
Pore surface engineering of covalent organic frameworks: structural diversity and applications
Harsh
Vardhan
a,
Ayman
Nafady
b,
Abdullah M.
Al-Enizi
b and
Shengqian
Ma
 *a
*a
aDepartment of Chemistry, University of South Florida, 4202 East Fowler Avenue, Tampa, Florida-33620, USA. E-mail: sqma@usf.edu; Fax: +1-813-974 3203; Tel: +1-813-9745217
bDepartment of Chemistry, College of Science, King Saud University, Riyadh 11451, Saudi Arabia
First published on 29th October 2019
Abstract
Connecting molecular building blocks by covalent bonds to form extended crystalline structures has caused a sharp upsurge in the field of porous materials, especially covalent organic frameworks (COFs), thereby translating the accuracy, precision, and versatility of covalent chemistry from discrete molecules to two-dimensional and three-dimensional crystalline structures. COFs are crystalline porous frameworks prepared by a bottom-up approach from predesigned symmetric units with well-defined structural properties such as a high surface area, distinct pores, cavities, channels, thermal and chemical stability, structural flexibility and functional design. Due to the tedious and sometimes impossible introduction of certain functionalities into COFs via de novo synthesis, pore surface engineering through judicious functionalization with a range of substituents under ambient or harsh conditions using the principle of coordination chemistry, chemical conversion, and building block exchange is of profound importance. In this review, we aim to summarize dynamic covalent chemistry and framework linkage in the context of design features, different methods and perspectives of pore surface engineering along with their versatile roles in a plethora of applications such as biomedical, gas storage and separation, catalysis, sensing, energy storage and environmental remediation.
 Harsh Vardhan | Harsh Vardhan received his Master of Science (M.Sc.) degree in Chemistry from the Indian Institute of Technology-Kharagpur, India. In 2016, he obtained his Ph.D. in Material Physics and Chemistry under the supervision of Prof. Francis Verpoort from the Laboratory of Organometallics, Catalysis, and Ordered Materials, Wuhan University of Technology, China. At present, he is a graduate student in the research group of Prof. Shengqian Ma at the University of South Florida, USA. His research interest is focused on developing novel covalent organic frameworks as organic scaffolds for heterogeneous catalysis, including C–H activation. |
 Ayman Nafady | Ayman Nafady is currently a Professor of Nanomaterials/Inorganic Electrochemistry at King Saud University, Riyadh, Saudi Arabia & Sohag University, Egypt. He is also an adjunct/visiting professor at RMIT University, Melbourne, Australia and Editor in Chief of the International Journal of Nanomaterials and Chemistry. Prof. Nafady obtained his Ph.D. (2000–2004) under the supervision of Prof. William Geiger at the University of Vermont, USA and has been a Research Fellow at Monash University (2005–2011), Australia working with Prof. Alan Bond. He has made significant contributions to the fields of inorganic/organometallic electrochemistry and nanomaterials science and has been a pioneer in the development of novel electrochemical and photochemical approaches for controlling the synthesis and fabrication of a wide range of metal–organic frameworks and nanostructured materials for applications in water splitting, supercapacitors, biosensors, catalysis and other energy-related applications. |
 Abdullah M. Al-Enizi | Abdullah M. Al-Enizi obtained his Ph.D. in 2013 jointly from King Saud University (KSA) and the University of Texas, Austin (USA) and then joined the Department of Chemistry at King Saud University as an Assistant Professor. His research interests are porous organic polymers, porous nanomaterials, metal–organic frameworks and electrocatalysis for applications in hydrogen production, sensors, supercapacitors, energy storage and bioapplications. He is a leading investigator in Advanced Polymers & Hybrid Nanomaterials Research and is also a lifetime member of International Scientific Societies. |
 Shengqian Ma | Shengqian Ma obtained his B.S. degree from Jilin University, China in 2003 and went on to obtain his Ph.D. from Miami University (Ohio) in 2008. After finishing a two-year Director's Postdoctoral Fellowship at Argonne National Laboratory, he joined the Department of Chemistry at the University of South Florida (USF) as an Assistant Professor in August 2010. He was promoted to an Associate Professor with early tenure in 2015 and to a Full Professor in 2018. His current research interest focuses on the development of functional porous materials including metal–organic frameworks (MOFs), covalent organic frameworks (COFs), and porous organic polymers (POPs) for energy, biological, and environmental-related applications. |
Introduction
The utilization of confined space for molecular purposes is one of the basic principles of living systems and is profoundly believed to be one of the vital steps in the origin of life. The biological process proceeds in the spatial confinement, commonly known as the enzyme pocket, to drastically influence the reactivity and selectivity of biochemical reactions. To mimic this, linking organic building blocks by strong covalent bonds to form crystalline materials is vital; however, one cannot neglect the possibility of thermodynamically favored amorphous disordered structures.1,2 In comparison to non-porous materials, porous materials have various characteristics such as a high surface area, nanometer-scale pores with peculiar properties and functions, tunable pore sizes, and chemical and thermal stability.3–11 Covalent organic frameworks (COFs) are porous, crystalline extended solids constructed from organic building units entirely composed of light elements (C, H, N, B, O) and connected by covalent bonds, which are robust and diverse in nature.12–19 The strength of covalent bonds is unambiguously reflected in the giant structure of Vitamin B12 and other solid networks existing in nature.20–22 In 2005, a revolution was made by successfully connecting boronic acid and catechol to form extended structures6 using the principle of dynamic covalent chemistry.23,24 Since then, the sustained expansion and development of COFs have been the answer to both long-lasting and newly recognized problems demanding new and evolving synthetic routes, characterization and applied studies.The development of elementary skeletons in frameworks primarily relies on the geometry of the building blocks. In 2D COFs, the building blocks are covalently bonded with a well-defined stacking of π-building units under the influence of π–π stacking interactions, whereas the lattice structure of 3D COFs is preserved by robust covalent bonds between precursor units using the principle of reticular chemistry.25–29 The uniquely confined nanometer-sized pores have practical implementation as an outstanding platform for future challenges. With this goal in mind, it is important to examine the peculiar properties of COFs, starting with crystallinity. Due to the ordered structure, crystallinity enables characterization using powder X-ray diffraction patterns (PXRD). The porous nature is a result of the crystalline structure and the uniform and periodic assembly of monomer units exhibiting surface areas as high as 4000 m2 g−1. Chemical/thermal stability (>400 °C) emerges from the robust and diverse nature of the covalent bonds, which resist severe conditions such as hydrolysis, oxidation, reduction, and pH range variations. Also, the low density, by virtue of uniform composition with lightweight elements allows a high gravimetric performance. De novo synthesis, to a lesser extent, suffers from various challenges such as active metal complexation, chemical conversion, tedious synthetic and purification procedures, etc. However, in order to address these challenges the tunable pore size of pristine frameworks must undergo pore surface engineering using established molecular organic chemistry to tune the structural and functional properties of COFs. In this review, we will briefly mention the extraordinary role of dynamic covalent chemistry in synthesizing COFs of specific pore size and broadly summarize the principle and examples of pore surface engineering in frameworks along with their role in a vast number of applications such as drug delivery, catalysis, environmental remediation, gas adsorption and separation, sensing and energy storage.
Dynamic linkages
Since the advent of the first COF in 2005, the last decade has observed substantial development in COFs from the perspective of design, construction and application. There are ranges of linkages, as shown in Fig. 1, which have been employed to construct extended frameworks of different pore structures and morphologies. Unlike traditional interactions, dynamic covalent bonds possess error-checking capabilities, and the robustness of reversible bonds bestow COFs with thermal stability, crystallinity, diversity, etc. The chemical stability and crystallinity are two peculiar properties that are dependent and come at the expense of others. In recent years, numerous thematic perspectives and reviews have discussed16,30–37 in great detail the role of dynamic covalent chemistry in constructing frameworks of different linkages under the umbrella of reticular chemistry, and their implementation in different pore sizes, pore volumes and dual pore frameworks. We are not trying to duplicate those aspects, however, we have briefly highlighted different linkages and pore geometries present in the COFs to create cohesion for a better understanding of the central theme of this review.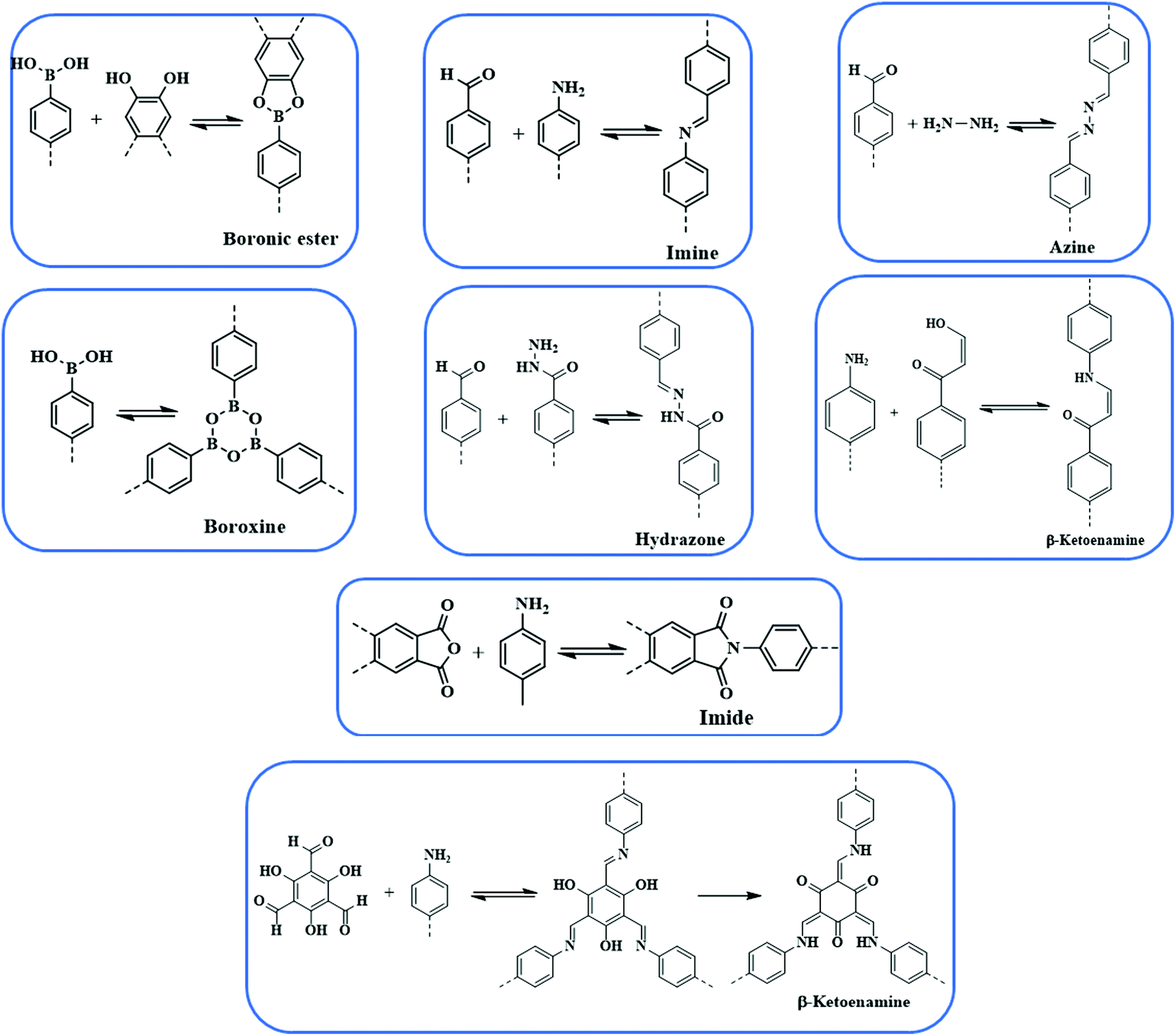 | ||
| Fig. 1 Different condensation reactions used for COFs synthesis. | ||
B–O linkages
Yaghi and co-workers reported the first class of boroxine-based frameworks6,38 (COF-1, COF-102) by the self-condensation of boronic acid-based building units, and the co-condensation of the same building blocks with catechol generates the boronate ester linkage framework (COF-5, COF-105). Due to the vulnerability of the B–O linkage, the frameworks hydrolyze in moisture. The Dichtel research group also highlighted the dissociation of HHTP-DPB COF in aqueous solution within minutes.39 To enhance the stability, Yaghi's group introduced the B–O–Si linkage in a borosilicate-linked framework (COF-202) prepared by the reaction of silanols with boronic acid, showing stability in moisture ranging from minutes to days.40 Additionally, the stability of frameworks in water and basic medium was visualized in spiroborate-linked ionic COFs by varying the hybridization of boron by incorporating alkali metal ions.41C–N linkages
Yaghi's research group designed COF-300 by using dynamic imine linkage, which in comparison to the boron-linked framework, exhibited higher stability in water, protic, aprotic solvents and entire pH ranges.42 To further elevate the stability of imine-based COFs, Banerjee and co-workers reported43 TpPa-1 and TpPa-2 by the reaction of 1,3,5-triformylphloroglucinol with p-phenylenediamine and 2,5-dimethyl-p-phenylenediamine, respectively. The stability mainly arose from aldimine condensation followed by irreversible tautomerism from enol–imine to keto–enamine.44–46 The Jiang research group reported47,48 an azine-linked framework (–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N–NC–) by the condensation reaction of azine and a formyl-substituted scaffold. In recent years, squaraine-based and viologen-based COFs were also reported to highlight the pivotal importance of C–N linkages.49
N–NC–) by the condensation reaction of azine and a formyl-substituted scaffold. In recent years, squaraine-based and viologen-based COFs were also reported to highlight the pivotal importance of C–N linkages.49
C–C linkages
The Hecht research group50 reported the first-ever C–C linked framework using the Ullmann coupling reaction and thereafter, Knoevenagel condensation was employed to construct the framework.51–55 Jiang and co-workers synthesized an sp2 carbon-conjugated framework by the condensation of nitrile-functionalized precursors such as 1,4-phenylenediactonirtrile with formyl substituted monomers.56 Recently, Yaghi's group reported a –CC– linked framework, with extraordinary stability in Brønsted acid or base, by the condensation of 2,4,6-trimethyl-1,3,5-triazine (TMT) and 4,4′-biphenyldicarbaldehyde (BPDA) via aldol condensation.57
Apart from these commonly used linkages in constructing COFs, borazine-, azodioxide- and hetero-linked frameworks were also reported.58–61 In addition to the linkage used to construct the framework, synthetic methods, such as solvothermal,38,42 ionothermal,62 microwave,63,64 mechanochemical,45,46 interfaces,65,66 and room temperature,67,68 also play a pivotal role in the synthesis and various properties such as crystallinity and the surface area of the synthesized frameworks.
Pore design
Reticular chemistry goes back to the work first highlighted by noble laureate Alfred Werner69 in the discovery of complexes such as β-Co(4-methylpyridine)4(NCS)2 and, later in 1959, the first crystalline coordination networks were reported with metal-neutral ligand linkages.70,71 Reticular chemistry is implemented in COFs from two standpoints, namely, the backbone of the framework, and the space comprised by the framework. These outlooks primarily depend on the linkage and symmetricity of building units, as shown in Fig. 2. The systematic pore network in COFs assists in the inclusion of various guest molecules under non-covalent interactions; therefore, the geometrical configuration of building blocks is of profound importance to translate the shape and size-selective ingress of molecules. Jiang's group underlined the relationship between the pore structure and building units in a topological diagram, where hexagonal, tetragonal, trigonal and rhombic structures have been widely employed.30,72 COF-6 exhibited the smallest pore (0.87 nm)73 whereas, PC-COF in 2016 showed the largest pore (5.8 nm) reported so far.74 Other significantly large pores that have been previously highlighted are HHTP-DPB COF39 (4.7 nm), BTP-COF64 (4.0 nm), and DTP-ANDI COF75 (5.3 nm).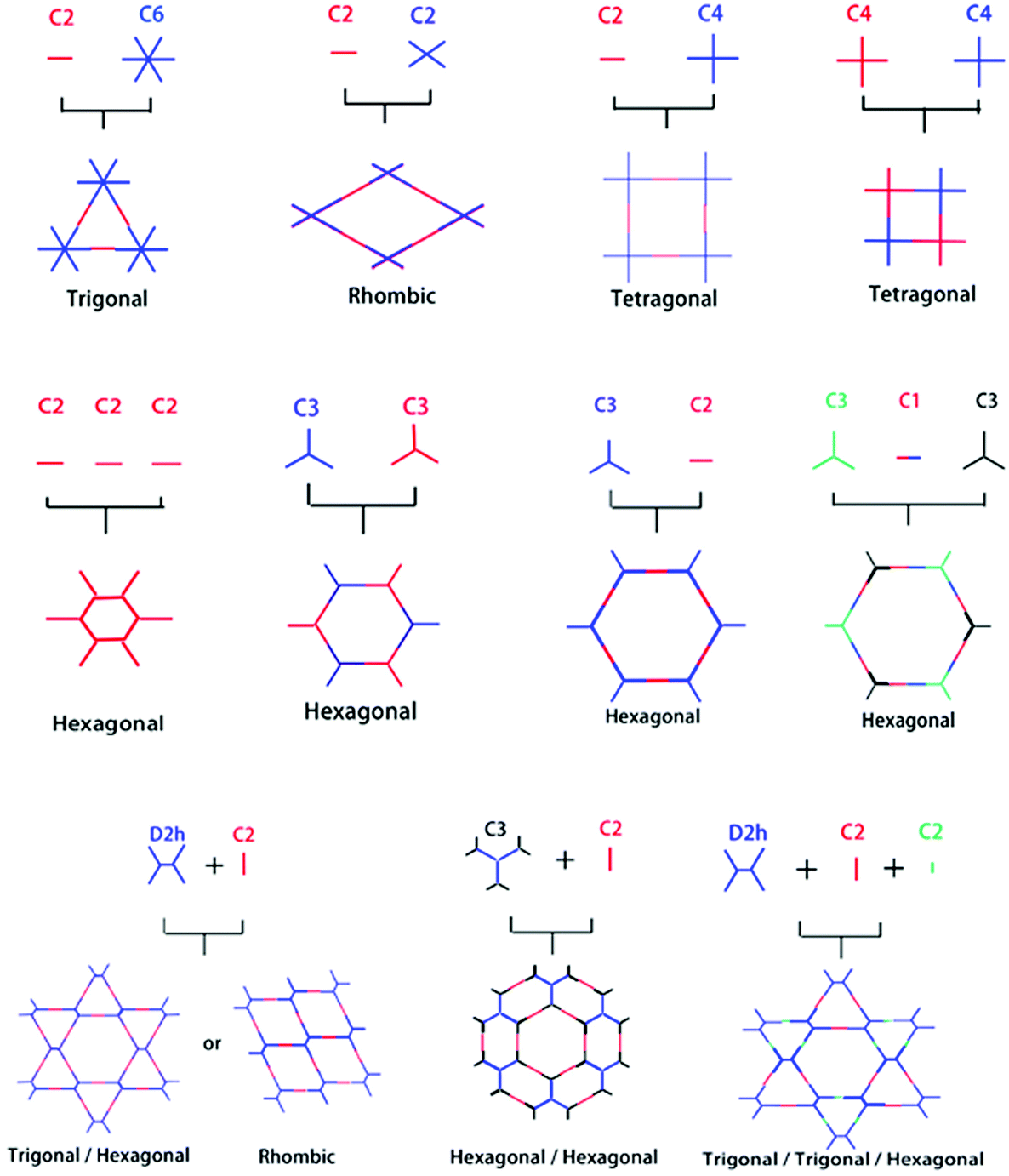 | ||
| Fig. 2 Organic building block geometries and the resulting COF structural patterns. Reproduced with permission from ref. 35 (Copyright © 2018, MDPI). | ||
The tunable pore size of the framework highlighted by the Lavigne research group76 by incorporating methyl, ethyl and propyl functionalities into frameworks experienced substantial decreases in surface area and pore size to 1.6 nm, 1.4 nm and 1.1 nm, respectively, thereby implying that the decrease in pore size is directly proportional to the size of the substituents (–Me < –Et < –Pr). Jiang and co-workers further highlighted alkenyl, alkynyl, and azide-decorated frameworks and observed a decrease in pore size in comparison to pristine frameworks.77 Moreover, the Zhao research group78 reported dual pores and three different kinds of pores by using the D2h symmetric 4,4′,4′′,4′′′-(ethane-1,1,2,2-tetrayl) tetraaniline building block (ETTA). Along with this, Yaghi, Wang, Bein and other research groups reported heterogeneous pore structures and variations in pore shape with the change in the building block concentration.17,79–84
Characterization
COFs synthesized by different methodologies are typically characterized using diverse physiochemical techniques to determine and understand their properties. The range of techniques widely used for the characterization of frameworks is briefly described, starting with powder X-ray diffraction (PXRD). PXRD is performed to determine the bulk crystallinity of frameworks. The structure identification can be done by comparing the experimental diffractogram of the frameworks with patterns calculated using the AA stacking and AB stacking models generated by computational modeling. The model structure undergoes Pawley refinement to optimize the lattice parameters iteratively until the RWP value converges, thereby providing crystallographic parameters such as lattice parameters and unit cells, and make it possible to distinguish between amorphous and crystalline materials. This is basic characterization, yet it is of profound importance and is extensively used to verify the synthesis of existing and novel COFs. Secondly, N2 adsorption isotherms at 77 K are used to determine various parameters such as nitrogen adsorption and desorption isotherms, pore-volume, pore-size distribution, and surface area. To investigate these textural parameters, the sample should be properly activated. In general, porous materials are usually activated by Soxhlet extraction, solvent exchange, supercritical drying and vacuum drying. Furthermore, the amount of sample is of key importance; to obtain infallible data, the weight of the sample (g) multiplied by the specific surface area (m2 g−1) should be equal to 100 m2 or more. In general, the physisorption isotherms are divided into six types of different shapes, which imply either micropores, mesopores or macropores. Thirdly, thermogravimetric analysis (TGA) measures the mass loss as a function of temperature in a controlled atmosphere. This technique helps in determining the thermal stability of the COFs; for instance, TGA data of imine-linked frameworks usually indicates thermal stability up to 350 °C. Generally, the weight loss of the COFs vs. temperature (°C) highlights solvent molecule loss at 50–100 °C and a second loss implies structural modification and the collapse of frameworks (350–450 °C). Fourthly, scanning electron microscopy (SEM) and transmission electron characterization (TEM) are used to examine the morphology, dispersion and mixture of phases, and the particle sizes of frameworks. The sample undergoes pretreatment by gold, coating the surface of conducting materials before SEM analysis. These microscopies can be coupled with Energy Dispersive Spectroscopy (EDS) or Energy Dispersive X-ray Analysis (EDX), which help in determining the elemental quantitative and qualitative composition of COFs. Fifthly, 13C cross-polarization magic-angle spinning (CP-MAS) is used to determine the purity of the COF and unreacted organic linkers. This helps in elucidating the structure, identity or chemical state of the frameworks; for instance, imine-linked frameworks showed –CN moieties at 159 ppm in 13C MAS NMR. These observations were further confirmed by using Fourier Transform-Infrared Spectroscopy (FT-IR), by the emergence of imine functionalities and the disappearance of carbonyl moieties. Lastly, the aqueous and chemical stability of COFs is determined. The stability is tested by soaking the COFs in water, protic, aprotic, acidic and basic solutions for the requisite amount of time. The solutions are left undisturbed at specific temperatures and time, followed by filtration and reactivation, and the material is then compared with pristine frameworks by means of PXRD, FT-IR and surface area measurements. Depending on the nature of the COFs, Ultraviolet–Visible Spectroscopy, Inductively Coupled Plasma Optical Emission Spectroscopy (ICP-OES) and photoluminescence spectroscopy techniques are also widely used.
Pore surface engineering
The robust interaction between uniformly distributed binding functionalities in COFs and metal ions such as Pd, V, Rh, Re, Cu under the principle of coordination chemistry is one of the common ways to impart catalytically active moieties in frameworks. COFs with bipyridine and bicatechol moieties are at the forefront of linking metal ions due to their donor ability to form σ dative bonds. It is worth pointing out that metal binding in between the nitrogen (Schiff base COFs) of the adjacent layers of the framework further helps to considerably increase the active catalytic sites. The functional groups directly linked to the organic scaffold provide anchimeric assistance along with imine nitrogens to combine with foreign species, which include well-known systems such as iminophenol, benzoyl hydrazone, salen, β-ketoenamine, etc. In addition, porphyrin-incorporated frameworks bestow inherent coordinative, redox and spectral features. The presence of four pyrrolic units allows binding with most elements present in the periodic table;85,86 Fe–porphyrin and Co–porphyrin-based COFs, for instance, showed peroxidase-like activity87 and electrocatalytic reduction of CO2,88 respectively. Planar triangular macrocycles, such as dehydrobenzoannulene derivatives (DBAs), are well-known to bind with low oxidation state transition metals.89 DBA, being neutral and possessing soft binding functionalities (alkyne) as compared to hard nitrogen atoms, has different reaction patterns under the HSAB rule; for instance, DBA-based COFs metalated with Ni(0) exhibited hybrid material with an exceptional uptake of ethylene and ethane.90Imine-linked frameworks are at the forefront in the study of pore surface engineering due to extraordinary thermal and chemical stability in moisture, protic and aprotic solvents. The Wang research group synthesized91 two-dimensional COF-LZU1 by the condensation of 1,3,5-triformylbenzene and p-phenylenediamine in the presence of acetic acid, demonstrating excellent crystallinity and surface area (410 m2 g−1). COF-LZU1 adopted the eclipsed structure with an adjacent layer distance of 0.37 nm and was susceptible to binding with palladium acetate, Pd/COF-LZU1 (Fig. 3). The well-preserved crystalline framework showed a small decrease in surface area and 0.7 eV negative shift in X-ray photoelectron spectroscopy (XPS), in comparison to 338.4 eV for free Pd(OAc)2. These results in totality affirm the pore surface engineering with a regular channel diameter of 1.8 nm; on exposure to the ingress of electronically and sterically bulky molecules, Pd/COF-LZU1 catalyzed Suzuki–Miyaura coupling reactions with extraordinary yields of 96% to 98% and excellent recyclability and reusability. Five years later, the Li research group used the same hybrid material, Pd/COF-LZU1 annealed at 500 °C to generate palladium nanoparticles encapsulated in the shell of nitrogen-doped hollow carbon material.92 The Pd(0) hybrid framework with similar morphology served as an efficient catalyst for the hydrogenation of nitroaromatics to amine, which is far superior to Pd(0) and other commercial carbon material.
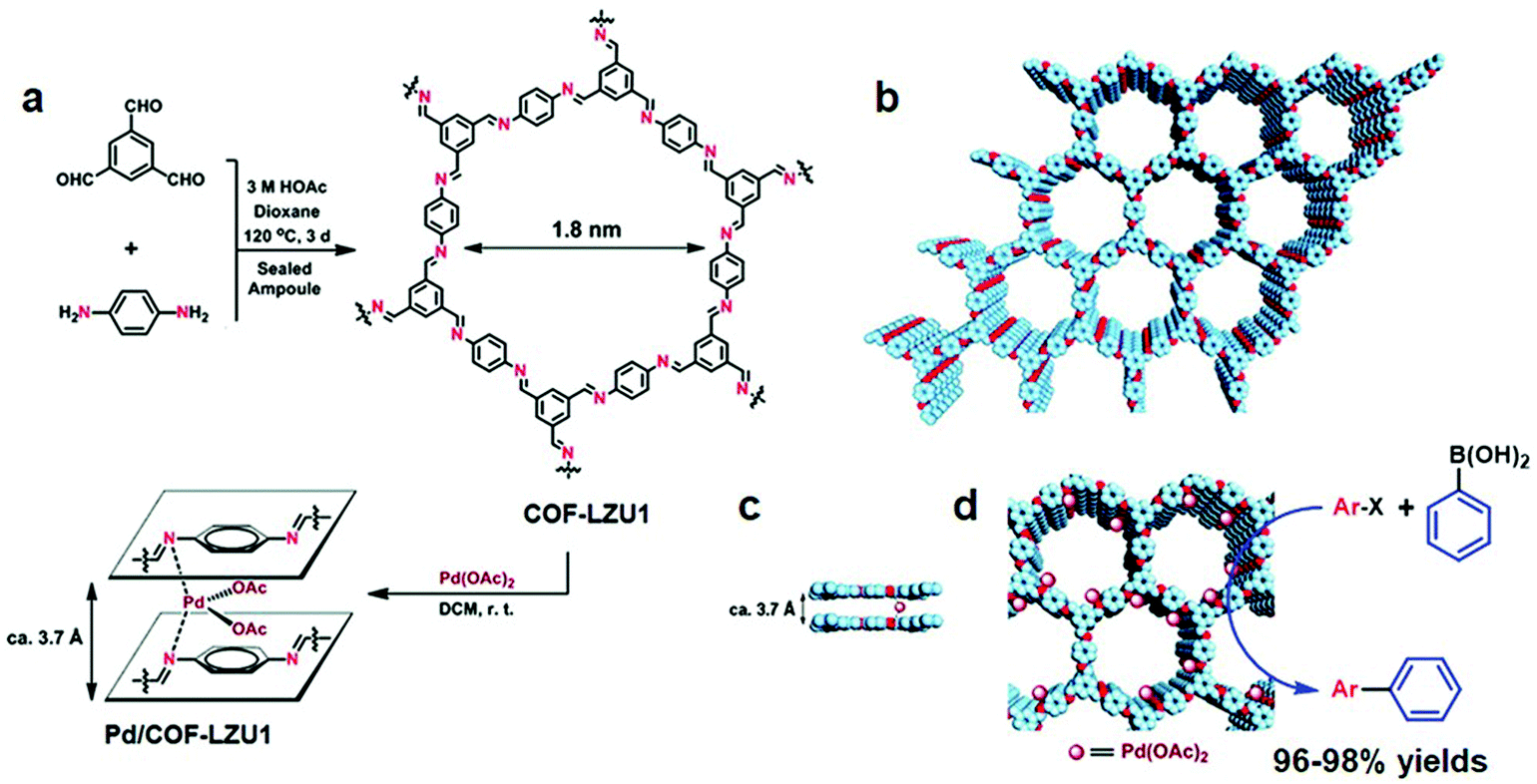 | ||
| Fig. 3 (a) COF-LZU1 and Pd/COF-LZU1 synthesis. (b–d) Proposed 2D COF-LZU1 and Pd/COF-LZU1 layered structures with Pd immobilization. Adapted with permission from ref. 91 (Copyright © 2011, American Chemical Society). | ||
In 2018, the Kim research group combined the chemistry of two porous materials, –NH2–functionalized MOF and COF-LZU1 with the impregnation of Pd(OAc)2. The XPS spectrum showed the binding of palladium with imine nitrogen, which was very similar to Pd/COF-LZU1; the hybrid Pd2+/MOF@COF material exhibited photocatalytic tandem dehydrogenation and hydrogenation reactions.93 Sticking to the pore surface engineering of COFs with palladium, the Jiang research group reported the successful immobilization of palladium into porphyrin-containing imine-linked COF and employed it in an effective Suzuki-coupling reaction under mild conditions with 97%–99% yield.94 Gao and co-workers demonstrated accuracy and precision control of the number and position of catalytically active centers. To highlight this control,95 Py-2,2′-BPyDCA COF was synthesized by the condensation of 4,4′,4′′,4′′′-(pyrene-1,3,6,8-tetrayl) tetraaniline (PyTTA) and 2,2′-bipyridine-5,5′-dicarbaldehyde (2,2′-BPyDCA)/4,4′-biphenyldialdehyde in different stoichiometric ratios. Using this planned synthetic method, the bulky Rh(COD)Cl (7.4 × 6.6 × 5.3 Å) was first coordinated with bipyridine functionalities, followed by palladium, resulting in the bimetallic RhI/PdII@BPy-COF for one-pot addition–oxidation cascade reactions (Fig. 4). Using the same principle, a similar bimetallic framework was prepared by docking MnCl2 followed by Pd(OAc)2 to Py-2,2′-BPyDCA COF to afford MnII/PdII@BPyCOF, with Mn(II) and Pd(II) content of 7.9 wt% and 0.8 wt%, respectively, which catalyzed the Heck reaction between styrene and iodobenzene and the subsequent epoxidation of trans-stilbene to trans-stilbene oxide.96
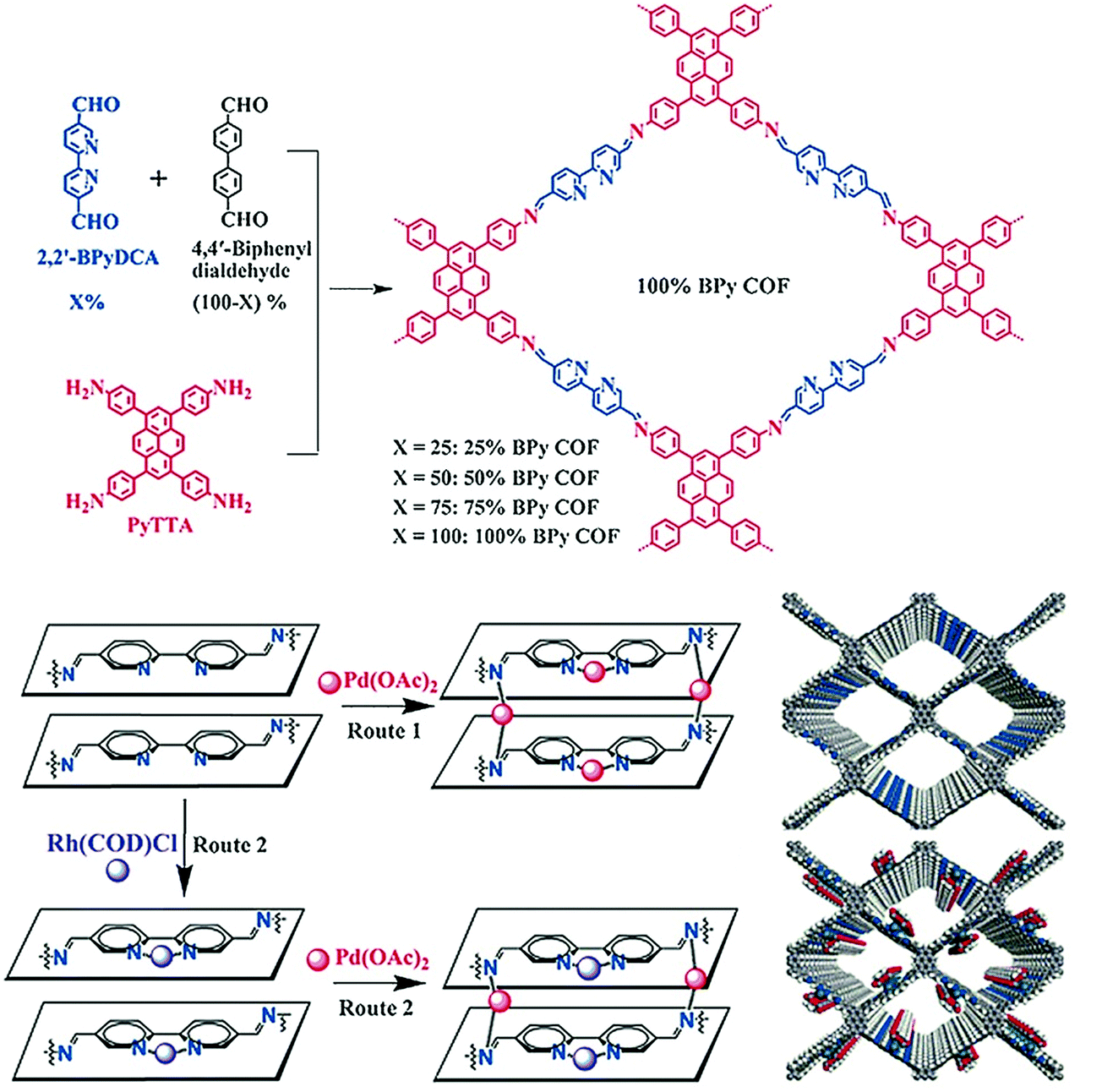 | ||
| Fig. 4 Synthesis of X%-BPy COFs and routes of pore surface engineering with rhodium and palladium. Reproduced with permission from ref. 95 (Copyright © 2016, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim). | ||
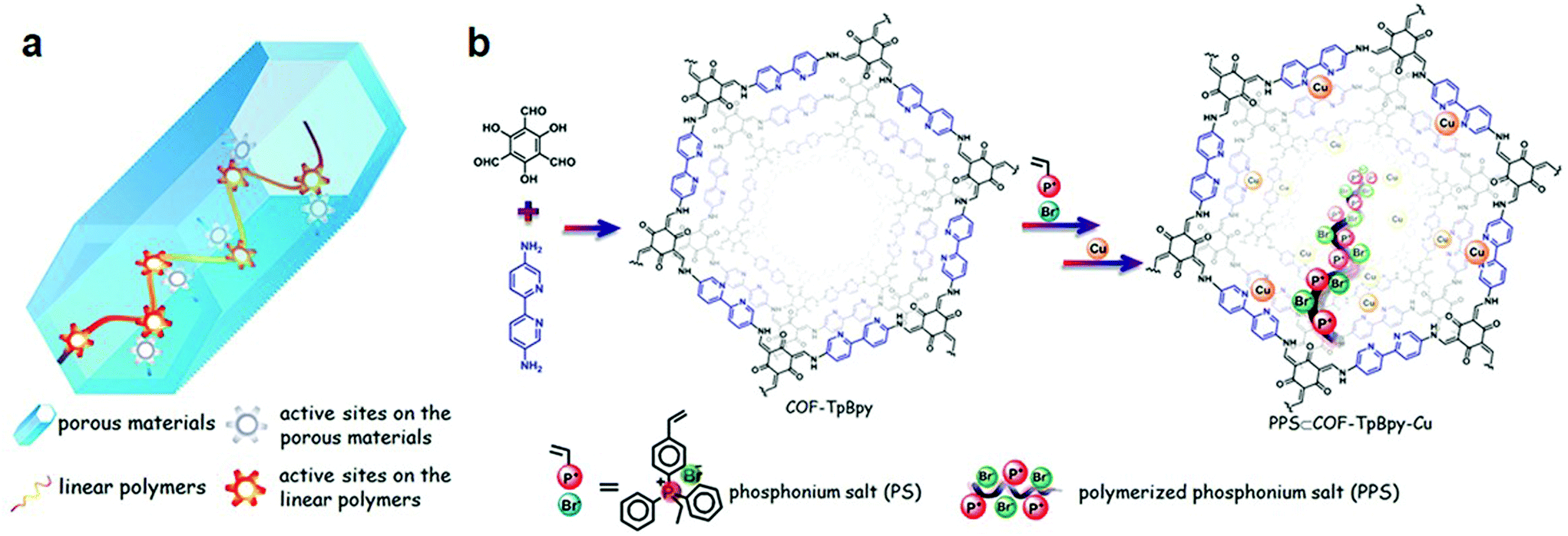
Pd(II)/COF-SDU1 was synthesized by the palladium-based pore surface engineering of the SDU1 framework prepared by the condensation of 1,4-phenylenediamine and tri(4-formacylphenoxy)-1,3,5-triazine. The hybrid framework was characterized using physiochemical analysis and was employed as an effective heterogeneous catalyst for the one-pot cross-coupling reaction between phenylsilane and a range of electronically substituted iodotoluenes with excellent recyclability.97 Pachfule et al. reported the in situ generation of Pd nanoparticle in the pores of β-ketoenamine TpPa-1 COF constructed by the condensation of 1,3,5-triformylphloroglucinol and 1,4-phenylenediamine. The hybrid Pd(0)@TpPa-1 catalyzed coupling reactions, the one-pot sequential Heck Sonogashira coupling reaction and intramolecular oxidative coupling.98 Iron, cobalt and nickel are among other transition metal ions incorporated into COFs through systematic pore surface engineering. RT-COF-1 was synthesized99 by the reaction of 1,3,5-tris(4-aminophenyl) benzene and 1,3,5-benzenetricarbaldehyde at ambient temperature in m-cresol. The treatment of RT-COF-1 with M(acac)n {M = Fe(III), Co(II), Ni(II); acac = acetylacetonate} in methanol at room temperature for 24 hours produced a metal-incorporated framework with different metal to nitrogen ratios. As shown by total X-ray fluorescence (TXRF), the atomic ratios of FeIII![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :N, CoII:N, and NiII:N were 1:280, 1:190 and 1:760, respectively, which are smaller as compared to Pd, Rh, and Mn-docked frameworks.100 The dramatic fluorescence quenching after metal docking due to metal-to-ligand charge transfer further affirmed the successful immobilization. The Banerjee research group reported101 cobalt immobilization in β-ketoenamine-linked COF-TpBpy prepared by the condensation reaction of 5,5′-diamino-2,2′-bipyridine and 1,3,5-triformylphloroglucinol. The solution-infiltration method of pore surface engineering imparts ∼12% cobalt content as investigated by ICP-OES. XPS, EDX, FT-IR analysis further confirmed the coordination of cobalt species to bipyridine moieties without any proof of linking with imine nitrogen. The crystalline Co-TpBpy framework with accessible surface area of 450 m2 g−1 acted as the OER catalyst for up to 1000 cycles. The same framework was used by our research group102 to highlight the role of the degrees of freedom within the pore channels of frameworks to accommodate linear ionic polymers and active Lewis acidic copper sites (Fig. 5). The two reactive centers in the hybrid PPS & COF-TpBpy-Cu worked together for the effective catalytic conversion of the sterically and electronically-substituted epoxide to the cyclic carbonate using CO2, in comparison to the pristine framework, ionic polymer or copper salt. The boost in cooperation remained active in the hybrid framework even after ten catalytic cycles without any drastic changes in activity and crystallinity.
:N, CoII:N, and NiII:N were 1:280, 1:190 and 1:760, respectively, which are smaller as compared to Pd, Rh, and Mn-docked frameworks.100 The dramatic fluorescence quenching after metal docking due to metal-to-ligand charge transfer further affirmed the successful immobilization. The Banerjee research group reported101 cobalt immobilization in β-ketoenamine-linked COF-TpBpy prepared by the condensation reaction of 5,5′-diamino-2,2′-bipyridine and 1,3,5-triformylphloroglucinol. The solution-infiltration method of pore surface engineering imparts ∼12% cobalt content as investigated by ICP-OES. XPS, EDX, FT-IR analysis further confirmed the coordination of cobalt species to bipyridine moieties without any proof of linking with imine nitrogen. The crystalline Co-TpBpy framework with accessible surface area of 450 m2 g−1 acted as the OER catalyst for up to 1000 cycles. The same framework was used by our research group102 to highlight the role of the degrees of freedom within the pore channels of frameworks to accommodate linear ionic polymers and active Lewis acidic copper sites (Fig. 5). The two reactive centers in the hybrid PPS & COF-TpBpy-Cu worked together for the effective catalytic conversion of the sterically and electronically-substituted epoxide to the cyclic carbonate using CO2, in comparison to the pristine framework, ionic polymer or copper salt. The boost in cooperation remained active in the hybrid framework even after ten catalytic cycles without any drastic changes in activity and crystallinity.
 | ||
| Fig. 5 (a) Porous heterogeneous concerted catalyst with flexible polymer and metal active sites. (b) Schematic representation of the PPS⊂COF-TpBpy-Cu synthesis from COF-TpBpy, PPS and Cu. Adapted with permission from ref. 102 (Copyright © 2016, American Chemical Society). | ||
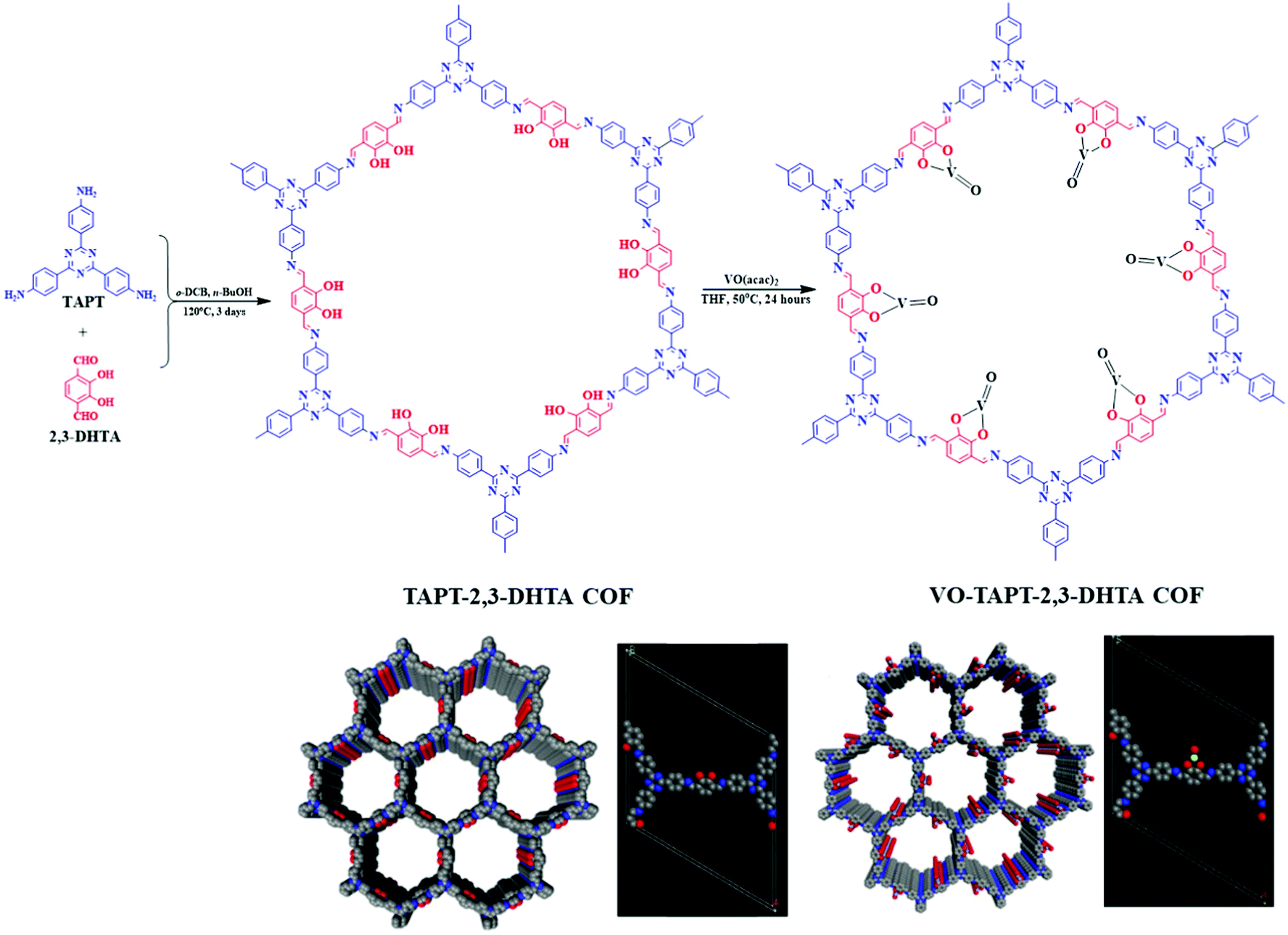
The pivotal role of bipyridine functionalities in imine-linked COFs for pore surface engineering is extraordinary. Similar attention was also drawn by the pendant hydroxyl groups attached to the building blocks distributed uniformly in the COFs. Chen and co-workers103 synthesized hydroxyl functionalized crystalline and porous frameworks by the dynamic imine linkage of 2,5-dihydroxyterephthalaldehyde and 1,3,5-tris(4-aminophenyl) triazine in o-dichlorobenzene, n-butanol and a catalytic amount of 3 M acetic acid. Due to the presence of pendant moieties and stacked structures, the authors further illustrated pore surface engineering using Cu(OAc)2; Cu(II) was coordinated by dangling hydroxyl groups and imine nitrogen with similar coordination environments to those extensively reported in homogeneous counterparts. Cu-COF was explored as a heterogeneous catalyst for the selective oxidation of alkene in the presence of hydroperoxide with more than 75% conversion and can be reusable for at least three catalytic cycles without any significant changes in crystallinity and morphology. Salen-based ligands have a special place in the field of coordination chemistry.104,105 Ding, Wang and co-workers illustrated the incorporation of these moieties into COFs by synthesizing Salen-COF via the condensation of ethylenediamine and C3 symmetric 1,3,5-tris[(5-tert-butyl-3-formyl-4-hydroxyphenyl)ethynyl] benzene. The crystalline nature of the framework was well-preserved in the entire pH range, and in protic and aprotic solvents. Furthermore, salen pockets in pore channels underwent pore surface engineering using Co(OAc)2, Cu(OAc)2, Ni(OAc)2, Zn(OAc)2 and Mn(OAc)2 to form metallo-salen-based COFs. Co/salen COF exhibited excellent catalytic activity for the Henry reaction using nitro alkane and aldehyde or ketone in the presence of base with recyclability and reusability for up to four catalytic cycles.106 Our research group demonstrated vanadium docking in a catechol functionalized TAPT-2,3-DHTA framework by the reaction of VO(acac)2 in THF at 50 °C (Fig. 6). Electron paramagnetic resonance (EPR) spectroscopy and XPS affirmed vanadium immobilization in VO-TAPT-2,3-DHTA COF with no changes in crystallinity and a surface area of 562 m2 g−1. The recyclable hybrid material exhibited catalytic activity for the Prins reaction (nopol formation) and substituted sulfide oxidation with 69% and 90% yield, respectively.107 Moreover, VO-TAPT-2,3-DHTA and VO-PyTTA-2,3-DHTA COFs demonstrated excellent catalytic efficiency for Mannich-type reactions with 75%–99% yield for Mannich bases formed from a range of electronically and sterically-substituted alcohols and 4-methylmorpholine N-oxide (NMO)/trimethylamine N-oxide (TMNO).108
 | ||
| Fig. 6 Synthesis and stacked structure of TAPT-2,3-DHTA COF and pore surface engineered VO-TAPT-2,3-DHTA COF. Reproduced with permission from ref. 107 (Copyright © 2019, American Chemical Society). | ||
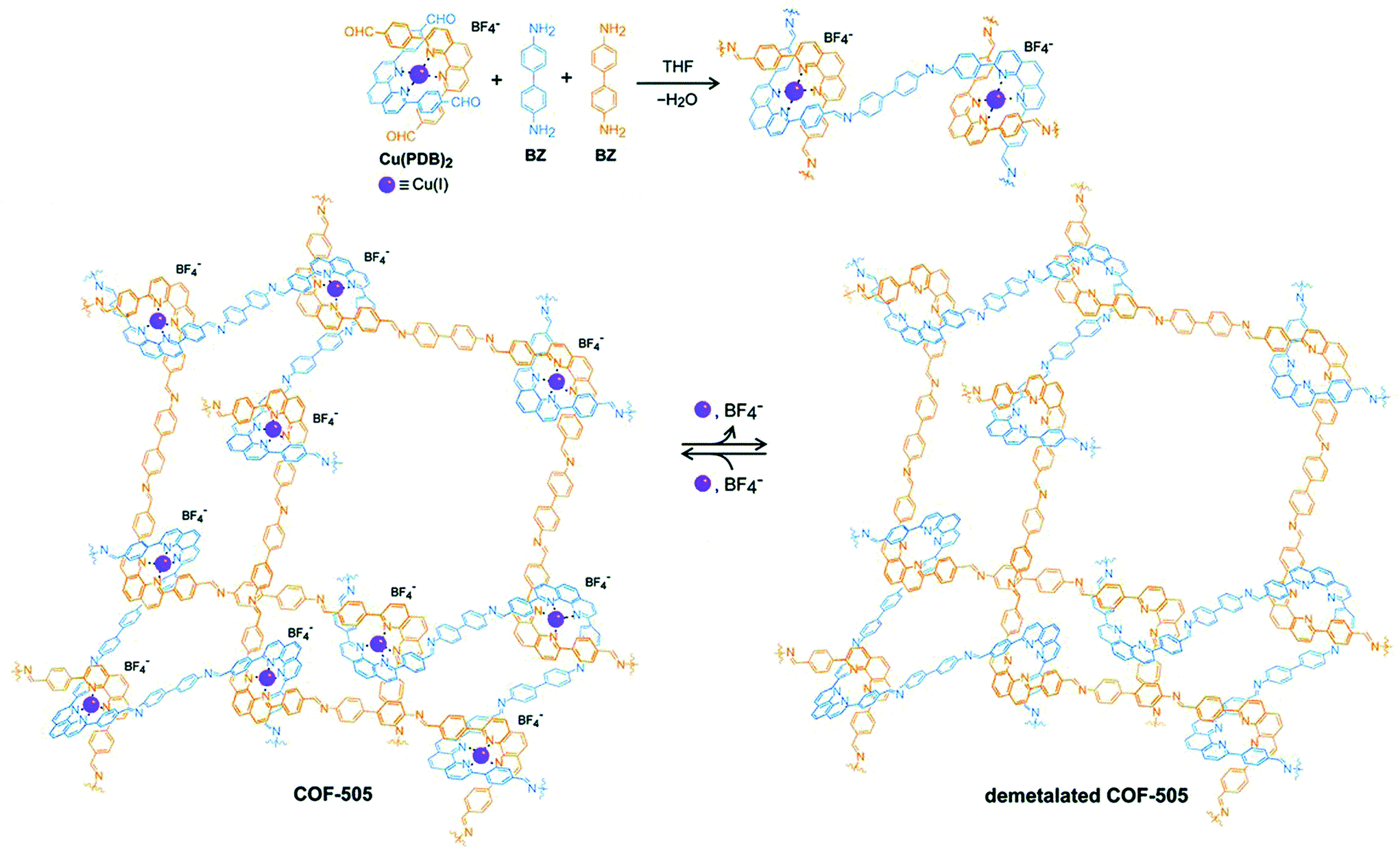
The planar and rigid nature of porphyrin building blocks with inherent abundantly functionalized nitrogen led to stabilized COFs due to π–π stacking interactions between adjacent layers. In 2017, the Star research group87 synthesized bifunctional Fe-DhaTph COF by two methods. The first involved the pre-metalation of 5,10,15,20-tetrakis(4-aminophenyl) porphyrin with FeCl2·2H2O, followed by condensation with 2,3-dihydroxyterephthalaldehyde; the second involved the post metalation of DhaTph COF with Fe(III). Recently, Marinescu and co-workers synthesized bimetallic frameworks both pre and post metalation strategies. COF-Re was prepared by dynamic imine linkage between Re-2,2′-BPyDCA and 5,10,15,20-tetrakis(4-aminophenyl) porphyrin, followed by pore surface engineering using cobalt and iron chloride, resulting in COF-Re_Co and COF-Re_Fe, respectively.88 The PXRD patterns illustrated that both bimetallic frameworks preserved their crystallinity during modification. Furthermore, XPS analysis revealed that peaks for both metals were present in the COFs highlighting the successful incorporation of metals with distinct properties. With 53.4 wt% of cobalt in COF-Re_Co, the framework exhibited CO2 reduction to CO with Faraday efficiency of 18%. In addition, phenanthroline moieties in copper(I)-bisphenanthroline tetrafluoroborate and benzidine afforded 3D COF-505 by mutual weaving at regular intervals.109 By definition, COFs are composed of organic building blocks; therefore, pore surface engineering is reflected in the removal of copper ions by heating COF-505 in KCN CH3OH–H2O solution, with approximately 92%–97% removal as determined by ICP analysis (Fig. 7). PXRD and SEM exhibited slight decreases in crystallinity and similar morphology in comparison to the pristine framework. Notably, the authors explained metalation and remetalation and, more importantly, this work provides alternatives to porphyrin, bipyridine, and hydroxyl moieties in the COFs community. 3D-metalated COFs restrict the accessibility challenges facing chemists in the production of symmetric monomers to form porous material with distinct properties. The McGrier research group reported crystalline DBA-3D-COF, by the solvothermal reaction of dehydrobenzoannulene (DBA) and tetra(4-dihydroxyborylphenyl) methane, with a uniformly distributed π-conjugated planar ring. The soft neutral π-ligands donated one-two electron pair per alkyne to form Ni-DBA-3D-COF with complete preservation of crystallinity. More importantly, XPS analysis exhibited nickel binding with the alkynyl units in the zero oxidation state without any interactions with boronate ester linkages. In comparison to the pristine framework, the nickel-metalated framework observed ethane and ethylene sorption of 0.07 mmol and 0.13 mmol at 295 K, respectively.90
 | ||
| Fig. 7 Synthesis of COF-505 from organic threads using Cu(I) as the template to design an extended weaving structure with reversible demetalation and remetalation. Reprinted with permission from ref. 109 (Copyright © 2016, American Association for the Advancement of Science). | ||
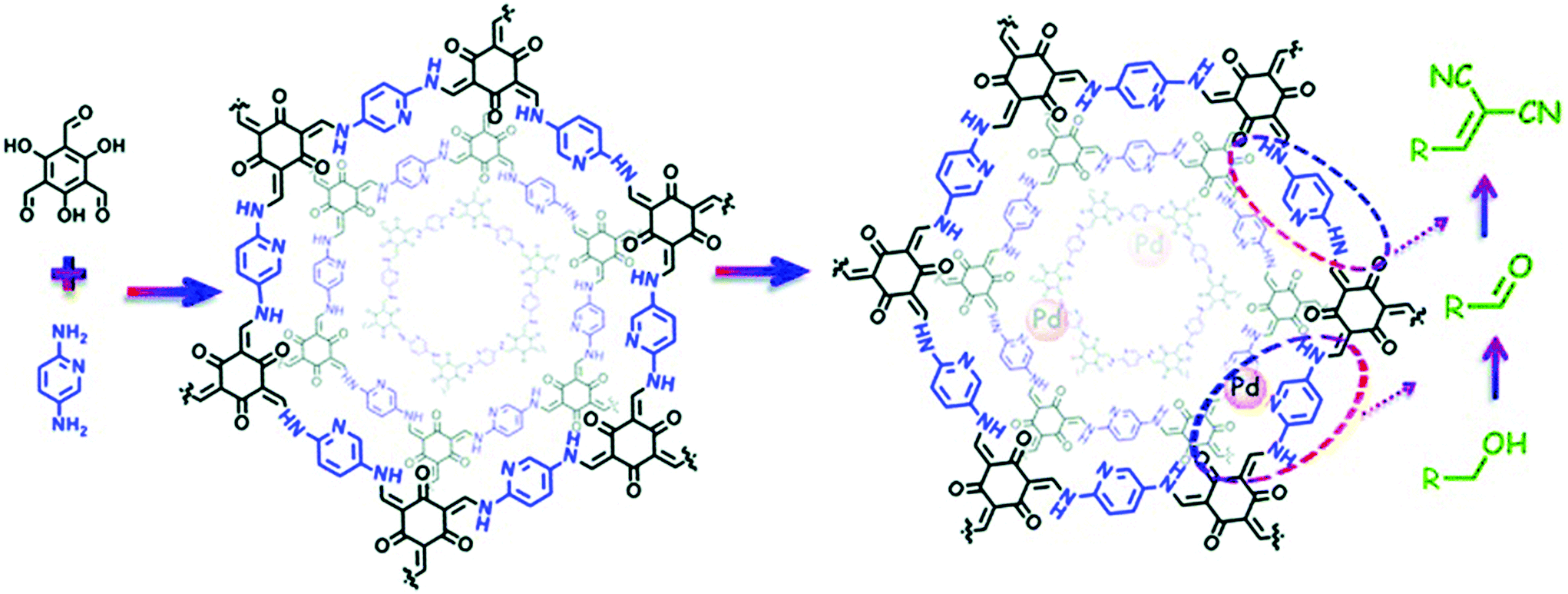
The extraordinary stability of β-ketoenamine COFs was extensively reported by Banerjee and co-workers and is usually prepared by the condensation of triformylphloroglucinol with a range of substituted amines. Shen, Zhang and co-workers used the structural and geometrical advantages of TpPa-2 COF43 for pore surface engineering with Ti4+ using Ti(SO4)2. The physicochemical analysis showed the complete retention of crystallinity and morphology with 33.49 wt% of titanium and the decorated TpPa-2-Ti4+ framework was used for phosphopeptide enrichment using tryptic digests of standard phosphoprotein β-casein.110 Our research group used the β-ketoenamine COF prepared by the condensation of triformylphloroglucinol and 2,5-diaminopyridine to examine the cascade catalysis after successful pore surface engineering by palladium using Pd(OAc)2 (Fig. 8). XPS revealed the palladium coordination with pyridinic and secondary amines of linkages in the framework with substantial weight percentage. The presence of two active palladium and basic nitrogen catalytic sites makes the hybrid framework suitable for the cascade catalyst for the oxidation–Knoevenagel condensation reaction from alcohols to unsaturated dinitriles with excellent recyclability.111 The Yuan and Zhu group extended the principle of pore surface engineering to study the gas adsorption behavior by synthesizing the carboxyl functionalized framework {(HOOC)x-COF, X = 17, 33, 50 and 100}. The TpPa-COOH framework was synthesized by the condensation of 2,5-diaminoterephthalic acid, 1,4-phenylendiamine and triformylphloroglucinol in different ratios, and compared with TpPa-1 prepared using 1,2-phenyldiamine. The simultaneous presence of proton-donating and proton-accepting groups for hydrogen bonding units reduced the ammonia uptake as compared to the pristine framework due to the decrease in pore size. Furthermore, the functionalized framework, on treatment with calcium chloride, manganese chloride and strontium chloride, led to the assimilation of the metal ions at carboxyl moieties, thereby serving as open metal sites. This synergism positively influenced the ammonia uptake of 14.3 mmol g−1, 19.8 mmol g−1 at 298 K and 273 K, respectively.112 In 2011, Yaghi and co-workers presented the first-ever report of hydrazone-linked, stable COFs;113 soon after, the Jiang research group exemplified the two-step bottom approach by the pore surface engineering of frameworks synthesized by the condensation of triformylphloroglucinol with terephthalohydrazide and substituted terephthalohydrazide using MoO2(acac)2.114 The strong salicylaldehyde benzoyl hydrazone coordination with molybdenum was shown in XPS, FT-IR and TGA analysis. ICP analysis revealed the 2.0 mmol g−1 density of molybdenum, which made the framework catalytically active for the epoxidation of cyclohexene in 99% conversion and up to 70% selectivity. Using the same principle, the Zhao research group used hydroxyl-functionalized COFs, synthesized by triformylphloroglucinol and 2,5-dihydroxyterephthalohydrazide or 2,3-dihydroxyterephthalohydrazide, to incorporate cobalt in the +2 oxidation state as evidenced by XPS and TEM-elemental mapping. These served as Lewis acidic sites for the cyanosilylation of electronically and sterically-substituted aldehyde to form cyanohydrin with excellent recyclability.115
 | ||
| Fig. 8 The Pd-based pore surface engineering of COF-TpPa-Py for one-pot cascade catalysis. Reproduced with permission from ref. 111 (Copyright © 2017, The Royal Society of Chemistry). | ||
The pendant functional groups in the well-defined pore channels of frameworks undergo conversion using established organic chemistry, especially the addition and substitution reactions, which are overcome by tedious chemical synthetic and purification procedures. The click reaction is predominantly used to introduce different functional groups such as ester, alcohols, thiols and others. The Jiang research group reported the first-ever example of the copper-catalyzed azide–alkyne cycloaddition click (CuAAC) reaction.77 The 2D mesoporous boronate ester-linked COF-5 with uniformly distributed pendant azide functionalities at the walls of hexagonal skeleton was used for pore surface engineering via the click reaction with a range of substituted alkynes to form five-membered substituted frameworks. Notably, the percentage of azide moieties can be altered by varying the stoichiometric ratio of three components in a system. The same research group widened the possibility of this strategy by the condensation of hydroxyl-functionalized Zn-phthalocyanine, azide functionalized 1,4-benzenediboronic acid and 1,4-benzenediboronic acid. The framework underwent pore surface engineering using CuAAC to covalently bind electron-acceptor60 fullerene moieties, thereby facilitating photoenergy conversion.116 The CuAAC reaction is relatively difficult to execute in B–O-linked frameworks, but imine-linked frameworks are more prone to a similar fashion of pore surface engineering. The Jiang research group synthesized alkyne-functionalized COFs by the condensation of porphyrin/Ni-porphyrin with 2,5-bis(2-propynyloxy)terephthalaldehyde and 2,5-dihydroxy terephthalaldehyde. By virtue of the CuAAC reaction, the electronic environment of –OH, –COOH, –COOMe, –NH2 functionalized frameworks117 drastically changed; furthermore, the same group also highlighted the assimilation of polyradicals in the pore channel to induce the redox nature in the framework, which is difficult to execute via de novo synthesis.118 To extend this study, the C3-symmetric 1,3,5-tris(4-aminophenyl) benzene organic scaffold with the same aldehyde monomers employed in different ratios afforded [HC![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C]x%-TBP-DMTP-COFs. Pore surface engineering via CuAAC using differently functionalized azides, including chiral pyrrolidine, thiol, uracil, and 2,2,6,6-tetramethyl-1-piperidinyloxy (TEMPO) radicals, formed the corresponding modified frameworks (Fig. 9); in particular, the chiral frameworks serve as catalysts for asymmetric Michael reactions with high activity and enantioselectivity.119 The nucleobase-modified framework exhibited the selective recognition of complementary nucleobases in aqueous solution.120 In addition, the thiol engineered COF showed the selective capture of Hg2+ from 10 mg L−1 to 1.5 mg L−1 within a few minutes, which is well below the acceptable limit for drinking water (2 mg L−1).121
C]x%-TBP-DMTP-COFs. Pore surface engineering via CuAAC using differently functionalized azides, including chiral pyrrolidine, thiol, uracil, and 2,2,6,6-tetramethyl-1-piperidinyloxy (TEMPO) radicals, formed the corresponding modified frameworks (Fig. 9); in particular, the chiral frameworks serve as catalysts for asymmetric Michael reactions with high activity and enantioselectivity.119 The nucleobase-modified framework exhibited the selective recognition of complementary nucleobases in aqueous solution.120 In addition, the thiol engineered COF showed the selective capture of Hg2+ from 10 mg L−1 to 1.5 mg L−1 within a few minutes, which is well below the acceptable limit for drinking water (2 mg L−1).121
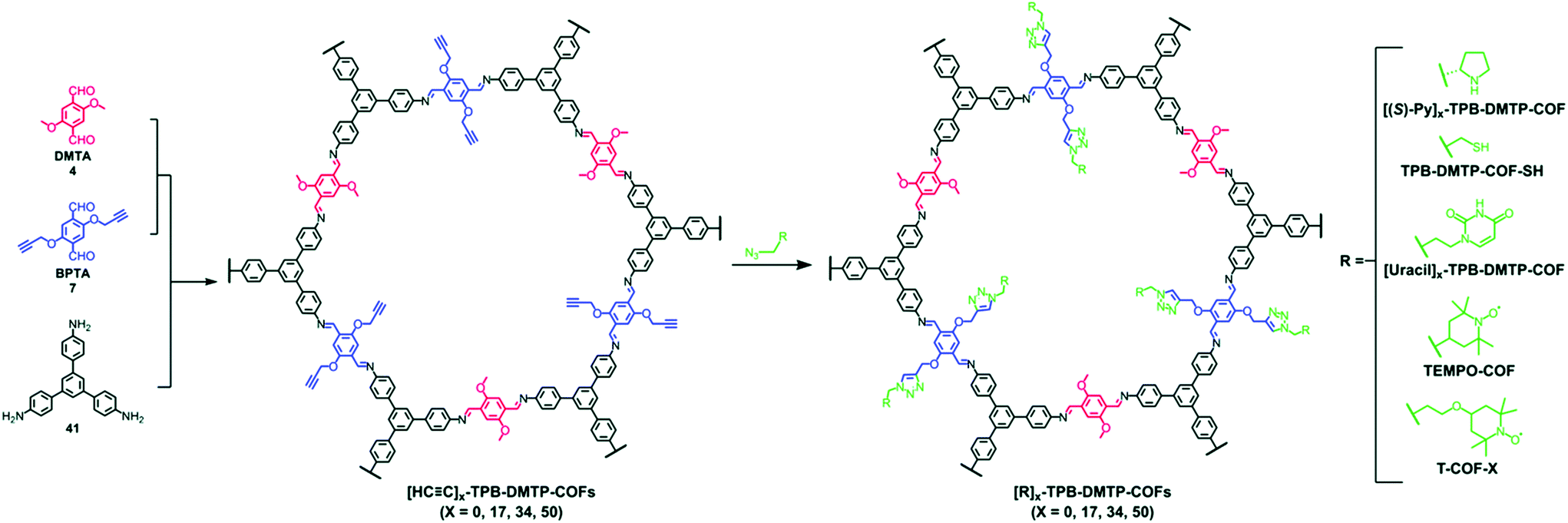 | ||
| Fig. 9 Pore surface engineering of [HCC]x-TPB-DMTP-COFs via click chemistry. Reprinted with permission from ref. 141 (Copyright © 2019, Royal Society of Chemistry). | ||
Sticking to the focal role of COFs as robust platforms for environmental remediation, the Dichtel research group reported a thiol–ene free radical addition reaction inside the pore channels of COFs.122 Our research group reported a 2D COF-V framework constructed by the dynamic imine condensation of 1,3,5-tris(4-aminophenyl) benzene and 2,5-divinylterephthalaldehyde, followed by pore surface engineering via the thiol–ene “click” reaction using 1,2-ethanedithiol and AIBN, with complete retention of crystallinity and porosity. Due to the higher degree of vinyl group participation, nearly 90% of the click chemistry led to the uniform distribution of binding thiol functionalities in COF-S-SH for mercury removal. The high mercury uptake of 1350 mg g−1 and 863 mg g−1 for Hg2+ and Hg0, respectively, was observed in aqueous solution. Fascinatingly, COF-S-SH exhibited a diffusion coefficient (Kd = 2.3 × 109 mL g−1) with high mercury uptake, from 5 ppm to 0.1 ppb, which is far less than the acceptable limit of drinking water (2 ppb). The flexible binding arms electronically and sterically adopt favorable conformations to exclusively interact with mercury via two sulphur atoms as confirmed by EXAFS, XPS, Raman spectra and other analytical techniques.123 In 2018, our research group highlighted the integration of superwettability using the same click reactions by grafting fluorinated compounds. The highly efficient absorption of oil along with various toxic solvents such as nitrobenzene with an uptake capacity up to 142 times of its mass was observed.124 This study addresses the growing ecological and environmental concerns such as oil spills, and materials such as activated carbon and zeolite have proven to be relatively ineffective. COFs, as a platform for environmental remediation studies, have been extended to uranium removal from seawater. Amidoxime moieties in organic scaffolds to construct frameworks via de novo synthesis present a profound challenge; our research group used cyano functionalized β-ketoenamine COF-TpDb prepared by the condensation of triformylphloroglucinol and 2-cyano-1,4-phenylenediamine. The cyano group uniformly distributed in the pore channel of the framework underwent pore surface engineering via the conversion of cyano functionalities to amidoxime (COF-TpDb-AO) by reaction with hydroxylamine in methanol (Fig. 10). COF-TpDb-AO, used as a uranium scavenger, reduced the uranium content in uranium-contaminated water samples from 1 ppm to 0.1 ppb in a short time; we also highlighted selectivity, adsorption affinities, capacities and kinetics.125 In addition to this conversion, the cyano group was partially hydrolyzed to amide in basic medium,126 and was completely hydrolyzed to carboxylic acid or reduced to amine as highlighted by Yan, Valtchev and co-workers.127 Apart from amidoxime functionalities, the introduction of –NH2 moieties is also demanding; however, amines bind with formyl groups to construct imine-linked COFs and therefore, the possibility of pendant –NH2 in COFs is low via the de novo method. The Dichtel research group reported128 pendant –NH2 functionalized COF via the pore surface engineering of (X% [N3]-COF) prepared by the condensation of 1,3,5-tris(4-aminophenyl) benzene with the appropriate ratio of azide-functionalized ethylene glycol terephthalaldehyde and terephthalaldehyde using PPh3 in CH3OH. The amine moieties in the pore channels of frameworks exhibited excellent crystallinity, a surface area of over 1000 m2 g−1 and fast uptake of GenX. The chiral framework reported by the Wang research group was obtained by the condensation of the chiral N-Boc-protected building blocks with 1,3,5-triformylbenzene or triformylphloroglucinol, then cleavage of BOC under acidic, basic or heating conditions, and served as organocatalysts for the asymmetric aldol reaction.129
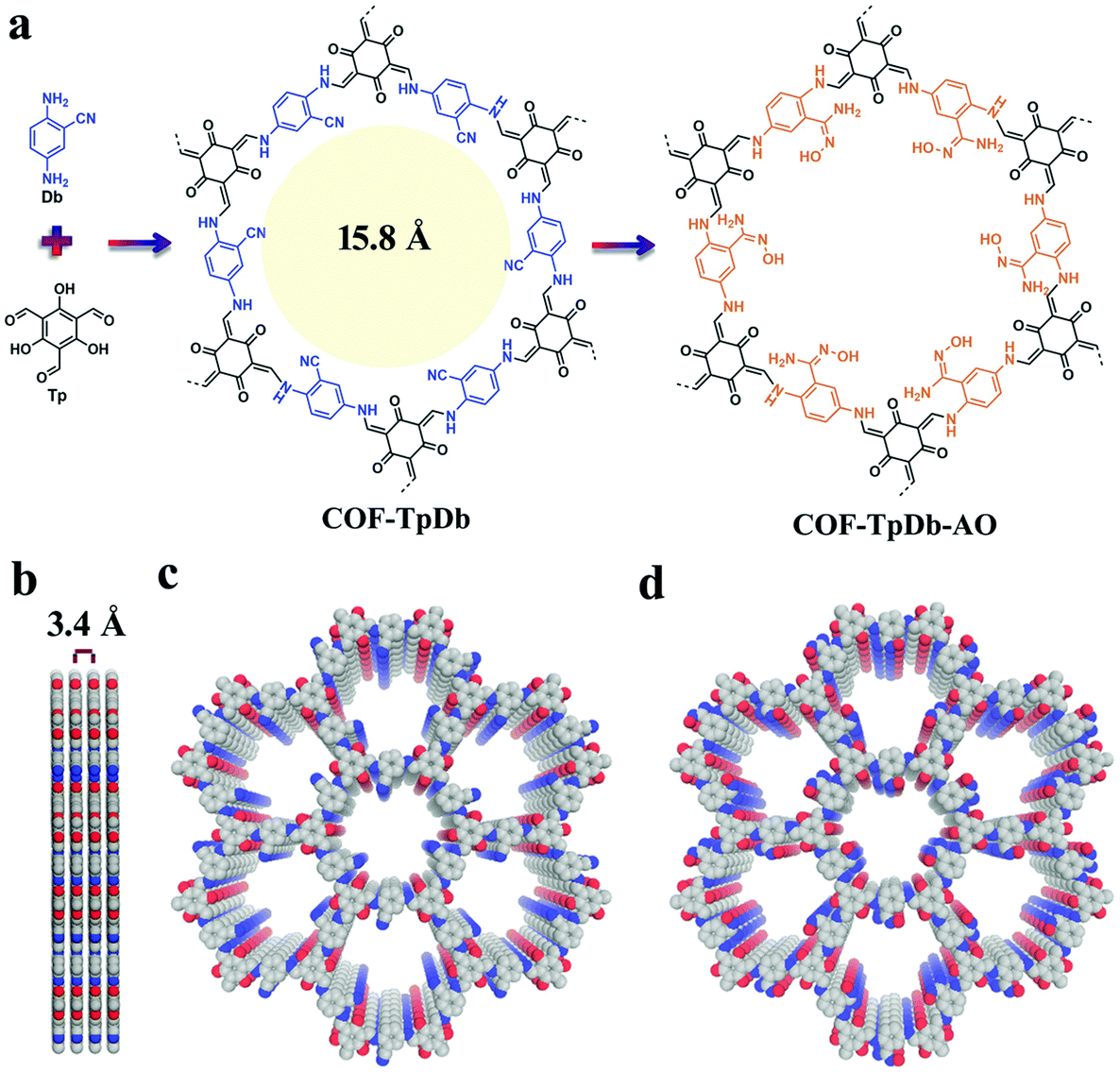 | ||
| Fig. 10 (a) COF-TpDb synthesis using dynamic imine linkage and corresponding pore surface engineering to amidoxime group (COF-TpDb-AO); (b–d) Graphic view of the eclipsed stacking structure of pristine and modified frameworks. Reproduced with permission from ref. 125 (Copyright © 2018, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim). | ||
Not all functionalities are difficult to uniformly decorate and modify in the frameworks; one of those functionalities is –OH. The Jiang research group reported COFs bestowed with the uniform distribution of hydroxyl functionalities by the reaction of 2,5-DHTA with the porphyrin scaffold. The [OH]x%-H2P-COFs (X = 25, 50, 75, 100) on reaction with succinic anhydride underwent a ring-opening reaction to form pendant carboxylic groups.130 The Gao research group reported modification by using the acylation reaction in the presence of a range of substituted acyl chlorides such as azobenzene and stilbene.131
The same research group employed hydroxyl-functionalized imine-linked frameworks [HO]x%-Py-COFs prepared by the condensation of PyTTA and 2,5-dihydroxyterephthalaldehyde and terephthalaldehyde in different ratios for pore surface engineering via the Williamson synthesis reaction using (2-bromoethyl) triethylammonium bromide. The modified frameworks [Et4NBr]x%-Py-COFs (X = 25, 50) catalyzed the N-formylation of amines with CO2 and PhSiH3 and, more importantly, provided a step towards metal-free catalysis (green catalysis).132 The same catalytic reaction was also highlighted by zwitterionic frameworks prepared by the surface functionalization of hydroxyl-decorated frameworks using the Williamson synthesis reaction by treatment with 2-((3-bromopropyl) dimethylammonio) acetate.133 Last year, the Fang and Valtchev research group synthesized a hydroxyl-functionalized 3D COF by the condensation of tetra(4-formylphenyl) methane and 3,3′-dihydroxybenzidine. The uniformly decorated hydroxyl groups on reaction with succinic anhydride afforded the pendant acyl carboxyl group, which in turn, showed selective binding of Nd3+ over Sr2+ and Fe3+ as proven by XPS and 13C NMR with excellent recyclability.134 Medina, Bein and co-workers also highlighted the reactivity of hydroxyl-functionalized COFs with fluorescent dyes under normal reaction conditions with no hindrance to crystallinity.135 They also highlighted a facile approach to reducing nitro-functionalized COFs to amine moieties via pore surface engineering in the presence of SnCl2·2H2O, which on further treatment with acetic anhydride, underwent aminolysis to form amide-functionalized COFs.136
The Zhou research group used the 2,2′-bipyridine-based TpBpy framework reported by Banerjee et al.137 to study Ni-based pore surface engineering using Ni(ClO4)2 as ion sources to form Ni-TpBpy. PXRD, XPS, FT-IR, UV-Vis, SEM, and HAADF-STEM affirmed the successful immobilization of nickel on the framework. The surface area and pore volume decreased from 973 m2 g−1 and 0.6 cm3 g−1 to 580 m2 g−1 and 0.4 cm3 g−1, despite an increase in the CO2 uptake for Ni-TpBpy, affirming the strong Lewis acid–base interaction between the nickel ions and carbon dioxide molecules. Based on the knowledge of the catalytic properties of Ni-linker complexes for CO2 reduction, the group investigated the photocatalytic CO2 conversion activity of Ni-TpBpy using [Ru(bpy)3]Cl2 (bpy = 2,2′-bipyridine) as the photosensitizer and triethanolamine (TEAO) as an electron donor. The evolution of CO and H2 from the Ni-TpBpy catalytic system in 5 h was 4057 and 170 μmol g−1, respectively, with a turnover number for CO evolution of 13.62 after 5 hours of irradiation. Moreover, control experiments were also performed to point out the key factors for CO2 to CO conversion and also investigate the negligible impact of TpBpy, Co2+, Fe3+, Zn2+ and Mn2+ immobilized frameworks. To gain mechanistic insight into the photocatalytic reduction of CO2 by using Ni-TpBpy, cyclic voltammetry and DFT calculations were also reported.138
TTB-COF, solvothermally synthesized by imine condensation of 2,5-bis(2-(ethylthio)ethoxy)terephthalohydrazide (BETH) and 1,3,5-triformylbenzene (TFB), was verified structurally by means of FT-IR, 13C-NMR and UV-Vis with a low surface area/pore volume, and the crystallinity was analyzed by PXRD. Due to the uniform distribution of pendant functionalities, Au ions were captured in over 98% within a few minutes (KSV = 4.46 × 105 M−1). In a solution mixture of Au3+, Fe3+, Ni2+, Co2+, Zn2+ and Cd2+ at concentration of 10 ppm, there was a high capture of Au ions, which was verified by XPS, and EDX via SEM showed the uniform distribution of gold within the framework.139 Recently, our group highlighted the immobilization of iridium by the reaction of Py-2,2′-BPyPh COF and [Ir(OMe)(1,5-cod)]2 at room temperature under an inert atmosphere. The iridium-decorated framework, (Ircod(I)@Py-2,2-BPyPh COF), exhibited complete retention of crystallinity, high thermal and chemical stability and surface areas of 859 m2 g−1. The iridium docking was further confirmed by 13C MAS NMR, XPS, EPR, SEM-EDS and was investigated as a heterogeneous catalyst for a C–H borylation reaction using bis(pinacolato)diboron (B2pin2) as the borylating agent.140 Mechanistically, Ir(I) on reaction with B2pin2 was converted into Ir(III), followed by oxidation addition to arene to form Ir(V), which in turn underwent reductive elimination to form the organoboron product along with the regeneration of catalyst.
Metal-free COFs functionalization
Pore surface engineering via metal coordination is widely studied and a similar principle can also extend to backbone modifications using established organic chemistry and the influence of non-covalent interactions to address various applications. The fundamental properties attached to the symmetric monomer and dynamic linkages present in the frameworks impart COFs with essential properties such as pore size, pore design, crystallinity, etc., which can be engineered to achieve the desired outcomes.141 Yaghi and co-workers reported the conversion of the imine linkage in COFs to amide functionalities in the presence of sodium chlorite, 2-methyl-2-butene, acetic acid in dioxane. The conversion and progress of the reaction were followed by Fourier-transform infrared spectroscopy (FT-IR), 13C cross-polarization magic angle spinning (CP-MAS) NMR spectroscopy and PXRD measurements.142 The Cui research group further extended this idea from 2D COFs to 3D COFs under the same reaction conditions.143 The difference between the stability of the imine and amide framework is prominent in both acidic and basic media; amide frameworks {(R,R)-CCOF6} in comparison to imine frameworks {(R,R)-CCOF5} exhibited superior activity as chiral stationary phases in high-performance liquid chromatography for the enantioseparation of racemic alcohol. Feng, Wang and co-workers highlighted the peculiar behavior of hydroxyl-functionalized crystalline DABH-TFP-COFs prepared by the condensation of 2,5-diamino-1,4-dihydroxybenzene dihydrochloride (DABH) and 1,3,5-triformylphloroglucinol (TFP). The framework was subjected to pore surface engineering to form benzoquinone-functionalized DABQ-TFP-COF by oxidation with O2 and NEt3, as exhibited by FT-IR and other physiochemical analyses, with faster kinetics of lithium storage. Interestingly, the condensation of 1,3,5-triformylphloroglucinol and 2,5-diamino-1,4-benzoquinone (DABQ) does not give same result, but forms an amorphous material instead (Fig. 11).144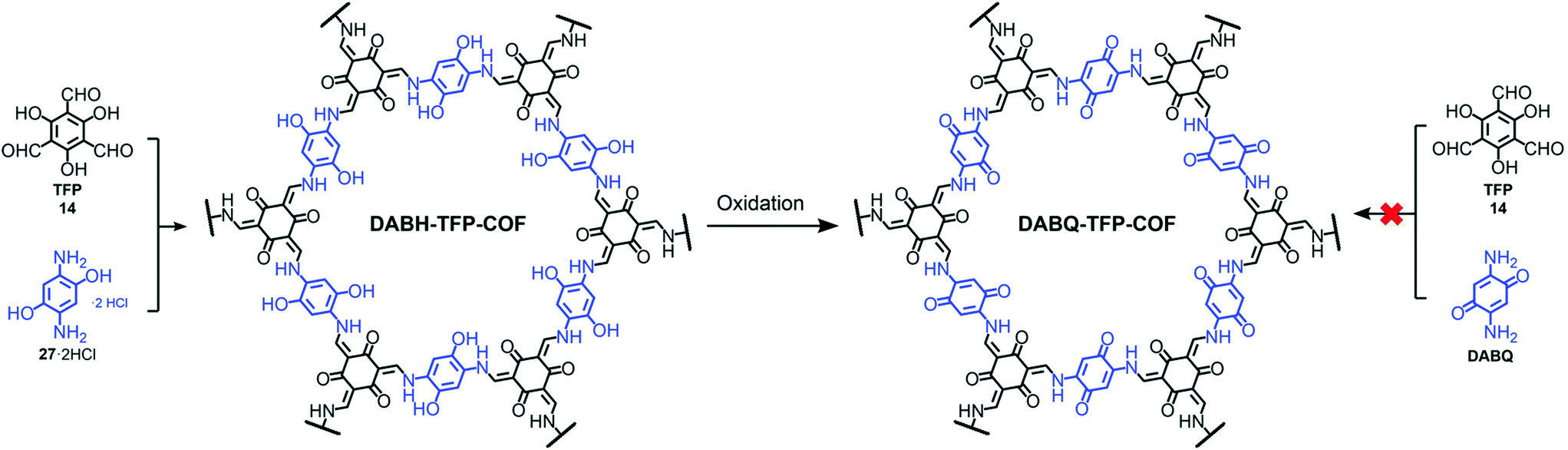 | ||
| Fig. 11 Chemical conversion of DABH-TFP-COF to DABQ-TFP-COF via the oxidation of hydroquinone units. Adapted with permission from ref. 141 (Copyright © 2019, Royal Society of Chemistry). | ||
Last year, the Lotsch research group, under harsh reaction conditions, exhibited the sulphur-abetted conversion of the imine-linked framework into the thiazole-based framework via two successive pore surface engineering steps (Fig. 12). Firstly, at an elevated temperature (155 °C), less viscous sulphur was mixed with the framework; secondly, the increase in the temperature up to 350 °C led to the conversion of imine to thioamide, which was finally oxidized to the thiazole ring. These sequential conversions inside the pore channel were confirmed by PXRD, 13C and 15N solid-state NMR spectroscopy.145 To a certain extent, similar imine conversion under relatively moderate conditions was achieved by the pore surface engineering of polarized imine-linked COF with substituted alkynes in the presence of BF3·OEt2. The Liu research group highlighted this cyclization by kinetically adjusting the imine bond with substituted acetylene via aza-Diels–Alder cycloaddition reactions. Notably, the quinoline-based framework is among of the most robust frameworks reported for COFs to date, and is able to withstand severe reaction conditions such as 12 M HCl, 14 M NaOH, KMnO4, NaBH4.146
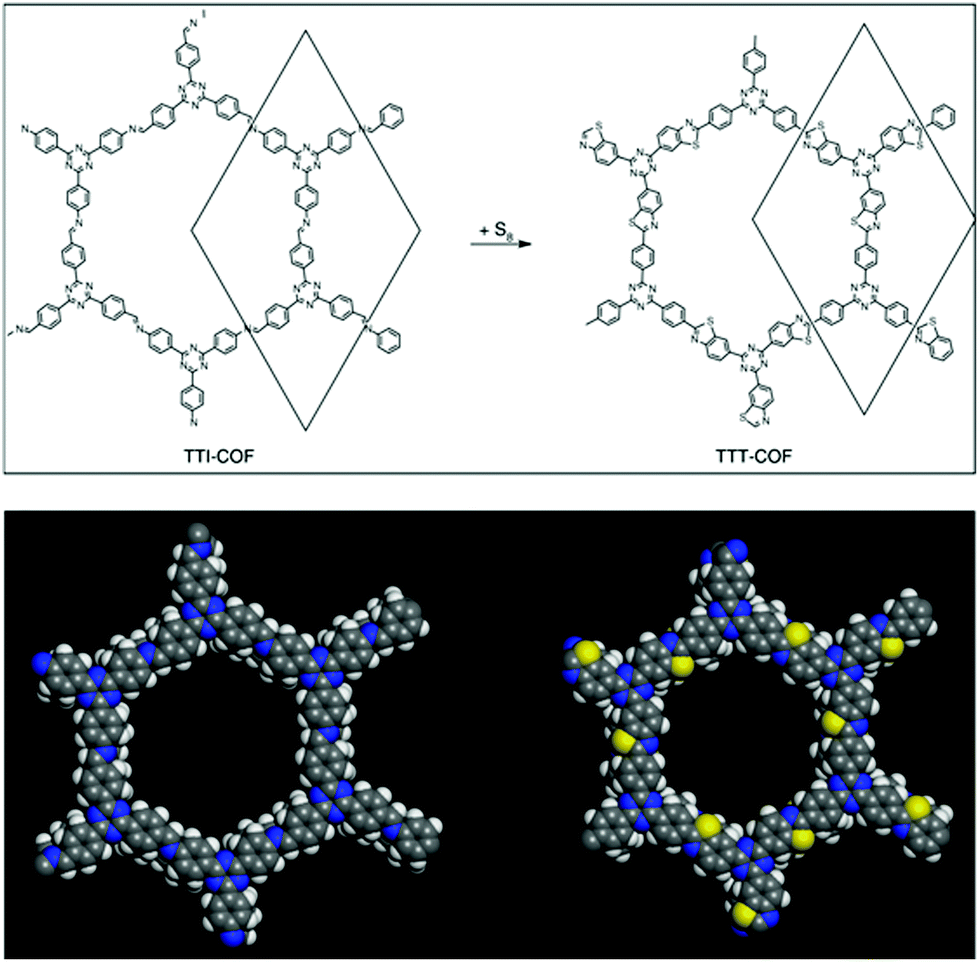 | ||
| Fig. 12 Conversion of imine-linked TTI-COF to thiazole TTT-COF using S8 with the space-filling model of one pore. Reprinted with permission from ref. 145 (Copyright © 2018, Nature Publishing Group). | ||
Recently, Xu, Chen and co-workers used vinyl-functionalized COF-V to immobilize sulphur within the pore channel via an inverse vulcanization strategy.147 After grinding the framework with elemental S8, the mixture was heated at an elevated temperature (155 °C) under an inert atmosphere to immobilize the molten sulphur and to ensure complete polymerization followed by heating at 200 °C. S-COF-V was used as the cathode for a Li–S battery, with the initial capacity of 1400 mA h g−1 that moderately declined to 959 mA h g−1 after 100 catalytic cycles; to the best of our knowledge, this is one of the best performances of the hybrid porous materials. Zhang, Awaga and co-workers148 used the inverse vulcanization strategy in alkyne-functionalized frameworks instead of the vinyl analogue. The formed hybrid material acted as the cathode for rechargeable lithium organic batteries with a capacity of 425 mA h g−1. The same research group used the aromatic nucleophilic substitution (SNAr) strategy via S-arylation for covalent linkage to sulphur in the backbone of the framework. COF-F synthesized by the condensation of 1,3,5-tris(4-aminophenyl) triazine and 2,3,5,6-terephthalaldehyde undergoes pore surface engineering in two steps. Firstly, S8 and the framework in a 3:1 weight ratio were heated at 160 °C for 15 hours to immobilize sulphur into the pore channels of the framework. Secondly, to assist the SNAr reaction, the temperature was increased to 350 °C for another 15 hours. COF-F-S exhibited high porosity and crystallinity with 60 wt% sulphur loading and served as a cathode material for lithium–sulphur batteries.149
COFs pore surface engineering achieved by backbone modification or metal immobilization is not the only way to tune the chemical and steric environment of pore channels. Tuning may also occur by taking advantage of bond dissociation and bond association under thermodynamic control. This way of conversion along with metal replacement is extensively reported in MOFs. The Zhao research group used TP-COF-BZ, prepared by the condensation of 1,3,5-triformylbenzene and benzidine, which underwent COF to COF transformation by monomer exchange using p-phenylenediamine to form TP-COF-DAB.150 To drive the equilibrium forward, p-phenylenediamine was added at higher concentrations to ensure the complete conversion in a short period of time (4 h). Notably, in comparison to TP-COF-DAB, TP-COF-BZ exhibited lower thermal stability and crystallinity. By this pivotal strategy, the crystallinity of frameworks can be substantially improved. The Horike research group employed two imine-linked COFs prepared by dynamic imine linkage between benzene-1,3,5-tricarbaldehyde with 1,4-diaminobenzene and 1,4-diaminonapthalene; they looked at both sides for building block exchange. Firstly, the benzene-based COF reacted with different equivalents of 1,4-diaminonapthalene, which led to the replacement of 1,4-diaminobenzene with 1,4-diaminonapthalene in different ratios. Interestingly, the modified framework exhibited improved crystallinity and a two-fold increment in surface area with similar morphology. Conversion was further confirmed by high-angle annular dark-field scanning transmission electron microscope (HAADF-STEM). Substituting the monomer in naphthalene-based COF with 1,4-diaminobenzene under same reaction conditions was enormously difficult due to robust π–π interactions (Fig. 13). It is worth pointing out that the electronic and steric environments of building blocks are not the only factors to affect monomer exchange; the chemical and thermal stability of the framework also draws from non-covalent interactions.151 The introduction of various functionalities such as amino groups in COFs via de novo synthesis is a profound challenge to the research community; however, pore surface engineering addresses such pivotal challenges without any hindrance to the frameworks’ crystallinity and morphology. With this approach in mind, the Yan research group used two frameworks prepared by condensation of 1,3,5-tris(4-formyl) triazine with 1,4-phenylenediamine and benzidine. To uniformly impart –NH2 functionalities in frameworks, the pristine COFs reacted with amino-substituted phenylenediamine/benzidine at 40 °C for 72 hours.152 Notably, the geometry of the monomer may be of vital importance as reflected in the reaction condition, which is far more moderate than the traditional way of constructing COFs.
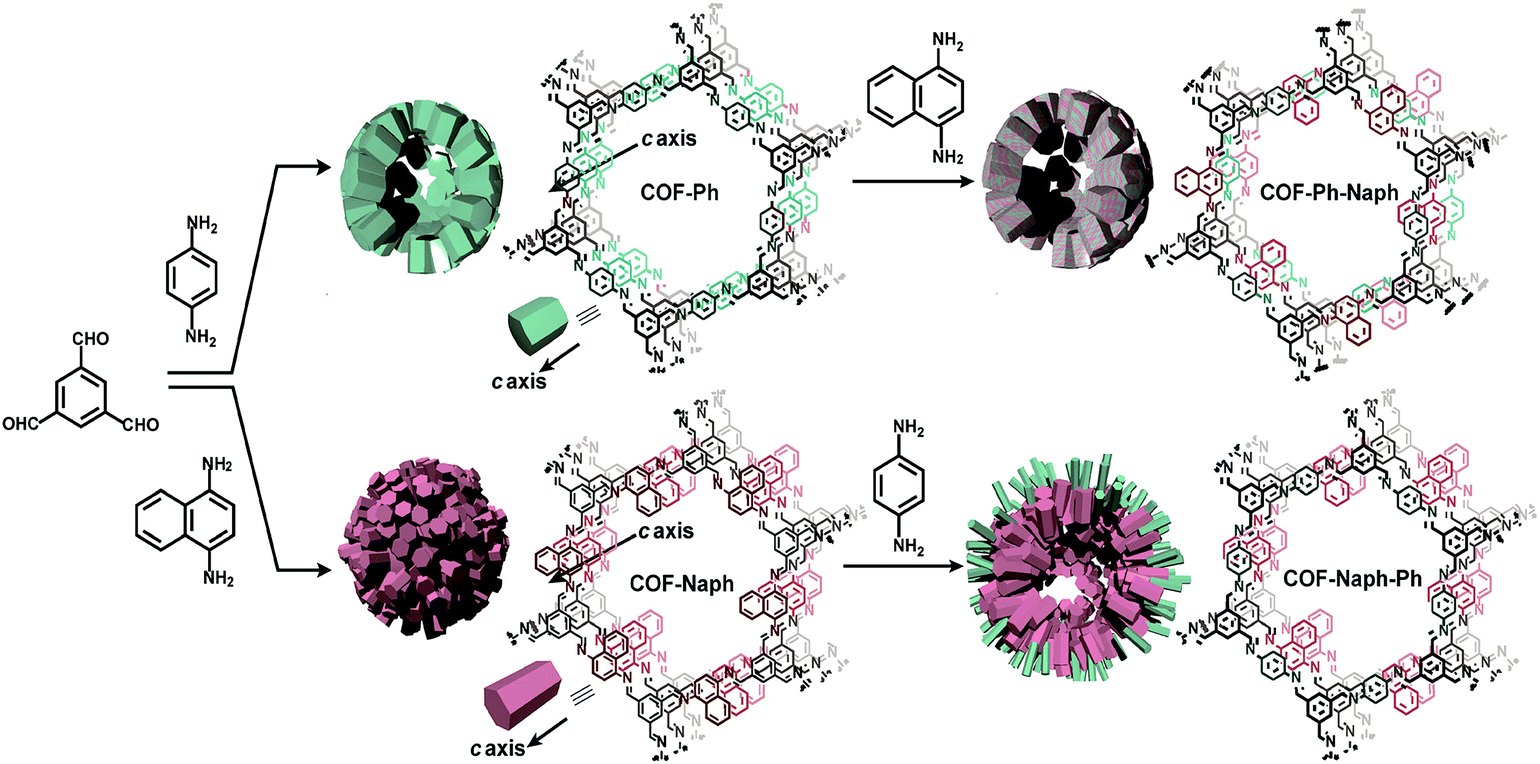 | ||
| Fig. 13 The COF-Ph, COF-Naph and COF-Naph-Ph syntheses via pre and post-synthetic processes. Reproduced with permission from ref. 151 (Copyright © 2018, American Chemical Society). | ||
Zhao et al. reported an approach to inducing amino functionalities at a high reaction rate, which was probably due to the electronic environment of the monomer. The building block exchange strategy positively influenced the stability of the frameworks via cyclization. The imine-linked IL-COF-1 synthesized by the condensation of 1,4-phenylenediamine and 1,3,6,8-tetrakis(4-formylphenyl) pyrene underwent linker exchange using 2,5-diaminobenzene-1,4-dithiol dihydrochloride and 2,5-diaminohydroquinone dihydrochloride followed by oxidation to form the stable oxazole-containing COF-921 and azole-containing LZU 192, respectively.153 Recently, the Dichtel research group synthesized β-ketoenamine-linked frameworks by building block exchange of 1,3,5-triformylbenzene with triformylphloroglucinol; as expected, the framework showed excellent crystallinity, stability and higher surface area.154
Applications
The high degree of crystallinity helps in establishing the structure–property relationship by offering numerous innovative ways to uniformly position a range of moieties including different functional groups. The sole presence of light elements (B, C, N, O) makes the framework less dense (COF-108 = 0.17 g cm−3) in comparison to other porous crystalline solids. In addition, high surface areas and pore volume along with uniform and periodic accessible pores added a remarkable advantage to COFs (COF-103 = 4210 m2 g−1 and 1.66 m3 g−1)38,73 The utilization of hydrogen bonding and the Michael addition–elimination route in addition to dynamic covalent linkage keep COFs resistant to thermal and chemical environments.155 This high resistance to hydrolysis, oxidation/reduction environments and a wide range of pHs is hardly evident in other porous materials; the open structural modularity via pore surface engineering, therefore, serves as an attractive and ideal platform for different applications.Biomedical
Yan and co-workers designed 3D polyimide frameworks (PI-COF-4 and PI-COF-5)156 by imidization between tetrahedral 1,3,5,7-tetraaminoadamantane (TAA) or tetra(4-aminophenyl) methane (TAPM) and pyromellitic dianhydride (PMDA) with a pore size of 1.3 nm and 1.0 nm. Ibuprofen (5 Å × 10 Å), a widely used drug with short biological half-life (t1/2 = 2 h) was trapped inside the pore channels of PI-COF-4 and PI-COF-5, which exhibited excellent release control. For both frameworks, slow drug delivery was observed (6 days) and reached 95% in comparison to the initial loading. Notably, PI-COF-5 demonstrated a relatively slower release rate due to its smaller pore size (1.0 nm) and interpenetrated structure; however, both frameworks showed similar delivery rates with captopril and caffeine. The in vivo biocompatibility and cytotoxicity of COFs have been less explored as compared to in vitro. The Zhao research group157 used two imine-linked frameworks, namely PI-2-COF and PI-3-COF for cell experiments. Both PI-2-COF and PI-3-COF showed high drug loading capacities for ibuprofen, captopril and 5-fluorouracil (5-FU), with release in 3 days.To substantially increase the biocompatibility, the Banerjee research group reported158 the salt-mediated scalable synthesis of TpAPH and TpASH frameworks followed by sequential pore surface engineering to form the required hybrid material (TpASH-FA). Specifically, essential sites required for cellular targeting agents were achieved by the modification of the hydroxyl-functionalized framework through the ring-opening of glycidol. The continuous release of 5-FU from the functionalized frameworks killed cancer cells via apoptosis. The interaction between COFs and the immobilized drug translates into better control of drug delivery. Lotsch and co-workers159 constructed TTI-COF by condensation of 1,3,5-tris(4-formyl) triazine and 1,3,5-tris(4-amino) triazine to study quercetin (3,3,4,5,7-pentahydroxyflavone) behavior toward human breast carcinoma MDA-MB-231 cells. Molecular dynamics simulations studies showed that the drug interacted with pore walls via C–H–π and H-bonding interactions. Quercetin decomposed slowly over a wide temperature range, therefore, TGA analysis failed to calculate the loading amount. Unfortunately, the drug is prone to oxidation and difficult to dissolve in buffer, which inhibited the in vitro drug release study. However, in comparison to the pristine framework, the drug-loaded framework showed moderate activity towards MDA-MB-231 cells. This study, even after a few limitations, unambiguously explained the material synthesis and the role of the framework as a drug delivery vehicle. Apart from COFs role as a drug delivery carrier, the Bhaumik research group highlighted160 the role of functional COFs as anticancer agents. EDTFP-1 COF was prepared by the condensation reaction of 1,3,5-triformylphloroglucinol and 4,4′-ethylenedianiline under an inert atmosphere and exhibited the acceleration of the ROS generation due to phloroglucinol derivatives leading to the apoptosis of cancer cells like HCT 116, HepG2, A549 and MIA-Paca2 (Fig. 14). Enzyme activity is often suppressed due to deactivation in certain temperature and pH ranges. This hindrance can be diminished by coupling the enzyme chemistry to porous materials chemistry, which primarily protects from deactivation and also provides a window of stability and recyclability. Banerjee and co-workers first reported COFs for enzyme encapsulation by using a mesoporous imine-linked framework synthesized by the condensation of 2,5-dihydroxyterephthaldehyde and 1,3,5-tris(4-aminophenyl) benzene, which exhibited excellent crystallinity with BET surface area and pore size of 1480 m2 g−1 and 3.7 nm, respectively. Trypsin (hydrodynamic size = 3.8 nm) immobilization was confirmed by confocal laser scanning microscopy and revealed that moderate activity (60% of free enzyme) was retained in the hydrolysis of N-benzoyl-L-arginine-4-nitroanilide to 4-nitroaniline.161 Our group, in collaboration with the Zhang and Chen research group, highlighted frameworks with pendant carboxylic groups that, on subsequent reaction with N-hydroxysuccinimide and amine-linked biomolecules such as peptides, afforded biomolecule-decorated pore channel COF (biomolecules & COFs), which serve as chiral stationary phases in both reverse and normal-phase high-performance liquid chromatography.162 Furthermore, we also investigated the enzymatic performance of lipase PS encapsulated in various mesoporous materials for the kinetic resolution of racemic 1-phenylethanol with vinyl acetate. More importantly, higher catalytic activity was observed for immobilized porous materials in comparison to free enzyme. We also studied the tunability of hydrophobic or hydrophilic environments inside pore channels of COFs by exposure of phenolic COF in basic medium (aq. NaOH) to form –ONa-functionalized COF, which in comparison to isoreticular COF with –OMe moieties showed significantly lower uptake of lipase.163
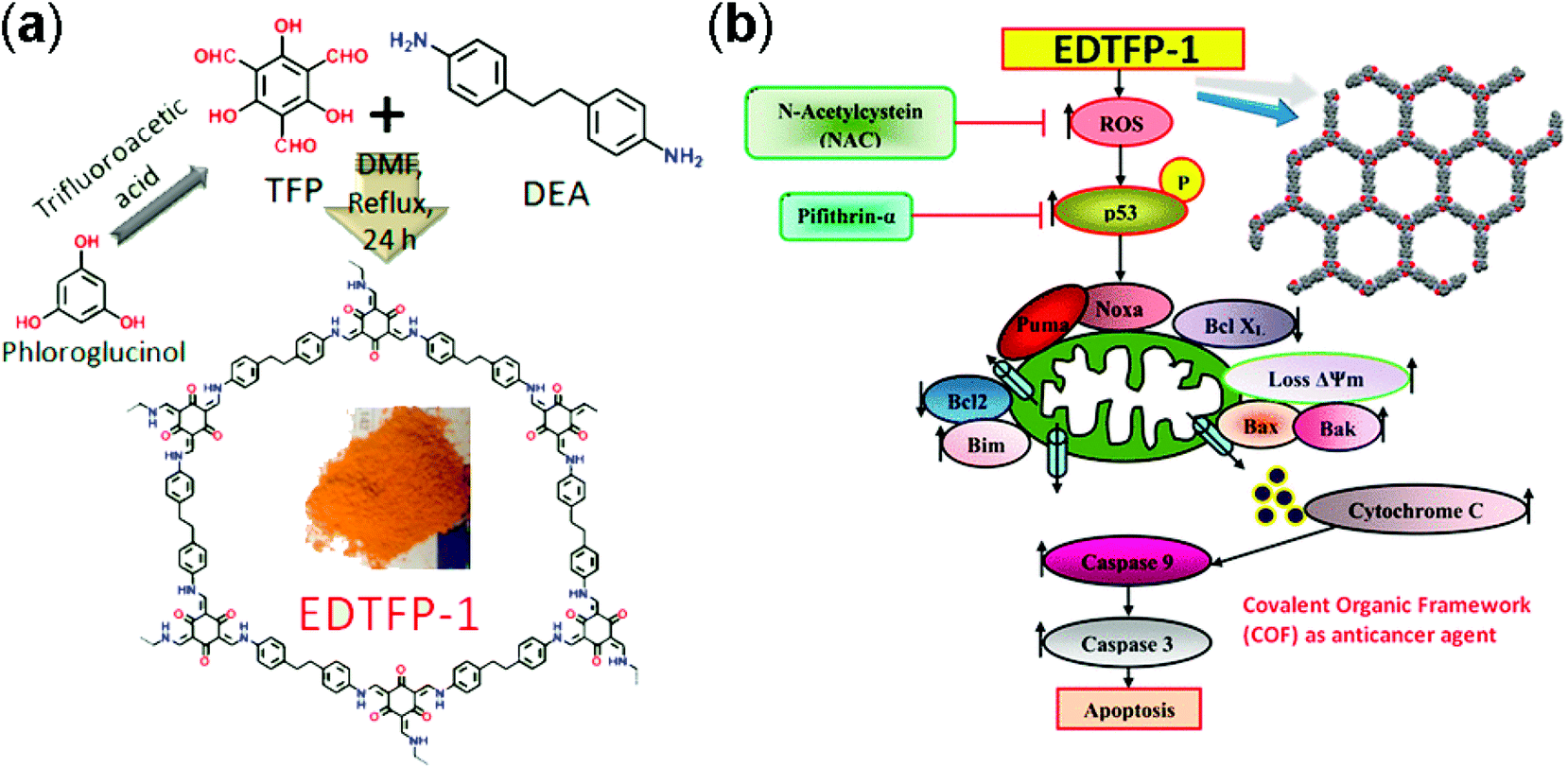 | ||
| Fig. 14 (a) Synthesis of EDTFP-1 from TFP and DEA, and (b) its induced apoptotic pathway. Adapted with permission from ref. 160 (Copyright © 2017, American Chemical Society). | ||
Photothermal therapy (PTT) was studied by the Guo research group through the deposition of an imine-linked framework on the surface of Fe3O4 nanoclusters. The resulting Fe3O4@COF not only elevated the temperature by 25 °C but also affected the photothermal conversion efficiency by 21.5%, which is comparable to some reported photosensitizers such as gold nano rods.164 Moreover, Xie and co-workers165 grew imine-based porphyrin COF on the surface of an amine-functionalized MOF with an average size of 176 nm. The hybrid UNM material produce singlet oxygen and showed significant toxicity toward HepG2 and HeLa upon irradiation using confocal laser scanning microscopy; however, no activity was observed without light. Antimicrobial applications of COFs are very sporadic in the literature. The Banerjee research group constructed self-exfoliated ionic covalent organic nanosheets by using guanidinium halide and 1,3,5-triformylphloroglucinol. The guanidinium moieties formed hydrogen bonds with phosphate anions, whereas positively charge CONs break the negatively charged phospholipid bilayers of bacteria. Antibacterial studies indicated that these nanosheets showed excellent antimicrobial activity against Gram-positive and Gram-negative bacteria.166
Catalysis
The continuous and sustainable advancement in the field of heterogeneous catalysis stems from environmental and economic challenges. This can be achieved by developing new hybrid materials to improve catalytic efficacy, lower cost, reduce waste, and facilitate recyclability and reusability. Within this context, the use of porous materials such as COFs with chemical/thermal stability, high surface area and great tunability along with characteristics from both the molecular and heterogeneous levels is pivotal. The regular pore channels in COFs provide well-defined coordination sites and environments for different catalytic centers without disturbing the crystallinity and morphology of the frameworks. In this subsection, we will summarize de novo synthesized COFs as heterogeneous catalysts.The Yan research group synthesized167 3D BF COFs by condensation of 1,3,5,7-tetraaminoadamantane (TAA) with 1,3,5-triformylbenzene (TFB) or triformylphloroglucinol (TFP). The extraordinary stability of the frameworks with surface area and pore size of 730 m2 g−1, 0.83 nm and 680 m2 g−1, 0.81 nm for BF-COF-1 and BF-COF-2, respectively, catalyzed Knoevenagel condensation between a range of substituted aldehydes with malononitrile and exhibited excellent conversion and size selectivity; for instance, benzaldehyde was converted to 95% condensation product whereas, p-tolualdehyde afforded <5% product. The Zhao research group used sulfonated decorated TFP-DABA COF as an effective catalyst for fructose dehydration to 5-hydroxymethylfurfural (HMF) with 100% conversion in 97% yield.168
The Yaghi research group reported COF-366-Co via the Schiff base condensation of 5,10,15,20-tetrakis(4-aminophenyl) porphinato cobalt [Co(TAP)] and terephthalaldehyde for the electrochemical reduction of CO2 to CO. The COF-366-Co showed 90% Faraday efficiency for CO in 24 hours with a turnover number (TON) of 34000. Furthermore, Co(TAP), on condensation of 4,4′-biphenyldicarbaldehyde, generated an expanded pore size COF, thereby positively influencing the reduction with TON of 48000, which is substantially higher as compared to COF-366-Co169 (Fig. 15). In addition, the conversion of the cycloaddition reaction between epoxide and CO2 to form cyclic carbonate was catalyzed by using an imine-linked hydroxyl-decorated framework prepared by the condensation of 2,3-dihydroxy terephthalaldehyde and 5,10,15,20-tetrakis(4-aminophenyl)-21H,23H-porphine.170
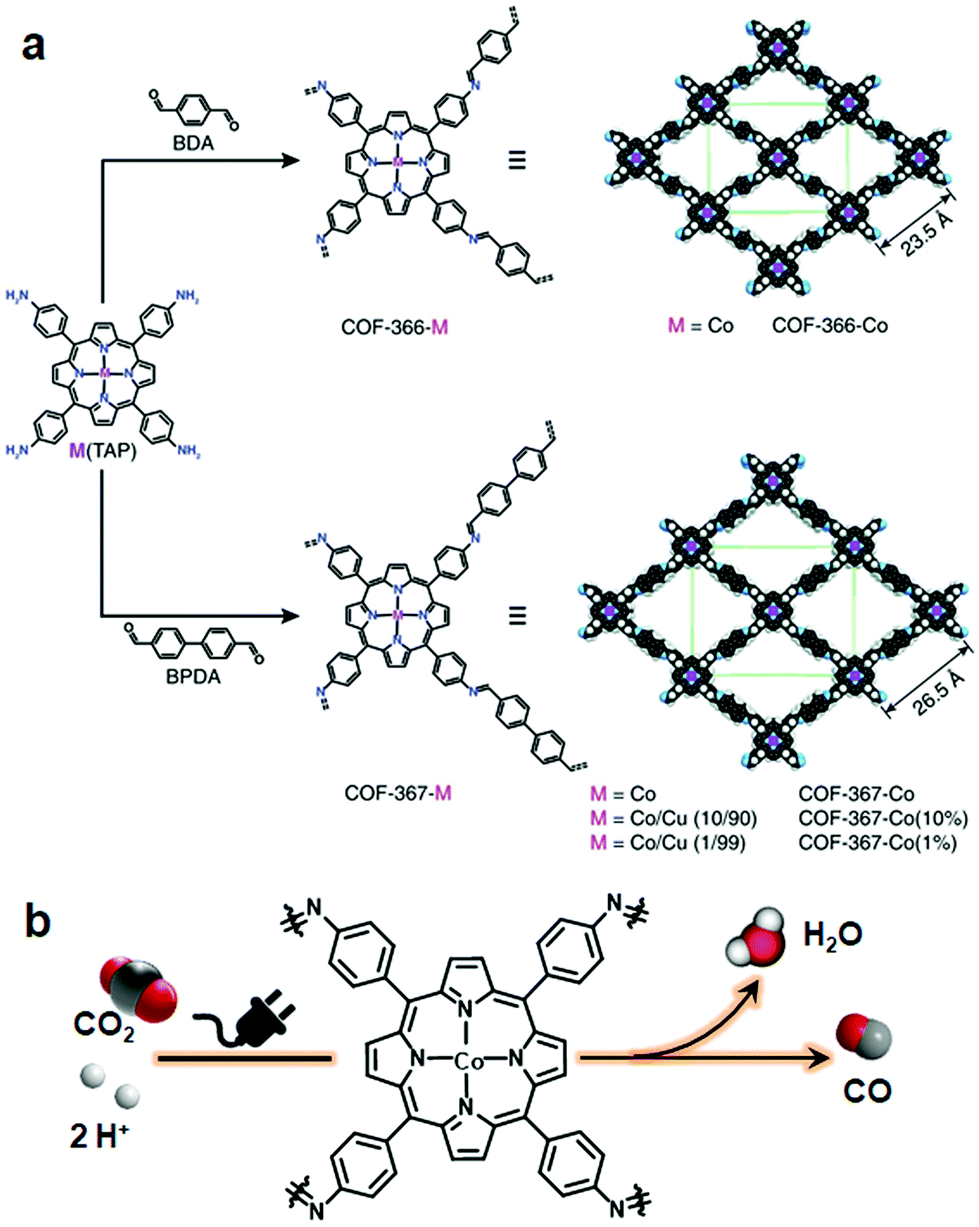 | ||
| Fig. 15 (a) Synthesis of metalloporphyrin-derived 2D COFs. (b) COF-366-Co-based electrocatalytic reduction of CO2 in water. Reprinted with permission from ref. 169 (Copyright © 2015, American Association for the Advancement of Science). | ||
The Cui research group reported chiral COF by the condensation of the chiral tetraaryl-1,3-dioxolane-4,5-dimethanol (TADDOL) unit aldehyde scaffold and 4,4′-diaminodiphenylmethane. The chiral framework, on treatment with Ti(OiPr)4, afforded CCOF/Ti as an effective catalyst for the asymmetric addition of diethyl zinc to aldehyde to form the alcohol in high percentage yield, outperforming the corresponding homogeneous analogue (TADDOL/Ti).171 The same research group reported a 3D framework by using the same chiral monomer and tetra(4-anilyl) methane in high-performance liquid chromatographic enantioseparation.144 Shinde et al. synthesized a framework bearing weak acidic and basic sites by the condensation reaction between 5,10,15,20-tetrakis(4-aminophenyl) porphyrin and 2,3-dihydroxyterephthalaldehyde as an effective catalyst for the domino reaction. The uniformly distributed porphyrin and imine moieties served as weak basic sites, whereas catechol groups acted as weak acid sites. The presence of weakly basic and acidic sites at vertices and edges, respectively, in the framework serve as heterogeneous catalysts for the cascade reaction of benzaldehyde dimethyl acetal and malononitrile in high yield.172 A similar tandem reaction was also highlighted by 3D frameworks prepared by the boroxine linkage between 1,3,5,7-tetraaminoadamantane and 4-formylphenylboronic acid/2-fluoro-4-formylphenylboronic acid. Our own group in collaboration with the Chen and Zhang research group synthesized squaramide-linked COF-SQ by the reaction of 1,3,5-triformylbenzene and 3,4-bis((4-aminophenyl) amino)-cyclobut-3-ene-1,2-dione as the organocatalyst for Michael addition under mild conditions173 (Fig. 16).
 | ||
| Fig. 16 Synthesis of the squaramide-linked organocatalyst COF-SQ. Reproduced with permission from ref. 173 (Copyright © 2019, Royal Society of Chemistry). | ||
The Lotsch research group reported COFs as an ideal platform for the construction of photoactive catalysts. The condensation reaction of hydrazine with triphenylarylaldehydes with the core ring having varying ratios of nitrogen atoms (0–3) afforded Nx-COFs (X = no. of nitrogens). The hydrogen evolution increased from 23, 90, 438 to 1703 μmol h−1 with the increase in the nitrogen concentrations of N0-COF, N1-COF, N2-COF and N3-COF, respectively after 8 h.174 Bein and co-workers synthesized stable BDT-ETTA COF by the condensation of 1,1′,2,2′-tetra-p-aminophenylethylene (ETTA) and benzo[1,2-b:4,5-b′]-dithiophene-2,6-dicarboxaldehyde (BDT) as the photoelectrode for photoelectrochemical water splitting in the absence of sacrificial agents and cocatalysts.175 Banerjee and co-workers reported176 the amalgamation of photocatalytically active CdS nanoparticles in COF for visible-light-driven hydrogen production. The extensive π-conjugation in the framework stabilized the nanoparticles and also prevented the recombination of electron and holes to facilitate charge transfer. In particular, a 30% increment in H2 evolution was observed for hybrid material in comparison to the pristine framework.
Environmental remediation
By virtue of high surface area, tailorable pore size and electronic environment, the potential of nuclear events and risks can be addressed using porous frameworks. To substantially increase the adsorptive capacity, kinetics and selectivity, a range of functionalities plays a pivotal role in battling pollution. The nature and virtue of host molecules is of prime importance. For instance, an amorphous host suffering from small pore size leads to the inaccessibility of grafting groups, whereas COFs with essential properties such as high pore size lead to the entrapment of heavy metal ions. More importantly, the presence of binding moieties uniformly distributed via the de novo strategy or pore surface engineering in the COFs further brand COFs at the forefront of these issues. There are a range of binding moieties known to entrap uranium; as proof-of-concept, β-ketoenamine frameworks decorated with amidoxime functionalities used for the sequestration of uranium far outperformed amorphous solids in the context of affinities, kinetics and absorption capacities.125 The decorated framework effectively removed uranium from water (408 mg g−1) and it diminished to 0.1 ppb, which is far less than limits set by the U.S. Environmental Protection Agency.Mercury is one of the important metals that cause public health problems. Explicitly, the selective capture of mercury or any other contamination in order to protect freshwater is the need of the hour. The Wang research group reported the hydrazine-linked fluorescent LZU8 COF, synthesized by the condensation of 1,3,5-triformylbenzene and a thioether-functionalized scaffold, with evenly and densely decorated thioether functionalities in 1D pore channels for high affinity and selectivity towards mercury ions (Fig. 17). LZU8 COF exhibited an uptake capacity of 300 mg g−1 with a detection limit as low as 25 ppb and excellent recyclability.177 In addition, the Jiang research group synthesized TAPB-BMTTPA-COF by the condensation reaction of 2,5-bis(methylthio)terephthalaldehyde (BMTTPA) and 1,3,5-tris(4-aminophenyl) benzene (TAPB) for mercury removal from aqueous solution. The thioether-functionalized framework, abundantly decorated with sulphur functionalities, led to the uptake capacity of 734 mg g−1, which is approximately 2.5 times higher than LZU8 COF.178 The Qiu research group synthesized two novel 3D-ionic-COF-1 and 3D-ionic-COF-2 by the condensation of tetrakis(4-formylphenyl) methane with diimidium bromide and ethidium bromide, respectively.179 Both ionic frameworks exhibited high crystallinity, porosity, stability and the potential for selective removal of permanganate. The observed kinetics with excellent recyclability supersedes signature materials such as PVBTAH-ZIF-8 and LDHs. Sticking to ionic frameworks, the Trabolsi research group reported for the first time viologen-linked COF using the Zincke reaction with excellent stability, porosity and controllable morphology. The condensation of 1,3,5-tris(4-aminophenyl) benzene and 1,1′-bis(2,4-dinitrophenyl)-[4,4′-bipyridine]-1,1′-diium dihydrochloride afforded a charged framework for efficiently capturing iodine from both solution and vapors.180
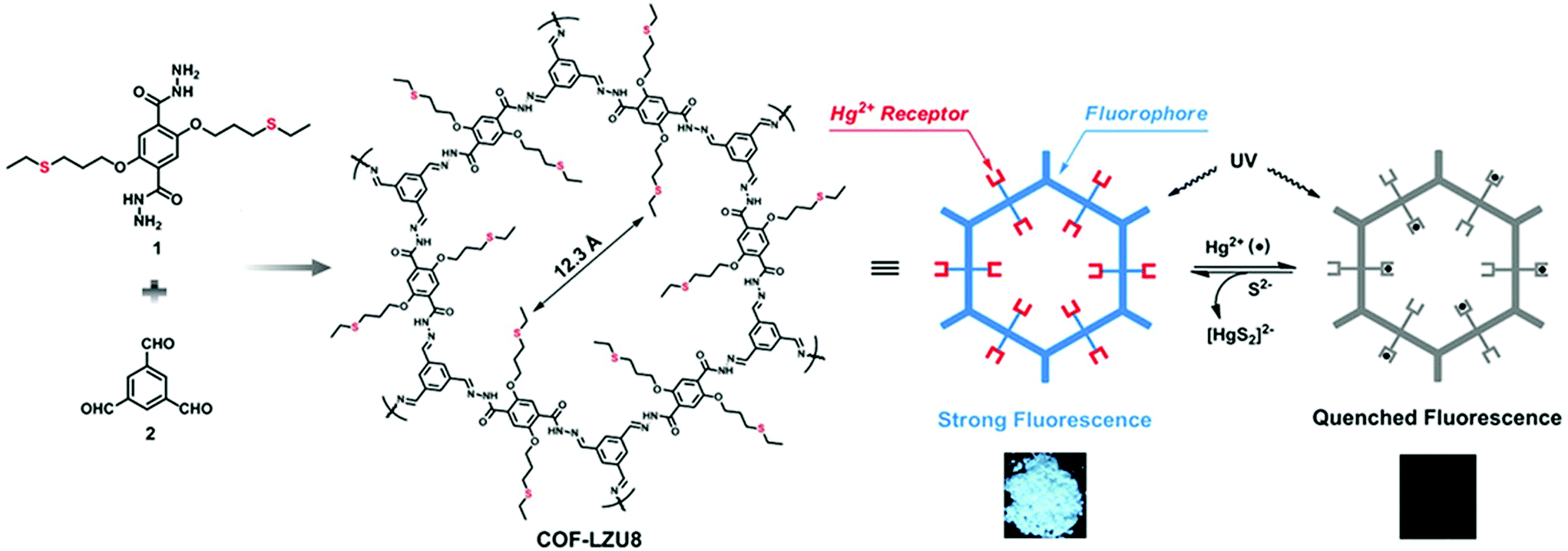 | ||
| Fig. 17 Synthesis of hydrazine-linked COF-LZU8 with thioether groups for the removal of Hg2+ ions. Photographs of COF-LZU8 under a UV lamp (λ = 365 nm) exhibiting changes in fluorescence after mercury adsorption. Adapted with permission from ref. 177 (Copyright © 2016, American Chemical Society). | ||
The Jiang research group reported the cationic PyTTA-BFBIm-iCOF synthesized by the dynamic imine linkage of PyTTA and ionic 5,6-bis(4-formylbenzyl)-1,3-dimethyl-benzimidazolium bromide (BFBIm) building blocks with BET surface area of 1532 m2 g−1. The decoration of benzimidazolium cations on either side of the pores due to the reverse AA stacking mode led to excellent uptake capacity of negatively charged methyl orange dye (553 mg g−1), which is the highest among various porous materials, probably due to the high degree of electrostatic interactions. Furthermore, excellent CO2 uptake capacities of 93 mg g−1 and 177 mg g−1 at 298 K and 273 K, respectively, were reported, which are significantly higher than neutral frameworks.181 The Loh research group synthesized salicylideneanilines-based COF (SA-COF) constructed by the condensation reaction of 1,3,5-tris(4-aminophenyl) benzene and triformylphloroglucinol, which exhibited BET surface area and pore size of 1588 m2 g−1 and 1.43 nm, respectively. Given the presence of reversible proton tautomerism and well-arranged pore channels, SA-COF was explored for size-dependent organic pollutant separation. The binding affinity of the framework with a range of dye molecules declined with the increase in molecular size and cannot entrap when the size of the guest molecule exceeds the host pore size. Notably, the nature of dye and pH sensitivity also plays a role in changing the pore environment and thereby, selectivity in charged molecule separation.182
Gas storage and separation
In the post-carbon future, hydrogen will be an important alternative source of energy, swiftly replacing nonrenewable sources of energy such as coal or petrol. This green alternative has been one of the first applications explored using COFs, the Yaghi research group reported183 boroxine and boronic ester-linked 2D and 3D COFs. It was found that medium-sized 3D COF-102 and COF-103 at 77 K showed hydrogen uptake values of 72.4 mg g−1 and 70.5 mg g−1, respectively, which are substantially higher than 2D COF-1 (14.8 mg g−1), COF-5 (35.8 mg g−1), COF-6 (22.6 mg g−1), COF-8 (35.0 mg g−1) and COF-10 (39.2 mg g−1). In addition to this, Qiu and co-workers highlighted imine- and boroxine-linked frameworks, denoted as DL-COF-1 and DL-COF-2, synthesized by condensation of 1,3,5,7-tetraaminoadamantane with 4-formylphenylboronic acid and 2-fluoro-4-formylphenylboronic acid, respectively. Both frameworks were tested for H2, CO2 and CH4 sorption behavior, DL-COF-1 showed 2.09 wt% H2 sorption at 77 K, 26.7 wt% CO2 sorption at 273 K, and 2.57 wt% for CH4 at ambient temperature, whereas DL-COF-2 exhibited 1.73 wt%, 21.8 wt%, 2.10 wt% sorption for H2 at 77 K, CH4 at 273 K, CO2 at 273 K, respectively.184 The improved hydrogen or carbon dioxide storage capacity of COFs was further improved by the spillover effect. Kalidindi et al. and the Zhao group summarized theoretical studies of H2 and CO2 capture in a detailed review.185,186Porous materials play profound roles in industrial ammonia transportation; COFs synthesized by boroxine or boronate ester are susceptible to Lewis acid and Lewis base interactions. The high density of boron atoms (Lewis acidic) in the framework trapped ammonia (Lewis base) through coordinate bonds. Boroxine-linked COF-10 was prepared by the dynamic linkage of hexahydroxytriphenylene and biphenyldiboronic acid, and showed ammonia uptake of 15 mol kg−1 at 25 °C, which is substantially higher than signature materials such as zeolites (9 mol kg−1) and amberlyst (11 mol kg−1). Notably, at elevated temperature (200 °C), adsorbed ammonia was evacuated and this adsorption/desorption cycling tested up to three runs without any significant variation in properties; however, a slight broadening and decrease in intensity was observed in PXRD during subsequent cycles187 (Fig. 18). The role of open metal sites in ammonia uptake is profoundly studied in MOFs, so enriching and combining the advantages with chemically and thermally stable COFs is far beyond notable. Zhu and co-workers decorated112 the walls of 2D imine linked frameworks with carboxyl functionalities under a multicomponent condensation strategy. The role of carboxyl moieties was found to be important with nearly 50% increment (9.34 mmol g−1) in comparison to the pristine framework (6.85 mmol g−1) at 298 K. The authors demonstrated that the binding of divalent metal ions such as calcium, strontium and manganese to carboxyl functionalities boosted ammonia uptake. As expected, the ammonia uptake was substantially higher in all three metal incorporated materials, for instance, the strontium-integrated framework had an equilibrium ammonia uptake of 14.30 mmol g−1. The Zhu research group188 constructed a microporous framework by the condensation of 1,2,4,5-tetrahydroxybenzene and TBPM for selective C2H6 and C2H4 adsorption over CH4.
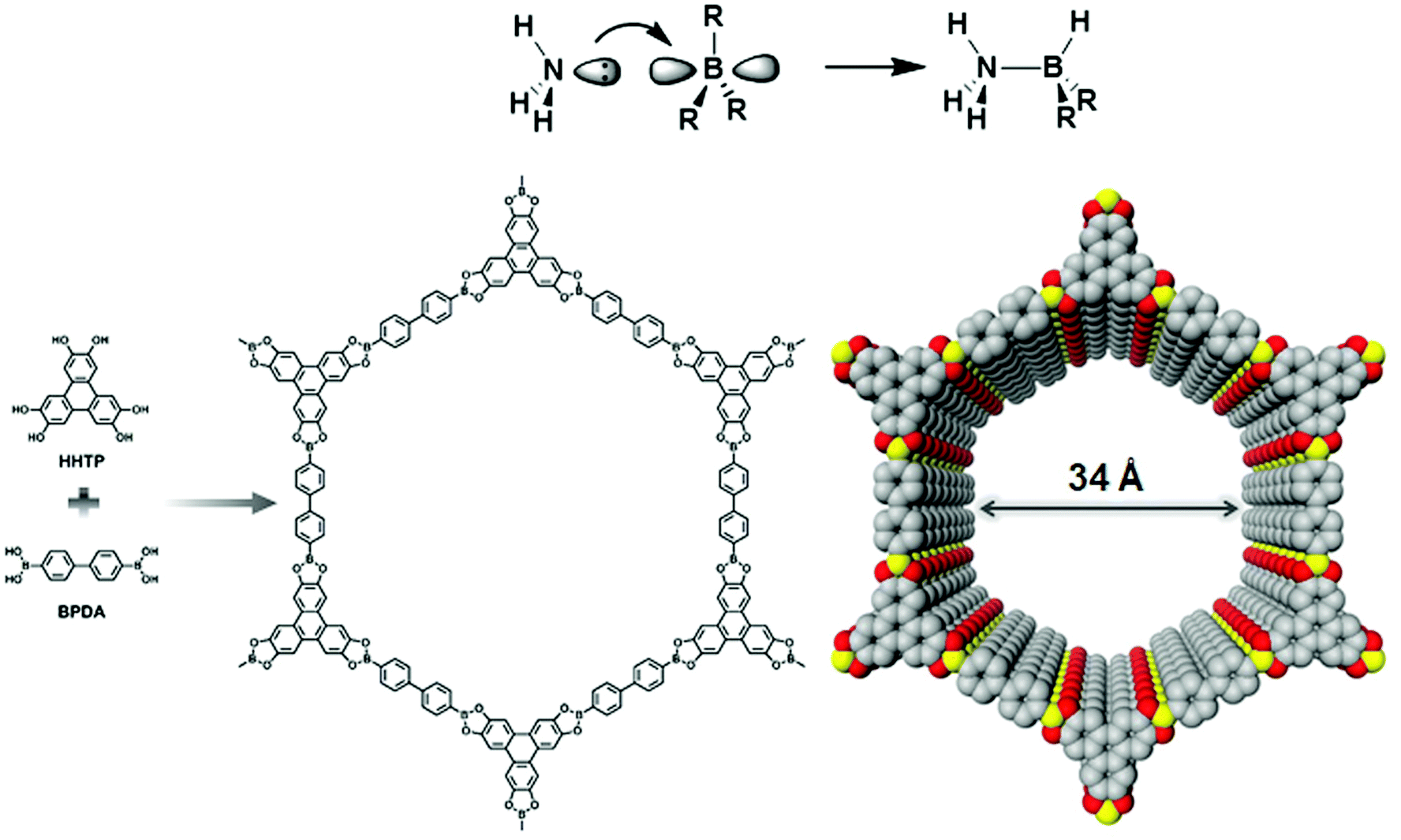 | ||
| Fig. 18 Lewis acid–base interaction between boron and ammonia, and the synthesis of COF-10 with an eclipsed stacking structure. Reproduced with permission from ref. 187 (Copyright © 2010, Nature Publishing Group). | ||
A negatively charged spiroborate framework constructed from trimethylborate and diol-functionalized macrocycles further introduced Li+ counter ions for high H2 and CH4 storage capacities at 3.11 wt% and 4.62 wt%, respectively. Furthermore, the nitrogen-rich nature of frameworks plays a favorable role in carbon dioxide adsorption. Cai et al. reported189 a brick-wall COF prepared by the condensation of PDA and 4,4′,4′′-(1H-benzo[d]imidazole-2,4,7-triyl) tribenzaldehyde with CO2 capacity of 3.95 wt% and 40.43 wt% at 273 K and 195 K, respectively. The central role of COFs in gas storage can be expanded for effective CO2 capture and conversion to value-added chemicals, which, in turn, mitigate environmental concerns. In 2009, the first report on CO2 adsorption was presented and thereafter, the Liu research group190 synthesized an azine-based framework (ACOF-1) by the condensation of 1,3,5-triformylbenzene and hydrazine with pore size of 0.94 nm. The high percentage of nitrogen on the pore channel walls and high surface area of 1176 m2 g−1 exhibited high carbon dioxide uptake (177 mg g−1), higher than various reported COFs. The Jiang research group synthesized an azide-decorated framework (x%N3-COF-5) by three-component condensation under an inert atmosphere followed by a click reaction to form triazole-linked modified frameworks. The decorated frameworks were investigated for the selective gas sorption of CO2 over N2 and a sixteen-fold increment in adsorption was observed from 25%PyTrz-COF-5 to 100%PyTrz-COF-5 as compared to the pristine framework.77
Sensing
The judicious choice of precursor units in constructing COFs with tunable pore size, electronic environment, and functional moieties along with stacking interactions plays a profound role in sensing under the shadow of non-covalent interactions. Dalapati et al. synthesized a stable azine-linked framework by condensation of 1,3,6,8-tetrakis(4-formylphenyl) pyrene and hydrazine. The high percentage of fused benzene rings and hydrogen bonding synthons in the framework serve as sensitive and selective sensors for 2,4,6-trinitrophenol.47 A similar principle was further extended for the selective fluorescence quenching of picric acid (75%, 20 ppm) using porous 3D-Py-COF prepared by imine linkage between tetra(p-aminophenyl) methane and 1,3,6,8-tetrakis(4-formylphenyl) pyrene.191An imide-linked thermally stable (>500 °C) PI framework192 constructed by condensation of 3,4,9,10-perylenetetracarboxylic dianhydride and tetra(4-aminophenyl) porphyrin showed high selectivity towards 2,4,6-trinitrophenol over the range of 0.5 to 10 μM with quenching constant and detection limit of 1 × 10−7 M−1 and 0.25 μM, respectively. 3PD and 3′PD ketoenamine frameworks155 constructed from Michael addition–elimination reactions exhibited quenching on treatment with various nitro- and peroxide explosives with a detection onset of 1 × 106 M for triacetone triperoxide, which probably originated from the oxidation of enamine units. Moreover, the sensing of various compounds including nitrobenzene, phenol and 2-nitrotoluene with effective fluorescence quenching was also reported by imine-linked TAT-COF-2 and the TfpBDH framework synthesized by the condensation of 2,7,12-triformyl-5,10,15-triethyltriindole with 2,7,12-triamino-5,10,15-triethyltriindole and pyromellitic-N,N’-bisaminoimide and 1,3,5-tris(4-formylphenyl) benzene, respectively.193,194 The selective sensing studies were not restricted nitro explosives, but also extended to the fluorescence quenching of ammonia. The boronic ester TPE-Ph framework81 synthesized by tetraphenylethylenetetraboronic acid and 1,2,4,5-tetrahydroxybenzene exhibited 30% fluorescence quenching upon the addition of ammonia, with kq value of 6.3 × 1014 M−1 s−1 and 1.4 × 1014 M−1 s−1 in toluene and cyclohexane, respectively. In addition, JLU-3 COF prepared by the condensation of hydrazine and hydroxyl functionalized triformylbenzene, served as a chelating agent for transition metal ions. The strong fluorescence quenching observed for Cu2+ ions among other transition metal ions such as Co2+, Fe3+ and Ni2+ ions with excellent recyclability is extraordinary,195 and is the first fluorescent framework for the selective and sensitive sensing of toxic metals.
Apart from metal ions and small explosive nitro molecules, DNA detection was highlighted by Zhang and co-workers who synthesized a [3 + 3] imine-linked framework196 by condensation of tris(4-formylphenyl) amine and tris(4-aminophenyl) amine, which possessed weak interlayer interactions due to the flexible nature of precursor units. Due to these core properties, the framework was readily exfoliated and exhibited excellent selectivity and sensitivity of DNA with a limit of 20 pM (Fig. 19). Moreover, the Ajayaghosh research group reported fluorescent cationic ultrathin 2D sheets of EB-TFP-iCONs by the self-exfoliation of the EB-TFP framework, given that the electrostatic interactions between the phosphate backbone of DNA and positively charged framework led to restacking as reflected in orange emission. Notably, this hybrid sensor material showed remarkable performance in distinguishing dsDNA from single-stranded ssDNA.197
 | ||
| Fig. 19 (a) Synthesis of the extended hexagonal structure of TPA-COF. (b) AFM image of TPA-COF NS; (c) CTF-corrected HRTEM image of the framework. (d) Illustration of a TPA-COF NS-based fluorescence sensor for the detection of DNA. Reprinted with permission from ref. 196 (Copyright © 2017, American Chemical Society). | ||
Energy storage
COFs with high stacking density of π-orbitals create opportunities in the field of carrier transport and mobilities in devices. The eclipsed stacked structure of COFs facilitates conduction via orbital interactions and also imparts exceptional thermal stability without undergoing any phase transition, which is frequently observed in other conducting materials. The Jiang research group reported the first-ever photoconductive eclipsed PPy-COF synthesized198 by the self-condensation of 2,7-pyrene diboronic acid. The long-range exciton delocalization was observed to be sandwiched between the Au and Al electrodes. The quick response along with the large on:off ratio as reflected by series of control experiments reflect the pivotal importance of stacked pyrene moieties in the framework. To further improve the photoconductive performance, the same research group incorporated the first-ever COF that integrated donors and acceptors in the building blocks as highlighted in the benzothiadiazole acceptor and triphenylene donor. The enhanced photoconductivity is attributed to ambipolar electron and hole conduction with vertically ordered p–n heterojunctions.199
The Bein research group reported the introduction of the fullerene electron acceptor [6,6]-phenyl-C61-butyric acid methyl ester (PCBM) into TT COF prepared by the co-condensation of thieno[3,2-b]thiophene-2,5-diyldiboronic acid and polyol HHTP under solvothermal conditions. The resultant hybrid composite was formed by soaking the framework in a PCBM chlorobenzene solution and showed substantial photoresponse and effective charge transfer.200 This infiltration strategy suffers from guest elusion from the pore channel; to overcome this obstacle, Jiang and co-workers incorporated covalently bound guest molecules into framework by a three component condensation reaction of 1,4-phenylenediboronic acid (BDBA), 2,5-bis(azidomethyl)-1,4-phenylenediboronic acid (N3-BDBA) and (2,3,9,10,16,17,23,24-octahydroxyphthalocyaninato) zinc (ZnPc[OH]8). The varying percentage of azide (X%N3-ZnPc-COFs, X = 0, 10, 25, 50) moieties in the framework with the covalently anchored C60 translated into charge separation, photoenergy conversion and photoinduced electron transfer.116 The Zhang research group reported ionic frameworks, ICOF-1 and ICOF-2 with sp3 hybridized boron anionic centers and positively charged (NMe2)+ and Li+ ions, respectively (Fig. 20). Both microporous frameworks exhibited surface areas of 1022 m2 g−1 and 1259 m2 g−1, respectively; however, they suffer from complex PXRD patterns, which led to the absence of simulated crystal packing. Given the high porosity and Li+ ion percentage, the frameworks showed conductivities of 3.05 × 10−5 S cm−1 at room temperature. Furthermore, the activation energy of the same framework was calculated to be 0.24 eV per atom, which is substantially higher than crystalline and polymer electrolytes.41 Similar lithium infiltrated and counter ion-based frameworks were reported by the Feng research group201 using γ-cyclodextrin and trimethylborate linked via tetrahedral tetrakis(spiroborate). The anionic framework with lithium counter ions exhibited the conductivity of 2.7 mS cm−1 at 30 °C. A polyelectrolyte-based framework was reported by Jiang and co-workers, which involved the infiltration of Li+ ions, using LiClO4, into the TPB-BMTP-COF, which was prepared by the condensation of 1,3,5-tris(4-aminophenyl) benzene (TPB) and 2,5-bis((2-methoxyethoxy) methoxy) terephthalaldehyde (BMTP). It facilitates the association with alkali metal ions to induce ionic conduction and transportation with more than 3 orders of magnitude relative to other systems.202
 | ||
| Fig. 20 (a) Synthesis of ICOF-1 and ICOF-2. (b) Proposed structure of ICOF-2. Adapted with permission from ref. 41 (Copyright © 2016, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim). | ||
The proton conduction in the framework should be studied under stable environments, which are primarily not only in protic and aprotic solvents but also in highly acidic and basic pH ranges. The Banerjee research group synthesized203 an azo-functionalized stable framework by the condensation of 4,4′-azodianiline (Azo) and triformylphloroglucinol (Tp) via β-ketoenamine-linkage. This strong linkage in the framework helped to maintain structural stability and crystallinity at 9 N HCl and 6 N NaOH. The simple infiltration of H3PO4 in the stable Tp-Azo COF anchored on azo moieties and counter ions (H2PO4−) was stabilized via hydrogen bonds. This strong anchoring was further proven by the authors by highlighting the crystal structure of H3PO4 linked to the azo center of 4-aminoazobenzene. With an uptake capacity of 5.4 wt%, PA@TpAzo COF showed a proton conductivity of 6.7 × 10−5 S cm−1 under inert conditions and 9.9 × 10−4 S cm−1 under 98% relative humidity. Polyoxometalates are well-known to exhibit proton conduction; however, their solubility causes key problems to their real-world applications. COFs as a decorating platform will provide assistance to overcome this barrier; for instance, the Zhu research group synthesized ionic EB-COF:Br by the condensation reaction of 1,3,5-triformylphloroglucinol and ethidium bromide (EB). The cationic monomer in COF imparts essential characteristics, which lead to ion exchange with H3PW12O40 and substantially improved proton conduction with 3.32 × 10−3 S cm−1 at 97% relative humidity. In comparison to the hybrid system, the H3PW12O40 and EB-COF:Br mixture exhibited smaller proton conduction (3.2 × 10−5 S cm−1), which is profoundly due to the uniform distribution of bromide anions204 (Fig. 21). In addition, the Dichtel research group reported the immobilization of 2,6-diaminoanthraquinone, a redox-active species in hydrostable 2D β-ketoenamines linked COF.205 The same research also highlighted the integration of the conductive polymer, poly(3,4-ethylenedioxythiophene) (PEDOT) within the 1D pore channel of the framework by the electropolymerization of 3,4-ethylenedioxythiophene (EDOT). The hybrid framework with 1 μm thickness exhibited considerably improved electrochemical performance. Most importantly, the hybrid material showed stable capacitance up to 10000 cycles.206 To further improve the electrical conductivity, Jiang and co-workers imitated graphene conductive material by synthesizing an sp2 carbon-conjugated COF (CC bond) under the principle of reticular chemistry.56 The π-conjugated crystalline material synthesized using tetrakis(4-formylphenyl) pyrene and 1,4-phenylenediacetonitirle exhibited semiconductor behavior with a band gap of 1.9 eV, which increased with the chemical oxidation of I2.
 | ||
| Fig. 21 (a) Schematic representation of EB-COF:Br synthesis (b) [PW12O40]3− doping in ionic framework. Reproduced with permission from ref. 204 (Copyright © 2016, American Chemical Society). | ||
COFs thin films are usually accessed by three prominent strategies. The first is solvothermal synthesis, where COFs are directly grown on a solid support; this method produces crystalline films with pores oriented perpendicular to the substrate, but suffers from various disadvantages such as difficult removal from the support, and scale-up. The second is interfacial polymerization, in which monomers or catalysts are dissolved separately in two immiscible solvents such that COF formation occurs at the interface with controllable thickness; however, poor crystals are often obtained, with thickness more than 50 nm. The third method is the exfoliation and reconstitution of COF powder. This strategy provides a direct route to the film morphology with precisely tuned dimensions. The Dichtel research group highlighted the first oriented thin COF films on substrate-supported single-layer graphene (SLG), which provided a suitable substrate for the COF growth via favourable π-interactions.207 Shortly thereafter, the same group reported large pore size frameworks based on large chromophore prepared as oriented thin films on substrate-supported SLG.208 Bein and co-workers incorporated mesoporous framework (BDT-COF) prepared by the condensation of benzodithiophene-containing diboronic acid (BDTBA) and 2,3,6,7,10,11-hexahydroxytriphenylene (HHTP) with various polycrystalline surfaces characterized by PXRD, SEM, TEM, and 2D grazing incidence diffraction (GID).209 The electron-donor/acceptor host–guest systems were obtained by infiltration of BDT-COF films with soluble fullerene derivatives such as [6,6]-phenyl C61 butyric acid methyl ester (PCBM), which is susceptible to electron transfer by photoluminescence quenching due to charge transfer between host and guest.
Wang and co-workers reported novel ways of synthesizing 2D COF thin films at the oil/water/hydrogel interfaces with controllable thickness from 4 to 150 nm and Young's modulus of 25.9 ± 0.6 GPa by AFM. The COF film-based hybrid material was used as a selective photoelectrochemical sensor for Ru3+ ions.210 Wang and co-workers reported the 2D NT-COF by the integration of electron-rich triphenyl amine (TPA) and electron-poor naphthalenediimide (NDI) in crystalline “bnn” topology, with 1.22 cm3 g−1 pore volume and 1276 m2 g−1 surface area. The successful synthesis was further verified by FT-IR, NMR, SEM and TEM. Due to the nature of monomers, the synergistic effect in the framework between intramolecular charge transfer from the TPA to the NDI and reversible electrochemical performance provide an essential basis for directing solar-to-electrochemical energy conversion/storage. The sheet-like layered structure of NT-COF serves as the cathode material in a solar Li+ ion battery under light irradiation and exhibited increased discharge voltage from 2.42 V to 2.96 V (0.5 V increase), decreased charge voltage by 0.5 V, from 3.04 V to 2.53 V, and more importantly 38.7% extra battery efficiency, from 78.3% to 117%.211
Wang and co-workers reported the synthesis of sp2 carbon-conjugated COF by the condensation of 5,10,15,20-tetrakis(4-benzaldehyde) porphyrin (p-Por-CHO) and 1,4-phenylenediacetonitrile (PDAN) via the Knoevenagel condensation reaction. The AA stacked structure was confirmed by PXRD pattern, structurally investigated by FT-IR and NMR, with thermal stability up to 250 °C and also under harsh conditions such as 9 M HCl and 9 M NaOH. The framework exhibited porosity with a BET surface area of 689 m2 g−1 and pore size of 1.8 nm. The metal-free olefin-linked heterogeneous catalyst serves as a photocatalyst for the visible-light-induced aerobic oxidation of amines to imines.212 The Zhao research group reported the highly conjugated imine-linked three-dimensional SP-3D-COF 1 and SP-3D-COF 2 frameworks by the condensation of 3,3′,6,6′-tetraamine-9,9′-spirobifluorene with terephthalaldehyde and 4,4′-biphenyldicarbaldehyde in o-DCB/n-BuOH/AcOH, respectively. SP-3D-COFs exhibited the P42/NNM space group with “dia” topology with a degree of interpenetration higher than 6 or 7 and was structurally verified using physicochemical analysis. The authors investigated the advanced photoelectric properties for solar energy harvesting by doping frameworks into the CH3NH3PbI3 perovskite layer with average power conversion efficiency (PCE) of 18.34% and maximum PCE of 19.07% for SP-3D-COF 1. However, an average PCE of 18.68% was obtained for SP-3D-COF 2, and more importantly, the average PCE over the reference undoped PSC was highly improved by 15.9% and 18.0% for SP-3D-COF 1 and SP-3D-COF 2 doping.213 Moreover, the mechanistic details of perovskite-SP-3D-COF interactions were determined from both experimental and computational studies, which in turn, helps in understanding the photoresponsive perovskite behaviour.
Conclusion
Covalent organic frameworks have been constructed by the dynamic covalent linkage of building block units using a range of functionalities under the principle of reticular chemistry. The extensive work carried out over a number of years in the field of porous materials, especially COFs, substantially developed the field from various viewpoints drawing the attention of different communities in chemistry or interdisciplinary fields. Fundamentally, peculiar properties associated with dynamic covalent chemistry are bestowed on designed frameworks due to robust covalent bonds that lead to thermal stability, which is by far one of the most important properties in addressing future challenges and industrialization. The geometric features associated with building blocks determine the topology, morphology, and the pore nature of frameworks. Most of the time, the introduction of specific properties in building blocks may be tedious or impossible but can be achieved through pore surface engineering. Pore surface engineering either in the context of metal complexation or chemical conversion in regular pore channels comes from well-established coordination chemistry or organic chemistry. Metal complexation plays a pivotal role in the field of heterogeneous catalysis, which overcomes the disadvantages attached to homogeneous catalysis such as residual waste, recyclability or reusability, and also addresses some of the key organic conversion and name reactions including Suzuki–Miyaura coupling, Prins reaction, C–H borylation, Henry reaction, CO2 reduction, CO2 insertion, and many more as discussed in this review. Pore surface engineering of functional groups plays a critical role in environmental remediation, such as selective uranium extraction via the introduction of amidoxime functionalities, or thiol functionalities to capture mercury ions in record uptake values (0.1 ppb), which is far less than the acceptable limit for drinking water (2 ppb) with excellent recyclability. This illustrates the impact of pore surface engineering, which originates from the signature properties of COFs, especially high porosity, thermal and chemical stability. The flexibility and potential of this approach have been extensively summarized in the functional group interconversion to bring structural diversity, which in turn broadens the role of the framework in rapidly growing challenges.The vast library of organic precursor units bearing different binding units is in itself illuminated in the structural and functional diversity of COFs. This is highlighted in gas adsorption and separation as demonstrated in 3D COF-102 and COF-103 with exceptional hydrogen uptake of 72.4 mg g−1 and 70.5 mg g−1, respectively. Moreover, the impact of the spillover effect, Lewis acid–base adducts (B–N) and the incorporation of ionic precursor units substantially affect selective gas adsorption such as CO2 over CH4. COFs porosity is also responsible for the high uptake of commercially available drugs. The favorable interactions between host and guest help in the release of various drug molecules (ibuprofen, captopril, caffeine and 5-fluorouracil) over days, instead of as a one-time shot. Furthermore, enzyme encapsulation in the framework induces the stability of enzymes, inhibiting denaturation and exhibiting catalytic reactions with excellent reusability without any changes in morphology or crystallinity. These essential characteristics along with the high density of hydrogen bonding synthons in COFs makes the porous materials an excellent selective sensor for nitro compounds over other compounds. The presence of chelating functionalities exhibited strong and selective quenching of Cu2+ and Hg2+ over other transition metal ions. The electrostatic and π–π stacking interactions in the frameworks help in distinguishing dsDNA from ssDNA and allows good energy storage. The development and fabrication of COFs architectures and structure control still remain a scientific challenge to research communities, along with scalable synthesis, controlled morphology, biocompatibility, and hydrolytic stability. A few other challenges are also mentioned below:
1. Building block exchange and sequential pore surface engineering of COFs to further embellish the backbone by innumerable functionalities play a central role in addressing a growing number of applications. This phase of COFs is still in its infancy; therefore, in-depth studies are of profound importance.
2. The library of metal-linked organic scaffolds to construct catalytically active COFs via de novo strategies using dynamic covalent linkage is very sporadic. Therefore, enriching the library of monomer units is of vital importance to widening the scope of COFs for catalytic applications.
3. 2D COFs have been successfully synthesized by using dynamic covalent chemistry; however, translating this enriched chemistry into 3D COFs is still in its early stages. One of the core challenges is crystallization, which emanates from imbalances between strong covalent bonds and reversible linkages.
4. Detailed mechanistic studies to form 3D COFs at room temperature are still unexplored. The organic scaffold to construct 3D frameworks is therefore limited, restricting the number of topologies that exist in this field.
Conflicts of interest
The author declares no conflicts of interest.Acknowledgements
The authors acknowledge the University of South Florida and NSF (CBET-1706025) for financial support. We also extend our appreciation to the Distinguished Scientist Fellowship Program (DSFP) at King Saud University for funding this work partially.References
- P. J. Waller, F. Gándara and O. M. Yaghi, Acc. Chem. Res., 2015, 48, 3053–3063 CrossRef CAS PubMed
.
- J. M. Lehn, Chem. – Eur. J., 1999, 5, 2455–2463 CrossRef CAS
- H. Li, M. Eddaoudi, T. L. Groy and O. M. Yaghi, J. Am. Chem. Soc., 1998, 120, 8571–8572 CrossRef CAS
- S. S. Chui, S. M. Lo, J. P. Charmant, A. G. Orpen and I. D. Williams, Science, 1999, 283, 1148–1150 CrossRef CAS PubMed
- H. Li, M. Eddaoudi, M. O'Keeffe and O. M. Yaghi, Nature, 1999, 402, 276–279 CrossRef CAS
- A. P. Côte, I. A. Benin, N. W. Ockwig, M. O'Keeffe and O. M. Yaghi, Science, 2005, 310, 1166–1170 CrossRef
- S. Das, P. Heasman, T. Ben and S. Qui, Chem. Rev., 2017, 117, 1515–1563 CrossRef CAS PubMed
- A. G. Slater and A. I. Cooper, Science, 2015, 348, 988–998 CrossRef CAS PubMed
- U. Diaz and A. Corma, Coord. Chem. Rev., 2016, 311, 85–124 CrossRef CAS
- K. E. Maly, J. Mater. Chem., 2009, 19, 1781–1787 RSC
- A. Thomas, Angew. Chem., Int. Ed., 2010, 49, 8328–8344 CrossRef CAS
- X. Feng, X. Ding and D. Jiang, Chem. Soc. Rev., 2012, 41, 6010–6022 RSC
- R. P. Bisbey and W. R. Dichtel, ACS Cent. Sci., 2017, 3, 533–543 CrossRef CAS PubMed
- F. Beuerle and B. Gole, Angew. Chem., Int. Ed., 2018, 57, 4850–4878 CrossRef CAS PubMed
- Y. Jin, Y. Hu and W. Zhang, Nat. Rev. Chem., 2017, 1, 0056 CrossRef
- C. S. Diercks and O. M. Yaghi, Science, 2017, 355, eaal1585 CrossRef
- P. J. Waller, F. Gandara and O. M. Yaghi, Acc. Chem. Res., 2015, 48, 3053–3063 CrossRef CAS PubMed
- S.-Y. Ding and W. Wang, Chem. Soc. Rev., 2013, 42, 548–568 RSC
- T. Ma, E. A. Kapustin, S. X. Yin, L. Liang, Z. Zhou, J. Niu, H. Li, Y. Wang, J. Su, J. Li, X. Wang, W. D. Wang, W. Wang, J. Sun and O. M. Yaghi, Science, 2018, 361, 48–52 CrossRef CAS PubMed
-
L. Pauling, The Nature of the Chemical Bond, Cornell University Press, New York, 2nd edn, 1940, pp. 7–10 Search PubMed
- R. B. Woodward, Pure Appl. Chem., 1973, 33, 145–178 CAS
- A. Eschenmoser and C. E. Wintner, Science, 1977, 196, 1410–1420 CrossRef CAS
- S. J. Rowan, S. J. Cantrill, G. R. L. Cousins, J. K. M. Sanders and J. F. Stoddart, Angew. Chem., Int. Ed., 2002, 41, 898–952 CrossRef
- Y. Jin, Q. Wang, P. Taynton and W. Zhang, Acc. Chem. Res., 2014, 47, 1575–1586 CrossRef CAS
- O. M. Yaghi, Mol. Front. J., 2019, 3, 1–18 CrossRef
- O. M. Yaghi, ACS Cent. Sci., 2019, 5, 1295–1300 CrossRef CAS PubMed
- Y. Liu, M. O'Keeffe, M. M. J. Treacy and O. M. Yaghi, Chem. Soc. Rev., 2018, 47, 4642–4664 RSC
- B. Rungtaweevoranit, C. S. Diercks, M. J. Kalmutzki and O. M. Yaghi, Faraday Discuss., 2017, 201, 9–45 RSC
- K. E. Cordova and O. M. Yaghi, Mater. Chem. Front., 2017, 1, 1304–1309 RSC
- N. Huang, P. Wang and D. Jiang, Nat. Rev. Mater., 2016, 1, 16068 CrossRef CAS
- J. Jiang, Y. Zhao and O. M. Yaghi, J. Am. Chem. Soc., 2016, 138, 3255–3265 CrossRef CAS
- S. Kandambeth, K. Dey and R. Banerjee, J. Am. Chem. Soc., 2019, 141, 1807–1822 CrossRef CAS
- C. S. Diercks, M. J. Kalmutzki and O. M. Yaghi, Molecules, 2017, 22, 1575–1580 CrossRef
- S. J. Lyle, P. J. Waller and O. M. Yaghi, Trends Chem., 2019, 1, 172–184 CrossRef
- F. Zhao, H. Liu, S. D. R. Mathe, A. Dong and J. Zhang, Nanomaterials, 2018, 8, 15–64 CrossRef
- Y. Song, Q. Sun, B. Aguila and S. Ma, Adv. Sci., 2019, 6, 1801410 CrossRef
- M. S. Lohse and T. Bein, Adv. Funct. Mater., 2018, 28, 1705553 CrossRef
- H. M. El-Kaderi, J. R. Hunt, J. L. Mendoza-Cortés, A. P. Côté, R. E. Taylor, M. O'Keeffe and O. M. Yaghi, Science, 2007, 316, 268–272 CrossRef CAS
- E. L. Spitler, B. T. Koo, J. L. Novotney, J. W. Colson, F. J. Uribe-Romo, G. D. Gutierrez, P. Clancy and W. Dichtel, J. Am. Chem. Soc., 2011, 133, 19416–19421 CrossRef CAS
- J. R. Hunt, C. J. Doonan, J. D. Levangie, A. P. Côté and O. M. Yaghi, J. Am. Chem. Soc., 2008, 130, 11872–11873 CrossRef CAS PubMed
- Y. Du, H. Yang, J. M. Whiteley, S. Wan, Y. Jin, S. H. Lee and W. Zhang, Angew. Chem., Int. Ed., 2016, 55, 1737–1741 CrossRef CAS
- F. J. Uribe-Romo, J. R. Hunt, H. Furukawa, C. Klock, M. O'Keeffe and O. M. Yaghi, J. Am. Chem. Soc., 2009, 131, 4570–4571 CrossRef CAS PubMed
- S. Kandameth, A. Mallick, B. Lukose, M. V. Mane, T. Heine and R. Banerjee, J. Am. Chem. Soc., 2012, 134, 19524–19527 CrossRef PubMed
- S. Chandra, S. Kandambeth, B. P. Biswal, B. Lukose, S. M. Kunjir, M. Chaudhary, R. Babarao, T. Heine and R. Banerjee, J. Am. Chem. Soc., 2013, 135, 17853–17861 CrossRef CAS PubMed
- B. P. Biswal, S. Chandra, S. Kandambeth, B. Lukose, T. Hein and R. Banerjee, J. Am. Chem. Soc., 2013, 135, 5328–5331 CrossRef CAS PubMed
- S. Karak, S. Kandambeth, B. P. Biswal, H. S. Sasmal, S. Kumar, P. Pachfule and R. Banerjee, J. Am. Chem. Soc., 2017, 139, 1856–1862 CrossRef CAS PubMed
- S. Dalapati, S. Jin, J. Gao, Y. Xu, A. Nagai and D. Jiang, J. Am. Chem. Soc., 2013, 135, 17310–17313 CrossRef CAS PubMed
- J. Guo, Y. Xu, S. Jin, L. Chen, T. Kaji, Y. Honsho, M. A. Addicoat, J. Kim, A. Saeki, H. Ihee, S. Seki, S. Irle, M. Hiramoto, J. Gao and D. Jiang, Nat. Commun., 2013, 4, 2736 CrossRef PubMed
- A. Nagai, X. Chen, X. Feng, X. Ding, Z. Guo and D. Jiang, Angew. Chem., Int. Ed., 2013, 52, 3770–3774 CrossRef CAS PubMed
- L. Grill, M. Dyer, L. Lafferentz, M. Persson, M. V. Peters and S. Hecht, Nat. Nanotechnol., 2007, 2, 687–691 CrossRef CAS PubMed
- M. O. Blunt, J. C. Russell, N. R. Champness and P. H. Beton, Chem. Commun., 2010, 46, 7157–7159 RSC
- C. R. Larrea and C. J. Baddeley, ChemPhysChem, 2016, 17, 971–975 CrossRef CAS PubMed
- T. Faury, S. Clair, M. Abel, F. Dumur, D. Gigmes and L. Porte, J. Phys. Chem. C, 2012, 116, 4819–4823 CrossRef CAS
- K. J. Shi, D. W. Yuan, C. X. Wang, C. H. Shu, D. Y. Li, Z. L. Shi, X. Y. Wu and P. N. Liu, Org. Lett., 2016, 18, 1282–1285 CrossRef CAS
- A. C. Marele, R. Mas-Balleste, L. Terracciano, J. Rodriguez-Fernandez, I. Berlanga, S. S. Alexandre, R. Otero, J. M. Gallego, F. Zamora and J. M. Gomez-Rodriguez, Chem. Commun., 2012, 48, 6779–6781 RSC
- E. Jin, M. Asada, Q. Xu, S. Dalapati, M. A. Addicoat, M. A. Brady, H. Xu, T. Nakamura, T. Heine, Q. Chen and D. Jiang, Science, 2017, 357, 673–676 CrossRef CAS
- H. Lyu, C. S. Diercks, C. Zhu and O. M. Yaghi, J. Am. Chem. Soc., 2019, 141, 6848–6852 CrossRef CAS
- K. T. Jackson, T. E. Reich and H. M. El-Kaderi, Chem. Commun., 2012, 48, 8823–8825 RSC
- D. Beaudoin, T. Maris and J. S. Wuest, Nat. Chem., 2013, 5, 830–834 CrossRef CAS PubMed
- X. Chen, M. Addicoat, E. Jin, H. Xu, T. Hayashi, F. Xu, N. Huang, S. Irle and D. Jiang, Sci. Rep., 2015, 5, 14650 CrossRef PubMed
- Y. Zeng, R. Zou, Z. Luo, H. Zhang, X. Yao, X. Ma, R. Zou and Y. Zhao, J. Am. Chem. Soc., 2015, 137, 1020–1023 CrossRef CAS PubMed
- P. Kuhn, M. Antonietti and A. Thomas, Angew. Chem., Int. Ed., 2008, 47, 3450–3453 CrossRef CAS PubMed
- N. L. Campbell, R. Clowes, L. K. Ritchie and A. I. Cooper, Chem. Mater., 2009, 21, 204–206 CrossRef CAS
- M. Dogru, A. Sonnauer, A. Gavryushin, P. Knochel and T. Bein, Chem. Commun., 2011, 47, 1707–1709 RSC
- J. W. Colson, A. R. Woll, A. Mukherjee, M. P. Levendorf, E. L. Spitler, V. P. Shields, M. G. Spencer, J. Park and W. R. Dichtel, Science, 2011, 332, 228–231 CrossRef CAS PubMed
- E. L. Spitler, J. W. Colson, F. J. Uribe-Romo, A. R. Woll, M. R. Giovino, A. Saldivar and W. R. Dichtel, Angew. Chem., Int. Ed., 2012, 51, 2623–2627 CrossRef CAS
- S. Y. Ding, X. H. Cui, J. Feng, G. Lu and W. Wang, Chem. Commun., 2017, 53, 11956–11959 RSC
- M. Matsumoto, R. R. Dasari, W. Ji, C. H. Feriante, T. C. Parker, S. R. Marder and W. R. Dichtel, J. Am. Chem. Soc., 2017, 139, 4999–5002 CrossRef CAS PubMed
- A. Werner, Z. Anorg. Allg. Chem., 1893, 3, 267–330 CrossRef
- Y. Kinoshita, I. Matsubara and Y. Saito, Bull. Chem. Soc. Jpn., 1959, 32, 1216–1221 CrossRef CAS
- Y. Kinoshita, I. Matsubara, T. Higuchi and Y. Saito, Bull. Chem. Soc. Jpn., 1959, 32, 1221–1226 CrossRef CAS
- N. Huang, L. Zhai, D. E. Coupry, M. A. Addicoat, K. Okushita, K. Nishimura, T. Heine and D. Jiang, Nat. Commun., 2016, 7, 12325 CrossRef
- A. P. Côté, H. M. El-Kaderi, H. Furukawa, J. R. Hunt and O. M. Yaghi, J. Am. Chem. Soc., 2007, 129, 12914–12915 CrossRef
- S.-B. Yu, H. Lyu, J. Tian, H. Wang, D.-W. Zhang, Y. Liu and Z.-T. Li, Polym. Chem., 2016, 7, 3392–3397 RSC
- S. Jin, K. Furukawa, M. Addicoat, L. Chen, S. Takahashi, S. Irle, T. Nakamura and D. Jiang, Chem. Sci., 2013, 4, 4505–4511 RSC
- R. W. Tilford, S. J. Mugavero, P. J. Pellechia and J. J. Lavigne, Adv. Mater., 2008, 20, 2741–2746 CrossRef CAS PubMed
- A. Nagai, Z. Guo, X. Feng, S. Jin, X. Chen, X. Ding and D. Jiang, Nat. Commun., 2011, 2, 536 CrossRef
- Z.-F. Pang, S.-Q. Xu, T.-Y. Zhou, R.-R. Liang, T.-G. Zhan and X. Zhao, J. Am. Chem. Soc., 2016, 138, 4710–4713 CrossRef CAS PubMed
- L. Ascherl, T. Sick, J. T. Margraf, S. H. Lapidus, M. Calik, C. Hettstedt, K. Karaghisoff, M. Dblinger, T. Clark, K. W. Chapman, F. Auras and T. Bein, Nat. Chem., 2016, 8, 310–316 CrossRef CAS
- Y. Zhu, S. Wan, Y. Jin and W. Zhang, J. Am. Chem. Soc., 2015, 137, 13772–13775 CrossRef CAS PubMed
- S. Dalapati, E. Jin, M. Addicoat, T. Heine and D. Jiang, J. Am. Chem. Soc., 2016, 138, 5797–5800 CrossRef CAS PubMed
- L. A. Baldwin, J. W. Crowe, M. D. Shannon, C. P. Jaroniec and P. L. McGrier, Chem. Mater., 2015, 27, 6169–6172 CrossRef CAS
- Y. P. Mo, X. H. Liu and D. Wang, ACS Nano, 2017, 11, 11694–11700 CrossRef CAS PubMed
- B. T. Koo, W. R. Dichtel and P. A. Clancy, J. Mater. Chem., 2012, 22, 17460–17469 RSC
-
J. W. Buchler,
in porphyrins, ed. D. Dolphin, Academic Press, New York, 1978, ch. 10, vol. 1, p. 389 Search PubMed
-
K. M. Kadish, K. M. Smith and R. Guilard, The Porphyrin Handbook, Academic Press, San Diego, CA, 2000, vol. 3 Search PubMed
- W. Seo, D. L. White and A. Star, Chem. – Eur. J., 2017, 23, 5652–5657 CrossRef CAS PubMed
- E. M. Johnson, R. Haiges and S. C. Marinescu, ACS Appl. Mater. Interfaces, 2018, 10, 37919–37927 CrossRef CAS PubMed
- K. Srinivasu and S. K. Ghosh, J. Phys. Chem. C, 2013, 117, 26021–26028 CrossRef CAS
- L. A. Baldwin, J. W. Crowe, D. A. Pyles and P. L. McGrier, J. Am. Chem. Soc., 2016, 138, 15134–15137 CrossRef CAS PubMed
- S.-Y. Ding, J. Gao, Q. Wang, Y. Zhang, W.-G. Song, C.-Y. Su and W. Wang, J. Am. Chem. Soc., 2011, 133, 19816–19822 CrossRef CAS PubMed
- L. Chen, L. Zhang, Z. Chen, H. Liu, R. Luque and Y. Li, Chem. Sci., 2016, 7, 6015–6020 RSC
- D. Sun, S. Jang, S.-J. Yim, L. Ye and D.-P. Kim, Adv. Funct. Mater., 2018, 28, 1707110 CrossRef
- Y. Hou, X. Zhang, J. Sun, S. Lin, D. Qi, R. Hong, D. Li, X. Xiao and J. Jiang, Microporous Mesoporous Mater., 2015, 214, 108–114 CrossRef CAS
- W. Leng, Y. Peng, J. Zhang, H. Lu, X. Feng, R. Ge, B. Dong, B. Wang, X. Hu and Y. Gao, Chem. – Eur. J., 2016, 22, 9087–9091 CrossRef CAS PubMed
- W. Leng, R. Ge, B. Dong, C. Wang and Y. Gao, RSC Adv., 2016, 6, 37403–37406 RSC
- S. Lin, Y. Hou, X. Deng, H. Wang, S. Sun and X. Zhang, RSC Adv., 2015, 5, 41017–41024 RSC
- P. Pachfule, M. K. Panda, S. Kandambeth, S. M. Shivaprasad, D. D. Díaz and R. Banerjee, J. Mater. Chem. A, 2014, 2, 7944–7956 RSC
- A. de la Peña Ruigómez, D. Rodríguez-San-Miguel, K. C. Stylianou, M. Cavallini, D. Gentili, F. Liscio, S. Milita, O. M. Roscioni, M. L. Ruiz-González, C. Carbonell, D. Maspoch, R. Mas-Ballesté, J. L. Segura and F. Zamora, Chem. – Eur. J., 2015, 21, 10666–10670 CrossRef
- J. Romero, D. Rodriguez-San-Miguel, A. Ribera, R. Mas-Ballesté, T. F. Otero, I. Manet, F. Licio, G. Abellán, F. Zamora and E. Coronado, J. Mater. Chem. A, 2017, 5, 4343–4351 RSC
- H. B. Aiyappa, J. Thote, D. B. Shinde, R. Banerjee and S. Kurungot, Chem. Mater., 2016, 28, 4375–4379 CrossRef CAS
- Q. Sun, B. Aguila, J. Perman, N. Nguyen and S. Ma, J. Am. Chem. Soc., 2016, 138, 15790–15796 CrossRef CAS PubMed
- M. Mu, Y. Wang, Y. Qin, X. Yan, Y. Li and L. Chen, ACS Appl. Mater. Interfaces, 2017, 9, 22856–22863 CrossRef CAS
- P. G. Cozzi, Chem. Soc. Rev., 2004, 33, 410–421 RSC
- T. P. Yoon and E. N. Jacobsen, Science, 2003, 299, 1691–1693 CrossRef CAS PubMed
- L.-H. Li, X.-L. Feng, X.-H. Cui, Y.-X. Ma, S.-Y. Ding and W. Wang, J. Am. Chem. Soc., 2017, 139, 6042–6045 CrossRef CAS PubMed
- H. Vardhan, G. Verma, S. Ramani, A. Nafady, A. M. Al-Enizi, Y. Pan, Z. Yang, H. Yang and S. Ma, ACS Appl. Mater. Interfaces, 2019, 11, 3070–3079 CrossRef CAS PubMed
- H. Vardhan, L. Hou, E. Yee, A. Nafady, A. M. Al-Enizi, Y. Pan, Z. Yang and S. Ma, ACS Sustainable Chem. Eng., 2019, 7, 4878–4888 CrossRef CAS
- Y. Liu, Y. Ma, Y. Zhao, X. Sun, F. Gándara, H. Furukawa, Z. Liu, H. Zhu, C. Zhu, K. Suenaga, P. Oleynikov, A. S. Alshammari, X. Zhang, O. Terasaki and O. M. Yaghi, Science, 2016, 351, 365–369 CrossRef CAS PubMed
- H. Wang, F. Jiao, F. Gao, Y. Lv, Q. Wu, Y. Zhao, Y. Shen, Y. Zhang and X. Qian, Talanta, 2017, 166, 133–140 CrossRef CAS PubMed
- Q. Sun, B. Aguila and S. Ma, Mater. Chem. Front., 2017, 1, 1310–1316 RSC
- Y. Yang, M. Faheem, L. Wang, Q. Meng, H. Sha, N. Yang, Y. Yuan and G. Zhu, ACS Cent. Sci., 2018, 4, 748–754 CrossRef CAS PubMed
- F. J. Uribe-Romo, C. J. Doonan, H. Furukawa, K. Oisaki and O. M. Yaghi, J. Am. Chem. Soc., 2011, 133, 11478–11481 CrossRef CAS PubMed
- W. Zhang, P. Jiang, Y. Wang, J. Zhang, Y. Gao and P. Zhang, RSC Adv., 2014, 4, 51544–51547 RSC
- T. Kundu, J. Wang, Y. Cheng, Y. Du, Y. Qian, G. Liu and D. Zhao, Dalton Trans., 2018, 47, 13824–13829 RSC
- L. Chen, K. Furukawa, J. Gao, A. Nagai, T. Nakamura, Y. Dong and D. Jiang, J. Am. Chem. Soc., 2014, 136, 9806–9809 CrossRef CAS PubMed
- N. Huang, R. Krishna and D. Jiang, J. Am. Chem. Soc., 2015, 137, 7079–7082 CrossRef CAS PubMed
- F. Xu, H. Xu, X. Chen, D. Wu, Y. Wu, H. Liu, C. Gu, R. Fu and D. Jiang, Angew. Chem., Int. Ed., 2015, 54, 6814–6818 CrossRef CAS PubMed
- H. Xu, J. Gao and D. Jiang, Nat. Chem., 2015, 7, 905–912 CrossRef CAS PubMed
- S. Royuela, E. García-Garrido, M. Martín Arroyo, M. J. Manche
![[small eta, Greek, tilde]](https://www.rsc.org/images/entities/char_e110.gif) o, M. M. Ramos, D. González-Rodríguez, A. Samoza, F. Zamora and J. L. Segura, Chem. Commun., 2018, 54, 8729–8732 RSC
o, M. M. Ramos, D. González-Rodríguez, A. Samoza, F. Zamora and J. L. Segura, Chem. Commun., 2018, 54, 8729–8732 RSC - L. Merí-Bofí, S. Royuela, F. Zamora, M. L. Ruiz-González, J. L. Segura, R. Muoz-Olivas and M. J. Mancheo, J. Mater. Chem. A, 2017, 5, 17973–17981 RSC
- D. N. Bunck and W. R. Dichtel, Chem. Commun., 2013, 49, 2457–2459 RSC
- Q. Sun, B. Aguila, J. Perman, L. D. Earl, C. W. Abney, Y. Cheng, H. Wei, N. Nguyen, L. Wojtas and S. Ma, J. Am. Chem. Soc., 2017, 139, 2786–2793 CrossRef CAS PubMed
- Q. Sun, B. Aguila, J. A. Perman, T. Butts, F.-S. Xiao and S. Ma, Chem, 2018, 4, 1726–1739 CAS
- Q. Sun, B. Aguila, L. D. Earl, C. W. Abney, L. Wojtas, P. K. Thallapally and S. Ma, Adv. Mater., 2018, 30, 1705479 CrossRef PubMed
- B. Zhang, M. Wei, H. Mao, X. Pei, S. A. Alshmimri, J. A. Reimer and O. M. Yaghi, J. Am. Chem. Soc., 2018, 140, 12715–12719 CrossRef CAS PubMed
- X. Guan, H. Li, Y. Ma, M. Xue, Q. Fang, Y. Yan, V. Valtchev and S. Qiu, Nat. Chem., 2019, 11, 587–594 CrossRef CAS
- W. Ji, L. Xiao, Y. Ling, C. Ching, M. Matsumoto, R. P. Bisbey, D. E. Helbling and W. R. Dichtel, J. Am. Chem. Soc., 2018, 140, 12677–12681 CrossRef CAS PubMed
- H.-S. Xu, S.-Y. Ding, W.-K. An, H. Wu and W. Wang, J. Am. Chem. Soc., 2016, 138, 11489–11492 CrossRef CAS PubMed
- N. Huang, X. Chen, R. Krishna and D. Jiang, Angew. Chem., Int. Ed., 2015, 54, 2986–2990 CrossRef CAS PubMed
- S. Zhao, B. Dong, R. Ge, C. Wang, X. Song, W. Ma, Y. Wang, C. Hao, X. Guo and Y. Gao, RSC Adv., 2016, 6, 38774–38781 RSC
- B. Dong, L. Wang, S. Zhao, R. Ge, X. Song, Y. Wang and Y. Gao, Chem. Commun., 2016, 52, 7082–7085 RSC
- Z.-J. Mu, X. Ding, Z.-Y. Chen and B.-H. Han, ACS Appl. Mater. Interfaces, 2018, 10, 41350–41358 CrossRef CAS PubMed
- Q. Lu, Y. Ma, H. Li, X. Guan, Y. Yusran, M. Xue, Q. Fang, Y. Yan, S. Qiu and V. Valtchev, Angew. Chem., Int. Ed., 2018, 57, 6042–6048 CrossRef CAS PubMed
- S. Rager, M. Dogru, V. Werner, A. Gavryushin, M. Götz, H. Engelke, D. D. Medina, P. Knochel and T. Bein, CrystEngComm, 2017, 19, 4886–4891 RSC
- M. S. Lohse, T. Stassin, G. Naudin, S. Wuttke, R. Ameloot, D. De Vos, D. D. Medina and T. Bein, Chem. Mater., 2016, 28, 626–631 CrossRef CAS
- D. B. Shinde, H. B. Aiyappa, M. Bhadra, B. P. Biswal, P. Wadge, S. Kandambeth, B. Garai, T. Kundu, S. Kurungot and R. Banerjee, J. Mater. Chem. A, 2016, 4, 2682–2690 RSC
- W. Zhong, R. Sa, L. Li, Y. He, L. Li, J. Bi, Z. Zhuang, Y. Yu and Z. Zou, J. Am. Chem. Soc., 2019, 141, 7615–7621 CrossRef CAS
- Z. Zou, W. Zhong, K. Cui, Z. Zhuang, L. Li, L. Li, J. Bi and Y. Yu, Chem. Commun., 2018, 54, 9977–9980 RSC
- H. Vardhan, Y. Pan, Z. Yang, G. Verma, A. Nafady, A. M. Al-Enizi, T. M. Alotaibi, O. A. Almaghrabi and S. Ma, APL Mater., 2019, 7, 101111 CrossRef
- J. L. Segura, S. Royuela and M. M. Ramos, Chem. Soc. Rev., 2019, 48, 3903–3945 RSC
- P. J. Waller, S. J. Lyle, T. M. Osborn Popp, C. S. Diercks, J. A. Reimer and O. M. Yaghi, J. Am. Chem. Soc., 2016, 138, 15519–15522 CrossRef CAS
- X. Han, J. Huang, C. Yuan, Y. Liu and Y. Cui, J. Am. Chem. Soc., 2018, 140, 892–895 CrossRef CAS
- S. Wang, Q. Wang, P. Shao, Y. Han, X. Gao, L. Ma, S. Yuan, X. Ma, J. Zhou, X. Feng and B. Wang, J. Am. Chem. Soc., 2017, 139, 4258–4261 CrossRef CAS PubMed
- F. Haase, E. Troschke, G. Savasci, T. Banerjee, V. Duppel, S. Dörfler, M. M. J. Grundei, A. M. Burow, C. Ochsenfeld, S. Kaskel and B. V. Lotsch, Nat. Commun., 2018, 9, 2600 CrossRef PubMed
- X. Li, C. Zhang, S. Cai, X. Lei, V. Altoe, F. Hong, J. J. Urban, J. Ciston, E. M. Chan and Y. Liu, Nat. Commun., 2018, 9, 2998 CrossRef PubMed
- Q. Jiang, Y. Li, X. Zhao, P. Xiong, X. Yu, Y. Xu and L. Chen, J. Mater. Chem. A, 2018, 6, 17977–17981 RSC
- Y. Wu, Z. Zhang, S. Bandow and K. Awaga, Bull. Chem. Soc. Jpn., 2017, 90, 1382–1387 CrossRef CAS
- D.-G. Wang, N. Li, Y. Hu, S. Wan, M. Song, G. Yu, Y. Jin, W. Wei, K. Han, G.-C. Kuang and W. Zhang, ACS Appl. Mater. Interfaces, 2018, 10, 42233–42240 CrossRef CAS PubMed
- C. Qian, Q.-Y. Qi, G.-F. Jiang, F.-Z. Cui, Y. Tian and X. Zhao, J. Am. Chem. Soc., 2017, 139, 6736–6743 CrossRef CAS PubMed
- G. Zhang, M. Tsujimoto, D. Packwood, N. T. Duong, Y. Nishiyama, K. Kadota, S. Kitagawa and S. Horike, J. Am. Chem. Soc., 2018, 140, 2602–2609 CrossRef CAS PubMed
- H.-L. Qian, Y. Li and X.-P. Yan, J. Mater. Chem. A, 2018, 6, 17307–17311 RSC
- P. J. Waller, Y. S. AlFaraj, C. S. Diercks, N. N. Jarenwattananon and O. M. Yaghi, J. Am. Chem. Soc., 2018, 140, 9099–9103 CrossRef CAS PubMed
- M. C. Daugherty, E. Vitaku, R. L. Li, A. M. Evans, A. D. Chavez and W. R. Dichtel, Chem. Commun., 2019, 55, 2680–2683 RSC
- M. R. Rao, Y. Fang, S. De Feyter and D. F. Perpichka, J. Am. Chem. Soc., 2017, 139, 2421–2427 CrossRef CAS PubMed
- Q. Fang, J. Wang, S. Gu, R. B. Kaspar, Z. Zhuang, J. Zheng, H. Guo, S. Qiu and Y. Yan, J. Am. Chem. Soc., 2015, 137, 8352–8355 CrossRef CAS
- L. Bai, S. Z. Phua, W. Q. Lim, A. Jana, Z. Luo, H. P. Tham, L. Zhao, Q. Gao and Y. Zhao, Chem. Commun., 2016, 52, 4128–4131 RSC
- S. Mitra, H. S. Sasmal, T. Kundu, S. Kandambeth, K. lllath, D. D. Díaz and R. Banerjee, J. Am. Chem. Soc., 2017, 139, 4513–4520 CrossRef CAS
- V. S. Vyas, M. Vishwakarma, I. Moudrakovski, F. Haase, G. Savasci, C. Ochsenfeld, J. P. Spatz and B. V. Lotsch, Adv. Mater., 2016, 28, 8749–8754 CrossRef CAS PubMed
- P. Bhanja, S. Mishra, K. Manna, A. Mallick, K. Das Saha and A. Bhaumik, ACS Appl. Mater. Interfaces, 2017, 9, 31411–31423 CrossRef CAS
- S. Kandambeth, V. Venkatesh, D. B. Shinde, S. Kumari, A. Halder, S. Verma and R. Banerjee, Nat. Commun., 2015, 6, 6786 CrossRef CAS PubMed
- S. Zhang, Y. Zheng, H. An, B. Aguila, C.-X. Yang, Y. Dong, W. Xie, P. Cheng, Z. Zhang, Y. Chen and S. Ma, Angew. Chem., Int. Ed., 2018, 57, 16754–16759 CrossRef CAS PubMed
- Q. Sun, C.-W. Fu, B. Aguila, J. Perman, S. Wang, H.-Y. Huang, F.-S. Xiao and S. Ma, J. Am. Chem. Soc., 2018, 140, 984–992 CrossRef CAS PubMed
- J. Tan, S. Namuangruk, W. Kong, N. Kungwan, J. Guo and C. Wang, Angew. Chem., Int. Ed., 2016, 55, 13979–13984 CrossRef CAS
- X. Zheng, L. Wang, Q. Pei, S. He, S. Liu and Z. Xie, Chem. Mater., 2017, 29, 2374–2381 CrossRef CAS
- S. Mitra, S. Kandambeth, B. P. Biswal, M. A. Khayum, C. K. Choudhury, M. Mehta, G. Kaur, S. Banerjee, A. Prabhune, S. Verma, S. Roy, U. K. Kharul and R. Banerjee, J. Am. Chem. Soc., 2016, 138, 2823–2828 CrossRef CAS PubMed
- Q. Fang, S. Gu, J. Zheng, Z. Zhuang, S. Qiu and Y. Yan, Angew. Chem., Int. Ed., 2014, 53, 2878–2882 CrossRef CAS PubMed
- Y. Peng, Z. Hu, Y. Gao, D. Yuan, Z. Kang, Y. Qian, N. Yan and D. Zhao, ChemSusChem, 2015, 8, 3208–3212 CrossRef CAS PubMed
- S. Lin, C. S. Diercks, Y.-B. Zhang, N. Kornienko, E. M. Nichols, Y. Zhao, A. R. Paris, D. Kim, P. Yang, O. M. Yaghi and C. J. Chang, Science, 2015, 349, 1208–1213 CrossRef CAS PubMed
- V. Saptal, D. B. Shinde, R. Banerjee and B. M. Bhanage, Catal. Sci. Technol., 2016, 6, 6152–6158 RSC
- X. Wang, X. Han, J. Zheng, X. Wu, Y. Liu and Y. Cui, J. Am. Chem. Soc., 2016, 138, 12332–12335 CrossRef CAS PubMed
- D. B. Shinde, S. Kandambeth, P. Pachfule, R. R. Kumar and R. Banerjee, Chem. Commun., 2015, 51, 310–313 RSC
- X. Li, Z. Wang, J. Sun, J. Gao, Y. Zhao, P. Cheng, B. Aguila, S. Ma, Y. Chen and Z. Zhang, Chem. Commun., 2019, 55, 5423–5426 RSC
- V. S. Vyas, F. Haase, L. Stegbauer, G. Savasci, F. Podjaski, C. Ochsenfeld and B. V. Lotsch, Nat. Commun., 2015, 6, 8505 CrossRef PubMed
- T. Sick, A. G. Hufnagel, J. Kampmann, I. Kondofersky, M. Calik, J. M. Rotter, A. Evans, M. Döblinger, S. Herbert, K. Peters, D. Böhm, P. Knochel, D. D. Medina, D. Fattakhova-Rohlfing and T. Bein, J. Am. Chem. Soc., 2018, 140, 2085–2092 CrossRef CAS PubMed
- J. Thote, H. B. Aiyappa, A. Deshpande, D. D. Díaz, S. Kurungot and R. Banerjee, Chem. – Eur. J., 2014, 20, 15961–15965 CrossRef CAS PubMed
- S.-Y. Ding, M. Dong, Y.-W. Wang, Y.-T. Chen, H.-Z. Wang, C.-Y. Su and W. Wang, J. Am. Chem. Soc., 2016, 138, 3031–3037 CrossRef CAS PubMed
- N. Huang, L. Zhai, H. Xu and D. Jiang, J. Am. Chem. Soc., 2017, 139, 2428–2434 CrossRef CAS PubMed
- Z. Li, H. Li, X. Guan, J. Tang, Y. Yusran, Z. Li, M. Xue, Q. Fang, Y. Yan, V. Valtchev and S. Qiu, J. Am. Chem. Soc., 2017, 139, 17771–17774 CrossRef CAS PubMed
- G. Das, T. Skorjanc, S. K. Sharma, F. Gándara, M. Lusi, D. S. S. Rao, S. Vimala, S. K. Prasad, J. Raya, M. Lusi, D. S. Han, R. Jagannathan, J.-C. Oslen and A. Trabolsi, J. Am. Chem. Soc., 2017, 139, 9558–9565 CrossRef CAS PubMed
- N. Huang, P. Wang, M. A. Addicoat, T. Heine and D. Jiang, Angew. Chem., Int. Ed., 2017, 56, 4982–4986 CrossRef CAS PubMed
- G.-H. Ning, Z. Chen, Q. Gao, W. Tang, Z. Chen, C. Liu, B. Tian, X. Li and K. P. Loh, J. Am. Chem. Soc., 2017, 139, 8897–8904 CrossRef CAS PubMed
- H. Furukawa and O. M. Yaghi, J. Am. Chem. Soc., 2009, 131, 8875–8883 CrossRef CAS PubMed
- H. Li, Q. Pan, Y. Ma, X. Guan, M. Xue, Q. Fang, Y. Yan, V. Valtchev and S. Qiu, J. Am. Chem. Soc., 2016, 138, 14783–14788 CrossRef CAS PubMed
- S. B. Kalidindi and R. A. Fischer, Phys. Status Solidi B, 2013, 250, 1119–1127 CrossRef CAS
- Y. Zeng, R. Zou and Y. Zhao, Adv. Mater., 2016, 28, 2855–2873 CrossRef CAS PubMed
- C. J. Doonan, D. J. Tranchemontagne, T. G. Glover, J. R. Hunt and O. M. Yaghi, Nat. Chem., 2010, 2, 235–238 CrossRef CAS PubMed
- H. Ma, H. Ren, S. Meng, Z. Yan, H. Zhao, F. Sun and G. Zhu, Chem. Commun., 2013, 49, 9773–9775 RSC
- S.-L. Cai, K. Zhang, J.-B. Tan, S. Wang, S.-R. Zheng, J. Fan, Y. Yu, W.-G. Zhang and Y. Liu, ACS Macro Lett., 2016, 5, 1348–1352 CrossRef CAS
- Z. Li, X. Feng, Y. Zou, Y. Zhang, H. Xia, X. Liu and Y. Mu, Chem. Commun., 2014, 50, 13825–13828 RSC
- G. Lin, H. Ding, D. Yuan, B. Wang and C. Wang, J. Am. Chem. Soc., 2016, 138, 3302–3305 CrossRef CAS PubMed
- C. Zhang, S. Zhang, Y. Yan, F. Xia, A. Huang and Y. Xian, ACS Appl. Mater. Interfaces, 2017, 9, 13415–13421 CrossRef CAS PubMed
- Y.-F. Xie, S.-Y. Ding, J.-M. Liu, W. Wang and Q.-Y. Zheng, J. Mater. Chem. C, 2015, 3, 10066–10069 RSC
- G. Das, B. P. Biswal, S. Kandambeth, V. Venkatesh, G. Kaur, M. Addicoat, T. Heine, S. Verma and R. Banerjee, Chem. Sci., 2015, 6, 3931–3939 RSC
- Z. Li, Y. Zhang, H. Xia, Y. Mu and X. Liu, Chem. Commun., 2016, 52, 6613 RSC
- Y. Peng, Y. Huang, Y. Zhu, B. Chen, L. Wang, Z. Lai, Z. Zhang, M. Zhao, C. Tan, N. Yang, F. Shao, Y. Han and H. Zhang, J. Am. Chem. Soc., 2017, 139, 8698–8704 CrossRef CAS
- A. Mal, R. K. Mishra, V. K. Praveen, M. A. Khayum, R. Banerjee and A. Ajayaghosh, Angew. Chem., Int. Ed., 2018, 57, 8443–8447 CrossRef CAS PubMed
- S. Wan, J. Guo, J. Kim, H. Ihee and D. Jiang, Angew. Chem., Int. Ed., 2009, 48, 5439–5442 CrossRef CAS
- X. Feng, L. Chen, Y. Honsho, O. Saengsawang, L. Liu, L. Wang, A. Saeki, S. Irle, S. Seki, Y. Dong and D. Jiang, Adv. Mater., 2012, 24, 3026–3031 CrossRef CAS
- M. Dogru, M. Handloser, F. Auras, T. Kunz, D. Medina, A. Hartschuh, P. Knochel and T. Bein, Angew. Chem., Int. Ed., 2013, 52, 2920–2924 CrossRef CAS
- Y. Zhang, J. Duan, D. Ma, P. Li, S. Li, H. Li, J. Zhou, X. Ma, X. Feng and B. Wang, Angew. Chem., Int. Ed., 2017, 56, 16313–16317 CrossRef CAS PubMed
- Q. Xu, S. Tao, Q. Jiang and D. Jiang, J. Am. Chem. Soc., 2018, 140, 7429–7432 CrossRef CAS PubMed
- S. Chandra, T. Kundu, S. Kandambeth, R. BabaRao, Y. Marathe, S. M. Kunjir and R. Banerjee, J. Am. Chem. Soc., 2014, 136, 6570–6573 CrossRef CAS PubMed
- H. Ma, B. Liu, B. Lin, L. Zhang, Y.-G. Li, H.-Q. Tan, H.-Y. Zang and G. Zhu, J. Am. Chem. Soc., 2016, 138, 5897–5903 CrossRef CAS PubMed
- C. R. DeBlase, K. E. Silberstein, T.-T. Truong, H. D. Abruna and W. R. Dichtel, J. Am. Chem. Soc., 2013, 135, 16821–16824 CrossRef CAS PubMed
- C. R. Mulzer, L. Shen, R. P. Bisbey, J. R. McKone, N. Zhang, H. D. Abruna and W. R. Dichtel, ACS Cent. Sci., 2016, 2, 667–673 CrossRef CAS PubMed
- J. W. Colson, A. R. Woll, A. Mukherjee, M. P. Levendorf, E. L. Spitler, V. B. Shields, M. G. Spencer, J. Park and W. R. Dichtel, Science, 2011, 332, 228–231 CrossRef CAS PubMed
- E. L. Spitler, J. W. Colson, F. J. Uribe-Romo, A. R. Woll, M. R. Giovino, A. Saldivar and W. R. Dichtel, Angew. Chem., Int. Ed., 2012, 51, 2623–2627 CrossRef CAS PubMed
- D. D. Medina, M. L. Petrus, A. N. Jumabekov, J. T. Margraf, S. Weinberger, J. M. Rotter, T. Clark and T. Bein, ACS Nano, 2017, 11, 2706–2713 CrossRef CAS PubMed
- Q. Hao, C. Zhao, B. Sun, C. Lu, J. Liu, M. Liu, L.-J. Wan and D. Wang, J. Am. Chem. Soc., 2018, 140, 12152–12158 CrossRef CAS
- J. Lv, Y.-X. Tan, J. Xie, R. Yang, M. Yu, S. Sun, M.-D. Li, D. Yuan and Y. Wang, Angew. Chem., Int. Ed., 2018, 57, 12716–12720 CrossRef CAS
- R. Chen, J.-L. Shi, Y. Ma, G. Lin, X. Lang and C. Wang, Angew. Chem., Int. Ed., 2019, 58, 6430–6434 CrossRef CAS
- C. Wu, Y. Liu, H. Liu, C. Duan, Q. Pan, J. Zhu, F. Hu, X. Ma, T. Jiu, Z. Li and Y. Zhao, J. Am. Chem. Soc., 2018, 140, 10016–10024 CrossRef CAS
| This journal is © The Royal Society of Chemistry 2019 |