
Synthesis and antiproliferative activities of OSW-1 analogues bearing 2-acylamino-xylose residues†
Lijun
Sun
 ab,
Ruina
Wang
c,
Xiaobo
Wang
c,
Yongjun
Dang
c,
Wei
Li
*bd and
Biao
Yu
*b
ab,
Ruina
Wang
c,
Xiaobo
Wang
c,
Yongjun
Dang
c,
Wei
Li
*bd and
Biao
Yu
*b
aDepartment of Chemistry, University of Science and Technology of China, 96 Jinzhai Road, Hefei, Anhui 230026, China
bState Key Laboratory of Bio-organic and Natural Products Chemistry, Center for Excellence in Molecular Synthesis, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, 345 Lingling Road, Shanghai 200032, China. E-mail: byu@sioc.ac.cn
cKey Laboratory of Metabolism and Molecular Medicine, the Ministry of Education, Department of Biochemistry and Molecular Biology, School of Basic Medical Sciences, Fudan University, Shanghai 200032, China
dDepartment of Medicinal Chemistry, China Pharmaceutical University, 24 Tong Jia Xiang, Nanjing, Jiangsu 210009, China. E-mail: wli@cpu.edu.cn
First published on 30th April 2019
Abstract
OSW-1 and SBF-1 are well studied saponins with exceptionally potent antitumor activities. Herein, we report the syntheses of a library of 38 C22-ester analogues bearing 2-acylamino xylose residues. SAR studies show that the introduction of the 2-acylamino xylose residues could further increase the antitumor activities as many as 40 folds than those of SBF-1 and the (1→3)-disaccharide linkage is crucial to the activities. A highly potent probe (3) bearing photoactivatable and clickable residues has been identified.
Introduction
OSW-1 (1), a naturally occurring steroidal saponin isolated from the bulbs of Ornithogalum saundersiae (Fig. 1),1,2 is known for its exceptionally potent antiproliferative activity against tumor cells. With IC50 values in nanomolar range, OSW-1 is 10 to 100 times more active than several clinical antitumor drugs, such as cisplatin.3 In addition, normal cell lines were found to be less sensitive to OSW-1,3,4 making it a potential candidate for developing antitumor drugs. Consequently, extensive efforts have been devoted into its synthetic and SAR studies.5–21 Among the numerous natural and synthetic analogues, it is noteworthy that SBF-1 (2), an artificial 22-ester analogue that can be readily prepared via a stereoselective aldol reaction, displays similar or even better antitumor activity.22 Therefore, SBF-1 (2) has been used as an accessible alternative in biological studies.23–26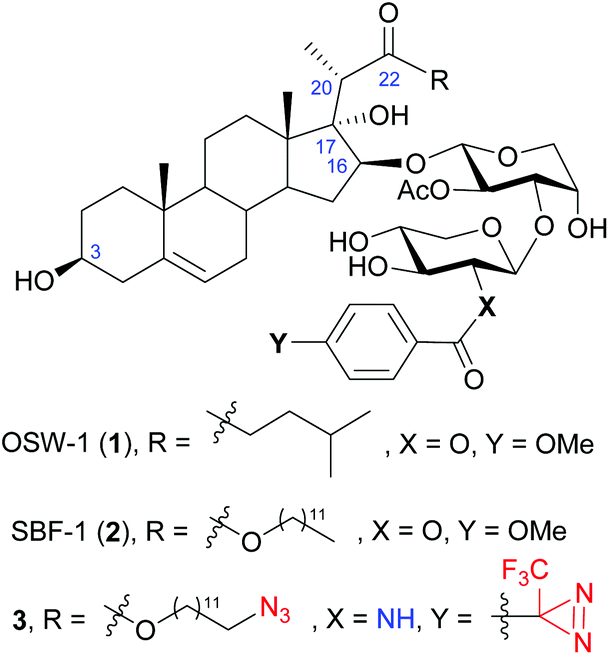 | ||
| Fig. 1 The structure of OSW-1 (1), synthetic analogue SBF-1 (2), and the designed probe 3 in the present work. | ||
Despite the tremendous efforts in the synthetic realm,27–33 the mechanism of the antitumor activity of OSW-1 still remains elusive. Applying a synthetic probe derived at the 3-OH of the xylose moiety to the pull-down assay, Shair and co-workers disclosed that oxysterol binding protein (OSBP) and its paralog ORP4L, both of which are usually related to sterol and lipid metabolism, could be targeted by OSW-1.34,35 Huang and co-workers found that the cytotoxic potency of OSW-1 relied on the homeostasis of calcium regulation, such as sodium–calcium exchange on the plasma membrane.4,36 Wu and co-workers suggested OSW-1 induced apoptosis related to Bcl-2 and caspase-8.37 Given these controversial results, it is highly possible that OSW-1 is able to target multiple proteins. Thus, development of new OSW-1 probes to identify other unknown binding proteins is of great interest. Recently, Sakurai and co-workers reported the preparation and cytotoxicity of several OSW-1 probes with the fluorescent or clickable functionalities installed at either 3-OH or 4-OH of the xylose moiety, some of which were equipped with diazirine residues for photoaffinity labeling.38–42 However, their application in the target identification has yet to be reported.
Based on SBF-1 (2) which is easier to prepare and similarly active compared to OSW-1, we envisioned a duo-labeled analogue (3) as a new probe of OSW-1 (Fig. 1). An azido group was attached at the end of the aliphatic side chain of the aglycone, which could couple with biotin or fluorescent groups via a click reaction; a diazirine group was introduced at the xylose residue, which could lead to conjugation to the binding proteins via a photoactivatable carbene insertion reaction. Previous SAR studies have disclosed the crucial necessity of the two acyl groups on the disaccharide for maintaining the potent antitumor activities,3,43–45 but a slight modification of the p-methoxybenzoyl (MBz) group at the C2 of the xylose moiety has been proved tolerable.3,42–44 Hence, C2 of the xylose residue would be an ideal position for the photoaffinity labeling by replacing the original MBz group with a diazirine-substituted benzoyl group.46 Due to the poor stability of the photosensitive diazirine group, its introduction should be scheduled at a late stage. To this end, an amino group was envisioned to replace the original 2-hydroxyl group on the xylose residue,47 it would be readily coupled with the commercially available diazirine-substituted benzoic acid at the final stage. In addition, this strategy would result in a series of OSW-1 analogues bearing 2-acylamino-xylose by coupling with different carboxylic acids. Herein, we report their chemical synthesis and antiproliferative activities against tumor cell lines.
Results and discussion
The synthesis commenced with the preparation of a sterol aglycone bearing a C22-ester chain with an azido group added at the terminal position, following modification of the previous approach to the synthesis of SBF-1 (2).22 Thus, 16α-OH ketone 4 was prepared from dehydroepiandrosterone via four steps with an overall yield of 60% (Scheme 1). 12-Azidododecyl propionate 5 was then attached to ketone 4 under the conditions of LiHMDS in THF at −10 °C, providing diol 6 in 53% yield with the resulting tertiary 17-OH and 21-methyl in the desired α- and S(C20)-orientations, respectively. Upon treatment of diol 6 with TPAP-catalyzed oxidation in the presence of NMO led to the corresponding ketone 7 in a satisfactory 80% yield; subsequent reduction with NaBH4 and CeCl3 in THF and H2O smoothly turned over the original 16α-OH of 6, furnishing the desired aglycone 8 with 16β-OH ready for glycosylation.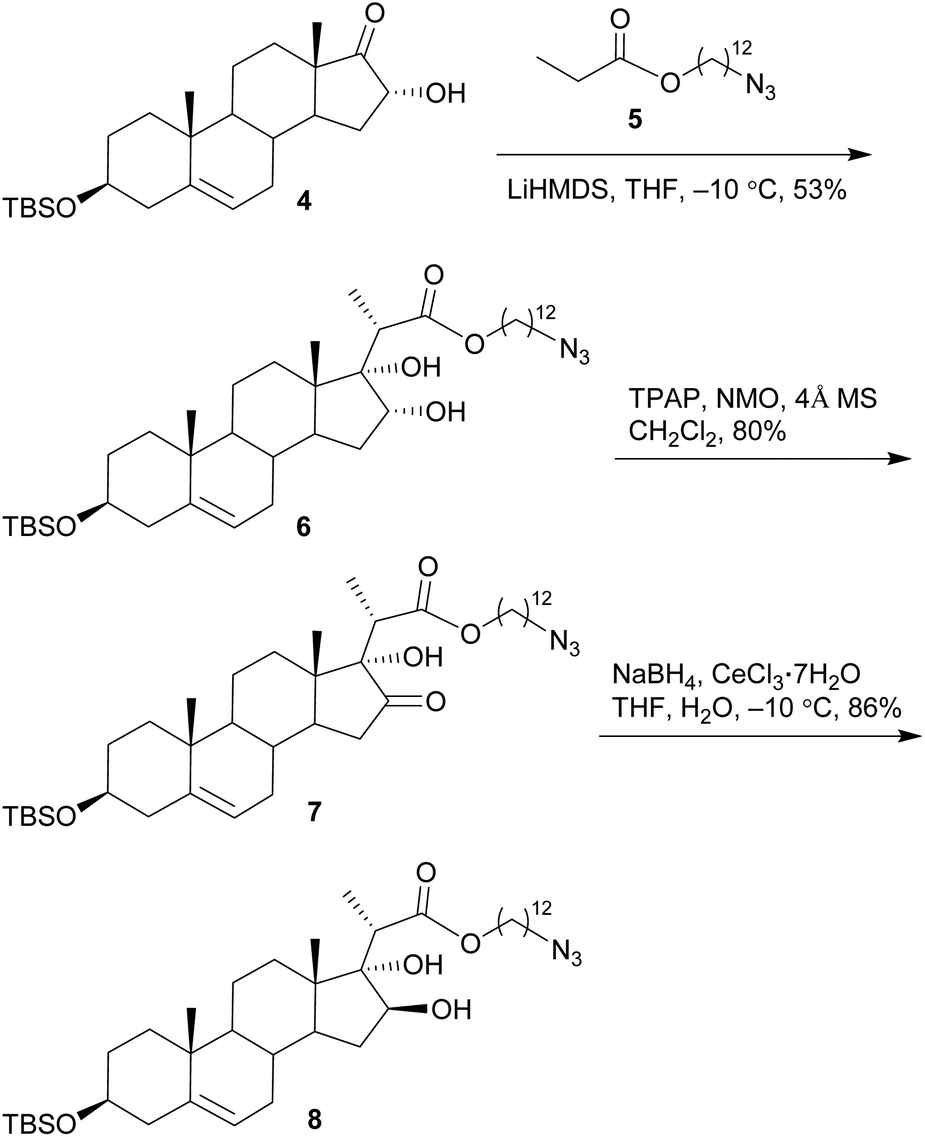 | ||
| Scheme 1 Synthesis of aglycone 8. | ||
On the other hand, we adopted an effective protocol to synthesize 2-amino-2-deoxy-D-xylose from D-xylose (Scheme 2). Treatment of D-xylose (9) with Ac2O and pyridine at 0 °C fully converted 9 into tetraacetyl xylose, which underwent bromination under the conditions of 33% HBr/AcOH in CH2Cl2; subjection of the resulting xylosyl bromide to a suspension of Zn and NH4Cl in CH3CN provided xylal 10 in an excellent 96% yield (based on 9).48,49 A successful introduction of an equatorial adizo group to C2 was realized by Lemieux's azidonitration of xylal 10 with CAN and NaN3 in CH3CN at −20 °C;47,50–52 the nascent anomeric nitro group was immediately replaced with an acetoxy group to provide 11 as a pair of inseparable anomers (α/β = 2.8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1).51 Next, acetates 11 were smoothly converted into thioglycosides 12 in 85% yield under the conditions of p-TolSH and BF3·OEt2 in CH2Cl2.53 Although both 11 and 12 were inseparable anomers, the α- and β-anomers of 13 obtained after deacylation could be partially separated and therefore their structures were unambiguously confirmed.
:1).51 Next, acetates 11 were smoothly converted into thioglycosides 12 in 85% yield under the conditions of p-TolSH and BF3·OEt2 in CH2Cl2.53 Although both 11 and 12 were inseparable anomers, the α- and β-anomers of 13 obtained after deacylation could be partially separated and therefore their structures were unambiguously confirmed.
 | ||
| Scheme 2 Synthesis of tolylthio 2-azido-2-deoxy-xylopyranoside 13. | ||
The anomeric thiol group of 13 could act as both protecting and leaving groups, due to its good stability toward acidic, basic and reductive conditions as well as the effective activation by specific reagents such as NIS. The selective protection for 2-NH2, 3-OH, and 4-OH, however, encountered difficulties. For the purpose of ensuring the β-selectivity in the later glycosylation, a protecting group capable of neighboring participation would be required at 2-NH2. Thus, a range of protecting groups was examined and phthalimido (Phth) group was found efficient to secure the β-selectivity. Other protecting groups such as Boc, Fmoc, Troc, and Teoc were excluded owing to the poor performance in either their installations or glycosylation of the arabinosyl acceptor. On the other hand, the choice of protecting groups for 3-OH and 4-OH was greatly constrained by the conditions of introducing Phth group that required basic DBU in heated toluene, wherein the widely-used TES groups in the previous synthetic work were easily removed. Therefore, thioglycosides 13 were treated with 2,3-butanedione and BF3·OEt2 in MeOH in the presence of CH(OMe)3 to give base-stable bisacetal 14 in a good 81% yield (Scheme 3).54,55 Reduction of the 2-azido group with Ph3P in THF and H2O afforded the desired 2-amino group on 15, which was subsequently protected with Phth group to afford phthalimide 16 in 90% yield under the conditions of phthaloyl dichloride and DBU in toluene at 120 °C.56 Similar to the anomers depicted in Scheme 2, the anomers of 14 were hardly separable, while those of 15 and 16 could be partially separated for the purpose of structural characterization. Meanwhile, counterparts of these compounds with the anomeric α-benzyl group were also synthesized (see ESI† for details) for the preparation of other types of xylosyl donors instead of the thioglycoside donor. Although all these counterparts were single α-anomers and their isolation and identification were much easier compared to the present thioglycosides, the Phth group encountered unexpected saturation during the hydrogenolysis of the anomeric benzyl group.
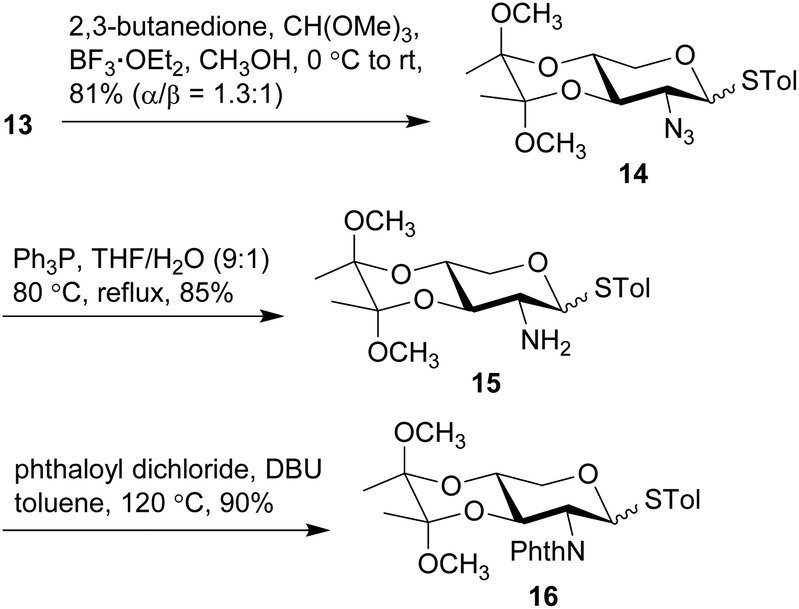 | ||
| Scheme 3 Synthesis of 2-amino-thioxyloside 16. | ||
With the 2-amino-thioxyloside donors 16 at hand, a regioselective glycosylation of arabinosyl diol 17 was conducted under the conditions of NIS and TMSOTf in the presence of 4 Å MS (Scheme 4).6 Surprisingly, unlike most of the previously reported glycosylation of diol 17 with xylosyl donors that mainly led to the (1→3)-linked disaccharide,32 the present glycosylation with 2-amino-xylosyl donors 16 led to (1→4)-linked disaccharide as the major product. Similar 4-OH selectivity has been reported in the previous synthesis of OSW-1 analogues,54,55,57,58 including a careful investigation conducted by Pakulski and co-workers. They attributed this unusual 4-OH selectivity to a novel equatorial 4-OH of arabinose with the 1C4 conformation constrained by the intramolecular hydrogen bonding between 3-OH and the cis-anomeric oxygen atom of arabinoside.55,58 However, this could not be used to explain the selectivity in our glycosylation because the anomeric oxygen atom on 17 is trans to 3-OH and no hydrogen bonding could be formed. Apparently, the regioselectivity of 3,4-diol arabinoside relies not only on arabinoside itself, but also on the xylosyl donors. Additionally, we found that the replacement of Phth group on 16 with Troc or Fmoc increased the 3-OH selectivity, but unfortunately trisaccharides from the bis-glycosylation of both 3-OH and 4-OH of 16 were also generated in a significant amount.
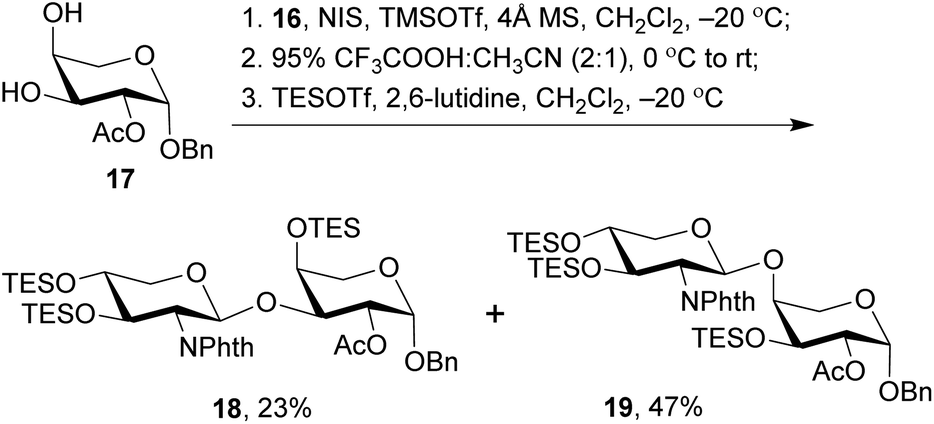 | ||
| Scheme 4 Synthesis of disaccharides 18 and 19. | ||
The (1→3)-linked and (1→4)-linked disaccharide products obtained from the glycosylation of 17 with 16 were only partially separable and both of them could be used in the preparation of OSW-1 analogues, so no further optimization was conducted. Our synthesis continued with the replacement of the butane-bisacetal group with TES groups, the removal of which would be much more reliable at a late stage.58 Thus, the mixture was treated with CF3COOH and CH3CN to cleave the acetal, and subsequent protection with TES groups under the conditions of TESOTf and 2,6-lutidine provided the separable (1→3)-linked and (1→4)-linked disaccharides 18 and 19 in 23% and 47% yield (three steps based on 16), respectively. Their structures were carefully characterized by 2D NMR, particularly HMBC spectra. Both 18 and 19 were employed in the following synthesis of OSW-1 analogues.
Hydrogenolysis of the anomeric benzyl group of disaccharide 18 with 10% Pd/C in EtOAc afforded the desired hemi-acetal 20 in 57% yield (Scheme 5), although a small part of the Phth group was also found saturated. After activation of 20 under the conditions of CCl3CN and DBU in CH2Cl2, subjection of the resulting imidate 21 to the glycosylation of sterol aglycone 8 in the presence of TMSOTf at −20 °C led to the desired 22 in 52% yield. Subsequent removal of the Phth group under the conditions of 85% N2H4·H2O in EtOH at 60 °C was accompanied by the partial cleavage of the acetyl group and therefore led to a mixture. Interestingly, TES groups that were usually considered as acid- and base-labile mainly remained intact under these conditions. The nascent free amino group in this mixture was selectively protected by a Fmoc group with FmocOSu, and the remaining free hydroxyl groups were re-acetylated to give 23 in 47% yield (based on 22). Next, Fmoc was removed with Et2NH in CH2Cl2 at rt, and the released amino group was then coupled with a wide range of carboxylic acids in the presence of HATU and DIPEA at rt. Final removal of all the silyl groups with CF3COOH in CH2Cl2 at rt furnished 25 OSW-1 analogues (3, 24–47) bearing (1→3)-linked 2-N-acyl-2-deoxyxylose residues, including the designed probe 3 with the aryl diazirine moiety for future photo-cross-linked reaction. Similar procedures were employed on disaccharide 19 to synthesize 13 (1→4)-linked analogues 48–60 bearing different acyl residues at the 2-amino group.
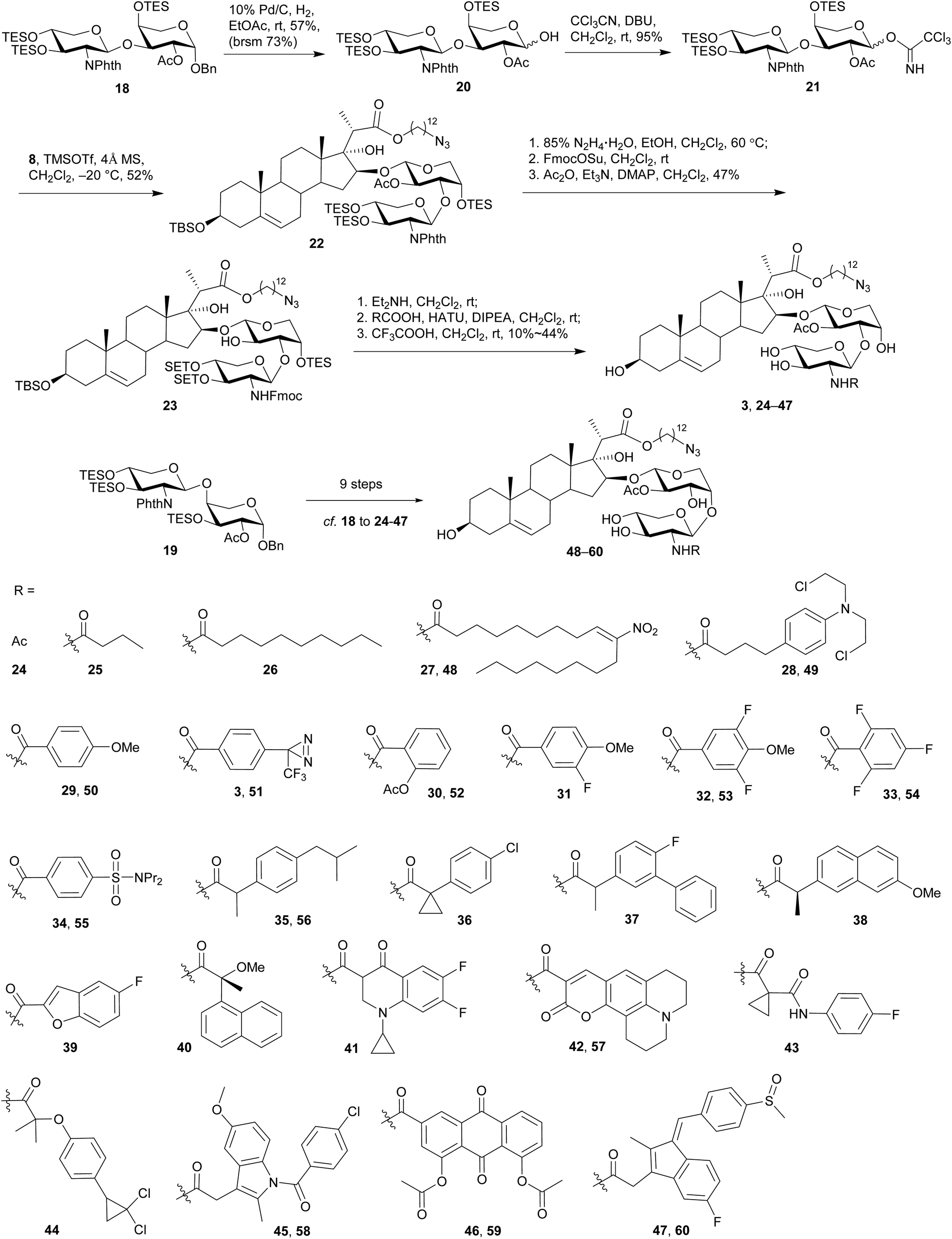 | ||
| Scheme 5 Synthesis of OSW-1 analogues bearing 2-N-acyl-2-deoxyxylose unit. | ||
Employing the CellTiter Glo Luminescent cell viability assay, we measured the antiproliferative activities of these newly synthesized OSW-1 analogues against three representative cell lines (Table 1), i.e., Jurkat (human leukemia cells) for suspension tumor cells, MDA-MB-231 (human breast cancer cells) for adherent tumor cells, and CRL1999 (human aorta smooth muscle cells) for normal cells. SBF-1 (2) and Taxol were used as positive controls in the evaluation. In general, most of the (1→3)-linked OSW-1 analogues bearing 2-N-acyl-2-deoxyxyloses (3, 24–47) displayed potent antiproliferative activities with IC50 values lower than 10 nM. Among them, compound 29 with the same MBz group as the natural OSW-1 showed the strongest activity and its IC50 reached as low as 0.11 nM against Jurkat T, which is 40 times more potent than SBF-1 (2) and taxol. In fact, all the (1→3)-linked analogues (3, 29–34) with benzoyl-type groups, including the designed photoaffinity probe 3, displayed better activities than SBF-1 (2). These comparisons clearly indicated that the replacement of the xylose residue with 2-amino-2-deoxyxylose would increase the antiproliferative activities. On the other hand, the introduction of short aliphatic acyl groups into the amino group, such as Ac (24) or butyryl group (25) rather than benzoyl group, could still retain the activities. But long aliphatic acyl groups on analogues 26 and 27 were found to increase the IC50 to around 100 nM. It is noteworthy that all the analogues (24–27) with aliphatic acyl groups showed stronger antiproliferative activities against MDA-MB-231 than Jurkat cells, whereas other analogues were usually more effective on Jurkat than MDA-MB-23 cells. These results suggest that the types of acyl groups on the xylose moiety could affect the antitumor selectivity, presumably related to the growth of tumors as suspension cells or adherent cells.
| Compounds | IC50 (nM) | Compounds | IC50 (nM) | ||||
|---|---|---|---|---|---|---|---|
| Jurkat | MDA-MB-231 | CRL1999 | Jurkat | MDA-MB-231 | CRL1999 | ||
| 24 | 6.56 | 2.43 | N/A | 25 | 5.57 | 0.85 | N/A |
| 26 | 162 | 24.8 | N/A | 27 | 153 | 136 | N/A |
| 28 | 7.10 | 27.0 | 12.0 | 29 | 0.11 | 0.51 | 0.40 |
| 3 | 1.80 | 3.70 | 11.0 | 30 | 7.26 | 0.88 | N/A |
| 31 | 0.59 | 0.75 | 0.72 | 32 | 0.44 | 0.76 | 1.50 |
| 33 | 0.46 | 1.40 | 0.68 | 34 | 1.00 | 4.10 | 4.40 |
| 35 | 164 | 33.7 | N/A | 36 | 8.60 | 11.0 | 56.0 |
| 37 | 29.0 | 69.0 | 220 | 38 | 2.50 | 7.90 | 11.0 |
| 39 | 0.75 | 0.64 | 1.40 | 40 | 24.0 | 56.0 | 130 |
| 41 | 3.50 | 6.00 | 9.00 | 42 | 6.80 | 6.90 | 23.0 |
| 43 | 3.50 | 8.50 | 28.0 | 44 | 83.0 | 260 | 2950 |
| 45 | 20.0 | 39.0 | 30.0 | 46 | 2.40 | 4.70 | 18.0 |
| 47 | 5.40 | 10.0 | 160 | 50 | 600 | 2400 | 3200 |
| 53 | 1300 | 5000 | 3900 | SBF-1 (2) | 4.30 | 15.0 | 17.0 |
| Taxol | 3.8 | 2.6 | 138 | ||||
Although all the (1→3)-linked OSW-1 analogues (3, 29–34) with benzoyl-type groups exhibited excellent activities against tumor cell lines, they were also comparably cytotoxic to normal cells CRL1999. Replacing the benzoyl groups with more complex acyl groups could result in several safer analogues to normal cell lines. For instance, analogues 44 and 47 are 30 times more potent against Jurkat than CRL1999 cells. This is similar to the antitumor selectivity found on taxol. More analogues and activity data are still required for further analysis of the SAR on the antitumor selectivity.
On the other hand, analogues bearing the (1→4)-linked disaccharides were found to be dramatically less active. The most potent one was compound 50 bearing the MBz group, however, its IC50 value even on the more sensitive Jurkat was as high as 600 nM, which is 6000 folds as much as 0.11 nM of its (1→3)-linked counterpart 29. This is consistent with the previous reports, wherein several (1→4)-linked analogues were synthesized but found inactive at a concentration of 10 μM.18,57 These results confirmed that the (1→3)-linkage in the disaccharide was essential to maintain the potent antiproliferative activities of OSW-1 analogues.
Conclusions
An effective route was established for the synthesis of OSW-1 analogues bearing 2-acylamino xylose residues. A library of 25 (1→3)-linked and 13 (1→4)-linked disaccharide analogues were prepared accordingly, including a photoaffinity and clickable probe 3. The evaluation of their antiproliferative activities indicated that the replacement with 2-aceylamino xylose residues could significantly increase the activities with the lowest IC50 value being down to 0.11 nM. In addition, the (1→3)-linkage of the disaccharide was confirmed to be crucial to retain the potent activities. Studies on target protein identification with probe 3 are in progress and the results will be reported in due course.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the financial support from the National Natural Science Foundation of China (21432012, 21621002 and 21672248), E-Institutes of Shanghai Municipal Education Commission (E09013), the Strategic Priority Research Program of the Chinese Academy of Sciences (XDB20020000), and the K. C. Wong Education Foundation.Notes and references
- S. Kubo, Y. Mimaki, M. Terao, Y. Sashida, T. Nikaido and T. Ohmoto, Phytochemistry, 1992, 31, 3969–3973 CrossRef CAS.
- V. L. Challinor and J. J. De Voss, Nat. Prod. Rep., 2013, 30, 429–454 RSC.
- Y. Mimaki, M. Kuroda, A. Kameyama, Y. Sashida, T. Hirano, K. Oka, R. Maekawa, T. Wada, K. Sugita and J. A. Beutler, Bioorg. Med. Chem. Lett., 1997, 7, 633–636 CrossRef CAS.
- Y. Zhou, C. Garcia-Prieto, D. A. Carney, R.-h. Xu, H. Pelicano, Y. Kang, W. Yu, C. Lou, S. Kondo, J. Liu, D. M. Harris, Z. Estrov, M. J. Keating, Z. Jin and P. Huang, J. Natl. Cancer Inst., 2005, 97, 1781–1785 CrossRef CAS PubMed.
- C. Guo and P. L. Fuchs, Tetrahedron Lett., 1998, 39, 1099–1102 CrossRef CAS.
- S. Deng, B. Yu, Y. Lou and Y. Hui, J. Org. Chem., 1999, 64, 202–208 CrossRef CAS PubMed.
- W. Yu and Z. Jin, J. Am. Chem. Soc., 2001, 123, 3369–3370 CrossRef CAS PubMed.
- J. W. Morzycki and A. Wojtkielewicz, Carbohydr. Res., 2002, 337, 1269–1274 CrossRef CAS PubMed.
- Q.-h. Xu, X.-w. Peng and W.-s. Tian, Tetrahedron Lett., 2003, 44, 9375–9377 CrossRef CAS.
- L. Deng, H. Wu, B. Yu, M. Jiang and J. Wu, Bioorg. Med. Chem. Lett., 2004, 14, 2781–2785 CrossRef CAS PubMed.
- B. Shi, P. Tang, X. Hu, J. O. Liu and B. Yu, J. Org. Chem., 2005, 70, 10354–10367 CrossRef CAS PubMed.
- J. W. Morzycki, A. Wojtkielewicz and S. Wołczyński, Bioorg. Med. Chem. Lett., 2004, 14, 3323–3326 CrossRef CAS PubMed.
- Y. Matsuya, S. Masuda, N. Ohsawa, S. Adam, T. Tschamber, J. Eustache, K. Kamoshita, Y. Sukenaga and H. Nemoto, Eur. J. Org. Chem., 2005, 803–808 CrossRef CAS.
- H.-J. Qin, W.-S. Tian and C.-W. Lin, Tetrahedron Lett., 2006, 47, 3217–3219 CrossRef CAS.
- A. Wojtkielewicz, M. Długosz, J. Maj, J. W. Morzycki, M. Nowakowski, J. Renkiewicz, M. Strnad, J. Swaczynová, A. Z. Wilczewska and J. Wójcik, J. Med. Chem., 2007, 50, 3667–3673 CrossRef CAS PubMed.
- P. Tang, F. Mamdani, X. Hu, J. O. Liu and B. Yu, Bioorg. Med. Chem. Lett., 2007, 17, 1003–1007 CrossRef CAS PubMed.
- J. Xue, P. Liu, Y. Pan and Z. Guo, J. Org. Chem., 2008, 73, 157–161 CrossRef CAS PubMed.
- D. Zheng, L. Zhou, Y. Guan, X. Chen, W. Zhou and P. Lei, Bioorg. Med. Chem. Lett., 2010, 20, 5439–5442 CrossRef CAS PubMed.
- Y. Guan, D. Zheng, L. Zhou, H. Wang, Z. Yan, N. Wang, H. Chang, P. She and P. Lei, Bioorg. Med. Chem. Lett., 2011, 21, 2921–2924 CrossRef CAS PubMed.
- J. Maj, J. W. Morzycki, L. Rárová, J. Oklešt'ková, M. Strnad and A. Wojtkielewicz, J. Med. Chem., 2011, 54, 3298–3305 CrossRef CAS PubMed.
- C. Liu, A.-p. Wang, L. Jin, Y. Guo, Y. Li, Z. Zhao and P. Lei, Tetrahedron, 2016, 72, 4091–4102 CrossRef CAS.
- B. Shi, H. Wu, B. Yu and J. Wu, Angew. Chem., Int. Ed., 2004, 43, 4324–4327 CrossRef CAS PubMed.
- W. Li, R. Song, X. Fang, L. Wang, W. Chen, P. Tang, B. Yu, Y. Sun and Q. Xu, Biochem. Pharmacol., 2012, 84, 172–181 CrossRef CAS PubMed.
- W. Li, Z. Ouyang, Q. Zhang, L. Wang, Y. Shen, X. Wu, Y. Gu, Y. Shu, B. Yu, X. Wu, Y. Sun and Q. Xu, Cell Death Dis., 2014, 5, e1581 CrossRef CAS PubMed.
- A. Elgehama, W. Chen, J. Pang, S. Mi, J. Li, W. Guo, X. Wang, J. Gao, B. Yu, Y. Shen and Q. Xu, Cancer Lett., 2016, 372, 82–88 CrossRef CAS PubMed.
- W. Chen, X. Qian, Y. Hu, W. Jin, Y. Shan, X. Fang, Y. Sun, B. Yu, Q. Luo and Q. Xu, J. Pharmacol. Sci., 2018, 138, 271–278 CrossRef CAS PubMed.
- J. W. Morzycki and A. Wojtkielewicz, Phytochem. Rev., 2005, 4, 259–277 CrossRef CAS.
- B. Yu, Y. C. Zhang and P. P. Tang, Eur. J. Org. Chem., 2007, 5145–5161, DOI:10.1002/ejoc.200700452.
- S. Lee, T. G. LaCour and P. L. Fuchs, Chem. Rev., 2009, 109, 2275–2314 CrossRef CAS PubMed.
- J. J. Forsman and R. Leino, Chem. Rev., 2011, 111, 3334–3357 CrossRef CAS PubMed.
- B. Yu, J. Sun and X. Yang, Acc. Chem. Res., 2012, 45, 1227–1236 CrossRef CAS PubMed.
- Y. Tang, N. Li, J.-a. Duan and W. Tao, Chem. Rev., 2013, 113, 5480–5514 CrossRef CAS PubMed.
- Y. Yang, S. Laval and B. Yu, Adv. Carbohydr. Chem. Biochem., 2014, 71, 137–226 CrossRef PubMed.
- A. W. G. Burgett, T. B. Poulsen, K. Wangkanont, D. R. Anderson, C. Kikuchi, K. Shimada, S. Okubo, K. C. Fortner, Y. Mimaki, M. Kuroda, J. P. Murphy, D. J. Schwalb, E. C. Petrella, I. Cornella-Taracido, M. Schirle, J. A. Tallarico and M. D. Shair, Nat. Chem. Biol., 2011, 7, 639–647 CrossRef CAS PubMed.
- L. Albulescu, J. R. Strating, H. J. Thibaut, L. van der Linden, M. D. Shair, J. Neyts and F. J. van Kuppeveld, Antiviral Res., 2015, 117, 110–114 CrossRef CAS PubMed.
- C. Garcia-Prieto, K. B. Riaz Ahmed, Z. Chen, Y. Zhou, N. Hammoudi, Y. Kang, C. Lou, Y. Mei, Z. Jin and P. Huang, J. Biol. Chem., 2013, 288, 3240–3250 CrossRef CAS PubMed.
- J. Zhu, L. Xiong, B. Yu and J. Wu, Mol. Pharmacol., 2005, 68, 1831–1838 CAS.
- K. Sakurai, T. Takeshita, M. Hiraizumi and R. Yamada, Org. Lett., 2014, 16, 6318–6321 CrossRef CAS PubMed.
- R. Yamada, T. Takeshita, M. Hiraizumi, D. Shinohe, Y. Ohta and K. Sakurai, Bioorg. Med. Chem. Lett., 2014, 24, 1839–1842 CrossRef CAS PubMed.
- R. Yamada, M. Hiraizumi, S. Narita and K. Sakurai, Asian J. Org. Chem., 2016, 5, 330–334 CrossRef CAS.
- M. Hiraizumi, R. Komatsu, T. Shibata, Y. Ohta and K. Sakurai, Org. Biomol. Chem., 2017, 15, 3568–3570 RSC.
- K. Sakurai, M. Hiraizumi, N. Isogai, R. Komatsu, T. Shibata and Y. Ohta, Chem. Commun., 2017, 53, 517–520 RSC.
- M. Kuroda, Y. Mimaki, A. Yokosuka, Y. Sashida and J. A. Beutler, J. Nat. Prod., 2001, 64, 88–91 CrossRef CAS.
- M. Kuroda, Y. Mimaki, A. Yokosuka, F. Hasegawa and Y. Sashida, J. Nat. Prod., 2002, 65, 1417–1423 CrossRef CAS.
- T. Tschamber, S. Adam, Y. Matsuya, S. Masuda, N. Ohsawa, S. Maruyama, K. Kamoshita, H. Nemoto and J. Eustache, Bioorg. Med. Chem. Lett., 2007, 17, 5101–5106 CrossRef CAS PubMed.
- M. Nassal, Liebigs Ann. Chem., 1983, 1510–1523 CrossRef CAS.
- Z. Jin, Y. Mei, L. Chen and A. Shah, US Patent, WO2017/100153A1, 2017 Search PubMed.
- H. Chen, T. Xian, W. Zhang, W. Si, X. Luo, B. Zhang, M. Zhang, Z. Wang and J. Zhang, Carbohydr. Res., 2016, 431, 42–46 CrossRef CAS PubMed.
- T. Taniguchi and K. Monde, Chem. – Asian J., 2007, 2, 1258–1266 CrossRef CAS PubMed.
- R. U. Lemieux and R. M. Ratcliffe, Can. J. Chem., 1979, 57, 1244–1251 CrossRef CAS.
- H. Hashimoto, K. Araki, Y. Saito, M. Kawa and J. Yoshimura, Bull. Chem. Soc. Jpn., 1986, 59, 3131–3136 CrossRef CAS.
- P. H. Seeberger, S. Roehrig, P. Schell, Y. Wang and W. J. Christ, Carbohydr. Res., 2000, 328, 61–69 CrossRef CAS PubMed.
- T. Ohtani, S. Sakai, A. Takada, D. Takahashi and K. Toshima, Org. Lett., 2011, 13, 6126–6129 CrossRef CAS PubMed.
- L. S. Khasanova, F. A. Gimalova, S. A. Torosyan, A. A. Fatykhov and M. S. Miftakhov, Russ. J. Org. Chem., 2011, 47, 1125–1129 CrossRef CAS.
- Z. Pakulski and P. Cmoch, Tetrahedron, 2015, 71, 4757–4769 CrossRef CAS.
- Y. Yang and B. Yu, Tetrahedron, 2014, 70, 1023–1046 CrossRef CAS.
- X. Ma, B. Yu, Y. Hui, D. Xiao and J. Ding, Carbohydr. Res., 2000, 329, 495–505 CrossRef CAS PubMed.
- K. Kuczynska, P. Cmoch, L. Rárová, J. Oklešt'ková, A. Korda, Z. Pakulski and M. Strnad, Carbohydr. Res., 2016, 423, 49–69 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9qo00462a |
| This journal is © the Partner Organisations 2019 |