
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
meta-C–H arylation of fluoroarenes via traceless directing group relay strategy†
Marc
Font
 ,
Andrew R. A.
Spencer
and
Igor
Larrosa
*
,
Andrew R. A.
Spencer
and
Igor
Larrosa
*
School of Chemistry, University of Manchester, Oxford Road, Manchester, M13 9PL, UK. E-mail: igor.larrosa@manchester.ac.uk
First published on 25th July 2018
Abstract
While several methods for the ortho selective arylation of fluoroarenes, meta-functionalisation has never been achieved. We report a new methodology, based on the traceless directing group relay concept, leading to the first meta-selective (hetero)arylation of fluoroarenes. In this strategy, CO2 is introduced as a transient directing group, to control a Pd-catalysed arylation meta to the fluoro functionality, prior to its release in a sequential, one-pot fashion. This method has shown compatibility with a number of functional groups and substitution patterns in both the fluoroarene core and aryl iodide coupling partners, and proceeds with complete meta-selectivity and mono vs. bis-arylation selectivity.
Introduction
Fluoroarenes are recurring structural motifs in pharmaceuticals, agrochemicals, biological imaging agents and organic materials.1 So much so that up to 30% of pharmaceuticals and 40% of agrochemical agents currently contain at least one fluorine atom, usually located at arene rings (Fig. 1).1l,2 Fluorine is a bioisostere of hydrogen of unique biological relevance that can provide compounds with enhanced metabolic stability, bioavailability, lipophilicity and binding affinity among other properties.2,3 Fluoro(hetero)biaryl motifs are a particularly important class within fluorinated compounds, with widespread presence in pharmaceuticals, including top selling rosuvastatin and atorvastatin (Fig. 1).1i,4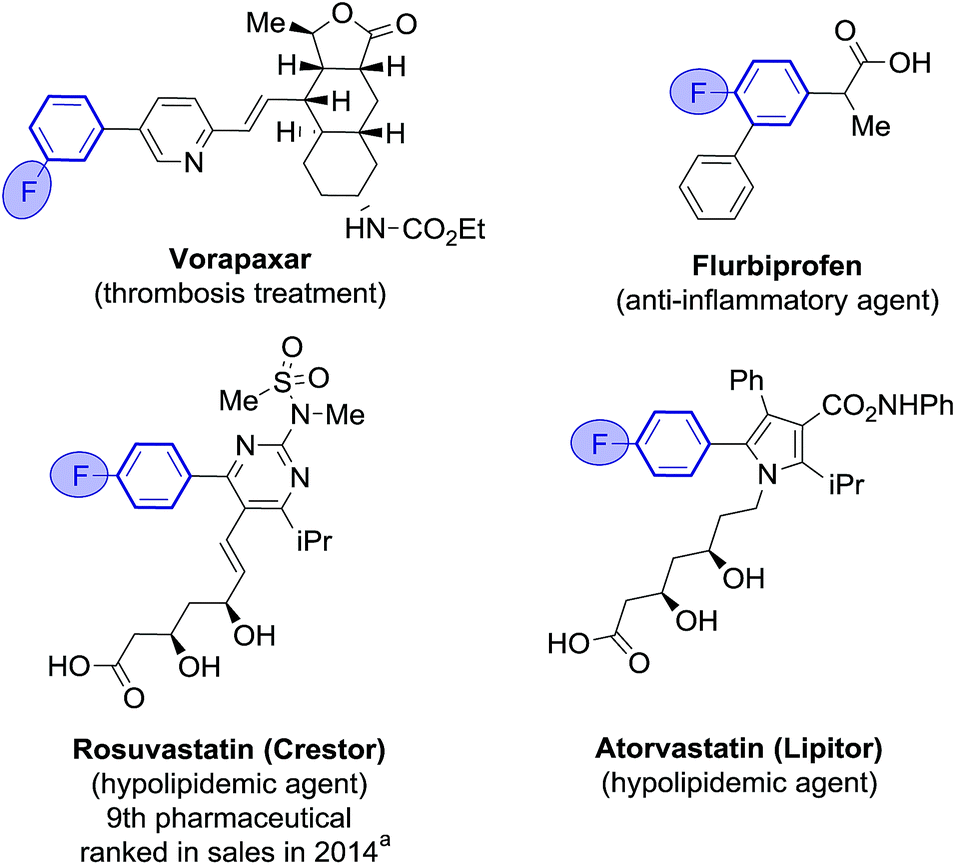 | ||
| Fig. 1 Examples of fluoro(heterobiaryl) motifs in pharmaceuticals. aSource: EvaluatePharma/Vantage. | ||
Fluoro(hetero)biaryl motifs can be accessed by assembly of the arene fragments via traditional cross-coupling or by late installation of the fluorine atom from suitably prefunctionalised precursors.5,6 However, precursors such as aryl halides and arylboronic acids have an elevated cost compared to their parent fluoroarenes and/or need to be synthesised, adding several steps to the overall process. Very recently, Ritter and co-workers have developed a general method for aromatic C–H fluorination, however restricted to the ortho and para positions of arene rings.7
On the other hand, methods for the C–H arylation of (mono)fluoroarenes are rare and again limited to ortho or para selective arylation. Furthermore, current methods require large excesses of the fluoroarene (often used as cosolvent) to bolster the activation of their relatively inert C–H bonds.8
Additional electron-withdrawing groups or π-complexation of a Cr(CO)3 fragment have been shown to result in improved reactivity towards ortho-arylation, allowing the use of these fluorobenzenes as limiting reagents.9,10 However, to date a methodology capable of overriding the inherent ortho and para reactivity of fluoroarenes, and affording general methods for the meta-selective arylation has been unattained.11 Thus, an effective and selective strategy for the meta-functionalisation of cheap and readily available fluoroarenes is highly desirable (Scheme 1a).
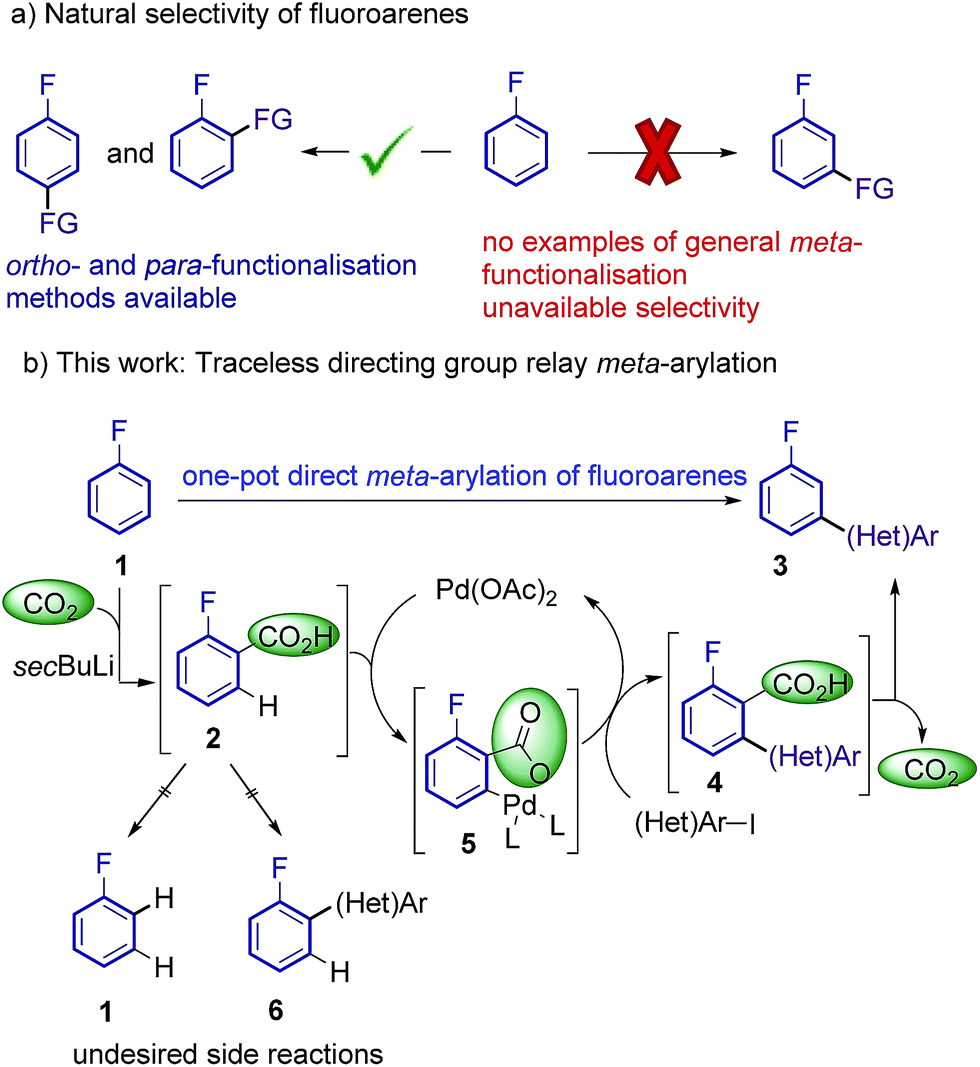 | ||
| Scheme 1 Proposed approach for regioselective meta-arylation of fluoroarenes. | ||
Current approaches for the selective meta C–H functionalisation of substituted arenes are scarce, generally relying on chelation-assistance and restricted to only a few classes of arenes bearing removable or non-removable directing groups.12,13 These strategies are not applicable to the fluoro functionality; hence an alternative approach is required. We envisioned that a traceless directing group relay strategy could be a suitable approach to provide the desired selectivity on fluoroarenes.14,15 Our working hypothesis (Scheme 1b) involved the installation of a temporary carboxylic acid group ortho-to-fluorine, which in turn would direct a palladium-catalyzed ortho-selective arylation16 (but meta relative to the fluorine group), prior to its cleavage. ortho-Carboxylation of fluorobenzenes has previously been achieved via directed ortho-metalation (DoM) with organolithiums and CO2.17
Indeed, one-pot DoM/cross-coupling sequences have proven successful for the rapid access to ortho-aryl fluorobenzenes.18–20 On the other hand, transition metal-catalysed protodecarboxylations have been extensively studied over the last decade and a vast repository of methods are available, making carboxylic acids ideal traceless directing groups.15d,21
The proposed approach poses several crucial challenges that have to be overcome in order to achieve the desired meta-selective arylation: (1) high yielding carboxylation reactions are needed. (2) Highly hindered adducts 4 need to readily undergo protodecarboxylation under the reaction conditions. (3) Undesired protodecarboxylation of benzoic acids 2 leading to fluoroarenes 1 has to be minimised. (4) Alternative decarboxylative ipso-arylation processes, which would lead to ortho-arylated fluoroarenes 6 have to be avoided.
Results and discussion
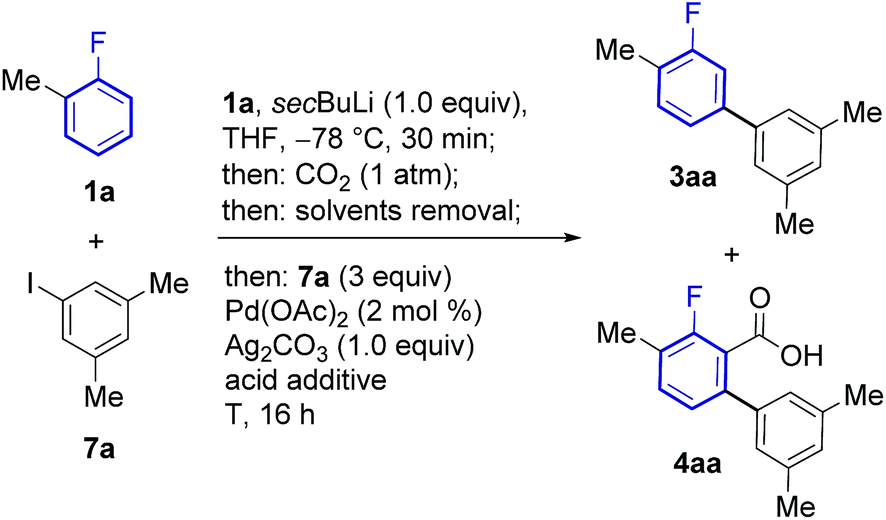
The feasibility of this strategy was first assessed for the coupling of 2-fluorotoluene (1a) with 5-iodo-meta-xylene (7a, Table 1). Optimisation of the lithiation/carboxylation step, revealed that quantitative formation of 3-methyl-2-fluorobenzoic acid could be achieved by using secBuLi and CO2 at atmospheric pressure. We then turned our attention to optimising the tandem arylation/protodecarboxylation process in order to obtain the desired meta-arylated fluoroarene products in a one-pot process. Examination of reaction conditions previously developed for the tandem arylation/decarboxylation of benzoic acids14c gave mixtures of the desired meta-arylation product 3aa and the corresponding non-decarboxylated arylation product 4aa even at high temperatures (entries 1 and 2). It has been shown that alkali carbonates can prevent protodecarboxylation in the ortho-arylation of benzoic acids.22 We hypothesised that the lithium benzoate formed in the lithiation/carboxylation step was responsible for the sluggish decarboxylation of 4aa. Replacement of acetic acid for isobutyric and pivalic acid afforded almost no 3aa product (entries 3 and 4). We then turned our attention to stronger carboxylic acids, which have proven beneficial in other Pd-catalysed protodecarboxylation reactions.23 Indeed, trifluoroacetic acid (TFA) led to 63% of the desired product 3aa, although still 19% of non-decarboxylated product 4aa was present after the reaction (entry 5). Gratifyingly, reducing the amount of TFA to only 2.5 equiv. led to 72% of the product with nearly complete decarboxylation of 4aa (entry 6).24 Importantly, the product was formed with complete meta-regioselectivity showing no traces of para- or ortho-arylation products. With the optimised conditions in hand we set out to investigate the generality of the methodology with respect to the aryl iodide coupling partner. The developed reaction conditions tolerate electron-donating and electron-withdrawing groups in the ortho, meta and para positions of the aryl iodide (Scheme 2).|
|
||||
|---|---|---|---|---|
| Entry | Acid additive (equiv.) | T (°C) | 3aa (%) | 4aa (%) |
| a Yields were determined by 19F NMR analysis using 4-bromofluorobenzene as an internal standard. | ||||
| 1 | AcOH (3.5) | 130 | 30 | 34 |
| 2 | AcOH (3.5) | 150 | 42 | 16 |
| 3 | iPrCOOH (3.5) | 130 | 3 | 51 |
| 4 | PivOH (3.5) | 130 | 5 | 53 |
| 5 | TFA (3.5) | 130 | 63 | 19 |
| 6 | TFA (2.5) | 130 | 72 | 1 |
| 7 | TFA (4) | 130 | 45 | 23 |
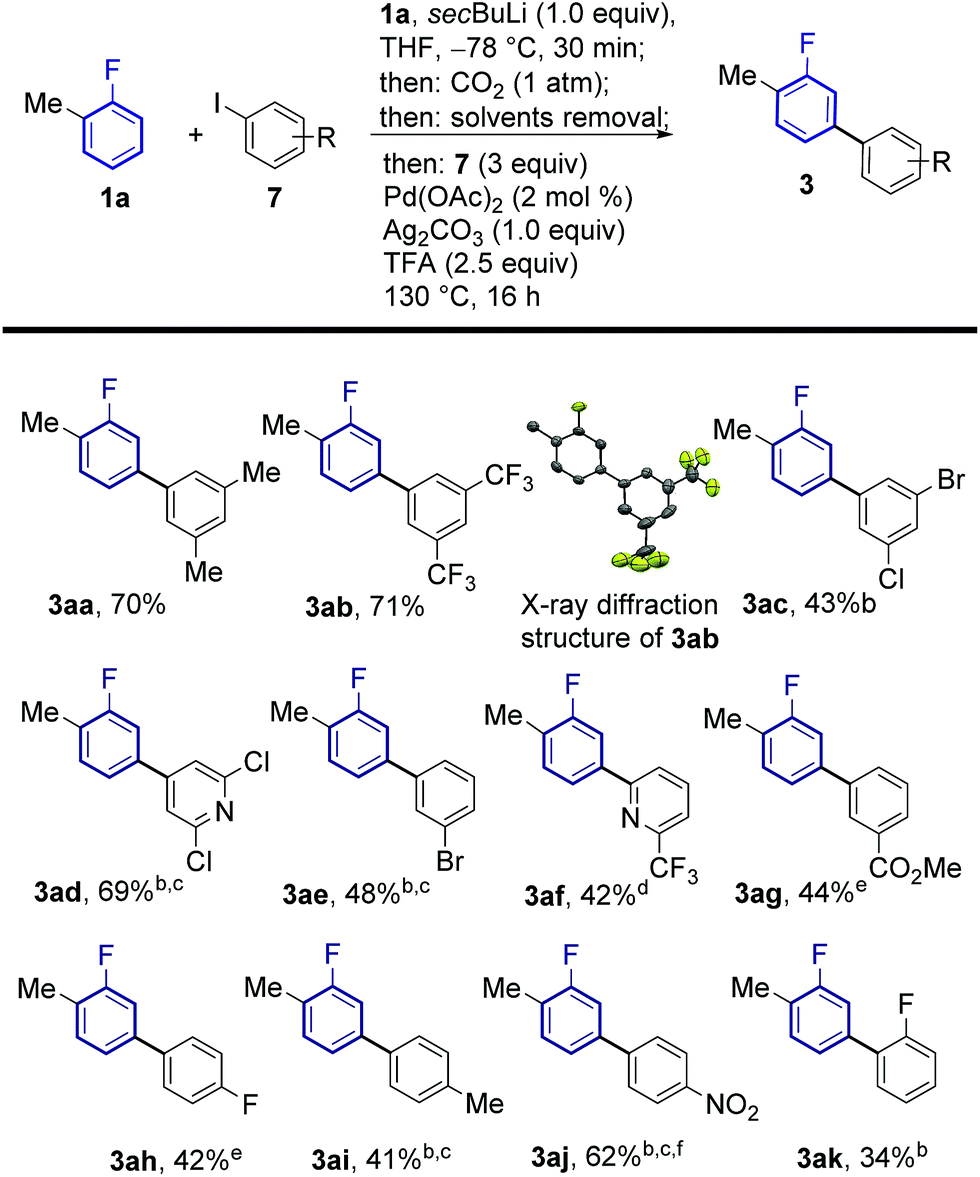 | ||
| Scheme 2 Substrate scope of the meta-arylation of fluoroarenes with aryl iodides. aYields are of pure isolated product. bAfter 16 h, 2 mL of DMSO were added and the reaction was stirred at 170 °C for 3 h. c1.5 equiv. of 7 employed. d0.5 equiv. of 7, 2 mol% Pd(OAc)2 and 0.5 equiv. of Ag2CO3 were employed. e2 equiv. of 7 employed for 10 more hours. fArylation/decarboxylation step run at 150 °C. | ||
The regiochemical outcome of the reaction was confirmed by X-ray diffraction analysis of compound 3ab. Aryl iodides bearing nitro, methyl, ester and trifluoromethyl functionalities were compatible with this methodology. Halogen substituents were also compatible (3ac, 3ad, 3ae) serving as handles for further functionalisation of the products. Remarkably, 2-substituted pyridyl iodides are suitable coupling partners affording the corresponding meta-(hetero)arylfluoroarene products 3ad and 3af. Pd-catalysed protodecarboxylation is usually disfavoured for electron-poor benzoic acids,23 accordingly, some of the more electron-poor arylated fluorobenzoic acids underwent sluggish decarboxylation. This issue could be resolved by adding DMSO and heating for additional 3 h, triggering a Ag-catalysed decarboxylation, which is favourable on these substrates.25 This led to complete protodecarboxylation of 4, thus providing the desired meta-fluorobiaryl products 3. We then explored the effect of substitution at the fluoroarene core (Scheme 3). Pleasingly, the one-pot meta-arylation protocol showed compatibility with ortho, meta and para substitution patterns in the fluoroarene ring, while maintaining complete meta-regioselectivity for the arylation. Furthermore, bis-arylation products, a common and generally undesired pathway in many meta-C–H functionalization protocols were completely suppressed. This one-pot method afforded the meta-aryl fluorobenzene 3ba in comparable yields to the reaction starting from fluorobenzoic acid.14c
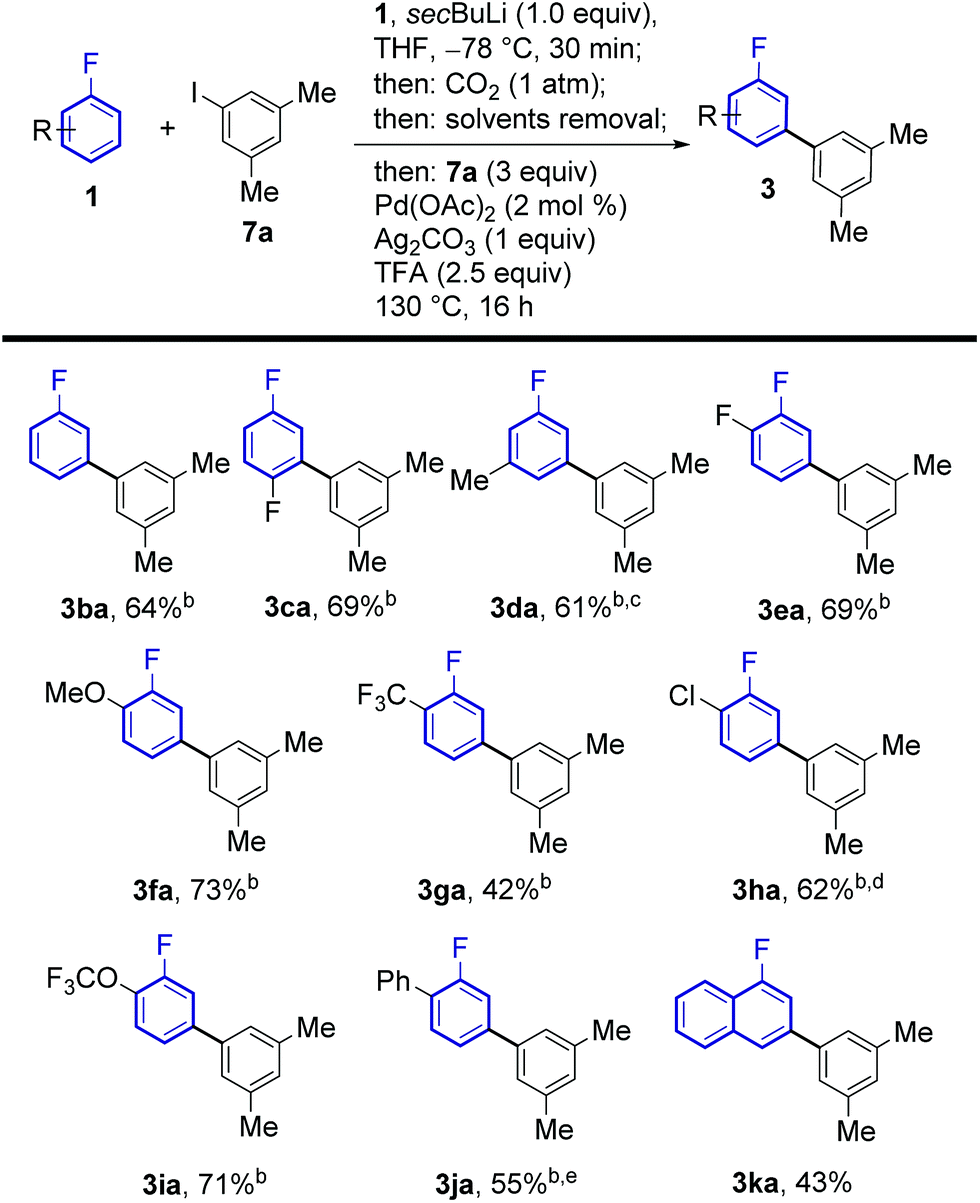 | ||
| Scheme 3 Substrate scope of the meta-arylation of fluoroarenes with aryl iodides. aYields are of pure isolated product. bAfter 16 h, 2 mL of DMSO were added and the reaction was stirred at 170 °C for 3 h. c0.5 equiv. 7a, 2 mol% Pd(OAc)2 and 0.5 equiv. of Ag2CO3 were employed. dAfter 6 h of arylation/decarboxylation reaction, an additional 2 mol% Pd(OAc)2 was added and the reaction was stirred at 130 °C for 10 more hours. e1.5 equiv. 7a. | ||
Fluoroarenes bearing other functional groups known to induce directed ortho-metalation (DoM) reactions15b,26 (3fa, 3ga, 3ia) afforded only the desired meta-to-fluorine products showing the prevalence of fluorine to direct the lithiation step over other functional groups. 1-Chloro-2-fluorobenzene afforded the corresponding desired product 3ha with no traces of dechlorinated side products. Our approach was also compatible with highly electron-poor fluoroarene rings (3ea, 3ga). The previously reported meta-arylated fluorobenzene (3ba) could also be prepared using this method.
Aryl ethers are useful, frequent intermediates in organic synthesis and are found in an impressive number of biologically active compounds and natural products.27 Pleasingly, we were also able to extend the scope of our protocol to this class of substrates (Scheme 4). After careful screening of reaction conditions for both the lithiation/carboxylation and arylation/decarboxylation steps, we found optimised conditions allowing the one-pot meta-arylation of anisoles, to form 9aa,14c and 9ba. Perfluorinated aryl alkyl ethers and acetals also showed good compatibility with our method and afforded the corresponding products 9ca and 9da with complete selectivity.
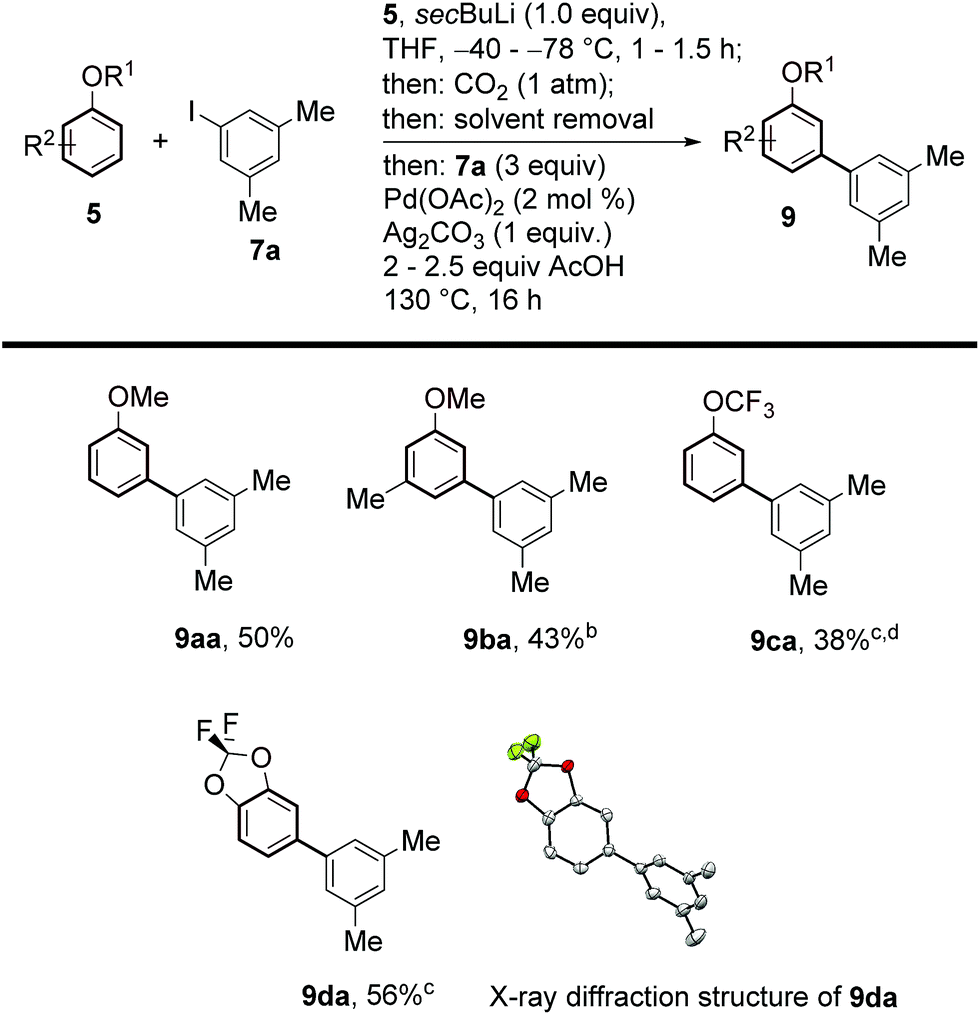 | ||
| Scheme 4 Substrate scope of the meta-arylation of substituted arenes with aryl iodides. aYields are of pure isolated product. b0.5 equiv. 7a, 2 mol% Pd(OAc)2 and 0.5 equiv. of Ag2CO3 were employed. cAfter 16 h, 2 mL of DMSO were added and the reaction was stirred at 170 °C for 3 h. c0.5 equiv. 7a, 2 mol% Pd(OAc)2 and 0.5 equiv. of Ag2CO3 were employed. dTFA used instead of AcOH. | ||
Since the developed method provides fast and selective access to meta-arylfluoroarenes, it can accelerate and lower the costs of synthesising compounds with such motifs. Indeed, the synthesis of γ-secretase inhibitor 11 (ref. 30) was accomplished in only three steps from 1-fluoro-2-(trifluoromethoxy)benzene 1ia with an overall 42% yield (Scheme 5). The key meta-arylation step afforded a 59% yield of 3ib, which upon subsequent alkylation29 and Suzuki coupling30 led to the desired product 12. The previous synthesis required nine steps starting from methyl 2-(3,5-dihydroxyphenyl)acetate (13) affording 11 in 12% overall yield.28
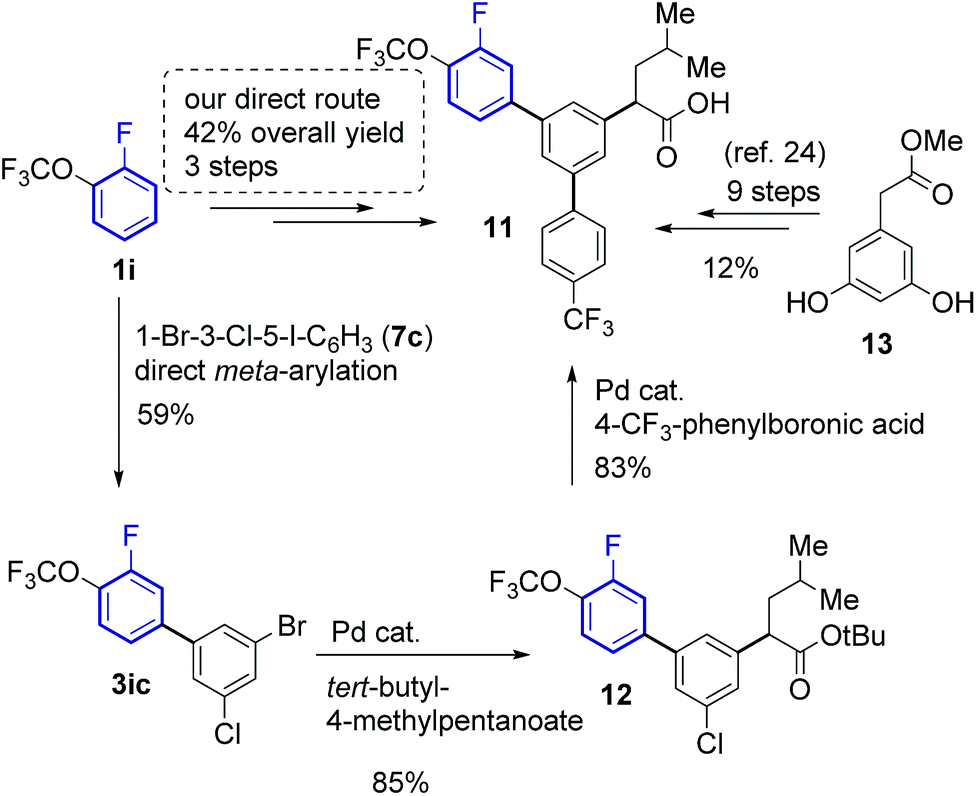 | ||
| Scheme 5 Synthesis of γ-secretase inhibitor 11. | ||
Conclusion
In summary, we have developed the first methodology for the direct meta-(hetero)arylation of fluoroarenes. Our method relies on the use of CO2 as traceless directing group. This one-pot protocol involving lithiation/carboxylation followed by tandem arylation/decarboxylation has proven compatible with different substituents and substitution patterns in both the fluoroarene and aryl iodide coupling partners. The method can be successfully applied to other classes of aryl ethers capable of directing selective ortho-lithiation events. It should be noted that most of the examples here presented cannot be currently directly made from the parent arenes via any other method.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge the Engineering and Physical Sciences Research Council (EPSRC, EP/L014017/2) and the Marie Skłodowska Curie actions (IF-656841 to MF) for funding. We thank Dr Iñigo J. Vitorica-Yrezabal (UoM) for X-ray crystallography.Notes and references
- (a) P. Kirsch and M. Bremer, Angew. Chem., Int. Ed., 2000, 39, 4216 CrossRef; (b) W. R. Dolbier, J. Fluorine Chem., 2005, 126, 157 CrossRef; (c) C. Isanbor and D. O'Hagan, J. Fluorine Chem., 2006, 127, 303 CrossRef; (d) F. Babudri, G. M. Farinola, F. Naso and R. Ragni, Chem. Commun., 2007, 1003 RSC; (e) M. Hird, Chem. Soc. Rev., 2007, 36, 2070 RSC; (f) D. O'Hagan, Chem. Soc. Rev., 2008, 37, 308 RSC; (g) P. Jeschke, Pest Manage. Sci., 2010, 66, 10 CrossRef PubMed; (h) R. Berger, G. Resnati, P. Metrangolo, E. Weber and J. Hulliger, Chem. Soc. Rev., 2011, 40, 3496 RSC; (i) J. Wang, M. Sánchez-Roselló, J. L. Aceña, C. del Pozo, A. E. Sorochinsky, S. Fustero, V. A. Soloshonok and H. Liu, Chem. Rev., 2014, 114, 2432 CrossRef PubMed; (j) E. A. Ilardi, E. Vitaku and J. T. Njardarson, J. Med. Chem., 2014, 57, 2832 CrossRef PubMed; (k) T. Fujiwara and D. O'Hagan, J. Fluorine Chem., 2014, 167, 16 CrossRef; (l) Y. Zhou, J. Wang, Z. Gu, S. Wang, W. Zhu, J. L. Aceña, V. A. Soloshonok, K. Izawa and H. Liu, Chem. Rev., 2016, 116, 422 CrossRef PubMed.
- V. V. Grushin, Acc. Chem. Res., 2010, 43, 160 CrossRef PubMed.
- (a) B. E. Smart, J. Fluorine Chem., 2001, 109, 3 CrossRef; (b) P. Jeschke, ChemBioChem, 2004, 5, 570 CrossRef PubMed; (c) K. Müller, C. Faeh and F. Diederich, Science, 2007, 317, 1881 CrossRef PubMed; (d) S. Purser, P. R. Moore, S. Swallow and V. Gouverneur, Chem. Soc. Rev., 2008, 37, 320 RSC; (e) W. K. Hagmann, J. Med. Chem., 2008, 51, 4359 CrossRef PubMed.
- N. A. Mcgrath, M. Brichacek and J. T. Njardarson, J. Chem. Educ., 2010, 87, 1348 CrossRef.
- (a) Metal-Catalyzed Cross-Coupling Reactions, ed. A. de Meijere and F. Diederich, Wiley-VCH, Weinheim, Germany, 2004 Search PubMed; (b) C. C. C. Johansson Seechurn, M. O. Kitching, T. J. Colacot and V. Snieckus, Angew. Chem., Int. Ed., 2012, 51, 5062 CrossRef PubMed.
- (a) T. Furuya, A. S. Kamlet and T. Ritter, Nature, 2011, 473, 470 CrossRef PubMed; (b) T. Liang, C. N. Neumann and T. Ritter, Angew. Chem., Int. Ed., 2013, 52, 8214 CrossRef PubMed; (c) M. G. Campbell and T. Ritter, Chem. Rev., 2015, 115, 612 CrossRef PubMed. For recent reviews on late-stage [18F]fluorination see: (d) A. F. Brooks, J. J. Topczewski, N. Ichiishi, M. S. Sanford and P. J. H. Scott, Chem. Sci., 2014, 5, 4545 RSC; (e) S. Preshlock, M. Tredwell and V. Gouverneur, Chem. Rev., 2016, 116, 719 CrossRef PubMed; (f) F. Buckingham and V. Gouverneur, Chem. Sci., 2016, 7, 1645 RSC.
- K. Yamamoto, J. Li, J. A. O. Garber, J. D. Rolfes, G. B. Boursalian, J. C. Borghs, C. Genicot, J. Jacq, M. van Gastel, F. Neese and T. Ritter, Nature, 2018, 554, 511 CrossRef PubMed.
- (a) M. Lafrance and K. Fagnou, J. Am. Chem. Soc., 2006, 128, 16496 CrossRef PubMed; (b) X. Wang, D. Leow and J.-Q. Yu, J. Am. Chem. Soc., 2011, 133, 13864 CrossRef PubMed; (c) Y.-X. Luan, T. Zhang, W.-W. Yao, K. Lu, L.-Y. Kong, Y.-T. Lin and M. Ye, J. Am. Chem. Soc., 2017, 139, 1786 CrossRef PubMed; (d) J. Kim and H. Hong, ACS Catal., 2017, 7, 3336 CrossRef.
- (a) T. Yan, L. Zhao, M. He, J.-F. Soule, C. Bruneau and H. Doucet, Adv. Synth. Catal., 2014, 356, 1586 CrossRef; (b) M. He, J.-F. Soule and H. Doucet, ChemCatChem, 2015, 7, 2130 CrossRef.
- (a) P. Ricci, K. Krämer, X. C. Cambeiro and I. Larrosa, J. Am. Chem. Soc., 2013, 135, 13258 CrossRef PubMed; (b) D. Whitaker, J. Bures and I. Larrosa, J. Am. Chem. Soc., 2016, 138, 8384 CrossRef PubMed; (c) D. Whitaker, M. Batuecas, P. Ricci and I. Larrosa, Chem.–Eur. J, 2017, 23, 12763 CrossRef PubMed.
- A meta-fluorobiaryl can be obtained, however, in the para-selective, cyano-controlled, direct arylation of 2-fluorocyano-benzene. See: R. Y. Peshkov, E. V. Panteleeva, L. N. Shchegoleva, I. Y. Bagryanskaya, T. V. Rybalova, N. V. Vasilieva and V. D. Shteingarts, Eur. J. Org. Chem., 2015, 4524 CrossRef.
- For recent reviews on transition metal-catalyzed meta-C–H functionalization see: (a) J. Yang, Org. Biomol. Chem., 2015, 13, 1930 RSC; (b) J. Lie, S. De Sarkar and L. Ackermann, Top. Organomet. Chem., 2016, 55, 217 CrossRef.
- (a) J. Zhang, R. Shrestha, J. F. Hartwig and P. Zhao, Nat. Chem., 2016, 8, 1144 CrossRef PubMed; (b) L. Huang, A. Biafora, G. Zhang, V. Bragoni and L. J. Gooßen, Angew. Chem., Int. Ed., 2016, 55, 6933 CrossRef PubMed; (c) N. Y. P. Kumar, A. Bechtoldt, K. Raghuvanshi and L. Ackermann, Angew. Chem., Int. Ed., 2016, 55, 6929 CrossRef PubMed; (d) H. J. Davis, M. T. Mihai and R. J. Phipps, J. Am. Chem. Soc., 2016, 138, 12759 CrossRef PubMed; (e) M. Bera, S. K. Sahoo and D. Maiti, ACS Catal., 2016, 6, 3575 CrossRef; (f) Y. Kuninobu, H. Ida, M. Nishi and M. Kanai, Nat. Chem., 2015, 7, 712 CrossRef PubMed; (g) X.-C. Wang, W. Gong, L.-Z. Fang, R.-Y. Zhu, S. Li, K. M. Engle and J.-Q. Yu, Nature, 2015, 519, 334 CrossRef PubMed; (h) Y. Zhang, H. Zhao, M. Zhang and W. Su, Angew. Chem., Int. Ed., 2015, 54, 3817 CrossRef PubMed; (i) N. Hofmann and L. Ackermann, J. Am. Chem. Soc., 2013, 135, 5877 CrossRef PubMed; (j) D. Leow, G. Li, T.-S. Mei and J.-Q. Yu, Nature, 2012, 486, 518 CrossRef PubMed; (k) H. A. Duong, R. E. Gilligan, M. L. Cooke, R. J. Phipps and M. J. Gaunt, Angew. Chem., Int. Ed., 2011, 50, 463 CrossRef PubMed; (l) O. Saidi, J. Marafie, A. E. Ledger, P. M. Liu, M. F. Mahon, G. Kociok-Köhn, M. K. Whittlesey and C. G. Frost, J. Am. Chem. Soc., 2011, 133, 19298 CrossRef PubMed; (m) R. J. Phipps and M. J. Gaunt, Science, 2009, 323, 1593 CrossRef PubMed.
- (a) J. Luo, S. Preciado and I. Larrosa, J. Am. Chem. Soc., 2014, 136, 4109 CrossRef PubMed; (b) J. Luo, S. Araromi, S. Preciado and I. Larrosa, Chem.–Asian J., 2016, 11, 347 CrossRef PubMed; (c) J. Cornella, M. Righi and I. Larrosa, Angew. Chem., Int. Ed., 2011, 50, 9429 CrossRef PubMed.
- For recent reviews on traceless directing groups see: (a) G. Rousseau and B. Breit, Angew. Chem., Int. Ed., 2011, 50, 2450 CrossRef PubMed; (b) F. Zhang and D. R. Spring, Chem. Soc. Rev., 2014, 43, 6906 RSC; (c) M. Pichette Drapeau and L. J. Gooßen, Chem.–Eur. J., 2016, 22, 18654 CrossRef PubMed; (d) M. Font, J. M. Quibell, G. J. P. Perry and I. Larrosa, Chem. Commun., 2017, 53, 5584 RSC.
- For recent examples on C–H functionalisation directed by carboxylic acids see: (a) A. S. Trita, A. Biafora, M. Pichette-Drapeau, P. Weber, L. J. Gooßen, Angew. Chem., Int. Ed., 2018, DOI:10.1002/anie.201712520; (b) P.-X. Shen, L. Hu, Q. Shao and J.-Q. Yu, J. Am. Chem. Soc., 2018, 140, 6545 CrossRef PubMed; (c) H. Li, Q. Jiang, X. Jie, Y. Shang, Y. Zhang, L. J. Gooßen and W. Su, ACS Catal., 2018, 8, 4777 CrossRef; (d) M. Simonetti, D. M. Cannas, A. Panigrahi, S. Kujawa, M. Kryjewski, P. Xie and I. Larrosa, Chem.–Eur. J., 2017, 23, 549 CrossRef PubMed; (e) A. Biafora, T. Krause, D. Hackenberger, F. Belitz and L. J. Gooßen, Angew. Chem., Int. Ed., 2016, 55, 14752 CrossRef PubMed; (f) R. Mei, C. Zhu and L. Ackermann, Chem. Commun., 2016, 52, 13171 RSC; (g) L. Huang and D. J. Weix, Org. Lett., 2016, 18, 5432 CrossRef PubMed.
- (a) V. Snieckus, Chem. Rev., 1990, 90, 879 CrossRef; (b) J. Clayden, in Organolithiums: Selectivity for Synthesis, J. E. Baldwin, and R. M. Williams, Pergamon, Amsterdam, 2002 Search PubMed.
- J. Board, J. L. Cosman, T. Rantanen, S. P. Singh and V. Snieckus, Platinum Met. Rev., 2013, 57, 234 CrossRef.
- M. A. Pena, J. Pérez Sestelo and L. A. Sarandeses, J. Org. Chem., 2007, 72, 1271 CrossRef PubMed.
- S. Roesner and S. L. Buchwald, Angew. Chem., Int. Ed., 2016, 55, 10463 CrossRef PubMed.
- (a) L. J. Gooßen, N. Rodríguez and K. Gooßen, Angew. Chem., Int. Ed., 2008, 47, 3100 CrossRef PubMed; (b) J. Cornella and I. Larrosa, Synthesis, 2012, 44, 653 CrossRef.
- C. Arroniz, A. Ironmonger, G. Rassias and I. Larrosa, Org. Lett., 2013, 15, 910 CrossRef PubMed.
- J. S. Dickstein, C. A. Mulrooney, E. M. O'Brien, B. J. Morgan and M. C. Kozlowski, Org. Lett., 2007, 9, 2441 CrossRef PubMed.
- Independent carboxylation and arylation/decarboxylation reactions were carried out to demonstrate that the one-pot protocol proceeds via carboxylation/arylation/decarboxylation. For full details see ESI.†.
- R. Grainger, J. Cornella, D. C. Blakemore, I. Larrosa and J. M. Campanera, Chem.–Eur. J., 2014, 20, 16680 CrossRef PubMed.
- E. Castagnetti and M. Schlosser, Eur. J. Org. Chem., 2001, 691 CrossRef.
- (a) F. R. Leroux, B. Manteau, J.-P. Vors and S. Pazenok, Beilstein J. Org. Chem., 2008, 4(13) DOI:10.3762/bjoc.4.13; (b) G. Evano, N. Blanchard and M. Toumi, Chem. Rev., 2008, 108, 3054 CrossRef PubMed.
- C. Y. Ho, International Patent Appl. WO 2009/052341 A1, Janssen Pharmaceutica N. V., 2009.
- W. A. Moradi and S. L. Buchwald, J. Am. Chem. Soc., 2001, 123, 7996 CrossRef PubMed.
- T. E. Barder, S. D. Walker, J. R. Martinelli and S. L. Buchwald, J. Am. Chem. Soc., 2005, 127, 4685 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1533208 and 1549121. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8sc02417k |
| This journal is © The Royal Society of Chemistry 2018 |