
Bismuthanylstibanes†
Katherine M.
Marczenko
and
Saurabh S.
Chitnis
 *
*
Department of Chemistry, Dalhousie University, 6274 Coburg Road, Halifax, Nova Scotia B3H 4R2, Canada. E-mail: Saurabh.chitnis@dal.ca
First published on 3rd February 2020
Abstract
Thermally-robust bismuthanylstibanes are prepared in a one-step, high yield reaction, providing the first examples of neutral Bi–Sb σ-bonds in the solid state. DFT calculations indicate that the bis(silylamino)naphthalene scaffold is well-suited for supporting otherwise labile bonds. The reaction chemistry of the Bi–Sb bond is debuted by showing fission using NH3BH3 and insertion of a sulfur atom, the latter providing the first example of a Bi–S–Sb motif.
Multiple bonding between heavy p-block elements (principle quantum number > 2) has been a topic of much research interest over the past several decades,1–3 gradually eroding the perception that heavy elements do not form π bonds. Indeed, numerous compounds containing homonuclear or heteronuclear multiple bonds have now been obtained, revealing important theoretical insights and new reactivity paradigms.1,2,4–6 Pursuing a program of developing new electronic structures and reactivity at Bi and Sb centres,7–9 we were surprised to note that although a thermally robust Bi–Sb π-bond (i.e. RBi = SbR) has been known in an isolable compound for two decades (A, Scheme 1),10 compounds containing the prototypical electron-precise σ-bond between these elements (i.e. R2Bi–SbR2, bismuthanylstibanes) have still not been isolated in the solid state. This is despite their presumed role as reaction intermediates10,11 and their potentially valuable chemistry as single source precursors for deposition of BiSb,12 which is a promising low-temperature n-type thermoelectric13–15 and a topological insulator.16,17
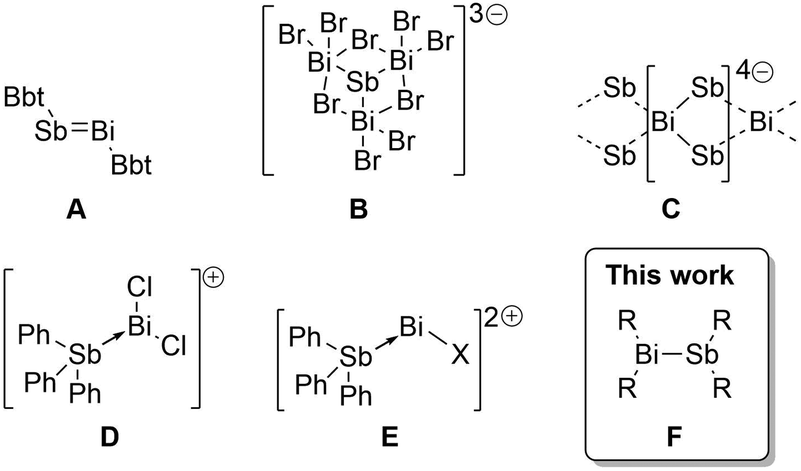 | ||
| Scheme 1 Isolated compounds containing Bi–Sb bonds. See text for references. Bbt = o,o-(CH(SiMe3)2)2-p-C(SiMe3)-Ph. | ||
This unusual gap is likely due to the kinetic lability of neutral Bi–Sb bonds – solution phase spectroscopic studies revealed that Me2BiSbMe2 undergoes rapid scrambling in solution to give combinations of dipnictanes at ambient temperature, precluding isolation of the heterobimetallic species.18 The introduction of molecular charge has nevertheless enabled characterization of four charged compounds exhibiting Bi–Sb interactions (Scheme 1): the [SbBi3Br9]3− cluster anion (B),19 the polymeric ribbon of [BiSb2]4− (C) found in the network solid Ba2BiSb2,20 and the molecular cations [Ph3SbBiCl2]+ (D) and [Ph3SbBiCl]2+ (E).21 These ions are likely persistent due to stabilization from lattice enthalpy and the high barrier to scrambling via associative interactions between similarly charged ions (coulombic repulsion). The successful isolation of these ionic examples encouraged us to seek the type of archetypal neutral σ-bond that is known for most element pairs in the p-block but remains as-yet unisolated between Bi and Sb centres.
Reactive functional groups at Bi and Sb centres have recently been stabilized using bulky and rigid bis(silylamino)naphthyl substituents.8,22,23 Here we show that these substituents also provide access to persistent bismuthanylstibanes (F in Scheme 1), which contain the first structurally characterized neutral Bi–Sb σ-bonds. Contrary to previous examples, the Bi–Sb bonds reported here are remarkably stable against redistribution. Density functional theory (DFT) calculations ascribe this robustness to a combination of inductive and dispersive effects inherent to the bis(silylamino)naphthalene scaffold. We also debut the reaction chemistry of the Bi–Sb functional group by revealing insertion of H+/H− and sulfur, evidencing, in the latter case, the first example of a Bi–S–Sb connectivity.
Attempts to form bismuthanylstibanes through traditional magnesium reduction or dehydrohalogenation reactions following Tokitoh's route to BbtSb = BiBbt10 were unsuccessful (Scheme 2a and b), yielding only intractable reaction mixtures containing metal deposits and traces of free ligands. We next envisioned the reaction of a preformed stibanide anion24,25 with a chlorobismuthane (Scheme 2c) but these attempts were also foiled – instead of yielding the anticipated stibanide, deprotonation of EtSbH with nBuLi immediately gave a nearly insoluble red precipitate, which was identified by X-ray crystallography as the polystibane (EtSb)4Sb8. The asymmetric unit of this compound contains four EtSb fragments connected by a tetracyclic Sb8 cage (Fig. 1a). As this motif has previously been observed by Breuing26 and Roesky,27 we did not investigate the mechanism of its formation in further detail. An analogous structure featuring As–Sb bonds was also observed by Hänisch.28
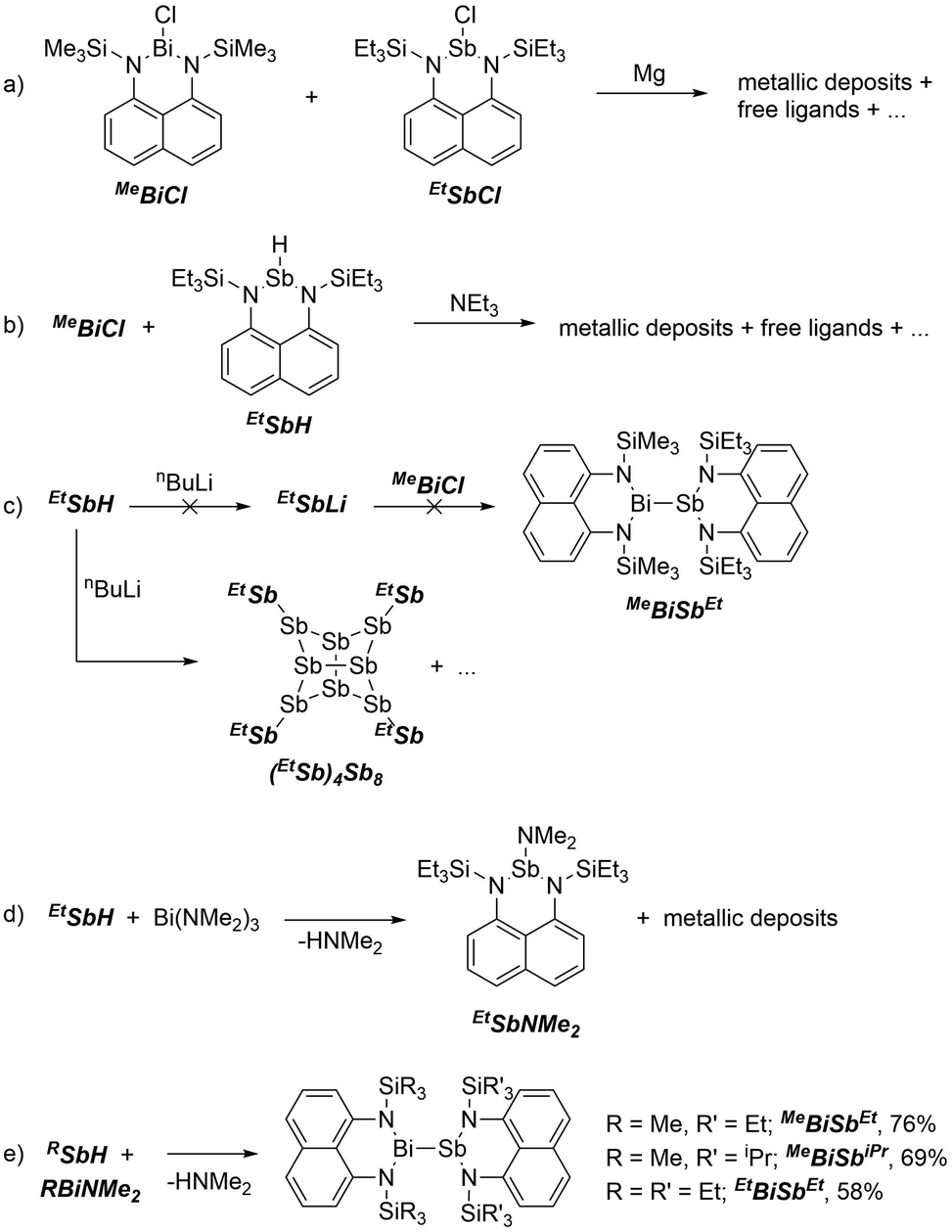 | ||
| Scheme 2 Synthesis of (EtSb)4Sb8 and derivatives of RBiSbR′. | ||
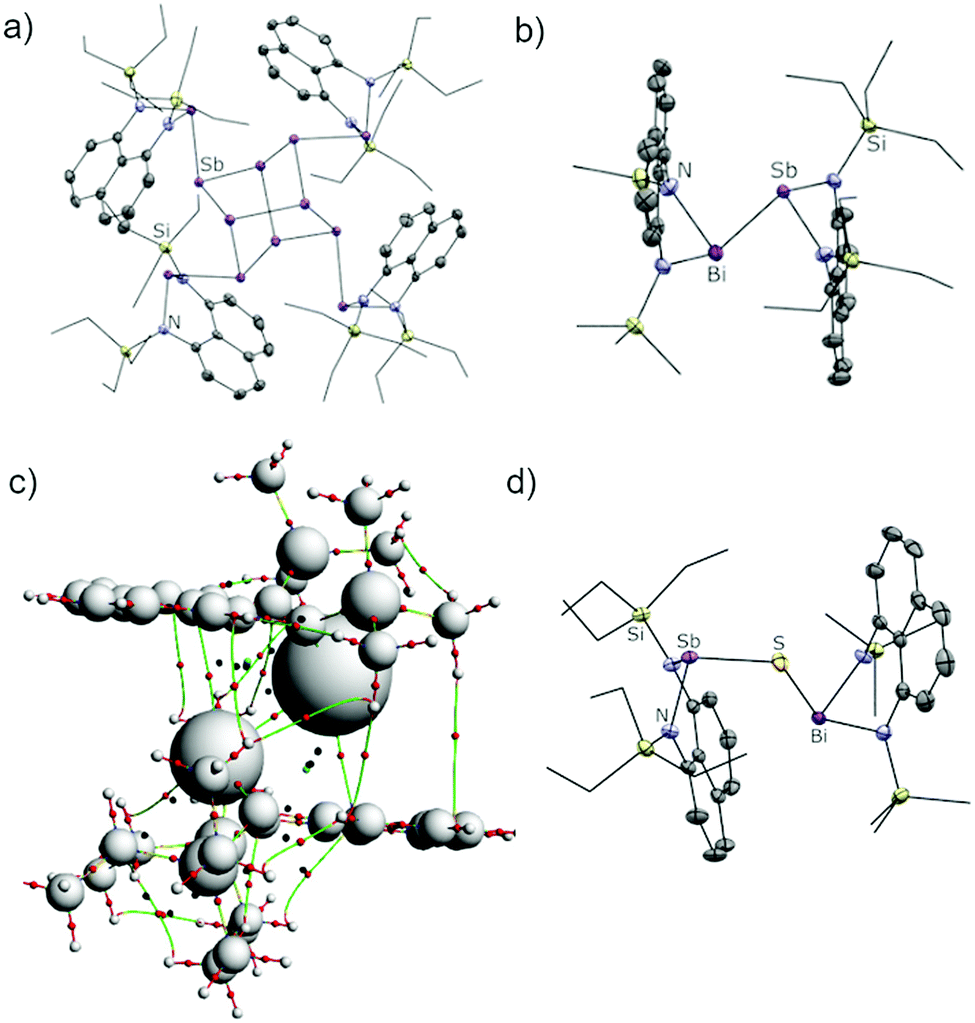 | ||
| Fig. 1 (a) Single-crystal X-ray structures of (EtSb)4Sb8. (b) Single-crystal X-ray structures of MeBiSbEt. Selected bond lengths (Å) and angles (°): Bi–Sb 2.9775(9), Bi–N 2.161(4) and 2.161(4), Sb–N 2.059(4) and 2.059(4), N–Bi–N 83.6(2), N–Sb–N 87.6(2) (c) AIM calculated bond paths and critical points for MeBiSbEt. White spheres denote the nuclear basins (atoms). The vertical green lines evidence bonding interactions between the ligands in the periphery of the Bi–Sb bond. (d) Single-crystal X-ray structures of MeBiSbEt. Selected bond lengths (Å) and angles (°): Sb–S 2.3838(8), Bi–S 2.5456(7), Bi–N 2.141(2) and 2.150(2), Sb–N 2.040(2) and 2.045(2), N–Bi–N 83.86(8), N–Sb–N 89.35(8). Thermal ellipsoids are shown at the 50% probability level. Hydrogens have been omitted and silyl groups are shown in wireframe for clarity. | ||
Next we attempted deaminative coupling between EtSbH and Bi(NMe2)3 (at 0 °C and at −78 °C), which gave a light-yellow solution and a metallic precipitate (Scheme 2d). Analysis of the crude mixture by 1H NMR spectroscopy showed formation of the antimony amide EtSbNMe2 (independently made from EtSbCl and LiNMe2, Fig. S10, ESI†) and HNMe2. We speculated that EtSbNMe2 could have formed via decomposition of a transient Bi–Sb bonded species by transfer of a NMe2 group from Bi to Sb.
Consistent with this hypothesis, tethering the amino groups at both Bi and Sb suppressed such decomposition and finally yielded the targeted bismuthanylstibanes (Scheme 2e). Dropwise addition of EtSbH to MeBiNMe2 at −30 °C gave a dark red solution over a ten-minute period. Concentration of the reaction mixture gave bright red crystals of MeBiSbEt in 76% isolated yield. The silane substituents were easily varied through use of different precursors to give MeBiSbiPr (69% isolated yield) and EtBiSbEt (58% isolated yield). The reactions are quantitative by 1H NMR spectroscopy and easily monitored by tracking the disappearance of the Sb–H resonance and formation of HNMe2. Upon completion, the 1H NMR spectra of bismuthanylstibanes show overlapping signals in the aromatic region due to the two distinct naphthalene diamine backbones (7.31–7.09 ppm). The alkyl region shows methylene, ethylene, and/or isopropyl signals with chemical shifts more upfield than the parent compounds.
The structures of all bismuthanylstibanes were confirmed by single-crystal X-ray diffraction (Fig. 1b and Fig. S1, ESI† for MeBiSbiPr and EtBiSbEt). The asymmetric units contain a bimetallic structure with trigonal pyramidal antimony and bismuth atoms. The lone pair sites on each of the metallic atoms are oriented in opposite directions along the a-axis achieving maximum spatial distance between the two distinct naphthalenediamine ligand frameworks. Due to the similarity of the silyl groups present, a Bi/Sb substitutional disorder was detected in EtBiSbEt, rendering the crystallographic data suitable only for connectivity information. The Bi–Sb, Sb–N, and Bi–N bond lengths (see Fig. 1 caption and deposited CIFs) in MeBiSbEt and MeBiSbiPr do not significantly vary when the silane substituents are changed, indicating that this type of “outer sphere” bulk has little influence on the immediate bond parameters at the Bi–Sb bond. The N–Bi–N angle is slightly more contracted than the N–Sb–N angle in both cases (see Fig. 1 caption and deposited CIFs). The Bi–Sb bonds in MeBiSbEt [2.9775(9) Å] and MeBiSbiPr [2.9764(7) Å] is comparable in length to the value in TbtBi = SbTbt [2.972(5) Å], which is in line with the reduced bond order in heavy-atom formal double bonds.6,29
To assess the stability of the Bi–Sb bonds, a sealed sample of MeBiSbEt was heated to 100 °C C6D6 for 72 h. No decomposition or redistribution was observed over this period, despite MeBiBiMe and EtSbSbEt being isolable compounds,8,22 in contrast to the aforementioned facile redistribution involving alkyl/aryl-substituted Bi–Sb bonds.18 To explore the specific influence of the bis(silylamino)naphthalene framework, we performed DFT calculations on MeBiSbEt, Ph2BiSbPh2, and (Me2N)2BiSb(NMe2)2. A Morokuma energy decomposition analysis (EDA)30 revealed that the Bi–Sb bonding interaction in MeBiSbEt (ΔEint = −72.03 kcal mol−1) is indeed intrinsically stronger than the corresponding interactions in the Ph2BiSbPh2 (ΔEint = −52.55 kcal mol−1) or (Me2N)2BiSb(NMe2)2 (ΔEint = −46.72 kcal mol−1).
As shown in Table 1, orbital (ΔEorb values) and electrostatic (ΔEelstat values) interactions are consistently more stabilizing in the amino-substituted compounds, because attachment to electronegative nitrogen atoms increases the partial charge (and therefore effective electronegativity) of the metal atoms. This lowers the energies of the interacting orbitals at the metals by inductive effects and simultaneously increases the extent of electrostatic bonding by making the metals stronger electron density acceptors. In particular, the rigid nature of the fused naphthalene backbone prevents effective overlap of the nitrogen lone pairs with the LUMO of the MeBiSbEt, further reducing electron density at the metal.8 This view is supported by the more positive Natural Bond Orbital (NBO) derived partial charges at Bi and Sb (qSb and qBi values in Table 1) when the naphthalene framework is used compared to the values in (Me2N)2BiSb(NMe2)2, where free rotation around the metal-nitrogen bonds allows overlap of the nitrogen lone pairs with metal-centred acceptor orbitals. Attractive dispersion forces31 between the bulky trialkylsilyl groups (known to be excellent dispersion donors)32 also play a significant role in stabilizing MeBiSbEt (ΔEdisp = −39.03 kcal mol−1). Consistently, Bader's Atom-In-Molecules (AIM)33 analysis detected numerous bond paths (Fig. 1c, vertical green lines) and bond critical points (red dots) between the ligands on each metal showing peripheral dispersive interactions between the bulky ligands. Thus, DFT calculations indicate that the bis(silylamino)naphthalene scaffold is uniquely suited to stabilize the otherwise weak Bi–Sb bond.
| Parameter | Me BiSb Et | (Me2N)2BiSb(NMe2)2 | Ph2BiSbPh2 |
|---|---|---|---|
| ΔEint | −72.03 | −46.72 | −52.55 |
| ΔEPauli | 329.36 | 238.66 | 224.41 |
| ΔEelstat | −126.07 | −88.05 | −96.26 |
| ΔEorb | −236.29 | −182.19 | −159.15 |
| ΔEdisp | −39.03 | −15.14 | −21.55 |
| ΔEprep | 0.81 | 6.12 | 0.68 |
| ΔE (−De) | −71.22 | −40.60 | −51.87 |
| d/Å | 3.003 | 2.981 | 2.940 |
| q Sb | 1.15 | 0.92 | 0.64 |
| q Bi | 1.12 | 0.93 | 0.68 |
The reactivity of MeBiSbEt towards a variety of unsaturated substrates was examined. No reaction between MeBiSbEt and azobenzene, phenylacetylene, or pyridine N-oxide was observed after several days in the presence of UV light or in refluxing C6D6 (Scheme 3a). Photochemical or thermal scrambling to homonuclear species was also not observed at any point of these reactivity studies, emphasizing the stability of the Bi–Sb bond. Heating solutions of MeBiSbEt and NH3BH3 resulted in fission of the metal–metal bond, giving EtSbH, borazine and a mixture of bis(trimethylsilylamino)naphthalene and bis(triethylsilylamino)naphthalene in a 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 ratio along with insoluble black metallic deposits (Scheme 2b). Based on the greater amount the trimethylsilyl-substituted ligand formed, we speculate that while EtSbH is a stable metal hydride,8 transiently formed MeBiH is thermally unstable34 and undergoes reductive elimination of bis(trimethylsilylamino)naphthalene and deposits metallic bismuth.
:2 ratio along with insoluble black metallic deposits (Scheme 2b). Based on the greater amount the trimethylsilyl-substituted ligand formed, we speculate that while EtSbH is a stable metal hydride,8 transiently formed MeBiH is thermally unstable34 and undergoes reductive elimination of bis(trimethylsilylamino)naphthalene and deposits metallic bismuth.
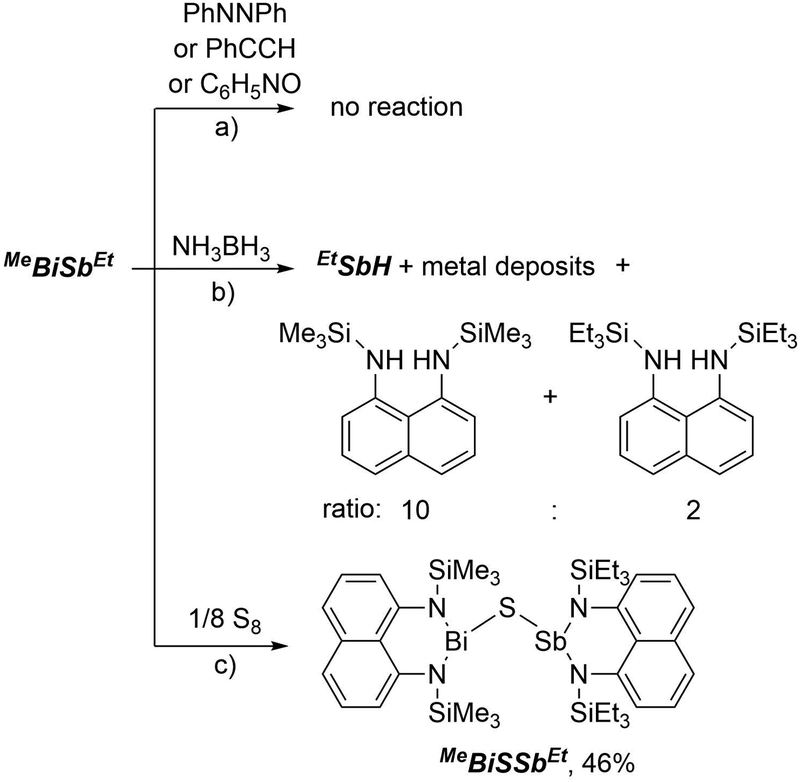 | ||
| Scheme 3 Reactivity of MeBiSbEt. | ||
In contrast, clean insertion of a sulfur atom into the Bi–Sb bond was achieved by heating a solution of MeBiSbEt and S8 in toluene at 100 °C for 1 hour (Scheme 2c). The resulting compound, MeBiSSbEt, formed quantitatively by NMR analysis and isolated in 46% crystalline yield, contains the first example of the Sb–S–Bi connectivity. Notably, MeBiSSbEt is also only the third structurally-characterized example of a molecular Sb–Z–Bi moiety, where Z is any element of the periodic table.35,36 Compound MeBiSSbEt was fully characterized and its structure was determined crystallographically (Fig. 1d). The Sb–S [2.3838(8) Å] and Bi–S [2.5456(7) Å] bond lengths are within range of mean E–S bond lengths observed for antimony sulfides and bismuth sulfides (Sb–S: 2.527 ± 0.173 Å; Bi–S: 2.791 ± 0.177 Å) and the Sb–S–Bi angle [116.06(3)°] is as expected for a bent geometry at sulfur. The N–Bi–S bonding angles [91.52(6), 91.15(6)°] are significantly more contracted than the N–Sb–S angles [101.50(6), 102.49(6)°].
In summary, we have reported the synthesis of thermally-robust bismuthanylstibanes in a one-step, high-yield reaction, providing the first examples of neutral compounds with Bi–Sb σ-bonds. DFT calculations indicate that the bis(silylamino)naphthalene scaffold is inherently well-suited for supporting otherwise labile bonds because it increases interaction energies through a combination of inductive effects and the dispersion donor effects. We also debuted the reaction chemistry of the Bi–Sb functional group by showing addition of H+/H− and insertion of a sulfur atom into the metal–metal bond. Further reactivity studies and the application of RBiSbR′ compounds as single-source precursors for depositing heterobimetallic phases are underway.
We acknowledge the Natural Sciences and Engineering Research Council (NSERC) of Canada, the Canada Foundation for Innovation (CFI), the Nova Scotia Research and Innovation Trust (NSRIT), and Dalhousie University for research funding. K. M. M. acknowledges the Vanier Canada Graduate Scholarships Program, the Killam Trusts, and the Walter C. Sumner Memorial Fellowships Program for funding. We thank Prof. Christian Hering-Junghans for valuable suggestions.
Conflicts of interest
There are no conflicts to declare.Notes and references
- E. Rivard and P. P. Power, Inorg. Chem., 2007, 46, 10047–10064 CrossRef CAS PubMed.
- P. P. Power, Acc. Chem. Res., 2011, 44, 627–637 CrossRef CAS PubMed.
- C. Präsang and D. Scheschkewitz, Chem. Soc. Rev., 2016, 45, 900–921 RSC.
- R. C. Fischer and P. P. Power, Chem. Rev., 2010, 110, 3877–3923 CrossRef CAS PubMed.
- P. P. Power, Nature, 2010, 463, 171–177 CrossRef CAS PubMed.
- H. B. Wedler, P. Wendelboe and P. P. Power, Organometallics, 2018, 37, 2929–2936 CrossRef CAS.
- M. B. Kindervater, K. M. Marczenko, U. Werner-Zwanziger and S. S. Chitnis, Angew. Chem., Int. Ed., 2019, 58, 7850–7855 CrossRef CAS PubMed.
- K. M. Marczenko, J. A. Zurakowski, K. L. Bamford, J. W. M. MacMillan and S. S. Chitnis, Angew. Chem., Int. Ed., 2019, 58, 18096–18101 CrossRef CAS PubMed.
- K. M. Marczenko, J. A. Zurakowski, M. B. Kindervater, S. Jee, T. Hynes, N. Roberts, S. Park, U. Werner-Zwanziger, M. Lumsden, D. N. Langelaan and S. S. Chitnis, Chem. – Eur. J., 2019, 25, 16414–16424 CrossRef CAS PubMed.
- T. Sasamori, N. Takeda and N. Tokitoh, Chem. Commun., 2000, 1353–1354 RSC.
- T. Sasamori, N. Takeda and N. Tokitoh, Phosphorus, Sulfur Silicon Relat. Elem., 2001, 169, 89–92 CrossRef.
- H. Zhang, J. S. Son, J. Jang, J.-S. Lee, W.-L. Ong, J. A. Malen and D. V. Talapin, ACS Nano, 2013, 7, 10296–10306 CrossRef CAS PubMed.
- G. E. Smith and R. Wolfe, J. Appl. Phys., 1962, 33, 841–846 CrossRef CAS.
- W. M. Yim and A. Amith, Solid-State Electron., 1972, 15, 1141–1165 CrossRef CAS.
- B. Lenoir, A. Dauscher, M. Cassart, Y. I. Ravich and H. Scherrer, J. Phys. Chem. Solids, 1998, 59, 129–134 CrossRef CAS.
- L. Fu and C. L. Kane, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 76, 045302 CrossRef.
- D. Hsieh, D. Qian, L. Wray, Y. Xia, Y. S. Hor, R. J. Cava and M. Z. Hasan, Nature, 2008, 452, 970–974 CrossRef CAS PubMed.
- A. J. Ashe, III and E. G. Ludwig, Jr., J. Organomet. Chem., 1986, 303, 197–204 CrossRef.
- B. Wahl, L. Kloo and M. Ruck, Angew. Chem., Int. Ed., 2008, 47, 3932–3935 CrossRef CAS PubMed.
- S. Ponou and T. F. Fässler, Inorg. Chem., 2004, 43, 6124–6126 CrossRef CAS PubMed.
- E. Conrad, N. Burford, R. McDonald and M. J. Ferguson, Chem. Commun., 2010, 46, 4598–4600 RSC.
- B. Nekoueishahraki, P. P. Samuel, H. W. Roesky, D. Stern, J. Matussek and D. Stalke, Organometallics, 2012, 31, 6697–6703 CrossRef CAS.
- B. Nekoueishahraki, S. P. Sarish, H. W. Roesky, D. Stern, C. Schulzke and D. Stalke, Angew. Chem., Int. Ed., 2009, 48, 4517–4520 CrossRef CAS PubMed.
- H. J. Breunig, I. Ghesner, M. E. Ghesner and E. Lork, Inorg. Chem., 2003, 42, 1751–1757 CrossRef CAS PubMed.
- H. J. Breunig, M. E. Ghesner and E. Lork, Z. Anorg. Allg. Chem., 2005, 631, 851–856 CrossRef CAS.
- H. Breunig, R. Rosler and E. Lork, Angew. Chem., Int. Ed. Engl., 1997, 36, 2237–2238 CrossRef CAS.
- C. Schoo, S. Bestgen, A. Egeberg, S. Klementyeva, C. Feldmann, S. N. Konchenko and P. W. Roesky, Angew. Chem., Int. Ed., 2018, 57, 5912–5916 CrossRef CAS PubMed.
- D. Nikolova and C. von Hänisch, Eur. J. Inorg. Chem., 2005, 378–382 CrossRef CAS.
- L. Zhao, S. Pan, N. Holzmann, P. Schwerdtfeger and G. Frenking, Chem. Rev., 2019, 119, 8781–8845 CrossRef CAS PubMed.
- K. Kitaura and K. Morokuma, Int. J. Quantum Chem., 1976, 10, 325–340 CrossRef CAS.
- J. P. Wagner and P. R. Schreiner, Angew. Chem., Int. Ed., 2015, 54, 12274–12296 CrossRef CAS PubMed.
- R. Pollice and P. Chen, Angew. Chem., Int. Ed., 2019, 58, 9758–9769 CrossRef CAS PubMed.
- R. F. W. Bader, Chem. Rev., 1991, 91, 893–928 CrossRef CAS.
- N. J. Hardman, B. Twamley and P. P. Power, Angew. Chem., Int. Ed., 2000, 39, 2771–2773 CrossRef CAS PubMed.
- A. Hinz, A. Schulz and A. Villinger, Chem. Commun., 2015, 51, 11437–11440 RSC.
- C. Ritter, B. Ringler, F. Dankert, M. Conrad, F. Kraus and C. von Hänisch, Dalton Trans., 2019, 48, 5253–5262 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed procedures and characterization data. CCDC 1975977–1975980 and 1980822. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0cc00254b |
| This journal is © The Royal Society of Chemistry 2020 |