | 122 | ✓28,29 | GIRWUA29 and KACPAH28 | |
| Tartaric acid | 75 | 348 | 110 | 132 | 72 | 74 | 126 | ✗ | ✓20 | LATRAB20 |
| Ethyl acetate | 339 | 124 | 185 | 183 | 246 | 198 | 149 | ✓ | ✓31,32 | TISJEL31 and TISJEL01 (ref. 32) |
| Hexanol | 155 | 40 | 164 | 60 | 365 | 138 | 158 | ✗ | ✓28 | AKEFEC28 |
| 1,4-Dioxane | 371 | 323 | 97 | 292 | 150 | 223 | 161 | ✓32 | YIVQUQ32 | |
| Heptanol | 147 | 29 | 163 | 54 | 386 | 139 | 163 | ✗ | ✓28 | AKEFIG28 |
| Octanol | 140 | 21 | 168 | 48 | 395 | 137 | 167 | ✗ | ✓28 | AKEFOM28 |
| Saccharin | 406 | 337 | 202 | 388 | 71 | 274 | 185 | ✓ | ✓20,43 | LATQOO20 |
| Picric acidb | 435 | 293 | 262 | 410 | 69 | 300 | 193 | ✓34 | CIKSEV34 | |
| 1,3-Diiodobenzene | 264 | 51 | 427 | 251 | 156 | 202 | 197 | ✓33 | RIMWAO33 | |
| Dichloromethane | 226 | 115 | 444 | 285 | 98 | 190 | 215 | ✓32 | YIVQIE32 | |
| 1,2,4,5-Tetrafluoro-3,6-di-iodobenzene | 248 | 39 | 434 | 231 | 249 | 238 | 255 | ✓33 | RIMJAB33 | |
| Toluenea | 224 | 75 | 450 | 257 | 212 | 233 | 267 | ✓32 | YIVQOK32 | |
| ε-Caprolactam | 230 | 336 | 328 | 363 | 126 | 276 | 306 | ✓37 | ZEYSAA37 | |
| Benzoic acid | 161 | 72 | 51 | 35 | 57 | 9 | 2 | ✓ | ✓20,42,43 | |
| 3-Hydroxybenzoic acid | 52 | 128 | 75 | 23 | 29 | 3 | 9 | ✓ | ||
| Gentisic acid | 64 | 136 | 162 | 62 | 20 | 21 | 27 | ✓ | ||
| G2 | ||||||||||
| 4-Aminobenzoic acid | 108 | 81 | 38 | 16 | 97 | 7 | 5 | ✗ | ✓43 | |
| Fumaric acid | 181 | 123 | 66 | 67 | 40 | 26 | 8 | ✗ | ✓20 | |
| Cinnamic acid | 190 | 44 | 76 | 40 | 175 | 43 | 23 | ✓43 | ||
| Gallic acid | 276 | 233 | 138 | 181 | 16 | 101 | 44 | ✗ | ✓43 | |
| Adipic acid | 66 | 69 | 32 | 8 | 330 | 44 | 59 | ✗ | ✓20 | |
| L-Mandelic acid | 88 | 270 | 141 | 119 | 47 | 60 | 78 | ✓43 | ||
| Oxalic acid | 153 | 248 | 305 | 222 | 4 | 130 | 150 | ✗ | ✓20,42,43 | |
| Malonic acid | 114 | 257 | 189 | 140 | 133 | 116 | 164 | ✗ | ✓20,43 | |
| Citric acid | 96 | 301 | 63 | 98 | 229 | 114 | 174 | ✗ | ✓43 | |
| Theophylline | 389 | 249 | 229 | 347 | 147 | 272 | 195 | Eutectic 21 | ||
| Sorbic acid | 174 | 55 | 384 | 165 | 187 | 160 | 196 | ✓20 | ||
| Uracil | 296 | 275 | 236 | 299 | 164 | 241 | 227 | ✓20 | ||
| Caffeine | 436 | 225 | 365 | 422 | 155 | 382 | 272 | Eutectic 21 | ||
| Propionamide | 8 | 247 | 242 | 116 | 264 | 152 | 279 | ✓20 | ||
| L-Proline | 203 | 296 | 318 | 310 | 228 | 291 | 344 | ✓20 | ||
| Urea | 145 | 350 | 279 | 280 | 236 | 269 | 357 | ✗ | ✓43 | |
| Glutamic acid | 85 | 442 | 326 | 335 | 378 | 396 | 448 | ✗ | ✓20 | |
| G3 | ||||||||||
| Hydroquinone | 115 | 90 | 165 | 68 | 17 | 23 | 18 | ✗ | ||
| Phloroglucinol | 213 | 188 | 143 | 134 | 3 | 61 | 32 | ✗ | ||
| Resorcinol | 116 | 173 | 181 | 106 | 5 | 38 | 37 | ✗ | ||
| Catechol | 137 | 197 | 160 | 113 | 9 | 45 | 39 | ✗ | ||
| Acetylsalicylic acid | 271 | 231 | 31 | 131 | 137 | 104 | 48 | ✗ | ||
| Orcinol | 113 | 230 | 169 | 122 | 12 | 52 | 50 | ✗ | ||
| Glycolic acid | 124 | 313 | 86 | 128 | 64 | 73 | 81 | ✗ | ||
| L-Malic acid | 49 | 291 | 102 | 91 | 75 | 48 | 84 | ✗20 | ||
| Succinic acid | 141 | 312 | 68 | 127 | 94 | 81 | 87 | ✗ | ✗20 | |
| L-Tartaric acid | 71 | 384 | 18 | 107 | 73 | 59 | 89 | ✗ | ✗20,43 | |
| Ferulic acid | 45 | 138 | 100 | 34 | 255 | 58 | 96 | ✗43 | ||
| Suberic acid | 47 | 35 | 78 | 7 | 391 | 64 | 102 | ✗20 | ||
| L-Ascorbic acid | 177 | 342 | 77 | 157 | 88 | 110 | 108 | ✗ | ✗20 | |
| DL-Malic acid | 16 | 297 | 147 | 99 | 76 | 56 | 120 | ✗ | ||
| Nicotinic acid | 358 | 112 | 320 | 290 | 100 | 196 | 130 | ✗ | ✗20 | |
| Stearic acid | 106 | 1 | 92 | 11 | 444 | 93 | 138 | ✗20 | ||
| Orotic acid | 260 | 212 | 277 | 256 | 54 | 162 | 144 | ✗20 | ||
| Cinnamamide | 4 | 80 | 252 | 51 | 287 | 83 | 190 | ✗20 | ||
| Benzamide | 58 | 117 | 259 | 88 | 243 | 107 | 191 | ✗ | ✗20 | |
| Vanillin | 243 | 215 | 297 | 265 | 139 | 199 | 209 | ✗ | ||
| Hippuric acid | 55 | 171 | 187 | 82 | 301 | 131 | 216 | ✗ | ★20,43 | |
| Nicotinamide | 312 | 113 | 296 | 228 | 265 | 251 | 226 | ✗ | ✗20 | |
| Theobromine | 423 | 220 | 311 | 395 | 168 | 346 | 241 | ✗ | ||
| Isonicotinamide | 319 | 131 | 337 | 287 | 257 | 290 | 260 | ✗ | ✗20 | |
| Piperazine | 274 | 318 | 263 | 337 | 160 | 273 | 269 | ✗20 | ||
| L-Arginine | 408 | 203 | 150 | 268 | 413 | 377 | 286 | ✗ | ✗20 | |
| Pyroglutamic acid | 225 | 277 | 364 | 346 | 140 | 266 | 297 | ✗20 | ||
| Succinamide | 83 | 160 | 270 | 123 | 358 | 186 | 304 | ✗20 | ||
| L-Aspartic acid | 14 | 263 | 317 | 156 | 238 | 169 | 326 | ✗ | ✗20 | |
| DL-Aspartic acid | 130 | 411 | 325 | 344 | 239 | 327 | 403 | ✗20 | ||
| Glycine | 301 | 430 | 302 | 423 | 293 | 427 | 416 | ✗20 | ||
| L-Alanine | 232 | 448 | 338 | 419 | 289 | 422 | 424 | ✗ | ||
| L-Valine | 217 | 433 | 354 | 413 | 323 | 428 | 437 | ✗20 | ||
| L-Glutamine | 22 | 429 | 340 | 293 | 394 | 384 | 449 | ✗ | ||
Groups G1 and G2 have been considered as the positive experimental results while molecules in group G3 are considered as the negative ones.
NVP co-crystals in the literature
There are several multicomponent forms containing NVP that are reported in the literature. Performing a search on the CSD database, nine solvates,28–32 eight co-crystals,20,21,33 and four salts34,35 containing the NVP molecule were found, besides the anhydrous26,32 and hemihydrate forms31,32 (Fig. 3). Also, found was a salt-hydrate structure with pentaiodide and water,36 and a crystalline inclusion complex.37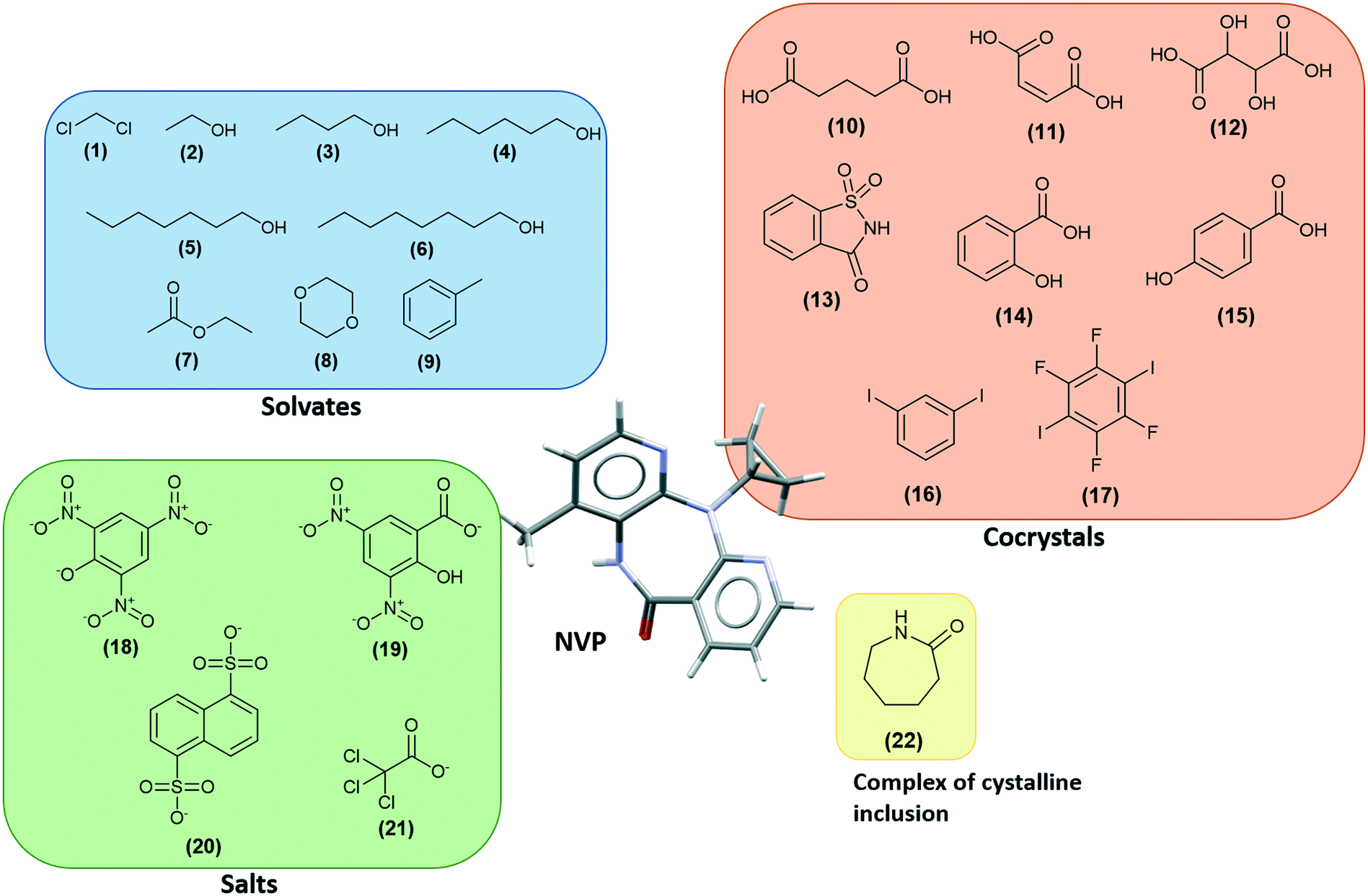 | ||
| Fig. 3 In the CSD, besides anhydrous and hydrate forms, it was found multi-component structures of NVP with (1) dichloromethane, (2) ethanol, (3) 1-butanol, (4) 1-hexanol, (5) 1-heptanol, (6) 1-octanol, (7) ethyl acetate, (8) 1,4-dioxane, (9) toluene, (10) glutaric acid, (11) maleic acid, (12) tartaric acid, (13) saccharin, (14) salicylic acid, (15) 4-hydroxybenzoic acid, (16) 1,3-diiodobenzene, (17) 1,2,4,5-tetrafluoro-3,6-diiodobenzene, (18) picrate, (19) 3,5-dinitrosalicylate, (20) naphthalene-1,5-disulfonate, (21) trichloro acetate, and (22) ε-caprolactam. | ||
The NVP molecule consists of a 7-membered ring flanked by two rings of aromatic pyridine. The central ring contains a rigid cis-amide group which, in most cases, forms a centrosymmetric synthon through a strong amide–amide hydrogen bonding interaction (Fig. 4). Therefore, NVP contains a unique HB-donor atom (the amide NH) and three possible strong HB-acceptor atoms (the amide C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and the two N in the pyridine rings). Besides, pyridine rings frequently form π–π stacking interactions. Observing the intermolecular potentials38,39 calculated around a central NVP molecule for the crystal structure of the anhydrous nevirapine (PABHIJ) showed that the strongest interaction is due to the π–π stacking interaction between the aromatic rings, followed by the amide–amide homodimer interaction (Fig. 4). Similarly, the full interaction maps (FIMs)40,41 generate the interaction landscape of the NVP molecule (Fig. 4) and it suggests that the amide–amide homodimer contact as the preferred interaction. Also, shown (in red, Fig. 4) are the preferred geometries of interaction with the pyridine nitrogens and regions (in brown) parallel to the pyridine rings indicate the possibility for a hydrophobic or π–π interaction. Using this information, we hypothesize that adequate co-formers would be molecules with delocalized double bonds or aromatic rings that can occupy the hydrophobic regions, and/or strong donor–acceptor groups such as carboxylic acids that may disrupt the amide dimer.16
O and the two N in the pyridine rings). Besides, pyridine rings frequently form π–π stacking interactions. Observing the intermolecular potentials38,39 calculated around a central NVP molecule for the crystal structure of the anhydrous nevirapine (PABHIJ) showed that the strongest interaction is due to the π–π stacking interaction between the aromatic rings, followed by the amide–amide homodimer interaction (Fig. 4). Similarly, the full interaction maps (FIMs)40,41 generate the interaction landscape of the NVP molecule (Fig. 4) and it suggests that the amide–amide homodimer contact as the preferred interaction. Also, shown (in red, Fig. 4) are the preferred geometries of interaction with the pyridine nitrogens and regions (in brown) parallel to the pyridine rings indicate the possibility for a hydrophobic or π–π interaction. Using this information, we hypothesize that adequate co-formers would be molecules with delocalized double bonds or aromatic rings that can occupy the hydrophobic regions, and/or strong donor–acceptor groups such as carboxylic acids that may disrupt the amide dimer.16
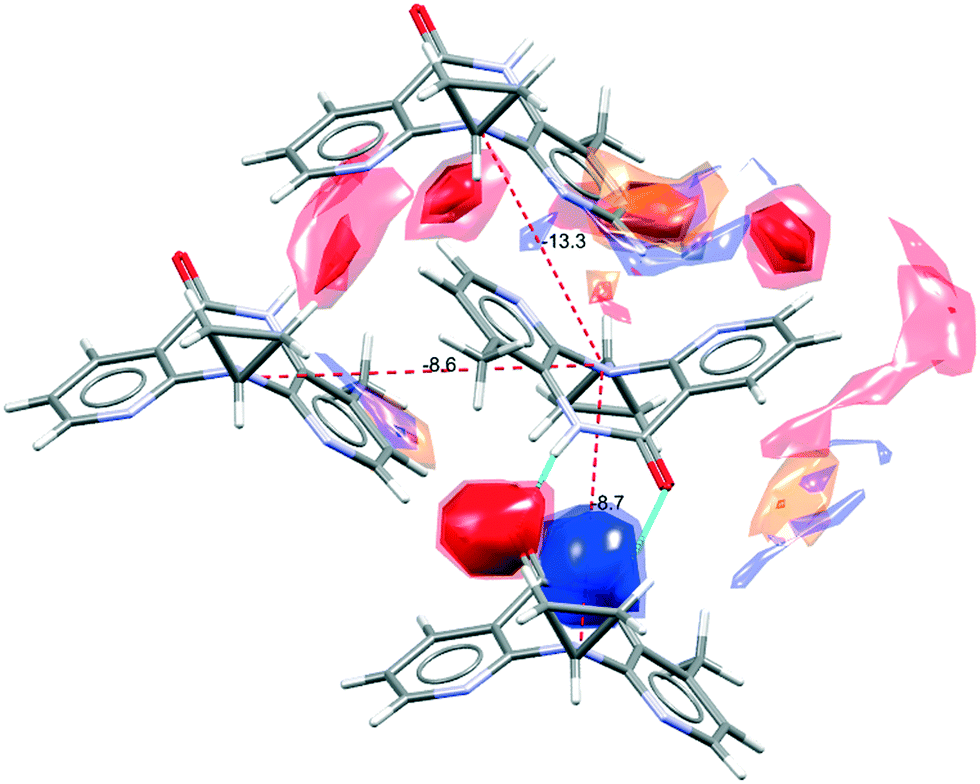 | ||
| Fig. 4 Representation of the full interaction maps (FIMs) and the intermolecular potentials (kcal mol−1) using the “UNI” force field from a centre molecule of NVP in its crystal structure (PABHIJ). | ||
Investigating the interactions exhibited by the multicomponent structures, the NVP amide–amide homodimer is disrupted only seven times. In four of these, a NVP–co-former heterodimer is formed through amide–carboxylic acid interactions (glutaric acid, maleic acid, tartaric acid, and 4-hydroxybenzoic acid), while in a further two, the homodimer is broken because of a water molecule, and in the remaining one, an amide–amide chain is observed (naphthalene-1,5-disulfonate). In the other fourteen structures with available coordinates, the homodimer is maintained. In many cases, a NVP–co-former interaction occurs with the pyridine N. Remarkably, all multicomponent structures exhibit a π–π NVP stacking interaction with just one exception (NVP–naphthalene-1,5-disulfonic acid).
These observations could corroborate our hypothesis that good co-formers must contain carboxylic groups and/or aromatic rings. However, looking at the molecules tested with NVP which have not formed multicomponents, (G3 in Tables 1 and S1†) or which have not been obtained yet, there is a high representation of carboxylic groups and aromatic rings. Therefore, it appears there is not an easy way to predict which molecules will form multicomponent structures.
New co-crystals of NVP
Experimental work to achieve new multicomponent forms of NVP was carried out at the same time that a new method to calculate the propensities of two molecules to crystallize together was developed. From the experimental screening, three molecules were potential candidates to form a NVP-multicomponent. And effectively, we achieve the cocrystallization of NVP–BZC, NVP–3HBZC, and NVP–GTS. The NVP–BZC co-crystal had already been reported as a positive hit in the literature,42,43 however, its crystalline structure had not been reported yet. Crystals obtained from the recrystallization of NVP and 3HBZC were shown to be a mix of crystals of pure NVP, pure 3HBZC, and the NVP–3HBZC multicomponent.The crystal structure of the compounds NVP–3HBZ and NVP–GTS are very similar. Both maintain the most frequent synthon of amide–amide interaction and the co-formers occupy a centre of symmetry and show disorder, but form a hydrogen bond (HB) between the OH in the carboxylic group (of the co-former) and the nitrogen of the NVP 4-methyl-pyridine (Fig. 5). Nevirapine molecules pack in the same molecular arrangement in both co-crystals, they are 3D isostructural. Also, their structures are similar to the structure of nevirapine with salicylic acid molecules (CSD refcode LATQUU). The three structures are triclinic P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) and their unit cell parameter are very similar (Table 2).
and their unit cell parameter are very similar (Table 2).
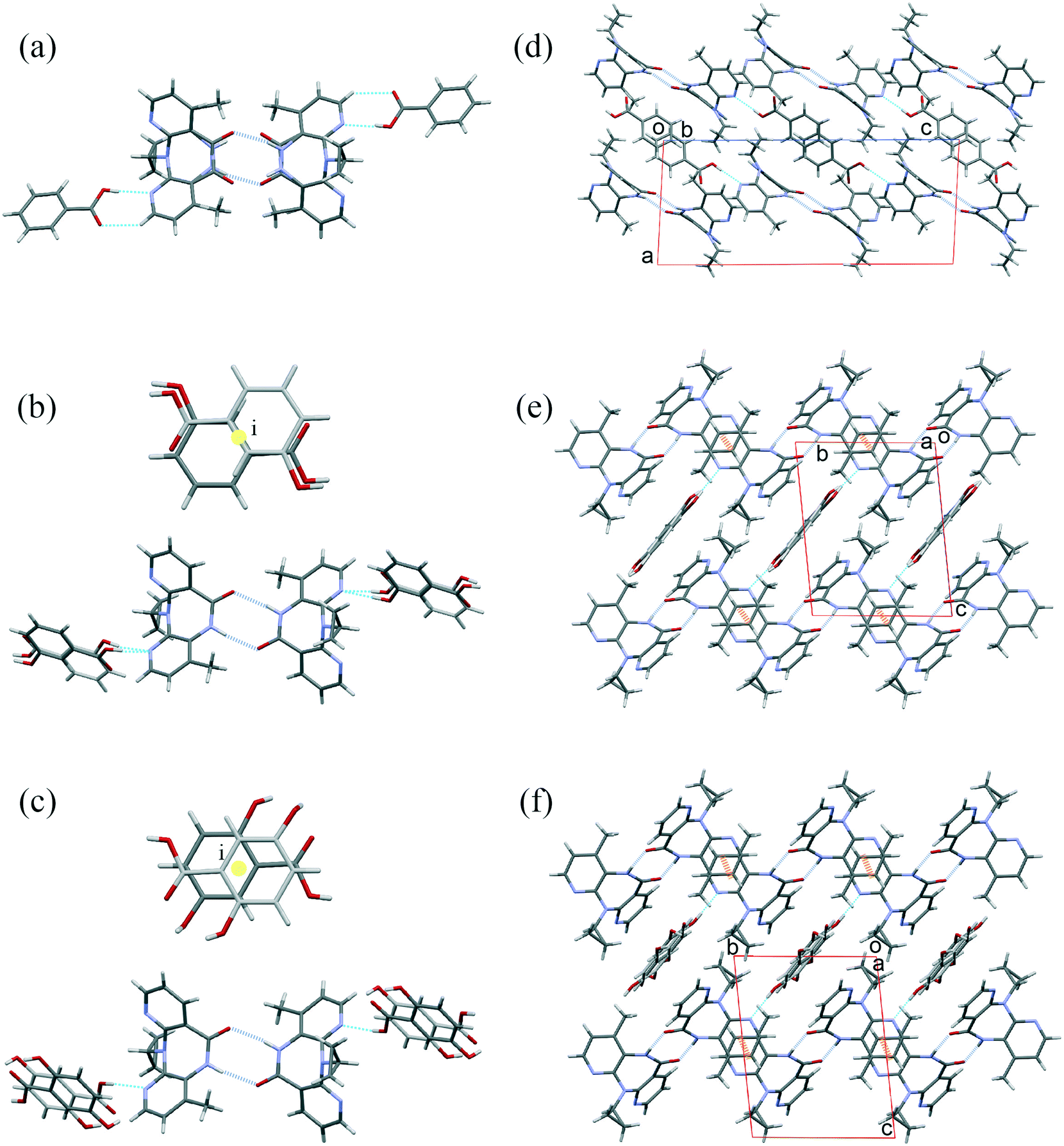 | ||
| Fig. 5 Structures of the compounds (a and d) NVP–BZC, (b and e) NVP–3HBZC and (c and f) NVP–GTS. Hydrogen bonding tetramers showing (a) the NVP disorder and (b and c) the co-formers disorder. On the right a perpendicular view of the crystal structure for (d) NVP–BZC, (e) NVP–3HBZC and (f) NVP–GTS showing in dashed orange lines the π⋯π interactions observed in the infinite pyridine stacking of the nevirapine molecules for the compounds NVP–3HBZC and NVP–GTS. | ||
| Name | Reference CSD-refcode | Space group | a, b, c (Å) | α, β, δ (°) | Volume (Å3) | Secondary structure |
|---|---|---|---|---|---|---|
| Benzoic acid | BZC | P21/c | 9.934(2), 8.360(2), 23.571(6) | 90, 92.977(8), 90 | 1954.9(8) | Amide–amide homodimer |
| Salicylic acid | LATQUU |
P |
7.1767(4), 9.6278(5), 11.9235(6) | 96.865(2), 93.039(2), 98.126(2) | 807.69(7) | Amide–amide homodimer |
| 3-Hydroxybenzoic acid | 3HBZC |
P |
7.2706(14), 9.5902(19), 11.881(2) | 96.11(3), 92.79(3), 98.34(3) | 813.3(3) | Amide–amide homodimer |
| 4-Hydroxybenzoic acid | POZCOZ | C2/c | 24.318(5), 7.5618(13), 23.242(5) | 90, 111.321(8), 90 | 3981.3(14) | Amide–COOH heterodimer |
| Gentisic acid | GTS |
P |
7.1010(3), 9.4938(4), 12.0005(5) | 95.619(2), 92.881(2), 99.035(2) | 793.37(6) | Amide–amide homodimer |
NVP–BZC crystallizes in a monoclinic system, P21/c. Nevirapine also displays the homodimers and, the benzoic acid molecule forms a hydrogen bond with the nitrogen in the 4-methyl-pyridine. While the co-former disorder is not observed the nevirapine molecules are disordered and present a pseudo plane perpendicular to the molecule that divides it into two halves. This structure is different to all nevirapine compounds observed before because it does not form any π⋯π parallel sandwich between pyridine rings. Only for the bis(nevirapinium) naphthalene-1,5-disulfonate salt (CSD refcode QUDWOD), are these NVP–NVP sandwiches replaced by NVP–naphthalene sandwiches. There is another hydroxyl benzoic nevirapine complex, the 4-hydroxy-benzoic acid (CSD refcode POZCOZ).21 This compound crystallizes forming heterodimers between the carboxylic acid and the amide chemical groups. This is the only one that disrupts the amide–amide interaction.
As we observed in a previous publication,21 infinite pyridine stacking (π⋯π interactions) of nevirapine molecules displayed in PABHIJ01 is also conserved in LATQUU (NVP–salicylic acid), POZCOZ (NVP–4-hydroxybenzoic acid), NVP–3HBZC and NVP–GTS cocrystals. While in LATQUU, NVP–3HBZC, and NVP–GTS cocrystals these chains grow into similar 3D structures as we have explained above; in POZCOZ, because of the heterodimers, only these 1D chains are conserved.
Method for statistical evaluation of frequency of interaction for multicomponent prediction
Based on the need for reliable predictive tools to reduce the number of co-formers to be tested, our group has evaluated and proposed a new methodology that modifies the existing tools. Three CCDC tools, H-bond propensity (HBP), coordination values (CV), and molecular complementarity (MC), in addition to the COSMOQuick software, have been used. In each tool, the methodology was applied to 450 co-formers and the complete results tables are available in the ESI† (Tables S3–S6). The modifications made and the results obtained are reported below.Results divided by quartiles
Quartiles division has been used with the goal of helping to explain the results obtained with the four techniques used, and to evaluate the ranking position of the 76 molecules that were attempted experimentally, reported in the literature or presented here; and classified as G1, G2 and G3. The 450 positions in each tool were divided into their quartiles: Q1 goes from position 1 to position 112, Q2 from 113 to 225, Q3 from 226 to 338, and the last one Q4 from 339 to the end. For each quartile, the number of molecules tested with NVP and belonging to each group G1, G2 and G3 have been counted and listed in Table 3.| A) Co-formers that multicomponent form with NVP has been reported (G1) | |||||||
|---|---|---|---|---|---|---|---|
| CV | MC | HBP | COSMO | Consensus A | Consensus B | Consensus C | |
| Q1 | 7 | 10 | 9 | 13 | 11 | 9 | 11 |
| Q2 | 7 | 7 | 10 | 5 | 4 | 11 | 11 |
| Q3 | 6 | 6 | 2 | 4 | 7 | 5 | 3 |
| Q4 | 5 | 2 | 4 | 3 | 3 | 0 | 0 |
| B) Co-formers that multicomponent form with NVP has been detected (G2) | |||||||
|---|---|---|---|---|---|---|---|
| CV | MC | HBP | COSMO | Consensus A | Consensus B | Consensus C | |
| Q1 | 6 | 4 | 5 | 5 | 5 | 6 | 6 |
| Q2 | 7 | 1 | 3 | 6 | 6 | 5 | 5 |
| Q3 | 2 | 9 | 7 | 5 | 4 | 4 | 3 |
| Q4 | 2 | 2 | 2 | 1 | 2 | 2 | 3 |
| C) Co-formers that do not form a multicomponent structure with NVP (G3) | |||||||
|---|---|---|---|---|---|---|---|
| CV | MC | HBP | COSMO | Consensus A | Consensus B | Consensus C | |
| Q1 | 12 | 5 | 9 | 13 | 11 | 17 | 13 |
| Q2 | 11 | 13 | 8 | 5 | 9 | 6 | 8 |
| Q3 | 8 | 9 | 14 | 11 | 8 | 5 | 8 |
| Q4 | 3 | 7 | 3 | 5 | 6 | 6 | 5 |
H-Bond propensity tool
The first tool employed was HBP analysis which evaluates the potential hydrogen bonding landscape for a target system. Although this tool has been available for multicomponent analysis in a previous version of the CSD system, it is now available only for polymorph assessment. This tool identifies all 2D functional group formulas in a system and calculates a propensity index for the formation of the hydrogen bond for all possible combinations of donor and acceptor pairs. The higher the index, the greater the propensity to observe an interaction between the chemical groups involved.This algorithm involves four stages: (1) data sampling where a search for structures from the CSD containing the same functional groups is performed; (2) extraction of data for the modelling step from structures in the training dataset. The data collected from the training dataset are whether a hydrogen bond exists between particular functional groups, and explanatory variables that include aromaticity, competition and steric density. Logistic regression is used in order to build a model that can predict the likelihood of a hydrogen bond forming given the behaviours observed, and the explanatory variables of the training dataset; (3) model validation, the logistic regression model is evaluated and is considered good enough to proceed when its area under receiver operating characteristic (ROC) curve is greater than 0.8; and (4) generation and presentation of the results table that contains the HB propensities for each possible donor–acceptor pair in the target structure.
A way to compare the stabilities of a multicomponent (AB) with respect to the structures of each component (A and B) is to compare the propensity index for heteromeric and homomeric interactions. Considering NVP as component A and the co-former as component B, we have identified the highest propensity to form an intermolecular interaction for the pure components (AA or BB) and the multicomponent (AB or BA). The multi-component score is calculated as the difference of the highest propensity (AB or BA) minus the highest propensity (AA or BB). Therefore, the multicomponent HBP score will be greater than zero for those structures with a good chance to co-crystallize, and it will be lower than zero for those systems where pure forms are more likely.
As expected, the co-formers with high multicomponent HBP scores are the ones that, in general, exhibit carboxyl, hydroxyl or sulfhydryl groups (Table S2†). The amino acids appear at the bottom of the table with low HBP values. It is important to note that the HB-pairs that provide the highest HBP scores is not always the HB-network observed in the solid-state of the structures already reported in the CSD. Low propensity interactions found at the expense of better donor–acceptor pairings can be related to a possible risk of polymorphism.44
Experimental structures show that the amide–amide interaction is disrupted only with carboxylic acid groups or water molecules. There are eight multicomponent structures where the co-former contains a carboxylic acid group, NVP–glutaric acid, NVP–maleic acid, NVP–tartaric acid, NVP–4-hydroxybenzoic acid, NVP–salicylic acid, NVP–BZC, NVP–3HBZC, and NVP–GTS; HBP in all of these cases provides a heteromeric carboxylic acid–amide interaction as more favourable than homomeric (Table S3†). However, the structures show that only in the first four compounds is this observed and that in the remaining four structures the amide–amide interaction is conserved. The HBP ranking range observed for the co-formers that interrupt the homomeric interaction is (33–117) while for those co-formers that do not break it, are found in the range (51–162).
Coordination values tool
Both HBP and CV calculations were performed using the same functional group definitions. Differently to the HBP tool, the CV tool19 calculates the likelihood to form 0, 1, 2, 3, 4, 5, and 6 bonds (contacts) by any donor or acceptor atom in a molecule or a target system. These values are derived from a statistical model using the organic structures in the CSD and are dependent on the atom type, the chemical group definition, the Gasteiger charge,45 the competition among the functional groups in the target system, and the 3D molecular shape that modifies the accessibility to the atoms. From these hydrogen-bond coordination likelihood values is possible to construct a hypothetical hydrogen bond arrangement for a molecule following Etter's rule “the strongest donors tend to interact with the strongest acceptors”.46 However, we want to go further and calculate what we have called the “donor and acceptor capacity” or the ability of a molecule to donate or accept HB contacts. It is defined in eqn (1) and calculates the total number of hydrogen bond contacts participating as either a donor (D) or an acceptor (A) that can form a molecule.It is expected that the stability of a system would depend on the balance of their donating and accepting ability, e.g., a pure molecule with D = 1.5 and A = 6 would form multicomponent systems easier than a molecule with D = 1.5 and A = 2. Because this imbalance is offset by the competition term behind the logic function, we have recalculated the models by removing this parameter from the statistical survey.
 | (1) |
Donor (D) and acceptor (A) capacities. Liken is the likelihood to form n bonds.
Therefore, a multicomponent would be expected to be preferred over its pure forms when the mismatch in its ability to donate and accept HB for the multicomponent system is less than that observed for the individual components. This idea of imbalance was explored before to predict hydration in organic crystals. Some authors47 point at a correlation with the donor/acceptor ratio in the organic molecule; however, we observed that while this ratio does not have a significant effect on the frequency of hydrate formation, the sum or the difference of the number of donor and acceptor atoms does.48 It has been quantified in a Coordination Value score (eqn (2)). The lower the CV score, the greater the propensity to obtain a cocrystal.
| CV score = |D–A|cocrystal − |D–A|NVP + |D–A|cocrystal − |D–A|coformer | (2) |
Calculation of the “comfort” of each pair of NVP and co-former to be together or separated.
The CV distribution, by quartiles, for the observed NVP–co-former structures in the CV ranking list is (7, 7, 5, 6) (Table 3A) which can be considered almost random. Furthermore, this distribution for those co-formers tested that do not form multicomponents is (12, 11, 8, 3) (Table 3C) showing an opposite trend to that expected.
Looking at the 112 structures of the first quartile in CV ranking in Table S4,† 25 molecules have been tested with NVP in experimental trials (Table 3). Half of them, thirteen molecules, have resulted in a multicomponent form with NVP, although their crystalline structure has been reported in only seven cases. The only observed cocrystal in the first 50 structures is the propionamide molecule that appears at position 8. On the other hand, among the twelve molecules that have not resulted in a multicomponent form with NVP, seven co-formers are found in the fifty highest ranked co-formers. The structures presented here NVP–BZC, NVP–3HBZC, and NVP–GTS are situated in CV positions of 161, 52, and 64, respectively, and the other hydroxyl derivates of the benzoic display positions of 109 for the NVP–salicylic acid and 56 for NVP–4-hydroxybenzoic acid.
Molecular complementarity tool
Developed by Fábián,14 it is a method to assess the likelihood of two molecules to form a co-crystal based on the shape and polarity of the molecules through the comparison of five molecular descriptors: M axis over L axis, S axis, S axis over L axis, dipole moment, and the fraction of N and O atoms over the total number of atoms in the molecule. S, M, and L are the short, medium, and long dimensions of a box that enclosed the van der Waals surface of the molecule and represent the dimensions of the molecule. Based on the observation that molecules that co-crystallize tend to have similar molecular properties,14 Fábián defined threshold values for the five molecular descriptors and, any molecule that differs from the target molecule outside the threshold, will be unlikely to co-crystallize with it. Three of the descriptors are based on the molecular shape, and are directly related to the molecular conformation. We have observed the case of the NVP–salicylic acid co-crystal.21 In this case, the MC result is dependent on a simple rotation in the H atom from the carboxylic group that can alter the S axis of the molecule and the molecule can either fail or pass.The NVP molecule exhibits the following values for each descriptor: M/L axes = 0.895, S axis = 6.667 Å, S/L axes = 0.611, μ = 1.972, and % of N and O = 0.25 (Table S5†). In our screening of 450 co-formers, the majority of the molecules that do not pass the molecular complementarity failed because the dimension of their box does not fit the ranges required for the descriptors (0.585 < axis ratio M/L < 1.0), (3.437 < S axis < 9.897), and (0.336 < axis ratio S/L < 1.0). This is the case, for example, of planar molecules (S axis too small) and molecules that contain a large aliphatic chain (L axis more than 1.7 times the M axis).
From the 25 NVP–co-former structures known, 11 fail the MC screening because of their box dimension descriptors, except the picric and tartaric acids which fail based on the percentage of N and O atoms.
To have a score and a ranking to add to the previously calculated HBP and CV scores, we need something else than a PASS/FAIL test. That is why we have proposed the normalization function in eqn (3). It uses the values calculated by the MC tool for each descriptor and the pre-established limits for the PASS/FAIL result. The lower the MC index, the more similar or complementary the pair of molecules are; and therefore, the greater the propensity to form a multicomponent.
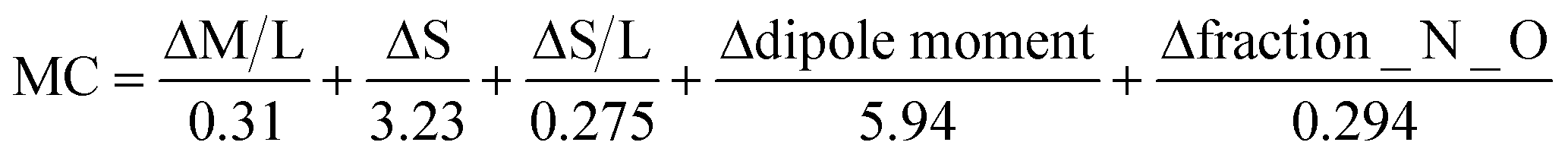 | (3) |
Normalization equation to calculate the MC score for each molecule.
Many of the molecules in our screening of 450 co-formers that fail because of their size parameters, using the MC as implemented in the Mercury program are at the top of the list when sorted by their MC ranking; and molecules at the bottom fail because the difference in the dipole moment value (Table S5†). Moreover, from the 25 NVP–co-former structures known, using the score obtained with the equation defined here, the fourteen molecules that pass the PASS/FAIL test got MC scores distributed by quartiles of (4, 5, 4, 1) and those that fail were distributed over the quartiles as follows (6, 2, 2, 1) and the ones that fail in the percentage of N and O atoms are those which obtain the worst MC scores (Table 3).
According to Fábián theory,14 if a molecule fails in at least one of the descriptors, it is possible to assume that this molecule will not form a multicomponent structure with the API. 44% of the structures reported for NVP multicomponents fail at least in one of the categories of descriptor. However, the MC score values obtained with our normalization function display good values for those molecules that fail the test but form multicomponent of NVP. Based on these observations, it is likely that the thresholds will need to be readjusted to make the three molecular descriptors based on the shape of the molecule more permissible.
COSMOQuick
The COSMOQuick tool11 is used to predict the tendency of cocrystal formation by calculating the excess enthalpy of formation (Hex) between NVP and the corresponding co-former relative to the pure components in a supercooled liquid phase. Hex is a rough approximation of the free energy of cocrystal formation ΔGcocrystal. Compounds with Hex < 0 show an increased probability of forming cocrystals.The COSMOQuick approach is based on the COSMO-RS theory. In the COSMO-RS theory,49,50 the σ-profile of a molecule can be calculated from a combination of σ-profile of different molecular fragments. Since this information, calculated from COSMO theory, is stored in a database, and due to a rigorous statistic, a consistent thermodynamic approximation can be obtained. Thus, the chemical potential and the free energy of a compound, as well as other physicochemical properties can be easily obtained.
Although the results are calculated over the excess enthalpy (Hex) of formation, the ranking is obtained over the function ffit. The ffit function is dependent on the Hex value, but also takes into account the number of rotatable bonds in the molecules.12 Thus, the ranking according to ffit gives improved results even if the co-former molecules have different numbers of rotatable bonds.12
The results obtained by COSMOQuick shown that there is a preference for NVP to co-crystallize with co-formers that exhibit hydroxyl and carboxyl functional groups (Table S6†). On the other hand, molecules that exhibit amide groups and the saccharides are those with less chance to co-crystallize with NVP, according to the thermodynamic calculation performed by COSMOQuick. This is an unexpected result, since the NVP itself exhibits an amide group, and it is expected that other amide groups could easily interact with it.
The distribution of the molecules for the 25 observed NVP–co-former structures by quartiles based on the COSMO ranking is (13, 5, 4, 3) (Table 3A) which is excellent if it was not similar to the distribution observed for those co-formers that do not form multicomponents (13, 5, 11, 5) (Table 3C). It is found that molecules containing aromatic rings substituted with –OH groups, e.g., phenol, catechol, resorcinol, and hydroquinone exhibit a good prediction to co-crystallize with NVP, according to COSMOQuick analysis (Tables 4 and S6†). However, after many tries to co-crystallize these co-formers with NVP, it was not possible to obtain these multicomponent forms. On the other hand, their equivalent molecules substituted with carboxyl and hydroxyl groups, benzoic acid, salicylic acid, 3-hydroxybenzoic acid, and 4-hydroxybenzoic acid could be co-crystallized with NVP, even though they obtained lower COSMOQuick rankings.
| Co-former | CV ranking | MC ranking | HBP ranking | Consensus ranking A | COSMO ranking | Consensus ranking B | Consensus ranking C | Tested by our team | Tested in literature | Molecular formula |
|---|---|---|---|---|---|---|---|---|---|---|
| Consensus ranking A is obtained by the sum of CV, MC, and HBP ranking positions. Consensus ranking B resulted by the addition of COSMO ranking position to the consensus ranking A. Consensus ranking C is obtained adding the ranking positions in MC, HBP, and COSMO rankings. | ||||||||||
| Benzoic acid | 161 | 72 | 51 | 35 | 57 | 9 | 2 | ✓ | ✓20,42,43 | C7H6O2 |
| Salicylic acid | 109 | 121 | 90 | 44 | 30 | 11 | 13 | ✓21 | ✓20 | C7H6O3 |
| 3-Hydroxybenzoic acid | 52 | 128 | 75 | 23 | 29 | 3 | 9 | ✓ | C7H6O3 | |
| 4-Hydroxybenzoic acid | 56 | 85 | 117 | 24 | 32 | 4 | 11 | ✓21 | ✓43 | C7H6O3 |
| Gentisic acid | 64 | 136 | 162 | 62 | 20 | 21 | 27 | ✓ | C7H6O4 | |
| 2,4-Dihydroxybenzoic acid | 65 | 114 | 14 | 10 | 15 | 1 | 1 | C7H6O4 | ||
| Gallic acid | 276 | 233 | 138 | 181 | 16 | 101 | 44 | C7H6O5 | ||
| Phenol | 17 | 120 | 227 | 65 | 23 | 22 | 42 | C6H6O1 | ||
| Catechol | 137 | 197 | 160 | 113 | 9 | 45 | 39 | ✗ | C6H6O2 | |
| Resorcinol | 116 | 173 | 181 | 106 | 5 | 38 | 37 | ✗ | C6H6O2 | |
| Hydroquinone | 115 | 90 | 165 | 68 | 17 | 23 | 18 | ✗ | C6H6O2 | |
| Phloroglucinol | 213 | 188 | 143 | 134 | 3 | 61 | 32 | ✗ | C6H6O3 | |
Consensus ranking
In each tool – HBP, CV, MC, and COSMOQuick – rankings of co-formers were obtained where the co-formers were ordered from the best to the worst probabilities to interact with the NVP molecule. Aiming to establish a unique result to assess the likelihood to obtain a multicomponent form of NVP, three different consensus rankings were calculated.These consensus rankings were generated from the sum of the positions in each of the rankings. The first consensus, or consensus ranking A, was obtained from the sum of the positions in HBP, CV, and MC rankings (Table S7†). The consensus ranking B was calculated adding to the consensus ranking A the position obtained in the COSMOQuick ranking (Table S7†). Due to the random values obtained for CV, a third consensus ranking was calculated omitting these values (HBP + MC + COSMO).
Thus, the co-formers were ordered from the highest to the lowest probabilities to form a multicomponent structure with the NVP. Examination of the distribution of the molecules tested by quartiles (Table 3) consensus ranking C seemed to be the most adequate for co-crystallization prediction.
Co-crystals of NVP with hydroxyl derivates of benzoic and phenol molecules
In the CSD there is recorded two multicomponents of NVP with benzoic acid derivatives (LATQUU and POZCOZ); and we are reporting in this manuscript two new structures, NVP–3HBZC and NVP–GTS besides the NVP–BZC structure.These five structures form a group of hydroxyl derivatives of benzoic acid. In order to probe the predictions, we have included in our experimental and theoretical work equivalent hydroxyl derivatives of the phenol molecule (Table 4): benzoic acid vs. phenol, salicylic acid vs. catechol, 3-hydroxybenzoic acid vs. resorcinol, 4-hydroxybenzoic acid vs. hydroquinone, and one more, phloroglucinol. While the five carboxylic acid compounds formed the multicomponent with the experimental techniques used, none of the hydroxyl compounds gave a different phase in our experiments.
Although all three statistical methods (HBP, CV, and MC) show better results for benzoic derivatives than for phenol derivatives, their values are distributed up to position 161 (reached with the CV's position for benzoic acid, NVP–BZC) of the 450 co-formers verified. With COSMOQuick, all these co-formers scored well in the first 57 positions, with hydroxyl compounds scoring better than the equivalent carboxylic acid derivatives (Table 4). With consensus ranking C, the carboxylic acid derivates are in the range (2–27) and the hydroxyl compound appears in the range (18–42).
Methodology validation
To validate the methodology used here is necessary to assembly all the information available in the literature concerning the attempts to find multicomponent forms of NVP in addition to the experiments realized in this work (Tables 1 and S1†). As we have explained before, a total of 76 molecules were tested to form multicomponent with NVP, and depending on the results they were grouped into three clusters, G1, G2 and G3.To evaluate the ranking position of the molecules that were attempted experimentally, the 450 positions in each tool (Table S7†) were divided into their quartiles. For each quartile, the number of molecules tested with NVP and belonging to each group G1, G2 and G3 have been counted and are shown in Table 3. For molecules in group G1, as was expected, there is a preference for the first and second quartiles for all the methods and the three consensus scores with exception of the distribution observed in the CV. The opposite tendency would be expected for the molecules in G3; however, while there is more representation of molecules in quartile Q4 the maximum is still in Q1 for all the methods. Therefore, it appears that the methods employed are not sufficiently discriminatory. Our opinion is that all the molecules selected to test with NVP have not been selected at random, and have been chosen with characteristics that suggest cocrystal formation is likely. And either they will never cocrystallize with NVP or their cocrystals have not been found yet. Table S8† contains the number and frequency of molecules containing some of the most frequent chemical groups in both samples: the 76 molecules tested to form multicomponents with NVP and the dataset of 450 molecules used in the theoretical screening. This distribution shows that the molecules selected to perform the NVP cocrystallization tests preferably have carboxyl and amide groups with a reduction of other common groups such as primary amine or hydroxyl. A higher phenyl ring frequency is also observed.
COSMO with 13 molecules of G1 in quartile Q1 could be identified as the best result but this method also presents the highest number of false positives with 13 molecules belonging to the group G3 also in quartile Q1.
Of the three consensuses ranking calculated, consensus C display the highest capacity for separation.
The performance of the methodology has been also evaluated by ROC curves. The “positive experimental results” include molecules in the groups G1 (green) and G2 (yellow) minus those in which we were unable to reproduce the multicomponents (Tables 1 and S1†). The “negative experimental results” included all molecules from G3 (red). ROC curves for each tool as well as for the consensus rankings A B and C were obtained (Fig. S1†). The overall performance was measured by the area under the curve (AUC) which gives a measure of the separability between classes. The higher the AUC, the better the predictive model is. AUC value must be higher than 0.5; if it is 0.5 the model has no discriminatory capacity. The AUC calculated for the ROC curves for HBP, CV, MC and COSMO are 0.56, 0.42, 0.65 and 0.59 respectively, and for the consensus A, B and C are 0.56, 0.56 and 0.62 respectively (Fig. S1†). According to these values MC has 65% chance to determine which molecules will form a multicomponent with NVP. Consensus C with 62% of predictive capacity (Fig. 6) improves compared to the other two consensuses. CV has an AUC of 0.42 what means that the model is not able to distinguish between positive and negative hits, it gives a random prediction.
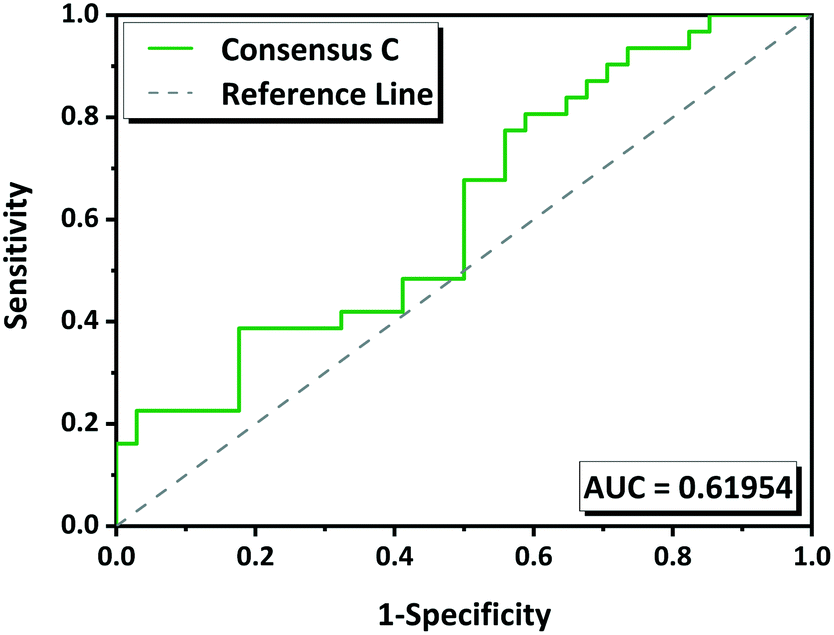 | ||
| Fig. 6 ROC curve for consensus ranking C. | ||
Conclusions
No one doubts that relationships exist between structure and function or structure and properties. This is the reason why great efforts are made trying to predict the structure of a molecule in its crystalline solid-state. However, the prediction of how a molecule will crystallize, or if it will exhibit polymorphs, whether it will form a hydrate, a solvate, a co-crystal, or in general, a multiple form, is not straightforward. Besides, for some molecular systems, it is quite complex.The present work has proposed a way to combine different tools aiming increase the strength of all of them individually. Even when the results are not very discriminatory, it is clear that MC, HBP and COSMO are as good as they are bad. None of them seems better than the other while the function developed to use CV scores generates what seems random results.
The strategy to combine results in a consensus ranking from different methods has been shown to have some utility, returning results that are more reliable. Furthermore, the new normalized function applied to the descriptors defined by Fábián in the molecular complementarity tool has resulted the best method for ordering the potential co-formers of nevirapine, based on the area under the ROC curve of 0.65.
The method used in HBP to evaluate multicomponents only uses the strongest interaction in the multicomponent (AB or BA) and the strongest contact in each component (AA or BB). The results obtained here show that a new strategy including more interactions could improve this method.
Experimentally, three new crystal structures (NVP–BZC, NVP–3HBZC, NVP–GTS) have been reported and further characterization of these co-crystals is underway and will be presented in future work. These three co-formers were the only ones that formed a new phase from a screening of 38 molecules and it corroborates our observation that NVP is likely to cocrystallize with molecules that contain carboxylic acid groups and aromaticity. However, the fact that none of the tools or its combinations were capable of identifying the negative hits is quite interesting and an explanation of this observation is attempted. First, we should keep in mind that, although these molecules have not formed multicomponent structures with NVP, with the LAG method, they may be obtained in the future by another method. Moreover, the thirteen molecules from group G3 (no cocrystals formed) that appear in quartile Q1 (predicted as good candidates to form multicomponents) by consensus C, are: suberic acid, malic acid, succinic acid, L-ascorbic acid, L-tartaric acid, ferulic acid, hydroquinone, resorcinol, phloroglucinol, catechol, orcinol, acetylsalicylic acid, and glycolic acid. It is possible to imply that all these molecules have been chosen following the criteria that they all have –OH and/or –COOH groups. Besides, some of them are, also, aromatic. That is, they had not been chosen randomly but the possible interactions with the NVP molecule were considered.
In addition all NVP structures indicate that the π–π interactions are important in NVP crystal packing; and probably none of the methods used adequately considers the stacking of molecules in their predictions of intermolecular interactions. Even the COSMO analysis, which considered the chemical potentials, is not capable to evaluate directly the impact of the aromatic rings in the multicomponent formation.
Thus, we could conclude that the characteristics of the API affected the methodology proposed. However, although the results were not as good as expected, our group considers that it is worth putting an effort to test this methodology in some other systems. And we would like to check the methods without bias in the selection of the co-formers, using molecules with the full range of characteristics in the experimental screening.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was conducted during scholarship supported by the Consejo Superior de Investigaciones Científicas (CSIC) (COOPA20094) and Red de Cristalografía y Cristalización “Factoría de Cristalización” (FIS2015-71928-REDC, MCIU). R. N. Costa would like to thank the support offered by CCDC during her stay in Cambridge as well as Richard Sykes for his support in the development of Python API scripts. This study was also financed in part by the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior – Brasil (CAPES) – Finance Code 001. D. Ch.-L. acknowledges funding by project no. PGC2018-102047-B-I00 (MCIU/AEI/FEDER, UE). R. N. Costa thanks CAPES for the Ph.D. scholarship. R. N. Costa would like to thank the support offered by Luana P. Meleiro in the obtention of ROC curves.Notes and references
- D. Braga, Chem. Commun., 2003, 2751–2754 RSC.
- A. Mukherjee, Cryst. Growth Des., 2015, 15, 3076–3085 CrossRef CAS.
- G. R. Desiraju, J. Chem. Sci., 2010, 122, 667–675 CrossRef CAS.
- G. R. Desiraju, J. Am. Chem. Soc., 2013, 135, 9952–9967 CrossRef CAS.
- G. R. Desiraju, Crystal Design: Structure and Function - Perspectives in Supramolecular Chemistry, John Wiley & Sons, Hyderabad, India, 2003, vol. 7 Search PubMed.
- S. Domingos, V. André, S. Quaresma, I. C. B. Martins, M. F. M. da Piedade and M. T. Duarte, J. Pharm. Pharmacol., 2015, 67, 830–846 CrossRef CAS.
- J. Aaltonen, M. Allesø, S. Mirza, V. Koradia, K. C. Gordon and J. Rantanen, Eur. J. Pharm. Biopharm., 2009, 71, 23–37 CrossRef CAS.
- N. Blagden, M. de Matas, P. T. Gavan and P. York, Adv. Drug Delivery Rev., 2007, 59, 617–630 CrossRef CAS.
- C. R. Taylor and G. M. Day, Cryst. Growth Des., 2018, 18, 892–904 CrossRef CAS.
- T. Grecu, C. A. Hunter, E. J. Gardiner and J. F. McCabe, Cryst. Growth Des., 2014, 14, 165–171 CrossRef CAS.
- Y. A. Abramov, C. Loschen and A. Klamt, J. Pharm. Sci., 2012, 101, 3687–3697 CrossRef CAS.
- C. Loschen and A. Klamt, J. Pharm. Pharmacol., 2015, 67, 803–811 CrossRef CAS.
- C. R. Groom, I. J. Bruno, M. P. Lightfoot and S. C. Ward, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2016, 72, 171–179 CrossRef CAS.
- L. Fábián, Cryst. Growth Des., 2009, 9, 1436–1443 CrossRef.
- P. T. A. Galek, L. Fábián, W. D. S. Motherwell, F. H. Allen and N. Feeder, Acta Crystallogr., Sect. B: Struct. Sci., 2007, 63, 768–782 CrossRef CAS.
- L. Infantes and W. D. S. Motherwell, Z. Kristallogr., 2005, 220, 333–339 CAS.
- L. Infantes and W. D. S. Motherwell, Chem. Commun., 2004, 1166–1167 RSC.
- J. Chisholm, E. Pidcock, J. van de Streek, L. Infantes, S. Motherwell and F. H. Allen, CrystEngComm, 2006, 8, 11–28 Search PubMed.
- P. T. A. Galek, J. A. Chisholm, E. Pidcock and P. A. Wood, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2014, 70, 91–105 CrossRef CAS.
- M. R. Caira, S. A. Bourne, H. Samsodien, E. Engel, W. Liebenberg, N. Stieger and M. Aucamp, CrystEngComm, 2012, 14, 2541–2551 RSC.
- R. N. Costa, A. L. Reviglio, S. Siedler, S. G. Cardoso, Y. G. Linck, G. A. Monti, A. M. G. Carvalho, J. A. L. C. Resende, M. H. C. Chaves, H. V. A. Rocha, D. Choquesillo-Lazarte, L. Infantes and S. L. Cuffini, Cryst. Growth Des., 2020, 20, 688–698 CrossRef CAS.
- R. Taylor, J. Cole, O. Korb and P. McCabe, J. Chem. Inf. Model., 2014, 54, 2500–2514 CrossRef CAS.
- J. C. Cole, O. Korb, P. McCabe, M. G. Read and R. Taylor, J. Chem. Inf. Model., 2018, 58, 615–629 CrossRef CAS.
- Bruker, 2016 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- P. W. Mui, S. P. Jacober, K. D. Hargrave and J. Adams, J. Med. Chem., 1992, 35, 201–202 CrossRef CAS.
- E. De Clercq, Int. J. Antimicrob. Agents, 2009, 33, 307–320 CrossRef CAS.
- N. Stieger, W. Liebenberg, J. C. Wessels, H. Samsodien and M. R. Caira, Struct. Chem., 2010, 21, 771–777 CrossRef CAS.
- C. C. P. da Silva, S. L. Cuffini, S. N. Faudone, A. P. Ayala and J. Ellena, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2008, 64, o292 CrossRef CAS.
- N. Stieger, M. R. Caira, W. Liebenberg, L. R. Tiedt, J. C. Wessels and M. M. De Villiers, Cryst. Growth Des., 2010, 10, 3859–3868 CrossRef CAS.
- B. G. Pereira, F. D. Fonte-Boa, J. A. L. C. Resende, C. B. Pinheiro, N. G. Fernandes, M. I. Yoshida and C. D. Vianna-Soares, Cryst. Growth Des., 2007, 7, 2016–2023 CrossRef CAS.
- M. R. Caira, N. Stieger, W. Liebenberg, M. M. De Villiers and H. Samsodien, Cryst. Growth Des., 2008, 8, 17–23 CrossRef CAS.
- M. A. Kryukova, A. V. Sapegin, A. S. Novikov, M. Krasavin and D. M. Ivanov, Crystals, 2019, 9, 71 CrossRef.
- W. T. A. Harrison, T. V. Sreevidya, B. Narayana, B. K. Sarojini and H. S. Yathirajan, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2007, 63, o3871 CrossRef CAS.
- S. Jin, H. Zhang, K. Xu, X. Ye, Y. Fang, Y. Zhang, L. Jin and D. Wang, J. Chem. Crystallogr., 2015, 45, 213–223 CrossRef CAS.
- M. A. Kryukova, A. V. Sapegin, A. S. Novikov, M. Krasavin and D. M. Ivanov, Z. Kristallogr., 2019, 234, 101–108 CAS.
- X. Yang, B. Yu, Z. Zhong, B. Guo and Y. Huang, Int. J. Pharm., 2018, 543, 121–129 CrossRef CAS.
- A. Gavezzotti and G. Filippini, J. Phys. Chem., 1994, 98, 4831–4837 CrossRef CAS.
- A. Gavezzotti, Acc. Chem. Res., 1994, 27, 309–314 CrossRef CAS.
- I. J. Bruno, J. C. Cole, J. P. M. Lommerse, R. S. Rowland, R. Taylor and M. L. Verdonk, J. Comput.-Aided Mol. Des., 1997, 11, 525–537 CrossRef CAS.
- P. A. Wood, T. S. G. Olsson, J. C. Cole, S. J. Cottrell, N. Feeder, P. T. A. Galek, C. R. Groom and E. Pidcock, CrystEngComm, 2013, 15, 65–72 RSC.
- P. P. Gujar, A. A. Kokil, P. S. Karekar, Y. A. Gurav and A. V. Yadav, Int. J. Pharm. Technol., 2013, 4, 4831–4842 CAS.
- Y. K. Nalte, V. A. Arsul, S. G. Shep and S. B. Bothara, J. Pharm. Res., 2015, 9, 556–561 CAS.
- P. T. A. Galek, F. H. Allen, L. Fábián and N. Feeder, CrystEngComm, 2009, 11, 2634–2639 RSC.
- J. Gasteiger and M. Marsili, Tetrahedron, 1980, 36, 3219–3228 CrossRef CAS.
- M. C. Etter, Acc. Chem. Res., 1990, 23, 120–126 CrossRef CAS.
- G. R. Desiraju, J. Chem. Soc., Chem. Commun., 1991, 426–428 RSC.
- L. Infantes, L. Fábián and W. D. S. Motherwell, CrystEngComm, 2007, 9, 65–71 RSC.
- A. Klamt, COSMO-RS from Quantum Chemistry to Fluid Phase Thermodynamics ad Drug Design, Elsevier, Amsterdam, 2005 Search PubMed.
- A. Klamt, V. Jonas, T. Bürguer and J. C. W. Lohrenz, J. Phys. Chem., 1998, 102, 5074–5085 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2012053–2012055. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0ce00948b |
| This journal is © The Royal Society of Chemistry 2020 |