
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInnovative omics-based approaches for prioritisation and targeted isolation of natural products – new strategies for drug discovery
Jean-Luc
Wolfender
 *a,
Marc
Litaudon
b,
David
Touboul
b and
Emerson Ferreira
Queiroz
a
*a,
Marc
Litaudon
b,
David
Touboul
b and
Emerson Ferreira
Queiroz
a
aSchool of Pharmaceutical Sciences, University of Geneva, University of Lausanne, CMU – Rue Michel Servet 1, 1211 Geneva 11, Switzerland. E-mail: Jean-Luc.Wolfender@unige.ch
bInstitut de Chimie des Substances Naturelles, CNRS-ICSN, UPR 2301, Université Paris-Saclay, 91198, Gif-sur-Yvette, France
First published on 10th May 2019
Abstract
Covering: 2013 to 2019
The exploration of the chemical diversity of extracts from various biological sources has led to major drug discoveries. Over the past two decades, despite the introduction of advanced methodologies for natural product (NP) research (e.g., dereplication and high content screening), successful accounts of the validation of NPs as lead therapeutic candidates have been limited. In this context, one of the main challenges faced is related to working with crude natural extracts because of their complex composition and the inadequacies of classical bioguided isolation studies given the pace of high-throughput screening campaigns. In line with the development of metabolomics, genomics and chemometrics, significant advances in metabolite profiling have been achieved and have generated high-quality massive genome and metabolome data on natural extracts. The unambiguous identification of each individual NP in an extract using generic methods remains challenging. However, the establishment of structural links among NPs via molecular network analysis and the determination of common features of extract composition have provided invaluable information to the scientific community. In this context, new multi-informational-based profiling approaches integrating taxonomic and/or bioactivity data can hold promise for the discovery and development of new bioactive compounds and return NPs back to an exciting era of development. In this article, we examine recent studies that have the potential to improve the efficiency of NP prioritisation and to accelerate the targeted isolation of key NPs. Perspectives on the field's evolution are discussed.
 Jean-Luc Wolfender | Jean-Luc Wolfender is a chemist and leads a natural product research group at the School of Pharmaceutical Sciences of the University of Geneva (Switzerland). He was closely involved with the introduction of LC-MS and LC-NMR for dereplication purposes in the 1990s. He is currently developing innovative MS- and NMR-based metabolomics strategies for natural product-based drug discoveries and chemical ecology projects. His research also examines modes of action of phytopharmaceuticals from a systems biology perspective. He is currently pursuing the promotion of metabolomics within the natural product community. |
 Marc Litaudon | Marc Litaudon received his PhD in Pharmaceutical Sciences in 1991 (University Paris-Sud, France) under the guidance of Dr M. Guyot and Prof. P. Potier. He then conducted postdoctoral research in marine chemistry with Prof. M. H. G. Munro and J. W. Blunt at the University of Canterbury (NZ). In 1994 he served in New Caledonia as the Head of “Laboratory of Medicinal Plants” (CNRS). In 2001, he joined the ICSN and since has led its phytochemical laboratory and the management and scientific development of the “ICSN Extracts Library”, one of the largest collections of natural extracts in France. |
 David Touboul | David Touboul is a former student of the Ecole Normale Supérieure of Cachan (1999–2003). He completed his PhD in Chemistry in 2006 and received a grant from Novartis US to complete a post-doctoral fellowship with Prof. R. Zenobi's research group at ETH Zurich (non-covalent complexes in the gas phase, 2006–2008). He was then recruited by the CNRS “Institut de Chimie des Substances Naturelles” as a research associate and has led the Mass Spectrometry group since January 2019. His team develops new analytical tools for the structural characterisation of natural products, including supercritical fluid chromatography, tandem mass spectrometry and bioinformatics tools (MetGem Software). |
 Emerson Ferreira Queiroz | Emerson F. Queiroz holds a PhD in pharmaceutical sciences from the University of Paris-Sud (France). In 1999 he joined Prof. Kurt Hostettmann's group at the University of Lausanne for post-doctoral training (1999–2006). From 2006 to 2011 he worked as the head of the research and development department of Aché Laboratories in Brazil. He is currently a Senior Scientist at the University of Geneva (Switzerland). His main research interests include the discovery of naturally occurring drugs, microbial biotransformation for the production of bioactive compounds and the development of innovative approaches to the isolation and rapid identification of novel natural products. |
1 Introduction
It is widely accepted in the scientific community that natural products (NPs) remain a major source of new chemical entities with potential applications for drug discovery (especially as anticancer and anti-infective agents). Standardised natural extracts are also widely used as botanicals for phytotherapy and notably for the management of chronic diseases. However, there has been a lack of interest from the pharmaceutical industry in investigating natural resources since the beginning of 2000.1 This is mainly attributable to the lengthy and tedious procedures involved in the isolation and characterisation of bioactive NPs, to the redundancy of NPs stored within libraries sourced for discovery, and to the limited compatibility of crude natural extracts (CNEs) with the high throughput of bioactivity screening campaigns on selected targets.2To address these issues, the scientific community has revitalised the discipline by: (I) rationally exploiting collections of CNEs and NPs representative of terrestrial and marine biodiversity while taking into account regulations for a fair and equitable sharing of benefits arising from the usage of genetic resources (Nagoya Protocol)3 and (II) developing advanced analytical protocols for the rapid identification of previously known NPs in CNEs (dereplication) to avoid their unnecessary re-isolation. For this latter aspect, the metabolite profiling of complex CNEs for the early identification of NPs (metabolite annotation) has evolved considerably over the last decade in line with the development of metabolomics in other fields of life sciences.4 In particular, the development of efficient workflows combining sophisticated analytical techniques and powerful data mining software has started to provide partial/full structure information of a large number of NPs in extracts. Such approaches allow for an in-depth exploration of the chemical space covered by metabolomes of micro- or macroscopic living organisms. They document the complexity/chemodiversity of such organisms and reveal new metabolic pathways through the concomitant exploration of genomics data and particularly in deciphering cryptic metabolic pathways. Such state-of-the-art metabolite profiling methods when used in a generic manner rarely allow for the full and unambiguous structure determination of detected NPs. They provide, however, invaluable estimations of the types of compounds present in given extracts and of their structural relationships. Their use has already led to notable successes in the discovery of new NPs and analogues.
This detailed compositional information can be complemented with semi-quantitative estimations made using orthogonal methods such as mass spectrometry (MS) (e.g., nuclear magnetic resonance (NMR), evaporative light scattering detector (ELSD) or charged aerosol detector (CAD)). Linking such data to bioactivity results acquired in screening campaigns and/or with previously reported pharmacological data on annotated NPs allows one to interpret screening results from a novel and holistic perspective. It also allows for the prioritisation of NPs and for the targeted isolation of new bioactive molecules of interest.
This highlight article does not claim to exhaustively list all methodologies developed and used by chemists and biologists for the discovery of new bioactive metabolites. We instead discuss the most relevant and/or innovative approaches based on their design bioactivity, chemical composition and/or genetic data mainly by means of chemometric/biochemometric/chemogenomic methods5,6 and show through select examples how they can accelerate the discovery of new bioactive NPs. We also discuss the most effective methods enabling the targeted isolation of NPs from metabolite profiling data for a rapid, efficient and complete characterisation of bioactive and/or novel NPs.
2 Design of NP libraries for prioritisation studies
The implementation of an efficient NP prioritisation pipeline must first rely on a well-designed collection of CNEs. One of the world's largest and most diverse screening libraries of CNEs was developed in the 1980s and 1990s by the National Cancer Institute (NCI). Approximately 65![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 plant specimens, 15000 marine specimens and 20000 fungal cultures have been processed to yield tens of thousands of CNEs with the collection being initially designed to facilitate anticancer drug discovery.7 Today, while it is probably not necessary to assemble such a large number of organisms to discover novel bioactive molecules, it is likely that chances of success are closely linked to the selection of certain types and number of initial samples. This can be done by maximising NP diversity in extract libraries prepared from plants, marine organisms, microorganisms and other organisms as long as the biodiversity of an ecosystem, a kingdom, a region or a given taxonomic group is well represented. A collection of extracts can be limited to a given genus or taxon or to specifically enriched extracts, for example, to allow for the study of a series of NP analogues with given scaffold types.
000 plant specimens, 15000 marine specimens and 20000 fungal cultures have been processed to yield tens of thousands of CNEs with the collection being initially designed to facilitate anticancer drug discovery.7 Today, while it is probably not necessary to assemble such a large number of organisms to discover novel bioactive molecules, it is likely that chances of success are closely linked to the selection of certain types and number of initial samples. This can be done by maximising NP diversity in extract libraries prepared from plants, marine organisms, microorganisms and other organisms as long as the biodiversity of an ecosystem, a kingdom, a region or a given taxonomic group is well represented. A collection of extracts can be limited to a given genus or taxon or to specifically enriched extracts, for example, to allow for the study of a series of NP analogues with given scaffold types.
The screening of CNEs is still a valid option when done with robust bioassays.8 In academic laboratories, the screening of collections of a few hundred extracts corresponding to a defined corpus is typical in focused studies.9–14 These small collections are easier to handle. Their size is compatible with concomitant metabolite profiling and mining and with the capacities of tools currently available for NP prioritisation studies as discussed below. However, to maximise the chances of discovering new bioactive NPs from complex mixtures, a systematic preliminary rapid enrichment step of CNEs using sequential extractions,15 solid phase extraction (SPE)16 or coarse generic semi-preparative HPLC fractionation17 seems necessary. Such enrichment procedures may improve biological and chemical profiling screening steps. In addition, the fractionation of extracts prior to bioassays or performed on selected bioactive extracts can ideally partly overcome potential synergistic or antagonistic effects observed within CNEs. This step also offers an undeniable advantage by increasing the constituent concentration in each fraction. In this context, the NCI in 2018 launched a new programme to produce a publicly accessible HTS amenable library of >1000000 fractions prepared from 125000 extracts.15 The project is intended to improve compatibility with HTS screening and to avoid pan-assay interference compounds (PAINS).18
3 Metabolite profiling of crude natural extracts
Since the early 2000s, advances in MS- and/or NMR-based approaches associated with bioinformatics tools have been decisive in the design and development of NP studies. These methods are used to investigate the molecular content of biological systems with unprecedented sensitivity and precision. They are extensively used for dereplication19 prior to performing bioactivity-guided isolation studies and have been to a greater extent used for the comprehensive metabolite profiling of CNEs.4 Most current metabolite profiling studies are performed with state-of-the-art high-resolution LC-MS20 tools that apply the (i) high resolution of ultra-high-performance liquid chromatography (UHPLC) for the chromatographic resolution of isomers and (ii) high-resolution MS methods for molecular formula assignment. While chromatography is usually performed using reverse phase (RP) liquid chromatography (LC), valuable alternative methods rely on the use of hydrophilic interaction liquid chromatography (HILIC) or supercritical fluid chromatography (SFC) for the profiling of very apolar or lipophilic NPs.21 Efforts have been made to predict the retention time behaviours of NPs to support metabolite ID,22 though due to the complexity of interactions further model development is needed to realise results comparable to what has been achieved with the Kovats retention index for GC.In classical pharmacognostic investigations, the dereplication step19 allows for the rapid identification of known NPs in CNEs and prevents their isolation when their bioactivity has already been described. In screening campaigns, it is particularly helpful to detect “frequent hitters”, which are also called PAINS.18 All of this ultimately leads to the prioritisation of extracts prior to isolation procedures.
Additional orthogonal detection methods such as photodiode array (PDA) or NMR methods are also important for metabolite annotation.23 PDA methods are still widely used in NP research as a complement to MS for the detection of characteristic chromophores of given NP scaffolds. NMR can be used for hyphenation24 but suffers from a low degree of intrinsic sensitivity compared to MS even when using the state-of-art LC-SPE-NMR system.25 Such methods are mainly used for the targeted identification of an LC-peak when complementary information is necessary for de novo identification. Detection within the low microgram range is possible with cryogenated NMR probes and particularly in conjunction with low detection volumes suitable for limited sample amounts.24
The direct NMR profiling of CNEs without prior chromatographic separations remains as the mostly widely used and complementary approach to LC-MS. NMR typically allows for detection in the order of a few tens of predominating NPs in extracts, which is significantly lower than the 1000s of features detected by MS. NMR, however, offers the advantage of providing an unbiased quantitative estimation of metabolite levels in CNEs.26 This presents a significant advantage when linking such profiles to bioactivity results with chemometrics methods, as such data are dependent on the concentration of active principles. The semi-quantitative estimation of metabolites levels is also possible by ELSD or CAD detection for LC27 in combination with MS although such approaches have not yet been widely adopted.4 The MS and NMR profiling methods are thus clearly complementary to better ascertaining the quality of metabolite annotation in CNEs. NMR, ELSD and CAD also offer the additional advantage of providing semi-quantitative estimations of main metabolites in extracts. New hybrid NMR/MS approaches are emerging for the identification of NPs in mixtures,28 and many advances in this direction are expected to emerge in the years to come.
4 Metabolite annotation, in silico databases and molecular networking
For early NP identification, recently developed approaches have relied on the combined use of high-resolution mass spectrometry (HRMS) for molecular formula assignment, on the comparison of fragmentation MS/MS spectra against databases (DBs) and on the visualisation of groupings of these spectra into a molecular network (MN).29In LC-MS metabolite profiling, two forms of MS/MS data acquisition are available: data independent or data dependent acquisition (DIA versus DDA).20 DDA is the standard method for running LC-MS/MS experiments and involved executing a short HRMS survey scan of currently eluting molecules followed by a series of MS/MS scans. DIA can generate a more comprehensive survey for the metabolite profiling of CNEs but is not yet widely used because informatics retreatment and DIA spectra libraries are still unavailable.
When hundreds to thousands of MS/MS spectra are collected by DDA, automatic classification is required. For this purpose, the Global Natural Product Social Molecular Networking (GNPS) web platform allows the generation of an MN.29 In an MN, two molecules with several structural similarities normally exhibit considerable similarities in their fragmentation patterns. Such spectral similarities are calculated from a cosine score used in the high-throughput identification of metabolites.30,31 A newly developed software program (MetGem) allows for the efficient and reliable calculation of cosine scores and plots using a GNPS-like format or t-distributed Stochastic Neighbour Embedding (t-SNE) algorithm.32 The procedure offers a complementary view of data clusters while preserving interactions between related groups of MS/MS spectra.
When using online experimental DBs, an MN can be marked with standards to annotate each molecular cluster. For this purpose, the use of spectral databases among other approaches constitutes a key element from which annotation becomes effective.33 While collections of pure NPs are available in many academic laboratories, large collections of spectra are still missing and efforts to share spectroscopic data should be made in the future. As an example, the efficiency of MNs combined with MS/MS searches through a restricted spectral library of monoterpene indole alkaloids for novel NP prioritisation has been demonstrated in the investigation of Apocynaceae species Geissospermum laeve.34
The quality and completeness of public or commercial MS/MS DBs are however still lacking with regard to the total number of NPs described to date. This is why in silico fragmentation tools that allow for a match between experimental and predicted spectra are increasingly used. In this context, an in silico MS/MS library generated by accessing the structures of most described NPs (>200000 MS/MS) is available.35 While these approaches can still be improved, the combination of MNs and in silico MS/MS DBs allows for the efficient early annotation of NPS in CNEs in a generic and high-throughput manner. To improve the accuracy of in silico predictions through the propagation of structural annotations even when there is no match to a MS/MS spectrum in spectral libraries, a new tool referred to as “Network Annotation Propagation” (NAP) has been introduced.36 NAP significantly improves the ranking of in silico annotations while recognising the fact that neighbour nodes in MN clusters are structurally related. The efficiency of this approach has been confirmed through the targeted isolation of dicoumarol neolignans from a stem extract of Sageretia theezans.37
The establishment of bridges between algorithmic and experimental approaches and notably via metabolite profiling therefore represents an innovative way to explore the complexities of CNEs. We thus describe below main strategies used for the prioritisation of NPs based on novelty and/or bioactivity.
5 Linking profiling data with other layers of information for NP prioritisation
Multiple NP prioritisation strategies have been developed in recent years and most often based on the combination of different sources of information (e.g., genomic, taxonomic, geographic, bioactivity and/or spectral data). These strategies can be quite different depending on the genetic resource studied (microorganisms, marine organisms, or plants). For an overview, we briefly describe below the most innovative procedures applied to both micro- and macro-organisms, which are grouped according to the profiling methods employed or based on approaches considered. The approaches are discussed separately for microorganism studies for which genetic information can often be integrated and for plant extracts for which links with genome information and metabolic pathways are more challenging to establish.5.1 Prioritisation procedures for the investigation of microorganisms
For microorganisms, pharmaceutical companies and academics have for decades devised means of prioritising strains in CNE libraries that involve morphology and 16S sequencing to prioritise specific genera for screening. More recently, strategies for evaluating the biosynthetic potential of such strains have been developed. In this context, a first example of bacterial strain prioritisation was studied to isolate novel NPs. For this study, 146 marine Salinospora and Streptomyces strains were profiled by LC-MS/MS in an MN to establish an exhaustive map of the chemical diversity of two bacterial genera.13 This study permitted a comprehensive evaluation of NP production according to media and culture conditions and extraction methods. Fifteen molecular families of NPs and analogues were identified from an MN constructed from approximately 1.8 million MS/MS spectra. These data document biosynthetic capacities of the strain collection and provide a means of comparing strains based on the variety and novelty of their constituents. In another non-targeted profiling study, 1000 randomly selected microorganisms were evaluated by applying a similar LC-MS/MS-MN approach to the GNPS platform.11 In this case, each strain was grown in two different media and three distinct types of extracts were prepared. To measure the biosynthetic potential of these strains, a principal component analysis (PCA) was applied to visualise different NPs clusters according to the type of extraction employed. This approach allowed for the annotation of 76 molecular families and highlighted strains showing unprecedented levels of metabolic production. In another study, LC-MS metabolic variations observed among 114 bacteria of genera Photorhabdus and Xenorhabdus strains were correlated with several abiotic factors (edaphic conditions). Machine learning was used to rank the importance of NPs in determining all given metadata for prioritisation.38In addition to MS-based approaches, NMR has been used for microorganism extract profiling. 2D NMR is particularly useful for resolving overlapping signals commonly found in profiling extracts. In this respect, to identify new antiplasmodial compounds from a collection of 119 unfractionated actinomycete extracts, an interesting HSQC-TOCSY 2D NMR fingerprinting approach was developed for the profiling of extracts containing polyketides (PKs) and peptides (PEPs).14 Such HSQC-TOCSY NMR experiments resolve TOCSY correlations (1H–1H correlation by a chain of couplings) into a carbon dimension through HSQCs (direct 1H–13C correlations). In this study, actinomycete extracts were prioritised based on bioactivity, high PK and PEP diversity levels and based on the uniqueness of HSQC-TOCSY fingerprints. While this approach suffers from the very low sensitivity of 2D NMR compared to MS, it proved very successful in detecting structural fragments specific to NPs produced by microorganisms and allowed for the characterisation of NP elaiophylin, which has been previously reported to exhibit antiplasmodial and antibiotic activity.
Rather than profiling metabolites, genome data can be used to evaluate the biosynthetic potential of a given organism in microbial research. This is the case for the HTS of bacterial genomes evaluated for their capacity to produce antibacterial compounds with potentially novel modes of action. In this context, a new web tool specialised for actinobacterial genomes called ARTS (Antibiotic Resistant Target Seeker) was developed.39 It facilitates the identification of gene clusters for compounds acting against a specific and novel target and allows for the use of multiple criteria and tools for the rapid exploration of potentially novel resistance targets.
In another study, a genomics-based approach was tested where cryptic pathways were stimulated by polymerase chain reactions (PCRs) to assess the biosynthetic potential of four classes of microbial NPs (aromatic polyketides (PKs), reduced PKs, non-ribosomal PEPs and diterpenoids) of a collection of 100 randomly selected Streptomyces strains.40 Here, the preparation of a pool of pathway-specific probes by a two-tiered PCR method allowed for a survey of all variants of biosynthetic machineries for the targeted class of NPs. The methodology was initially validated on 16 “talented” representative strains known to have the potential to produce all classes of NPs. The study of a collection of 100 strains ultimately allowed for the prioritisation of a specific strain of Streptomyces griseus and led to the characterisation of four novel non-ribosomal PEPs along with known diterpenoids and PKs.
In contrast to purely genomic approaches, more studies have combined genome and metabolite profiling methods. In this respect, a systematic bioinformatics framework under which MS data are used to verify NP gene cluster family designations and to demonstrate utility for de novo correlations of NPs and biosynthetic genes has been proposed. These studies have highlighted the strong potential of such a roadmap for NP discovery based on large-scale genomics and metabolomics used in microbial research.41
Two recent studies9,42 have combined a genomic method with an MN approach to explore the biosynthetic potential of cyanobacteria that serve as prolific NP producers. In the first study, PCR screening was performed on 61 strains belonging to different genera to detect the presence of genes encoding non-ribosomal peptide synthetases (NRPS) and polyketide synthases (PKS) to target their adenylation and ketosynthase domains.9 In the second study, three species of genus Moorea were subjected to genome sequencing to identify their biosynthetic genes (NRPS and PKS).42 In both studies, LC-MS/MS-MN analyses allowed for the detection of novel NPs and showed that both PKS and NRPS genes were present in the majority of cyanobacterial strains and that the majority of NPs were strain-specific. The combination of two orthologous techniques proved well-adapted to the prioritisation of cyanobacterial strains for the discovery of novel compound classes. Another approach was applied to endophytes taken from various organs of medicinal plant Rhoeo spathacea43 to identify those capable of producing bioactive antibacterial PKs and PEPs. In this study, bioactivity-based screening was used in combination with genetic screening for PKS and NRPS genes to select the most promising strain among 10 fungal endophytic strains. The results indicate that a positive correlation was established between the presence of PKS and/or NRPS encoding genes in endophytes and the bioactivity of their respective organic extracts.
Algorithms are currently being developed to connect genome information with the presence of molecular families to accelerate the discovery of NPs and to better understand their ecological roles from the relationship between specialised metabolism and functions. This is notably being executed through a consortium designed to facilitate studies of a large set of samples.44
These examples demonstrate that many optimal analytical and computer-based approaches allow for the prioritisation of microbial strains in measuring optimal levels of chemical diversity. Relatively few studies, however, integrate biological assays in their workflows. The combination of biological screening with metabolite profiling data through chemo-informatics, however challenging, has the potential to become the most effective approach NP discovery to date. A proof of concept study on 10 cyanobacteria species belonging to the Symploca genus was performed using integrated an LC-MS/MS-MN cancer cell cytotoxicity assay.12 This approach allowed for the targeted isolation of samoamide A, a new cytotoxic NP, from a cluster representative of a novel cyanobacterial chemotype. While such a strategy involves a preliminary fractionation step, no iterative bioassay steps were required to isolate the desired compound.
For prioritisation, the type of bioassays used is also crucial and for this purpose high content screening methods are of special interest. Several approaches connecting high-content datasets to biological annotations although highly challenging are now effective for the early prioritisation of extracts and compounds with unique biological properties. The main untargeted profiling methodologies applied indifferently for mammalian, yeast or bacterial strain cell screening or in vivo screening in zebrafish have been recently reviewed.45 Employed strategies, which can be numerous, rely on various methodologies such as image- or biomap-based screening or gene profiling methods, most of which focus on comprehensive biological characterisation or allow for phenotypic annotations of natural extracts to investigate those with unique or atypical biological profiles.
As an example, the BioMAP high-throughput platform providing detailed biological characterisations of antibacterial properties of CNEs by applying activity profile matching to a set of 15 clinically relevant strains has been designed to discover novel antibiotics. Applied to a set of 3120 prefractionated extracts, the strategy is successful at accurately predicting the presence of known antibiotics and at identifying arromycin, a structurally unique naphthoquinone antibiotic.46 In a more recent study, the phenotype-guided NP discovery tool established the cytological profiling of HeLa cells of a collection of 5304 pre-fractionated extracts taken from marine-derived Actinobacteria.47 In using innovative computational software to facilitate the interpretation of image-based screening data, the study allowed for the selection of one promising extract with the searched antimitotic phenotype. Further chemical investigations have led to the targeted isolation of an antimitotic-active diketopiperazine. In parallel pyrone analogues showing cytotoxic activity through the modulation of calcium ion channel functions were identified. The creation of a compound activity mapping platform in another study allowed for phenotypic screening information taken from the cytological profiling assay of a library of 234 extracts to be connected to untargeted metabolomics data.48 This new tool, which can be used to identify novel bioactive constituents and which provides predictions on compound modes of action directly from primary screening data has led to the identification of 11 known compound families and four new compounds (quinocinnolinomycins A–D inducing endoplasmic reticulum stress).
5.2 Prioritisation procedures used in plant extract studies
For most of the microorganism studies discussed above, plant extract prioritisation has rarely applied stages of genomics mining. For this purpose, a ‘phytochemical genomics’ initiative aims at linking the genomics basis of the synthesis and functions of plant metabolites and an IS particularly based on advanced metabolomics.49 This approach has been successfully used to predict the presence of various NPs in plants.50 However, thus far its usage has been limited to a small number of model plants such as Arabidopsis thaliana, Oryza sativa and Catharanthus roseus.For studies of CNEs of plant origin, tools for incorporating other layers of information have been developed. Compared to microorganisms, the 1H-NMR profiling of plant extracts is more frequently used and serves as an efficient means to obtain convoluted fingerprints of signals of main constituents.4 Here, 2D NMR fingerprinting strategies can additionally be employed to better deconvolute signals, and such information is useful for the isolation of structural-specific NPs. In this way, the 1H and 1H–13C HSQC NMR metabolite profiling of 39 extracts from Endiandra and Beilschmiedia plant species allows for the prioritisation of leaf extracts of B. ferruginea, from which 11 new endiandric acids of three different structural types have been identified, among which three exhibits strong levels of antiapoptotic activity.51
In line with the use of 1H-NMR in metabolomics, specific statistical methods for the analysis of complex spectroscopic data have been developed to detect multiple NMR peaks from the same molecule based on the multi-collinearity of their intensities in a set of NMR spectra (e.g., statistical total correlation spectroscopy (STOCSY)).52 In this respect, heterocovariance (HetCA)-based metabolomics have been found to serve as a powerful tool.53
In a proof of concept study, the variance of extract constituents was first assessed in plants of different families and correlations with tyrosinase or 5-lipoxygenase enzymes inhibition were calculated.53 Correlations between spectral features and activity levels were, for example, examined by tracking the efficiency of a single species to identify the structural characteristics of bioactive components. In this case, the covariance between NMR and activity data was plotted for the most active plant species. Spectral lines showing strong correlations with activity were found to be indicative of specific molecular scaffolds, demonstrating that the HetCA approach can provide structural information prior to purification and showing that in the case of Dorycnium hirsutum, activity is correlated with the presence of kaempferol 3-O-(4-O-acetylrhamnosyl)-7-O-rhamnoside. This also shows that spectral data reflecting the concentration differences of components of an extract can correlate statistically with measurable dose-dependent properties of bioassays. To limit extract complexity, which can hinder analysis, HetCA has also been applied to relatively coarse fractions obtained from an extract that may reveal the concentration variance of the same compound in subsequent fractions. This has led to the identification of a series of 1H NMR signals across fractions that correlate with activity. In addition, similar correlations has been recorded in MS and have been correlated with NMR by statistical heterospectroscopy (SHY).54 The integration of HetCA STOCSY and SHY allows for the identification of stilbene derivatives as active tyrosinase inhibitors in Morus alba without the need for full purification.53
Based on a similar premise, SHY MS-NMR correlations have been established between features of micro-fractions obtained by preparative-HPLC capillary 1H-NMR (CapNMR) and LC-MS profiles associated with a cancer chemo-preventive assay successfully deconvoluting bioactive compounds as their reconstituted pseudo 1H NMR spectra.55
Several approaches also rely on LC-MS-based profiling for plant extract prioritisation. To identify new inhibitors of dengue virus replication, approximately 3500 plant extracts were screened using a dengue NS5 polymerase assay (DENV-NS5). The results were examined from a phylogenetic perspective via regression residual analysis, leading to the selection of 80 Diospyros extracts from the Ebenaceae family, which showed the highest residual score. Metabolomics profiling using MZmine 2 software was applied to the selected extracts, leading to the isolation of eight bioactive lupane- and ursane-type triterpenoids from D. glans.56
A metabolomics tool based on PCA and LC-MS was designed to select from a set of 278 Malaysian CNEs those containing structurally new photosensitisers.57 The authors hypothesised that discriminant clusterisation between photocytotoxic extracts containing known photosensitisers against those producing uncommon photosensitisers would emerge from PCA analyses of their LC-MS profiles. With this LC-MS-PCA approach and in applying molecular mass and UV-visible absorption profile filters, two new photosensitisers with a cyclic tetrapyrrolic structure were identified. The LC-MS-PCA approach was found to be an efficient and rapid method capable of prioritising the 10% most promising extracts for further investigation.
Another metrics-based prioritisation strategy has recently been developed.16 The methodology is based on the use of a dedicated tool integrated with the LC-HRMS database allowing for the novelty, complexity and diversity of samples to be ranked according to various calculated metric scores. It has been applied to a set of eight marine sponges and to six tunicate samples previously fractionated by solid-phase extraction (SPE).16 Although the study presents some limitations, the proposed strategy's efficiency was demonstrated through the isolation and characterisation of two new eudistomins and two new nucleosides.
In this context, biochemometrics approaches, which rely on the use of statistical modelling tools to correlate metabolite profiles with biological datasets, are very useful for assigning a biological activity to a particular compound detected from complex mixtures. The advantages and disadvantages of three of these used in metabolomics (partial least-squares (PLS), S-plot and selectivity ratios) were critically assessed and the latter was found best identify MS features correlated to bioactivity.58 More recently, selectivity ratio models have been applied to inactive botanical mixtures spiked with known antimicrobial compounds to assess how biochemometrics may highlight the active constituents of CNEs. This shows that data-processing approaches (data transformation and model simplification tools using a variance cut-off) have significant impacts on model capacities to detect active constituents in CNEs.6 Applications of such methods reveal their interesting potential as well as their current limitations. This also highlights the fact that they must be validated prior to implementation in prioritisation workflows to assess complex botanical mixtures.
Other approaches combining LC-MS/MS-MN with the integration of various information layers (Fig. 1) have recently been successful in the prioritisation of CNEs. In this respect, an innovative LC-MS/MS-MN-based approach has been developed for the screening of a focused library of Euphorbiaceae plant species.10,59,60
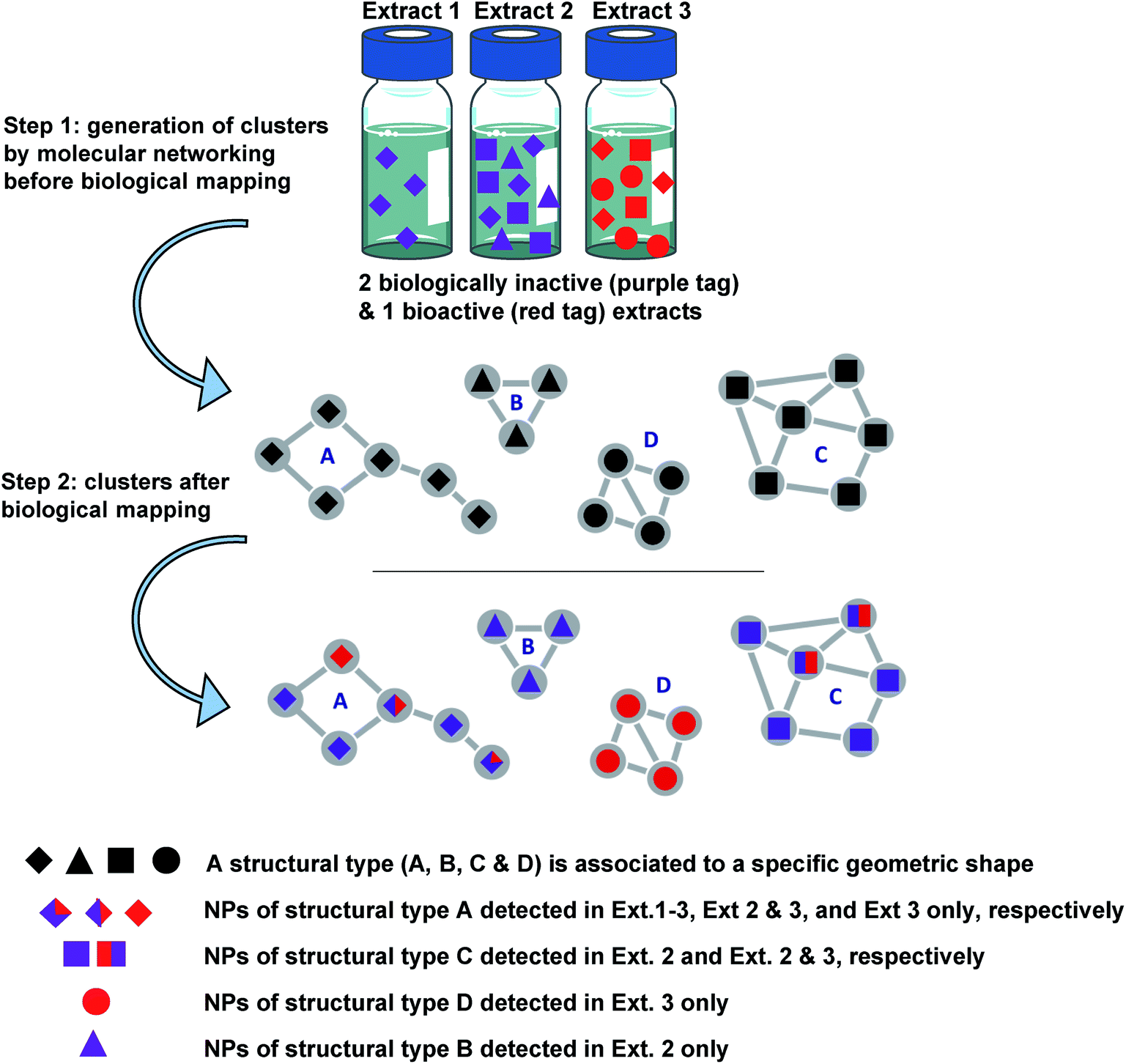 | ||
| Fig. 1 Concept of NP prioritisation. Step (1) generation of clusters (MN) according to spectral similarity of compounds detected in 3 different extracts 1, 2 and 3. Step (2) colour tags are associated with compounds of the clusters according to whether they come from bioactive extracts (red) or inactive extracts (purple). Compounds coloured in purple, red and purple, and red were detected in inactive, inactive and bioactive, and bioactive extracts, respectively. Clusters of red and purple diamonds or squares correspond to compounds of types A or C detected both in bioactive and bio-inactive extracts. They are not selected for further investigation. The cluster of purple triangles is associated with compounds of the structural type B, detected only in inactive extracts and is not selected for further investigation. The cluster of red circles is associated with compounds of the structural type D, detected only in a bioactive extract and should be selected first. It should be noted that the bioactivity of extract 3 could also be associated with the sole presence of the compound represented by an entirely red diamond (not detected in other inactive extracts). Thus, it is important to carry out the purification of a representative number of molecules within a considered cluster. A similar approach is possible using different colour tags for taxonomy mapping, leading to genus or sample-specific clusters. The combination of biological and taxonomical mapping leads to more stringent targeting. | ||
For this purpose, various layers of information such as biological results and/or taxonomic data have been used to discriminate specific ion clusters to allow for the in fine targeting the isolation of structurally new bioactive NPs.10 Results of the biological screening of extracts using a chikungunya virus (CHIKV) cell-based assay and an oncogenic Wnt signalling assay were mapped to the MN, and colour tags were applied depending on the IC50 or EC50 values obtained. This step was followed by MN mapping with taxonomic information. From 88000 nodes distributed across 7840 clusters obtained from MS/MS data on 293 plant extracts, the application of filters to the massive MN allowed for the selection of 21 clusters composed of putative inhibitors of the Wnt signalling pathway and for the targeting of no more than 5% of clusters for their anti-chikungunya (anti-CHIKV) potential. The principles of this approach are summarised in Fig. 2.
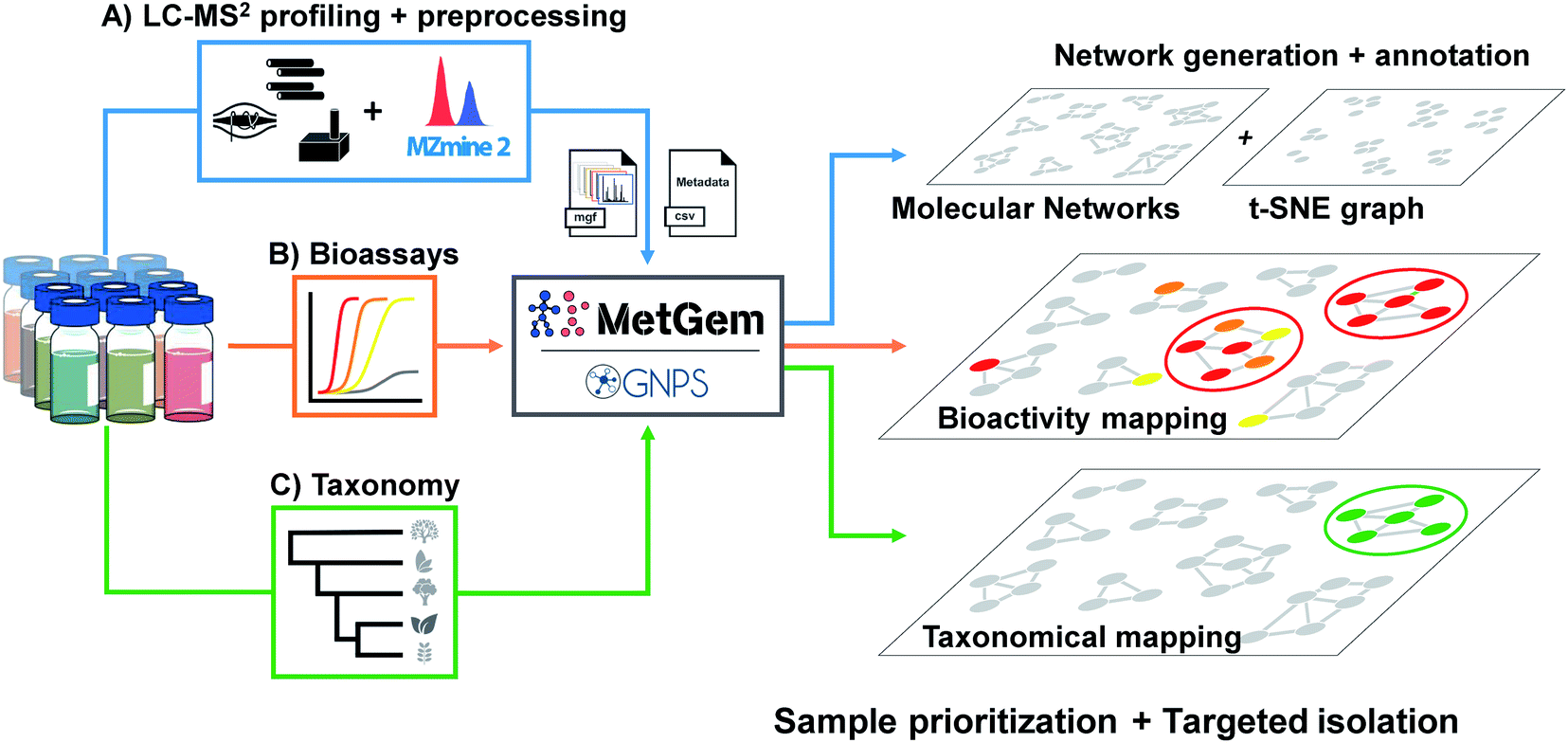 | ||
| Fig. 2 General workflow for the generation of molecular networks and group mapping. Samples prepared at the same concentration are first injected in LC-MS/MS (A). Data are acquired using the data-dependent acquisition (DDA) mode, then converted through a MZmine 2 software processing. The final .mgf file is then used for the generation of the MN by GNPS web-platform or MetGem software. In parallel, bioassays (B) are conducted to record activity and taxonomic information are tabulated (C). Finally, the MNs are annotated by the interrogation of public or private databases, and bioactivity together with taxonomy can be mapped on the MNs for sample prioritisation and targeted isolation. Figure adapted and reprinted with permission from ref. 60 2019. Copyright 2019 American Chemical Society. | ||
From two selected clusters, the application of an MS-directed isolation approach led to the characterisation of four new Wnt pathway inhibitors and one new anti-CHIKV compound. It should be noted that this approach did not require the use of any bio-guided isolation procedure and that structure/activity relationships can quickly be drawn when a representative number of compounds are isolated within the considered cluster. The LC-MS/MS profiles of chromatographic fractions of pre-selected bioactive extracts were additionally combined with data on the initial MN in a second step to allow for the enrichment of clusters of interest with potentially undetected minor NPs during the initial investigation of CNEs. In a second study, from the same set of extracts, the samples59 were analysed on another MS platform and the data were processed via the MZmine 2 software.61 Here prioritisation was based on taxonomic considerations rather than biological ones (CHIKV, DENV and ZIKV assays) to find new or uncommon structures. Taxonomic mapping involved overlaying the 20 Euphorbiaceae genera found in the extract collection and detecting genus- or sample-specific clusters. From 1231 clusters containing ≥three nodes, 82 genus-specific ones were selected, among which one was sample-specific and resulted in the characterisation of five unprecedented tetracyclic chlorinated monoterpenyl quinol-4-one, two new daphnane diterpenoid orthoesters possessing potent anti-CHIK activities, and two new 1,4-dioxane-fused phenanthrene dimers, of which one was the first natural dual inhibitor of dengue and Zika NS5 characterised to date.60
When using such taxonomical layouts within a homogeneous set of samples, family-, genus- and species-specific compounds can be more easily distinguished from other ubiquitous NPs in an MN, significantly increasing chances of discovering structurally new lead compounds. To further target bioactive NPs within complex mixtures, a bioactive MN workflow integrating an algorithm that predicts putative bioactive molecules present in fractions has recently been added to GNPS methods.62 This involves the calculation of a bioactivity score from the Pearson correlation between feature intensity levels across fractions and bioactivity levels associated with these fractions in ways similar to those previously discussed in NMR correlation studies. Bioactivity scoring results were directly visualised in the MN and molecules with significant predicted bioactivity scores could in turn be targeted more accurately. The main advantage of this procedure lies in the fact that minor bioactive compounds can be detected relatively easily within enriched fractions. However, the approach involves the coarse fractionation of bioactive extracts.
6 Rapid targeted isolation of NPs from CNEs using prioritisation information
As is described above, the various analytical methods used for metabolite profiling in conjunction with bioassay data are designed to putatively identify structurally novel NPs in extracts with a high probability of being bioactive. To validate such results the NPs identified in extracts need to be isolated for complete biological and structural characterisation. This procedure must be efficient and yield enough of the targeted pure NP for full de novo NMR characterisation and for an evaluation of its bioactivity.In the case of LC-MS metabolite profiling, the feature(s) highlighted through the prioritisation process (a given m/z value for a given retention time) allow(s) for the precise localisation of the NP of interest in the ion map (see Fig. 3A). For this final characterisation, sub-mg quantities of each pure NP are required. They are generally sufficient for the extensive identification by 2D NMR measurements with NMR equipped with cryoprobes and for activity potency determination with sensitive bioassays.24
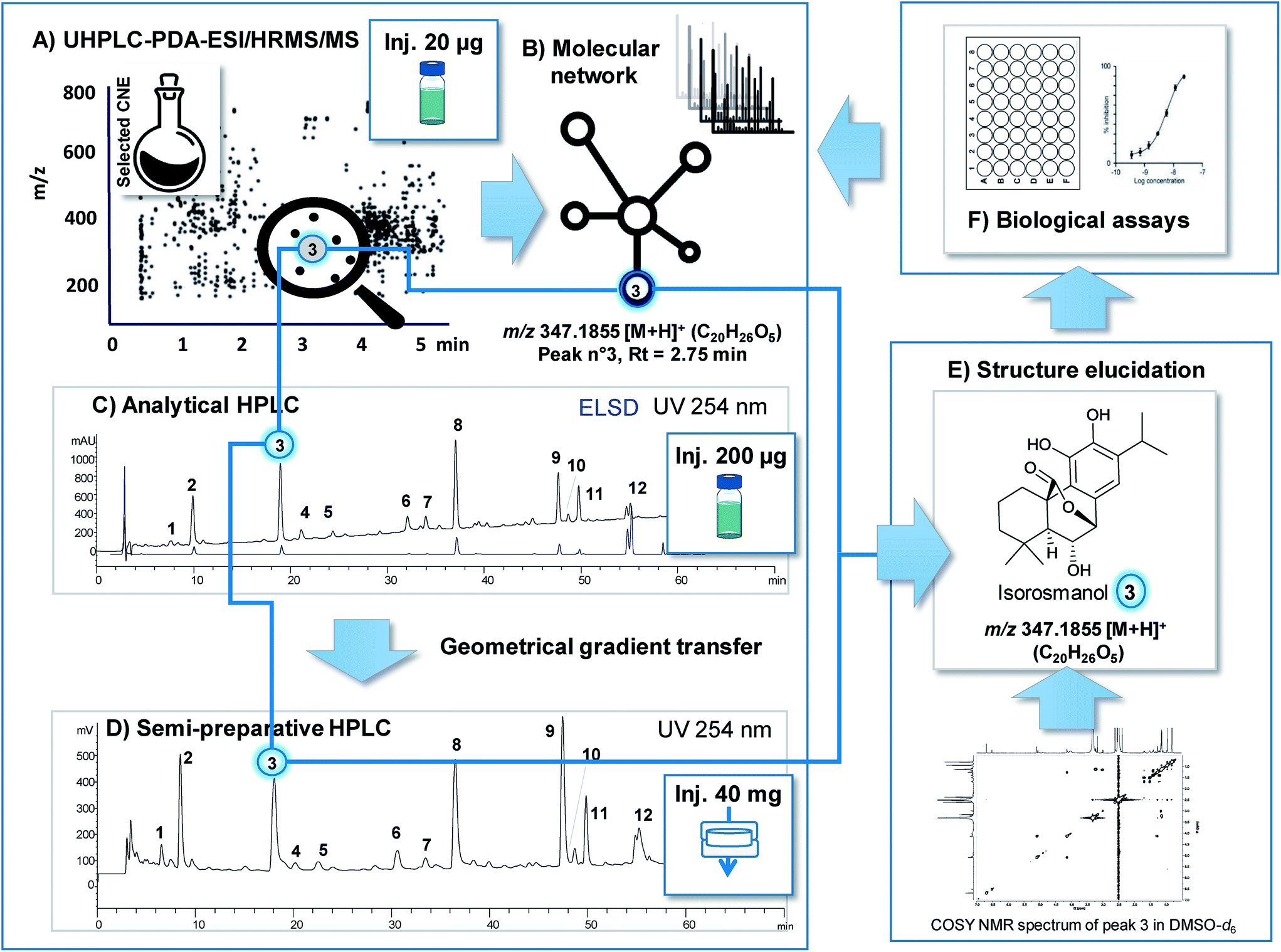 | ||
| Fig. 3 Efficient link between feature highlighted by prioritisation in LC-MS profiles and obtaining of the corresponding NP for final structural and biological characterisation. The methanolic extract of S. officinalis was used as a typical plant extract to illustrate how a highlighted feature (m/z @ RT) can be readily obtained in one-step by targeted isolation at the semi-preparative level with appropriate chromatographic gradient transfer and generate sufficient amount of bioassay assessment and 2DNMR. The methanolic CNE was first analysed by UHPLC-PDA-ESI-HRMS/MS (A). Combination of bioactivity screening results (F) and MN (B) permitted the identification a given feature (compound 3m/z 347.1855 [M + H]+ @ RT). Gradient transfer between the analytical conditions (Xbridge C18 column (250 × 4.6 i.d, mm, 5 μm, 1 ml min−1)) (C) and the semi-preparative separations with the same phase chemistry (Xbridge C18 column (250 × 19 i.d, mm, 5 μm, 17 ml min−1)) (D) yielded similar profiles and precise RT prediction. ELSD detection enable semi-quantitative estimation of respective amounts in the CNE for estimation of the isolation yields (C). Sample introduction by dryload (40 mg of CNE (D)) instead of classical injection was key to maximise resolution. Using this approach compound 3 was isolated directly from the CNE in one-step with high purity and identify by 2DNMR (E) and submitted to bioassay (F) for final assessment. | ||
Over recent years developments have been made to achieve efficient isolation directly from CNEs at the preparative chromatographic scale by maintaining chromatographic selectivity and resolution levels very similar to those achieved during metabolite profiling at the analytical scale. We view this step as crucial since NP isolation involves tedious multiple chromatographic steps applied on a large scale whereby the monitoring of a prioritised feature is difficult to achieve.
To this end, one solution is to perform multiple injections of the CNE using analytical separation conditions and to collect the LC-peak of interest by trapping on solid phase extraction (SPE) cartridges. The content of the SPE can then be directly eluted via NMR using state-of-the-art LC-SPE-NMR systems.24 Some bioassays can already be performed at this scale as demonstrated for the identification of protein tyrosine phosphatase inhibitors of Vietnamese plants.25 While this approach may integrate MS detection prior to trapping and offers the advantage of being automatised, it also suffers from limitations in SPE trapping efficiency and from a need for multiple injections to achieve sufficient levels of enrichment.24
Another approach to the rapid and direct isolation of NPs from CNEs at the μg mg−1 scale at the highest LC resolution is that of semi-preparative HPLC. For this method, efficient chromatographic gradient transfers (from analytical to semi-preparative HPLC) can be calculated using the same column stationary phase to maintain exactly the same level of selectivity at both scales (see Fig. 3C and D).63 To achieve the best chromatographic resolution, the optimal introduction of a large amount of CNEs to a semi-preparative HPLC column serves as a critical step. Due to the solubility issues related to CNEs and notably with RP separation, these samples are often diluted and injected in large volumes of organic solvents that significantly compromise resolutions. Newly developed dry load injection methods compatible with semi-preparative separation constitute an excellent means to avoid the compromises that generally need to be made between high loading and sample solubilisation.64 Thus, as is shown in Fig. 3, the targeted isolation of a prioritised feature from a typical plant CNE can be efficiently performed at the semi-preparative level with almost no loss of resolution and while yielding high enough volumes for full de novo characterisation and bioactivity assessment. Through such a process ELSD detection in addition to MS and/or UV are key to estimating the scale at which isolation must be applied to obtain a sufficient volume of the NP of interest.65,66
In our opinion, even if preparative chromatographic techniques are well established, their optimised use by strictly following rules established for fundamental chromatography is key to efficient isolation.63,64 This point is often neglected in the NP community and results in inefficient and lengthy purification protocols. Since the prioritisation procedures presented here are designed to highlight valuable NPs in CNEs, their isolation from complex mixtures merits special attention to efficiently complete the entire process.
7 Conclusion
Most prioritisation strategies highlighted in this paper aim at efficiently isolating NPs exhibiting significant levels of bioactivity and/or novel and chemo-diverse chemical entities mainly for drug discovery purposes. This is achieved by using genomic, metabolomic, or taxonomic information with or without relations to bioassay screening data to highlight valuable NPs. These different efforts aim at replacing lengthy classical bioactivity-guided isolation procedures with a potential cherry picking of targeted NPs to gather multidimensional information at hand for such selection.In our view, strategies that ideally integrate all levels of information (genomes, metabolomes, and relevant bioassays) in conjunction with well-designed libraries (enriched CNEs free of interfering constituents) will pave the way to rational NP discovery. Such efforts must also be made in good agreement with newly enforced ABS legislation.3
To achieve such goals, developments are still needed in various fields. Key to the whole process is the design of CNE libraries and this may vary according to final objectives. All data associated with such libraries should be of high quality and acquired in formats that would ease their mining with advanced bioinformatics tools. Efficient means of standardisation and dissemination and the open sharing of relevant datasets are prerequisites to advancing in this field.67 In this vein, initiatives for sharing metabolite profiling data with data-driven social-networking platforms such those of GNPS provide a means to collectively translate big data into shared knowledge across the entire NP research community.29 This is central to rendering natural extract metabolome data of a similar level as genome data, which are also decisive for prioritisation.
In this context, in NP research, massive metabolite annotation with high levels of confidence remains a major challenge. Here, the methodical approaches vary and cannot be standardised to the level of proteomics and genomics. Having at hand massive and partly redundant MS datasets serves as one way to gain confidence for annotation especially when contextualising such data (e.g., by taxonomy or genetics) is done carefully. NMR remains a key complement to MS for unambiguous identification. In this respect, new initiatives for the community sharing of raw NMR data in public repositories are emerging.68 All these efforts of mutual data sharing29,68 should finally yield results of higher quality and integrity and are also well in line with the requirements of data management plans of funding agencies.
In our opinion, obtaining high quality and semi-quantitative compositional information on natural extracts is essential to prioritisation-driven studies. Given the high chemodiversity of bioactive NPs, strategies that integrate several levels of orthogonal information, each of which is associated with a score, should be developed, as is the case for MS. Such scores can be associated with (i) chemotaxonomy (phylogeny, genetics, and structural relations in metabolomes) and (ii) analytical parameters such as matches with predicted retention time and/or collision cross sections (in ion mobility) based on structure integration and NMR information for main CNEs constituents. Methods that integrate all of these scores, each of which is individually imperfect, should ultimately support more confidence in automated NP annotation. Efforts made in this direction must be continued and intensified. For quantitative estimations, protocols for extract profiling should more systematically involve the additional recording of profiles with detectors to provide semi-quantitative estimations of metabolites such as ELSD and/or CAD as well as LC-MS/NMR studies of both ID and quantification aspects. The mastery of this information should ultimately lead to the generation of detailed metabolome information on a large number of natural organisms, which can support evaluations of traits of CNE composition and reveal novel bioactive NPs.
In regard to bioassays, high-content phenotypic screens appear to be well adapted to screening CNEs and to directly predicting the compound mode of action from primary screening data. For example, cytological profiling using image-based phenotypic screens associated with untargeted HR-MS-based metabolomics appears to be among the most advanced means to rapidly and efficiently target and identify constituents with unique biological properties.45
In addition to this method, the multi-informational MN approach embedding various information layers appears to be a powerful and efficient approach to the prioritisation and targeted isolation of novel bioactive compounds.10 The combination of phenotypic-based profiling methodologies with such a multi-informative MN approach would likely prove to be one of the smarter and most effective means to identify new drug leads. Efficient biochemometric tools for the co-correlation of bioassay and composition data are needed and must be carefully evaluated.58 Adequate algorithms should enable one to efficiently highlight spectroscopic features of large datasets to finally guide the targeted isolation of NPs of interest.
To be effectively implemented by the NP community, all necessary statistical and bioinformatics tools of prioritisation workflows should ideally be user-friendly and intuitive. However, such tools should provide a good understanding of how the data are processed to avoid biases. For pipelines integrating complex multilayer information, the use of validated bioactive reference standards spiked in extracts is highly recommended to evaluate the efficiency of the prioritisation strategy envisioned.6
When prioritisation is not directly connected to given bioactivity results but aims at finding novel chemodiverse NPs, the virtual screening of scaffolds (docking, modelling chemical space, etc.) obtained may represent an alternative means to valorise the new chemical entities discovered.69 However, in silico valorisation pipelines strongly depend on the use of advanced bioinformatics tools. To explore large volumes of aggregated data including chemical, biological, taxonomical and genomics information more efficiently and to maximise chances of successfully tracking new and/or active natural products, the use of systems that can correctly interpret external data such those of artificial intelligence (AI) will become a necessity.70
The various prioritisation protocols described finally aim at identifying single bioactive constituents from CNEs for an in-depth investigation of their bioactivity and pharmacological profiles. In many instances, however, CNEs may exhibit remarkable activities that cannot be retrieved after the isolation of their single constituents and it is not uncommon to sacrifice bioactivity when performing classic activity-guided isolation protocols. Such an effect can be attributed to the degradation or loss of the active principle during isolation or to synergistic effects between individual constituents that may emerge from specific target(s) or more likely in phenotypic screens. While synergistic and/or antagonistic effects are difficult and time consuming to investigate and especially given the rapid pace of screening campaigns, they are still worth studying. The use of semi-quantitative compositional data on CNEs in parallel with screening results allows one to document bioactivity losses during fractionation from changes in composition or to investigate trends of common compositional features of bioactive CNEs. Such information may highlight a co-occurrence of NPs in extracts of a given ratio and reveal potential synergistic mechanisms that must then be assessed with a combination of single NPs. This aspect is however still extremely challenging to address. In this context, synergy maps71 that provide a global visualisation of compound combination effects could be used with CNE profiling data of closely related samples to identify constituent patterns putatively involved in synergetic effects. Using a detailed metabolite profile of active NPs can also allow for the analysis of potential NP loss or degradation during fractionation and design strategies for recovering bioactive NPs. On the other hand, the characterisation of the bioactivity of isolated NPs is not always straightforward, and final purity assessments are essential at this stage since impurities (residual complexity) can be responsible for bioactivity.72
The acquisition of high-quality analytical data combined with results of biological activities using relevant biochemometric tools should provide detailed information on the metabolomes of a large number of natural organisms, allowing for the efficient identification of novel bioactive NPs. The comprehensive integration of these dimensions has begun and will continue in the future to induce a paradigm shift in NP research for a beneficial renewal of pharmacognosy in the digital era. Such a change is necessary and we are convinced that it will bring NPs back to the forefront of drug discovery programs by recapturing the pre-eminent role they once played in this field.
8 Conflicts of interest
There are no conflicts to declare.9 Acknowledgements
JLW is grateful to the Swiss National Science Foundation (SNF) for supporting its natural product metabolomics projects (grants no. 310030E-164289, 31003A_163424 and 316030_164095). This work has benefited from an “Investissement d'Avenir” grant managed by Agence Nationale de la Recherche (CEBA, ref ANR-10-LABX-25-01). We acknowledge Tomas Knoop and CombineDesign from the Noun Project (https://thenounproject.com/) and for the icons used in the Fig. 3. The authors acknowledge Mr Adriano Rutz and Dr Laurence Marcourt for their help in the realisation of Fig. 3, and Florent Olivon for Fig. 1 and Dr Pierre-Marie Allard for fruitful discussions in the preparation of the manuscript.10 References
- J. D. McChesney, S. K. Venkataraman and J. T. Henri, Phytochemistry, 2007, 68, 2015–2022 CrossRef CAS PubMed.
- B. David, J.-L. Wolfender and D. A. Dias, Phytochem. Rev., 2015, 14, 299–315 CrossRef CAS.
- B. David, Phytochem. Rev., 2018, 17, 1211–1223 CrossRef CAS.
- J. L. Wolfender, J. M. Nuzillard, J. J. J. van der Hooft, J. H. Renault and S. Bertrand, Anal. Chem., 2019, 91, 704–742 CrossRef CAS PubMed.
- E. Maréchal, S. Roy and L. Lafanechère, Chemogenomics and Chemical Genetics, Springer-Verlag, Berlin, Heidelberg, 2011 Search PubMed.
- L. K. Caesar, J. J. Kellogg, O. M. Kvalheim and N. B. Cech, J. Nat. Prod., 2019 DOI:10.1021/acs.jnatprod.9b00176.
- T. G. McCloud, Molecules, 2010, 15, 4526–4563 CrossRef CAS PubMed.
- M. S. Butler, F. Fontaine and M. A. Cooper, Planta Med., 2014, 80, 1161–1170 CAS.
- A. Brito, J. Gaifem, V. Ramos, E. Glukhov, P. C. Dorrestein, W. H. Gerwick, V. M. Vasconcelos, M. V. Mendes and P. Tamagnini, Algal Res., 2015, 9, 218–226 CrossRef.
- F. Olivon, P. M. Allard, A. Koval, D. Righi, G. Genta-Jouve, J. Neyts, C. Apel, C. Pannecouque, L. F. Nothias, X. Cachet, L. Marcourt, F. Roussi, V. L. Katanaev, D. Touboul, J. L. Wolfender and M. Litaudon, ACS Chem. Biol., 2017, 12, 2644–2651 CrossRef CAS.
- D. J. Floros, P. R. Jensen, P. C. Dorrestein and N. Koyama, Metabolomics, 2016, 12, 145 CrossRef.
- C. B. Naman, R. Rattan, S. E. Nikoulina, J. Lee, B. W. Miller, N. A. Moss, L. Armstrong, P. D. Boudreau, H. M. Debonsi, F. A. Valeriote, P. C. Dorrestein and W. H. Gerwick, J. Nat. Prod., 2017, 80, 625–633 CrossRef CAS.
- M. Crusemann, E. C. O'Neill, C. B. Larson, A. V. Melnik, D. J. Floros, R. R. da Silva, P. R. Jensen, P. C. Dorrestein and B. S. Moore, J. Nat. Prod., 2017, 80, 588–597 CrossRef CAS PubMed.
- L. Buedenbender, L. J. Habener, T. Grkovic, D. I. Kurtboke, S. Duffy, V. M. Avery and A. R. Carroll, J. Nat. Prod., 2018, 81, 957–965 CrossRef CAS.
- C. C. Thornburg, J. R. Britt, J. R. Evans, R. K. Akee, J. A. Whitt, S. K. Trinh, M. J. Harris, J. R. Thompson, T. L. Ewing, S. M. Shipley, P. G. Grothaus, D. J. Newman, J. P. Schneider, T. Grkovic and B. R. O'Keefe, ACS Chem. Biol., 2018, 13, 2484–2497 CrossRef CAS PubMed.
- J. N. Tabudravu, L. Pellissier, A. J. Smith, K. Subko, C. Autreau, K. Feussner, D. Hardy, D. Butler, R. Kidd, E. J. Milton, H. Deng, R. Ebel, M. Salonna, C. Gissi, F. Montesanto, S. M. Kelly, B. F. Milne, G. Cimpan and M. Jaspars, J. Nat. Prod., 2019, 82, 211–220 CrossRef CAS.
- F. Gueritte, T. Sevenet, M. Litaudon and V. Dumontet, in Chemogenomics and Chemical Genetics, ed. E. Maréchal, S. Roy and L. Lafanechère, Springer-Verlag, Berlin, Heidelberg, 2011 Search PubMed.
- J. B. Baell, J. Nat. Prod., 2016, 79, 616–628 CrossRef CAS.
- J. Hubert, J. M. Nuzillard and J. H. Renault, Phytochem. Rev., 2017, 16, 55–95 CrossRef CAS.
- T. Kind, H. Tsugawa, T. Cajka, Y. Ma, Z. Lai, S. S. Mehta, G. Wohlgemuth, D. K. Barupal, M. R. Showalter, M. Arita and O. Fiehn, Mass Spectrom. Rev., 2018, 37, 513–532 CrossRef CAS PubMed.
- A. Grand-Guillaume Perrenoud, D. Guillarme, J. Boccard, J.-L. Veuthey, D. Barron and S. Moco, J. Chromatogr. A, 2016, 1450, 101–111 CrossRef CAS.
- M. A. Samaraweera, L. M. Hall, D. W. Hill and D. F. Grant, Anal. Chem., 2018, 90, 12752–12760 CrossRef CAS.
- N. G. M. Gomes, D. M. Pereira, P. Valentao and P. B. Andrade, J. Pharm. Biomed. Anal., 2018, 147, 234–249 CrossRef CAS.
- N. Bohni, K. Ndjoko-Ioset, A. S. Edison and J.-L. Wolfender, in Liquid Chromatography, ed. S. Fanali, P. R. Haddad, C. F. Poole and M.-L. Riekkola, Elsevier, 2nd edn, 2017, pp. 479–514, DOI:10.1016/B978-0-12-805393-5.00020-8.
- B. T. D. Trinh, A. K. Jager and D. Staerk, Molecules, 2017, 22, 1228 CrossRef.
- C. Simmler, J. G. Napolitano, J. B. McAlpine, S.-N. Chen and G. F. Pauli, Curr. Opin. Biotechnol., 2014, 25, 51–59 CrossRef CAS PubMed.
- M. Ligor, S. Studzinska, A. Horna and B. Buszewski, Crit. Rev. Anal. Chem., 2013, 43, 64–78 CrossRef CAS.
- K. Bingol and R. Bruschweiler, Curr. Opin. Biotechnol., 2017, 43, 17–24 CrossRef CAS.
- M. Wang, J. J. Carver, V. V. Phelan, L. M. Sanchez, N. Garg, Y. Peng, D. D. Nguyen, J. Watrous, C. A. Kapono, T. Luzzatto-Knaan, C. Porto, A. Bouslimani, A. V. Melnik, M. J. Meehan, W. T. Liu, M. Crusemann, P. D. Boudreau, E. Esquenazi, M. Sandoval-Calderon, R. D. Kersten, L. A. Pace, R. A. Quinn, K. R. Duncan, C. C. Hsu, D. J. Floros, R. G. Gavilan, K. Kleigrewe, T. Northen, R. J. Dutton, D. Parrot, E. E. Carlson, B. Aigle, C. F. Michelsen, L. Jelsbak, C. Sohlenkamp, P. Pevzner, A. Edlund, J. McLean, J. Piel, B. T. Murphy, L. Gerwick, C. C. Liaw, Y. L. Yang, H. U. Humpf, M. Maansson, R. A. Keyzers, A. C. Sims, A. R. Johnson, A. M. Sidebottom, B. E. Sedio, A. Klitgaard, C. B. Larson, C. A. Boya, D. Torres-Mendoza, D. J. Gonzalez, D. B. Silva, L. M. Marques, D. P. Demarque, E. Pociute, E. C. O'Neill, E. Briand, E. J. N. Helfrich, E. A. Granatosky, E. Glukhov, F. Ryffel, H. Houson, H. Mohimani, J. J. Kharbush, Y. Zeng, J. A. Vorholt, K. L. Kurita, P. Charusanti, K. L. McPhail, K. F. Nielsen, L. Vuong, M. Elfeki, M. F. Traxler, N. Engene, N. Koyama, O. B. Vining, R. Baric, R. R. Silva, S. J. Mascuch, S. Tomasi, S. Jenkins, V. Macherla, T. Hoffman, V. Agarwal, P. G. Williams, J. Dai, R. Neupane, J. Gurr, A. M. C. Rodriguez, A. Lamsa, C. Zhang, K. Dorrestein, B. M. Duggan, J. Almaliti, P. M. Allard, P. Phapale, L. F. Nothias, T. Alexandrov, M. Litaudon, J. L. Wolfender, J. E. Kyle, T. O. Metz, T. Peryea, D. T. Nguyen, D. VanLeer, P. Shinn, A. Jadhav, R. Muller, K. M. Waters, W. Shi, X. Liu, L. Zhang, R. Knight, P. R. Jensen, B. O. Palsson, K. Pogliano, R. G. Linington, M. Gutierrez, N. P. Lopes, W. H. Gerwick, B. S. Moore, P. C. Dorrestein and N. Bandeira, Nat. Biotechnol., 2016, 34, 828–837 CrossRef CAS PubMed.
- K. X. Wan, I. Vidavsky and M. L. Gross, J. Am. Soc. Mass Spectrom., 2002, 13, 85–88 CrossRef CAS PubMed.
- J. Watrous, P. Roach, T. Alexandrov, B. S. Heath, J. Y. Yang, R. D. Kersten, M. van der Voort, K. Pogliano, H. Gross, J. M. Raaijmakers, B. S. Moore, J. Laskin, N. Bandeira and P. C. Dorrestein, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, E1743–E1752 CrossRef CAS PubMed.
- F. Olivon, N. Elie, G. Grelier, F. Roussi, M. Litaudon and D. Touboul, Anal. Chem., 2018, 90, 13900–13908 CrossRef CAS PubMed.
- S. Böcker, Curr. Opin. Chem. Biol., 2017, 36, 1–6 CrossRef PubMed.
- A. E. F. Ramos, C. Alcover, L. Evanno, A. Maciuk, M. Litaudon, C. Duplais, G. Bernadat, J. F. Gallard, J. C. Jullian, E. Mouray, P. Grellier, P. M. Loiseau, S. Pomel, E. Poupon, P. Champy and M. A. Beniddir, J. Nat. Prod., 2017, 80, 1007–1014 CrossRef.
- P. M. Allard, T. Peresse, J. Bisson, K. Gindro, L. Marcourt, V. C. Pham, F. Roussi, M. Litaudon and J. L. Wolfender, Anal. Chem., 2016, 88, 3317–3323 CrossRef CAS PubMed.
- R. R. da Silva, M. Wang, L. F. Nothias, J. J. J. van der Hooft, A. M. Caraballo-Rodriguez, E. Fox, M. J. Balunas, J. L. Klassen, N. P. Lopes and P. C. Dorrestein, PLoS Comput. Biol., 2018, 14, e1006089 CrossRef PubMed.
- K. B. Kang, E. J. Park, R. R. da Siva, H. W. Kim, P. C. Dorrestein and S. H. Sung, J. Nat. Prod., 2018, 81, 1819–1828 CrossRef CAS.
- N. Tobias, C. Parra-Rojas, Y.-N. Shi, Y.-M. Shi, S. Simonyi, A. Thanwisai, A. Vitta, N. Chantratita, E. A. Hernandez-Vargas and H. B. Bode, BioRxiv, 2019 DOI:10.1101/535781.
- M. Alanjary, B. Kronmiller, M. Adamek, K. Blin, T. Weber, D. Huson, B. Philmus and N. Ziemert, Nucleic Acids Res., 2017, 45, W42–W48 CrossRef CAS.
- P. F. Xie, M. Ma, M. E. Rateb, K. A. Shaaban, Z. G. Yu, S. X. Huang, L. X. Zhao, X. C. Zhu, Y. J. Yan, R. M. Peterson, J. R. Lohman, D. Yang, M. Yin, J. D. Rudolf, Y. Jiang, Y. W. Duan and B. Shen, J. Nat. Prod., 2014, 77, 377–387 CrossRef CAS PubMed.
- J. R. Doroghazi, J. C. Albright, A. W. Goering, K. S. Ju, R. R. Haines, K. A. Tchalukov, D. P. Labeda, N. L. Kelleher and W. W. Metcalf, Nat. Chem. Biol., 2014, 10, 963–968 CrossRef CAS.
- K. Kleigrewe, J. Almaliti, I. Y. Tian, R. B. Kinnel, A. Korobeynikov, E. A. Monroe, B. M. Duggan, V. Di Marzo, D. H. Sherman, P. C. Dorrestein, L. Gerwick and W. H. Gerwick, J. Nat. Prod., 2015, 78, 1671–1682 CrossRef CAS PubMed.
- A. Alvin, J. A. Kalaitzis, B. Sasia and B. A. Neilan, J. Appl. Microbiol., 2016, 120, 1229–1239 CrossRef CAS.
- M. H. Medema, mSystems, 2018, 3, e00182 CrossRef.
- K. L. Kurita and R. G. Linington, J. Nat. Prod., 2015, 78, 587–596 CrossRef CAS.
- W. R. Wong, A. G. Oliver and R. G. Linington, Chem. Biol., 2012, 19, 1483–1495 CrossRef CAS PubMed.
- J. L. Ochoa, W. M. Bray, R. S. Lokey and R. G. Linington, J. Nat. Prod., 2015, 78, 2242–2248 CrossRef CAS.
- K. L. Kurita, E. Glassey and R. G. Linington, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 11999–12004 CrossRef CAS.
- K. Saito, Curr. Opin. Plant Biol., 2013, 16, 373–380 CrossRef CAS.
- T. Tohge, L. P. de Souza and A. R. Fernie, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2014, 966, 7–20 CrossRef CAS.
- C. Apel, C. Geny, V. Dumontet, N. Birlirakis, F. Roussi, P. Van Cuong, M. Huong Doan Thi, N. Van Hung, C. Van Minh and M. Litaudon, J. Nat. Prod., 2014, 77, 1430–1437 CrossRef CAS PubMed.
- D. J. Crockford, E. Holmes, J. C. Lindon, R. S. Plumb, S. Zirah, S. J. Bruce, P. Rainville, C. L. Stumpf and J. K. Nicholson, Anal. Chem., 2006, 78, 363–371 CrossRef CAS PubMed.
- N. Aligiannis, M. Halabalaki, E. Chaita, E. Kouloura, A. Argyropoulou, D. Benaki, E. Kalpoutzakis, A. Angelis, K. Stathopoulou, S. Antoniou, M. Sani, V. Krauth, O. Werz, B. Schütz, H. Schäfer, M. Spraul, E. Mikros and L. A. Skaltsounis, ChemistrySelect, 2016, 1, 2531–2535 CrossRef CAS.
- O. Cloarec, M. E. Dumas, A. Craig, R. H. Barton, J. Trygg, J. Hudson, C. Blancher, D. Gauguier, J. C. Lindon, E. Holmes and J. Nicholson, Anal. Chem., 2005, 77, 1282–1289 CrossRef CAS PubMed.
- S. Bertrand, A. Azzollini, A. Nievergelt, J. Boccard, S. Rudaz, M. Cuendet and J.-L. Wolfender, Molecules, 2016, 21, 259 CrossRef PubMed.
- L. A. Peyrat, V. Eparvier, C. Eydoux, J. C. Guillemot, D. Stien and M. Litaudon, Fitoterapia, 2016, 112, 9–15 CrossRef CAS PubMed.
- N. Samat, P. J. Tan, K. Shaari, F. Abas and H. B. Lee, Anal. Chem., 2014, 86, 1324–1331 CrossRef CAS.
- J. J. Kellogg, D. A. Todd, J. M. Egan, H. A. Raja, N. H. Oberlies, O. M. Kvalheim and N. B. Cech, J. Nat. Prod., 2016, 79, 376–386 CrossRef CAS.
- F. Olivon, C. Apel, P. Retailleau, P. M. Allard, J. L. Wolfender, D. Touboul, F. Roussi, M. Litaudon and S. Desrat, Org. Chem. Front., 2018, 5, 2171–2178 RSC.
- F. Olivon, S. Remy, G. Grelier, C. Apel, C. Eydoux, J. C. Guillemot, J. Neyts, L. Delang, D. Touboul, F. Roussi and M. Litaudon, J. Nat. Prod., 2019, 82, 330–340 CrossRef CAS PubMed.
- F. Olivon, G. Grelier, F. Roussi, M. Litaudon and D. Touboul, Anal. Chem., 2017, 89, 7836–7840 CrossRef CAS PubMed.
- L. F. Nothias, M. Nothias-Esposito, R. da Silva, M. X. Wang, I. Protsyuk, Z. Zhang, A. Sarvepalli, P. Leyssen, D. Touboul, J. Costa, J. Paolini, T. Alexandrov, M. Litaudon and P. C. Dorrestein, J. Nat. Prod., 2018, 81, 758–767 CrossRef CAS PubMed.
- D. Guillarme, D. T. T. Nguyen, S. Rudaz and J.-L. Veuthey, Eur. J. Pharm. Biopharm., 2008, 68, 430–440 CrossRef CAS PubMed.
- E. F. Queiroz, A. Alfattani, A. Afzan, L. Marcourt, D. Guillarme and J.-L. Wolfender, J. Chromatogr. A, 2019 DOI:10.1016/j.chroma.2019.03.042.
- J. Pazourek and K. Smejkal, Molecules, 2016, 21, 1495 CrossRef PubMed.
- J. Liigand, R. de Vries and F. Cuyckens, Rapid Commun. Mass Spectrom., 2019, 33, 314–322 CrossRef CAS PubMed.
- P.-M. Allard, J. Bisson, A. Azzollini, G. F. Pauli, G. A. Cordell and J.-L. Wolfender, Curr. Opin. Biotechnol., 2018, 54, 57–64 CrossRef CAS PubMed.
- J. B. McAlpine, S. N. Chen, A. Kutateladze, J. B. MacMillan, G. Appendino, A. Barison, M. A. Beniddir, M. W. Biavatti, S. Bluml, A. Boufridi, M. S. Butler, R. J. Capon, Y. H. Choi, D. Coppage, P. Crews, M. T. Crimmins, M. Csete, P. Dewapriya, J. M. Egan, M. J. Garson, G. Genta-Jouve, W. H. Gerwick, H. Gross, M. K. Harper, P. Hermanto, J. M. Hook, L. Hunter, D. Jeannerat, N. Y. Ji, T. A. Johnson, D. G. I. Kingston, H. Koshino, H. W. Lee, G. Lewin, J. Li, R. G. Linington, M. M. Liu, K. L. McPhail, T. F. Molinski, B. S. Moore, J. W. Nam, R. P. Neupane, M. Niemitz, J. M. Nuzillard, N. H. Oberlies, F. M. M. Ocampos, G. Pan, R. J. Quinn, D. S. Reddy, J. H. Renault, J. Rivera-Chavez, W. Robien, C. M. Saunders, T. J. Schmidt, C. Seger, B. Shen, C. Steinbeck, H. Stuppner, S. Sturm, O. Taglialatela-Scafati, D. J. Tantillo, R. Verpoorte, B. G. Wang, C. M. Williams, P. G. Williams, J. Wist, J. M. Yue, C. Zhang, Z. R. Xu, C. Simmler, D. C. Lankin, J. Bisson and G. F. Pauli, Nat. Prod. Rep., 2019, 36, 35–107 RSC.
- A. A. Lagunin, R. K. Goel, D. Y. Gawande, P. Pahwa, T. A. Gloriozova, A. V. Dmitriev, S. M. Ivanov, A. V. Rudik, V. I. Konova, P. V. Pogodin, D. S. Druzhilovsky and V. V. Poroikov, Nat. Prod. Rep., 2014, 31, 1585–1611 RSC.
- T. Rodrigues, Org. Biomol. Chem., 2017, 15, 9275–9282 RSC.
- R. Lewis, R. Guha, T. Korcsmaros and A. Bender, J. Cheminf., 2015, 7, 36 Search PubMed.
- G. F. Pauli, S. N. Chen, J. B. Friesen, J. B. McAlpine and B. U. Jaki, J. Nat. Prod., 2012, 75, 1243–1255 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2019 |